Il ciclo dell'azoto è un ciclo biogeochimico con il quale l'azoto si muove
principalmente tra l'atmosfera, il terreno e gli esseri viventi.
Questo ciclo viene definito gassoso poiché il pool di riserva, cioè il
serbatoio di questo elemento chimico, è appunto l'atmosfera, dove l'azoto
occupa circa il 78 % del volume totale.
L'importanza del ciclo per gli organismi viventi è dovuta alla loro
necessità di assimilare azoto per la formazione di composti organici vitali,
quali le proteine e gli acidi nucleici, ma, ad eccezione di particolari
batterii, l'azoto atmosferico non può essere direttamente assorbito dagli
organismi e ciò rappresenta spesso un fattore limitante per lo sviluppo
forestale.
Le piante, però, possono assimilare l'azoto tramite l'assorbimento di
alcuni composti azotati (nitriti, nitrati e sali d'ammonio) che, disciolti
nell'acqua, giungono fino alle loro radici Una volta organicato nella
fitomassa, l'azoto viene quindi trasferito agli organismi eterotrofi, come
gli animali, mediante la catena alimentare. La decomposizione dei resti
organici restituisce al terreno l'elemento, che può ritornare nell'atmosfera
grazie all'azione di alcuni batteri specializzati.
Questo ciclo risulta molto complesso proprio perché l'atomo di azoto può
entrare a far parte di un elevato numero di molecole: azoto molecolare,
ammoniaca e sali d'ammonio, nitriti, nitrati ed azoto organico. I processi
chimici coinvolti per la loro formazione possono essere suddivisi in
quattro tipi: azotofissazione, ammonificazione, nitrificazione e
denitrificazione
La concentrazione di aa. nel plasma (espressa come N
amminico) è pari a 4-6 mg/dl
Sono in gran parte di origine alimentare (dalla digestione delle
proteine e dall’assorbimento intestinale) e dal catabolismo
delle proteine endogene).
L’amminoacidemia risente essenzialmente dello stato di
funzionalità epatica.
normalità: N ureico/N aminico > 2
epatopatie: N ureico/N aminico < 2
Ma per la diagnosi è molto più importante l’amminoaciduria
(eliminazione urinaria degli aa).
L’escrezione urinaria di aa aumenta in alcune condizioni
patologiche.
Assorbimento intestinale degli amino acidi
Vengono
assorbiti
dall’intestino
con
un
trasporto
“attivo”simile a quello visto per il glucosio
Specifici trasportatori fanno entrare gli amino acidi
insieme a ioni Na+:
La pompa di Na+ implica l’idrolisi di ATP
Amino acidi essenziali e non
Per la sintesi proteica devono essere disponibili tutti e 20
gli amino acidi naturali
Alcuni amino acidi non possono essere
sintetizzati e
devono essere introdotti con la dieta (aa essenziali)
La fenilalanina è necessaria per la sintesi degli ormoni
tiroidei e dell’adrenalina
Cosa succede se mancano gli aa essenziali?
In caso di mancanza di aa essenziali l’organismo è costretto
a demolire alcune sue proteine
Durante il digiuno prolungato il fegato può perdere il 50%
delle proprie proteine, il muscolo scheletrico il 30% e il cuore
solo il 3%
Alcune proteine del fegato e del muscolo scheletrico
possono quindi servire da “riserva” di amino acidi
Catabolismo degli aminoacidi
La funzione primaria degli amino acidi è la loro utilizzazione
per la sintesi proteica
Gli amino acidi in eccesso rispetto alla richiesta della
sintesi proteica vengono catabolizzati a scopo energetico
Gli amino acidi delle proteine rappresentano
praticamente le uniche sostanze trasformabili in glucosio da
parte dell’organismo. (ricordarsi perché invece non possiamo
convertire gli acidi grassi in glucosio)
IL METABOLISMO DEI COMPOSTI
AZOTATI
pag. 33
Animali ammoniotelici:
la maggior parte dei
vertebrati
acquatici
così come i teleostei e
le larve degli anfibi
Animali ureotelici: molti
vertebrati terrestri e gli
squali
Animali Uricoltelici:
uccelli e rettili
35
IL DESTINO METABOLICO DEGLI AMINOACIDI NEL FEGATO
PROTEINE
sintesi
proteica
CORPI chetogenesi
CHETONICI
proteolisi
20 AMINOACIDI
ORDINARI
gluconeogenesi
GLUCOSO
transaminazione
(interconversione)
(vit. B6PLP)
GLUTAMMATO
deaminazione
decarbossilazion
e
PURINE
PIRIMIDINE
AMMINE BIOGENE
(istamina, adrenalina,
dopamina, serotonina)
NH3
ALFA-CHETOGLUTARATO
(intermedio ciclo di Krebs)
EME
CICLO
DELL’UREA
AMMINOACIDI
METABOLISMO del
GRUPPO AMMINICO
IL METABOLISMO DEGLI AMINOACIDI: LA TRANSAMINAZIONE
Le reazioni di transaminazione, catalizzate da transaminasi PLP - dipendenti,
consentono l’interconversione di 18 aminoacidi su 20 (lisina e treonina) e
comportano tutte come reagenti il glutamato e l’alfa-chetoglutarato.
Esempio: elevata concentrazione di alanina, bassa concentrazione di
aspartato:
Alanina + alfa-chetoglutarato
1°AA
GPT
1° chetoacido
glutamato + ossalacetato
aspartato
piruvato
+
glutamato
2° chetoacido
GOT
2°AA
alfa-chetoglutarato +
Queste reazioni sono reversibili, hanno Keq prossime all’unità e quindi la loro
direzione si sposta dipendentemente dalle concentrazioni dei reagenti.
La presenza fissa della coppia alfa-chetoglutarato – glutamato consente di
spostare il gruppo aminico distribuendolo fra i vari aminoacidi e mantenendo
così equilibrato, per le necessità cellulari, il “pool” aminoacidico.
Transaminazione
E’ il processo con cui un amino acido perde il
gruppo aminico, tappa fondamentale verso il
catabolismo
Il gruppo aminico viene trasferito (reversibilmente)
da un amino acido ad un a- cheto acido
Il processo è catalizzato dalle transaminasi o
aminotransferasi
Compito di questi due enzimi è di
incanalare i gruppi amminici verso
due composti acido glutammico ed
aspartico .
Il
primo
andrà
incontro
a
desaminazione
ossidativa
con
produzione di potere riducente
NAD(P)H ed il secondo donerà il
gruppo amminico alla citrullina nel
ciclo dell’urea
Transaminazione
NH2
|
HOOC - CH - R1
O
+
||
HOOC - C - R2
O
||
HOOC - C - R1
NH2
+
|
HOOC - CH - R2
VITAMINA B6
Assorbimento
Viene assorbita per diffusione passiva.
Prima dell’assorbimento il gruppo 5-fosfato di ciascuna forma di vitamina
B6 viene idrolizzato da fosfatasi specifiche.
Nel citosol delle cellule epiteliali intestinali la vit B6 viene nuovamente
fosforilata e lì trattenuta.
Viene trasporta dal plasma, legata all’albumina e dai globuli rossi, legata
all’emoglobina.
La maggior parte viene immagazzinata nel fegato e successivamente
trasportata ai tessuti periferici via plasma, in forma defosforilata.
Il PL in eccesso viene irreversibilmente convertito in 4-acido piridossico,
escreto con le urine
VITAMINA B6
assorbimento
La piridossina viene trasformata in piridossale e/o
piridossamina, che sono i precursori delle rispettive
forme fosforilate biologicamente attive:
il piridossalfosfato (PLP)
la piridossamina fosfato (PMP).
Questi sono le forme coenzimatiche di numerose
reazioni metaboliche degli AA e delle amine.
VITAMINA B6
CONVERSIONE NELLA FORMA ATTIVA
Tutte le reazioni in cui interviene la vitamina B6 avvengono previo legame del gruppo
aminico con il gruppo aldeidico del PLP formando un “base di Schiff”.
Mediante lo spostamento del doppio legame della base di Schiff verso il legame da scindere
si hanno reazioni diverse quali:
transaminazione, decarbossilazione degli AA
Deaminazione ossidativa delle amine
Deidratazione della serina distacco dello zolfo dalla cisteina
Racemizzazione enzimatica (porta all’interconversione degli L- e D-aminoacidi )
Metabolismo del triptofano (trasformazione dell’ac. xanturenico in ac. nicotinico)
Trasformazione ac. Linoleico in ac. arachidonico
Formazione sfingolipidi della guaina mielinica
Sintesi neurotrasmettitori
Formazione ac. δ-amino-levulinico, precursore della sintesi dell’EME
Il PLP è presente nell’enzima glicogeno-fosforilasi.
Partecipa al metabolismo del frammento monocarbonioso (trasformazione della serina in
glicina ad opera dell’idrossimetiltrasferasi )
VITAMINA B6
RUOLO METABOLICO
VITAMINA B6
metabolismo della metionina
FONTI ALIMENTARI
• In natura è presente sia negli alimenti di origine vegetale (cereali, legumi,
ortaggi ) che animale e derivati (fegato, rene, uova, latte, formaggio ).
• E' sintetizzata anche dalla flora intestinale.
• Viene assorbita nell’intestino tenue.
• FABBISOGNO Il livello di assunzione, calcolato in base a un apporto
proteico del 15% con la dieta, è di 1,5 mg per 100 g di proteine alimentari.
• L'ipervitaminosi determina una neuropatia sensoriale periferica.
CARENZE
Il deficit di vitamina B6 è raro e può causare disturbi al sistema nervoso,
caratteristici soprattuto nel bambino
•
irritabilità,
•
stato depressivo,
•
convulsioni
•
astenia
•
depressione
altre manifestazioni: crampi muscolari e lesioni mucose (glossite), lesioni
della cute attorno agli occhi al naso e alla bocca (dermatite), anemia
microcitica
Transaminazione
Gli a-cheto acidi che partecipano al processo sono: achetoglutarato, piruvato e ossalacetato
Glutammato-ossalacetato transaminasi o GOT
Glutammato alanina aminotrasferasi o GTP
Transaminazione
Nel loro insieme le transaminazioni tendono a convogliare il
gruppo aminico degli amino acidi sull’a-chetoglutarato per
formare glutammato
Il glutammato perde poi il gruppo aminico nella deaminazone
ossidativa per rigenerare alfa-chetoglutarato
Importanza diagnostica (elevata concentrazione nel
sangue indice della lesione di organo e rilascio in circolo)
GOT per il miocardio
…
GTP per il fegato
Visione generale del
metabolismo epatico e
muscolare
Deaminazione
Il definitivo distacco del gruppo aminico degli amino
acidi avviene nel processo di deaminazione, che
può essere:
Ossidativo
Non ossidativo
Glutammato deidrogenasi
Costituisce il principale processo di formazione
dell’ammoniaca (nei mitocondri):
Deaminazione non ossidativa
Decarbossilazione degli aminoacidi
La terza via del catabolismo degli amino acidi consiste
nella loro decarbossilazione nelle corrispondenti amine:
Catalizzate da specifiche decarbossilasi
Vengono prodotte alcune amine importanti come
istamina
(vasodilatatoria),
serotonina
(vaso
costrittore), dopamina (precursore dell’adrenalina)
Poliamine
Le poliammine sono composti organici aventi due o più gruppi amminici,
le più comuni sono putresceina, cadaverina, spermidina, e spermina, che
sono fattori di crescita per cellule eucariotiche e procariotiche. Le
poliammine sono sintetizzate solo da cellule eucariotiche attraverso una
via biochimica altamente regolata, la loro funzione attuale non è ancora
del tutto chiara anche se sembrano avere funzioni stimolanti la
proliferazione cellulare e certamente giocano un ruolo fondamentale nel
superavvolgimento della catene del DNA nel nucleo cellulare. Le
poliammine sono a pH fisiologico dei policationi, ed è proprio questa
caratteristica chimica che conferisce ad esse grande affinità per il DNA,
che è un polianione ma anche per altri cationi fisiologici come , lo ione
Mg++ o Ca++. Se la sintesi delle poliammide viene bloccata la crescita
cellulare viene bloccata o profondamente rallentata.
Recenti test
farmacologici hanno evidenziato la capacità delle poliammine di agire
come potenti modulatori di vari recettori canale come i recettore per il
glutammato NMDA e alcuni recettori canale come quello per il potassio.
Il Ciclene è la più rappresentativa poliammina ciclica.
La putresceina (detta anche putrescina o putrescene) è un composto
chimico organico di formula NH2(CH2)4NH2 (1,4-diamminobutano o
butano-1,4-diammino) che scaturisce dalla carne in putrefazione e ne reca
il caratteristico odore fetido.
È in diretta relazione con la cadaverina; entrambi sono prodotti dalla
rottura degli aminoacidi negli organismi, un processo comune anche agli
organismi vivi ma evidente per quelli morti.
Infatti, la putresceina è sintetizzata dalle cellule vive e sane, per azione
dell‘ ornitina decarbossilasi.
Le poliammine, delle quali la putresceina è una delle più semplici, è uno
dei fattori di crescita essenziali per la divisione cellulare.
La famiglia dei composti dall'odore nauseabondo include il metantiolo e
l'acido butirrico.
La cadaverina, una diammina fetida, è un prodotto di degradazione delle
proteine, in particolare è il prodotto di decarbossilazione
dell’aminoacido lisina. Si sviluppa nei processi di putrefazione dei
tessuti animali, insieme alla putrescina e ad altre poliammine. La
cadaverina presenta una certa tossicità, ha formula bruta
NH2(CH2)5NH2 ed è molto simile alla putresceina dalla quale si
differenzia per la catena alchilica formata da cinque CH2 anziché quattro
. Il suo nome IUPAC è 1,5-diamminopentano.
La cadaverina è anche riscontrabile in piccole quantità nel liquido
seminale e in alcuni organismi marini.
La spermina appartiene alla classe delle poliammine ed è coinvolta nel
metabolismo cellulare, è presente in tutte le cellule eucariotiche. Si
forma a partire dalla spermidina per addizione di un amminopropilico ad
opera dell'enzima spermina sintasi. Si riscontra in numerosi organi e
tessuti ed è un fattore di crescita essenziale per molti batteri. A tutti i
valori di pH superiori inferiori a 10 la spermina ha almeno un azoto
protonato, a pH fisiologico ha tutti gli azoti protonati ed è dunque un
policatione.
Un processo importante cui vanno incontro gli
aminoacidi è la sintesi delle amine biogene:
Dalla tirosina
dopamina
Dal triptofano
serotonina
Dall’ istidina
istamina
Dall’acido glutamico
acido γ-amino butirrico
(GABA)
L'istamina viene sintetizzata a partire dall'istidina ad opera di un enzima
chiamato istidina-decarbossilasi.L’istamina, assieme ad altre sostanze
quali la tiramina, la cadaverina, la putrescina, sono composti azotati,
chiamati anche “ammine biogene”, che possono essere presenti in vari
tipi di alimenti a seguito dell’azione di microrganismi. Le più importanti
sindromi di origine alimentare causate dall’ingestione di ammine biogene
sono l’avvelenamento da istamina (sindrome sgombroide) e
l’intossicazione da tiramina (sindrome del formaggio), documentate da
numerosi studi epidemiologici.
L'istamina come ormone tessutale gioca un ruolo come messaggero in
tanti processi: immunitari (infiammazione), neurovegetativi / organici,
cardiaci.
H COOH
H
N
H
H
CH2
Istidina
NH
N
H
H
N
HN
H
HN
N
H
H N
N
pI = 7.6. A Ph fisiologico l’imidazolo è protonato e funge da accettore
E donatore di protoni.
È immagazzinata in vescicole citoplasmatiche che si trovano in cellule
specializzate come i granulociti basofili, le piastrine, i mastociti, i primi due
circolanti nel sangue, l'ultimo intersperso nel tessuto connettivo intorno ai vasi
sanguiferi, nella cute nelle mucose del canale digerentee dell'apparato
respiratorio. L’istamina è contenuta nei granuli delta delle piastrine, nei granuli dei
mastociti e dei basofili. I mastociti sono cellule residenti nel tessuto connettivo
che possiedono sulla superficie recettori per le IGE, immunoglobuline rilasciate
nell'organismo a seguito di una reazione allergica. i recettori sulla superficie dei
mastociti si legano alle IgE circolanti e tutto questo complesso si lega
all'allergene (che ha scatenato la reazione). Questo attacco provoca una serie di
reazioni all'interno dei mastociti che provocano il rilascio finale di molte sostanze,
tra cui appunto l'istamina. L'effetto dell'istamina, che accompagna la
vasodilatazione, è di alterare l'equilibrio idrico tra l'acqua contenuta nel sangue e
l'acqua contenuta nel tessuto connettivo: il risultato è un accumulo di acqua nel
tessuto connettivo che provoca gonfiore, il quale può costituire un rischio serio
se si sviluppa nella gola o nei bronchi. L'acqua in eccesso viene solitamente
eliminata attraverso gli occhi e il naso nel corso dell'allergia. Una seconda
esposizione all'allergene può avere effetti molto violenti perché in quel caso gli
anticorpi (immunoglobuline) IgE sono già presenti sul mastocita. L'istaminasi, un
enzima che neutralizza l'istamina, è contenuta nei granuli dei globuli bianchi
eosinofili, che vengono richiamati da sostanze (chiamate chemiotattiche) liberate
anche dagli stessi mastociti.
La tiramina è una ammina derivata dall'amminoacido tirosina. È una feniletilamina
sostituita. Ampiamente presente nell'organismo degli esseri viventi, viene
sintetizzata per decarbossilazione della tirosina in seguito a processi fermentativi
o di decomposizione batterica. Cibi ricchi di tiramina sono i formaggi, le carni
lavorate, la salsa di soia e altri prodotti alimentari fermentati. Pesce, cioccolato e
bevande alcoliche, tra le quali il vino rosso, sono un'altra considerevole fonte di
tiramina. Essa è anche una delle principali sostanze a cui sono imputati gli effetti
legati all'ubriachezza e al conseguente mal di testa. È anche una molecola
responsabile di allergie alimentari.
Il triptofano è un amminoacido poco polare, la sua molecola è chirale.
L'enantiomero L è uno dei 20 amminoacidi ordinari, il suo gruppo laterale è un
indolile.
Negli esseri umani è essenziale, cioè va assunto tramite l'alimentazione, dato che
l'organismo umano non è in grado di sintetizzarlo.
Il triptofano è anche un precursore della serotonina (un neurotrasmettitore) e della
melatonina.
La serotonina (5-idrossitriptamina, 5-HT) è un neurotrasmettitore monoaminico
sintetizzato nei neuroni serotoninergici nel sistema nervoso centrale, nonché
nelle cellule enterocromaffini nell'apparato gastrointestinale.
Le più alte concentrazioni di 5-HT si trovano in tre diversi siti corporei:
Nella parete intestinale. Le cellule enterocromaffini contengono circa il 90% della
quantità totale di 5-HT presente nell'organismo: queste sono cellule derivate dalla
cresta neurale, simili a quelle della midollare del surrene, e mescolate alle cellule
mucosali, principalmente nello stomaco e nell'intestino tenue.
Nel sangue. La 5-HT è presente in elevate concentrazioni nelle piastrine, che la
accumulano dal plasma attraverso un sistema di trasporto attivo e la rilasciano in
seguito all'aggregazione che si verifica nei siti di danno tissutale.
Nel sistema nervoso centrale. La 5-HT è un importante trasmettitore del SNC ed è
presente in elevate concentrazioni in specifiche aree del mesencefalo.
La biosintesi della 5-HT endogena segue una via simile a quella della
noradrenalina , con la differenza che l'aminoacido precursore è il
triptofano, invece della tirosina.Il triptofano viene convertito in 5idrossitriptofano grazie all'azione della triptofano-idrossilasi. Il 5idrossitriptofano così prodotto iene decarbossilato a 5-HT, a opera
dell'aminoacido decarbossilasi. Le piastrine accumulano la 5-HT durante
il loro passaggio attraverso la circolazione intestinale, dove la
concentrazione locale è relativamente alta. La 5-HT viene spesso
immagazzinata nei neuroni e nelle cellule enterocromaffini come cotrasmettitore insieme con vari rmoni di natura peptidica, come la
somatostatina, la sostanza P, e il polipeptide vasoattivo intestinale.
La degradazione della 5-HT avviene principalmente attraverso una
deaminazione ossidativa, catalizzata dalle monoaminossidasi, seguita
dall'ossidazione ad acido 5-idrossiindolacetico (5-HIAA).
La fenilalanina è un amminoacido non polare che partecipa alla
costituzione delle più comuni proteine alimentari. La sua molecola è
chirale.
L'enantiomero L è uno dei 20 amminoacidi ordinari, il suo gruppo
laterale è un gruppo benzile.
Nell'organismo umano, la fenilalanina è un amminoacido essenziale.
Può essere convertito nella tirosina che a sua volta può venire
trasformata nell'L-DOPA, nell'epinefrina e nella norepinefrina.
La malattia genetica della fenilchetonuria è dovuta all'incapacità di
metabolizzare la fenilalanina. Nei neonati e fino alla pubertà, l'accumulo
di fenilalanina nel sangue, nelle urine e nei tessuti, può provocare un
mancato sviluppo del sistema nervoso centrale che si traduce in un
ritardo neuromotorio e psichico. Se la malattia viene identificata alla
nascita, un trattamento precoce e ben seguito rende possibile uno
sviluppo normale e previene la compromissione del sistema nervoso
centrale.
Infine, la fenilalanina è parte della composizione dell'aspartame, un
dolcificante usato anche nell'industria alimentare, in special modo nelle
bevande gassate.
Normalmente la fenilalanina viene ridotta a tirosina
mediante l'intervento di una monossigenasi e di altri
coenzimi tra i quali la tetraidrobiopterina. Un ossigeno
viene inserito in para e dall'altro si forma una molecola
d'acqua. I due idrogeni sono forniti dalla tetrabiopterina
che non può permamere nello stadio ossidato e per
questo
motivo
viene
ridotta
dal
NADPH.
Amine
ormone tiroideo, catecolamine (adrenalina, noradrenalina); precursore comune
tirosina.
Aumentata produzione di calore;
Tetraiodiotironina (T4)
mantenimento del metabolismo del
glucosio e di altri «carburanti»; ampi
effetti sull'espressione genica e
Tiroide
sull'induzione di sintesi enzimatiche
Triidiotironina (T3)
Epinefrina
Midolla
surrenale
Mastociti
Aumento delle pulsazioni e della
pressione sanguigna; contrazione
della maggior parte dei muscoli lisci;
glicogenolisi nel fegato e nel
muscolo; idrolisi dei lipidi del
tessuto adiposo
Norepinefrina
Istamina
Contrazione
diminuzione
periferica.
delle
arteriole;
della circolazione
Dilatazione dei piccoli vasi
sanguigni
La dopamina (o dopammina) è una ammina biogena naturalmente
sintetizzata dal corpo umano.
All'interno del cervello la dopamina funziona da neurotrasmettitore,
tramite l'attivazione di recettori specifici D1, D2 e D3 subrecettori.
La dopamina è anche un neuro ormone rilasciato dall'ipotalamo. La sua
principale funzione come ormone è quella di inibire il rilascio di
prolattina da parte del lobo anteriore dell'ipofisi.
La dopamina non può essere utilizzata come farmaco ma viene
comunemente somministrato un suo precursore: la L-dopa
(profarmaco), che subisce decarbossilazione ad opera dell'enzima
decarbossilasi degli amminoacidi aromatici. La dopamina agisce sul
Sistema nervoso simpatico causando l'accelerazione del battito
cardiaco e l'innalzamento della pressione sanguigna.
Gli antagonisti dopaminergici sono farmaci che trovano ampio utilizzo
come neurolettico in ambito psichiatrico, mentre agonisti dopaminergici
sono usati sia come terapia di prima scelta nel morbo di Parkinson, sia -in
misura minore- come antidepressivi.
La biosintesi della dopamina avviene, a livello centrale, a partire da Ltirosina che viene idrossilata a L-dopa. La successiva decarbossilazione
porta alla dopamina. Successivi passaggi biosintetici portano prima alla
noradrenalina e poi all'adrenalina. La dopamina viene rilasciata a livello
centrale dalla substantia nigra e la sua azione è mirata a modulare
l'attività inibitoria dei neuroni GABAergici. Dopo aver interagito con i suoi
recettori, la dopamina viene metabilizzata da due enzimi diversi:
dalle MAO B (Mono-Amino-Ossidasi) ad acido 3,4-diidrossi-fenilacetico;
dalle COMT (Catecol-O-Metil-Transferasi) ad 3-metossi-tiramina.
L'acido γ-amminobutirrico (GABA) è il principale amminoacido inibitorio
del sistema nervoso centrale. Al contrario, il glutammato rappresenta il
più importante neurotrasmettitore eccitatorio del cervello.
Viene rilasciato da neuroni dei circuiti locali presenti nel cervello, i quali
presentano un piccolo corpo neuronale e arborizzano a breve distanza
formando principalmente sinapsi asso-assoniche con i neuroni di
proiezione (eccitatori). Esistono 3 tipi di recettore per il GABA
SINTESI E METABOLISMO DEL GABA:
Il GABA si forma per decarbossilazione
dell'acido
glutammico
Questa reazione è catalizzata dalla GAD (Glutamico Acido
Decarbossilasi), enzima altamente specifico che ha come cofattore il
piridossal-fosfato (PLP o Vit B6 (*)) ed è inibito da diversi antagonisti del
piridossal-fosfato, come isoniazide, tiosemicarbazide, ecc che bloccano il
gruppo aldeidico della Vit B6 sotto forma di derivati idrazonici.
L'accumulo vescicolare del GABA è effettuato da un trasportatore
specifico che utilizza, come fonte di energia, sia il gradiente elettrico che
di pH presente tra lume vescicolare e citoplasma e generato dalla H+ATPasi (pompa protonica) vescicolare.
La CREATINA (dal greco kreas = carne) o metil-glico-ciamina è un
componente del metabolismo intermedio che viene formata nel fegato,
reni e pancreas. In quantità quasi costante, secondo una reazione che
coinvolge gli aminoacidi GLICINA, ARGININA e ORNITINA e viene
depositata per circa il 95% nei muscoli.
Dopo la produzione è trasportata
muscoli (95%), cervello e cuore,
Presente nella dieta, soprattutto
carne e pesce.
La creatina viene convertita nell’organismo a fosfocreatina (all’interno del
muscolo 70% della creatina viene convertita in fosfocreatina) Durante la
contrazione muscolare ATP si trasforma in ADP liberando un radicale
fosforico che fornisce energia. La fosfocreatina riforma ATP a partire
dall’ADP. Durante l'attività motoria di intensità massimale e di breve
durata, la disponbilità dinamica di ATP è ottenuta quasi esclusivamente a
mezzo del processo anaerobico alattacido che si realizza mediante la
defosforilazione della fosfocreatina, con il conseguente passaggio
dell'ADP allo stato di ATP, atto a liberare energia per la contrazione
muscolare, mediante la seguente reazione reversibile pH-dipendente:
secondo cui la creatina viene poi rifosforilata durante il periodo di
riposo
La tossicità dell’ammoniaca
I meccanismi coinvolti nella tossicità dell’ammoniaca, in
particolare l’encefalopatia, non sono ben definiti. E’ tuttavia
chiaro che quando la concentrazione dell’ammoniaca nel
sangue e negli altri liquidi biologici aumenta, questa sostanza
diffonde nelle cellule attraverso la barriera ematoencefalica.
L’aumento della concentrazione di ammoniaca stimola la
sintesi di glutammato dall’alfa-chetoglutarato come pure la
sintesi della glutammina. Benchè questa sia una normale
reazione cellulare di detossificazione, la concentrazione
dell’ammoniaca è notevolmente aumentata la disponibilità di
alfa-chetoglutarato nelle cellule del sistema nervoso può
venire meno, con conseguente inibizione del ciclo degli acidi
tricarbossilici e della produzione di ATP.
ENCEFALOPATIA PORTO-SISTEMICA
(Encefalopatia epatica; Coma epatico)
Sindrome neuropsichiatrica secondaria ad un'epatopatia e generalmente
associata a uno shunt venoso porto-sistemico. Il termine "encefalopatia
porto-sistemica" descrive la situazione fisiopatologica meglio di
"encefalopatia epatica" o di "coma epatico", ma clinicamente i 3 termini
vengono usati indifferentemente.
Eziologia
L'encefalopatia porto-sistemica si può verificare nell'epatite fulminante
causata da virus, farmaci o tossine, ma, più comunemente, si verifica
nella cirrosi o in altri processi cronici quando, in seguito all'ipertensione
portale, si è sviluppata un'estesa rete di anastomosi porto-sistemiche. La
sindrome è anche secondaria alle derivazioni porto-cavali o ad altre
anastomosi chirurgiche porto-sistemiche.
Nei pazienti con un'epatopatia cronica, l'encefalopatia è, di solito,
scatenata da cause specifiche, potenzialmente reversibili (p. es.,
sanguinamento GI, infezione, squilibrio elettrolitico (specialmente
ipokaliemia), abuso di alcol) o da cause iatrogene (tranquillanti, sedativi,
analgesici, diuretici).
Patogenesi
Il fegato metabolizza e disintossica i prodotti originati dalla digestione e
provenienti dall'intestino attraverso la vena porta. Nelle epatopatie, tali
prodotti raggiungono inalterati il circolo sistemico, o perché il sangue
portale non viene a contatto con gli epatociti o perché la funzionalità di
queste cellule è gravemente compromessa. Gli effetti tossici che ne
derivano sul sistema nervoso centrale sono responsabili della sindrome
clinica.
Le sostanze tossiche non sono del tutto conosciute e la sindrome è
verosimilmente multifattoriale. L'ammoniaca, che è un prodotto della
digestione delle proteine, probabilmente riveste un ruolo importante, ma
anche le amine biologiche, gli acidi grassi a catena corta o altri prodotti di
origine intestinale possono essere responsabili o possono agire insieme
all'ammoniaca. Solitamente, i livelli sierici degli aminoacidi aromatici
sono elevati e quelli degli aminoacidi a catena ramificata sono bassi, ma
non è detto che questo rappresenti un fattore causale.
Anche la patogenesi del quadro tossico a livello dell'encefalo non è
chiara. Le alterazioni della permeabilità cerebrovascolare e dell'integrità
cellulare possono svolgere un ruolo, specialmente nell'epatite fulminante.
Il sistema nervoso centrale dei pazienti epatopatici appare
particolarmente sensibile alle alterazioni metaboliche. È possibile che si
verifichino delle alterazioni a carico del metabolismo energetico cerebrale
e una qualche inibizione nei processi di trasmissione degli impulsi
nervosi da parte delle ammine tossiche che agiscono come falsi
trasmettitori. Decisive evidenze chiamano in causa anche l'acido gaminobutirrico (GABA), il principale neurotrasmettitore cerebrale di tipo
inibitorio; sembra che la sua sintesi sia aumentata e che vi possa essere
un'alterazione sia dei recettori cerebrali del GABA che di quelli delle
benzodiazepine endogene correlate.
Le alterazioni anatomopatologiche solitamente sono limitate all'iperplasia
degli astrociti con un danno neuronale nullo o trascurabile, anche se
nell'epatite fulminante è comune il riscontro di un edema cerebrale.
Le modificazioni della personalità (p. es., comportamenti inappropriati,
alterazioni dell'umore, senso critico alterato), rappresentano le comuni
manifestazioni iniziali che possono precedere la comparsa di chiare
alterazioni dello stato di coscienza.
Sintesi del glutammato
Reazione già vista in precedenza, catalizzata
glutammato deidrogenasi, percorsa in senso opposto:
dalla
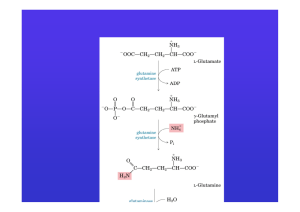
SINTESI E DEMOLIZIONE DELLA GLUTAMMINA
La reazione catalizzata dalla glutammina sintetasi è ATP
dipendente:
Si forma soprattutto nei muscoli e viene utilizzata
soprattutto dai reni, ricchi di glutamminasi
La glutammina è l’amminoacido più utilizzato a scopi
biosintetici (basi puriniche e pirimidiniche, amminozuccheri)
Glutamminasi
E’un enzima mitocondrialeche deamina idroliticamente la glutamina in
NH3 ed acido glutammico:
Formazione della glutammina per azione della glutammina sintetasi (1) nel
muscolo e sua deaminazione idrolitica per azione della glutamminasi(2).
Questa è particolarmente attiva nei reni; la NH3 che si forma incorpora
l’eccesso di H+ per formare NH4 che viene eliminato con le urine,
contribuendo primariamente a diminuire l’acidosi.
Destino metabolico dell’ammoniaca
Un accumulo di NH3 risulterebbe particolarmente tossico soprattutto
per il tessuto nervoso
L’ammoniaca viene quindi incorporata in composti atossici ed
eliminata come urea
I tre processi di incorporazione dell’NH3 sono:
Aminazione dell’a-chetoglutarato in Glutammato
amidazione del glutammato in glutammina
sintesi del carbamilfosfato
LA SINTESI DEL CARBAMIL FOSFATO
- La sintesi dell’enzima Carbamil Fosfato sintasi (CPS I) che prende parte
alla sintesi dell’urea, si trova nei mitocondri, prevalentemente nel fegato.
- Un altro enzima la CPS II, è localizzato nel citoplasma e si trova in tutti i
tessuti.
- Benchè il prodotto di entrambi gli enzimi sia lo stesso, il carbamil
fosfato, essi sono codificati da geni diversi e funzionano in due vie
metaboliche diverse: l’uno nella sintesi dell’urea (CPS I) e l’altro nella
biosintesi delle pirimidine (CPS II).
I due enzimi si differenziano inoltre per la provenienza dell’azoto che
utilizzano come substrato, NH3 nel caso della CPS I, al contrario della CPS
II, richiede N-acetilglutammato per il suo funzionamento.
In circostanze normali i due enzimi funzionano in modo indipendente
l’uno dall’altro e in compartimenti cellulari diversi; tuttavia qualora si
verifichi un blocco del ciclo dell’urea, per una carenza di ornitina
transcarbamilasi, il carbamil fosfato che si accumula nel mitocondrio si
riversa nel citoplasma e può stimolare eccessivamente la sintesi di
pirimidine. Questo si riflette in un aumento della concentrazione di acido
orotico nel sangue e nell’urina.
Sintesi del carbamil fosfato
Viene sintetizzato da:
NH3
CO2proveniente dal ciclo di Krebs …
Pi (ceduto
dall’ATP)
Avviene nella matrice mitocondriale
Catalizzata dalla carbamilatosintetasi
Ciclo dell’urea (o dell’ornitina)
☻E’ il processo catalitico nel quale l’ammoniaca, attivata
come carbami-fosfato, viene incorporata nell’urea
☻Nei mitocondri il carbamil-fosfatoreagisce con l’ornitina a
dare la citrullina:
Ciclo dell’urea
La citrullina lascia il mitocondrio, entra nel citoplasma e
viene trasformata in
sintetasi:
argino succinato dall’opportuna
Ciclo dell’urea
L’argino succinato viene demolito in arginina e fumarato in
una reazione di â-eliminazione catalizzata dall’arginato
liasi:
Ciclo dell’urea
L’arginina viene quindi idrolizzata in urea e ornitina per azione
della arginasi:
L’urea, rilasciata in circolo, viene escreta con le urine.
☺Eliminiamo in 24 ore circa 10-25 g di urea
Energetica
Il processo di formazione dell’urea è molto dispendioso
energeticamente
Ad ogni ciclo vengono consumate 3 molecole di ATP!
Necessario data l’elevata tossicità dell’NH3
Ciclo dell’urea (riassunto)
Enzima
Reazione
catalizzata
Commenti
Carbamilfosfato
sintetasi
Formazione di carbamil fosfato
da ammoniaca e CO2
Fissa ammoniaca originata dagli aminoacidi,
specialmente glutammina, usa 2 molecole di
ATP, si trova nel mitocondrio, la carenza
provoca l’innalzamento della concentrazione
d’ammoniaca nel sangue, con conseguente
tossicità.
Ornitinatranscarbamilasi
Formazione di citrullina da
ornitina e carbamil fosfato.
Libera Pi un esempio di transferasi, localizzata
nel mitocondrio, la carenza provoca l’aumento
della concentrazione di ammoniaca e acido
orotico nel sangue perché il carbamil fosfato
è dirottato nella biosintesi delle pirimidine.
Argininosuccinato
sintasi
Formazione di
argininosuccinato da arginina e
aspartato
Richiede ATP che viene scisso in AMP e PPi è
un esempio di ligasi localizzata nel citosol la
carenza provoca l’aumento della
concentrazione ematica di ammoniaca e
citrullina.
Argininosuccinasi
Scissione di argininosuccinato
in arginina e fumarato
Un esempio di ligasi localizzata nel citoslo la
carenza provoca l’aumento della
concentrazione ematica di ammoniaca e
citrullina.
Arginasi
Scissione dell’arginina in
ornitina e urea
Esempio di idrolasi localizzata nel citosol e in
prevalenza nel fegato la carenza provoca un
aumento moderato dell’ammoniemia e un
aumento notevole della concentrazione di
arginina nel sangue
Regolazione del ciclo dell’urea
Il principale meccanismo di regolazione del ciclo dell’urea sembra basato
sul controllo della concentrazione di N-acetilglutamato, l’attivatore
allosterico della CPS, la cui formazione è favorita dall’abbondanza di
arginina.
Inoltre la concentrazione degli enzimi del ciclo urogenetico aumenta o
diminuisce in risposta all’apporto più o meno elevato di proteine con la
dieta.
L’acidosi può intervenire nella regolazione riducendo la sintesi e
l’escrezione di urea e aumentando quella di ammoniaca.
La carenza di uno qualsiasi degli enzimi del ciclo dell’urea provoca
conseguenze gravi. I neonati con un difetto in uno dei primi quattro enzimi del
ciclo sembrano normali alla nascita, ma presto diventano letargici, ipotermici e
manifestano problemi respiratori. Si osserva un rapido aumento della
concentrazione di ammoniaca nel sangue, seguito da edema cerebrale.
I sintomi sono molto gravi quando sono interessate le prime reazioni del
ciclo ureogenico. I deficit di attività di questi enzimi possono produrre
gravi conseguenze causando iperammoniemia che produce edema
cerebrale, coma e morte. L’enzima ornitina transcarbamilasi è l’enzima
frequentemente più colpito dal deficit genetico che è il tipo congenito
legato al cromosoma X
Gli altri difetti enzimatici ereditari associati al ciclo dell’urea sono di tipo.
autosomico-recessivo. Una carenza di arginasi, l’ultimo enzima del ciclo,
provoca sintomi meno gravi, ma si associa ad un aumento della
concentrazione ematica di arginina e anche ad una moderata
iperammoniemia. I soggetti interessati da un innalzamento della
concentrazione di ammoniaca nel sangue devono essere sottoposti a
emodialisi, a cui frequentemente si associa un trattamento con sodio
benzoato e fenillattato per via endovenosa.
Queste sostanze possono condensarsi, rispettivamente, con la glicina e
la glutammina, formando composti solubili che sequestrano l’ammoniaca
in una forma atossica e che possono essere eliminati con l’urina.
Metabolismo di proteine e aminoacidi nel muscolo
L’equilibrio tra proteolisi e sintesi determina la crescita, l’ipertrofia e
l’atrofia del muscolo
L’equilibrio è anche importante per l’omeostasi generale del corpo
date le dimensioni del tessuto muscolare (reservoir)
Es. nelle prime fasi del digiuno gli aminoacidi (aac) del muscolo
sono utilizzati per fare gluconeogenesi epatica e per produrre
energia a bassi livelli di glucosio
Vie di degradazione delle proteine nel muscolo
Sistema ubiquitina-proteasoma è la via principale di degradazione delle
proteine citosoliche nel muscolo
Proteasoma: macrocomplesso multiproteico che degrada proteine a cui
è stata attaccata (da ligasi) residui singoli o a catena del peptide
ubiquitina.
Vie di degradazione delle proteine nel muscolo
Lisosomi: funzione principale nel muscolo normale è
degradazione di proteine di membrana o endocitate; funzione
cambia in patologie
Caspasi: esecutori di apoptosi (morte cellulare programmata)
Peptidasi calcio-dipendenti (necrosi)
Metodi di studio
Marcatura con aac radioattivi con cannulazione di arteria e vena e
perfusione controllata
Misurazione di secrezione urinaria di N-metil-istidina che è istidina
metilata post-traduzionalmente, presente quasi esclusivamente in
actina, non degradabile né incorporabile in nuove proteine
Su muscoli isolati: rilascio di tirosina che non è sintetizzata né
catabolizzata in muscolo
Metabolismo degli aminoacidi nel muscolo
Contrariamente a quanto si crede, il metabolismo degli aac non
avviene solo nel fegato
Nel muscolo si ha sintesi di alanina e glutamina
Il muscolo è importante catabolizzatore di aac ramificati (leucina,
isoleucina, valina) e di aac non-essenziali (alanina, glutamina,
aspartato)
Aminoacidi che non sono catabolizzati nel muscolo: lisina, serina,
treonina, triptofano
Metabolismo degli aminoacidi nel muscolo
Ossidazione degli aminoacidi ramificati (aac-r)
Il muscolo è importante organo di catabolismo di aac-r per i quali
l’uptake epatico è scarso
L’ossidazione degli aac-r avviene anche in tessuto adiposo, in
cervello e rene
Funzione dell’ossidazione degli aac-r varia con tessuto
Es. in muscolo leucina ossidata rapidamente e produce Co2 e energia
In tessuto adiposo in condizioni anaerobiche serve alla produzione di
acetilCoA utilizzato per sintesi trigliceridi
Meccanismo di degradazione aac-r
Transaminasi converte aac-r in a-ketoacido
a-ketoacido decarbossilato da a-ketoacido-deidrogenasi (enzima
esclusivamente mitocondriale)
La decarbossilazione produce 3 mol di acetil-CoA utilizzati dal ciclo di
Krebs
Regolazione positiva della decarbossilazione degli aac-r nel tessuto
muscolare
•Glucocorticoidi
•digiuno (ATP fosforila e inibisce deidrogenasi; riduzione ATP in
digiuno stimola catabolismo aac-r e loro utilizzazione per produrre
energia)
Contributo degli aac al metabolismo energetico del muscolo
Leucina come fonte di energia
E’ la fonte alternativa di energia durante il digiuno
A digiuno la concentrazione di leucina aumenta nel sangue e nel
muscolo
Quindi aumenta la sua degradazione nel muscolo e viene inibita la
degradazione di molecole gluconeogenetiche (piruvato)
La quantità di ATP prodotta a partire da leucina durante digiuno pareggia
la quantità non prodotta per mancanza di glucosio
La leucina
•inibisce ossidazione di glucosio e piruvato del 40%
•stimola il rilascio di lattato
•non altera glicolisi o uptake di glucosio
Influenza di leucina e aac-r sul turnover di proteine
muscolari
Aac-r, in particolare leucina, stimolano la sintesi proteica e riducono il
catabolismo (doping? Uso nell’anziano per prevenire atrofia? Attenzione
al carico epatico e renale)
La stimolazione della sintesi proteica avviene a livello traduzionale:
aumento polisomi
Effetti mediati da via di trasduzione che è presente già nei batteri:
Nutrient-sensing pathway
La via coinvolge trasduttori intermedi mTOR e Akt (target di rapamicina,
immunosoppressore) che sono utilizzati anche dal segnale insulinico
Ciclo dei nucleotidi purinici e effetti dell’esercizio fisico
L’esercizio provoca:
↑ la degradazione delle proteine
↑ uptake di aac-r
↑ l’ossidazione della leucina
↑ la produzione di NH3
Attivazione del ciclo dei nucleotidi purinici durante esercizio da
degradazione di leucina
NH2 da aac-r viene trasferito a ossalocitrato →
→ aspartato → si combina con inosina-mono-fosfato (IMP) → adenil
succinato →
→ degradato a fumarato ( ciclo TCA) e AMP
AMP degradato a NH3 e IMP che promuove glicogenolisi e glucolisi
Il consumo di AMP stimola la formazione di ATP (equilibrio ATP/AMP)
Produzione e rilascio di alanina
L’accumulo di NH2 da degradazione di aac-r può essere tossico
Nel muscolo manca il sistema enzimatico di trasformazione di NH3 in
urea
I residui NH2 provenienti dalla degradazione degli aac-r e aspartato sono
utilizzati per fare alanina e glutamina
La produzione di alanina e glutamina è accoppiata alla
degradazione degli aac-r
Sintesi di alanina: NH2 proviene da aac-r, il piruvato dal glucosio
La sintesi di alanina non produce glucosio, ma l’ossidazione della
leucina e la ridotta degradazione di piruvato consentono di risparmiare
glucosio durante il digiuno
La produzione di alanina e glutamina è accoppiata alla degradazione
degli aac-r
La produzione di alanina nel muscolo è importante per la
glicemia
Alanina è l’aac più importante nel fegato per la gluconeogenesi
Porta al fegato NH2 per eliminazione con urea
Ormoni e metabolismo muscolare
Ormoni e metabolismo muscolare
Insulina e glucosio
Insulina stimola la traslocazione in membrana dei trasportatori di aac
Aumenta la velocità di sintesi proteica
Inibisce la proteolisi
IGF-1
Il muscolo produce due isoforme: IGF-1 e MGF (mechanogrowth factor)
L’attività è modulata a legame a IGFBPs di cui esistono diverse isoforme
modulate indipendentemente
IGF-1 e MGF agiscono via PI3kinasi e via di mTOR/Akt
Ormoni e metabolismo muscolare
Ormoni tiroidei
•Aumentano proteolisi ad alte dosi
•Probabilmente per aumento espressione sistema proteasomaubiquitina
•Ormoni e metabolismo muscolare
Glucocorticoidi
Effetti dipendenti da assunzione di nutrienti, altri ormoni e dose
In alimentato: inibizione sintesi proteica, no effetti su degradazione
In digiunante: lisi proteica per fare gluconeogenesi
Effetto mediato da modulazione trascrizionale di sistema ubiquitinaproteasoma e inibito da IGF-1
Effetti antagonizzati da insulina
Catecolamine e Testosterone
Riducono proteolisi
Aumentano sintesi proteica
Fattori nutrizionali e metabolismo proteico muscolare
•Digiuno breve (2-3 giorni): riduzione della sintesi proteica e aumento
della proteolisi
Effetti mediati da insulina (assenza) e da attivazione di sistema ubiquitinaproteasoma e lisosomale
•Digiuno lungo (> 4 giorni)
La necessità di utilizzare glucosio è ridotta perché il cervello usa corpi
chetonici per fare ATP
Proteolisi muscolare ridotta
•Dieta ipoproteica
Si riduce la sintesi ma anche la degradazione delle proteine: equilibrio
Attività e metabolismo proteico muscolare
Work-induced growth
Aumenta la sensibilità all’insulina e ai suoi effetti trofici
perché vi è
regolazione trascrizionale positiva di diversi
componenti della via di segnalazione insulinica, compresi m-TOR e
Akt
e non
L’attività muscolare aumenta la sintesi di proteine muscolari
modifica la proteolisi
Atrofia da denervazione
Legata a aumentata espressione genica dei componenti del
sistema ubiquitina-proteasoma
Stretch
I geni di molte proteine muscolari rispondono allo stretch:
stretch-responsive elements?
Citochine e la regolazione del metabolismo proteico nel
muscolo
Grave riduzione di massa muscolare in sepsi e cachessia neoplastica
Ruolo di citochine che stimolano proteolisi ubiquitina-dipendente e
attivazione lisosomale
IL-2 e TNF-a.