Assorbimento e vie di
somministrazione dei farmaci
ASSORBIMENTO: passaggio del farmaco dal sito di somministrazione alla
circolazione sistemica (o all’interstizio regionale in caso di somm. topica)
Enerali
Sistemiche
Parenterali
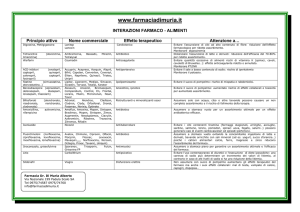
Vie di somministrazione
Topiche
Vie enterali
Orale
Sublinguale
Rettale
(per os)
Vie
parenterali
sistemiche
Intravascolare
Intramuscolare
Cutanea
Endovenosa (e.v.), intracardiaca, intrarteriosa
(i.m.)
Sottocutanea (s.c.) e intradermica (anche topica)
Altre vie
D’Organo
Intracavitaria
Transcutanea
Transmucosale
Intratecale, intrarticolare e inalatoria (anche sistemica)
Intraperitoneale e intrapleurica
Topica, ma anche regionale o sistemica (es. cerotti)
Oculare, vaginale, etc.
NB: la distinzione tra vie sistemiche e topiche non è assoluta (per es. la via s.c. è “sistemica” x
l’insulina, “locale” per gli anestetici locali)
La scelta della via di somministrazione deve tenere conto di:
caratteristiche fisico-chimiche del farmaco:
- un farmaco molto idrofilo, poco assorbito per via enterale, deve essere
somministrato per via parenterale se si desidera un effetto sistemico)
- un farmaco lipofilo, che attraversa bene le membrane, anche se somministrato per
via topica potrà essere assorbito dando luogo ad effetti sistemici
strategia terapeutica:
- rapidità e durata dell’effetto (per es. e.v. e via i.m.; via sublinguale per la NTG)
- per terapie croniche si preferisce la via meno traumatica per il paziente (per os),
mentre le vie parenterali sono preferite nei pazienti non collaborativi (per es. pz
comatosi, neonati, etc.)
- alcuni regimi terapeutici sono seguiti con maggiore accuratezza dal paziente se la
via di somministrazione è più complessa (per es. vie parenterali)
LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
Ad esclusione della via e.v., tutte le altre vie di somministrazione richiedono il passaggio
del farmaco attraverso cellule e/o membrane e la diffusione in spazi intercellulari prima
di arrivare al sangue.
La velocità di assorbimento dipende sia dalla via di somministrazione che da una serie
di variabili legate alle caratteristiche del farmaco e/o al tessuto.
LE VARIABILI CHE INFLUENZANO LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
DIPENDONO:
- dal farmaco o dalla preparazione farmaceutica (coefficiente di ripartizione, dissolubilità)
- dalla superficie assorbente (estensione, permeabilità, vascolarizzazione)
COEFFICIENTE DI RIPARTIZIONE:
Influenza l’assorbimento attraverso quelle vie (enterali, transcutanea, inalatoria, etc.)
che prevedono la presenza di barriere cellulari (epitelio intestinale, cute, epitelio
alveolare, etc.)
I farmaci più lipofili passano le barriere con maggiore facilità.
Il farmaco deve anche attraversare gli spazi intercellulari acquosi per cui deve
possedere anche un certo grado di idrofilia.
Per farmaci molto idrofili si sceglie una via enterale (e.v.)
DISSOLUBILITA’ DEL FARMACO:
DISSOLUBILITA’ DEL FARMACO:
La dissolubilità del farmaco è indispensabile per il suo assorbimento efficace.
DISSOLUBILITA’: capacità della preparazione farmaceutica di sciogliersi
completamente nell’ambiente da cui deve essere assorbita.
Per farmaci in forma solida o in sospensione: se la velocità di dissoluzione è più
lenta di quella di assorbimento, il passaggio in circolo del farmaco è regolato dalla
velocità con cui la preparazione si dissolve. In alcuni casi (preparazioni ritardo)
l’assorbimento lento e prolungato è desiderato.
La velocità di dissoluzione di una preparazione farmaceutica dipende da:
- superficie della preparazione
- solubilità della preparazione
- temperatura
- pH
- composizione degli eccipienti (rilascio razionale del farmaco mediante l’uso di
particolari polimeri – drug delivery)
- presenza di eventuali rivestimenti protettivi (per es. compresse insolubili
nell’ambiente acido gastrico)
LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
Ad esclusione della via e.v., tutte le altre vie di somministrazione richiedono il passaggio
del farmaco attraverso cellule e/o membrane e la diffusione in spazi intercellulari prima
di arrivare al sangue.
La velocità di assorbimento dipende sia dalla via di somministrazione che da una serie
di variabili legate alle caratteristiche del farmaco e/o al tessuto.
LE VARIABILI CHE INFLUENZANO LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
DIPENDONO:
- dal farmaco o dalla preparazione farmaceutica (coefficiente di ripartizione, dissolubilità)
- dalla superficie assorbente (estensione, permeabilità, vascolarizzazione)
LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
Ad esclusione della via e.v., tutte le altre vie di somministrazione richiedono il passaggio
del farmaco attraverso cellule e/o membrane e la diffusione in spazi intercellulari prima
di arrivare al sangue.
La velocità di assorbimento dipende sia dalla via di somministrazione che da una serie
di variabili legate alle caratteristiche del farmaco e/o al tessuto.
LE VARIABILI CHE INFLUENZANO LA VELOCITA’ DI ASSORBIMENTO DEI FARMACI
DIPENDONO:
- dal farmaco o dalla preparazione farmaceutica (coefficiente di ripartizione, dissolubilità)
- dalla superficie assorbente (estensione, permeabilità, vascolarizzazione)
ESTENSIONE DELLA SUPERFICIE ASSORBENTE:
Maggiore è la superficie assorbente, più rapido è l’assorbimento di un farmaco (per es.
intestino vs stomaco).
PERMEABILITA’ DELLA SUPERFICIE ASSORBENTE:
La permeabilità degli epiteli di rivestimento dipende dal loro spessore e dal loro grado
di corneificazione.
La velocità di assorbimento di un farmaco è > attraverso l’epitelio intestinale
(monostratificato) rispetto a quello faringeo (stratificato non corneificato) e alla cute
(epitelio stratificato corneificato).
La permeabilità può variare in condizioni patologiche (per es. farmaci applicati su
abrasioni o ulcere cutanee possono passare rapidamente in circolo).
Per le vie di somministrazione parenterali (non e.v. o i.a.) il farmaco deve superare la
barriera endoteliale capillare (permeabile a sostanze idrofile anche di grosse
dimensioni) => la velocità di assorbimento dipende dal grado di perfusione ematica al
sito di inoculazione e, in minima parte, dallo stato di idratazione del tessuto
connettivale circostante (varia tra tessuti e con l’età).
VASCOLARIZZAZIONE
La velocità di assorbimento è maggiore nelle zone di assorbimento più vascolarizzate
e con maggiore flusso ematico.
La vascolarizzazione dipende da:
- Tipo di tessuto (i.m. > s.c.)
- Stati infiammatori locali
- Richieste funzionali (attività fisica nel muscolo, riscaldamento della cute, etc)
- Sostanze vasocostrittrici (v. per es. VC + anestetici locali)
VIE ENTERALI
Vie enterali
Orale
Sublinguale
Rettale
(per os)
Vie
parenterali
sistemiche
Intravascolare
Intramuscolare
Cutanea
Endovenosa (e.v.), intracardiaca, intrarteriosa
(i.m.)
Sottocutanea (s.c.) e intradermica (anche topica)
Altre vie
D’Organo
Intracavitaria
Transcutanea
Transmucosale
Intratecale, intrarticolare e inalatoria (anche sistemica)
Intraperitoneale e intrapleurica
Topica, ma anche regionale o sistemica (es. cerotti)
Oculare, vaginale, etc.
Vantaggi: la somministazione è semplice, non provoca dolore e non richiede
l’intervento di personale specializzato.
VIA DI SOMMINISTRAZIONE ORALE
Sangue
F -> Dissoluzione nel tubo digerente -> mucosa GI -> sist. portale -> fegato -> sangue
BIODISPONIBILITA’ ORALE: la percentuale della dose somministrata che entra nella
circolazione sistemica ed è in grado di distribuirsi a tutto l’organismo.
La biodisponibilità orale e la velocità dell’assorbimento dipendono da: stato funzionale
dell’apparato digerente, stato fisico e composizione del contenuto, velocità di transito
GI, attività della flora intestinale, eventuale metabolizzazione epatica del F.
L’assorbimento dal lume GI al sangue avviene principalmente per diffusione passiva
(=> è influenzato dal CR e dall’eventuale grado di ionizzazione del F).
L’assorbimento avviene soprattutto a livello del piccolo intestino per:
- elevata superficie assorbente (ca. 200 m2)
- elevata vascolarizzazione (flusso ematico ca. 1L/min)
- permeabilità elevata per molecole di piccole dimensioni anche se dotate di CR
relativamente basso (x molti F la velocità di assorbimento dipende soprattutto dal
flusso ematico intestinale)
Il tempo di transito nel piccolo intestino è di ca. 3 ore => variazioni del transito
intestinale possono modificare l’assorbimento dei farmaci.
Poiché in genere l’assorbimento a livello gastrico è molto basso, lo stomaco funziona
solo da organo di passaggio/deposito. Il tempo di svuotamento gastrico influenza la
velocità di assorbimento (da parte dell’intestino) e la rapidità dell’insorgenza
dell’effetto.
Farmaci o situazioni che alterano la motilità gastrica possono modificare
l’assorbimento di altri farmaci assunti per via orale (es: AD triciclici, atropina, ulcera
gastrica, emicrania rallentano lo svuotamento gastrico, mentre la metoclopramide e
il digiuno aumentano la motilità gastrica)
Sali di Ca2+ o Al3+ (antiacidi) rallentano lo svuotamento gastrico => riducono
l’assorbimento di beta-bloccanti e di altri farmaci. I sali di Mg, invece, accelerano lo
svuotamento gastrico.
Farmaci che alterano
l’assorbimento
Effetto
sull’assorbimento
Farmaci il cui assorbimento è
alterato
Anticolinergici
Ridotto
Aumentato
Paracetamolo, Tetracicline,
Fenilbutazone, Litio (preparazioni
ritardo)
Digossina
Analgesici narcotici
Ridotto
Alcool (morfina)
AD triciclici
Ridotto
Aumentato
Fenilbutazone
Ac. para-aminosalicilico
Metoclopramide
Ridotto
Aumentato
Digossina
Aspirina, Levodopa, Paracetamolo,
Tetracicline, Litio (preparaz ritardo)
FATTORI CHE INFLUENZANO LO SVUOTAMENTO GASTRICO:
A digiuno: lo svuotamento gastrico di liquidi, e quindi dei farmaci in essi disciolti, può
essere molto rapido. Lo stomaco presenta un’intensa contrazione espulsiva ogni ca.
30 min => il farmaco sciolto nei succhi gastrici passa all’intestino (se la dissoluzione
non è completa, il farmaco può aderire alla parete gastrica e non essere espulso).
A stomaco pieno: contrazioni regolari e discrete provvedono a mescolare e a triturare
il contenuto gastrico; attraverso il piloro passano solo particelle < 2mm (le forme
farmaceutiche solide devono dissolversi in particelle più piccole di 2 mm). Tempo di
svuotamento gastrico = ca. 7h (> pasti grassi, < pasti leggeri).
Presenza e tipo di cibo: la presenza e il tipo di cibo a livello intestinale influenzano il
flusso ematico splancnico (> con cibi ricchi di proteine, < con i carboidrati) e quindi la
velocità di assorbimento.
pH: l’aumento del pH accelera lo svuotamento a digiuno (le forme tamponate di ASA
sono assorbite più rapidamente). Farmaci che aumentano il pH: antiacidi o inibitori
della pompa protonica (omeprazolo).
EFFETTO DI PRIMO PASSAGGIO
I farmaci assorbiti a livello intestinale
passano attraverso il fegato, prima di
raggiungere il sangue => a livello
epatico possono essere
metabolizzati.
Se il metabolismo durante il primo
passaggio attraverso il fegato è tale
da ridurre o annullare l’attività
farmacologica (<<< biodisponibilità),
si parla di effetto di primo passaggio.
Farmaci sensibili all’effetto di primo passaggio:
Amitriptilina, Desipramina (AD triciclici)
Diltiazem, Nifedipina, Verapamil (Ca-antagonisti)
5-Fluorouracile, Mercapropurina (chemioterapici)
Isoproterenolo (agonista beta-adrenergico)
Lodocaina (anestetico locale)
Metoprololo, Propanololo (beta-bloccanti)
Morfina (analgesico oppiaceo)
Neostigmina (inibitore colinesterasi)
Testosterone (ormone sessuale)
Nitroglicerina (antianginoso) => somm. sublinguale (o buccale)
Effetto della contemporanea assunzione di cibo
sull’assorbimento di alcuni farmaci somministrati per os
Assorbimento ridotto
Assorbimento aumentato
Ampicillina
Amoxicillina
Rifampicina
Aspirina
Isoniazide
Levodopa
Griseofulvina
Carbamazepina
Propanololo
Metoprololo
Spironolattone
Idralazina
NB: la biodisponibilità orale del propanololo è influenzata dalla presenza di cibo
poiché i processi di cattura epatica del farmaco sono saturati da substrati presenti
negli alimenti => l’effetto di primo passaggio del F è ridotto e la conc plasmatica > se
assunto durante i pasti.
REAZIONI CHIMICHE A LIVELLO DEL LUME GI:
Nel lume GI il farmaco può reagire con i cibi, con altri farmaci, con l’HCl gastrico o può
essere trasformato dalla flora batterica intestinale => alterazioni della biodisponibilità.
Reazione
Farmaco
Effetto
Formazione di
complessi
insolubili
Tetracicline
Ridotto assorbimento dei
complessi con Ca2+ (latte) o
Al3+ (antiacidi)
Solfoconiugazione
Glucuronazione
Isoproterenolo
Salicilamide
Perdita di attività
farmacologica
Idrolisi acida
Penicillina G, Eritromicina
Idrolisi enzimatica
Aspirina
Perdita di attività
farmacologica
Formazione di ac. salicilico,
attivo
Riduzione (flora
intestinale)
Sulfasalazina
Formazione di ac. 5aminosalicilico attivo
Vantaggi della somministrazione orale:
- Somministrazione facile, economica, poco rischiosa
- Possibile modulazione della velocità di assorbimento (in base alla forma
farmaceutica)
- Possibile assorbimento di “complessi” sia per trasporto attivo che per pinocitosi
Svantaggi della somministrazione orale
- Irritazione => vomito
- Inattivazione per idrolisi enzimatica (insulina)
- Inattivazione per l’acidità gastrica (penicillina G)
- Metabolizzazione epatica (effetto di primo passaggio)
- Distruzione della flora intestinale (antibiotici a largo spettro)
Per farmaci che vengono disattivati nel lume GI o che subiscono effetto di primo
passaggio si preferiscono le vie sublinguale o rettale (bocca e plessi emorroidari
-> vena cava -> ventricolo dx)
VIA DI SOMMINISTRAZIONE SOTTOLINGUALE
CARATTERISTICHE DEL CAVO ORALE:
-
pH = 6
-
superficie limitata
-
intensa irrorazione (la NTG viene assorbita rapidamente!)
CARATTERISTICHE DEI F ASSORBITI X QUESTA VIA:
-
elevato CR olio/acqua (es. NTG)
-
elevata potenza (attivo a basse dosi)
-
rapida solubilizzazione
VANTAGGI:
-
assorbimento rapido => azione immediata (v. NTG)
-
passaggio diretto nel ventricolo dx (no effetto di 1° passaggio)
SVANTAGGI:
-
sostanze sgradevoli o irritanti non possono essere somministrate x questa via
-
la ridotta superficie di assorbimento limita l’uso di tale via ai pochi farmaci dotati di
CR elevato
VIA DI SOMMINISTRAZIONE RETTALE
La maggior parte del F assorbito non passa attraverso il fegato. Solo la vena
emorroidaria superiore si immette nel circolo portale mentre la vena emorroidaria
media e la inferiore si immettono nella vena cava inferiore -> cuore
La via rettale viene utilizzata in caso di:
-irritazione gastrica provocata dal farmaco
-chirurgia GI
-vomito protratto
-pazienti incapaci di deglutire o non cooperanti (es. bambini)
SVANTAGGI:
-modesta area di assorbimento
-assorbimento non costante => biodisponibilità imprevedibile
-assorbimento più lento e prolungato=> consente il matenimento di conc efficaci di
alcuni F antinfiammatori o boncodilatatori durante le ore notturne
-facile irritazione -> possibile espulsione
-inattivazione locale da parte dei batteri
VIE PARENTERALI SISTEMICHE
Vie enterali
Orale
Sublinguale
Rettale
(per os)
Vie
parenterali
sistemiche
Intravascolare Endovenosa (e.v.), intracardiaca, intrarteriosa
Intramuscolare (i.m.)
Cutanea
Sottocutanea (s.c.) e intradermica (anche topica)
Altre vie
D’Organo
Intracavitaria
Transcutanea
Transmucosale
Intratecale, intrarticolare e inalatoria (anche sistemica)
Intraperitoneale e intrapleurica
Topica, ma anche regionale o sistemica (es. cerotti)
Oculare, vaginale, etc.
VIA INTRAVASCOLARE
VANTAGGI:
- rapidità di intervento (terapia d’urgenza*)
- controllo rapido dell’efficacia del farmaco (utile per farmaci a basso IT)
- biodisponibilità = 100% (anche per farmaci ad alto PM, es. emoderivati, o per
farmaci rapidamente metabolizzati prima o durante l’assorbimento)
- possibilità di somministrare farmaci irritanti (somm dolorosa x via s.c. o i.m.)
- somministrazione in unico bolo o per infusione continua (fleboclisi, anche x volumi
elevati)
La via endoarteriosa, meno usata, è sfruttata in quelle situazioni in cui il F deve
raggiungere conc. elevate in un organo specifico (es. chemioterapici o angiografia).
SVANTAGGI/COMPLICANZE:
- una volta immesso nel torrente circolatorio il F non è più recuperabile
- la somministrazione rapida può indurre danni a carico degli organi + perfusi (cuore
e cervello) => è consigliabile somm. la dose e.v. molto lentamente (ca. 2 min),
anche per avere il tempo di interrompere l’iniezione in seguito a comparsa di effetti
tossici acuti.
- infezioni causate dall’utilizzo di materiale e/o composti non sterili (es.
tossicodipendenti)
- embolismo causato dalla somministrazione di sostanze oleose (non destinate alla
somm e.v.), aria o gas, precipitati presenti nella soluzione, soluzioni ipertoniche (->
aggregati di globuli rossi) o ipotoniche (-> emolisi).
VIA INTRAMUSCOLARE
La sede più utilizzata è il muscolo gluteo (anche deltoide) che consente di iniettare
anche volumi elevati (> 5ml). Negli anziani e nei bambini: attenzione alle lesioni del
nervo sciatico.
L’assorbimento dipende da:
- sito dell’iniezione (la velocità di assorbimento è > nel deltoide rispetto al gluteo)
- irrorazione del muscolo (> con l’attività fisica, VD e massaggio)
- CR del F
- volume e osmolarità della soluzione iniettata
- presenza di tessuto adiposo (deposito di sostanze lipofile => assorb + lento)
Le molecole piccole passano direttamente nel sangue, mentre grosse proteine vi
arrivano dopo essere passate dal circolo linfatico.
Farmaci in soluzione acquosa: assorbimento in 10-30 min
Farmaci insolubili al pH interstiziale o sospesi in soluzione oleosa: assorbimento lento
(preparazioni ritardo, per es. il sale di procaina della penicillina G è assorbito in 24 h,
mentre il sale sodico in 2-3 h).
SVANTAGGI/COMPLICAZIONI:
• Dolore (x distensione e irritazione del tessuto muscolare)
• Necrosi tissutale locale (soluzioni alcaline)
• Lesioni vascolari o nervose (dovute ad imperizia)
• Contaminazione batterica (ascessi)
VIA CUTANEA
1) Intradermica: piccoli volumi (0,1-0,2 ml) vengono iniettati nel connettivo dermico
sottostante l’epidermide (usata solo per l’introduzione di allergeni a scopo
diagnostico)
2) Sottocutanea: il F è iniettato nel connettivo sottocutaneo (vol. fino a 2 ml) a livello
dell’avambraccio o dell’addome.
La velocità di assorbimento dipende principalmente dal flusso ematico locale:
-
Il flusso ematico cutaneo è < di quello muscolare => l’assorbimento è più lento
-
VC => rallenta l’assorbimento sistemico, aumenta e prolunga l’efficacia a livello
locale (es. adrenalina + anestetici locali)
VANTAGGI:
-
Si possono somm. farmaci con bassa biodisponibilità orale (es. eparina, insulina,
etc.)
-
Effetti locali e/o sistemici
-
Possibilità di impianto di forme a lento rilascio (deposito di farmaci relativamente
insolubili e/o sospesi in un eccipiente semisolido che non dissolve rapidamente nel
tessuto sottocutaneo). Es. ormoni steroidei, insulina-zinco, etc.
SVANTAGGI:
-
Dolore, ascesso o necrosi (evitare la somm di preparazioni irritanti o non sterili)
-
Più dolorosa rispetto alla via i.m. (+ fibre nocicettive)
- La velocità di assorbimento è piuttosto variabile (dip dal flusso ematico locale)
ALTRE VIE
Vie enterali
Orale
Sublinguale
Rettale
(per os)
Vie
parenterali
sistemiche
Intravascolare
Intramuscolare
Cutanea
Endovenosa (e.v.), intracardiaca, intrarteriosa
(i.m.)
Sottocutanea (s.c.) e intradermica (anche topica)
Altre vie
D’Organo
Intracavitaria
Transcutanea
Transmucosale
Intratecale, intrarticolare e inalatoria (anche sistemica)
Intraperitoneale e intrapleurica (poco usate)
Topica, ma anche regionale o sistemica (es. cerotti)
Oculare, vaginale, etc.
VIA INALATORIA
VANTAGGI:
- Uso topico (es aerosol: broncodilatatori, mucolitici, cortisonici, etc) o sistemico (es
gas: anestetici generali)
- L’assorbimento a livello alveolare è molto rapido (superficie ca. 100 m2 + stretta
vicinanza fra epitelio alveolare ed endotelio capillare)
L’assorbimento è regolato dalla profondità e dalla frequenza degli atti respiratori:
1° fase (lavaggio polmonare): equilibrio fra aria inspirata e aria alveolare
2° fase (fase circolatoria): equilibrio fra aria alveolare, plasma e tessuti
AEROSOL
Farmaci in forma solida o liquida non volatile possono essere sospesi in aria o altri gas e
somministrati in forma di aerosol, ovvero particelle di dimensioni così ridotte da rimanere
a lungo sospese e sedimentare lentamente.
La > parte delle particelle si deposita sulle pareti dell’albero respiratorio per impatto.
La percentuale di deposizione dipende da:
- velocità di sedimentazione (proporzionale al diametro delle particelle)
- velocità di diffusione
- precipitazione inerziale (tendenza di una particella a proseguire verso la stessa
direzione quando il flusso d’aria cambia, per es. biforcazioni bronchiali)
- volume respiratorio (se > il vol resp, > transito, < deposizione nelle vie aeree superiori,
mentre è favorita la deposizione nelle vie bronchiali profonde)
- velocità di flusso (la probabilità che una particella si depositi aumenta con la riduzione
del flusso => la deposizione di particelle medio-piccole è > nei bronchioli terminali e
negli alveoli dove la velocità di flusso è minima)
- pervietà delle vie respiratorie (es. malattie polmonari ostruttive croniche)
- caratteristiche fisiche dell’aerosol (stabilità, tensione superficiale, etc)
=> L’ASSORBIMENTO PER VIA INALATORIA E’ MOLTO VARIABILE!!!
Nel caso degli areosol, il destino delle particelle
dipende dalle loro dimensioni:
Ø > 20 mm
tendono a depositarsi nelle vie aeree superiori =>
inutili x patologie bronchiali o alveolari
1 mm < Ø < 10 mm
si depositano nell’albero tracheo-bronchiale.
Quelle solubili sono assorbite e possono produrre
azione locale (x affezioni bronchiali o alveolari),
quelle insolubili sono rimosse dai movimenti delle
ciglia, portate in faringe e quindi ingerite
Ø < 1 mm
raggiungono gli alveoli dove potrebbero essere
assorbite, ma in genere vengono in buona parte
esalate senza essere trattenute.
SVANTAGGI DELLA VIA INALATORIA:
- Possibili irritazioni locali
- Scarsa possibilità di regolare la dose di farmaco somministrata
- Assorbimento variabile
VIA INTRARTICOLARE
Consiste nell’iniettare farmaci (antinfiammatori, anestetici locali, antibiotici) all’interno
delle capsule articolari per la cura di patologie locali.
Somministrazioni ripetute e frequenti possono produrre effetti lesivi.
VIA INTRATECALE
Consiste nella somministrazione diretta nel liquido cerebrospinale (nello spazio
subaracnoideo per iniezione lombare o nelle cavità ventricolari)
Utile per ottenere effetti rapidi a livello delle meningi e delle radici dei nervi spinali. Utile
anche per trattare infezioni acute (penicillina nella meningite acuta) o tumori cerebrali o
spinali o per introdurre mezzi di contrasto per indagini radiografiche. Per l’anestesia
spinale si preferisce la via epidurale.
Consente di superare l’ostacolo della BEE
Via dolorosa e rischiosa
VIA TRANSCUTANEA (O DERMICA)
Oli, creme, unguenti, paste, polveri dispersorie, lozioni, spray, linimenti
Usata generalmente per ottenere azioni locali o topiche, ma anche per farmaci ad
azione sistemica x evitare l’effetto di primo passaggio (es. gliceril-trinitrato per la
terapia cronica dell’angina pectoris, cerotti per il mal di moto)
Potenziali siti di accesso (≠ permeabilità):
1) strato corneo (può anche rappresentare un sito di deposito)
2) ghiandole sudoripare
3) follicoli piliferi (x gli steroidi è la via principale)
FATTORI CHE INFLUENZANO LA BIODISPONIBILITA’:
- Condizioni della cute (abrasioni, lesioni ulcerative e ustioni => discontinuità dello strato
corneo => rapida diffusione del F al connettivo sottostante e quindi al sangue)
- Grado di vascolarizzazione (flogosi, > temperatura o agenti irritanti => ↑assorbimento
freddo, lacci emostatici, VC => ↓assorbimento)
- Stato di idratazione (lo strato corneo del neonato è più sottile e idratato dell’adulto; le
medicazioni occlusive aumentano l’idratazione dello strato corneo => ↑ assorbimento)
- Spessore e permeabilità cutanei (dipende dalla regione: plantare<avambraccio<cuoio
capelluto<scroto<faccia posteriore del padiglione auricolare)
- Caratteristiche del F (CR, farmaci poco liposolubili vengono assorbiti meglio se sospesi
in unguenti o creme grasse)
- Caratteristiche del veicolo in cui e disciolto il F (propilenglicole e DMSO alterano lo
stato di idratazione dello strato corneo)
Nella zona retroauricolare la cute è sottile e poco esposta ad agenti atmosferisci o
esterni che indurrebbero variazioni della vascolarizzazione locale.
ALTRE VIE
Vie enterali
Orale
Sublinguale
Rettale
(per os)
Vie
parenterali
sistemiche
Intravascolare
Intramuscolare
Cutanea
Endovenosa (e.v.), intracardiaca, intrarteriosa
(i.m.)
Sottocutanea (s.c.) e intradermica (anche topica)
Altre vie
D’Organo
Intracavitaria
Transcutanea
Transmucosale
Intratecale, intrarticolare e inalatoria (anche sistemica)
Intraperitoneale e intrapleurica (poco usate)
Topica, ma anche regionale o sistemica (es. cerotti)
Oculare, vaginale, etc.
VIE MUCOSALI
Mucose nasali, congiuntivali, oro-faringee, vaginali e uretrali
Assorbimento consistente per mancanza di strato corneo e ridotta stratificazione
epiteliale (=> possibilità di effetti sistemici)
La via nasale è frequentemente usata per farmaci decongestionanti (vasocostrittori); ma
l’applicazione frequente può danneggiare e ulcerare la mucosa (es. cocainomani)
La via nasale può anche essere sfruttata per avere effetti sistemici rapidi (per es. spray
di ormoni peptidici come la calcitonina)
Via oculare: assorbimento attraverso la congiuntiva e attraverso la mucosa nasale (dopo
drenaggio dal dotto lacrimale)
CINETICHE DI ASSORBIMENTO
L’assorbimento dei farmaci somministrati per via orale o parenterale generalmente
segue una cinetica di I ordine (flusso proporzionale alla concentrazione).
CINETICA DI PRIMO ORDINE: la quantità di F assorbita per unità di tempo è una %
costante di quella che rimane da assorbire.
Kass
EMIVITA DI ASSORBIMENTO
(t1/2ASS): tempo necessario a
dimezzare la concentrazione del
farmaco nel compartimento
assorbente
Linea tratteggiata: cinetica di
assorbimento di ordine zero =>
Vass non dipende dalla [F], ma è
costante (es. trasporto attivo)
La cinetica è definita da una costante di assorbimento (frazione assorbita per unità di
tempo), ovvero da una costante di tempo dell’assorbimento t = 1/Ka
(orale, i.m., s.c., etc.)
La concentrazione plasmatica (Cp) dipende dall’equilibrio fra assorbimento ed eliminazione:
- all’inizio: assorbimento massimo, eliminazione nulla
- con il passare del tempo: ↓assorbimento (<conc al sito di somm), ↑ eliminazione (> con ai
siti di eliminazione)
ASS > ELIM
ASS=ELIM
ASS < ELIM
La velocità di assorbimento determina il livello del picco massimo plasmatico del
farmaco e il tempo necessario per raggiungerlo:
Uno stesso F è somministrato in due
muscoli con irrorazione diversa (A>B
=> VassA>VassB)
Le diverse velocità di assorbimento
influnzano sia i flussi di eliminazione
che le conc plasmatiche (C e D)
Il tempo di picco (Vass = Velim) è
ritardato ed ha un valore minore
quando l’assorbimento è più lento (D).
In tempi successivi al picco, Cp è > se
l’assorbimento è + lento.
La velocità di assorbimento determina il livello del picco massimo plasmatico del
farmaco e il tempo necessario per raggiungerlo:
Uno stesso F è somministrato in due
muscoli con irrorazione diversa (A>B
=> VassA>VassB)
1) Più lento è l’assorbimento,
Le diverse velocità di assorbimento
piùinflunzano
basso sia
e iritardato
è
flussi di eliminazione
che le conc plasmatiche (C e D)
il picco massimo di concentrazione plasmatica
2) Il valore e il tempo del piccoIl tempo
massimo
di piccodipendono
(Vass = Velim) è
dall’efficienza dei
ritardato ed ha un valore minore
processiquando
di eliminazione
l’assorbimento è più lento (D).
In tempi successivi al picco, Cp è > se
l’assorbimento è + lento.
La velocità di assorbimento dipende dalla via di somministrazione
Somministrando una stessa dose di F:
CONC. PICCO: e.v.>i.m.>s.c.>enterale
VELOCITA’ ASS.: i.m.>s.c.>enterale
via intramuscolare
Conc. dopo picco: enterale>i.m.>e.v.
I livelli plasmatici dipendono dalla via di somministrazione
A parità di via di somministrazione, i livelli plasmatici del farmaco dipendono dalla
dose somministrata (raddoppiando la dose, si raddoppia la quantità assorbita, salvo
saturazione dei meccanismi di assorbimento).
Sono disponibili diverse formulazioni che rilasciano il F con cinetiche controllate
(tecnologia del “drug delivery”) per consentire:
- protezione del F dalla degradazione e stabilizzazione (es. capsule
gastroresistenti)
- preparazioni di F (biodisponibili solo per via parenterale) somministrabili per os,
per via cutanea o inalatoria
- prolungamento della permanenza in circolo del F (es. cerotti transdermici)
- indirizzamento del F a specifici organi, tessuti o tipi cellulari (es. liposomi con
tropismo cutaneo, polimeri bio-adesivi)
- rilascio regolato del F in funzione di condizioni locali o attraverso stimoli
elettrici/magnetici (es.ionoforesi)