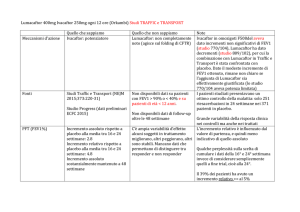
Studi TRAFFIC e TRANSPORT - Wainwright et al. Lumacaftor–Ivacaftor in Patients with
Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015 Jul 16;373(3):220-31.
a cura di Roberto Buzzetti
Report e commenti
1
Si tratta di due studi randomizzati di fase 3, in doppio cieco, controllati con placebo,
disegnati per valutare l'efficacia e la sicurezza di lumacaftor in combinazione con ivacaftor nei
pazienti CF con età di 12 anni e oltre, omozigoti per la mutazione F508del.
Ognuno dei 2 studi ha reclutato due gruppi di trattamento attivo:
lumacaftor 600 mg una volta al giorno + ivacaftor 250 mg ogni 12 ore
lumacaftor 400 mg ogni 12 ore + ivacaftor 250 mg ogni 12 ore
e un gruppo placebo.
I pazienti nello studio hanno continuato i loro trattamenti in corso con le terapie standard.
1.108 soggetti in tutto hanno ricevuto almeno una dose del farmaco, suddivise nei due studi
(549 TRAFFIC, 559 TRANSPORT) in circa 200 centri di sperimentazione clinica in tutto il Nord
America, Europa e Australia.
L'endpoint primario è stata la variazione assoluta, rispetto al basale, del FEV1 predetto al
termine del periodo di trattamento di 24 settimane. Per valutare tale endpoint è stato
considerato l’effetto medio delle settimane 16 e 24.
Analisi statistica con modello misto per Misure ripetute (MMRM). Sulla base del disegno dello
studio, che comprendeva diversi bracci di trattamento all'interno di ogni studio, la
significatività statistica per ciascun braccio contro placebo era basata su un valore di p< o
=0.025.
Oltre alle analisi di ognuno dei due studi, era stata pre-pianificata una analisi congiunta dei 2
studi insieme (POOL).
Tutti i pazienti che hanno completato lo studio di 24 settimane, indipendentemente dal
trattamento assegnato, hanno avuto la possibilità di iscriversi in uno studio di rollover. Dopo la
fine del periodo di somministrazione di 24 settimane, più di 1.000 pazienti ha scelto di entrare
nello studio di rollover e continuare a ricevere la terapia di combinazione.
Diversi sono gli elementi che rendono complicata la comprensione dei risultati:
due studi, praticamente identici (TRAFFIC e TRANSPORT), più la loro somma (POOL)
due livelli di trattamento più il placebo
la coesistenza, nella presentazione dei risultati, di absolute changes e relative changes
(vedere oltre)
Efficacia - Risultati - Lung Function (ppFEV1):
Sono stati osservati miglioramenti medi della funzione polmonare statisticamente significativi
per tutti e quattro i gruppi di trattamento, sia entro gruppo che contro placebo, in tutte le
rilevazioni all'interno dello studio (settimane 2, 4, 8, 16 e 24). Il miglioramento assoluto con i
due farmaci (media tra 20 e 24 settimane) era compreso tra 2,6 e il 4% (P<0,001),
corrispondente a miglioramenti relativi compresi tra il 4,3% e il 6,7% (P<0,001).
Efficacia. Risultati - endpoint secondari:
Nell’analisi congiunta dei due studi, la proporzione di pazienti con un miglioramento di FEV1
uguale o superiore al 5% (relativo) è del 39% e del 46% rispettivamente per Lumacaftor 600
X 1 e per Lumacaftor 400 X 2. Anche il 22% dei trattati con il placebo raggiunge tale soglia (da
questi risultati, vedi oltre, è possibile calcolare il NNT)
Nell’analisi congiunta dei due studi si sono registrate, durante il periodo di trattamento di 24
settimane, riduzioni statisticamente significative dei tassi di esacerbazioni polmonari (definite
come peggioramento sintomatologico della malattia che richiede un trattamento con
antibiotici) del 30 (RR=0,70) e 39 (RR=0,61) per cento rispetto al placebo; e miglioramenti
statisticamente significativi dell'indice di massa corporea.
La media degli incrementi relativi di FEV1 è potenzialmente ingannevole. Si dà una
certa enfasi, nell’articolo, all’incremento relativo maggiore/uguale al 5% / 10% del FEV1 %
predetto rispetto al baseline, ma non si trovano tutte le altre analisi promesse sulla
proporzione di soggetti che vanno oltre queste soglie per quanto riguarda all’incremento
assoluto del FEV1 % predetto rispetto al baseline (neppure nell’appendice supplementare)
Vale la pena ricordare come le variazioni relative siano totalmente inaffidabili: esempio: se un
paziente passasse da 60 a 90 e un altro da 90 a 60, la media delle variazioni assolute sarebbe
pari a zero. Le variazioni relative invece sarebbero rispettivamente +50% e -33,3%, con una
media di circa 8,3%. La stessa discrepanza si osserva anche tra due variazioni entrambe
positive o entrambe negative, con verso della differenza variabile secondo i casi. Non va
dimenticato che nei due studi in questione tra i trattati poco più della metà hanno variazioni
positive e poco meno della metà variazioni negative.
Data l’elevatissima variabilità dell’effetto (le variazioni rispetto al valore di baseline sono
dell’ordine di 0,5 se espresse come errori standard, ma le corrispondenti deviazioni standard
hanno valori di circa 7 – 8), sarebbe fondamentale poter valutare la distribuzione delle
responses, promessa ma non presentata: “bar charts showing the percentage of subjects by
response categories (<-10, ≥-10 to < -7.5, ≥-7.5 to <-5, ≥-5 to <-2.5, ≥-2.5 to <0, ≥0 to
<2.5, ≥2.5 to <5, ≥5 to <7.5, ≥7.5 to <10, ≥10, in percentage points) of average absolute
change from baseline in percent predicted FEV1 at Week 16 and at Week 24 will be plotted by
treatment groups”.
Ancora più importante sarebbe dare un senso clinico a questa variabilità: vi sono soggetti che
con il trattamento migliorano, anche parecchio, e altri che peggiorano, anche parecchio. In
termini di change assoluto (quello relativo è poco affidabile per le ragioni già esposte) si passa
da miglioramenti dell'ordine di +10 a peggioramenti di -10, in poche settimane (Appendix pag
13). Questo sia per i trattati che per i controlli, con i primi che vanno un po' meglio dei
secondi.
2
E’ importante, al di là dell’effetto medio, modesto sia pur significativo rispetto al placebo, che
comunque necessita di una conferma sul lungo periodo, poter identificare i responder e i
non responder. Un’ipotesi potrebbe essere studiare la predittività della risposta immediata (2
o 4 sett) sulla risposta a 24 sett e oltre. Sembrerebbe importante ottenere un supplemento di
informazione, per capire se la risposta a 4 settimane sia predittiva della risposta a lungo
termine: servono per questo i dati longitudinali di ogni paziente. Il poter testare
predittivamente i responders farebbe risparmiare una grande quantità di risorse.
3
Sicurezza:
I farmaci sono stati generalmente ben tollerati (risultati aggregati per ogni braccio di dosaggio
di entrambi gli studi).
Gli eventi avversi più comuni, indipendentemente dal gruppo di trattamento, sono stati:
riacutizzazione infettiva polmonare, tosse, mal di testa e aumento dell'espettorato; si sono
verificati più frequentemente nei pazienti trattati rispetto a quelli che avevano ricevuto il
placebo.
Il 4,2 per cento di tutti i pazienti trattati, indipendentemente dal gruppo di dosaggio, ha
interrotto il trattamento a causa di eventi avversi, rispetto al 1,6 per cento del gruppo placebo.
Aumento degli enzimi epatici (superiore a tre volte il limite superiore della norma) è stato
osservato nel 5,2 per cento dei trattati rispetto al 5,1 per cento del gruppo placebo.
Eventi avversi gravi (rilevante innalzamento dei test di funzionalità epatica) sono stati
registrati in 7 pazienti trattati rispetto a nessun paziente del gruppo placebo. Dopo la
sospensione o interruzione del trattamento di combinazione, i test sono tornati ai valori basali
per sei dei sette pazienti, e il settimo è migliorato senza tornare ai valori basali.
Considerazioni finali, sul costo del trattamento.
Per valutare la rilevanza clinica dei risultati anche in termini di costi, si può fare riferimento al
numero necessario da trattare per avere un paziente che risponde al trattamento
(NNT). Dalla appendice on line dell’articolo del NEJM si può calcolare l’NNT per quello che gli
AA definiscono un outcome rilevante, cioè un incremento della FEV1 di almeno il 5% o il 10%.
L’NNT varia, rispettivamente, tra 4-6 e 7-9 per i due parametri. Il costo stimato del farmaco e’
intorno a 250.000 dollari/anno/paziente. In base a quanto detto per l’NNT il costo del farmaco
dovrebbe essere moltiplicato per 4-6 o 7-9 secondo che si consideri l’incremento del 5% o del
10%, rispettivamente. In pratica bisogna considerare un costo annuo variante tra 1-1,5
milioni, o tra 1,75-2,25 milioni perché una persona tragga un beneficio. Nel calcolo della
variabilità andrebbe tenuto in conto anche il problema della variabilità dell’NNT e dei
conseguenti costi (espressa dagli intervalli di confidenza). La tabella che segue può dare
un’idea riassuntiva dei costi (i calcoli sono dedotti dall’appendice e sono abbastanza precisi,
anche se andrebbero verificati sui dati originali).
limiti interv di
NNT
confidenza 95%
Incremento FEV1
almeno del 5%
Incremento FEV1
almeno del 10%
da
a
costo $
(250.000
$/anno x
paz)
limiti interv di
confidenza 95%
da
A
4
LUM 600 mg qd + IVA
250 mg q 12 h
4
3,3
5,8
1.000.000
825.000
1.450.000
LUM 400 mg q 12 h + IVA
250 mg q 12 h
6
4,2
9,6
1.500.000
1.050.000
2.400.000
LUM 600 mg qd + IVA
250 mg q 12 h
7
5,9
12
1.750.000
1.475.000
3.000.000
LUM 400 mg q 12 h + IVA
250 mg q 12 h
9
6
18,4
2.250.000
1.500.000
4.600.000
Va specificato che l’NNT è calcolabile per i soggetti inclusi nello studio, che per definizione
(pena l’esclusione) hanno caratteristiche omogenee. Non sappiamo quale sarà il beneficio nella
pratica clinica, quando utilizzeremo il farmaco su soggetti che hanno caratteristiche diverse.
Solo da studi osservazionali prospettici di fase quarta si potranno dedurre i benefici per
pazienti diversi da quelli arruolati nell’RCT, soprattutto sulla diminuzione delle riaccensioni
infettive polmonari, parametro la cui rilevanza non e’ deducibile nello studio per il basso
numero di riaccensioni del gruppo controllo. Si potrebbe adottare quanto proposto per la
Dornase, provando il farmaco per un periodo di tempo da stabilire verificando l’effetto sul
raggiungimento di una FEV1 da stabilire e continuando il farmaco solo ai soggetti nei quali si
dimostri il beneficio.
E’ anche ipotizzabile che, presso l’AIFA, come già fatto per altri farmaci, si possa discutere di
accettare l’uso del farmaco in risk sharing con l’industria che sarebbe tenuta a rimborsare i
costi al SSN laddove il farmaco non funzionasse.