Glicina
Un amminoacido
Chimotripsina
Una proteina globulare
- In teoria un numero enorme di differenti catene polipeptidiche
potrebbe essere sintetizzato con i 20 amminoacidi standard.
204 = 160.000 differenti peptidi con 4 amminoacidi
20300 differenti proteine con 300 amminoacidi
- Solo un numero limitato delle possibili catene polipeptidiche
può assumere una singola conformazione tridimensionale stabile.
- Le proteine che non hanno una conformazione stabile non sono
“utili” e vengono eliminate dalla selezione naturale
- Le proteine sono state “costruite” in un modo così preciso che il
cambio di pochi atomi di un amminoacido può talvolta
distruggere la struttura di una proteina e comprometterne la
funzione .
Diversi conformeri della molecola dell’etano
Livelli di struttura nell’architettura delle proteine
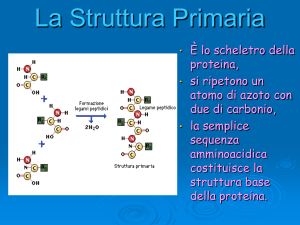
Struttura primaria:
Struttura secondaria:
Sequenza degli amminoacidi e posizione
dei ponti disolfuro.
Insieme dei legami covalenti.
Disposizione nello spazio dei residui
amminoacidici adiacenti nella
sequenza lineare.
Disposizioni particolarmente stabili dei
residui amminoacidici che danno
origine a organizzazioni ricorrenti
Tipiche e più comuni strutture secondarie: α elica, foglietto β,
ripiegamento β
Struttura secondaria di una proteina = distribuzione di α eliche,
foglietti β e turns lungo la catena proteica.
Livelli di struttura nell’architettura delle proteine (b)
Struttura terziaria:
Disposizione nello spazio dei residui
amminoacidici lontani tra loro nella
sequenza lineare.
Descrive tutti gli aspetti del
ripiegamento tridimensionale di un
polipeptide
Struttura quaternaria:
In proteine con più di una catena
polipeptidica (subunità) si riferisce alla
distribuzione nello spazio di queste
subunità e alla natura dei contatti che
esistono tra loro.
Le interazioni chimiche che stabilizzano la conformazione di
una proteina sono:
Ponti disolfuro (nelle proteine extracellulari)
Legami non covalenti:
Legami idrogeno
Interazioni ioniche
Interazioni di van der Waals
Interazioni idrofobiche
La conformazione di una proteina con la minore energia libera
(cioè la più stabile) è generalmente quella con il maggior numero di
interazioni deboli
Il ripiegamento (o folding) delle proteine
1) I residui idrofobici devono essere localizzati all’interno della
proteina, lontano dal contatto con l’acqua.
2) Il numero di legami idrogeno deve essere massimo.
Il ripiegamento (o folding) delle proteine (b)
La maggior parte della variazione di energia libera che si ha
durante il folding di una proteina è dovuta all’aumento di
entropia della soluzione acquosa.
Si ha aumento dell’entropia dell’acqua quando le catene laterali
degli amminoacidi idrofobici tendono a raggrupparsi all’interno
della proteina
Si ha aumento dell’entropia dell’acqua quando si generano
legami idrogeno intramolecolari o interazioni ioniche tra gruppi
carichi della molecola
Il numero di molecole d’acqua negli strati ordinati è
proporzionale all’area superficiale del soluto
idrofobico
Interazioni idrofobiche
Molecole non polari in un ambiente acquoso tendono a raggrupparsi.
L’aggreagazione delle molecole non polari in acqua porta ad un aumento
dell’entropia del sistema dovuto alla riduzione della superficie esposta al
solvente ed alla conseguente riduzione delle molecole d’acqua negli “strati
ordinati”.
Queste associazioni di molecole non polari in ambiente acquoso sono dette
Interazioni (o attrazioni) idrofobiche
Molecole non polari in una soluzione acquosa vengono tenute insieme non tanto
perché hanno affinità tra loro ma per ridurre la superficie esposta all’acqua.
Il ripiegamento (o folding) delle proteine (c)
La scomparsa degli strati di molecole di acqua altamente
ordinate rappresenta la forza entropica trainante che guida
l’avvolgimento di una proteina
IL legame amidico C-N in un peptide (1,32 Å) ha una lunghezza intermedia tra
quella di un singolo legame C-N (1,49 Å) e di un doppio legame C=N (1,27 Å)
(Pauling and Corey, 1930)
Nel legame peptidico vi è parziale ridistribuzione delle 2 coppie di elettroni tra
l’atomo di ossigeno carbonilico e l’atomo di azoto amidico.
Il legame C-N ha caratteristiche parziali di doppio legame ed è pertanto rigido.
L’idrogeno del gruppo aminico sostituito è quasi sempre trans (opposto)
rispetto all’ossigeno del gruppo carbonilico.
Il legame peptidico
Legami peptidici trans e cis
La forma trans è fortemente favorita perché nella forma cis vi
sono impedimenti sterici.
Legami X-Pro cis e trans
Le energie di queste due forme sono paragonabili perchè
impedimenti sterici sono presenti in entrambe le configurazioni
La prolina può partecipare anche a legami peptidici di tipo cis
Tipiche distanze di legame in una unità peptidica
I legami peptidici sono strutture rigide e planari
Gruppo peptidico
(rigido)
Ciascuna unità peptidica può ruotare intorno a 2 legami
N - Cα
e Cα - C’
L’angolo di rotazione intorno al legame N - Cα = φ (phi)
L’angolo di rotazione intorno al legame Cα − C’ = ψ (psi)
φ e ψ possono teoricamente assumere tutti i valori compresi
tra -180 e + 180
C’è libertà di rotazione intorno ai legami che uniscono i gruppi
peptidici agli atomi di carbonio α.
Catena principale
(Scheletro covalente)
Catena laterale
Piano amidico
Carbonio α
Gruppo laterale
Piano amidico
Per convenzione, gli angoli φ e ψ sono definiti pari a 0o quando i due legami
peptidici che fiancheggiano un atomo di carbonio α sono sullo stesso piano e
nella posizione qui mostrata.
In una proteina questa conformazione non è consentita per le sovrapposizioni
steriche tra l’atomo di ossigeno carbonilico e l’atomo di idrogeno α-amminico
Interferenze steriche tra due gruppi peptidici adiacenti.
Una rotazione può portare a una conformazione in cui l’atomo di
idrogeno amidico e l’atomo di ossigeno carbonilico del residuo
successivo sono più vicini delle loro distanze di van der Waals
B) Visione lungo il legame tra l’atomo di azoto e il carbonio α che mostra
come si misura φ
C) Visione lungo il legame tra il carbonio α e il cabonio carbonilico che
mostra come si misura ψ
Una rotazione in senso orario intorno a ciascun legame del gruppo più
lontano in una visione frontale come quelle qui mostrate, corrisponde a un
valore positivo; una rotazione in senso antiorario corrisponde a un valore
negativo
Un residuo di una catena polipeptidica non può avere
qualunque coppia di valori degli gli angoli φ e ψ, perché certe
combinazioni non sono possibili per interferenze steriche tra gli
atomi dello scheletro del polipeptide e quelli delle catene
laterali.
I valori permessi di questi angoli possono essere riportati in un
grafico in cui ψ viene analizzato in funzione di φ, un’indagine
che va sotto il nome di grafico di Ramachandran.
Un grafico di Ramachandran per residui di L-alanina
- Conformazioni ulteriori sono possibili per la glicina (in rosso)
perché la sua catena laterale è piccola (H).
α-elica
Nella struttura ad α-elica lo scheletro del polipeptide è avvolto
intorno all’asse longitudinale della molecola e le catene laterali dei
residui amminoacidici sporgono verso l’esterno
α-elica
L’α elica è stabilizzata da legami idrogeno che si formano tra il
gruppo C’= O di un residuo n ed il gruppo N-H del residuo n+4
Tutti i gruppi C’= O ed N-H, eccetto il primo N-H ed il primo
C=O, sono impiegati in legami idrogeno
Sono presenti 3,6 residui per giro.
α elica
I piani dei
legami
peptidici rigidi
sono paralleli
all’asse
dell’elica.
= catene laterali R
Modello spaziale dell’α elica, in cui si può osservare come sono
ammassati gli atomi
C
N
C
N
L’avvitamento dell’elica è più comunemente destrorso con angoli
φ e ψ compresi tra -600 e -500.
L’avvolgimento dell’α elica consente la
formazione di interazioni tra la catena
laterale di un amminoacido e quella del
residuo distante 3 (a volte 4) residui in
entrambe le direzioni dell’elica.
Spesso
gli
amminoacidi
carichi
positivamente si trovano distanziati di 3
residui
da
amminoacidi
carichi
negativamente, in modo da formare
interazioni ioniche.
Due amminoacidi aromatici possono
anch’essi essere distanziati di 3 residui in
modo
da
generare
un’interazione
idrofobica.
Elica affossata
nella molecola
= residuo idrofobico
= residuo polare
= residuo carico
Elica parzialmente
esposta al solvente
Elica completamente
esposta al solvente
Residuo di prolina impegnato in un legame
peptidico
La presenza di un residuo di prolina limita la formazione di un α-elica
1) L’atomo di azoto imminico di un residuo di prolina fa parte di un anello
rigido e quindi non è possibile rotazione intorno al legame N - Cα (φ).
2) L’atomo di azoto di un residuo di prolina coinvolto in un legame peptidico
non ha l’atomo di idrogeno sostituente che è necessario per generare un
legame idrogeno con altri residui.
In ogni legame peptidico esiste
un piccolo dipolo elettrico
I
dipoli
si
sommano
attraverso i legami idrogeno
presenti nell’elica e quindi il
dipolo aumenta con la
lunghezza dell’elica
In quattro amminoacidi alle due estremità di un’elica non partecipano completamente
alla formazione di legami idrogeno. Le cariche parziali negative e positive del dipolo
delle eliche risiedono sui gruppi N-H e C=O dei legami peptidici, vicino all’estremità
ammino- e carbossi-terminale dell’elica.
Amminoacidi carichi negativamente sono spesso presenti vicino all’estremità amminoterminale di un elica, dove possono generare interazioni stabilizzanti con la carica
positiva del dipolo dell’elica. Residui carichi positivamente nella stessa posizione sono
destabilizzanti.
Il contrario accade all’estremità carbossi-terminale.
Tre modi differenti di rappresentare una α-elica:
a) modello a palle e bastoncini; b) modello a nastro; c) modello a cilindro
La ferritina: una proteina che serve per accumulare il ferro, ha
una struttura costituita prevalentemente da α-eliche.
Conformazione β
E’ la conformazione più estesa delle catene polipeptidiche.
Lo scheletro della catena polipaptidica è esteso con un andamento
a zig-zag
Conformazione β
Si formano legami igrogeno tra i gruppi C=O di un filamento e i
gruppi N-H di un filamento adiacente.
Conformazione β
Catene adiacenti di una struttura β possono avere direzioni
opposte (foglietto β antiparallelo):
C
N
N
C
Coppie di legami idrogeno ravvicinate si alternano a coppie a
maggiore distanza. I legami idrogeno sono perpendicolari alla
direzione dei filamenti.
Conformazione β
Catene adiacenti di una struttura β possono avere la stessa
direzione (foglietto β parallelo):
parallelo
N
C
N
C
Le coppie di legami idrogeno si presentano regolarmente
distanziate e non sono perpendicolari alla direzione dei filamenti.
Foglietto β di tipo misto:
I gruppi R di residui amminoacidici adiacenti sporgono al di fuori della
struttura a zig-zag, alternativamente da una parte o dall’altra del piano
La catena principale forma tutti i legami idrogeno possibili tranne nel caso dei
due filamenti esterni che, trovandosi ai due lati del foglietto, sono affiancati da
un solo filamento β.
A) Foglietti β antiparalleli
B) Foglietti β paralleli
Modi differenti di rappresentare una struttura β:
a) Un modello a palle e bastoncini
b) Un modello schematico
b) Rotazione di 90o della
rappresentazione
schematica per mettere in
evidenza la torsione dei
diversi filamenti β.
La struttura di una proteina che lega gli acidi grassi,
costituita quasi esclusivamente da foglietti β
La fibroina della seta
La fibroina della seta è
costituita da strati di foglietti
β antiparalleli ricchi di
residui di Alanina e Glicina.
Catene laterali di
Alanina
Catene laterali di
Glicina
Sequenza
(Gly-Ser-Gly-Ala-Gly-Ala)n
Le catene laterali piccole di
questi residui consentono un
avvicinamento perfetto degli
strati di foglietti.
Fotografia colorata al microscopio elettronico di fili
di fibroina che escono dalla filiera di un ragno
Ripiegamento β (β turn)
Nelle proteine globulari, circa un terzo dei residui sono presenti in
ripiegamenti o anse a livello dei quali la catena polipeptidica
modifica la sua direzione
I ripiegamenti che uniscono due segmenti consecutivi di un
foglietto β antiparallelo sono detti ripiegamenti β.
La struttura è un ripiegamento di circa 180o, che comprende 4
residui amminoacidici
La struttura è stabilizzata da un legame idrogeno tra il gruppo
carbonilico del primo amminoacido (i) e il gruppo amminico del
quarto residuo (i+3). Il secondo e il quarto legame peptidico non
partecipano alla formazione di legami.
Ripiegamento β
Ripiegamento β (β turn)
Nei ripiegamenti β sono spesso presenti residui di Glicina e di
Prolina.
Glicina : amminoacido piccolo e flessibile
Prolina : il legame peptidico con l’azoto imminico della
prolina assume facilmente la configurazione cis,
una forma che si adatta bene ad un cambio di
direzione molto stretto.
Legami X-Pro cis e trans
Le energie di queste due forme sono paragonabili perchè
impedimenti sterici sono presenti in entrambe le configurazioni
La prolina può partecipare anche a legami peptidici di tipo cis
la configurazione cis si adatta bene ad un cambio di direzione
molto stretto.
Gly
Loop (Anse) sulla superficie di una
proteina
I β turn sono frequentemente
localizzati sulla superficie delle
proteine dove i gruppi peptidici
dei due residui centrali della
struttura possono formare legami
idrogeno con le molecole di acqua
Una parte di una molecola di un anticorpo. Sono evidenziati i loop sulla
superficie che sono coinvolti nell’interazione con altre molecole
I valori degli angoli φ e ψ per tutti gli amminoacidi, eccetto le
glicine nell’enzima piruvato chinasi da coniglio
Probabilità relative della presenza di un dato amminoacido nei tre
tipi più comuni di struttura secondaria