Capitolo 16 • Atresia polmonare con difetto interventricolare
143
Capitolo 16
Atresia polmonare
con difetto interventricolare
Descrizione della lesione
Una certa percentuale di pazienti (~10%) con tetralogia di Fallot presenta atresia polmonare anziché stenosi polmonare. Oltre al difetto interventricolare (DIV) non restrittivo e alla deviazione antero-superiore del setto
infundibolare, c’è assenza di comunicazione diretta tra la cavità ventricolare destra e il tronco polmonare. La mancata comunicazione può essere a
livello sottovalvolare/muscolare (più comune) o valvolare. Un’altra caratteristica di questa patologia è la complessa alterazione del letto vascolare
polmonare e della sua irrorazione. Questa patologia è stata definita come
“atresia polmonare con difetto interventricolare”, ma si tratta di un termine ampio che può includere anche la trasposizione delle grosse arterie, la
trasposizione congenitamente corretta e la doppia entrata ventricolare. Tali
patologie sono però differenti dalla “tetralogia di Fallot con atresia polmonare” e non saranno trattate in questo capitolo.
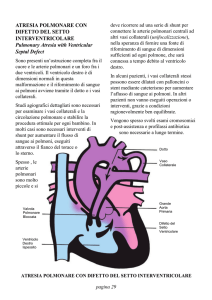
L’anatomia del letto vascolare polmonare e il flusso polmonare possono
essere di tre tipi: una è descritta come circolazione unifocale, due come
multifocali (Fig. 16.1). In una circolazione unifocale tutte le arterie intrapolmonari sono connesse a delle arterie polmonari confluenti e non stenotiche la cui fonte di flusso è data da un dotto arterioso pervio (Fig.
16.1b). Quando invece diversi segmenti polmonari sono irrorati da più
fonti di flusso si parla di circolazione multifocale. Nel primo tipo le arterie polmonari sono confluenti ma tendenzialmente ipoplasiche, con fonte
di flusso da collaterali sistemico-polmonari (Fig. 16.1c). Nell’altro, la fonte di flusso polmonare è data sempre da collaterali sistemico-polmonari (o
MAPCAs, major aorto-pulmonary collateral arteries), ma le arterie polmonari non sono confluenti (Fig. 16.1d).
Queste collaterali originano generalmente dall’aorta discendente toracica opposta all’origine delle arterie intercostali e si estendono fino al punto di partenza delle arterie intrapolmonari a livello segmentario presso l’ilo polmonare. Nei modelli multifocali, le resistenze vascolari polmonari
totali (RVP) sono difficili da quantizzare in quanto spesso ogni segmento
polmonare è irrorato da differenti fonti arteriose. La malattia vascolare
ostruttiva polmonare può svilupparsi in un singolo segmento polmonare
esposto a una pressione sistemica attraverso una collaterale sistemico-polmonare non stenotica. Stenosi naturali delle collaterali, tuttavia, possono
144 Cardiopatie congenite
a
b
AO
AO
AP
AP
AS
AS
AD
AD
VS
VS
VD
VD
Normale
c
Atresia polmonare con DIV
e arterie polmonari confluenti,
irrorate da un dotto arterioso pervio
d
APD
AO
APS
AP
Arterie polmonari confluenti
con irrorazione da collaterali
sistemico-polmonari
Arterie pomonari non confluenti
con irrorazione da collaterali
sistemico-polmonari
Figura 16.1 I tre modelli di anatomia delle arterie polmonari più comuni in pazienti con
tetralogia di Fallot e atresia polmonare. a Normale. b Circolazione unifocale ove tutte le arterie intrapolmonari sono connesse a delle arterie polmonari non ostruite e confluenti, irrorate da un dotto arterioso.Questo è il modello più frequente (85% dei casi).c Circolazione multifocale con arterie polmonari confluenti ma ipoplasiche e irrorate da collaterali sistemicopolmonari. d Circolazione multifocale con flusso ematico da collaterali sistemico-polmonari
e con arterie polmonari non confluenti. AO, aorta; AP, arteria polmonare; AD, atrio destro; AS,
atrio sinistro: VD, ventricolo destro; VS, ventricolo sinistro; APD, arteria polmonare destra: APS,
arteria polmonare sinistra; DIV, difetto interventricolare
proteggere i polmoni dalla pressione sistemica e quindi dallo sviluppo di
vasculopatie polmonari segmentarie. Tipiche sedi di stenosi sono:
• all’origine della collaterale presso l’aorta (forma più comune);
• a livello dell’anastomosi con l’arteria polmonare;
• a livello arteriolare polmonare.
Capitolo 16 • Atresia polmonare con difetto interventricolare
145
Incidenza ed eziologia
L’atresia polmonare con DIV rappresenta circa l’1-2% delle cardiopatie
congenite. Questa malformazione può essere parte della sindrome 22q11
velo-cardiaca (anomalie del viso e dell’orecchio, palatoschisi, ritardo di
sviluppo mentale).
Manifestazioni e decorso in età pediatrica
La diagnosi è formulata nell’infanzia attraverso i sintomi di cianosi, scompenso e dispnea da sforzo. L’ecocardiografia, la risonanza magnetica
nucleare e il cateterismo cardiaco confermano la diagnosi. In generale la
prognosi non è buona, ma variabile in base alla stabilità e all’adeguatezza
del flusso polmonare. Una sopravvivenza prolungata senza chirurgia è rara.
• Se il flusso polmonare è dotto-dipendente, la cianosi e la sintomatologia peggiorano
alla chiusura del dotto. L’infusione di prostaglandine E1 mantiene il dotto arterioso
pervio fino a quando il cateterismo cardiaco e la chirurgia possono essere effettuate.
• I pazienti con adeguato ma non eccessivo flusso polmonare possono sopravvivere fino
all’età adulta senza chirurgia. Questa circolazione bilanciata si sviluppa raramente.
• Più comunemente i pazienti sono stabili ma con una certa inadeguatezza del flusso polmonare. La sopravvivenza a lungo termine di questi pazienti è in relazione
alla chirurgia mirata a incrementare il flusso polmonare.
Esame fisico
• Pazienti non corretti
– Cianosi.
– Assenza di soffio o soffio continuo da dotto arterioso pervio o da flusso attraverso collaterale sistemico-polmonare ai polmoni.
– Secondo tono singolo.
• Pazienti corretti
– In presenza di un condotto valvolato, presenza di soffio sistolico da eiezione polmonare con o senza soffio da insufficienza.
– Segni di scompenso cardiaco destro che suggeriscono una ostruzione a livello
del condotto o delle arterie polmonari.
– Insufficienza aortica (secondaria alla dilatazione dell’anello aortico).
Indagini strumentali utili
• ECG: dilatazione atriale destra e ipertrofia ventricolare destra.
• Radiografia del torace: silhouette cardiaca a zoccolo e solitamente
ridotta vascolarità polmonare.
146 Cardiopatie congenite
• Ecocardiografia: simile alla tetralogia di Fallot ma con assenza di flusso
diretto fra ventricolo destro (VD) e arteria polmonare (AP). Valutare la
funzione del condotto (in pazienti operati), la funzione del VD, la dilatazione dell’anello aortico e l’eventuale presenza di insufficienza aortica.
• Cateterismo cardiaco, risonanza magnetica o TAC: per determinare le
dimensioni e la confluenza delle arterie polmonari, la fonte del flusso
polmonare e le resistenze vascolari polmonari.
Gestione chirurgica
I pazienti sottoposti a procedure chirurgiche palliative e i pazienti non
operati possono essere trattati in maniera conservativa se stabili. Se la sintomatologia peggiora, la chirurgia riparativa può essere presa in considerazione a patto che non sia presente malattia vascolare polmonare irreversibile e che l’anatomia polmonare si presenti favorevole.
Gli obiettivi della chirurgia riparativa sono: chiudere il DIV e ricostruire il tratto di uscita del VD e l’anatomia polmonare. Il raggiungimento di tali obiettivi dipende dall’anatomia.
• L’obiettivo è facilmente raggiungibile quando le dimensioni delle arterie polmonari sono adeguate (≥50% rispetto alla norma) e l’architettura polmonare è preservata (circolazione unifocale). L’approccio chirurgico è la chiusura del DIV e il ripristino di una continuità tra il VD e l’AP con un patch o con un condotto valvolato
(homo- o heterograft).
• In presenza di collaterali o arterie polmonari ipoplasiche la correzione durante un
unico intervento chirurgico non è possibile. Una o più procedure chirurgiche palliative vengono eseguite al fine di stimolare la crescita delle arterie polmonari. Sono
possibili tre opzioni.
– Uno shunt sistemico-polmonare (es.: shunt di Blalock-Taussig).
– Ricostruzione del tratto di efflusso del ventricolo destro lasciando il DIV aperto
o quantomeno fenestrato per stimolare una crescita più uniforme delle arterie
polmonari.
– Uno shunt centrale tra l’aorta ascendente e l’arteria polmonare. Questo shunt
deve essere della misura corretta per stimolare la crescita polmonare senza sottoporre i polmoni a un eccessivo flusso ematico.
Una volta che le arterie polmonari hanno raggiunto dimensioni accettabili e il flusso
polmonare è adeguato, può essere presa in considerazione la correzione completa.
Questa comprende la chiusura del DIV e la ricostruzione del tratto di efflusso del VD.
La correzione non può essere eseguita se le resistenze vascolari polmonari e la pressione sistolica del VD rimangono significativamente elevate.
• Il gruppo di pazienti più difficile da sottoporre a correzione si presenta con arterie
polmonari piccole e non confluenti, con svariate collaterali sistemico-polmonari
che irrorano differenti regioni dei polmoni. Un possibile approccio a questa complessa anatomia è dato da:
– ottenimento del massimo numero di segmenti polmonari perfusi da una arteria
polmonare centrale creata chirurgicamente connettendo le collaterali a un’unica fonte di flusso polmonare (unifocalizzazione) che, a sua volta, può richiedere
Capitolo 16 • Atresia polmonare con difetto interventricolare
147
o meno l’inserimento di uno shunt sistemico-polmonare. Questo approccio può
rendere necessari svariati interventi chirurgici;
– in seguito queste neo-arterie polmonari verranno connesse al VD attraverso un
condotto e il DIV verrà chiuso.
Le procedure di emodinamica interventistica includono l’occlusione, la
dilatazione e lo stent di arterie polmonari e/o di collaterali. La valvuloplastica con palloncino per stenosi del condotto o della sua valvola è generalmente inefficace.
Circa il 25-50% dei pazienti è adatto a questo approccio chirurgico riparativo. Gli altri non necessitano di procedure chirurgiche oppure, in proporzione limitata, si possono considerare candidati per un trapianto cuorepolmone, anche se i risultati di tale procedura sono generalmente scarsi.
Reinterventi
Le sequele a lungo termine variano in funzione del tipo di chirurgia palliativa
o correttiva. Generalmente il 10-15% dei pazienti necessita di un reintervento
nei primi venti anni. La sostituzione di un condotto polmonare è l’evento più
frequente (la libertà dal reintervento per questi pazienti è del 55% a 10 anni e
del 32% a 20 anni). Il reintervento potrebbe rendersi necessario per:
• Revisione del tratto di efflusso del VD
– Stenosi infundibolare residua: resezione aggiuntiva o posizionamento di un nuovo condotto tra il VD e l’AP quando la pressione sistolica nel VD è >75% della pressione sistemica, specialmente in presenza di disfunzione del VD.
– Sostituzione di un condotto valvolato in presenza di ostruzione o
insufficienza della valvola con progressiva dilatazione delle sezioni
destre del cuore.
– Meno frequentemente aneurisma del tratto di efflusso del VD.
• Sostituzione della valvola aortica per insufficienza aortica: un’insufficienza aortica progressiva può svilupparsi con maggiore frequenza nella tetralogia di Fallot con atresia polmonare più che nella tetralogia di
Fallot con stenosi polmonare.
• Plastica della valvola tricuspide per insufficienza dovuta alla progressiva
dilatazione delle sezioni destre del cuore. Ciò è generalmente associato a
ostruzione significativa del tratto di efflusso del VD o alla sua disfunzione.
• DIV residuo se causa di concomitante sovraccarico di volume delle
sezioni sinistre del cuore.
• Aritmie atriali refrattarie che possono richiedere ablazione chirurgica
con radiofrequenza o procedura MAZE. Generalmente la chirurgia delle aritmie viene eseguita quando c’è indicazione elettiva al reintervento.
Complicanze tardive
Le cause di morte in questa tipologia di pazienti sono prevalentemente
cardiache e includono:
148 Cardiopatie congenite
•
•
•
•
chirurgia cardiaca (43%);
aritmie;
chirurgia non cardiaca;
scompenso cardiaco cronico (flusso polmonare eccessivo, aumentate resistenze vascolari polmonari, disfunzione del VD, insufficienza aortica);
• emottisi;
• morte improvvisa;
• endocarditi;
• aumento della cianosi (riduzione del flusso polmonare da stenosi delle
collaterali, stenosi dell’AP, o aumento delle resistenze vascolari polmonari).
È interessante notare come la chirurgia cardiaca sia la prima causa di
sopravvivenza e miglioramento della cianosi in questi pazienti ma al tempo stesso la prima causa di mortalità.
Nonostante la presenza di forme molto complesse con anomale fonti di
flusso polmonare, se i presupposti emodinamici sono favorevoli (DIV
chiuso, corretta ricostruzione del tratto di efflusso del ventricolo destro e
resistenze vascolari polmonari vicine alla norma) la sopravvivenza dei
pazienti sottoposti a chirurgia correttiva è in genere simile a quella della
tetralogia di Fallot.
La sopravvivenza scende a livelli molto più bassi per le forme più complesse e per le forme con risultati chirurgici non ottimali (sopravvivenza
del 61% a 20 anni per pazienti sottoposti a procedure palliative). Il trapianto cuore-polmone può essere considerato una opzione quando le altre
opzioni chirurgiche falliscono, ma occorre ricordare che questa procedura
è estremamente difficile dal punto di vista tecnico in presenza di numerose collaterali sistemico-polmonari.
Raccomandazioni per il follow-up
I pazienti con tetralogia di Fallot e atresia polmonare dovrebbero essere
seguiti con regolarità da cardiologi esperti in cardiopatie congenite dell’adulto. Eventuali variazioni della sintomatologia con dispnea, aumento della cianosi, variazioni all’auscultazione, scompenso cardiaco o aritmie
necessitano di particolare attenzione.
Raccomandazioni per l’endocardite
La profilassi dell’endocardite è raccomandata in tutti questi pazienti lungo tutta la vita.
Attività fisica
Nei pazienti con eccellente emodinamica persiste un certo grado di limitazione delle capacità fisiche. I pazienti con una emodinamica non otti-
Capitolo 16 • Atresia polmonare con difetto interventricolare
149
male sono soggetti a limitazioni fisiche importanti. Per questi ultimi sono
sconsigliate attività fisiche “estreme” e competitive.
Gravidanza
Il rischio in gravidanza è basso per pazienti sottoposte a chirurgia, con
buona emodinamica e assenza di aritmie. Il rischio aumenta in presenza
di ipossiemia (saturazione di ossigeno <85%), ipertensione polmonare,
disfunzione ventricolare, segni di scompenso cardiaco e aritmie. Le pazienti con sindrome di DiGeorge dovrebbero essere sistematicamente controllate prima di una gravidanza.
Elementi clinici chiave
• Nonostante l’atresia della polmonare con DIV assomigli per molti aspetti alla tetralogia di Fallot, la complessità delle anomalie delle arterie polmonari e del flusso polmonare la rendono una malformazione molto più complessa da gestire.
• La sopravvivenza è scarsa senza intervento chirurgico.
• La gestione chirurgica varia in funzione della complessità della circolazione polmonare. Può limitarsi al semplice impianto di un condotto tra VD e AP o può richiedere svariate procedure chirurgiche per connettere differenti segmenti polmonari
a un’unica fonte di flusso arterioso (unifocalizzazione) prima dell’impianto di un
condotto tra il VD e l’AP.
• I condotti tra VD e AP dovranno essere sostituiti.
• La mortalità e la morbilità sono in relazione a:
– complessità anatomica;
– completezza nella correzione;
– funzionalità del ventricolo destro.
Letture consigliate
Bull K, Somerville J, Ty E & Spiegelhalter D (1995) Presentation and attrition in
complex pulmonary atresia. Journal of the American College of Cardiology, 25,
491-499
Cho JM, Puga FJ, Danielson GK et al (2002) Early and long-term results of the
surgical treatment of tetralogy of Fallot with pulmonary atresia, with or
without major aortopulmonary collateral arteries. Journal of Thoracic and
Cardiovascular Surgery, 124, 70-81
Clarke DR & Bishop DA (1995) Ten year experience with pulmonary allografts in
children. Journal of Heart Valve Disease, 4, 384-391
Dearani JA, Danielson GK, Puga FJ et al (2003) Late follow-up of 1095 patients
undergoing operation for complex congenital heart disease utilizing pulmonary ventricle to pulmonary artery conduits. Annals of Thoracic Surgery, 74,
399-411
150 Cardiopatie congenite
Leonard H, Derrick G, O’Sullivan J & Wren C (2000) Natural and unnatural
history of pulmonary atresia. Heart, 84, 499-503
Murthy KS, Rao SG, Naik SK, Coelho R, Krishnan US & Cherian KM (1999)
Evolving surgical management for ventricular septal defect, pulmonary atresia,
and major aortopulmonary collateral arteries. Annals of Thoracic Surgery, 67,
760-764
Reddy VM, McElhinney DB, Amin Z et al (2000) Early and intermediate outcomes after repair of pulmonary atresia with ventricular septal defect and major
aortopulmonary arterial collateral arteries. Circulation, 101, 126-137