Hereditary NonPolyposis Colorectal Cancer
(HNPCC): aspetti clinici
Registro tumori ereditari
del colon retto
a
uide
m
for
g
le
CROin
Picco
Registro tumori ereditari
del colon retto
Hereditary NonPolyposis Colorectal Cancer
(HNPCC): aspetti clinici
Mara Fornasarig
Alessandra Viel
SOC Gastroenterologia - SOC Oncologia Sperimentale I
Centro di Riferimento Oncologico di Aviano 2010
CROinforma. Piccole guide
Serie INFORMAZIONI SCIENTIFICHE
1
Prima edizione 2010
© Centro di Riferimento Oncologico di Aviano (CRO Aviano)
Collana curata dalla Direzione Scientifica - Biblioteca
Responsabile Scientifico
Paolo De Paoli (Direttore Scientifico CRO)
Coordinamento editoriale e di redazione
Ivana Truccolo (Responsabile Biblioteca CRO)
Elena Giacomello (Biblioteca CRO)
Impaginazione grafica
Nancy Michilin (Direzione Scientifica - Biblioteca CRO)
Testi di
Dr.ssa Mara Fornasarig
SOC Gastroenterologia
Direttore: Dr. Renato Cannizzaro
Centro di Riferimento Oncologico, IRCCS - Aviano
Dr.ssa Alessandra Viel
SOC Oncologia Sperimentale I
Direttore: Dr.ssa Roberta Maestro
Centro di Riferimento Oncologico, IRCCS - Aviano
Con il patrocinio di DANIELI SpA
Indice
Prefazione ............................................................................................ pag. 6
Introduzione ....................................................................................... pag. 7
1. Hereditary NonPolyposis Colorectal Cancer (HNPCC):
diagnosi clinica e molecolare ...................................................pag. 9
2. Aspetti della storia familiare da approfondire
con la diagnosi molecolare .....................................................pag. 15
A) Carcinoma endometriale .................................................pag. 15
B) Cancro colo-rettale giovanile in assenza di storia
familiare compatibile con HNPCC ................................pag. 18
C) Tumori multipli ...................................................................pag. 24
3. Famiglie HNPCC con tumori stabili (MSS) .........................pag. 32
4. Legenda .......................................................................................pag. 36
5. Bibliografia ..................................................................................pag. 37
Prefazione
Cari Colleghi,
è con piacere che Vi presentiamo il libro “Registro tumori ereditari del
colon retto: Hereditary nonpolyposis colorectal cancer (HNPCC): aspetti clinici” a cura delle Dr.sse Mara Fornasarig e Alessandra Viel.
Il libro è il seguito del volume “Registro dei Tumori Ereditari del colonretto, 12 anni di esperienza”.
La presentazione dei quadri clinici multiformi nell’HNPCC, corredati
da una ricca serie di esempi di alberi genealogici, rende agevole la
lettura del volume.
Grazie all’ormai ventennale esperienza del team traslazionale che si
occupa di tumori del colon-retto eredo-familiari, al Centro di Riferimento Oncologico gli autori sono riusciti a rendere chiaro e comprensibile un argomento complesso e particolarmente attuale.
Il cancro colo-rettale è uno dei tumori che maggiormente colpisce la
popolazione delle nostre regioni e lo screening, ormai in piena attuazione, permetterà la riduzione di morbilità e mortalità.
Nell’ambito dello screening sarà sempre più frequente venire a contatto con le problematiche genetiche che necessitano di un inquadramento clinico in Centri di Riferimento riconosciuti con gruppi di
lavoro consolidati ed esperti.
Ci auguriamo che le informazioni ed i consigli contenuti in questo
testo possano essere utili nell’attività quotidiana che svolgete, non
solo nell’assistenza diretta al paziente e alla sua famiglia, ma più in
generale nei confronti dell’intera comunità in cui si sviluppa la vostra
professione.
Dr. Paolo De Paoli
Direttore Scientifico
indice
Dr. Renato Cannizzaro
Direttore SOC Gastroenterologia
Introduzione
A seguito della pubblicazione dei dati del Registro dei Tumori Ereditari del Colon-retto del 2007, abbiamo riscontrato che persiste una
obiettiva difficoltà a individuare gli elementi dell’anamnesi familiare
che inducono a sospettare una forma di predisposizione ereditaria al
cancro colo-rettale tipo Hereditary NonPolyposis Colorectal Cancer
(HNPCC) o sindrome di Lynch.
Lo scopo di questa pubblicazione è di dare degli strumenti utili per:
• individuare le famiglie che possono trarre beneficio da un approfondimento diagnostico molecolare e dal test genetico, qualora la
storia familiare non presenti gli aspetti tipici per la diagnosi clinica
di HNPCC;
• programmare la sorveglianza in famiglie con storia familiare compatibile con l’HNPCC, ma in assenza di difetti genetici noti.
indice
Hereditary NonPolyposis
Colorectal Cancer (HNPCC):
diagnosi clinica e molecolare
L’HNPCC è una forma di predisposizione
ereditaria al cancro colo-rettale a trasmissione
autosomica dominante e causata da una alterazione
genetica a carico di uno dei quattro geni (MLH1,
MSH2, MSH6, PMS2) del mis-match repair (MMR),
deputati a correggere gli errori di duplicazione del
DNA che si formano durante la divisione cellulare.
La storia naturale del cancro colo-rettale, associato
a questa predisposizione ereditaria, è diversa da
quella del cancro sporadico perché dallo sviluppo
del polipo adenomatoso alla cancerizzazione
trascorre solo un breve periodo (2-3 anni). Per
effettuare una diagnosi precoce c’è la necessità
di una sorveglianza specifica che prevede la
ripetizione della colonscopia ogni uno o due anni.
L’individuazione delle famiglie e dei soggetti a
rischio viene effettuata con la diagnosi clinica
ricostruendo la storia familiare e applicando
dei criteri, chiamati criteri di Amsterdam (1) e
Amsterdam modificati (fig 1.1 e 2.1), secondo i
quali:
1) la famiglia deve presentare almeno tre casi di
cancro colo-rettale istologicamente identificati
o di tumori appartenenti allo spettro tumorale
dell’HNPCC (2) (endometrio, tenue, vie biliari);
2) i casi diagnosticati devono essere distribuiti in
almeno due generazioni;
3) un familiare affetto deve essere di primo grado
rispetto agli altri due e almeno un caso di
cancro deve essere diagnosticato prima dei 50
anni.
indice
Fig. 1.1: Criteri di Amsterdam
colon 64 endometrio 51
retto 45
colon 35
•almeno tre casi di CRC
• distribuiti in almeno due generazioni
• un membro affetto deve essere familiare di primo
grado rispetto agli altri due
• almeno un caso deve essere diagnosticato prima
dei 50 anni
indice
10
10
Fig. 2.1: Criteri di Amsterdam
modificati
endometrio 50
endometrio 45
uretere 68
tenue 58
•almeno tre casi di tumori delleo spettro tumorale
del HNPCC (endometrio tenue vie urinarie)
• distribuiti in almeno due generazioni
• un membro affetto deve essere familiare di
primo grado rispetto agli altri due
• almeno un caso deve essere diagnosticato
prima dei 50 anni
indice
11
11
La diagnosi clinica e la consulenza genetica
permettono di selezionare le famiglie a rischio di
HNPCC da inviare alla diagnosi molecolare che
confermerà se una aggregazione familiare di tumori
sia determinata geneticamente.
La diagnosi molecolare si svolge in due tempi
(fig. 3.1).
A) Analisi del tessuto tumorale di un familiare
affetto:
• studio dell’instabilità genetica dei microsatelliti
(3,4). I microsatelliti sono delle sequenze
ripetute di DNA che non hanno funzione
codificante. Confrontando il DNA estratto da
tessuto sano e da quello tumorale si evidenzia
se queste sequenze sono modificate per
numero di ripetizioni. Se le sequenze sono
uguali il tumore è considerato stabile (MSS),
aspetto questo tipico dei tumori sporadici.
Se le sequenze sono modificate il tumore è
instabile (MSI-H), aspetto molecolare tipico dei
tumori correlati all’HNPCC;
• immunoistochimica per la valutazione delle
proteine codificate dai quattro geni MMR.
Se nel tumore è assente una delle proteine
codificate dai geni MMR significa che quel gene
non è funzionante.
indice
12
Fig. 3.1: Algoritmo diagnostico
molecolare in HNPCC
SOSPETTO CLINICO DI
HNPCC
DISPONIBILITA’ DI
TESSUTO TUMORALE
TEST MSI
MSS
PROGRAMMA DI
SORVEGLIANZA
MSI-H
IHC PER
PROTEINE MMR
ANALISI
MUTAZIONALE
14
13
indice
Quando nel tumore si repertano l’instabilità
genetica dei microsatelliti (MSI-H) e l’assenza
di una delle proteine codificate dai geni MMR,
si procede al secondo tempo della diagnosi
molecolare.
B) Il test genetico vero e proprio che viene
effettuato, tramite un prelievo di sangue venoso,
estraendo il DNA dai linfociti circolanti per
l’analisi del gene risultato non funzionante
all’immunoistochimica.
La diagnosi molecolare, qualora sia analizzabile il
tessuto tumorale, è indicata anche in famiglie in
cui non sono rappresentati tutti i criteri clinici
di Amsterdam, ma sono invece presenti alcuni
parametri clinico-patologici peculiari dell’HNPCC:
• esordio in età precoce del cancro colo-rettale,
• localizzazione del cancro al colon destro;
• tumori sincroni e/o metacroni;
• neoplasie extracoliche (2) appartenenti
allo spettro tumorale dell’HNPCC (cancro
dell’endometrio, ovaio, stomaco, vie urinarie,
vie biliari e tenue).
indice
14
Aspetti della storia familiare
da approfondire con la diagnosi
molecolare
A) Carcinoma endometriale
Il cancro più frequentemente diagnosticato
nell’HNPCC dopo quello colo-rettale è il
cancro dell’endometrio. Infatti, il programma di
prevenzione nei soggetti portatori di mutazioni
prevede un protocollo per la sorveglianza
dell’utero a partire dai 35 anni con visita
ginecologica, ecografia trans-vaginale e PAP test
ogni due anni.
Talvolta nelle famiglie riscontriamo pochi casi
di cancro colo-rettale, ma sono prevalenti i
cancri dell’endometrio. Le nostre prime famiglie
HNPCC registrate (5) sono state diagnosticate
partendo proprio da pazienti affette da cancro
dell’endometrio.
I dati clinici che ci devono far sospettare l’HNPCC
sono:
•insorgenza di carcinoma endometriale in età
fertile;
•assenza dei tipici fattori di rischio come ad
esempio l’obesità.
indice
15
Nella famiglia riportata (Fig. 1.2), il nostro caso
indice è una donna che a 52 anni, ancora fertile,
normopeso e con 5 gravidanze fu affetta da cancro
dell’endometrio. Il padre era deceduto per un
cancro del retto in età relativamente giovane, la
storia familiare relativa agli zii paterni e nonni non
era verificabile.
La storia familiare quindi non presentava tutti
i criteri di Amsterdam essendo verificabili solo
due casi di cancro (il cancro dell’endometrio
del caso indice ed il cancro del retto nel padre).
Infatti, due erano i parametri assenti per poter
definire HNPCC la famiglia: il numero di casi e la
giovane età. Le caratteristiche cliniche del nostro
caso indice ci hanno guidato all’approfondimento
molecolare. Il test genetico nella signora ha
evidenziato una mutazione a carico del gene MLH1.
In seguito, due delle cinque figlie, nel corso del
loro programma di sorveglianza hanno sviluppato
una iperplasia ghiandolare atipica borderline con
l’adenocarcinoma dell’endometrio. Una nipote,
individuata portatrice della mutazione, già al primo
controllo endoscopico presentava un adenoma
cancerizzato del colon destro.
indice
16
Fig. 1.2: Famiglia HNPCC,
mutazione in MLH1
retto 51
endometrio 52
3
endometrio
+ ovaio 41
endometrio 39
colon trasverso 26
18
indice
17
B) Cancro colo-rettale giovanile
in assenza di storia familiare
compatibile con HNPCC
Il cancro colo-rettale diagnosticato in giovane età
(sotto i 45 anni) è da imputare ad un processo
cancerogenetico accelerato in cui sono implicati
prevalentemente fattori genetici piuttosto che
fattori ambientali, ma raramente questi tumori in
assenza di una familiarità tipica sono da imputare
all’HNPCC. Infatti, nella nostra casistica solo
due casi su 115 casi giovanili (età< 45 anni) sono
HNPCC correlati.
indice
18
Le linee guida (6) consigliano lo studio
dell’instabilità genetica dei microsatelliti nei tumori
di pazienti affetti da cancro colo-rettale sotto i 50
anni in assenza di storia familiare; perché qualora il
tumore sia stabile (MSS), il follow-up endoscopico
sarà meno impegnativo e, soprattutto, il grado di
rischio di cancro colo-rettale sarà inferiore nei
familiari di I grado.
indice
19
In Fig. 2.2, il nostro caso indice aveva sviluppato
a 34 anni un cancro del colon sinistro. L’istotipo
dell’adenocarcinoma era mucinoso e proprio la
presenza di mucina ci aveva fatto sospettare una
causa genetica.
Per quanto riguarda la famiglia, la madre vivente
non è portatrice della mutazione in MSH2
riscontrata alla figlia ed il padre era deceduto per
un cancro spino-cellulare dell’esofago (tipo di
tumore che non appartiene allo spettro tumorale
dell’HNPCC).
I fratelli non risultano portatori della mutazione.
indice
20
Fig. 2.2: Caso giovanile con
mutazione in MSH2
wt
esofago 73
wt
wt
wt
colon 34
wt = wild type: assenza
della mutazione
21
indice
21
Il caso riportato in Fig. 3.2, casualmente sempre
a 34 anni, aveva sviluppato un cancro del colon
ascendente. Il padre, fumatore, era deceduto per
una neoplasia polmonare; madre e fratello non
erano portatori della mutazione identificata in
MLH1.
Nel primo caso riportato, oltre alla giovane età,
l’istotipo mucinoso ci aveva indirizzato all’analisi
molecolare, in questo caso, invece, alla sede del
tumore (colon ascendente).
indice
22
Fig. 3.2: Caso giovanile con
mutazione in MLH1
4
wt = wild type: assenza
della mutazione
polmone 74
wt
colon 34
wt
23
indice
23
C) Tumori multipli
La diagnosi di più tumori in un paziente è un
evento infrequente, quando questo accade
dobbiamo sospettare l’HNPCC. I tumori però
sono quelli dello spettro dell’HNPCC (stomaco,
vie urinarie e vescica, endometrio, ovaio, tenue, vie
biliari).
In Fig 4.2, il nostro caso indice era stato affetto
da cancro gastrico con uno stadio avanzato,
endometriale, mammario e da tumori colici
sincroni multipli (tre cancri: ascendente e doppia
neoplasia del sigma). Non riportava storia familiare,
ma, alla ricostruzione dell’albero, padre e tre
zii paterni erano in realtà stati affetti da tumori
gastro-intestinali la cui sede non era stata verificata.
indice
24
Fig. n. 4.2: Caso con tumori
multipli, mutazione in MSH2
KSU 65
KSU 70
KSU 70
stomaco 49
endometrio 55
mammella 65
ascendente +
discendente 68
sigma 69
KSU = sede del
cancro non verificata
25
indice
25
Nella Fig. 5.2, come nella precedente, la storia
familiare non comprende tutti i criteri di
Amsterdam, ma la storia personale del nostro
caso indice (cancro dell’endometrio in età fertile),
associata alla storia del padre affetto da tumori
multipli (neoplasia gastrica, colica, vescica, bacinetto
renale) e deceduto per un cancro polmonare,
avevano indotto a un approfondimento molecolare
che evidenziò, in effetti, una mutazione in MLH1.
indice
26
Fig. 5.2: Famiglia con tumori multipli,
mutazione in MLH1
stomaco 40
colon 52
vescica 55
rene sx 60
polmone70
endometrio 48
27
indice
27
L’albero della Fig. 6.2 fa rilevare ancora una volta
l’importanza dei tumori multipli metacroni come
indicatori di HNPCC. Il nostro caso indice aveva
sviluppato 6 tumori, ma i suoi genitori non erano
stati affetti da tumori tipici dell’HNPCC anche se il
cancro prostatico e mammario ricorrono spesso in
queste famiglie.
indice
28
Fig. 6.2: Caso con tumori multipli,
mutazione in MSH2
prostata 72
mammella 70
utero 51
retto 58
vescica 62
sigma 63
ascendente 65
tenue 70
29
indice
29
In Fig. 7.2 abbiamo una famiglia in cui padre e figlia
presentano una storia personale simile, entrambi
avevano sviluppato cancri colo-rettali metacroni.
In questa storia familiare siamo in assenza di uno
dei criteri di Amsterdam: il numero di casi, non
avendo notizie certe sui nonni.
La giovane età della figlia e la doppia diagnosi,
in entrambi i casi, hanno indirizzato allo studio
molecolare.
indice
30
Fig. n. 7.2: Famiglia con tumori
multipli colici, mutazione in MLH1
mammella 77
wt
colon 47
retto 69
colon 21
colon 40
31
indice
31
Famiglie HNPCC con tumori
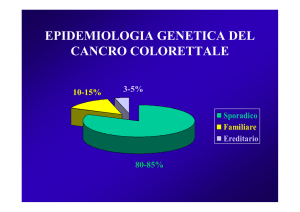
stabili (MSS)
Le famiglie, quando sono rappresentati tutti i
criteri clinici di Amsterdam, sono definite HNPCC
indipendentemente dalla causa genetica. Infatti,
ci possiamo trovare di fronte a famiglie con una
importante storia familiare di cancro colo-rettale,
ma l’analisi molecolare evidenzia che i tumori sono
stabili (MSS) (capitolo 1) e quindi non causati da
una mutazione nei geni responsabili dell’HNPCC.
Queste famiglie HNPCC sono definite da qualche
autore famiglie di tipo X, riservando il termine
HNPCC o sindrome di Lynch per quelle in cui
viene accertata la mutazione MMR.
La storia familiare è importante e quindi senz’altro
sono implicati dei geni che, al momento attuale,
non sono stati identificati.
indice
32
Ci sono delle lievi differenze cliniche fra queste
famiglie e quelle con il riscontro di mutazioni (7):
• il rischio di cancro nei familiari è lievemente
inferiore, ma ancora non quantificabile;
• l’età media di insorgenza è tra i 50-60 anni
(ma abbiamo anche casi molto giovanili come
nel nostro esempio);
• la sede prevalente del cancro è al colon
sinistro;
• la trasformazione maligna dei polipi è più lenta;
• il rischio di cancro endometriale non è
aumentato rispetto alla popolazione generale;
• così pure il rischio di altri cancri;
• i tumori presentano la stabilità genetica dei
microsatelliti (MSS) e le proteine dei geni
MMR sono espresse, ma possono presentare
modificazioni reversibili del DNA
(per esempio ipometilazione del DNA).
• La sorveglianza (8), raccomandata nei familiari
di I grado, viene effettuata con la colonscopia,
da iniziarsi dieci anni prima dell’età del parente
più giovane affetto da cancro colo-rettale e
con una frequenza quinquennale.
indice
33
L’esempio riportato (fig. 1.3) è peculiare: nella
storia familiare sono rappresentati tutti i criteri
di Amsterdam (numero di casi, più generazioni
affette, I grado di parentela fra i casi e l’età
giovanile), ma i tumori diagnosticati erano stabili
e quindi non abbiamo completato la diagnosi
molecolare con il test genetico.
La storia familiare fa emergere come sia
importante la valutazione clinica del singolo
soggetto in una famiglia, dato che l’ultimo caso
di cancro colo-rettale è stato diagnosticato a 21
anni, quindi 22 anni prima del caso più giovane.
I programmi di sorveglianza ci suggeriscono
quando iniziare e con quale frequenza effettuare
un esame ma vanno applicati solo a soggetti
asintomatici. Qualora siano presenti dei sintomi
l’iter diagnostico deve essere intrapreso
indipendentemente dall’età e dai risultati
molecolari.
indice
34
Fig 1.3: Famiglia HNPCC, tumori MSS
sigma52
retto 50
sigma 43
discendente 21
35
indice
35
Legenda
LEGENDA:
Maschio non affetto
Femmina non affetta
colon 52
Cancro verificato
Sede ed età della diagnosi
Matrimonio
Deceduto
Caso indice
indice
36
Bibliografia
1. Vasen HF, Watson P, Mecklin JP et al. New clinical criteria for
hereditary nonpolyposis colorectal cancer (HNPCC, Lynch sindrome)
proposed by the International Collaborative Group on HNPCC.
Gastroenterology 116:1453-6, 1999.
2. Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis
colorectal cancer. Cancer 71:677-85; 1993.
3. Boland CR, Thibodeau SN, Hamilton SR et al. National Cancer
Institute Workshop on microsatellite instability for cancer detection
and familial predisposition: development of international criteria for
determination of microsatellite instability in colorectal cancer. Cancer
Res 58:5248-57; 1998.
4. Capozzi E, Della Puppa L, Fornasarig M et al. Evaluation of
the replicazion error phenotype in ralation to molecular and
clinicopathological features in hereditary nonpolyposis colorectal
cancer. Eur J Cancer 35:289-95; 1999.
5. Fornasarig M, Campagnutta E, Talamini R et al. Risk factors for
endometrial carcinoma according to familial susceptibility. Int J Cancer
77:29-32; 1998.
6. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines
for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and
microsatellite instability. J Natl Cancer Inst 96:261-8; 2004.
7. Lindor MN. Familial colorectal cancer typeX: the other half of
hereditary nonpolyposis colorectal cancer syndrome. Surg Oncol Clin
N AM 18:637-45; 2009.
8. Dove-Edwin I, de Jong AE, Adams J et al. Prospective results of
surveillance colonoscopy in dominant familial colorectal cancer with
or without Lynch sindrome. Gastroenterology 130:1995-2000; 2006.
indice
37
Appunti
indice
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
38
Appunti
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
_________________________________________
39
indice
CROinforma. Piccole guide
Serie
LA RICERCA
CHE CURA
1
Dalla biologia
alla medicina. Perché
la ricerca è necessaria
per curare i tumori.
Serie
INFORMAZIONI
SCIENTIFICHE
1
2
indice
Registro tumori
ereditari del colon
retto. Hereditary
nonpolyposis colorectal
cancer (HNPCC): aspetti
clinici.
La predisposizione
ereditaria allo sviluppo
di tumori della
mammella e dell’ovaio.
Informazioni e suggerimenti
per famiglie a elevato
rischio genetico.
Serie
PERCORSI
DI CURA
1
Dopo il cancro: aspetti
psicosociali e qualità di vita.
La Chemioterapia
ad Alte Dosi
con reinfusione
di cellule staminali
emopoietiche
ovvero l’a-b-c-d del
Trapianto di Cellule
Staminali Autologhe
(ASCT) in press
prima parte
a) Cenni storici e note
introduttive.
seconda parte
b) Raccolta di cellule
staminali.
terza parte
c) Il trapianto.
quarta parte
d) Dalla dimissione alla
gestione ambulatoriale
del follow up.
2
Serie
ISTRUZIONI
ALL’USO DI...
1
Guida ai servizi
della Biblioteca
Scientifica e per
i Pazienti del CRO.
in press
Serie
AREA GIOVANI
1
2
Area Giovani. Brochure
informativa.
in press
Serie
CIFAV
INFORMAZIONE
SUL FARMACO
1
La mucosite orale
(stomatite). Guida pratica
per limitare i disturbi del
cavo orale (bocca, gola)
che si possono manifestare
durante la terapia
oncologica.
2
Conosciamo
e utilizziamo bene
gli antibiotici.
Radio Trolla. Favola per
i bambini in trattamento
radiante.
in press
indice
Ottobre 2010
CROinforma. Serie INFORMAZIONI SCIENTIFICHE
1
Piccole guide