Tesina di Neurofisiologia Cellulare.
Arianna Racca, Irene Coppola, Federica Cordella.
Autofagia: meccanismo molecolare e ruolo nel morbo di Alzheimer
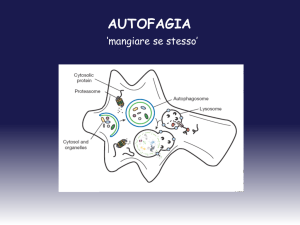
L’autofagia
Il corretto equilibrio tra sintesi e degradazione delle biomolecole e degli organelli è essenziale per il
differenziamento, la crescita e la sopravvivenza delle cellule. Nelle cellule eucariotiche le due
principali vie di degradazione e riciclo dei componenti citoplasmatici sono l’autofagia e il sistema
ubiquitina-proteasoma. Quest’ultimo degrada le proteine danneggiate e gli aggregati proteici
etichettati dall’ubiquitina. L’autofagia invece è un processo catabolico lisosomiale, ubiquitario ed
evolutivamente conservato, deputato non solo alla degradazione di proteine e aggregati proteici ma
anche di interi organelli intracellulari (Moreira et al., 2010; Hu et al. 2014).
Esistono tre diverse forme di autofagia: la microautofagia, l’autofagia mediata da chaperon (CMA) e
la macroautofagia. Durante la microautofagia, il materiale citoplasmatico da rimuovere è incluso nel
lisosoma tramite invaginazione o protrusione della membrana di questo organello (Nikoletopoulou et
al., 2015). Nell’autofagia mediata da chaperon, le proteine da degradare sono riconosciute dai
chaperon grazie ad una specifica sequenza amminoacidica e poi sono traslocate attraverso la
membrana lisosomiale. Invece la macroautofagia prevede la formazione di una cisterna a singola
membrana detta fagoforo che, mano a mano che include i substrati da eliminare, si ingrandisce fino
a sigillarsi alle due estremità formando una vescicola a doppia membrana detta autofagosoma.
Quando è maturo l’autofagosoma ha un diametro di circa 0,5-1 µm e si fonde con il lisosoma
esponendo alle idrolasi il proprio contenuto costituito da proteine e interi organelli. Gli amminoacidi
e i lipidi così ottenuti sono rilasciati nel citoplasma e sono riutilizzati dalla cellula per la biosintesi di
nuove macromolecole o per ottenere energia (Feng et al., 2014; Hu et al., 2014; Xilouri & Stefanis,
2015). L’autofagosoma può seguire due strade per arrivare al lisosoma: può direttamente fondersi
con tale organulo e formare un autolisosoma, oppure prima di raggiungerlo può unirsi a un endosoma
e formare un anfisoma. La macroautofagia può essere un processo selettivo, poiché i substrati possono
essere riconosciuti grazie a recettori specifici che mediano il sequestro di organuli danneggiati, degli
aggregati proteici e di patogeni (Parzyc & Klionsky, 2014; Feng et al., 2014).
La macroautofagia (o più semplicemente autofagia) è espressa in maniera costitutiva in tutte le cellule
eucariotiche, dove agisce nel turnover di proteine e organuli contribuendo al mantenimento
dell’omeostasi cellulare. Questo processo può anche essere stimolato in risposta a segnali ormonali e
fattori di crescita o in situazioni di stress cellulare, come la deprivazione di nutrienti, lo stress
ossidativo, l’accumulo di proteine danneggiate o l’irradiazione. In mancanza di nutrienti
l’eliminazione di proteine ed organelli non essenziali e il riciclo delle loro componenti rappresenta
un’importante possibilità di sopravvivenza per le cellule. Oltre all’adattamento al digiuno, l’autofagia
svolge molteplici funzioni: è coinvolta nel rimodellamento durante lo sviluppo embrionale e nel
differenziamento cellulare ed è stata proposta come meccanismo anti-invecchiamento, in quanto è
coinvolta nell’eliminazione di membrane, proteine e organelli danneggiati dalle specie reattive
dell’ossigeno che si accumulano con l’età. Inoltre l’autofagia agisce come meccanismo di immunità
innata nel controllo dell’infezione da parte di batteri o virus (Piacentini, 2014).
Una deregolazione dell’autofagia porta ad un’insufficiente o eccessiva degradazione dei costituenti
citoplasmatici e ciò può intaccare la stabilità e l’integrità cellulare (Ariosa & Klionsky, 2016). In
particolare, le cellule neuronali, a causa della loro peculiare morfologia e della loro natura postmitotica, risultano essere molto sensibili all’accumulo di aggregati proteici o di organelli
disfunzionali (Tooze & Schiavo, 2008). Negli ultimi anni sempre più lavori hanno mostrato una
connessione tra deregolazioni dell’autofagia e diverse malattie neurodegenerative, come l’Alzheimer,
il Parkinson, la Corea di Huntington e la sclerosi laterale amiotrofica (SLA) (Hu et al., 2014). Inoltre,
1
nel sistema nervoso l’autofagia svolge un ruolo anche in processi altamente specializzati come la
mechanisms of disease
plasticità sinaptica e la sazietà (Tang et al., 2002; Hernandez et al., 2012).
Initiation
Vesicle elongation
Maturation
Docking and fusion
Vesicle breakdown
and degradation
Lysosome
Lysosomal
acid hydrolases
Isolation membrane
(phagophore)
Autophagosome
Autolysosome
Figure 1. Phases of the Autophagic Pathway.
The autophagic
pathway
proceeds through
including initiation (formation of a preautophagosomal structure leading to
Fig.
1 Gli stadi
dell’autofagia
(Choiseveral
et al.,phases,
2013).
an isolation membrane, or phagophore), vesicle elongation, autophagosome maturation and cargo sequestration, and autophagosome–
OLOR FIGURE
lysosome fusion. In the final stage, autophagosomal contents are Cdegraded
by lysosomal acid hydrolases and the contents of the autolysoDraft 1
1/11/2013
some are released for metabolic recycling.
Author
Choi_ra1205406
Il macchinario molecolare dell’autofagia
Il processo della macroautofagia può essere diviso in diversi stadi: induzione e nucleazione del
fagoforo, espansione e maturazione dell’autofagosoma, legame e fusione dell’autofagosoma con il
During infection, autophagy assists in the imF unc
ions of Au t oph
lisosoma (Fig.
1).tL’attuazione
di agy
ciascuno mune
di questi
stadi
è garantita
da proteine
codificate da un
response
by degrading
intracellular
bacteAUTHOR PLEASE NOTE:
7,14,20
Autophagy
acts as a survival
mechanism
under
ria (autophagy-related
and viruses (i.e., xenophagy).
Autophagy
gruppo di
geni altamente
conservati
detti
ATG
genes).
conditions
stress, maintaining
cellular
integ- contributes
to the suppression
of inflammation,
Induzione.
Nei of
mammiferi
l’inizio
dell’autofagia
è mediato
dal complesso
della chinasi ULK1 che
rity by regenerating metabolic precursors and including the down-regulation of both interferon
induce la
formazione
fagoforo
reclutando
altre proteine
ATG.andQuesto
complesso è formato da
1-3 This process
clearing
subcellulardel
debris.
con- responses
to viral infection
proinflammatributes to basal cellular
homeostasis,
tory cytokine
responses(con
to invading
pathogens
due protein-chinasi,
ULK1andetissue
ULK2,
dalla proteina
RB1CC1
funzione
strutturale), da ATG13
as
well
as
developmental
regulation
in
higher
orand
the
inhibition
of
inflammasome-dependent
(con funzione regolatoria) e dalla proteina accessoria ATG101 (Hara et al., 2008; Hosokawa et al.,
ganisms, and can affect pathogenesis (Fig. 3 and maturation and secretion of proinflammatory
2009 a; Mercer
et al.,
2009;). Ilhave
sensore
dei
mTORC1
(complesso
1 della chinasi target della
Table 2). Recent
investigations
identified
se-nutrienti
cytokines (e.g.,
interleukin-1β
and interleukin-18),
lectivity
the recognitionè of
autophagy
sub- through
the preservation
of mitochondrial
funcrapamicina
deiinmammiferi)
uno
dei principali
attori
nella regolazione
dell’autofagia.
In condizioni
strates (previously considered to occur through tion.33-35 Autophagic proteins may also regulate
di disponibilità
di nutrienti, mTORC1 mantiene l’autofagia ad un livello basale poiché fosforila
nonspecific sequestration of cytosol), which is an unconventional pathway for secretion of cyto27 Autoph36
ATG13 orchestrated
e ULK1/2,
inibendo lafactors.
capacità
di autofosforilazione
delle
due chinasi. Durante condizioni
by cargo-specific
kines (e.g., interleukin-1β).
agy
participates
in
the
turnover
of
mitochondria
Autophagy
can
also
play
crucial
roles
in
di scarsità di nutrienti, mTORC1 è rapidamente rimosso dal complesso della
chinasi
ULK1 e ciò porta
(through the selective process of mitophagy) and adaptive immune responses such as antigen
alla defosforilazione
di
entrambe
le
proteine
ULK1/2
e
alla
loro
attivazione.
In
questo
stato attivato
other organelles (e.g., endoplasmic reticulum presentation and lymphocyte development.14,20
27,28
ULK1/2andfosforilano
RB1CC1
stesse e ciò
il proseguimento degli stadi
peroxisomes). ATG13,
Furthermore,
autophagyeis se
Autophagosomes
can permette
fuse with major-histocominvolved
in the clearance(Hosokawa
of polyubiquitinated
pro- 2009
patibility-complex
(MHC)
class II loading comsuccessivi
dell’autofagia
et al.,
b; Jung
et
al.,
2009).
tein aggregates (i.e., aggrephagy), which accu- partments.37 In addition, autophagy can faciliNucleazione.
L’induzione dell’autofagia comporta innanzitutto la formazione del fagoforo. L’origine
mulate during stress, aging, and disease owing tate the generation of a self-tolerant T-cell
29 repertoire.
dei lipidito perturbations
necessari per
assemblare
vescicola
nonHigh
è chiara:
potrebbero
derivare
dai mitocondri,
in protein
structure ortale
folding.
constitutive
expression of
auAutophagy
also beendalla
implicated
as a regula- plasmatica
tophagy in thymic
epithelial
cells endoplasmatico.
delivers endall’apparato
delhasGolgi,
membrana
o
dal
reticolo
Dati recenti
tor of lipid metabolism (i.e., lipophagy).30
dogenous proteins to MHC class II molecules
suggeriscono
un contributo
principale
da parte
del RE,toinCD4+
cuiT-cell
il fagoforo
sembra formarsi da una
Autophagy
primarily acts as
a protective mechand contributes
selection.38 These
particolare
regione
questo
detta examples
omegasoma
et al., 2008;
Yla-Anttila
et al., 2009).
anism
that maydiprevent
cell organello
death. Interaction
suggest(Axe
that autophagy
can affect
the
between
regulatory
elements
of
both
autophagy
regulation
of
inflammation
and
immune-system
Il processo di nucleazione e assemblaggio del fagoforo è regolato da due componenti principali: il
and apoptosis (e.g., the inhibitory interaction function.
complesso
della
fosfatidilinositolo
3 chinasi di classe III (PI3K3) e la chinasi ULK1. Il complesso
between
BCL-2
and Beclin 1 and the interaction
31,32
between
LC3B andil Fas)
however,
della PI3K3
produce
lipide suggests,
fosfatidilinositolo
(3,4,5)
-trifosfato
(PIP3),
Au t oph
agy in Dise
a se che è il segnale necessario
complex cross-talk
between
these two
processes.
al reclutamento
di altre
proteine
ATG.
Nei Cancer
mammiferi esistono almeno tre tipi di complessi della
Although neither excessive autophagy nor imPI3K3: paired
i complessi
e influence
BENC1-PIK3C3-PIK3R4autophagy hasBENC1-PIK3C3-PIK3R4-ATG14-AMBRA1
been proved to be a direct Autophagy may exert a multifactorial
cause of cell death, che
both may
be associated
with on agendo
the initiation
progression
cancer,
as
UVRAG-SH3GLB1
attivano
l’autofagia
in and
diversi
stadiofdel
processo
e il complesso
3
apoptosis
in
some
model
systems.
well
as
on
the
effectiveness
of
therapeutic
interBENC1-PIK3C3-PIK3R4-UVRAG-RUBCN che invece inibisce l’autofagia (Fig. 2) (Kametaka et
al., 1998; Kihara et al., 2001; Furuya
al.,nejm.org
2005).february
La proteina
beclina 1 (BECN1) agisce da653
adattatore
n engl j medet
368;7
14, 2013
molecolare permettendo l’interazione
di
diverse
proteine
ATG
con
la
chinasi
PIK3C3
e la sua
The New England Journal of Medicine
Downloaded from nejm.org at UNIVERSITY OF TENNESSEE - KNOXVILLE on February 18, 2013. For personal use only. No other uses without permission.
proteina regolatoria PIK3R4,
in modo che si possa formare uno dei due complessi attivatori o quello
Copyright © 2013 Massachusetts Medical Society. All rights reserved.
inibitorio. In condizioni di disponibilità di nutrienti l’autofagia è inibita, perché la proteina antiapoptotica BCL2 interagisce con BECN1, impedendogli di associarsi al complesso della PI3K3. Al
contrario, in condizioni di carenza di nutrienti BCL2 si dissocia da BECN1, permettendo a
quest’ultimo di svolgere la sua funzione nella formazione del complesso PI3K3. La chinasi ULK1
partecipa alla regolazione del processo di nucleazione poiché rende disponibile la proteina AMBRA1,
Fig #
Title
1
Autophagy in Human
Health and Disease
DE
ME
Artist
Pub Date
Longo
Forti
Williams
2/14/2013
Figure has been redrawn and type has been reset
Please check carefully
2
permettendo la formazione del complesso attivatore BENC1-PIK3C3-PIK3R4-ATG14-AMBRA1
(Ariosa & Klionsky, 2016).
Espansione. L’espansione del fagoforo dipende da due reazioni di coniugazione simili
all’ubitiquitinazione. La prima reazione coniuga ATG12 a ATG5 attraverso l’azione di ATG7 e di
ATG10 (rispettivamente, enzimi simili a E1 ed E2), dopo di che il complesso ATG12-ATG5 si
associa a ATG16L e funge da ligasi (enzima simile a E3) nella seconda reazione di coniugazione
(Tanida et al., 2001; Mizushima et al., 2003; Ariosa & Klionsky, 2016). Quest’ultima prevede la
coniugazione della proteina LC3 con il lipide di membrana fosftatidiletanolammina (PE). Prima di
essere coniugata al PE il residuo di arginina al C-terminale di LC3 deve essere rimosso, in modo che
venga esposto il1220residuo di glicina necessario per il legame con J ilMol Med
fosfolipide.
La proteina così
(2016) 94:1217–1227
modificata vieneFig. 2riconosciuta
e processata dall’enzima di attivazione simile a E1 (ATG7) e
The yeast Atg1 induction
a Nutrient-rich
machinery. a Under nutrient-rich
Atg1
dall’enzima simile
a
E2
(ATG3).
Il legame di PE e LC3 è catalizzato
dal complesso simile a E3
P
conditions, TORC1 actively
P
Autophagy
TORC1
phosphorylates Atg13, thereby
Atg13di coniugazione.
(ATG12-ATG5-ATG16)
prodotto
in
seguito
alla
prima
reazione
Sebbene la
inhibiting Atg1 activity. b During
P P
nitrogen starvation, TORC1’s
presenza di questi
sistemi
di coniugazione sia essenziale per l’autofagia, la loro azione nel processo
kinase
function is suppressed,
Atg17
allowing for Atg13 to become
Atg29
di maturazione del
fagoforo
2016). La proteina ATG9
partially dephosphorylatednon è ancora stata chiarita (Ariosa & Klionsky,
Atg31
resulting in the
Atg1. In
sembra svolgere autophosphorylation
un ruolo ofchiave
nell’ulteriore
espansione del fagoforo, infatti è stato ipotizzato che
b Starvation
this induced form, Atg1 serves as
P
a master
regulator and cues in
questa proteina sia
responsabile
del
trasporto
dei
lipidi verso il fagoforo
(Young et al., 2006; Webber
Atg1
other Atg proteins to localize to
P
the
growing
phagophore.
et al., 2007).
Autophagy
TORC1
Atg13
Inhibitory and stimulatory
P
are shown in
P P
Fusione. Anche phosphorylations
se
il
meccanismo
attraverso
il
quale
l’autofagosoma
si
fonde
con il lisosoma non è
Atg17
pink and green, respectively
Atg29
Atg31
ben conosciuto, lo sono le proteine che accompagnano tale stadio.
Queste includono le proteine
P
SNARE, il complesso HOPS e membri della famiglia ESCRT e sono coinvolte anche in altri
meccanismi di trasporto che terminano nel lisosoma.
Degradazione ed efflusso.
Una voltaBCL2che PIK3C3
la PIK3R4
fusione è avvenuta, la membrana interna
a
BECN1
BECN1
dell’autofagosoma e il suo contenuto
sono
degradati
da
varie idrolasi.
I metaboliti prodotti da questo
PIK3C3 PIK3R4
BCL2
Starvation
BECN1
MAPK8,
DAPK
processo includono amminoacidi e lipidi che sono attivamente
pompati nel citoplasma e riutilizzati
BCL2
dalla cellula.
P
P
P
P
b
P
BECN1
PIK3C3 PIK3R4
c
P
ATG14
AMBRA1
AMBRA1
BECN1
PIK3C3
ATG14
PIK3R4
P
Autophagy
SH3GLB1
UVRAG
BECN1
PIK3C3 PIK3R4
P
BECN1
PIK3C3 PIK3R4
UVRAG
SH3GLB1
UVRAG
UVRAG
P
BECN1
PIK3C3 PIK3R4
RUBCN
Fig. 3 The class III PtdIns3K complex and the regulation of
autophagosome nucleation. PtdIns3P is an important signaling molecule
that allows for the nucleation and expansion of the phagophore. In
mammals, BECN1 is a key regulatory player in the activation of
PtdIns3P synthesis carried out by PIK3C3 and its regulatory partner
protein, PIK3R4. a Under non-inducing conditions, BECN1 is associated
with BCL2 preventing its localization to the PAS. Upon nitrogen
P
RUBCN
Autophagy
BECN1
PIK3C3 PIK3R4
starvation, BECN1 and BCL2 become phosphorylated by DAPK and
MAPK8/JNK1, respectively, allowing them to dissociate. b, c When
autophagy is induced, BECN1 is released from BCL2 and is then free
to bind to a wide array of proteins that can suppress or stimulate autophagy. BECN1 bound to ATG14, AMBRA1, and UVRAG–SH3GLB1 results in autophagy activation whereas when bound to UVRAG-RUBCN,
autophagy is inhibited
Fig. 2 Formazione dei tre complessi della PI3K3 (Ariosa & Klionsky, 2016)
L’autofagia nei neuroni e nel sistema nervoso
I neuroni presentano una peculiare architettura cellulare in cui si identifica un corpo cellulare o soma,
dal quale si estroflettono corti prolungamenti detti dendriti e un lungo prolungamento detto assone.
La funzionalità dei neuroni si basa principalmente sull’efficienza del trasporto attivo delle
macromolecole dal corpo cellulare agli assoni e ai dendriti, perciò queste cellule sono molto sensibili
all’accumulo di organelli danneggiati e aggregati citoplasmatici che può anche portare all’insorgenza
di numerose malattie neurodegenerative (Tooze & Schiavo, 2008).
In risposta alla loro complessità morfologica i neuroni hanno dovuto escogitare alcuni meccanismi
peculiari per poter eseguire l’autofagia in maniera efficiente ed efficace. Innanzitutto gli
autofagosomi si possono formare nelle terminazioni assonali per poi essere trasportati verso il corpo
3
cellulare lungo i microtubuli (trasporto retrogrado a lunga distanza). Durante questo movimento
processivo e unidirezionale gli autofagosomi si fondono con i lisosomi che incontrano lungo la strada.
In caso sia necessario eliminare il surplus di proteine e organelli in maniera molto più rapida,
l’autofagia può anche avvenire interamente alla periferia dei neuroni. Inoltre, la mito-autofagia
(autofagia di mitocondri disfunzionali) può avvenire anche in maniera transcellulare; infatti questi
organuli possono essere accumulati a livello degli assoni, dove formano delle protrusioni sempre più
pronunciate che alla fine vengono inglobate e degradate dagli astrociti vicini. Infine meccanismi di
regolazione ancora poco chiari fanno si che l’autofagia nei neuroni sia un processo spazialmente
compartimentalizzato, poiché impediscono agli autofagosomi che hanno raggiunto il soma o che si
sono formati nel soma di dirigersi nell’assone. E¢ da sottolineare che questi processi possono non
avvenire contemporaneamente e che possono essere impiegati dal neurone in risposta a diversi stimoli
di sviluppo o di stress (malattie o danni cellulari) (Ariosa & Klionsky, 2016).
Studi recenti hanno dimostrato che l’autofagia nei neuroni è attiva a livello basale e partecipa al
mantenimento dell’omeostasi cellulare e che, quando invece è stimolata in risposta a stress cellulari,
oltre ad avere effetti citoprotettivi, può determinare la morte cellulare. Infatti in presenza di gravi
danni neuronali l’eccessiva induzione dell’autofagia porta alla morte cellulare, detta morte cellulare
programmata di tipo II (PCD II) o morte cellulare autofagica (ACD). La dicotomia del ruolo svolto
dall’autofagia è il risultato della complessa interazione tra il pathway autofagico e quello apoptotico
(Hu et al., 2014).
Autofagia e malattie del sistema nervoso
I neuroni presentano una spiccata sensibilità all’accumulo di strutture cellulari danneggiate e di
aggregati proteici che, se non sono prontamente rimossi, possono indurre citotossicità e necrosi
(Komatsu et al., 2006; Frake et al., 2015; Kiriyama et al., 2015). Quindi l’autofagia è essenziale per
il mantenimento dell’omeostasi funzionale dei neuroni in generale e degli assoni in particolare, infatti
una sua deregolazione può portare a degenerazione e distrofia assonale (Komatsu et al., 2007).
L’aumento della concentrazione di aggregati proteici è principalmente causato da alterazioni del
processo autofagico oltre ad essere dovuto a una risposta cellulare, in cui le proteine mutate vengono
tagliate e i peptidi risultanti si associano fra loro (Martinez-Vicente & Cuervo, 2007). L’accumulo di
aggregati proteici e di strutture cellulari è alla base di molte patologie neurodegenerative, come il
morbo di Parkinson, la Corea di Huntington e il morbo di Alzheimer. Gli aggregati proteici
presentano una diversa composizione in base al tipo di malattia e la loro presenza è correlata anche a
disfunzioni del processo autofagico, ad esempio nell’Alzheimer si ha la perdita dell’attività del
lisosoma, mentre il morbo di Parkinson è legato a difetti della mitofaga e nella SLA si hanno difetti
nel traffico vescicolare.
Autofagia e Alzheimer
Il morbo di Alzheimer è la causa più comune di demenza senile. Questa patologia neurodegenativa è
caratterizzata da un’iniziale perdita di memoria che progredisce con il totale deterioramento delle
capacità cognitive. Una delle caratteristiche neuropatologiche associate a questa malattia è la perdita
diffusa di neuroni causata dall’accumulo a livello extracellulare di placche amiloidi, che derivano dal
clivaggio della proteina precursore dell’amiloide (APP) e dalla presenza di aggregati neurofibrillari
intracellulari composti da isoforme iperfosforilate di tau, una proteina associata ai microtubuli che
normalmente è solubile. Inoltre la malattia è accompagnata anche da una forte diminuzione della
produzione e rilascio di acetilcolina, che conduce i neuroni alla morte e che nel complesso porta ad
una progressiva atrofia del cervello (Moreira et al., 2010).
Il precursore dell’amiloide (APP) è una proteina trans-membrana a funzione ignota. La maggior parte
dell’APP prodotto viene degradato durante il suo trasporto verso la superficie cellulare ad opera di
tre enzimi: l’α-secretasi, la β-secretasi e la ϒ-secretasi. Quando la ϒ-secretasi taglia l’APP insieme
all’α-secretasi genera un peptide innocuo, chiamato p3. Invece, quando la ϒ-secretasi agisce insieme
alla β-secretasi porta alla formazione di due peptidi neurotossici di 42 e 40 amminoacidi,
4
rispettivamente Aβ40 e Aβ42. Quest’ultimo è considerato il più tossico tra i due poiché tende ad
aggregarsi più velocemente a livello extracellulare, formando il componente chiave delle cosiddette
placche amiloidi. Diversi studi hanno rivelato che in soggetti sani la degradazione dell’APP è a carico
principalmente dell’α-secretasi, mentre in pazienti affetti da MA la proteasi maggiormente attiva è
proprio la β-secretasi. Se il peptide Aβ42 non viene prontamente rimosso, si accumula nel neurone e
ne induce l’apoptosi. Alla morte del neurone, questi peptidi vengono rilasciati nell’ambiente
extracellulare dove tendono ad accumularsi in aggregati neurofibrillari insolubili che via via
divengono più grandi fino ad essere definiti placche amiloidi (Moreira et al., 2010).
Nella genesi del morbo di Alzheimer ha un ruolo chiave anche l’autofagia poiché è convolta nel
metabolismo di tau e Aβ. Studi recenti mostrano che nei cervelli di pazienti affetti da MA sono
presenti accumuli di vescicole autofagiche (Nixon et al., 2005; Boland et al., 2008). All’inizio si
pensava che questo accumulo fosse dovuto ad un’intensa attività autofagica dei neuroni, ma studi
recenti hanno evidenziato che invece è il risultato di un’alterazione nella clearance degli
autofagosomi. Questa alterazione è il risultato di una disfunzione nel processo di maturazione
lisosomiale che genera lisosomi non acidificati e perciò incapaci di fondersi con gli autofagosomi. La
presinilina1 (PS1) è un’aspartato-proteasi molto importante nel pathway autofagico poiché è
indirettamente implicata nella maturazione dei lisosomi. Infatti questa proteina regola l’attività della
ϒ-secretasi che, oltre a produrre i peptidi Aβ, facilita la glicosilazione della subunità V0a1 della pompa
H+-ATPasi del lisosoma che è necessaria all’acidificazione dell’ambiente intra-vescicolare. Nei
pazienti affetti da MA familiare sono state riscontrate mutazioni del gene codificante per la PS1, che
quindi promuovono la deposizione delle placche amiloidi sulla superficie cerebrale sia perché
causano un aumento dell’attività della ϒ-secretasi, sia perché portano ad una mancata acidificazione
del lisosoma inducendo l’accumulo degli autofagosomi a livello neuronale.
Diversi studi eseguiti sul tessuto cerebrale di pazienti affetti dal MA hanno riportato una diminuzione
dei livelli della beclina1 (BECN1), una proteina importante per la formazione dell’autofagosoma
(Small et al., 2005; Pickford et al., 2008). Inoltre è stato osservato che nei topi transgenici la mancanza
di questa proteina causa la deposizione dei peptidi Aβ, mentre una sua over-espressione riduce
l’accumulo di tali peptidi (Spencer et al., 2009). Altri studi invece riportano che l’autofagia modula
anche i livelli della proteina tau. Da questi risultati emerge la possibilità che una stimolazione
dell’autofagia potrebbe avere effetti positivi sul MA andando a diminuire i livelli dei peptidi Aβ e
della proteina tau che caratterizzano questa patologia (Hu et al., 2014).
Quindi il morbo di Alzheimer è una malattia multifattoriale che sembra essere associata anche alla
diminuzione dell’attività autofagica dovuta al rallentamento della clearance degli autofagosomi.
Questo rallentamento è il risultato di una disfunzione del processo di maturazione dei lisosomi e
anche di una minor formazione delle vescicole autofagosomiche causata da una carenza della proteina
beclina1. Il punto di collegamento tra morbo di Alzheimer e autofagia è il ruolo svolto da questo
processo nel metabolismo dei peptidi Aβ e anche in quello della proteina tau.
Conclusioni
L’autofagia è un processo catabolico lisosomiale, ubiquitario nelle cellule eucariotiche, che agisce
nel mantenimento dell’omeostasi cellulare sia in condizioni fisiologiche, sia in condizioni di stress
cellulare. L’autofagia svolge una funzione anche nei processi di differenziamento cellulare e sviluppo
embrionale, nel sistema immunitario e nella plasticità neurale.
Nel 2016 gli studi sui meccanismi molecolari dell’autofagia hanno valso il Premio Nobel per la
Medicina al biologo cellulare giapponese Yoshinori Ohsumi. Infatti diverse patologie, come
cardiomiopatie, alcuni tipi di tumore e malattie neurodegenerative risultano essere connesse a
deregolazioni dell’autofagia. In particolare sempre più evidenze sottolineano il legame tra una
disfunzione del processo autofagico nei neuroni e il morbo di Alzheimer. L’importanza dell’autofagia
va di pari passo con la sua complessità molecolare, che deve essere ancora chiarita nel dettaglio e che
nei neuroni raggiunge il suo massimo grado. Nel complesso la ricerca sui meccanismi molecolari
dell’autofagia potrebbe fornire un nuovo approccio nella prevenzione e nella cura di molte malattie.
5
Bibliografia
Ariosa, A. R., & Klionsky, D. J. (2016). Autophagy core machinery: overcoming spatial barriers
in neurons. Journal of Molecular Medicine, 94(11), 1217-1227.
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., ... &
Ktistakis, N. T. (2008). Autophagosome formation from membrane compartments enriched in
phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. The
Journal of cell biology, 182(4), 685-701.
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., & Nixon, R. A. (2008).
Autophagy induction and autophagosome clearance in neurons: relationship to autophagic
pathology in Alzheimer's disease. The Journal of Neuroscience, 28(27), 6926-6937.
Choi, A. M., Ryter, S. W., & Levine, B. (2013). Autophagy in human health and disease. New
England Journal of Medicine, 368(7), 651-662.
Feng, Y., He, D., Yao, Z., & Klionsky, D. J. (2014). The machinery of macroautophagy. Cell
research, 24(1), 24-41.
Frake, R. A., Ricketts, T., Menzies, F. M., & Rubinsztein, D. C. (2015). Autophagy and
neurodegeneration. The Journal of clinical investigation, 125(1), 65-74.
Furuya, N., Yu, J., Byfield, M., Pattingre, S., & Levine, B. (2005). The evolutionarily conserved
domain of Beclin 1 is required for Vps34 binding, autophagy, and tumor suppressor function.
Autophagy, 1(1), 46-52.
Hara, T., Takamura, A., Kishi, C., Iemura, S. I., Natsume, T., Guan, J. L., & Mizushima, N.
(2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in
mammalian cells. The Journal of cell biology, 181(3), 497-510.
Hosokawa, N., Sasaki, T., Iemura, S. I., Natsume, T., Hara, T., & Mizushima, N. (2009).
Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy, 5(7), 973979. (a).
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., ... & Guan, J. L.
(2009). Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex
required for autophagy. Molecular biology of the cell, 20(7), 1981-1991. (b).
Itakura, E., Kishi-Itakura, C., Koyama-Honda, I., & Mizushima, N. (2012). Structures
containing Atg9A and the ULK1 complex independently target depolarized mitochondria at
initial stages of Parkin-mediated mitophagy. J Cell Sci, 125(6), 1488-1499.
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., ... & Kim, D. H. (2009).
ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery.
Molecular biology of the cell, 20(7), 1992-2003.
Kametaka, S., Okano, T., Ohsumi, M., & Ohsumi, Y. (1998). Apg14p and Apg6/Vps30p form a
protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. Journal of
Biological Chemistry, 273(35), 22284-22291.
6
Kihara, A., Noda, T., Ishihara, N., & Ohsumi, Y. (2001). Two Distinct Vps34
Phosphatidylinositol 3–Kinase complexes function in autophagy and carboxypeptidase Y
Sorting inSaccharomyces cerevisiae. The Journal of cell biology, 152(3), 519-530.
Kiriyama, Y., & Nochi, H. (2015). The function of autophagy in neurodegenerative diseases.
International journal of molecular sciences, 16(11), 26797-26812.
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J. I., Tanida, I., ... & Tanaka, K. (2006).
Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature,
441(7095), 880-884.
Martinez-Vicente, M., & Cuervo, A. M. (2007). Autophagy and neurodegeneration: when the
cleaning crew goes on strike. The Lancet Neurology, 6(4), 352-361.
Mercer, C. A., Kaliappan, A., & Dennis, P. B. (2009). A novel, human Atg13 binding protein,
Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy, 5(5), 649-662.
Mizushima, N., Kuma, A., Kobayashi, Y., Yamamoto, A., Matsubae, M., Takao, T., ... &
Yoshimori, T. (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic
isolation membrane with the Apg12-Apg5 conjugate. Journal of cell science, 116(9), 16791688.
Moreira, P. I., Santos, R. X., Zhu, X., Lee, H. G., Smith, M. A., Casadesus, G., & Perry, G.
(2010). Autophagy in Alzheimer’s disease. Expert review of neurotherapeutics, 10(7), 12091218.
Nikoletopoulou, V., Papandreou, M. E., & Tavernarakis, N. (2015). Autophagy in the
physiology and pathology of the central nervous system. Cell Death & Differentiation, 22(3),
398-407.
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., & Cuervo, A. M.
(2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron
microscopy study. Journal of Neuropathology & Experimental Neurology, 64(2), 113-122.
Orsi, A., Razi, M., Dooley, H. C., Robinson, D., Weston, A. E., Collinson, L. M., & Tooze, S.
A. (2012). Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane
integration, are required for autophagy. Molecular biology of the cell, 23(10), 1860-1873.
Parzych, K. R., & Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism,
and regulation. Antioxidants & redox signaling, 20(3), 460-473.
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., ... & WyssCoray, T. (2008). The autophagy-related protein beclin 1 shows reduced expression in early
Alzheimer disease and regulates amyloid β accumulation in mice. The Journal of clinical
investigation, 118(6), 2190-2199.
7
Small, S. A., Kent, K., Pierce, A., Leung, C., Kang, M. S., Okada, H., ... & Kim, T. W. (2005).
Model-­‐guided microarray implicates the retromer complex in Alzheimer's disease. Annals of
neurology, 58(6), 909-919.
Spencer, B., Potkar, R., Trejo, M., Rockenstein, E., Patrick, C., Gindi, R., ... & Masliah, E.
(2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative
pathology in α-synuclein models of Parkinson's and Lewy body diseases. The Journal of
Neuroscience, 29(43), 13578-13588.
Tanida, I., Tanida-Miyake, E., Ueno, T., & Kominami, E. (2001). The human homolog of
Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates
including human Apg12p, GATE-16, GABARAP, and MAP-LC3. Journal of Biological
Chemistry, 276(3), 1701-1706.
Tooze, S. A., & Schiavo, G. (2008). Liaisons dangereuses: autophagy, neuronal survival and
neurodegeneration. Current opinion in neurobiology, 18(5), 504-515.
Webber, J. L., Young, A. R., & Tooze, S. A. (2007). Atg9 trafficking in mammalian cells.
Autophagy, 3(1), 54-56.
Xilouri, M., & Stefanis, L. (2015). Chaperone mediated autophagy to the rescue: a new-fangled
target for the treatment of neurodegenerative diseases. Molecular and Cellular Neuroscience,
66, 29-36.
Ylä-Anttila, P., Vihinen, H., Jokitalo, E., & Eskelinen, E. L. (2009). 3D tomography reveals
connections between the phagophore and endoplasmic reticulum. Autophagy, 5(8), 1180-1185.
Young, A. R., Chan, E. Y., Hu, X. W., Köchl, R., Crawshaw, S. G., High, S., ... & Tooze, S. A.
(2006). Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and
endosomes. Journal of cell science, 119(18), 3888-3900.
Hu, Z., Yang, B., Mo, X., & Xiao, H. (2015). Mechanism and regulation of autophagy and its
role in neuronal diseases. Molecular neurobiology, 52(3), 1190-1209.
Sitografia
Piacentini M. (2014). Regolazione dell'autofagia e sue applicazioni in patologia.
http://www.accademiamedicadiroma.it.
8