La diversità genetica umana: la fallacia di Lewontin.
Autore: A.W.F. Edwards
Anno: 2003.
1
1. Sommario.
In un famoso articolo che minimizza le differenze genetiche fra popolazioni umane, viene
spesso affermato che circa l’85% della variabilità genetica è dovuta a differenze fra individui
della stessa popolazione e solo il 15% è dovuto a differenze fra diverse popolazioni o gruppi
etnici. Questo ha quindi suggerito che la divisione dell’Homo Sapiens in questi gruppi non
trova giustificazione nei dati genetici. Questa conclusione, ad opera R.C. Lewontin nel 1972,
è totalmente inesatta perché viene ignorato il fatto che la maggior parte delle informazioni
che distinguono le popolazioni è nascosta nella struttura della correlazione dei dati e non
semplicemente nella variabilità di fattori individuali. La logica alla base di ciò è stata discussa
nei primi anni dell’ultimo secolo, viene qui presentata grazie all’uso di un esempio genetico
semplice.
<<Quando un grande numero di individui (di qualunque tipologia di organismo) viene
misurato in più caratteristiche fisiche, peso, colore, densità ecc, è possibile descrivere con
un certo grado di precisione la popolazione globale. Gli individui analizzati nel nostro
esperimento possono essere considerati come un campione di questa popolazione. Allo
stesso modo è possibile distinguere diversi tipi di popolazioni che divergono per origine
genetica o circostanze ambientali. Quindi vi possono essere razze locali molto diverse fra
loro, nonostante la classificazioni di certi individui possa essere non univoca. >>
(R. A. Fisher – 1925, vedi rif 1.).
<<E’ chiaro che la percezione di grandi differenze fra razze umane e sottogruppi sono
distorte, se comparati alla variabilità all’interno di questi gruppi. Se pensiamo alle differenze
genetiche, le razze umane e le popolazioni sono molto simili le une alle altre. La maggior
parte della variabilità umana si riferisce alle differenze fra gli individui. La classificazione in
razze non ha alcun valore sociale ed è assolutamente destabilizzante per le relazioni sociali
ed umane. Dal momento che questa classificazione razziale è evidentemente priva di senso
né dal punto di vista genetico né dal punto di vista tassonomico, non vi è alcuna
giustificazione per continuare ad utilizzarla.>>.
(R. C. Lewontin – 1972, vedi rif 2.).
2
<<Lo studio della variabilità genetica nell’Homo Sapiens mostra che c’è molta più variabilità
genetica all’interno della stessa popolazione che fra diverse popolazioni. Questo significa
che prendendo a caso 2 individui da un dato gruppo si hanno quasi le stesse differenze che
prendendo a caso 2 individui dal mondo intero. Nonostante possa essere facile vedere
differenze esteriori fra gruppi di persone, è più difficile distinguere questi gruppi da un punto
di vista genetico dal momento che la maggior parte della variabilità genetica è all’interno dei
gruppi.>>.
(Nature – 2001, vedi rif 3.).
3
2. Introduzione
In famosi articoli che minimizzano le differenze genetiche fra le diverse popolazioni umane
viene spesso affermato, di solito senza alcuna fonte, che circa l’85% della variabilità
genetica umana è dovuta a differenze individuali all’interno delle popolazioni e solo il 15%
è dovuto a differenze fra le diverse popolazioni o gruppi etnici. Si è quindi suggerito che la
suddivisione dell’Homo Sapiens in questi gruppi non trova giustificazione nei dati genetici.
Le persone di tutto il mondo sarebbero molto più simili geneticamente di quanto lo sembrino
in apparenza. Quindi un articolo del New Scientist afferma che nel 1972 Richard Lewontin
della Harvard University
<<scoprì che circa l’85% della diversità genetica umana è dovuta a differenze individuali
all’interno di una singola popolazione. In altre parole, due individui sono diversi perché sono
individui distinti, non perché appartengono a razze diverse>>.
Nel 2001, l’edizione di Nature dedicata al genoma umano comprendeva un compact disc
che affermava un concetto analogo a quello prima citato. Queste frasi sembrano tutte riferirsi
ad un articolo del 1972 scritto da Lewontin nel suo articolo
“Biologia Evoluzionista”.
Lewontin analizzò i dati da 17 “luoghi polimorfici” (ovvero luoghi in cui un gene può avere
un gran numero di possibili sequenze, ndt), inclusi i maggiori gruppi sanguigni e le 7 “razze”
(Caucasici, Africani, Mongoloidi, Aborigeni dell’Asia del Sud, Amerindi, Oceanici ed
Aborigeni Australiani). La frequenza genetica venne data alle 7 razze ma non alle
popolazioni individuali che le comprendevano, nonostante l’analisi finale citò la varianza
all’interno della popolazione.
<< I risultati sono decisamente degni di nota. Le diversità all’interno della specie sono dovute
a varianza fra le popolazioni per l’85,4%... Meno del 15% della differenza genetica umana
è dovuta a differenze fra i gruppi umani! Inoltre la differenza fra popolazioni all’interno di una
stessa razza conta per un 8,3% ulteriore, quindi solo il 6,3% è dovuto alla classificazione
razziale>>.
4
Lewontin concluse affermando
<<Dal momento che questa classificazione razziale è evidentemente priva di senso né dal
punto di vista genetico né dal punto di vista tassonomico, non vi è alcuna giustificazione per
continuare ad utilizzarla>>.
Lewontin continuò a sostenere questa teoria nel suo libro del 1974 “Le basi genetiche del
cambiamento evolutivo”.
<<Viene data un’importanza sproporzionata alla divisione tassonomica della specie umana
in razze, dal momento che spiega solo una piccola parte della diversità umana. Il fatto che
scienziati ed anche non scienziati continuano ad evidenziare queste differenze genetiche
minori e trovano nuove giustificazioni “scientifiche” per questo è un indicatore del potere
dell’ideologia basata su criteri socio-economici a scapito dell’obiettività della conoscenza>>.
5
3. La fallacia
Queste conclusioni si basano sul vecchio errore statistico di analizzare i dati come se le
variabili non fossero fra loro correlate; così facendo si traggono conclusioni esclusivamente
dal risultato di questa analisi. Il “significato tassonomico” dei dati genetici spesso è dovuto
alla correlazione fra le diverse variabili, perché senza questa informazione non si riesce a
spiegare correttamente la variabilità fra i diversi gruppi. Cavalli-Sforza e Piazza coniarono
la parola “treeness” per descrivere il modo in cui fra le correlazioni fra le frequenze genetiche
è nascosta una struttura ad albero. L’analisi superficiale di Lewontin ignora questo aspetto
della struttura dei dati e porta inevitabilmente alla conclusione che i dati non hanno questa
struttura. È un cane che si morde la coda. Un’analisi che contraddiceva quella di Lewontin
usando dati molti simili fu quella presentata da Cavalli-Sforza ed Edwards nel 1963 al
Congresso Internazionale di Genetica. Senza fare alcuna assunzione a priori sull’esistenza
della struttura ad albero, derivarono un albero evolutivo per tutte e 15 le popolazioni che
studiarono. Lewontin, nonostante avesse partecipato al Congresso, non si riferì a questa
analisi.
Il problema statistico era stato compreso almeno dall’epoca della discussione sul
coefficiente di Pearson applicato alla classificazione razziale negli anni ’20. Esso è citato in
tutte le edizioni dei Metodi Statistici per i Ricrecatori, ad opera di Fisher, fin dal 1925. Un
articolo utile è quello di Gower, in un libro sulla conferenza del 1972 “La Valutazioni delle
somiglianze delle popolazioni umane”. Come osservò,
<<… la mente umana distingue fra gruppi differenti perché ci sono caratteri correlati
all’interno di questi gruppi.>>.
L’originale discussione coinvolse dati antropometrici, ma è possibile far notare la fallacia
anche usando terminologia genetica moderna. Si considerino due popolazioni di aploidi
(Aploide =corredo cromosomico aploide, il numero di cromosomi (n) caratteristico delle
cellule germinali mature ( gameti ) degli organismi diploidi ed equivalente a metà di quello
delle cellule somatiche, ndt), ognuno di numerosità “n”. Si consideri “p” la frequenza di un
gene, detto “+” al contrario di “-” in un singolo loco diallelico, nella popolazione 1. Sia detta
“q” la stessa frequenza nella popolazione 2, p + q =1 (per definizione).
6
Ogni popolazione manifesta una variabilità di tipo binomiale, e la variabilità media è
aumentata dalle differenze nelle medie. Il metodo naturale per analizzare la variabilità è
l’analisi della varianza, dalla quale si scopre che il rapporto fra la della varianza all’interno
della varianza totale è semplicemente 4*p*q.
Considerando p = 0,3 e q = 0,7, il rapporto è 0,84; l’84% della variabilità è quella all’interno
dei gruppi, percentuale molto vicina ai dati di Lewontin. La probabilità di errata
classificazione di un individuo basandosi sul suo gene è p, in questo caso 0,3. È difficile che
i geni in un singolo locus diano grandi informazioni sulla popolazione alla quale
appartengono i loro portatori. Supponiamo ora ci siano k loci simili, tutto con frequenza
genetica = p nella popolazione 1 e frequenza genetica = q nella popolazione 2. Il rapporto
della variabilità totale è ancora l84% per ogni locus. Il numero totale di geni “+” in un
individuo avrà una distribuzione binomiale con media = k*p e varianza = k*p*q in entrambi i
casi. Continuiamo con la stessa frequenza dei geni e poniamo k = 100, ovvero poniamo che
ci siano 100 loci. Le medie sono rispettivamente 30 e 70, la varianza è di 21 e la deviazione
standard è pari a 4,58. Con una differenza fra le medie di 40 ed una deviazione standard di
meno di 4,6 non vi è praticamente alcuna sovrapposizione fra le 2 distribuzioni, e la
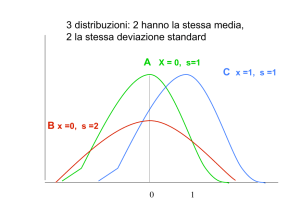
probabilità di errata classificazione è infinitesimale. La fig.1 mostra come decresce questa
probabilità se si considerano da 1 a 20 loci.
Fig 1. Grafico che mostra come la probabilità di errata classificazione diminuisce all’aumentare del numero di loci per il primo esempio
descritto nell’articolo. La proporzione della variabilità all’interno dei gruppi è l’84% come nei dati di Lewontin, ma la probabilità di errata
classificazione diventa irrisoria.
7
Un metodo per leggere questo risultato è apprezzare come il numero di geni “+” è come il
primo componente in un’analisi delle componenti principali (vedi Box 1). Per questa
componente la varianza fra le popolazioni è molto maggiore della varianza all’interno delle
popolazioni. Per le altre componenti è il contrario, ovvero in media la varianza fra le
popolazioni è solo una piccola proporzione della varianza totale (in questo esempio il 16%).
Tuttavia questo non deve ingannarci facendoci pensare che le due popolazioni non sono
separabili quando è evidente che lo sono. Ogni locus addizionale contribuisce allo stesso
modo alla varianza fra le popolazioni ed alla varianza all’interno delle popolazioni. La loro
proporzione quindi resta invariata ma, allo stesso tempo, ogni locus ci dà informazioni
aggiuntive sulla classificazione, che è cumulativa fra i loci in quanto la loro frequenza
genetica è correlata.
8
4. Box 1: Analisi delle Componenti Principali.
L’Analisi delle Componenti Principali (ACP) è uno strumento che permette di tirar fuori le
maggiori informazioni da dati multivariati, la cui alta dimensione rende impossibili
rappresentazioni grafiche semplici. La procedura può essere facilmente compresa anche
con solo due variabili, anche se il suo uso in questo caso può non essere inutile. Prendendo
un esempio dall’antropometria (disciplina in cui l’ACP è nata), possiamo avere dati con la
lunghezza e la larghezza di un numero di ossa umane. Ogni osso può essere rappresentato
come un punto in un diagramma i cui 2 assi sono “lunghezza” e “larghezza”. Dal momento
che lunghezza e larghezza saranno sicuramente in qualche misura correlati, la nuvola di
punti tenderà a seguire una certa direzione, andando da “scarsa lunghezza e scarsa
larghezza” (ossa piccole) ad “elevata larghezza ed elevata larghezza (ossa grandi).
L’ACP definisce questa direzione in modo preciso. Con l’ACP si identifica l’asse fattoriale
nella direzione di massima variabilità della nube dei punti-unità, in modo da deformare il
meno possibile la distanza reciproca tra punti: si minimizza la somma delle distanze dei
punti dall’asse (AB), che equivale a massimizzare la somma delle proiezioni dei punti
sull’asse (OA) (Teorema di Pitagora).
9
In altre parole, la variabilità dei dati è stata divisa in due componenti, una delle quali, lungo
questa linea, è detta Prima Componente Principale in quanto include tutta la variabilità che
può essere rappresentata in una dimensione. La Seconda Componente, analogamente,
include la variabilità restante, che è sicuramente molto minore.
Queste due componenti possono essere usate come assi del grafico. A volte la Prima
Componente avrà un significato ovvio, come potrebbe essere il caso delle ossa, nel quale
è chiaro che essa corrisponde in qualche modo alla “dimensione”.
Analogamente la Seconda Componente corrisponde in qualche modo alla “struttura”,
perché un osso i cui punti-dati sono lontani dalla linea della Prima Componente sarà sia più
lungo che più stretto della norma, o più corto e più largo.
La procedura si può generalizzare a qualunque numero di variabili e le Prime, Seconde, …
N-sime Componenti Principali sono quindi direzioni mutualmente ortogonali che dividono la
variabilità totale in quote decrescenti. La Prima Componente Principale spiega una quota
maggiore di variabilità della Seconda, che spiega una quota maggiore di variabilità della
Terza e così via. Il grafico delle prime 2 componenti principali rappresenta il massimo delle
informazioni che si possono avere usando due dimensioni.
10
5. Classificazione.
Si potrebbe supporre, anche se sarebbe sbagliato, che in questo esempio vi è l’assunzione
a monte che l’appartenenza all’una o all’altra popolazione è conosciuta in anticipo e che in
ogni locus ci sia la stessa popolazione che ha la maggiore frequenza del gene “+”. Tuttavia
nei fatti l’unico vantaggio di questa assunzione era che grazie ad essa è ancora più evidente
che il numero totale dei geni “+” è la migliore discriminanti fra le due popolazioni.
Per fugare questi dubbi, si consideri lo stesso esempio ma con “+” e “-“ scambiati in ogni
locus con probabilità 0,5 ciascuno. Si supponga che non vi è alcuna informazione a priori
sull’appartenenza degli individui all’una o l’altra popolazione. Chiaramente, il numero totale
dei geni “+” posseduti da un individuo non è più una discriminante, perché il numero atteso
è ora lo stesso per ogni gruppo. Sarà necessaria una Cluster Analysis (Analisi dei Gruppi)
per scoprire i gruppi; un criterio che conviene è ancora basato sull’analisi della varianza con
il metodo introdotto da Edwards e Cavalli-Sforza. Qui la divisione in due clusters massimizza
la varianza fra i cluster oppure minimizza la varianza all’interno dei cluster (il che è
esattamente la stessa cosa).
Come evidenziato da questi autori, è molto facile calcolare queste varianze dai dati binari,
perché tutte le informazioni sono contenute nella matrice delle distanze a coppie fra gli
individui e per ogni locus la distanza è 0 (“match”) dove vi è una perfetta corrispondenza dei
geni ed 1 (“mismatch”) se non vi è alcuna corrispondenza genetica. Dal momento che
scambiare “+” e “-“ non fa alcuna differenza sul numero di “match” e “mismatch”, è chiaro
che il cambiamento casuale di cui si parlava è irrilevante.
Proseguendo con l’esempio simmetrico, la probabilità di avere un match è p^2+q^2 se i due
individui appartengono alla stessa popolazione e 2*p*q se appartengono a popolazioni
diverse. Con k loci, quindi, la distanza fra due individui della stessa popolazione avrà una
distribuzione binomiale con media = k*(p^2+q^2) e varianza = k*(p^2+q^2)*(1-p^2-q^2). La
distanza fra due individui di popolazioni diverse avrà una distribuzione binomiale di media =
2*p*q e varianza = 2*k*p*q(1-2*p*q). Queste varianze saranno in ogni caso uguali.
11
Se prendo p = 0.3, q = 0.7 e k = 100, le medie saranno rispettivamente 58 e 42, con una
differenza di 16. Le varianze sono 24.36 e quindi le deviazioni standard entrambe 4.946
(ndt, la deviazione standard è la radice quadrata della varianza, per definizione, ed è una
misura di variabilità molto importante perché confrontabile con la media). Le medie
differiscono quindi di più del triplo delle deviazioni standard. Le componenti della matrice
delle distanze a coppie saranno quindi divise in due gruppi con una sovrapposizione molto
piccola. Sarà quindi possibile identificare i due gruppi con un rischio di errata classificazione
che tenderà a zero all’aumentare dei loci considerati. Analogamente all’esempio
precedente, è molto probabile che un calcolo delle frequenze di base del DNA in 4 tratti
omologhi di un genoma potrebbe rivelarsi una discriminante molto forte dal punto di vista
statistico per la classificazione di individui in gruppi.
12
6. Conclusione
Non c’è niente di sbagliato nell’analisi della varianza di Lewontin dal punto di vista statistico.
L’errore sta nel credere che il risultato non sia rilevante per la classificazione.
Non è vero che “la classificazione razziale non ha alcun significato genetico né
tassonomico”. Non è vero che, come veniva scritto sulla rivista “Nature”, che “due individui
presi a caso da qualunque gruppo sono diversi fra loro quanto due individui presi a caso dal
mondo intero”. Non è vero nemmeno che, come veniva scritto sul New Scientist, “due
individui sono diversi in quanto sono individui e non perché appartengono a razze diverse”,
né tantomeno che “non si può capire la razza di qualcuno dai suoi geni”. Queste frasi
potrebbero essere vere solo se tutti i caratteri studiati fossero fra loro incorrelati, e non è
vero.
Lewontin usò la sua analisi della varianza esclusivamente per sferrare un attacco
ingiustificato alla classificazione, soltanto perché lui è contrario ad essa per motivi ideologici
e sociali. Fu proprio lui a scrivere <<infatti l’intera storia del problema della variabilità
genetica è un chiaro esempio del ruolo che i presupposti ideologici radicati giocano nel
determinare le “verità scientifiche” e la direzione della ricerca scientifica>>.
In un articolo del 1970 intitolato “Razza ed Intelligenza” aveva scritto <<Proverò a spiegare
il ragionamento del professor Jensen in questo articolo, per mostrare che è costruito
artificiosamente per arrivare a certe conclusioni che corrispondono alle sue opinioni
personali. Queste sue opinioni lo portano a conclusioni errate.>>.
Una corretta analisi dei dati negli umani ci dà informazioni importanti sulle differenze
genetiche. Parlare di ciò che questo implica è un altro discorso. Tuttavia un errore pericoloso
è presupporre che l’eguaglianza degli esseri umani dal punto di vista morale implichi
uguaglianza dal punto di vista genetico e biologico perché la differenza, se scoperta, allora
diventa un pretesto per la diseguaglianza morale.
Fisher, nella sua opera “Metodi Statistici di Ricerca Scientifica”, scrive che <<Le migliori
causi tendono ad attrarre a loro supporto le peggiori argomentazioni. Questo sembra valere
sia a livello intellettuale che a livello morale>>.
13
7. Epilogo
Questo articolo potrebbe e forse dovrebbe essere stato scritto subito dopo il 1974. Ovvero
da quando sono state fatte molte scoperte sia per quanto riguarda la genetica sia per quanto
riguarda l’applicazione delle tecniche statistiche che facilitano lo studio delle differenze fra
popolazioni a partire dai dati genetici. Il libro magistrale di Cavalli Sforza, Menozzi e Piazza,
”La storia e la Geografia dei Geni Umani”, del 1994, e da allora molti altri studi hanno
confermato la validità dell’approccio. Studi molto recenti hanno trattato gli individui allo
stesso modo in cui Cavalli Sforza ed Edwards trattarono le popolazioni nel 1963, cioè
sottoponendo le loro informazioni genetiche ad una Cluster Analysis che rivelarono affinità
sorprendenti fra la genetica, la geografia, la lingua e la cultura. Gli autori di questi studi
dissero <<Fu solo accumulando piccole differenze fra frequenze di alleli in molti loci che la
struttura della popolazione venne scoperta.>>.
14
8. Bibliografia
1. Fisher RA. Statistical Methods for Research Workers. Edinburgh: Oliver
and Boyd. 1925.
2. Lewontin RC. The apportionment of human diversity. In: Dobzhansky T,
Hecht MK, Steere WC, editors. Evolutionary Biology 6. New York:
Appleton-Century-Crofts. 1972. p 381–398.
3. The Human Genome. Nature 2001;409:following p 812.
4. Ananthaswamy A. Under the skin. New Scientist 2002;174:34–37.
5. Lewontin RC. The Genetic Basis of Evolutionary Change. New York:
Columbia University Press. 1974.
6. Cavalli-Sforza LL, Piazza A. Analysis of evolution: evolutionary rates,
independence and treeness. Theor Pop Biol 1975;8:127–165.
7. Cavalli-Sforza LL, Edwards AWF. Analysis of human evolution. Proc. 11th
Internat. Congr. Genetics, The Hague 1963, Genetics Today 3. Oxford:
Pergamon. 1965. p 923–933.
8. Pearson K. On the coefficient of racial likeness. Biometrika 1926;18:
105–117.
9. Gower JC. Measures of taxonomic distance and their analysis. In: Weiner
JS, Huizinga J, editors. The Assessment of Population Affinities in Man.
Oxford: Clarendon. 1972. p 1–24.
10. Edwards AWF, Cavalli-Sforza LL. A method for cluster analysis.
Biometrics 1965;21:362–375.
11. Lewontin RC. Race and intelligence. Bulletin of the Atomic Scientists.
March 1970;2–8.
12. Fisher RA. Statistical Methods and Scientific Inference. Edinburgh: Oliver
and Boyd. 1956.
13. Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of
Human Genes. Princeton University Press. 1994.
14. Pritchard JK, Stephens M, Donnelly P. Inference of population structure
using multilocus genotype data. Genetics 2000;155:945–959.
15. Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky
LA, Feldman MW. Genetic structure of human populations. Science
2002;298:2381–2385.
15