26
L'ossidazione degli aminoacidi
Il pool plasmatico di aminoacidi deriva, oltre che dalla dieta, dalla degradazione delle proteine
intracellulari (proteine tissutali).
A differenza dei lipidi che possono essere conservati in quantità notevoli nel tessuto adiposo e dei
glucidi che, se in eccesso, possono essere conservati solo nei pochi mg di glicogeno contenuti nel
muscolo e nel fegato oppure eliminati con le urine, gli aminoacidi che eccedono il fabbisogno delle
sintesi di proteine o di altre
biomolecolecole non possono
essere né conservati né
escreti.
Il destino degli aminoacidi in
eccesso è quindi quello di
essere indirizzati verso il
metabolismo energetico previa
rimozione dei gruppi αamminici. Lo scheletro
carbonioso che rimane, un αchetoacido, entra, in qualche
modo, nei cicli ossidativi che
abbiamo studiato. (ciclo di
Krebs)
L'utilizzazione degli aminoacidi
nel catabolismo avviene
ovviamente anche se le necessità energetica non è soddisfatta da altri nutrienti ed in questo caso sono
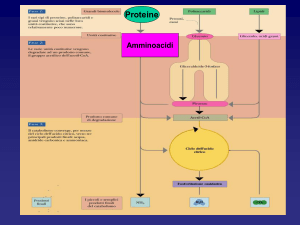
le proteine tissutali ad essere degradate per fornire aminoacidi. Lo schema mostra gli aspetti generali
del metabolismo degli aminoacidi. E' interessante sapere che il catabolismo degli aminoacidi
contribuisce al fabbisogno energetico per oltre il 15% del totale.
La prima tappa del catabolismo aminoacidico è dunque la rimozione del gruppo α-amminico tramite
enzimi altamente specializzati chiamati aminotransferasi o, più comunemente, transaminasi. Le
transaminasi non operano
una vera e propria perdita
dei gruppi amminici, ma un
trasferimento su un
chetoacido, generando un
nuovo aminoacido.
Nella maggior parte degli
aminoacidi il gruppo amminico viene trasferito all'atomo di carbonio in α dell'a-chetoglutarato con
formazione dell'a-chetoacido corrispondente all'aminoacido che trasferisce il gruppo amminico.
Come si vede nell'esempio di transaminazione in figura, il gruppo amminico viene trasferito dall'alanina,
un aminoacido, all'a-chetoglutarato, un chetoacido che diviene così glutammato (acido glutammico, un
importante aminoacido). L'alanina, a sua volta, perdendo il gruppo amminico, forma il chetoacido
piruvato. Sia il piruvato che l'a-chetoglutarato li abbiamo già incontrati nel metabolismo energetico.
Questa reazione è catalizzata da una transferasi, e precisamente l'alanina aminotrasferasi, ALAT,
chiamata anche glutammico piruvico transaminasi (GPT). il distacco del gruppo amminico avviene perché
le transaminasi contengono, come coenzima, il piridossalfosfato (PLP), derivato dalla piridossina, la
vitamina B6. Il meccanismo di catalisi delle transaminasi si chiama meccanismo a ping-pong. L'aminoacido
27
si lega all'enzima, cede al PLP il gruppo amminico e si allontana come chetoacido. Il sito attivo del PLP
può così legare il chetoacido e cedere il gruppo amminico rimosso precedentemente.
Molti aminoacidi vengono transaminati cedendo il gruppo amminico all'a-chetoglutarato. E' evidente
quindi che il glutammato che si forma è un aminoacido importante nel metabolismo dei gruppi
amminici (-NH2, NH3, NH4+) raccogliendoli, per transaminazione sull'a-chetoglutarato, da molti
aminoacidi. Il glutammato,
citosolico, entrerà nei
mitocondri epatici dove cederà
il gruppo amminico sotto forma
di ione ammonio NH4+ che
entrerà nel ciclo di produzione
dell'urea ed eliminato con le
urine. La rimozione
mitocondriale dei gruppi
amminici del glutammato è
operata, tramite una deaminazione ossidativa, da un enzima che si chiama glutammato deidrogenasi
(GDH), come si vede nella figura. Come si vede la GDH è un enzima allosterico, modulato positivamente
dall'ADP ed inibito dalla GTP.
L'azione combinata delle transaminazione e della deaminazione ossidativa, convoglia, nel citosol, i
gruppi amminici nel glutammato dal quale verranno in sede mitocondriale definitivamente rimossi.
Nei tessuti extraepatici, oltre alla transaminazione, sono attivi meccanismi ossidativi di rimozione che
liberano un gruppo amminico netto che si converte in ammoniaca, NH3, molecola estremamente
neurotossica alterando, probabilmente, il pH intracellulare.
L'organo deputato alla eliminazione dell'NH3 è il fegato. Non potendo essere esportata nel sangue, per
la sua tossicità, l'ammoniaca viene, nei tessuti extraepatici, cervello compreso, convertita in un
composto non tossico, la glutammina.
Come si vede in figura, la glutammina, si forma per addizione
enzimatica, catalizzata dalla glutammina sintetasi, di un
gruppo amminico al glutammato preventivamente esterificato
con un gruppo fosfato su un gruppo acido.
Si ha così la trasformazione del glutammato, polare, in
glutammina che, essendo neutra, può facilmente attraversare
le membrane cellulari e, passata nel sangue, viene trasportata
al fegato. Il suo azoto ammidico verrà rilasciato sotto forma
di ammoniaca solo nei mitocondri epatici ad opera di un enzima
chiamato glutamminasi, ed entrerà nel ciclo di produzione
dell'urea, molecola solubile che sarà escreta con le urine. Si
realizza così l'eliminazione dell'N amminico degli aminoacidi. Il
loro scheletro carbonioso, un a-chetoacido, entrerà nel
metabolismo energetico. [vedi destino degli scheletri
carboniosi]
28
L'alanina è un aminoacido chiave nel trasporto di gruppi amminici al fegato in forma non tossica. Nel
muscolo si ha la transaminazione del glutammato sul piruvato. L'alanina passa nel sangue per raggiungere
il fegato nel quale subirà una nuova transaminazione sull'a-chetoglutarato (reazione inversa della
precedente) cedendo così il gruppo NH2 e riproducendo glutammato che lo convoglierà tramite la GDH,
nel ciclo dell'urea. il piruvato può, tramite la gluconeogenesi produrre
glucosio da immettere in circolo ed esportare nel tessuto muscolare, che tramite la glicolisi, lo
riconverte in piruvato. (ciclo del glucosio-alanina) L'ammoniaca liberata dalla deaminazione del
glutammato (GDH) e dalla glutamminasi converge nel ciclo dell'urea.