Prof. ELENA LURASCHI
ANTIVIRALI
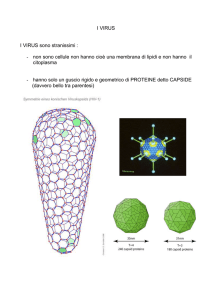
COS E UN VIRUS?
• E una particella infettante costituita da una molecola di acido
nucleico (DNA o RNA), inserita in un capside proteico che può
a sua volta essere rivestito da un envelope , costituito da un
bilayer lipidico;
• data la sua estrema semplicità, un virus può replicarsi solo
all interno di una cellula ospite;
• la replicazione del virus nella cellula ospite può avvenire:
§ mediante ciclo litico, in seguito al quale si ha rottura della
cellula ospite e rilascio delle particelle virali;
§ mediante ciclo lisogeno, in seguito al quale il cromosoma
virale s integra in un cromosoma della cellula ospite, dove
si replica come provirus, insieme al genoma che lo ospita.
POSSIBILI BERSAGLI DEI
FARMACI ANTIVIRALI
• inattivazione del virus prima del riconoscimento o della
penetrazione nella cellula ospite;
• inibizione dell interazione con i recettori per il virus, presenti
sulla membrana della cellula ospite;
• inibizione dell uncoating virale;
• inibizione dell integrazione del DNA virale nel genoma dell ospite;
• inibizione dell espressione di mRNA e proteine virali;
• alterazione delle varie fasi di formazione del capside e del
rilascio del virus.
Difficoltà nell uso degli antivirali
• agiscono per lo più a livello della replicazione degli acidi nucleici
virali e dell uncoating;
• poiché i virus condividono molti dei processi metabolici delle
cellule infettate, è difficile trovare farmaci con azione selettiva
sull agente patogeno;
• la maggior parte degli agenti antivirali è efficace soltanto
quando il virus è in replicazione;
• la diagnosi clinica di queste patologie può essere effettuata solo
quando il processo di replicazione è di solito già molto avanzato
e, conseguentemente, l intervento terapeutico è problematico.
Vaccinazione
E il miglior modo per controllare un infezione virale. Si tratta di
una immunizzazione attiva fatta mediante somministrazione di
vaccini costituiti da:
• virus attenuati (vaccino del vaiolo, morbillo, parotite, rosolia…);
• virus inattivi (vaccino antipoliomielitico, antinfluenzale…);
• subunità virali (vaccino antiepatite B, costituito dall antigene
di superficie dell epatite B).
Esistono quindi due tipi di immnunizzazione:
• immunizzazione attiva che evoca una risposta immnunitaria
specifica dopo la somministrazione di un immunogeno;
• immnunizzazione passiva che utilizza immunoglobuline per
modificare il decorso della malattia.
CLASSIFICAZIONE DEI VIRUS IN
BASE AL PROPRIO GENOMA
VIRUS
DNA
CLASSE I
DNA doppia elica
RNA
C L A S S E II
DNA (+) singola elica
C L A S S E III
RNA doppia elica
C L A S S E IV
RNA (+) singola elica
CLASSE V
RNA (-) singola elica
C L A S S E VI - RETROVIRUS
RNA (+) singola elica
VIRUS A DNA
Esempi sono dati da herpes virus (herpes e varicella),
adenovirus (mal di gola e congiuntivite) e poxvirus (vaiolo).
Il genoma virale può essere costituito da un singolo filamento
positivo di DNA oppure da un doppio filamento di DNA di cui
uno positivo e l altro negativo.
• Il filamento positivo codifica per le proteine;
• il filamento negativo funge da stampo per la sintesi del
filamento positivo. L mRNA è trascritto dal filamento negativo.
Ciclo di replicazione dell herpes
simplex virus, un virus a DNA e
possibili siti d azione degli antivirali
FARMACI CONTRO I VIRUS A DNA
ANALOGHI STRUTTURALI DELLE PURINE
-Modificazioni della baseACICLOVIR
N
N
O
HO
Meccanismo d azione
Analogo della guanosina, penetra nella cellula
infettata dal virus dove subisce una prima
O
fosforilazione ad aciclovir monofosfato ad opera di
una timidina kinasi virale e successivamente
convertito alle forme di fosfato e trifosfato da kinasi
NH
della cellula ospite. L aciclovir
trifosfato inibisce la DNA-polimerasi virale:
N
NH2 incorporato, nel DNA virale, essendo privo
dell ossidrile in 3 dello zucchero, impedisce
l ulteriore allungamento della catena. Per mezzo di un
meccanismo di inattivazione suicida , lo stampo interrotto
di DNA si lega alla DNA-polimerasi virale inattivandola in
maniera irreversibile. L’aciclovir viene attivato selettivamente
all’interno di cellule infettate, poichè possiede un’affinità per la
timidina chinasi virale circa 200 volte maggiore rispetto a quella
per la chinasi cellulare.
Impieghi terapeutici
Viene utilizzato nel trattamento delle infezioni da virus herpes simplex oltre che nel trattamento o
nella profilassi dell herpes zoster, riducendo la durata delle infezioni ed alleviando i sintomi
generali. Inoltre può essere utilizzato in soggetti trapiantati e malati di AIDS per curare eventuali
infezioni cutanee; nella cura di cefaliti erpetiche, infezioni genitali e come profilassi in pazienti
che utilizzano farmaci immunosoppressivi.
Mostra, invece, resistenza a infezioni da citomegalovirus per mutazioni di geni virali codificanti la timidina
kinasi o mutazioni della polimerasi provocando polmoniti, encefaliti ed infezioni mucocutanee.
Farmacocinetica
L aciclovir può essere somministrato per via topica sotto forma di crema o pomata, per via
endovenosa, oppure per via orale sotto forma di compresse. Tuttavia, il suo assorbimento dopo
somministrazione orale è basso e variabile e la sua biodisponibilità é intorno al 15-30%.
Si distribuisce ad un ampia gamma di tessuti raggiungendo concentrazioni nel liquor
pari a circa il 50% di quelle nel plasma.
Ha un’emivita di circa 2-3 ore ed è eliminato principalmente per escrezione renale.
Tossicità
Sono stati osservati lievi effetti locali come eruzioni e disturbi gastrointestinali.
VALACICLOVIR
Il valaciclovir è l estere dell aciclovir con la L-valina, quindi può essere considerato come un
profarmaco: infatti, dopo essere stato assorbito, viene convertito ad aciclovir per
metabolizzazione enzimatica di primo passaggio nel fegato.
Impieghi terapeutici
Viene utilizzato nel trattamento dell herpes zoster e di infezioni da herpes simplex.
Nelle
encefalopatie, inoltre diminuisce del 50% la mortalità e ne riduce le conseguenze
neurologiche.
Farmacocinetica
Viene somministrato per via orale alla dose di 250 mg, quattro volte al giorno. La sua
biodisponibilità per via orale è di gran lunga superiore a quella dell aciclovir e ciò
permette di
ottenere concentrazioni plasmatiche di farmaco più elevate e quindi di somministrarlo
meno
frequentemente.
Tossicità
Gli effetti collaterali sono lievi e simili a quelli dell aciclovir.
PENCICLOVIR E FAMCICLOVIR
O
N
N
NH
N
Penciclovir
NH2
Meccanismo d azione
Inibiscono la sintesi del DNA virale, dopo attivazione
alla forma di nucleoside trifosfato, competendo con
la DNA-polimerasi virale (in misura minore rispetto
all’aciclovir, ma permanendo più a lungo nelle cellule
infettate), senza necessariamente provocare
l’interruzione della catena.
HO
HO
Il penciclovir è un analogo della guanosina.
Impieghi terapeutici
Si sono dimostrati efficaci nella cura d infezioni da herpes virus, varicella zoster
virus ed herpes genitale, riducendo il tempo di guarigione ed abbreviando la
durata dell eliminazione del virus e la durata del dolore.
Se utilizzati in soggetti immunodepressi si osserva un alleviamento dei sintomi.
Farmacocinetica
Viene somministrato per via endovenosa in quanto presenta una biodisponibilità
orale limitata.
Il famciclovir è il diacetil-estere del penciclovir (somministrato per via orale
500-750 mg 3volte/die), è ben assorbito e rapidamente metabolizzato a penciclovir.
Hanno una emivita piuttosto lunga, permettendo di ridurre la frequenza di
somministrazione e/o la durata della terapia.
Vengono eliminati principalmente per escrezione renale e per lo più in forma
immodificata.
Tossicità
Possibili effetti collaterali sono cefalea, diarrea e nausea.
GANCICLOVIR
O
N
N
NH
N
NH2
O
HO
HO
Meccanismo d azione
Il ganciclovir è un nucleoside sintetico analogo della guanosina.
Come l aciclovir deve essere attivato a nucleoside trifosfato e,
in questa forma, compete con la guanosina trifosfato per
l incorporazione nel DNA virale. Sopprime la duplicazione del
DNA virale, ma al contrario dell aciclovir non blocca
l allungamento della catena e non viene rapidamente degradato,
persistendo nelle cellule infettate da citomegalovirus anche per
18-20 ore. La fosforilazione iniziale del ganciclovir è effettuata
da una proteina kinasi codificata dal citomegalovirus umano.
Il citomegalovirus differisce da altri virus erpetici umani perché
non codifica per una timidina kinasi virus-specifica.
Impieghi terapeutici
Viene utilizzato per il trattamento di infezioni gravi da citomegalovirus, che possono
essere fatali o possono portare a cecità, in pazienti immunodepressi.
Inoltre viene utilizzato in soggetti trapiantati come prevenzione per ridurre il rischio di
infezioni da citomegalovirus.
Farmacocinetica
Viene, di solito, somministrato per via endovenosa data la sua scarsa biodisponibilità
per via orale.
Ha una emivita di 4 ore e viene eliminato principalmente per escrezione renale, di cui
il 91% della dose somministrata viene escreto immodificato entro 24 ore.
Tossicità
Forte mielodepressione che ne limita l utilizzo; l’alto rischio di neutropenia nei pazienti
che ricevono simultaneamente ganciclovir e zidovudina preclude l’uso di
quest’associazione nell infezioni da HIV.
-Modificazione del residuo glucidicoVIDARABINA
NH2
N
N
HO
O
HO
OH
N
N
Meccanismo d azione
E un analogo dell adenosina con al posto del ribosio il suo
epimero arabinosio.
Viene fosforilata dalle kinasi cellulari a nucleoside trifosfato,
che agisce come inibitore relativamente selettivo per la
DNA-polimerasi virale competendo con la deossiadenosinatrifosfato.
Impieghi terapeutici
Inizialmente fu utilizzata come farmaco di prima scelta nelle
cefaliti erpetiche con un miglioramento della malattia di circa
il 70%, e nei pazienti resistenti all aciclovir.
E , invece, un farmaco di seconda scelta nelle infezioni da
virus herpes simplex e varicella zoster virus.
Mostra inoltre una certa attività sul poxvirus e sul virus
dell epatite B.
Farmacocinetica
A causa della sua bassa solubilità viene somministrata in grandi volumi di liquidi mediante
infusione endovenosa lenta e continua in un arco di tempo di 12 ore; può essere anche
applicata localmente. E , infatti, disponibile come preparazione oftalmica topica. Presenta una
buona distribuzione nell organismo, con una concentrazione nel liquido cerebrospinale pari a
circa il 30-50% di quella plasmatica. Ha una emivita di 3-5 ore; viene escreta principalmente nell’urina,
per la maggior parte sotto forma di metabolita deaminato, meno attivo, che raggiunge concentrazioni
plasmatiche più elevate di quelle del farmaco progenitore.
Tossicità
Gli effetti indesiderati includono nausea, vomito e diarrea.
Nelle infezioni gravi ha mostrato tossicità gastrointestinale ed in pazienti con insufficienza
renale un accentuarsi del danno renale.
Nella terapia ad alte dosi, in fase tardiva, si può anche manifestare neurotossicità,
solitamente reversibile.
Ad alte dosi sono state anche riportate soppressione del midollo osseo ed alterazione
dell emopoiesi.
Inoltre la vidarabina può essere mutagena e cancerogena.
Per tutte queste ragioni è stata soppiantata dall aciclovir.
ANALOGHI STRUTTURALI DELLE PIRIMIDINE
-Modificazioni della base
IDOXIURIDINA
O
I
HN
O
HO
N
O
OH
Meccanismo d azione
E un analogo della timidina che inibisce la replicazione dei
virus a DNA.
Viene fosforilata da kinasi cellulari e il nucleoside trifosfato
è incorporato sia nel DNA virale sia in quello cellulare,
inibendo la captazione di timidina nel DNA.
Questo le conferisce proprietà mutagene sconsigliandone
l’uso sistemico.
Impieghi terapeutici
Il suo utilizzo principale è nel trattamento delle infezioni oculari da herpes simplex e
varicella zoster.
E anche attiva contro i citomegalovirus e gli adenovirus.
Inoltre viene utilizzata nella cheratite erpetica nell’uomo, per via cutanea (poiché per via orale può
causare danno epatico e depressione midollare), favorendone la guarigione spontanea.
Non viene invece utilizzata nel trattamento di cheratiti stromali perché richiederebbe dosi troppo
elevate con conseguente tossicità.
Farmacocinetica
Benché sia stata somministrata per via endovenosa, oggi è considerata inefficace e troppo
tossica perché giustifichi la somministrazione per questa via. Di conseguenza l idoxiuridina
viene usata soltanto per via topica.
Per la somministrazione deve essere disciolta in dimetilsulfossido, un solvente che può
causare irritazione.
Tossicità
E una sostanza irritante, cancerogena e teratogena.
Può provocare dermatite da contatto.
TRIFLURIDINA
O
HN
O
HO
F
F
F
N
Meccanismo d azione
E un analogo della 2 -deossiuridina con un gruppo
trifluorometilico sul C5 dell anello pirimidinico.
Agisce inibendo gli enzimi che intervengono nella
sintesi del DNA.
Impieghi terapeutici
E’ attiva contro il virus herpes simplex e viene usata
come preparazione oftalmica nel trattamento della
cheratite erpetica.
O
OH
Farmacocinetica e Tossicità
Viene, in genere, somministrata per via topica
poiché essendo fortemente mielodepressiva
risulta scarsamente tollerata se somministrata
sistemicamente.
-Modificazione del residuo glucidico-
SORIVUDINA
O
H
Br
HN
O
N
H
Meccanismo d azione
E un analogo dell uridina con al posto del ribosio
il suo epimero arabinosio.
Presenta anche una modificazione della base con
un gruppo bromovinilico sul C5 dell’anello
pirimidinico.
Viene trifosforilata ad opera di una timidina chinasi
virale ed agisce selettivamente in cellule infettate
da herpes virus competendo con la
deossitimidina-trifosfato.
HO
O
HO
OH
Impieghi terapeutici
Somministrata sia per via endovenosa che per
via orale, risulta più attiva dell’aciclovir a dosi
elevate nel trattamento dell’herpes zoster in
pazienti immunodepressi.
Farmacocinetica
La sorivudina viene ben assorbita per via orale.
Il tempo medio di dimezzamento plasmatico è di 5-7 ore; negli anziani tale tempo
aumenta fino al 30%.
La maggior parte della sorivudina si ritrova inalterata nelle urine: solo il 5% viene
escreta come bromoviniluridina (BVU), un prodotto del metabolismo.
Il legame alle proteine plasmatiche è molto alto (98%), ma non se ne conosce il
significato clinico.
Tossicità
In generale, la terapia orale a breve termine è ben tollerata dal paziente. Gli
effetti collaterali riportati con più frequenza sono disturbi gastrointestinali
caratterizzati da nausea e vomito, diarrea e cefalea.
Talvolta si possono manifestare aumenti dei livelli degli enzimi epatici.
DERIVATI INORGANICI
FOSCARNET
O
NaO
P
ONa
COONa
Meccanismo d azione
E un analogo sintetico non nucleosidico del pirofosfato
inorganico, che, data la similarità di struttura, inibisce
la DNA-polimerasi virale, legandosi direttamente ai siti
di legame del pirofosfato in maniera selettiva e non
competitiva. In questo modo impedisce la liberazione
del pirofosfato dal deossinucleotide trifosfato.
Mostra un’affinità per la DNA-polimerasi virale cento
volte maggiore rispetto a quella per la DNA-polimerasi
cellulare.
Impieghi terapeutici
Viene utilizzato nel trattamento delle infezioni da citomegalovirus, quali
retiniti, e delle infezioni da herpes simplex resistenti all’aciclovir negli
individui immunodepressi.
Farmacocinetica
Somministrato per via endovenosa, viene segregato nel tessuto osseo e
lentamente rilasciato.
Viene eliminato principalmente per escrezione renale per lo più in forma
immodificata.
Tossicità
E un farmaco non ben tollerato. Può infatti presentare gravi effetti
nefrotossici oltre ad anemia, ipercalcemia ed iperfosfatemia, ma è
stata anche osservata ipocalcemia sintomatica.
VIRUS A RNA
Esempi sono dati da : Orthomyxovirus
(influenza),Paramyxovirus (morbillo e
parotite),Rubivirus(rosolia) e Picornavirus
(raffreddore,meningite e poliomielite)
Il genoma virale può essere costituito da un doppio
filamento oppure da un singolo filamento o positivo o
negativo di RNA.Quest ultimo non è in grado di funzionare
da messaggero e deve essere quindi trascritto da una
RNApolimerasi-RNA dipendente nel filamento
complementare che fungerà da mRNA.
CLASSIFICAZIONE DEI VIRUS IN
BASE AL PROPRIO GENOMA
VIRUS
DNA
CLASSE I
DNA doppia elica
RNA
C L A S S E II
DNA (+) singola elica
C L A S S E III
RNA doppia elica
C L A S S E IV
RNA (+) singola elica
CLASSE V
RNA (-) singola elica
C L A S S E VI - RETROVIRUS
RNA (+) singola elica
Virus dell influenza
I virus dell influenza sono virus a
RNA, con genoma segmentato (8
segmenti ciascuno associato ad
una RNA-polimerasi) ed
appartengono alla classe degli
Ortomyxovirus. Sono classificati
in tre sottotipi (A, B, C) che si
differenziano per le loro proteine
interne.
Il tipo A è più diffuso e produce
una severa epidemia, il tipo B è
meno diffuso, ma può comunque
produrre epidemie, il tipo C,
infine, è responsabile della forma
più lieve dell influenza.
I peplomeri sono costituiti da due
glicoproteine, l emoagglutinina
e la neuraminidasi,codificate da
due distinti geni virali.
Il virus dell'influenza A normalmente penetra nella
cellula legandosi alle molecole di acido sialico, per poi
essere endocitato. Nella vescicola endocitica, grazie alla
presenza della proteina canale M2 che pompa H+ al suo
interno, si crea un ambiente acido che modifica la
struttura dell’emoagglutinina. Questa è infatti composta
da due subunità, H1 e H2, collegate tra loro da un
ponte disolfuro. H1 è l’antirecettore virale, mentre H2
(che è nascosta da H1) è la proteina fusogena. Il pH
acido serve a rompere il ponte disolfuro e quindi a
liberare la proteina fusogena che permette appunto la
fusione. Bloccando M2 non può avvenire la liberazione
dell'RNA virale e il processo infettivo è interrotto.
Replicazione del virus dell influenza
L infezione inizia quando l emoagglutinina si lega in modo non covalente alla neuramina,
un residuo di acido sialico, presente sull estremità di alcune glicoproteine di membrana,
che svolgono il ruolo di recettori specifici cellulari.
In seguito, la neuraminidasi, una proteina integrale di membrana, con attività
enzimatica, diffusa in diversi virus e microrganismi, elimina il residuo di acido sialico per
permettere il rilascio delle nuove particelle virali, affinché non restino impigliate, dopo la
loro uscita, sulle cellule infette. Tale enzima è importante anche per l ingresso del virus
nella cellula ospite, liberandolo così dal legame con il recettore. La neuraminidasi infine
promuove anche il movimento del virus attraverso il muco delle vie respiratorie,
costituito prevalentemente da polimeri contenenti acido sialico.
La funzione dell emoagglutinina è di rendere il virus in grado di adsorbirsi ai recettori
glicoproteici presenti sulla superficie di vari tipi di cellule e globuli rossi.
Ogni peplomero di emoagglutinina è formato da tre identici polipeptidi, da tre
monomeri. Ogni monomero è sintetizzato come singola catena polipeptidica e in
seguito associato alle altre.
Durante il processo di maturazione l emoagglutinina viene tagliata da proteasi cellulari
in due polipeptidi distinti, denominati HA1 e HA2, i quali sono tenuti insieme da ponti
disolfurici. Il taglio permette alla emoagglutinina di assumere una conformazione tale
da rendere infettiva la particella virale, in quanto è in questa conformazione che è in
grado di legarsi al residuo di acido sialico presente sul recettore glicoproteico della
cellula ospite.
Il legame con il recettore permette la fusione dell involucro lipoproteico virale con la
membrana della cellula e consente in questo modo la penetrazione del nucleocapside
nel citoplasma. Quindi l Ortomyxovirus entra nella cellula per endocitosi mediata da
recettori e poi viene internalizzato nell endosoma. La proteina virale M2, che funziona da
canale ionico, è importante in due stadi della replicazione virale all interno della cellula
ospite:
1) fusione della membrana virale con la membrana dell endosoma;
2) stadio di assemblaggio e di rilascio di nuovi virioni dalla superficie della cellula ospite.
Il basso pH dell endosoma (circa 5) provoca un cambio conformazionale
dell emoagglutinina che in seguito a ciò espone siti idrofobici che si inseriscono nel
doppio strato lipidico dell endosoma, permettendo la fusione della membrana virale con
quella dell endosoma e l uscita dell acido nucleico nel citosol.
Nella fase di liberazione, durante la quale si verifica la formazione dell envelope, il
nucleocapside si associa alla membrana plasmatica dove sono presenti le proteine
virali, che si raccolgono attorno ad esso, escludendo dalla formazione dell envelope
le proteine di membrana dell ospite.
Il virus dell influenza viene trasmesso attraverso le secrezioni delle vie aeree (goccioline
di saliva emesse da soggetti infetti attraverso tosse e starnuti). Il virus è favorito da
umidità e temperature basse. L affollamento e le condizioni ambientali tipiche
dell inverno (luoghi chiusi e scarsa ventilazione) favoriscono pertanto la trasmissione.
Uno degli aspetti più importanti del virus dell influenza è la capacità di
modificarsi (emoagglutinina e neuraminidasi) e generare ceppi varianti. Questo
fenomeno spiega perché l influenza possa ricolpire lo stesso individuo e causare
epidemie annuali
Ruolo dell emogglutinina e della sialidasi
La reazione catalizzata dall enzima sialidasi è la scissione del legame α-chetosidico tra
l acido sialico terminale e un residuo saccaridico di glicoproteine, glicolipidi e oligosaccaridi
presenti sulla membrana della cellula ospite. Questa reazione distrugge il recettore
dell emoagglutinina, che aveva legato l acido sialico del recettore della cellula ospite, in
modo da permettere il rilascio della progenie virale della cellula infetta.
Replicazione del virus a RNA (influenza): possibili
siti d azione degli antivirali
Ciclo di replicazione di un virus a DNA (Hsv) un e
possibili siti d azione degli antivirali
FARMACI CONTRO I VIRUS A RNA
-Primi inibitori della sialidasi-
DANA e FANA
I primi studi per lo sviluppo di farmaci diretti contro i virus a RNA, si sono rivolti alla
ricerca di inibitori della neuraminidasi.
Poiché essa come enzima possiede uno stato di transizione, si sono sintetizzati farmaci
in grado di mimare tale stato di transizione: gli analoghi acido 2,3 di deidro N-acetil
neuraminico (DANA) e l acido 2,3 di deidro N-trifluoro acetil neuraminico (FANA).
Tuttavia tali composti si mostrarono privi di alcuna selettività e mostrarono in vivo una debole
attività antivirale. Il problema risiedeva nel tipo di sostituenti introdotti sull anello diidropiranico.
Si vide, infatti, che vi erano sostituenti energeticamente favoriti che conferivano al composto
una maggiore selettività per l enzima.
ZANAMIVIR
Si è passati quindi a studiare il composto GG167 (o Zanamivir), il quale presenta un gruppo
carbossilico sul C2 dell anello ed un gruppo guanidinico basico sul C4 al posto di un ossidrile.
OH
O
H
O
HO
OH
OH
HN
NH2
NH
H3C
O
NH
Tutti i sostituenti forniscono contributi essenziali al legame e non è
possibile rimuoverli senza una significativa riduzione dell attività inibitoria.
Relazioni struttura-Attività (SAR)
L interazione tra gg167 (Zanamivir) e sialidasi, determinata dalla cristallografia a raggi x ha
messo in luce l importanza di ciascuno dei sostituenti del diidropirano.
C-2: l acido carbossilico forma 3
ponti salini ed è essenziale per
l attività biologica. Infatti esteri o
ammidi sono più deboli inibitori,
forse perché non possono formare
interazioni elettrostatiche con i 3
residui di Arginina. Inoltre non c è
spazio per sostituenti addizionali.
C-4: il gruppo basico guanidinico
non sostituito è il sostituente
preferito a questa posizione.
Forma legami salini con residui di
ASP 151 e GLU 227 e legami
idrogeno con il residuo carbonilico
del TRP 178. Gli unici composti,
con attività che supera quella del
DANA, sono quelli che contengono
un piccolo residuo basico.
C-5: l ammide sembra essere
importante per l attività grazie
alla formazione di un legame
idrogeno con un residuo di ARG
152. La completa rimozione di
questo sostituente riduce
l attività di circa 105. L optimum
è fornito dal residuo acetamidico
che si posiziona in una piccola
tasca lipofila formata dalle
catene laterali di ILE e TRP (è
importante la spinta idrofobica
perché favorisce l interazione).
C-6: i gruppi OH in posizione
C-8 e C-9 del residuo di glicerolo
formano dei legami idrogeno
con GLU 276. La rimozione
sequenziale dei gruppi
idrossimetilici dal residuo del
glicerolo rivela una graduale
risoluzione nell attività inibitoria.
Anello diidropiranico: un analogo cicloesene ha mostrato attività biologica equivalente al
corrispondente derivato diidropiranico. Una ulteriore saturazione dell anello indebolisce l attività.
Meccanismo d azione
Queste sostituzioni portavano ad un incremento dell interazione della molecola con
l enzima per formazione di ponti salini con residui di glutammato, presenti nel sito
attivo dell enzima.
Il fine è quello di impedire che le particelle virali escano dalla membrana della
cellula ospite.
Infatti, bloccando l enzima, i virioni vengono trattenuti sulla superficie della cellula
con conseguente riduzione del propagarsi dell infezione.
Impieghi terapeutici
Tale composto si è pertanto rivelato estremamente selettivo nei confronti della neuraminidasi
virale ed ha mostrato attività antivirale in numerosi modelli animali.
Farmacocinetica
Deve essere somministrato per via inalatoria; la somministrazione per via orale è impedita dalla
presenza del gruppo guanidinico che essendo carico attraverserebbe con difficoltà le membrane
cellulari lipofile.
Tossicità
Non ha presentato effetti tossici né collaterali.
AMANTADINA, RIMANTADINA E
TROMANTADINA CLORIDRATO
O
H3C
Amantadina
Rimantadina
CH3
+
H3C
NH2
NH2
H3C
Cl
N
NH
O
CH3
Tromantadina cloridrato
La classe di farmaci in grado di interferire sull uncoating virale è rappresentata dagli adamantani,
il cui capostipite è l aminoadamantano cloridrato, una amina primaria triciclica idrosolubile scoperta
agli inizi degli anni 60 in quanto inibitore della replicazione del virus dell influenza di tipo A.
Inizialmente fu utilizzata nel trattamento del Morbo di Parkinson; solo successivamente fu
identificata la sua azione antivirale.
Benché il composto più conosciuto sia l’amantadina, la rimantadina è il farmaco che ha il profilo
farmaco-terapeutico migliore tra i vari composti di questa classe.
Meccanismo d azione
L amantadina e la rimantidina inibiscono una fase iniziale del processo di replicazione
virale detta di uncoating (spogliazione dell involucro esterno).
Il sito di legame è rappresentato dalla proteina M2, proteina della membrana virale con
funzioni di canale e necessaria per acidificare l interno del virus, evento da cui dipende la
dissociazione delle ribonucleoproteine dalla membrana virale e quindi il trasferimento del
codice virale nel citoplasma della cellula.
Il farmaco si accumula a livello endo- e lisosomiale innalzando il pH di questi organelli.
E probabile che ciò ostacoli la fusione della membrana virale con quella della cellula
ospite.
Inoltre l alterato funzionamento della proteina M2 e il disturbo del pH all interno degli
organelli citoplasmatici sono probabilmente anche responsabili di alterazioni nel corso
della sintesi e della maturazione dell emaglutinina virale neosintetizzata e quindi
dell assemblaggio del capside virale.
L’effetto finale è rappresentato dal blocco della replicazione virale.
Impieghi terapeutici
Amantadina e rimantadina sono approvate per la prevenzione ed il trattamento delle
infezioni causate dal virus dell influenza A. La profilassi stagionale eseguita con
entrambi i farmaci assicura protezione contro l influenza di tipo A nel 70-90% dei casi.
La profilassi dovrebbe essere iniziata dopo l’identificazione del virus dell’influenza all’interno di una
comunità o di una regione, e dovrebbe essere protratta per tutto il periodo di rischio (4-8 settimane),
poiché gli effetti protettivi vengono persi alcuni giorni dopo l’interruzione della somministrazione dei
farmaci.
In alternativa, l’assunzione di questi farmaci può iniziare congiuntamente con l’immunizzazione e
continuare per due settimane fino a quando si sviluppa la risposta immunitaria protettiva.
Sono stati riportati diversi casi di ceppi resistenti agli adamantini. La resistenza che si
sviluppa è crociata con tutti i farmaci appartenenti a questa classe e è trasmissibile da ceppo
a ceppo.
Tale resistenza è stata associata con variazioni di un singolo nucleotide che comportano
sostituzioni di aminoacidi nella regione transmembrana della proteina M2.
Farmacocinetica
L amantadina e la rimantadina sono ben assorbite per via orale, tuttavia sono anche
disponibili preparazioni per somministrazioni per via aerosolica. Vengono somministrate
ad una dose di 100mg due volte al giorno. Entrambi presentano un ampio volume di
distribuzione.
Attraversano la barriera ematoencefalica.
L amantadina viene eliminata quasi per intero in forma immodificata con le urine. La sua
eliminazione dipende fortemente dalla funzionalità renale per cui l emivita, che in soggetti
giovani è pari a circa 12-18 ore, aumenta di gran lunga negli anziani e può raggiungere
valori anche più elevati in presenza di insufficienza renale.
Al contrario, la rimantadina viene estesamente metabolizzata con una emivita compresa tra
le 24 e le 36 ore.
Una quantità inferiore al 15% della dose somministrata viene escreta immodificata con le
urine e il 20% o più è eliminato nelle urine sottoforma di metaboliti idrossilati.
Tossicità
I più comuni effetti collaterali dipendono dalla dose (disturbi gastrointestinali minori e del
sistema nervoso centrale) e sono molto meno frequenti con l impiego di rimantadina.
L’amantadina è teratogenica.
FARMACI CONTRO VIRUS A DNA,
RNA, RETROVIRUS
RIBAVIRINA
O
H2N
N
N
HO
N
O
OH OH
E un analogo nucleosidico delle purine con una base
modificata e un D-ribosio. Inibisce la replicazione di
molti virus a DNA e a RNA, quali orthomyxovirus,
paramyxovirus, herpes virus, adenovirus e
retrovirus.
Meccanismo d azione
Il meccanismo alla base dell azione antivirale della ribavirina non è completamente
definito, tuttavia sembra essere collegato con l alterazione dei pool di nucleotidi
cellulari e con l inibizione della sintesi dell mRNA virale.
La fosforilazione intracellulare della ribavirina a derivato mono, di- e trifosfato è
mediata dall azione di enzimi della cellula ospite.
La ribavirina monofosfato inibisce competitivamente l enzima cellulare inosina-5 fosfato deidrogenasi e interferisce con la sintesi della guanosina trifosfato e, più in
generale, con la sintesi degli acidi nucleici. Inoltre la ribavirina trifosfato inibisce in
maniera competitiva la reazione GTP-dipendente di capping in 5 dell RNA
messaggero virale e, nel caso del virus dell influenza, interferisce specificamente
con l attività trascrizionale.
La ribavirina sembra avere molteplici siti d azione e alcuni di questi (ad esempio,
inibizione della sintesi del GTP) possono potenziare gli altri (ad esempio, inibizione
degli enzimi GTP-dipendenti).
Impieghi terapeutici
La ribavirina somministrata per aerosol (20 mg/ml per 18 ore di esposizione al giorno)
è approvata per il trattamento delle bronchioliti e delle polmoniti presenti tra i bambini
ospedalizzati, riducendo la gravità della malattia, migliorando l ossigenazione arteriosa
e, in misura variabile, limitando la diffusione del virus.
Anche una terapia a dosi elevate per un periodo ridotto (60 mg/ml per 2 ore, tre volte/
die) sembra essere efficace.
In alcuni casi, la ribavirina per via endovenosa e/o per aerosol è stata impiegata per
trattare le infezioni gravi da virus dell influenza e nel trattamento di pazienti
immunodeficienti e di infezioni da virus parainfluenzali e da virus del morbillo.
In pazienti infettati dall’HIV, la somministrazione orale cronica di ribavirina a dosi tollerabili non
fornisce effetti benefici sicuri.
La comparsa di resistenza alla ribavirina non è certa, ma è stato possibile selezionare cellule che
non fosforilano questo farmaco e quindi non permettono la formazione delle forme attive.
Farmacocinetica
Viene somministrata principalmente per via endovenosa avendo una biodisponibilità
orale piuttosto scarsa.
Può essere anche utilizzata sottoforma di aerosol, raggiungendo livelli plasmatici elevati,
soprattutto nelle secrezioni delle vie respiratorie, che aumentano con la durata
dell esposizione.
La ribavirina trifosfato si concentra negli eritrociti, dove persiste con un tempo di
dimezzamento di circa 40 giorni.
La cinetica di eliminazione della ribavirina è complessa.
In una fase iniziale l emivita è di circa 2 ore, ma è anche presente una fase terminale
prolungata con una emivita di 18-36 ore.
La clearance renale è pari al 40% della dose di ribavirina somministrata, la quota
restante viene metabolizzata ed escreta per via epatica.
Tossicità
La ribavirina assunta per aerosol è stata ben tollerata ma può causare irritazioni
congiuntivali, eruzioni cutanee e deterioramento reversibile della funzionalità
polmonare.
La ribavirina per via sistemica provoca, invece, un'anemia dose-dipendente dovuta a
emolisi extravasale e soppressione dose-dipendente del midollo osseo.
Durante la somministrazione orale a breve termine si manifestano aumenti reversibili
delle concentrazioni plasmatiche di bilirubina, di ferro e di acido urico.
Nei pazienti infettati dall HIV, la terapia orale cronica è associata con linfopenia,
disturbi
gastrointestinali e del SNC: tutti sintomi dose-dipendente.
La ribavirina, infine, risulta teratogena, embriotossica, oncogena e, probabilmente,
gonadotossica.
RETROVIRUS
Il virione dei retrovirus ha forma sferica ed è formato da tre componenti
morfologiche:
• un involucro esterno, costituito da una membrana lipidica;
• un nucleo interno (capside) formato da proteine e RNA;
• un nucleoide, contenente l RNA nonché gli enzimi necessari
per la replicazione del virus.
Il genoma è costituito da due frammenti identici di RNA lineare a
singolo filamento, uniti per complementarietà di basi e legami
idrogeno.
Il ciclo di replicazione
dell HIV-1, un esempio di
retrovirus.
Il primo passaggio del processo di infezione si basa
sull interazione tra le glicoproteine dell involucro
virale e alcune zone specifiche della membrana
della cellula ospite.
1)
Seguono la fusione dell involucro virale con la
membrana cellulare e il rilascio del nucleo virale
nel citoplasma. Le proteine del capside vengono
rimosse enzimaticamente.
2)
A questo punto, l RNA virale viene copiato in DNA
ad opera di un enzima chiamato trascrittasi
inversa (una DNA polimerasi RNA-dipendente).
Viene così a formarsi una molecola di DNA a
singolo filamento, complementare a quella di RNA.
3)
Quest ultima funge quindi da stampo per la
sintesi
dell elica complementare di DNA,
sempre ad opera della trascrittasi inversa.
4)
Il DNA a doppia elica s integra nel genoma della cellula
ospite, dando luogo alla formazione del provirus.
5)
I geni provirali sono trascritti in molecole di
mRNA.
6)
La traduzione nel citoplasma di tali molecole di
mRNA del provirus serve a fornire sia le proteine
virali sia gli RNA a singolo filamento che andranno
a formare il genoma dei nuovi virioni.
7) Le proteine virali, compresa la trascrittasi inversa,
migrano verso zone specifiche della membrana
citoplasmatica della cellula ospite, nelle quali sono
presenti glicoproteine virali. A livello di questi siti
della membrana cellulare avviene l assemblaggio
dei virus.
8) Infine, tali virus vengono rilasciati per
gemmazione.
I retrovirus non uccidono la cellula ospite, sicchè la
replicazione ed il rilascio di nuovi virus possono
continuare indefinitamente.
FARMACI CONTRO I RETROVIRUS
INIBITORI NUCLEOSIDICI DELLA TRASCRITTASI INVERSA
-Analoghi del substratoZIDOVUDINA (ZDV, AZT)
O
CH3
HN
O
N
HO
O
H
H
N3
Meccanismo d azione
È un didesossinucleotide analogo della timidina, attivo
contro HIV-1 e HIV-2. È il farmaco di riferimento nel
trattamento dei pazienti con infezione da HIV.
È un profarmaco, infatti viene fosforilato a livello
intracellulare da kinasi cellulari alla forma trifosfato attiva.
Il suo meccanismo d azione pertanto è basato
sull inibizione dell incorporazione della timida nel DNA
virale ad opera della trascrittasi inversa, interrompendo in
questo modo l allungamento delle catene di DNA virale.
Impieghi terapeutici
La zidovudina ritarda la progressione della malattia verso l AIDS e riduce le infezioni
opportunistiche, aumentando la sopravvivenza dei pazienti con infezione da HIV,
nello stadio avanzato.
Dopo esposizione di lungo periodo al farmaco, spesso può insorgere resistenza in
seguito a mutazioni entro il gene per la trascrittasi inversa.
Tuttavia, la zidovudina è l unico farmaco che si sia dimostrato capace di prolungare la
sopravvivenza nell infezione da HIV ed è uno dei farmaci antiretrovirali più potenti.
Per la sua buona penetrazione nella barriera ematoencefalica è importante
nel prevenire lo sviluppo dell encefalopatia da AIDS.
Farmacocinetica
La zidovudina mostra una biodisponibilità di circa il 60%, dopo somministrazione per via
orale. Raggiunge concentrazioni plasmatiche massime dopo 0.5 - 1.5 ore.
L emivita plasmatica è breve (circa 1 ora), ma quella intracellulare del suo metabolita
trifosfato è ben più lunga e tale da consentire un regime posologico di tre somministrazioni
al giorno.
La zidovudina penetra nel liquido cerebrospinale.
L eliminazione avviene per glucuronazione a livello epatico e successiva escrezione renale.
Tossicità
Il più rilevante effetto tossico della zidovudina è la depressione del midollo osseo e tossicità
epatica. In un piccolo numero di pazienti possono anche insorgere miopatia, effetti avversi
gastrointestinali e cefalee. Occorre pertanto un accurato monitoraggio clinico qualora la
zidovudina fosse somministrata insieme ad altri farmaci mielosoppressivi (ganciclovir).
Alcuni farmaci (ad esempio, il trimetoprim) fanno aumentare le concentrazioni plasmatiche
di zidovudina, poiché inibiscono la glucuronazione epatica e/o poiché interferiscono con la
sua escrezione tubulare renale.
DIDANOSINA (didesossinosina; ddl)
O
N
NH
HO
N
O
H
N
Meccanismo d azione
È un didesossinucleotide analogo dell adenina.
Agisce sulla trascrittasi inversa di HIV-1, HIV-2
e HIV-1 zidovudina resistente.
È stato osservato sinergismo in vitro tra
didanosina e zidovudina.
H
Impieghi terapeutici
La didanosina ha la stessa efficacia clinica globale della zidovudina. Tuttavia, si è osservata
una maggiore efficacia della zidovudina nelle prime settimane di terapia e un miglioramento
dell efficacia nel successivo passaggio alla didanosina.
Dopo esposizione di lungo periodo al farmaco, può insorgere resistenza in seguito a
mutazioni a livello del codone 74 del gene per la trascrittasi inversa.
Farmacocinetica
La didanosina è acido-labile, ragione per la quale viene somministrata con un tampone
a stomaco vuoto. Il queste condizioni la sua biodisponibilità è compresa tra il 20 e il
40%.
Come la zidovudina, presenta un emivita plasmatica breve (0.2 - 2 ore), ma una
maggiore emivita intracellulare del suo metabolica attivo, tale da consentire un regime
posologico di 2 volte al giorno.
La didanosina subisce un ampio metabolismo (forma trifosfato, acido urico e/o
metabolici purinici), ma circa il 30-60 % viene escreto immodificato nell urina.
Tossicità
A differenza della zidovudina, la didanosina ha una minima tossicità ematologia.
I principali, ma rari effetti avversi sono la neuropatia periferica e la pancreatite.
Le preparazioni di didanosina contenenti magnesio e/o alluminio come antiacidi
(tampone) possono far diminuire le concentrazioni plasmatiche di alcuni chinolonici
e di alcune tetracicline.
ZALCITABINA (didesossicitidina, ddC)
NH2
N
N
HO
O
Meccanismo d azione
O
H
H
È un analogo del nucleoside desossicitidina che, dopo
aver subito la conversione intracellulare nel suo
metabolica trifosfato attivo, inibisce la replicazione
dell HIV.
Come la zidovudina e la didanosina, la zalcitabina
agisce inibendo la trascrittasi inversa e terminando le
catene del DNA virale. È attiva contro l HIV-1, l HIV-2
e le varianti zidovudina-resistenti.
Esercita un azione additiva e/o sinergica, se
somministrata in associazione a zidovudina.
Impieghi terapeutici
L efficacia clinica è minore di quella della zidovudina e il suo uso principale è nella terapia
d associazione.
La resistenza alla zalcitabina può svilupparsi più lentamente rispetto a quella alla zidovudina.
Farmacocinetica
La biodisponibilità orale è circa del 70-80%.
Le concentrazioni plasmatiche massime si registrano 1 - 2 ore dopo la somministrazione.
La zalcitabina penetra nel liquor in misura minore rispetto alla zidovudina.
Viene eliminata principalmente per escrezione renale.
Tossicità
I principali effetti avversi sono neuropatia periferica, stomatite ed eruzioni.
Nell 1% dei pazienti si è registrata pancreatite e ancor più raramente tossicità ematologia.
STAVUDINA (d4T)
O
CH3
HN
HO
N
O
O
H
H
Meccanismo d azione
È un analogo nucleosidico (diedro-didesossitimidina) usato
nelle infezioni da HIV.
Agisce come inibitore della trascrittasi inversa.
È efficace sia contro l HIV-1, sia contro l HIV-2, compresi
i ceppi resistenti alla zidovudina.
Impieghi terapeutici
Il passaggio dalla zidovudina alla stavudina è associato ad un miglioramento,
rispetto alla prosecuzione della somministrazione di zidovudina.
L entità dell effetto della zidovudina e quella della stavudina è simile in individui
che precedentemente non hanno ricevuto terapia antiretrovirale.
Farmacocinetica
La biodisponibilità orale è abbastanza alta (>80%).
Viene eliminata per escrezione renale.
Benché l emivita plasmatica sia di sole 0.8 - 1.6 ore, la forma trifosfato attiva del
farmaco ha un emivita intracellulare simile a quella della zidovudina.
Tossicità
Il principale effetto avverso che limita la dose di stavudina è la neuropatia periferica.
Altri effetti avversi meno frequenti sono anemia e aumento delle transaminasi.
INIBITORI NON NUCLEOSIDICI DELLA
TRASCRITTASI INVERSA
Nevirapina, Delavirdina, Efavirenz
›› Meccanismo d'azione
Gli inibitori non nucleosidici della trascrittasi inversa si legano
sulla trascrittasi inversa virale in un sito vicino ma diverso
rispetto agli inibitori nuclseosidici della trascrittasi inversa.
Questi inibitori non nucleosidici non competono (a differenza
dei nucleosidici) con altri nucleosidi trifosfato antivirali, e non
necessitano di attivazione per fosforilazione.
Il legame col sito attivo blocca l'azione di DNA e RNA
polimerasi virale.
Caratteristiche e impieghi
Impiegati contro virus HIV-1.
Farmacocinetica
Somministrati per via orale (con buona biodisponibilità),
legano le proteine plasmatiche, tra cui l'albumina, e hanno
lunga emivita, sono metabolizzati nel fegato dal CyP450,
di cui Delavirdina è inibitore e Efavirenz induttore. Escreti
per via urinaria.
›› Interazioni
Interazioni con altri farmaci metabolizzati dal CyP 450.
›› Effetti collaterali e avvertenze
Nausea, rash cutanei anche mortali,.mal di testa, diarrea,
Efavirenz è anche teratogeno.
INIBITORI DELLA PROTEASI
-Analoghi dello stato di transizione-
Per la replicazione virale sono necessari solo tre geni virali:
• gag, che codifica per le proteine nucleo-specifiche situate nel
nucleocapside;
• pol, che codifica per la trascrittasi
inversa;
• env, che codifica per le proteine
dell involucro.
L ordine di questi geni, in direzione 5 -3 è gag-pol-env in tutti i retrovirus. Il trascritto virale
contiene un sito di taglio vicino al gene gag e un sito accettore subito prima del gene env. Il
prodotto del gene env è tradotto da un mRNA tagliato in corrispondenza dei due siti e risaldato,
mentre i geni gag e pol sono tradotti da un mRNA delle dimensioni dell intero genoma.
L HIV contiene una aspartato-proteasi peculiare che si attiva al momento della gemmazione
virale, scindendo il grande polipeptide virale derivato dal gene gag nelle proteine costituenti
(proteine strutturali, enzimi virali quali la trascrittasi inversa, l integrasi e la proteasi stessa).
SAQUINAVIR, INDINAVIR,
RITONAVIR e NELFINAVIR
Mediante studi di modellizzazione molecolare, è stato possibile disegnare diversi inibitori
della proteasi che mimano strutturalmente i substrati peptidici nello stadio di transizione.
Altri inibitori interagiscono con i residui catalitici e rimuovono una molecola d acqua
strutturale.
I più rilevanti inibitori della proteasi sono:
O
NH
O
H
N
N
N
H
O
O
N
H
OH
H
NH2
saquinavir
N
OH
OH
H
N
N
N
H2O
O
HN
O
indinavir
O
N
S
O
H
N
N
S
N
H
N
H
O
OH
ritonavir
O
N
S
O
O
NH
O
H3C
HO
N
H
N
H
S
OH
O
OH
H
Nelfinavir (AG1343)
Meccanismo d azione
Gli inibitori non competitivi non si sono dimostrati efficaci, percui la maggior parte dei
farmaci sono inibitori competitivi che imitano gli stadi transizionali della proteasi.
Impieghi terapeutici
Questi inibitori della proteasi bloccano la maturazione virale e quindi sono attivi sia nella
fase acuta sia nella fase cronica dell infezione cellulare.
Sono privi di attività contro le proteasi di mammifero.
La terapia di associazione con analoghi didesossinucleosidici e inibitori della proteasi si è
dimostrata sinergica in vitro, producendo un miglioramento di media entità.
Dopo un anno di trattamento, circa il 50% dei pazienti mostra resistenza, generalmente
di grado moderato (da 3 a 10 volte meno sensibili).
Sono stati descritti potenti effetti antivirali, associati ad una riduzione di 10-100 volte del
titolo plasmatico di RNA virale.
Sono in corso alcuni studi clinici per valutare l efficacia di terapie di combinazione tra
diversi inibitori della proteasi ed altri farmaci, compresi gli analoghi nucleosidici.
Farmacocinetica
Benché la loro biodisponibilità orale sia bassa (il saquinavir, ad esempio, 4%), di solito
viene assorbita una quantità di farmaco sufficiente per produrre concentrazioni
plasmatiche superiori alle concentrazioni inibitorie minime, in vitro.
In vivo, alcuni di questi inibitori della proteasi hanno scarsa attività a causa di un
legame molto esteso alle proteine plasmatiche, in particolare all alfa-1 glicoproteina
acida, della bassa biodisponibilità e/o di un emivita plasmatica molto breve.
Vengono metabolizzati nel fegato, dal citocromo P450 e nel tratto gastrointestinale.
Effetti collaterali: Iperglicemia, lipodistrofia, ipercolesterolemia, possibile aumento di episodi
emorragici in pazienti emofiliaci
Tossicità
Questi farmaci, dei quali si sta proseguendo lo sviluppo, sono privi di tossicità apprezzabile.




![Lezione 15 Virus [modalità compatibilità]](http://s1.studylibit.com/store/data/000771737_1-84b1cca561c5813066d1b76125338a98-300x300.png)