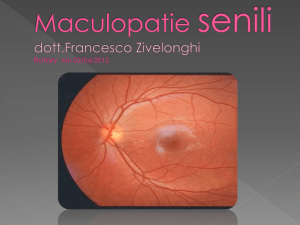
LA DEGENERAZIONE MACULARE SENILE PUO'
ESSERE CURATA CON "ACETILCARNITINA"
Il pericolo comincia a sessant'anni. Sintomi? Uno solo: la
riduzione del campo visivo. Nella degenerazione maculare
senile c'è una macchia che copre la parte centrale di ciò che
si sta guardando. Restano nitidi i contorni. Gli oculisti
portano l'esempio di piazza San Pietro, a Roma.
L'anziano colpito da degenerazione maculare senile, dicono,
non vede la basilica ma solo una parte del colonnato del
Bernini. Ciò provoca serie conseguenze: diventa
difficilissimo leggere e ancora più difficile guidare
l'automobile. Questo perchè la macula (che è la pèarte
centrale e più nobile della retina) è seriamente
compromessa. Questa è una malattia tipic della terza età
(dato che si vive sempre più a lungo i casi sono in aumento):
negli ottantenni supera la soglia del 30%, nei settantenni si
ferma al venti. E' quindi indispensabile una diagnosi
precoce, certamente favorita da frequenti controlli
dell'acuità visiva. Chi avverte difficoltà deve subito
consultare uno specialista per sapere se si tratta di
degenerazione maculare senile, di cataratta o di altre
patologie. Nel primo caso è urgente correre ai rimedi.
I ricercatori di tutto il mondo cercano da anni un rimedio
sicuro per fermare l'avanzata di questa patologia, che
rappresenta la prima causa di cecità dell'anziano. molte
scoperte sono ancora sotto esame: servono, infatti, conferme
affidate a studi controllati eseguiti in doppio cieco.
Una novità arriva dall'Ungheria, nazione in cui l'oculista
Janos Feher ha condotto uno studio clinico in
collaborazione con Sigma - Tau Health Science su cento
pazienti di età compresa tra i 55 e i 70 anni, tutti affetti da
degenerazione maculare secca, ma fortunatamente nella
fase iniziale. Cinquanta di essi sono stati trattati con un
placebo, cinquanta con un mix di acetilcarnitina, acidi grassi
polinsaturi e coenzima Q10, che protegge dall'ossidazione i
mitocondri.
Dice il professor Feher, visiting professor all' università
romana La Sapienza: " Lo studio è durato un annoma già al
quinto - sesto mese abbiamo ottenuto non solo la
decelerazione che ci aspettavamo, ma adirittura una
magiore acuità visiva nei cinquanta soggetti che avevano
assunto per via orale il nuovo preparato. Ho presentato lo
studio un mese fa, inb Florida, al congresso internazionale
dell'Arvo". Feher sostiene di aver curato fino ad oggi più di
cinquemila pazienti, ma di aver capito che si stava aprendo
una nuova possibilità per la terapia della degenerazione
maculare soltanto con questo trial,destinato ad entrare nella
stria di questa brutta malattia.
da "IL GIORNALE"
NUOVI TRAGUARDI NELLA RICERCA
DEGENERAZIONE MACULARE UMIDA
Si cercano pazienti per una nuova sperimentazione clinica.
All'esame un medicamento del quale si intende accertare la
non nocività e l'efficacia rispetto al trattamento con la
terapia fotodinamica
La Alcon Laboratories ha in corso di effettuazione una
sperimentazione clinica multipla di fase 3 per persone che
presentano perdita della vista a causa della forma "umida"
della Degenerazione Maculare Senile (DMS) derivata dalla
neovascolarizzazione della coroide (NVC), cioè la crescita di
vasi sanguigni anormali nella parte posteriore dell'occhio.
Questi vasi sanguigni spesso sanguinano, perdendo liquido,
fenomeno al quale la DMS "umida" deve il suo nome. La
sperimentazione clinica, che al momento "arruola" pazienti,
paragonerà la non nocività e l'efficacia di un medicamento
denominato "Anecortave acetato" rispetto al trattamento
con la terapia fotodinamica Visudyne, approvata dalla Food
And Drug Administration (FDA) per l'inibizione della
crescita di vasi sanguigni nella parte posteriore dell'occhio,
salvando la vista delle persone che sono state colpite dalla
DMS umida. Nel corso di due anni, i pazienti del gruppo
Anecortave acetato riceveranno quattro iniezioni (una ogni
sei mesi) di Anecortave acetato dietro l'occhio, mentre i
pazienti del gruppo Visudyne riceveranno fino ad otto
trattamenti con Visudyne. Inoltre tutti i pazienti verranno
sottoposti gratuitamente ad esami fisici ed oculistici. I
pazienti ignoreranno ovviamente se assumeranno il farmaco
sperimentale Anecortave acetato o il Visudyne e per essere
ammessi alla sperimentazione devono prima sottoscrivere
una dichiarazione di consenso con la quale riconoscono di
essere stati informati circa i trattamenti cui verranno
sottoposti e i rischi correlati. La sperimentazione clinica è
stata offerta a cittadini statunitensi affetti da DMS umida, di
almeno 50 anni di età. Per maggiori informazioni,
consultare il sito della "Alcon Laboratories":
www.alconic.com
Tratto da "LUMEN", numero 30/2003
da Newsletter, inverno 2002, organo di Fighting Blindness Irlanda. Traduzione e adattamento di Carlo Parolini
CHE COSA SONO LE SPERIMENTAZIONI
CLINICHE?
Le sperimentazioni cliniche sono studi atti a stabilire la non
nocività e l'efficacia di terapie sperimentali in soggetti
ammalati e rappresentano la fase finale di ricerca prima che
la FDA dia l'approvazione perché un trattamento possa
essere utilizzato su esseri umani. La prova è normalmente
attuata in tre fasi, ciascuna delle quali implica in successione
un numero crescente di persone. Gli studi della fase 1 sono
principalmente rivolti ad accertare la non nocività del
farmaco e a determinarne il dosaggio massimo tollerato.
Questa fase iniziale della prova su esseri umani si svolge su
un numero piccolissimo di volontari. Talvolta vi sono
sottoposti adulti sani privi della malattia; a volte lo sono
invece pazienti all'ultimo stadio. Ad esempio, nelle
sperimentazioni della fase 1 iniziale, che tendono a provare
soltanto la non nocività dei trapianti di cellule retiniche,
sono implicati unicamente pazienti già non vedenti: in
questo modo non si mettono a rischio di peggioramento
della funzione visiva pazienti che presentano ancora un
residuo visivo. Una volta che un farmaco sia trovato non
nocivo, il passo successivo è di testarne l'efficacia in uno
studio di fase 2, la cui durata dipende largamente dalla
natura del trattamento in sperimentazione e dalla patologia
specifica. Gli studi di fase 2 possono interessare grandi
numeri di pazienti presso diversi Centri medici in tutta la
nazione, i quali partecipano alla sperimentazione. Quando le
terapie approvate dall'FDA già esistono per una determinata
patologia, si richiedono studi della fase 3 per verificare il
nuovo trattamento nei confronti della terapia standard al
fine di stabilire quale sia la più efficace. Gli studi della fase 3
consentono inoltre ai ricercatori di monitorare gli effetti
collaterali di un medicamento su una vasta popolazione di
pazienti affetti e questa verifica su vasta scala fornisce
all'industria farmaceutica e all'FDA una comprensione più
approfondita sia della validità del farmaco in esame, sia dei
suoi benefici e della gamma di possibili reazioni
indesiderate. Queste fasi delle sperimentazioni cliniche sono
state originariamente studiate per l'utilizzo nella ricerca sul
cancro, quali strumenti di aiuto per sperimentare nuovi
trattamenti in modo sicuro e sistematico. Tuttavia, le varie
fasi di una sperimentazione vengono talvolta combinate: ad
esempio, le sperimentazioni della fase 2 e della fase 3 sono
spesso combinate quando non esistano già terapie standard.
In altri casi sono le sperimentazioni della fase 1 e della fase 2
ad essere combinate quando siano ben compresi la non
nocività e il dosaggio di un farmaco.
Tratto da "LUMEN", numero 30/2003
CURA DELLA RETINITE PIGMENTOSA ALLO STATO
AVANZATO
Un nuovo tipo di trapianto dai risultati tutti da verificare
Al professor Valter Gualandi è stato richiesto di commentare
una sperimentazione che sembrerebbe aver avuto esiti
promettenti.
LA NOTIZIA: Il trapianto di una porzione di retina di occhi
di feti abortiti sembra avere notevolmente migliorato la
visione di due persone, sulle quattro sottoposte a questo
intervento, tutte affette da retinite pigmentosa a uno stadio
avanzato. Il miglioramento è stato attestato soggettivamente
dagli stessi due pazienti, i quali hanno affermato di riuscire
a vedere, dopo l'intervento, particolari del viso di altre
persone, anche se poste a distanza. La sperimentazione è
stata condotta da Robert Aramant e Magdalene Seller,
dell'Istituto Oculistico Dehany di Los Angeles, che, alla luce
dei risultati ottenuti, hanno chiesto l'autorizzazione a
continuare questi studi sperimentali. Il tessuto trapiantato
ha una superficie di due millimetri quadrati e comprende
due strati istologici.
IL COMMENTO del Prof. Valter Gualandi: "Anzitutto
precisiamo che la retina ha per l'appunto una struttura
istologica a strati: lo strato più esterno si chiama epitelio
pigmentato ed è formato da cellule ricche di pigmento.
Sopra di esso, ovvero verso il cristallino, sono stratificati lo
strato dei coni e dei bastoncelli, ossia le cellule che
propriamente ricevono la stimolazione della luce e hanno la
proprietà di trasformarla in segnale nervoso, e poi lo strato
delle cellule nervose che accolgono impulsi nervosi
trasmettendoli al cervello. Il trattamento sperimentale delle
malattie degenerative della retina prevede da diversi anni il
trapianto di cellule fetali; la loro finalità dichiarata non è
però quella di sostituire il tessuto retinico in via di
degenerazione, bensì quello di salvare le cellule retiniche
ancora biologicamente attive. Questo salvataggio avverrebbe
tramite la produzione di fattori di crescita, ovvero di
sostanze chimiche che sostengono lo stato trofico delle
cellule-bersaglio. La novità della sperimentazione messa in
atto dai due ricercatori statunitensi, sta nell'avere
trapiantato una porzione di retina che ha due precipue
caratteristiche:
1) proviene da feti abortiti. Quindi, è verosimile che le cellule
che la costituiscono non abbiano ancora sviluppato gli
antigeni di superficie che condizionano il rigetto. In tale
condizione il rigetto non si verificherebbe sensibilmente e i
pazienti non necessiterebbero del trattamento
immunodepressivo.
2) il tessuto trapiantato comprende lo stato epiteliale
pigmentato e anche quello delle cellule sensoriali e nervose.
Queste ultime. Pertanto, sarebbero supportate fisicamente e
nutrite dallo strato sottostante, per cui potrebbero
sopravvivere e stabilire una connessione con le altre
strutture retiniche. Si realizzerebbe così non solo l'arresto
della malattia (il salvataggio) ma anche una vera e propria
sostituzione del tessuto trapiantato con quello in corso di
degenerazione.
Questa sperimentazione comprende purtroppo tanti
condizionali e non si può escludere che il risultato ottenuto
sia appunto ascrivibile all'effetto salvataggio e sia pertanto
transitorio. Sta il fatto che i pazienti dichiarano "un sensibile
miglioramento" nella capacità visiva, un miglioramento di
rapida realizzazione, ma non "un arresto" nella progressione
della malattia. Esprimere un giudizio definitivo su questa
sperimentazione non appare pertanto possibile, in quanto si
pone infatti la necessità di verificare:
a) se il miglioramento è duraturo e se evolve in qualche
maniera.
b) se si possono cogliere segni obiettivi (per esempio
elettrofunzionali o che altro) sulla situazione che si instaura
nella retina nella quale è avvenuto il trapianto. Diversi
clinici, esperti in questo specifico campo, pur riconoscendo
il danno che conseguirebbe a generare false o troppo
anticipate speranze, ritengono comunque giustificato che gli
esperimenti di tale natura continuino anche per altre
patologie degenerative della retina, come la degenerazione
maculare senile".
Tratto da "LUMEN", numero 30/2003
RETINITE PIGMENTOSA: CELLULE DAI
CADAVERI
LONDRA - Cellule prelevate dagli occhi dei morti
potrebbero un giorno essere utilizzate per curare la cecità
nei vivi. Secondo quanto riportato dalla rivista britannica
New Scientist infatti, un'équipe di scienziati dell'università
di Toronto è riuscita ad isolare delle cellule staminali
intorno all'iride. Le cellule, iniettate poi negli occhi di topi
appena nati, hanno generato a loro volta altre cellule
retiniche tra cui coni e bastoncelli sensibili alla luce,
accendendo così la speranza di nuove cure per alcuni tipi di
cecità. Le cellule staminali sono state prelevate da occhi
umani donati alla Banca dell'Occhio del Canada. Ogni occhio
ha fornito circa 10.000 cellule staminali, da ciascuna delle
quali potevano essere cresciute altre 15.000 cellule «figlie».
Presto Derek van der Kooy, direttore della ricerca, ripeterà
l'esperimento iniettando le cellule negli occhi di topi adulti
affetti da retinite pigmentosa.
da "LA GAZZETTA DI PARMA"
STUDI SULL'INNOCUITA' DELLA PRIMA TERAPIA
MEDICA PER LA CURA DELLA RETINITE
PIGMENTOSA
Da questo autunno sono in corso le sperimentazioni di fase 1
Grazie ad una nuova tecnologia individuata dalla Neurotech,
si spera di riuscire ad introdurre attraverso la barriera
ematica i promettenti farmaci FCNT, che hanno la capacità
di ritardare la morte dei fotorecettori retinici. La
statunitense Foundation Fighting Blindness ha di recente
appreso che la FDA ha approvato una richiesta da parte
della società di biotecnologia Neurotech per iniziare già da
questo autunno una sperimentazione clinica di fase 1 sugli
esseri umani per verificare l’innocuità di uno strumento di
somministrazione contenente un farmaco per il trattamento
di pazienti affetti da retinite pigmentosa allo stadio
terminale. Una delle sfide maggiori per il trattamento delle
affezioni retiniche era rappresentata dalla difficoltà di
somministrare farmaci terapeutici direttamente nella retina.
La Neurotech, con sede in Francia e Rhode Island, ha
sviluppato la tecnologia della cellula incapsulata (TCI), che
permette la somministrazione continua controllata, nel
lungo periodo, di un farmaco denominato fattore ciliare
neurotrofico (FCNT). La Foundation Fighting Blindness è
stata uno dei primi sostenitori dell’uso di questa tecnologia
per le malattie retiniche e il Dottor Gerald Chader, che ne
dirige il settore scientifico, ha dichiarato al riguardo che
«l’annuncio è del genere che noi tutti da tempo attendevamo
con ansia. Le terapie con l’FCNT e con altri farmaci hanno
di-mostrato di essere promettenti in un ampia gamma di
modelli animali affetti da retinite pigmentosa. Tuttavia,
nessuno di questi farmaci può attraversare la barriera
ematica retinica, rendendo inefficace la loro
somministrazione con iniezioni sistemiche o pillole. La
tecnologia di impianto introdotta dalla Neurotech si spera
possa finalmente superare questo enorme ostacolo».
UNA PROMETTENTE CAPSULA
La TCI è una minuscola capsula contenente cellule
dell’epitelio pigmentato retinico, geneticamente modificate
per produrre l’FCNT. La capsula possiede minuscoli pori che
permettono la diffusione di ossigeno e di altri elementi
nutritivi a sostegno delle cellule dell’epitelio pigmentato
retinico, permettendo altresì la diffusione dell’FCNT
proteggendo le cellule del sistema immunitario del corpo.
L’FCNT è stato scelto per la sua notevole capacità di
ritardare, negli studi sugli animali, la morte delle cellule dei
fotorecettori retinici e l’esperimento clinico della prima fase
su pazienti preselezionati affetti da retintite pigmentosa
all’ultimo stadio servirà ad attestare l’innocuità sia del
dispositivo TCI sia del farmaco. In questo modo si potrà
valutare l’innocuità del trattamento senza rischio per il
residuo visivo esistente e se, come si spera, tutto andrà bene
in questo studio di fase 1, la sperimentazione di fase 2
servirà a comprovare la capacità del trattamento di
preservare la vista sui pazienti RP che ancora ci vedono. Non
è ancora dato sapere il calendario delle future
sperimentazioni e al mo-mento non risulta che si stiano
ri-cer-cando nuovi pazienti. Il TCI e altri strumenti di
somministrazione dei farmaci potrebbero comunque aprire
la porta ai numerosi fattori di sopravvivenza che, come
l’FCNT, hanno dimostrato di essere promettenti nel
trattamento dell’intera gamma di affezioni degenerative
retiniche. Speriamo di vedere presto altre terapie
farmacologiche, oltre all’FCNT, in sperimentazioni cliniche:
c’è ancora molto lavoro da portare avanti per chi si è
dedicato a queste ricerche, ma gli sforzi della FFB stanno
chiaramente dando i primi promettenti frutti.
da "LUMEN" (Ottobre 2003)
DOMANI LA RETINA SI POTRA’ RIGENERARE
La Retinite Pigmentosa è una malattia genetica incurabile
che rende progressivamente cieche un milione e mezzo di
persone. Essa causa la degenerazione delle cellule che
formano la retina, la parte dell’occhio che risponde alla luce.
EREDITARIA.
Nell’ultimo decennio si è tentato di trapiantare vari tipi di
cellule della retina, ma venivano rigettate. Ora Robert
Aramant del Doheny Eye Institute di Los Angeles, ha
trapiantato dietro la retina in degenerazione di quattro
malati, un doppio strato di cellule retiniche fetali: due
millimetri quadrati di tessuto contenente lo strato di
supporto delle cellule epiteliali e lo strato superiore delle
cellule che “sentono” la luce: coni e bastoncelli. Le epiteliali
hanno una funzione di supporto, e dovrebbero fermare la
malattia, mentre coni e bastoncelli dovrebbero rimpiazzare
quelli perduti. Due pazienti su quattro dicono di aver notato
miglioramenti, ma i ricercatori non si sbilanciano. “È troppo
presto per dire qualsiasi cosa” dice Aramant.
da “FOCUS” (Dicembre 2003)
RETINITE PIGMENTOSA: SINDROME DI USHER
Scoperto un nuovo gene responsabile della Sindrome di
Usher; fino ad oggi ne erano stati scoperti 7 geni
La Sindrome di Usher è una malattia genetica, caratterizzata
dalla presenza contemporanea di Retinite Pigmentosa e di
un difetto uditivo. A seconda dell’epoca di insorgenza e della
gravità del deficit uditivo si distinguono tre forme di
Sindrome di Usher (il tipo 1 in cui la sordità è profonda,
presente fin dalla nascita e tale da non consentire lo
sviluppo del linguaggio, il tipo 2 con deficit uditivo più
tardivo e lieve e il tipo 3 in cui la perdita dell’udito è rapida e
progressiva).
Recentemente presso il Dipartimento di Neurobiologia
dell’UCLA Jules Stein Eye Institute è stato identificato nei
topi un gene, denominato “SLC4A7”, responsabile di una
forma di Sindrome di Usher. Il medesimo gene è presente
sul cromosoma 3 nell’uomo e ciò rende il gene SLC4A7 uno
dei geni candidati per la sindrome di Usher di tipo 2
nell’uomo. L’importanza di avere a disposizione un modello
animale che riproduce una malattia presenta nell’uomo offre
la possibilità di studiare la malattia in modo più
approfondito e rapido, con una maggiore possibilità di
valutare la scoperta e l’efficacia di una terapia che miri a
curare la malattia stessa.
da "RETINITIS:IT"
CONTRO LA RETINITE PIGMENTOSA
di Enrica Strettoi
La Retinite Pigmentosa comprende una famiglia di malattie
ereditarie caratterizzate dalla progressiva degenerazione
delle cellule fotosensibili della retina, ossia dei bastoncelli e
dei coni (i fotorecettori). A tutt’oggi non c’è cura per la
Retinite Pigmentosa; esistono, tuttavia, promettenti
strategie terapeutiche in fase di attiva sperimentazione. Gli
scienziati che studiano la RP sono consapevoli del fatto che
questa famiglia di patologie ha un impatto molto elevato
sulla qualità della vita dei pazienti che ne sono affetti, e già
negli ultimi 5 anni si sono riscontrati notevoli avanzamenti
nell’ambito di vari approcci terapeutici.
Le più promettenti strategie di trattamento della RP
comprendono: a) la terapia genica, che permette la
sostituzione del gene difettoso con uno appropriato,
mediante l’uso di virus non patogeni. Questa strategia è
attualmente applicata con successo in vari modelli animali
di retinite pigmentosa. Il successo maggiore è stato ottenuto
nel 2001 da un’equipe di scienziati americani, che hanno
restituito la vista a dei cani nati ciechi e affetti da una
patologia denominata “amaurosi congenita di Leber”, affine
alla retinite pigmentosa). b) il trapianto di cellule
multipotenti, in grado di attecchire nella retina e di
differenziarsi come fotorecettori, rimpiazzando, quindi, le
cellule che nella RP sono andate perdute; c) l’impianto di
protesi elettroniche contenenti elementi fotosensibili, che
dovrebbero stimolare direttamente gli strati interni della
retina, scavalcando così i fotorecettori danneggiati.
Recentemente, vari pazienti hanno ricevuto l’impianto di
“retine al silicone”, sia negli Stati Uniti che in Europa.
Queste promettenti terapie si basano su un unico
fondamento, e cioè che la retina interna, quella situata “a
valle” dei fotorecettori, e contenente cellule fondamentali
per la funzione visiva, sia assolutamente intatta, indenne
dagli effetti della degenerazione dei bastoncelli e dei coni,
pronta a ricevere cellule trapiantate, a formare connessioni,
a essere stimolata elettricamente da protesi siliconiche.
Tuttavia, non si deve dimenticare che la retina è una vera e
propria “fettina di cervello”; come nel cervello, le cellule
della retina sono connesse le une alle altre in circuiti
complessi. E’ probabile che la morte di un numero elevato di
cellule (i fotorecettori) abbia effetti a cascata sugli altri
elementi ad essi collegati. Questi effetti devono essere
descritti e studiati, per essere eventualmente prevenuti,
perché potrebbero rendere vani i tentativi terapeutici sopra
descritti.
Pochi ricercatori si sono interessati finora agli effetti che la
morte dei fotorecettori produce sulle altre cellule della
retina. Il nostro laboratorio, invece, sta studiando proprio
queste cellule, con l’idea di conoscere meglio gli effetti della
RP sulla retina “residua”. Infatti, è proprio conoscendo cosa
accade nella retina residua, che si possono disegnare meglio
eventuali terapie di trattamento della RP. La retina
“residua” è l’oggetto su cui si fondano molte terapie
possibili, compresa quella della stimolazione con protesi
bioniche. I nostri studi hanno dimostrato, finora, che le
cellule della retina interna reagiscono in modo piuttosto
imponente alla scomparsa dei coni e dei bastoncelli. La
reazione è tanto più evidente e precoce tanto più è
aggressiva la forma di RP considerata. I neuroni della retina
che abbiamo studiato perdono progressivamente i contatti
con le altre cellule e, a un certo punto, muoiono. Tuttavia, lo
fanno in maniera graduale, e, in qualche modo, prevedibile.
Questo fa pensare che, se la terapia è disegnata
precocemente, gli effetti secondari da noi descritti possano
essere evitati. Attualmente, il nostro laboratorio è
impegnato a cercare di capire i meccanismi cellulari per cui
questi effetti secondari si innescano, per cercare di
prevenirli o di combatterli con efficacia.
Dott.ssa ENRICA STRETTOI
Primo Ricercatore - Istituto di Neuroscienze del CNR di Pisa
LA RICERCA IN BREVE
Genetica
La ricerca genetica é il primo passo critico per lo sviluppo di
trattamenti e cure per le malattie degenerative della retina. I
ricercatori della statunitense Foundation Fighting Blindness
(FFB) hanno isolato più di cinquanta geni con mutazioni che
causano le degenerazioni retiniche.
TERAPIA GENICA
I ricercatori finanziati dalla FFB hanno ridato la vista a cani
nati ciechi e su questa base sono stati preparati programmi
di terapia genica per l’intero spettro delle degenerazioni
retiniche.
TRAPIANTO DI CELLULE RETINICHE E CELLULE
STAMINALI
Mediante il trapianto di cellule retiniche sane i ricercatori
cercano di porre rimedio alla perdita della vista. La recente
scoperta di cellule staminali retiniche adulte sta preparando
la strada alla sostituzione delle retine ammalate, restituendo
la vista.
IMPIANTO DI RETINA ARTIFICIALE
Retine artificiali impiantabili ottenute da silicone hanno
terminato le prove cliniche. La chirurgia di trapianto
retinico mostra di promettere la restituzione della vista
perduta a pazienti di DMS.
NUTRIZIONE E STILE DI VITA
Prove cliniche hanno provato che certi nutrienti possono
rallentare la perdita della vista. Risultati di studi
epidemiologici stanno scoprendo fattori che influenzano il
rischio e l’andamento della malattia.
MEDICAMENTI
Per lo più, le terapie a base di medicamenti non raggiungono
la retina. Ricercatori della FFB stanno lavorando allo
sviluppo di sistemi innovativi tesi alla penetrazione della
barriera ematica retinica per il passaggio di sostanze
farmacologiche.
TERAPIA FARMACOLOGICA
I ricercatori della FFB hanno scoperto vari medicamenti che
rallentano in modo sostanziale la perdita della vista. IL
GENE "RPGR" NEL FUNZIONAMENTO DELLA
RETINA
Progetto di ricerca coordinato dal Dottor Alfredo
Ciccodicola, Istituto di Genetica e Biofisica “Adriano Buzzati
- Traverso”, Centro nazionale delle ricerche di Napoli
La diagnosi della retinite pigmentosa rappresenta spesso
una sfida difficile per i genetisti molecolari: sono infatti oltre
trenta i geni noti per essere causa della malattia e altri
restano ancora da scoprire. Già nel 1996 il gruppo di ricerca
coordinato dal Dottor Alfredo Cic-codicola aveva identificato
il gene RPGR, responsabile di una delle forme più gravi di
retinite pigmentosa chiamata RP3. Dato che il gene si trova
su uno dei due cromosomi sessuali, il cromosoma X, la
forma RP3 colpisce quasi esclusivamente i maschi, che ne
possiedono una sola copia. Le femmine possono, invece,
compensare iI difetto con una copia normale dello stesso
cromosoma e non si ammalano, ma possono risultare
portatrici sane. Tuttavia, contrariamente alle aspettative, il
gene RPGR risultò alterato solo in un numero molto piccolo
di pazienti affetti da RP3, mentre la maggior parte dei casi
rimaneva senza spiegazione. Studiando più a fondo il
problema, i ricercatori napoletani e i loro colleghi hanno ora
scoperto che la maggior parte delle mutazioni che causano la
RP3 si concentra in una “zona calda” del gene di cui finora si
ignorava l’esistenza: «Abbiamo scoperto che il gene RPGR è
più grande di quanto si pensasse, contiene infatti una
regione che viene “letta“, cioè trascritta in RNA messaggero,
soltanto nelle cellule della retina e che finora non era mai
stata esplorata» spiega Ciccodicola.
Una scoperta di grande importanza scientifica
Analizzando il DNA di 47 pazienti affetti da RP3, e per i
quali i precedenti esami molecolari avevano dato esito
negativo, i ricercatori hanno scoperto che oltre la metà di
essi possedevano mutazioni proprio nella porzione appena
individuata: «Grazie a questi risultati potremo allargare le
possibilità di diagnosi pre e post-natale fino a comprendere
circa il 70 per cento dei casi di retinite pigmentosa legata al
cromosoma X» conclude il Dottor Alfredo Ciccodicola. Un
altro importante risvolto della ricerca riguarda il ruolo del
gene RPGR nel funzionamento della retina, un aspetto
tuttora poco conosciuto. La sequenza di basi nucleotidiche
della regione identificata è risultata infatti molto simile in
tutte le specie esaminate, dall’uomo ai pesci, suggerendo che
si tratti di una porzione particolarmente importante proprio
perché conservata nel corso dell’evoluzione e nella quale
potrebbe esserci la chiave per comprendere i meccanismi di
funzione del gene. Attualmente, il progetto di ricerca si
propone l’analisi funzionale della proteina RPGR, la
caratterizzazione degli elementi di regolazione del
promotore e l’isolamento di nuovi geni coinvolti in altre
forme di patologie retiniche, il che consentirà di aumentare
le conoscenze sul ruolo svolto da RPGR nella fisiologia della
retina. L’analisi degli elementi trascrizionali del promotore
permetterà di analizzare la regolazione dell’espressione di
RPGR, dando inizio a nuove ricerche nell’ambito della
trascrizione tessuto specifica della retina. Inoltre, l’analisi di
nuovi geni consentirà di approfondire le conoscenze sulla
eterogeneità genetica della malattia.
IL GENE DELLA CECITA' SENILE
L'identificazione del gene CFH potrebbe condurre verso
nuovi trattamenti
Una variazione di un singolo gene potrebbe essere
responsabile della metà di tutti i casi di degenerazione
maculare senile. Lo sostengono tre diversi gruppi di
ricercatori, la cui scoperta potrebbe condurre verso nuovi e
migliori trattamenti per quella che è la principale causa di
cecità in età avanzata.
La degenerazione maculare senile provoca il deterioramento
della retina dell'occhio, danneggiando così la vista. Finora
non esistono cure efficaci, anche se un farmaco di recente
approvazione pare in grado di rallentare il disturbo in alcuni
pazienti. Gli scienziati hanno ora scoperto che le persone
con una mutazione nel gene CFH (complement factor H),
coinvolto in un componente del sistema immunitario che
regola l'infiammazione, hanno maggiori probabilità di
sviluppare la malattia.
Anche se la mutazione di un solo gene non può essere
l'unico fattore della degenerazione maculare, la scoperta
potrà aiutare a identificare gli individui a rischio e a
comprendere il processo di degenerazione. I tre gruppi di
ricercatori, guidati da Albert Edwards del Southwestern
Medical Center dell'Università del Texas, da Josephine Hoh
della Scuola di Medicina dell'Università di Yale, e da
Margaret Pericak-Vance del Medical Center della Duke
University, hanno pubblicato separatamente i propri
risultati sulla rivista "Science".
da "Le Scienze"
DAL SILICIO E DALLE STAMINALI LE SPERANZE
PER RECUPERARE LA VISTA
Affascina e inquieta l'immagine ormai realistica di un corpo
bionico: quello che non molto tempo fa era retaggio della
fantascienza, si sta facendo strada nella vita reale. E
l'ingegneria biologica alimenta ora anche le aspettative di
chi ha subito dei danni, come quelli alla vista. Negli ultimi
anni, negli Stati Uniti, hanno fatto progressi importanti gli
studi sull'occhio elettronico. Grazie alle ricerche della
University of Southern California, si sono sperimentate delle
retine artificiali, per pazienti affetti da malattie
degenerative. Così, è stata messa a punto una videocamera
montata su un paio di occhiali, che trasmette i segnali a
degli elettrodi posti nella stessa retina. I test hanno mostrato
come i pazienti sottoposti alla ricerca riescano almeno a
riconoscere la presenza di una fonte di luce. Nel frattempo,
la società Optobionics, in Illinois, ha cercato di realizzare
una retina in silicio dotata di sensori, evitando l'uso della
telecamera esterna. Non del tutto dissimile è il cosiddetto
Implantable Miniature Telescope: una piccola sfera che va a
sostituire il cristallino dell'occhio, concepita dalla società
californiana VisionCare Ophthalmic Technologies. Anche in
questi casi i dati visivi fanno leva sulla parte non
danneggiata della retina. C'è poi il metodo Brain Implant,
messo a punto dal centro Dobelle, che trasmette le immagini
riprese dalla telecamera a un impianto di elettrodi posto
direttamente sulla corteccia cerebrale. I danni della retina e
del nervo ottico sono in tal modo scavalcati. Nei mesi scorsi,
all'occhio bionico si è affiancata anche la sperimentazione
con le cellule staminali provenienti dall'embrione: se finora
si è proceduto sugli animali, fra poco più di un anno
potrebbero cominciare i test sull'uomo. In prima linea ci
sono i ricercatori della Advanced Cell Technology di
Chicago. «Non solo possiamo prevenire ulteriori perdite
della vista, ma le staminali sembrano in grado di
ricostruire l'itero bulbo oculare», afferma Robert Lanza,
scienziato e autore, con altri, di un recente studio,
disponibile online
(www.liebertpub.com/media/content/clo92204.pdf).
Ma dal fronte dell'occhio elettronico arrivano ora delle
novità. Nel corso del convegno annuale della Association for
Research in Vision and Ophthalmology (www.arvo.org),
appena tenutosi in Florida, ne sono stati ribaditi i progressi,
corroborati da qualche numero. Gli scienziati della
University of Southern California e dell'affiliato Doheny Eye
Institute hanno constatato la fertilità delle loro ricerche sulla
retina artificiale, tanto da presagire una sua disponibilità sul
mercato. Si tratta dell'impianto di una minigriglia con 16
elettrodi. Una microcamera wireless, montata sugli occhiali,
trasmette prima le informazioni visive a un chip posizionato
dietro l'orecchio dei pazienti: esso le trasforma in impulsi
elettrici, facendole poi arrivare agli elettrodi della retina
mediante un cavo posto sottopelle. Gli elettrodi stimolano i
fotorecettori che il danno impediva di funzionare: il segnale
proveniente dalla telecamera può così raggiungere il nervo
ottico e infine il cervello. «I nostri pazienti sono ciechi
perché non hanno fotorecettori», osserva Mark Humayun,
professore di oftalmologia e ingegneria biomedica alla
University of Southern California. Il sistema, battezzato
Argus, funziona infatti solo su soggetti che hanno perso la
vista a causa di patologie che alterino i fotorecettori della
retina (coni e bastoncelli), come la retinite pigmentosa. Sono
escluse le persone col nervo ottico danneggiato o con altre
forme di cecità. «L'impianto riesce a mettere in moto le
cellule rimanenti della retina, creando una vera simbiosi con
la telecamera», aggiunge lo scienziato, che presto comincerà
a testare un sistema da 60 elettrodi, in grado di
incrementare la qualità dell'occhio bionico. La
sperimentazione procede su 6 pazienti, all'inizio
completamente ciechi: essi sono ora in grado di percepire la
luce e di coglierne i movimenti. Secondo quanto dichiarato
da Humayun alla rivista Wired, il sistema Argus sarà messo
in commercio non prima del 2008 dalla società Second
Sight Medical Products (www.2sight.com); il costo dei
dispositivi potrebbe oscillare tra i 30 mila e i 50 mila dollari.
di Andrea Rustichelli de "La Repubblica"
RETINITE PIGMENTOSA: TERAPIA INNOVATIVA
Uno studio americano ha evidenziato che l’acido
docosexanoico contribuisce a rallentare la degenerazione
retinica di chi soffre di Retinite Pigmentosa.
Lo studio, condotto secondo i dettami della moderna
sperimentazione clinica, prevedeva la possibilità di
confrontare gruppi di pazienti sottoposti a diversi regimi
terapeutici, con lo scopo di valutare gli effetti dell’acido
docosexanoico sulla progressione della Retinite Pigmentosa.
È stato evidenziato che dopo quattro anni di terapia i
pazienti che avevano iniziato ad assumere per la prima volta
15.000 U.I. di Vitamina A palmitato associata a 1.200 mg al
giorno di acido docosexanoico presentavano un
rallentamento della progressione della malattia. Lo studio
ha anche messo in evidenza che una dieta ricca di acidi
grassi Omega-3 riduce il peggioramento del campo visivo
nei pazienti che già assumevano la Vitamina A palmitato da
almeno due anni.
Questo articolo è un ulteriore esempio della necessità di
sostenere la ricerca scientifica, adesso non solo con la
donazione dei somme di denaro ma anche con il vostro
impegno civile e consapevole. Il 12 e 13 Giugno prossimi Vi
invitiamo ad esprimere un Vostro diritto civile e
democratico: SOSTIENI LA RICERCA CONTRO
TUTTE LE GRAVISSIME MALATTIE EREDITARIE
CHE COLPISCONO MIGLIAIA DI BAMBINI E
PERSONE ADULTE. ESPRIMI UN "SI" PER OGNI
SCHEDA. LA RICERCA E’ ANCHE LIBERTA’ E
DEMOCRAZIA!
COME SI FORMA IL NERVO OTTICO
In assenza di segnali, i neuroni primitivi sono programmati
per costruire solo la retina. Quando le cellule nervose
primitive dell'embrione del topo cominciano a formare un
occhio, sono inizialmente programmate per costruire solo
una retina. Ma la capacità di vedere dipende dalla
connessione della retina al cervello attraverso il nervo ottico.
A meno che queste cellule embrionali non ricevano il
segnale giusto al momento giusto, esse formeranno un
gigantesco occhio consistente solamente di una retina e
privo di nervo ottico.
La scoperta che la retina rappresenta il "setting di default"
per lo sviluppo nell'occhio dell'embrione è stata fornita da
una ricerca del neurobiologo Greg Lemke e colleghi al Salk
Institute for Biological Studies di La Jolla, in California,
pubblicata sulla rivista "Genes & Development". Gli
scienziati hanno utilizzato topi in laboratorio come modello
della biologia umana.
"I nostri risultati - spiega Lemke - suggeriscono che la retina
rappresenti effettivamente il percorso di default per lo
sviluppo degli occhi nei mammiferi". Gli autori hanno
dimostrato che due segnali chimici (proteine segnalatrici)
devono essere presenti al momento giusto e all'istante giusto
per arrestare questo processo di default e consentire al
nervo ottico di svilupparsi. La scoperta ha importanti
conseguenze, in quanto il controllo del destino delle cellule
staminali trapiantate nel cervello è fondamentale se si
intende usare queste cellule in maniera sicura ed efficace
nelle terapie su esseri umani. "È probabile - commenta
Lemke - che ci siano anche altre aree del cervello il cui
sviluppo si basa sull'arresto di una tendenza delle staminali
a trasformarsi nello stesso tipo di cellula di quelle vicine".
da "Le Scienze"
LA DEGENERAZIONE MACULARE NELLE
SCIMMIE
La malattia è simile nell'uomo e nei macachi.
La degenerazione maculare senile (AMD) è la principale
causa di cecità in età avanzata, eppure i ricercatori ignorano
ancora molti dei fattori che causano questa malattia
incurabile. Ora, però, alcuni scienziati negli Stati Uniti e in
Germania affermano che un legame genetico fra i macachi
“resi” e gli esseri umani che soffrono di AMD potrebbe
svelare indizi importanti sulle prime fasi della malattia,
quando la perdita della vista può ancora essere impedita.
Gli autori hanno analizzato una regione cromosomica e
alcuni marcatori genetici della degenerazione maculare negli
esseri umani e nei macachi. L'associazione della malattia
delle scimmie con quella degli uomini consentirà agli
scienziati di studiare come progredisce negli animali e di
giungere a trattamenti migliori e forse addirittura a una
cura.
A differenza di quasi tutti gli altri animali, gli occhi dei
macachi resi presentano l'identica struttura complessa di
quelli umani, rendendoli un perfetto modello per la ricerca.
William W. Dawson dell'Università della Florida e colleghi
hanno studiato sette siti genetici nelle scimmie i cui siti
corrispondenti nei cromosomi umani sono associati alla
malattia maculare. Una di queste aree contiene geni che
predicono lo sviluppo della malattia. Gli autori sospettavano
da tempo che il disturbo negli uomini e nelle scimmie fosse
molto simile, ma i risultati, pubblicati online sulla rivista
"Experimental Eye Research", finalmente lo confermano.
da "Le Scienze"
L'EQUILIBRIO NEURONALE DELLA VISTA
Alcuni gruppi di neuroni collaborano per escludere le
informazioni non
essenziali.
Scavando sempre più in profondità nell'intricata
architettura del cervello, alcuni ricercatori del Salk Institute
for Biological Studies di La Jolla, in California, hanno
scoperto come due differenti tipi di cellule nervose, o
neuroni, agiscono insieme per trasmettere esattamente la
giusta quantità e il giusto tipo di informazione sensoriale. Lo
studio, pubblicato online sulla rivista "Nature
Neuroscience", spiega come specifici gruppi di neuroni
inibitori nella corteccia visiva del cervello di un topo sono
collegati - e "parlano" - con singoli neuroni eccitatori.
Questa "conversazione", che serve a mantenere il corretto
equilibrio di segnali chimici, spesso esclude i neuroni
circostanti.
"I neuroni inibitori - commenta il neurobiologo Ed
Callaway, co-autore dello studio insieme a Yumiko
Yoshimura dell'Università di Nagoya - non sono
semplicemente dei freni ma agiscono anche da timone". Nel
sistema della vista, per esempio, le risposte inibitorie nella
corteccia visiva aiutano a concentrarsi sulle cose che si
desiderano vedere, ignorando tutto il resto. Lo studio
contribuisce a chiarire il quadro
dell'organizzazione del cervello in network "intelligenti" ed
efficienti, e i ricercatori sperano che un giorno questi
dettagli possano chiarire le radici di disturbi neurologici
come la schizofrenia.
da "Le Scienze"
LA RETINA ARTIFICIALE
Lo sviluppo delle biotecnologie apre ogni giorno nuovi
campi di ricerca. È dagli inizi degli anni ’90 che gruppi di
ricercatori, in tutto il mondo, sono impegnati nelle
realizzazione di retine artificiali cioè elettroniche.
Una elevata percentuale (oltre il 50 % nel mondo civilizzato)
di handicap visivo o cecità consegue a patologie che
compromettono la funzionalità della retina. A tutt’oggi non
esistono sussidi medici o chirurgici in grado di ripristinare il
tessuto retinico compromesso. Prospettiva ma ancora nulla
di realistico proviene dalle cellule staminali.
È così comprensibile come, seppure sporadicamente
praticamente, in tutto il mondo gruppi di ricercatori sono
impegnati, fin dall’inizio degli anni ’90, nella realizzazione di
retine artificiali cioé elettroniche; veri e propri pezzi di
ricambio per retine usurate e compromesse. L’enorme
evoluzione delle nanotecnologie rappresenta una concreta
incentivazione per tali ricerche. Permangono allo stato
attuale problematiche complesse che risulteranno meglio
comprensibili illustrando quali sono le funzioni fisiologiche
della retina e quali le strategie seguite per realizzare un
supporto elettronico sostitutivo.
La retina è la membrana interna bulbare che ha la
caratteristica di trasformare l’impulso fotonico, cioè la luce
che giunge dal mondo esterno, in impulso bioelettrico.
Questo processo, indicato come trasduzione si realizza
grazie a cellule specializzate dette fotorecettori dotate di
pigmento (derivato dalla vitamina A) che, assorbendo i
fotoni, attivano canali ionici della membrana cellulare
innescandone l’eccitamento. Queste cellule sono
estremamente sensibili quando si consideri che un singolo
fotone è già in grado di indurre l’attivazione di membrana.
L’eccitamento è quindi trasmesso ad un complesso sistema
di neuroni (cellule nervose) intraretiniche (cellule bipolari e
cellule orizzontali) dove subisce codificazioni che traducono
il mondo esterno in punti luminosi a diverso contrasto,
colore, orientamento e movimento. Le cellule nervose
intermedie sono connesse ad un altro strato di cellule dette
ganglionari ottiche che codificando il segnale in
modulazione di frequenza attraverso il loro assone e lo
inviano al corpo genicolato, prima stazione centrale, che lo
smista alle cortecce visive. Va precisato che ciascun punto
della retina comunica con una corrispondente area corticale
(V 1), formandosi quindi una sorta di calco retinico
(retinotopico) a livello cerebrale e che la retina periferica è
abilitata a trasmettere immagini in movimento mentre la
retina centrale dà la percezione dei particolari (capacità
discriminativa) e dei colori.
La retina artificiale è finalizzata a sostituire integralmente il
ruolo dei fotorecettori e parzialmente del sistema cellulare
interposto fra fotorecettori e cellule ganglionari ottiche.
In talune patologie ed in particolare nella retinite
pigmentosa la lesione di base coinvolge fotorecettori ed
epitelio pigmentato e solo tardivamente le cellule nervose
più interne. In tale situazione un dispositivo elettronico
capace di captare la luce e di convertirla in stimolo elettrico
permette un ripristino della funzione retinica cioè la
percezione della luce. Sono questi i concetti che hanno
ispirato all’inizio degli anni ’90 ricercatori dell’Università
dell’Illinois a Chicago alla impostazione della prima retina
artificiale assemblando microfotodiodi fra loro connessi da
resistori ed inserendoli in una sorta di tasca realizzata
chirurgicamente tra retina nervosa ed epitelio pigmentato.
Per dare l’idea del grado di miniaturizzazione precisiamo
che in un area di 2 millimetri sono inseriti 3500 diodi. Gli
studi successivi condotti anche dalla scuola tedesca (Prof.
Eberard Zrenner Tubinga) hanno portato a costruire
microfotodiodi capaci di indurre polarizzazioni positive o
negative con voltaggi di stimolo e distanze tra elettrodi
collimanti con le caratteristiche bioelettriche e con la densità
delle cellule retiniche interfacciate. Si è inoltre provveduto a
dotare la protesi di superfici porose tali da permetterne un
connessione ottimale con le cellule retiniche. La biocompatibilità al livello sperimentale risulta assai
soddisfacente.
Realmente adottabile quindi al livello clinico la retina
artificiale? A riguardo sussistono notevoli riserve
concernenti innanzitutto le capacità di sopravvivenza del
tessuto retinico nervoso quando venga a mancare il
supporto della coriocapillare e del epitelio pigmentato. Un
possibile effetto coadiuvante nel migliorare la vitalità
neuronale è delineato dall’uso dei fattori di crescita in
particolare le neurotrofine quali BDNF.
A livello sperimentale è stata documentata un’azione diretta
sulla vitalità dei neuroni retinici di cui è in grado di inibire
l’apoptosi e stabilizzarne le sinapsi genicolate. Riserve,
inoltre, sul rendimento visivo cioè sull’entità del recupero
fin qui verificato. Le percezioni realmente ottenibili sono
limitate a sensazioni di luce-ombre. Prospettive decisamente
meno allettanti derivano dalle protesi studiate dai
ricercatori del Boston Harvard Medical School in
collaborazione con il Massachussets Istitute of Tecnology e
da un altro team di ricerca coordinato dal Prof. Rolf
Eckmiller dell’Università di Bonn che hanno messo a punto
protesi concettualmente diverse in quanto sfruttano un
apparato esterno all’occhio che converte le immagini di una
fotocamera in impulsi laser che vanno ad attivare un chip
interno all’occhio situato sulla faccia interna della retina. Il
chip a mezzo di appositi elettrodi trasferisce l’impulso alle
cellule ganglionali.
RETINA: NUOVE IMMAGINI CATTURATE PER LA
PRIMA VOLTA
Presso il Center for Visual Science dell'Università di
Rochester (Stati Uniti) sono state catturate per la prima
volta delle immagini esclusive della retina. Le immagini,
pubblicate su Journal of Neuroscience sono molto diverse e
per questo molto sorprendenti. Sorpendenti perchè gli
studiosi non immaginavano che le migliaia di cellule
responsabili nel rilevare i colori nella parte più profonda
dell'occhio, fossero così diverse da individuo ad individuo.
Tutti gli individui, tranne chi ha problemi di visione, vedono
i colori nello stesso modo. La retina è una sottile membrana
che riveste quasi tutta la parte interna dell'occhio. Si tratta
di una struttura estremamente complessa formata da
milioni di cellule (fotorecettori) sensibili alla luce che
trasformano gli stimoli luminosi in impulsi elettrici. I
fotorecettori sono di due tipi: i coni e i bastoncelli. I coni
sono cellule nervose che funzionano in condizioni di piena
illuminazione e il loro compito è quello di produrre
immagini molto dettagliate e a colori. I bastoncelli, invece,
sono responsabili della visione notturna o comunque in
condizioni di scarsa illuminazione.
Lo strato dei fotorecettori si trova nella parte più profonda
della retina e appoggia su uno strato detto epitelio
pigmentato. Finora si riteneva che in media ogni individuo
possiede circa sette milioni di coni in una retina, il 64% dei
quali sono rossi, il 32% verdi e il 2% blu, i colori che servono
per raccogliere la luce dell'intero spettro luminoso. Le nuove
fotografie della retina realizzate mostrano che da persona a
persona c'è una differenza nel numero dei coni dei vari
colori che giunge addirittura al 40%. Il dott. Joseph Carrol,
uno dei ricercatori dell'Università di Rochester racconta: -In
un primo momento ci siamo chiesti se in conseguenza a
questa variabilità le persone vedessero i colori in modo
differente. Ma non è così. Allora si deduce che il cervello
interviene in modo diverso in ciascun individuo nel
compensare il numero dei coni al fine di offrire ad ogni
persona la visione dei medesimi colori. Ora bisognerà capire
come fanno diversi cervelli a lavorare per dare ad ogni
individuo la medesima tonalità di colore o al più una piccola
differenza. I ricercatori sono riusciti in questi intento
utilizzando 'ottiche adattative' che correggono nelle
macchine fotografiche usate i difetti presenti nell'occhio così
da ottenere immagini ad altissima risoluzione.
Le ottiche adattative sono utilizzate dagli astronomi che
vogliono osservare oggetti molto lontani e che appaiono
sfuocati dopo che la loro luce ha attraversato l'atmosferaspiega il prof. David Williams, direttore del Center for Visual
Science. La tecnica sarà ora utilizzata per studiare le
malattie che colpiscono la retina, le cui ricerche erano fino
ad ora ostacolate proprio dall'impossibilità di avere una
visione precisa della parte più profonda dell'occhio.
Dell'Università di Rochester
da "Occhio.it"
UNA NANO-BATTERIA NELL'OCCHIO
In futuro potrebbe alimentare una retina artificiale e
contribuire a curare certe forme di cecità. Il progetto
inaugurerà un nuovo centro di ricerca statunitense dedicato
alla nanomedicina.
Albuquerque (USA) - Un team di ricercatori del Sandia
National Laboratories, insieme a scienziati di altri istituti di
ricerca americani, sta sviluppando una batteria nanometrica
che in futuro potrebbe essere impiantata in un occhio per
alimentare una retina artificiale. Il progetto verrà condotto
in un nuovo centro di ricerca di prossima apertura, il
National Center for Design of Biomimetic Nanoconductors
(NCDBN), la cui costruzione è stata finanziata dal National
Eye Institute of the National Institutes of Health (NIH) per
sviluppare e sperimentare nuove nanotecnologie per la
medicina.
L'NCDBN, con sede nell'università Urbana-Champaign
dell'Illinois, controllerà l'intero ciclo di sviluppo, che va
dall'ideazione fino alla produzione, dei dispositivi medici
basati sulle nanotecnologie. Il primo obiettivo del nuovo
istituto di ricerca sarà quello di progettare una nuova classe
di dispositivi capaci di generare elettricità. Il fabbisogno di
elettricità è da considerare primario nel caso in cui non ci sia
possibilità di alimentare tramite rete elettrica le
apparecchiature. Inoltre, la possibilità di fornire dispositivi
che vengano integrati all'interno di un sistema vivente, detta
la necessità di trovare fonti diverse di alimentazione. Per
questo motivo, le batterie che alimenteranno tutte le
apparecchiature che il centro di ricerca svilupperà, saranno
delle bio-batterie, capaci di immagazzinare elettricità
direttamente dal corpo umano. Infatti, il progetto della
retina artificiale non potrebbe essere completo se non vi
fosse un'alimentazione di tipo biologico.
Insieme alle retine artificiali, le nano-batterie potrebbero
contribuire a risolvere certi tipi di cecità causati dalla
degenerazione maculare. Il gruppo di scienziati del Sandia si
occuperà in modo particolare di progettare al computer
modelli tridimensionali della batteria molecolare, e simulare
la sua interazione con la retina artificiale e l'occhio. Ma i
campi di applicazione della bio-batteria potrebbero essere
ben più ampi: questa potrebbe infatti alimentare un'ampia
varietà di microscopici chip in grado di curare o alleviare
certe malattie e infermità. Per giungere alla progettazione di
questi dispositivi, Susan Rempe, responsabile del gruppo di
ricerca, afferma quanto sia importante l'apporto fornito dai
programmi di modellazione grafica. "I nostri esperti di
modellazione ci facilitano il compito mostrandoci come le
strutture riescano a lavorare assieme. Le informazioni che
riceviamo da questi programmi ci fanno capire quanta
energia è necessaria per lo spostamento di determinate
componenti e quindi di quale tipo di microbatteria è
necessaria". La grande utilità dei software di modellazione e
previsione è dimostrata anche "dalla possibilità di
visualizzare su schermo ciò che si riesce ad immaginare e
quindi, successivamente, a progettare".
Il team della Rempe conta di riuscire presto a sviluppare
nuovi dispositivi impiantabili capaci di sopperire alle
funzioni biologiche che risultano lese o addirittura mancanti
in alcuni soggetti. Interessante notare che tutti i software di
grafica sui quali lavorano i ricercatori del centro di ricerca di
Sandia utilizzano sistemi operativi basati su Linux.
L'NCDBN è solamente uno dei tasselli del grande mosaico
che, con un fondo di 43 miliardi di dollari, fa parte del
programma di ricerca sulle nanotecnologie inaugurato nel
2003 dagli USA con il progetto di ricerca medica. Un
progetto che presto si arricchirà di altri due sedi presso
l'University of California a San Francisco e la Columbia
University di New York.
da "Punto Informatico"
...RETINA ARTIFICIALE
La rivista “Proceedings of the National Academy of
Sciences” ha divulgato la notizia della creazione di un nuovo
prototipo di processore impiantabile da parte di un team di
ricercatori della scuola di medicina dell'Università di
Stanford. Questo chip ha una doppia funzione: può aiutare
pazienti affetti dalla cosiddetta cecità della vecchiaia in
quanto può essere adattato come retina prostetica; può
fungere da somministratore di farmaci per patologie
neurodegenerative come il morbo di Parkinson.
Questo piccolo processore non utilizza l'elettricità come
stimolatore dei nervi, come fanno gli altri chip, ma fa si che
le cellule vengano stimolate da piccole quantità di sostanze
chimiche. Una funzione peculiare se si tiene conto che le
cellule solitamente interagiscono fra loro mediante i
neurotrasmettitori, che sono sostanze chimiche.
Questo chip, che è stato creato nei laboratori dello Stanford
Ophthalmic Tissue Engineering dal dottor Harvey A.
Fishman, potrebbe assolvere a delicatissimi interventi su
tessuti estremamente sensibili come quelli dell'occhio e delle
aree cerebrali. Esso ha la capacità di poter liberare
piccolissime quantità di sostanze chimiche, servendosi
dell'elettro-osmosi, e di controllare i neuroni. In caso
estremo, il chip ha pure la possibilità di ritirare i fluidi onde
evitare accumuli di sostanze che potrebbero risultare
tossiche.
di Massimo Bertolucci da “ecplanet.com”
Istituzione scientifica citata nell'articolo: Stanford
University School of Medicine
N.B. Gli eventuali indirizzi di recapito presenti nell'articolo
possono cambiare senza che la redazione di atritoscana.it ne
venga a conoscenza.
ARRIVA L'OCCHIO ARTIFICIALE
Microelettronica e nanotecnologia potrebbero presto portare
un po' di luce ad alcuni non vedenti. Il punto sulla ricerca, le
sfide da raccogliere.
Sin dagli inizi dell'era dell'elettronica, scienziati e autori di
fantascienza (si veda ad esempio il film italiano Nirvana del
1997) hanno sognato di poter sostituire un occhio non
funzionante con un apparato artificiale e ridare così una
visione (almeno parziale) ai ciechi. Oggi questo ambizioso
obiettivo sembra essere a portata di mano - anche se le
soluzioni attualmente in sperimentazione saranno
disponibili sul mercato solo fra qualche anno e potranno
risolvere solo alcune specifiche patologie.
Sostituire la retina artificiale.
In molti laboratori sono in corso ricerche focalizzate sullo
sviluppo di una retina artificiale, per sostituire l'organo
umano che trasforma la luce in impulsi elettrici da trasferire
poi al cervello attraverso il nervo ottico. Il primo
esperimento di impianto di un sistema di visione artificiale
in un essere umano risale ormai all'anno 2000. Il sistema,
composto da una microtelecamera incorporata in speciali
occhiali, accoppiata ad un sensore a ultrasuoni, trasmette i
segnali a un piccolo computer tascabile, che elabora
l'informazione, la ritrasmette a un altro computer e di qui a
una rete di 68 elettrodi posti nella superficie del cervello.
Questo impianto high-tech è riuscito a ridare un poco di
vista a un paziente cieco da 36 anni. I risultati, se pur
interessanti, sono solo un primo passo verso una buona
soluzione: il paziente ha recuperato una capacità visiva pari
a quella di una persona molto miope, e l'apparato è
relativamente scomodo. Sono dunque partiti parecchi
progetti destinati a costruire un occhio artificiale più
performante e portatile. In Europa, solo per citare alcuni
esempi, è stato attivo il progetto comunitario OPTIVIP con
l'obiettivo di realizzare una protesi in grado di stimolare
direttamente il nervo ottico, è in corso una ricerca da parte
dell'ospedale oftalmologico di Colonia e avanzate
sperimentazioni sono condotte dal pioniere Claude Veraart.
La ricerca ha prodotto risultati e i primi prototipi di retina
artificiale europea sono già stati impiantati, con risultati
interessanti. Anche questo sistema si basa su un apparato
esterno collegato però al nervo ottico: dovrebbe essere
quindi applicabile anche a pazienti con la retina totalmente
inattiva, ma che abbiano un nervo ottico funzionante. Si
prevede che il prodotto potrebbe essere reso disponibile al
pubblico entro il 2010, a un costo attorno ai 20.000 euro.
Al lavoro anche i laboratori nucleari.
Negli Stati Uniti si è addirittura mobilitato l'establishment
militar-nuclear-industriale, coinvolgendo enti come il
Sandia National Laboratories, un laboratorio chiave per la
ricerca nucleare a fini bellici statunitense o l'Argonne
National Laboratory, laboratorio che fu parte fondamentale
del progetto Manhattan (legato, come noto, alla costruzione
della prima bomba atomica). Ancora una volta, aziende ed
enti seguono il principio "piatto ricco mi ci ficco" e cercano
di godersi una fetta dei sostanziosi stanziamenti messi a
disposizione dal National Institutes of Health (e meno male
che ogni tanto si mettono i brillanti cervelli dei ricercatori a
lavorare su progetti benefici per l'umanità). Almeno un paio
di aziende statunitensi sono già a un discreto punto della
sperimentazione su pazienti umani, chi usando device
connessi a hardware esterni, chi passando invece a impianti
interni come nel caso della retina artificiale (ASR), un chip
di un paio di millimetri di diametro e più sottile di un
capello, da impiantare all'interno dell'occhio. Questo
particolare chip contiene 5.000 fotosensori in grado di
convertire la luce in impulsi elettrochimici e con questi
stimolare le cellule della retina del paziente ancora in grado
di funzionare (il che dovrebbe rendere inutile l'apparato nel
caso di pazienti con la retina totalmente compromessa).
In cerca della nanobatteria.
Questo nuovo tipo di dispositivi elettronici pongono agli
scienziati tutta una serie di problemi, in parte inediti.
Seguendo la tradizione consolidata dell'elettronica, a ogni
successiva generazione il prodotto rischia di diventare più
vorace di energia. Ci si potrebbe dunque trovare dinnanzi al
problema di dover dipendere da una qualche forma di
energia esterna o di accumulatore impiantato nel corpo. In
realtà il microchip ASR si alimenta da solo, sfruttando la
luce che lo colpisce- ma questa soluzione rischia di metterlo
in difficoltà in situazioni di scarsa luce ambientale, il che
limiterebbe la sua utilità. Sul fronte delle fonti energetiche
impiantabili si sta dunque muovendo un consorzio di enti e
aziende americane, che ha intrapreso il lavoro di ricerca su
una batteria che dovrebbe produrre elettricità imitando i
processi biologici degli organismi viventi. Una batteria tanto
piccola da poter trovare posto nell'occhio insieme alla retina
artificiale, per arrivare ad una soluzione del tutto interna e
quindi più comoda ed "accettabile" per il paziente . L'altro
grande problema è garantire il funzionamento di un
dispositivo delicato come un microchip in un ambiente così
aggressivo come l'interno del corpo umano, proteggendo al
contempo il delicato corpo umano da possibili effetti
collaterali del chip impiantato nell'occhio. Una soluzione a
questo problema sta per essere individuata attraverso un
sofisticato rivestimento, basato sull''applicazione di uno
strato ultrananocristallino composto da cristalli di diamante
del calibro di 5 milionesimi di millimetro.
Piccoli passi verso l'uomo bionico.
Anche se queste soluzioni, ancora ai primi passi,
rappresentano una possibile soluzione solo per alcune forme
di cecità, sembra si possa essere ottimisti, almeno per i
ciechi del mondo occidentale, in grado di permettersi (anche
grazie a una mutua o assicurazione sanitaria) il costo di
apparato ed operazione. È dunque probabile che a medio
termine questo tipo di impianti ridaranno almeno
parzialmente la vista a un certo numero di non vedenti. Nel
lungo periodo, conoscendo come funzionano gli esseri
umani e tenendo in conto le probabili evoluzioni
tecnologiche, non mi sorprenderebbe diventasse comune
farsi sostituire occhi perfettamente funzionanti con occhi
bionici, capaci di vedere più lontano, funzionanti in assenza
di luce o in grado di captare radiazioni non visibili, essendo
in grado di "vedere" l'infrarosso o l'ultravioletto.
di Roberto Venturini
NELLA MUTAZIONE DI DUE GENI LA
PRINCIPALE CAUSA DI MACULOPATIA
La scoperta è stata fatta dai ricercatori del Columbia
University Medical Center
Tre casi su quattro di maculopatia, la principale causa di
cecità dopo i 60 anni, sono legati a mutazioni a carico di
due geni che producono proteine del sistema immunitario.
L'importantissima scoperta è merito degli studi coordinati
da Rando Allikmets del Columbia University Medical
Center e potrebbe consentire lo sviluppo di terapie
preventive o che arrestino l'inesorabile decorso di questa
malattia della retina.
La notizia è stata riportata sulla rivista Nature Genetics e i
due geni coinvolti nel 75% dei casi di degenerazione
maculare senile sono quelli che producono il fattore B e il
fattore H, due proteine del sistema di difesa
dell'organismo con un ruolo chiave nel controllo dei
processi infiammatori.
La degenerazione maculare senile è una grave malattia
degenerativa che colpisce il centro della retina, la macula,
rendendo progressivamente meno nitida la visione fino a
deteriorarla in modo rovinoso se non si interviene
tempestivamente per bloccarne il decorso.
La maculopatia è un problema che interessa soprattutto gli
anziani ed è in costante aumento nel mondo occidentale.
Oggi non ci sono strategie per prevenirla se non la
raccomandazione di seguire stili di vita sani ed adottare
una alimentazione corretta che prediliga verdure e frutta,
centellinando invece il consumo di fritti e altre
preparazioni meno salutari.
Le sue cause sono sicuramente complesse e di certo
coinvolgono, oltre che fattori ambientali, fattori ereditari
come numerosi screening genetico hanno dimostrato in
passato.
I genetisti Usa avevano dimostrato solo pochi mesi fa in un
precedente studio il possibile coinvolgimento del sistema
immunitario nella malattia ed individuato i fattori
ereditari coinvolti.
In particolare i ricercatori avevano pubblicato la scoperta
del coinvolgimento del gene per il Fattore H, una molecola
che controlla la risposta infiammatoria ad agenti patogeni
penetrati nell'organismo. Gli esperti avevano visto che
mutazioni a carico di questo gene erano presenti in un
caso su due della malattia. Persone con mutazioni sul gene
per il Fattore H hanno un'eccessiva reazione
infiammatoria a piccole infezioni, inoltre difetto del fattore
H si traducono in incapacità di sopire la reazione
infiammatoria dopo che l'infezione è stata eliminata,
quindi quando la reazione infiammatoria non serve più.
Ma poiché si tratta di una malattia complessa, la
maculopatia non può essere spiegata con difetti su un solo
gene. Questa considerazione ha indotto i genetisti Usa a
cercare ancora, concentrando l'attenzione su altri geni
legati ai processi infiammatori.
Con l'analisi genetica di 1300 pazienti ed individui di
controllo, i genetisti hanno quindi trovato che un'altra
molecola immunitaria, il fattore B, è il principale agente
modificatore della malattia. Questa molecola ha un'attività
opposta (induce i processi infiammatori e immunitari) a
quella inibitrice del fattore H, quindi, hanno dichiarato i
due esperti, sembra logico che entrambi i geni siano
coinvolti nella maculopatia e le loro mutazioni rafforzino a
vicenda il rischio di ammalarsi.
I ricercatori, infatti, hanno visto che circa tre pazienti su
quattro, (74% dei pazienti), hanno uno o entrambi questi
geni difettosi. «Non conosco nessuna malattia complessa
(ossia in parte di natura genetica in parte ambientale) ha
dichiarato con estremo entusiasmo Allikmets in cui sia
stato identificato il 75% della causa genetica».
Il sistema immunitario deve avere un ruolo principe nella
genesi della malattia, ha concluso Allikmets, per questo
ora bisogna andare alla ricerca di quegli agenti esterni, per
esempio infezioni, che premono il grilletto facendo
scoppiare la maculopatia nelle persone geneticamente
predisposte.
da "Corriere.com"
NEI TESTICOLI TROVATE CELLULE SIMILI ALLE
EMBRIONALI
GERMANIA - Risultato sorprendente, e destinato a
provocare scossoni nel mondo scientifico internazionale. Nei
testicoli di alcuni topi di laboratorio sono state isolate cellule
staminali del tutto simili a quelle embrionali, sul cui utilizzo
nella ricerca si scontrano buona parte degli scienziati da un
lato, e diversi schieramenti politici dall'altro. Anche in Italia.
La scoperta è stata realizzata dai ricercatori dell'università di
Goettingen, in Germania, secondo cui “estraendo queste
staminali dai testicoli umani, tramite una semplice biopsia,
si potrà avere a disposizione una fonte alternativa di cellule
'factotum' da utilizzare all'occorrenza per uso terapeutico”.
Lo studio è pubblicato sulla rivista "Nature".
Una scoperta "epocale", che "vale 10" in una ipotetica
classifica di importanza per gli scienziati. e che " e' destinata
a relegare la disputa epica e politica sull'utilizzo nella ricerca
delle cellule staminali embrionali a dibattito datato e
sterile". Ruggero De Maria, ricercatore dell'Istituto
Superiore di Sanità, si congratula apertamente con gli
scienziati tedeschi che hanno trovato e isolato staminali
simili alle embrionali nei tessuti dei testicoli di topi di
laboratorio adulti.
da "Adnkronos"
A SECONDA VISTA: ARRIVA L'OCCHIO
ARTIFICIALE
Microelettronica e nanotecnologia potrebbero presto portare
un po' di luce ad alcuni non vedenti. Il punto sulla ricerca, le
sfide da raccogliere. Sin dagli inizi dell'era dell'elettronica,
scienziati e autori di fantascienza (si veda ad esempio il film
italiano Nirvana del 1997) hanno sognato di poter sostituire
un occhio non funzionante con un apparato artificiale e
ridare così una visione (almeno parziale) ai ciechi. Oggi
questo ambizioso obiettivo sembra essere a portata di mano
- anche se le soluzioni attualmente in sperimentazione
saranno disponibili sul mercato solo fra qualche anno e
potranno risolvere solo alcune specifiche patologie.
Sostituire la retina artificiale.
In molti laboratori sono in corso ricerche focalizzate sullo
sviluppo di una retina artificiale, per sostituire l'organo
umano che trasforma la luce in impulsi elettrici da trasferire
poi al cervello attraverso il nervo ottico. Il primo
esperimento di impianto di un sistema di visione artificiale
in un essere umano risale ormai all'anno 2000. Il sistema,
composto da una microtelecamera incorporata in speciali
occhiali, accoppiata ad un sensore a ultrasuoni, trasmette i
segnali a un piccolo computer tascabile, che elabora
l'informazione, la ritrasmette a un altro computer e di qui a
una rete di 68 elettrodi posti nella superficie del cervello.
Questo impianto high tech è riuscito a ridare un poco di
vista a un paziente cieco da 36 anni. I risultati, se pur
interessanti, sono solo un primo passo verso una buona
soluzione: il paziente ha recuperato una capacità visiva pari
a quella di una persona molto miope, e l'apparato è
relativamente scomodo. Sono dunque partiti parecchi
progetti destinati a costruire un occhio artificiale più
performante e portatile. In Europa, solo per citare alcuni
esempi, è stato attivo il progetto comunitario OPTIVIP con
l'obiettivo di realizzare una protesi in grado di stimolare
direttamente il nervo ottico, è in corso una ricerca da parte
dell'ospedale oftalmologico di Colonia e avanzate
sperimentazioni sono condotte dal pioniere Claude Veraart.
La ricerca ha prodotto risultati e i primi prototipi di retina
artificiale europea sono già stati impiantati, con risultati
interessanti. Anche questo sistema si basa su un apparato
esterno collegato però al nervo ottico: dovrebbe essere
quindi applicabile anche a pazienti con la retina totalmente
inattiva, ma che abbiano un nervo ottico funzionante. Si
prevede che il prodotto potrebbe essere reso disponibile al
pubblico entro il 2010, a un costo attorno ai 20.000 euro.
Al lavoro anche i laboratori nucleari.
Negli Stati Uniti si è addirittura mobilitato l'establishment
militar-nuclear-industriale, coinvolgendo enti come il
Sandia National Laboratories, un laboratorio chiave per la
ricerca nucleare a fini bellici statunitense o l'Argonne
National Laboratory, laboratorio che fu parte fondamentale
del progetto Manhattan (legato, come noto, alla costruzione
della prima bomba atomica). Ancora una volta, aziende ed
enti seguono il principio "piatto ricco mi ci ficco" e cercano
di godersi una fetta dei sostanziosi stanziamenti messi a
disposizione dal National Institutes of Health (e meno male
che ogni tanto si mettono i brillanti cervelli dei ricercatori a
lavorare su progetti benefici per l'umanità).
Almeno un paio di aziende statunitensi sono già a un
discreto punto della sperimentazione su pazienti umani, chi
usando device connessi a hardware esterni, chi passando
invece a impianti interni come nel caso della retina
artificiale (ASR), un chip di un paio di millimetri di
diametro e più sottile di un capello, da impiantare all'interno
dell'occhio. Questo particolare chip contiene 5.000
fotosensori in grado di convertire la luce in impulsi
elettrochimici e con questi stimolare le cellule della retina
del paziente ancora in grado di funzionare (il che dovrebbe
rendere inutile l'apparato nel caso di pazienti con la retina
totalmente compromessa).
In cerca della nanobatteria.
Questo nuovo tipo di dispositivi elettronici pongono agli
scienziati tutta una serie di problemi, in parte inediti.
Seguendo la tradizione consolidata dell'elettronica, a ogni
successiva generazione il prodotto rischia di diventare più
vorace di energia. Ci si potrebbe dunque trovare dinnanzi al
problema di dover dipendere da una qualche forma di
energia esterna o di accumulatore impiantato nel corpo. In
realtà il microchip ASR si alimenta da solo, sfruttando la
luce che lo colpisce- ma questa soluzione rischia di metterlo
in difficoltà in situazioni di scarsa luce ambientale, il che
limiterebbe la sua utilità. Sul fronte delle fonti energetiche
impiantabili si sta dunque muovendo un consorzio di enti e
aziende americane, che ha intrapreso il lavoro di ricerca su
una batteria che dovrebbe produrre elettricità imitando i
processi biologici degli organismi viventi. Una batteria tanto
piccola da poter trovare posto nell'occhio insieme alla retina
artificiale, per arrivare ad una soluzione del tutto interna e
quindi più comoda ed "accettabile" per il paziente .
L'altro grande problema è garantire il funzionamento di un
dispositivo delicato come un microchip in un ambiente così
aggressivo come l'interno del corpo umano, proteggendo al
contempo il delicato corpo umano da possibili effetti
collaterali del chip impiantato nell'occhio. Una soluzione a
questo problema sta per essere individuata attraverso un
sofisticato rivestimento, basato sull''applicazione di uno
strato ultrananocristallino composto da cristalli di diamante
del calibro di 5 milionesimi di millimetro.
Piccoli passi verso l'uomo bionico.
Anche se queste soluzioni, ancora ai primi passi,
rappresentano una possibile soluzione solo per alcune forme
di cecità, sembra si possa essere ottimisti, almeno per i
ciechi del mondo occidentale, in grado di permettersi (anche
grazie a una mutua o assicurazione sanitaria) il costo di
apparato ed operazione. È dunque probabile che a medio
termine questo tipo di impianti ridaranno almeno
parzialmente la vista a un certo numero di non vedenti. Nel
lungo periodo, conoscendo come funzionano gli esseri
umani e tenendo in conto le probabili evoluzioni
tecnologiche, non mi sorprenderebbe diventasse comune
farsi sostituire occhi perfettamente funzionanti con occhi
bionici, capaci di vedere più lontano, funzionanti in assenza
di luce o in grado di captare radiazioni non visibili, essendo
in grado di "vedere" l'infrarosso o l'ultravioletto.
di Roberto Venturini de "Apogeonline"
RIVEDERE LA LUCE GRAZIE AD UN'ALGA
I primi esperimenti su topi geneticamente affetti da
degenerazione retinica
C'è ancora molto lavoro da fare, ma i risultati di un
esperimento condotto da Zhuo-Hua Pan, della Wayne State
University fanno sperare che grazie a una nuova strategia
terapeutica sia in futuro possibile restituire la vista a chi
soffre di alcune patologie degenerative della retina. Un
primo risultato è stato già ottenuto in un ceppo di topi che
soffre di una deficienza di funzionalità dei fotorecettori
analoga a quella che si manifesta nelle persone affette da
retinite pigmentosa. Utilizzando un virus innocuo, ZhuoHua Pan ha infatti introdotto nei neuroni che formano lo
strato interno della retina, che normalmente non sono
fotosensibili, un gene che codifica una proteina sensibile alla
luce. Come riferisce Zhuo-Hua Pan in un articolo apparso
sul numero odierno di Neuron, la nuova proteina prodotta
dalla cellula - una forma di rodopsina (ChR2) comunemente
presente in un'alga verde - ha fatto sì che i neuroni trattati si
attivassero in presenza di luce, inviando un segnale alla
corteccia visiva dell'animale. Inoltre, la proteina non viene
degradata subito dagli enzimi presenti nella cellula nervosa
e persiste in essa a lungo. Uno dei problemi che restano da
affrontare è la determinazione dell'intensità del segnale che
arriva alla corteccia cerebrale, dato che la proteina dell'alga
è meno sensibile di quelle che vengono sfruttate da coni e
bastoncelli. Inoltre, osserva Zhuo-Hua Pan, " le patologie
degenerative della retina sono eterogenee e sono necessari
ulteriori studi per stabilire quali tipi di esse siano
eventualmente trattabili". Rispetto ad altre tecniche
sperimentali perseguite in questo campo, quella ideata da
Zhuo-Hua Pan offre il vantaggio di non richiedere l'impianto
di alcuna protesi o chip elettronico all'interno dell'occhio,
fonte di possibili reazioni negative da parte dell'organismo
ricevente.
da "Le Scienze"
LA MOLECOLA CHE BLOCCA L'ANGIOGENESI
Ricerca approvata in USA. Laser e farmaci neutralizzano la
crescita dei vasi sanguigni.
USA - Gli anziani possono perdere la propria indipendenza
nelle attività quotidiane a causa della degenerazione
maculare legata all'età, che può rendere difficile o
impossibile leggere o guidare. Ma esistono anche forme
cosiddette giovanili della malattia, una delle quali, la più
diffusa, è quella miopica. In questo caso, i vasi sanguigni
indeboliti dalla miopia favoriscono i processi di
degenerazione maculare. In entrambi i casi il trattamento
prevede l'uso di specifici farmaci o laser con particolari
lunghezze d'onda.
Scopo della terapia farmacologica è quella di neutralizzare
con apposite sostanze le proteine Vegf (Vascular endothelial
growth factor) che stimolano la crescita dei vasi anomali,
principali responsabili della malattia. È stato recentemente
pubblicato sull'American e sul British Journal of
Ophthalmology che le statine, utilizzate per abbassare i
livelli di colesterolo, possano essere utili anche a mitigare
l'infiammazione cronica che porta alla produzione di Vegf e
alla malattia.
Ifarmaci di nuovissima generazione, testati in
sperimentazioni cliniche internazionali, combattono
direttamente i fattori di crescita vascolare che portano alla
formazione dei vasi sanguigni patologici nella degenerazione
maculare legata all'età. Il primo di questi farmaci ( per il
quale abbiamo partecipato al Board europeo) ha avuto
recentemente l'approvazione della Fda ( Food and drug
administration), l'organismo americano di riferimento per
farmaci e cibi, e da poco anche la pre approvazione
dell'omologa europea Emea ( European medicine evaluation
agency). Occorrono però ancora mesi perché sia in
commercio in Italia. Tali farmaci devono essere posti a
contatto della macula, e per questo occorre un piccolo
intervento chirurgico che veicoli i farmaci sulla macula
dall'interno o dall'esterno dell'occhio con speciali iniezioni.
Per questo si stanno mettendo a punto e sperimentando
particolari procedure chirurgiche mini invasive.
I risultati dell'efficacia di questi farmaci, promettenti ma
ancora non utilizzati su una grande scala, sono stati discussi
a Chicago lo scorso ottobre durante il Congresso
dell'American Academy of Ophthalmology. E farmaci simili
sono in fase finale di sperimentazione ed entreranno presto
in commercio.
La terapia laser utilizza particolare lunghezze d'onda. Il
raggio laser può andare direttamente a distruggere i vasi
anomali, o indirettamente interagendo con una sostanza
fotosensibile preventivamente iniettata in vena e affine ai
vasi anomali. Quest'ultima è nota come terapia
fotodinamica, e spesso sono necessarie più sedute prima di
giungere alla chiusura definitiva dei vasi anomali.
Negli ultimi due tre anni si è però constatato che tale
terapia, seppur utile in molti casi, non è sempre efficace. Per
questo sono stati avviati studi sperimentali - tuttora in corso
- che associano le terapie laser con i nuovi farmaci
antiproliferativi.
Allo studio la possibilità di associare la « cura »
fotodinamica ai nuovi antiproliferativi
di Claudio Azzolini de "Il Sole 24 Ore"
RENEURON
(traduzione dall’originale di Serena Greci Green)
ReNeuron annuncia primi dati preclinici ottenuti con il suo
programma ReN003 con cellule staminali retiniche e firma
un accordo di collaborazione con lo Schepens Eye Research
Institute.
Guildford, Regno Unito - il Gruppo ReNeuron plc (LSE:
RENE.L) annuncia oggi alcuni primi dati sull'efficacia di
sopravvivenza relativi al suo programma ReN003 di terapia
con cellule staminali per le patologie della retina. La ricerca
congiunta, guidata dai Prof. John Greenwood e Stephen
Moss dell'Istituto UCL di Oftalmologia di Londra, ha
evidenziato un'espansione delle cellule progenitrici retiniche
umane con marker di fotorecettori su molteplici raddoppi di
popolazione. Queste progenitrici hanno mostrato una
capacità ad impiantarsi nello strato fotorecettore della retina
ed a proteggerlo dalla degenerazione in un modello
distrofico retinico. La ricerca è stata finanziata da una
sovvenzione per la ricerca strategica sulle cellule staminali
conferita dal Medical Research Council (il Consiglio
nazionale britannico per la ricerca medica), ed è stata
presentata all'assemblea annuale dell'Associazione per la
Ricerca Oftalmica e sulla Vista (Association for Research in
Vision and Ophthalmology - ARVO) che si è tenuta a Fort
Lauerdale, in Florida, dal 30 aprile al 4 maggio, 2006.
Per portare avanti il suo programma ReN003 sulle cellule
staminali della retina, ReNeuron ha anche annunciato di
aver firmato un accordo di collaborazione di ricerca con lo
Schepens Eye Research Institute della Harvard Medical
School di Boston (USA). La ricerca condotta da questa
collaborazione si svolgerà presso i laboratori del Dott.
Michael Young, e si propone di stabilire le condizioni chiave
per coltivare linee di cellule staminali retiniche che possano
essere sviluppate per ottenere una terapia dimensionabile,
efficace e sicura che utilizzi la tecnologia di espansione cmycERTAM di proprietà di ReNeuron. L'obiettivo è
sviluppare queste linee di cellule staminali per trattare
importanti malattie causa di cecità, quali la degenerazione
maculare legata all'età, la retinite pigmentosa e la
retinopatia diabetica, che tutte assieme rappresentano una
rilevante necessità medica tuttora non soddisfatta.
Dice il dott. John Sinden, Capo Funzionario Scientifico della
ReNeuron: "Sono molto felice che la ReNeuron stia
lavorando così strettamente sia con l'Istituto di Oftalmologia
UCL e con l'Istituto Schepens, ovvero con due dei centri
clinici e di ricerca più importanti del mondo nel campo delle
patologie della retina. La nostra nuova collaborazione con lo
Schepens permetterà di unire la sua importante tecnologia
brevettata e le sue conoscenze alla versatile piattaforma di
cellule staminali della ReNeuron: l'obiettivo è generare
nuove terapie con cellule staminali per queste gravi
patologie della retina. Future collaborazioni con entrambe
queste istituzioni offrono la possibilità di portare queste
terapie alla pratica clinica nella maniera più efficiente
possibile".
ReNeuron Group plc
ReNeuron, collocata nel Regno Unito, è un'azienda leader
nelle terapie che utilizzano cellule staminali adulte. La
società sta applicando le sue innovative tecnologie di
piattaforma di cellule staminali per sviluppare terapie
d'avanguardia che utilizzino le cellule staminali per
necessità cliniche importanti ma ancora poco o affatto
soddisfatte. ReNeuron ha usato la sua tecnologia cmycERTAM per generare linee di staminali neuronali
geneticamente stabili. Questa piattaforma tecnologica è
coperta da brevetti internazionali ed è pienamente regolata
mediante un interruttore di sicurezza indotto chimicamente.
La crescita delle cellule può dunque venire completamente
arrestata prima dell'impianto in vivo.
Altre applicazioni della ricerca ReNeuron
La Re001, la sua principale terapia con staminali contro
l'invalidità cronica a seguito di ictus, è in fase avanzata di
sviluppo preclinico. Se la sperimentazione preclinica verrà
completata con successo, la società intende richiedere
quest'anno l'autorizzazione a procedere alle prime
sperimentazioni cliniche per l'ictus, ed avviare tali prove
appena possibile.
Mediante la sua terapia ReN005 con cellule staminali, la
società ha anche generato dati sull'efficacia preclinica per il
morbo di Huntington, una rara e mortale malattia genetica
neurodegenerativa che colpisce circa una persona su
100.000. Questo programma è in fase di sviluppo preclinico.
In aggiunta ai suoi programmi per l'ictus e il morbo di
Huntington, la ReNeuron sta sviluppando terapie con cellule
staminali contro il morbo di Parkinson, il diabete di Tipo 1 e
le malattie della retina.
ReNeuron ha anche spinto le sue tecnologie a staminali in
aree non terapeutiche: si tratta della sua gamma ReNcell di
linee di cellule per l'utilizzo in applicazioni tese alla scoperta
di farmaci nell'industria farmaceutica.
Ulteriori informazioni su ReNeuron ed i suoi prodotti sono
disponibili su www.reneuron.com.
L'Istituto UCL di Oftalmologia
L'Istituto UCL di Oftalmologia (IO) è uno dei più grandi
istituti di ricerca al mondo consacrati al progresso della
comprensione delle malattie della vista e dell'occhio. La sua
missione è portare nuove terapie innovative alla pratica
clinica a beneficio dei pazienti in tutto il mondo. L'IO fa
parte della UCL Biomedicine, uno dei maggiori aggregati di
scienze biomediche del mondo, ed è stata valutata 5* (la
valutazione più alta possibile) negli ultimi due esercizi di
valutazione della ricerca. I 40 scienziati che fanno parte
dell'organico dell'IO coprono un ampio spettro di talenti, da
chi indaga sui processi cellulari fondamentali agli scienziati
clinici che eseguono le prove cliniche. L'IO collabora con la
Fondazione NHS dell'Ospedale Oftalmico di Moorfields ed
ha forti legami con altri ospedali oftalmici nel Regno Unito
ed in Europa. La biologia delle cellule staminali ha un ruolo
importante: sono quattro i progetti finanziati dal Medical
Research Council attualmente in corso, uno dei quali
sostiene la collaborazione descritta sopra. Per trattare
pazienti con malattie della superficie oculare viene usato un
centro clinico per le cellule staminali recentemente
accreditato. L'IO è anche leader mondiale nel campo delle
scoperte e delle terapie genetiche relative alle patologie
dell'occhio.
La varietà di malattie dell'occhio studiate dal personale
dell'IO è ampia, ma particolare attenzione viene dedicata a,
fra l'altro, la degenerazione della retina, compresa quella che
colpisce i giovani, alla degenerazione maculare legata all'età
(AMD) ed al glaucoma. L'AMD è la causa più comune di
cecità non curata presente nel mondo industrializzato.
Lo Schepens Eye Research Institute
Fondato nel 1951, lo Schepens Eye Research Institute è il
maggiore centro indipendente di ricerca oftalmica presente
nelle Americhe. Affiliato alla Scuola di Medicina di Harvard,
l'istituto ha avuto un notevole impatto sulla pratica
oftalmica a livello internazionale. I suoi ricercatori hanno
pubblicato oltre 4.000 relazioni scientifiche e formato più di
600 scienziati della vista e specialisti in oftalmologia negli
USA e in più di 40 altri paesi nel mondo. Gli scienziati dello
Schepens fanno parte di équipes di ricerca interattive i cui
obiettivi sono di sviluppare metodi più potenti per la
diagnosi non invasiva delle malattie dell'occhio e di creare
nuovi trattamenti basati sulla terapia genetica oculare, sul
trapianto di cellule retiniche e staminali, su apparecchi
adiuvanti e riabilitazione per ipovedenti, sul trapianto della
cornea, sulla bioingegneria e sull'ingegneria dei tessuti.
(Il presente annuncio contiene dichiarazioni prospettive
sulla situazione finanziaria, sui risultati dell'attività e
sull'andamento/performance economica della ReNeuron,
come anche ad alcuni piani ed obiettivi della dirigenza
ReNeuron. Tali dichiarazioni possono generalmente, ma
non sempre, essere identificate dall'uso di parole quali
"dovrebbe", "prevede", "stima", "ritiene" od altre espressioni
analoghe. Questo annuncio contiene altresì dichiarazioni
prospettive attribuite ad alcuni terzi relativamente alle loro
stime circa la crescita dei mercati e della domanda di
prodotti. Per loro stessa natura, le dichiarazioni prospettive
implicano rischi ed incertezze perché rispecchiano le attuali
aspettative e supposizioni di ReNeuron in merito a
circostanze ed eventi futuri che potrebbero rivelarsi inesatte.
Diversi fattori potrebbero far sì che la reale situazione
finanziaria, i risultati delle sui risultati dell'attività e
sull'andamento/performance economica della ReNeuron
differiscano materialmente dalle stime fatte od implicite in
tali dichiarazioni prospettive; di conseguenza, non bisogna
fare affidamento su tali dichiarazioni.
I termini "ReNeuron" e "la Società" si riferiscono a
ReNeuron Group plc e le sue attività sussidiarie.
LUCENTIS: LA RICERCA VA AVANTI
E’ allo studio un nuovo farmaco contro la forma essudativa
della degenerazione maculare legata all’età – il suo nome è
LUCENTIS. Anche in questo caso il farmaco si
somministra mediante iniezioni direttamente dentro
l’occhio. Il Lucentis (ranibizumap) è un anticorpo che
inibisce il VEGF, una proteina che gioca un ruolo critico
nella angiogenesi ed è direttamente coinvolta nella
formazione di nuovi vasi. Il Lucentis bloccherebbe quindi
la formazione della neovascolarizzazione che è la causa
principale di perdita della funzione visiva nei pazienti
affetti dalla forma essudativa della degenerazione
maculare.
Novartis ha appena annunciato che si è concluso il primo
di uno degli studi di Fase III e i risultati sono decisamente
confortanti. Il 95% dei pazienti trattati con iniezioni
intraoculari di Lucentis manteneva o migliorava la vista
(definita come perdita di meno di 15 lettere) a distanza di
un anno dal trattamento, contro il 62% dei pazienti nel
gruppo di controllo. Gli effetti collaterali oculari erano
inferiori all’1%.
Al momento Lucentis non si trova ancora in commercio in
quanto deve passare il vaglio di ulteriori sperimentazioni
cliniche e della approvazione da parte della Food and Drug
Administration (FDA).
da "Occhioallaretina.it"
UN FATTORE DI CRESCITA PER IL NERVO
OTTICO
È attivo, in vitro, anche su altre cellule nervose.
BOSTON - Ricercatori del Children's Hospital di Boston
hanno scoperto un fattore di crescita naturale che stimola la
rigenerazione degli assoni lesi all'interno del sistema
nervoso centrale. In condizioni normali gran parte degli
assoni presenti nel sistema nervoso centrale non sono in
grado di ricrescere dopo essere stati danneggiati. La
scoperta di questo nuovo fattore di crescita nervoso,
battezzato oncomodulina, è descritta nell'ultimo numero
della versione on line di Nature Neuroscience. Lo studio è
stato condotto sul nervo ottico, che connette la retina ai
centri cerebrali della visione. Quando l'oncomodulina è stata
addizionata al brodo di coltura in cui era conservato uno di
questi neuroni lesionati, la velocità di ricrescita è
raddoppiata, evidenziando una capacità di stimolazione
della sostanza molto superiore a quella di qualsiasi altro
fattore di crescita noto. Gli autori della scoperta sperano che
l'oncomodulina possa essere in futuro utilizzata per riparare
danni subiti dal nervo ottico in seguito a glaucoma, tumori o
insulti traumatici. L'oncomodulina ha inoltre mostrato di
essere attiva, quanto meno in vitro, su almeno un altro tipo
di cellula nervosa, inducendo i ricercatori a iniziare ulteriori
ricerche in vista di una sua possibile utilizzazione anche nel
caso di danni conseguenti a ictus o a lesioni del midollo
spinale.
da "Le Scienze"
VISTA DA LINCE GRAZIE ALL'RNA
Nuova arma per colpire la proteina VEGF, responsabile
dello sviluppo della degenerazione maculare senile
USA. Minare le basi della cecità che colpisce molte persone
con l'avanzare dell'età servendosi di minuscoli pezzetti di
RNA (acido ribossinucleico, il 'cugino' del più noto Dna)
diretti contro i geni che scatenano la malattia. è questo
l'oggetto della prima sperimentazione sull'uomo di una
terapia genica che sfrutta un processo naturale di
regolazione dell'espressione dei geni da parte di piccoli
frammenti di Rna; i risultati sono stati presentati la scorsa
settimana in occasione di un incontro tenutosi a Baltimora,
organizzato dalla società americana che si occupa di terapia
genica.
Lo studio ha valutato in via preliminare l'efficacia di un
farmaco chiamato Bevasiranib su pazienti affetti da
degenerazione maculare senile, una malattia sempre più
frequente che può portare a cecità irreversibile per la
crescita di vasi sanguigni che ricoprono la parte posteriore
dell'occhio. Le terapie disponibili oggi, infatti, hanno
un'efficacia limitata e non rallentano la progressione della
malattia.
I ricercatori della Acuity Pharmaceuticals di Philadelphia si
sono quindi concentrati sul meccanismo che porta alla sovra
produzione di vasi sanguigni nella retina, elemento centrale
per lo sviluppo della malattia. Il protagonista di questo
fenomeno è una proteina detta VEGF, dimostratasi
necessaria e sufficiente allo sviluppo della degenerazione
maculare se presente in alta quantità e che rappresenta un
bersaglio riconosciuto non solo in campo oftalmico ma
anche in quello oncologico.
Per colpire questa proteina, Bevasiranib si avvale di piccoli
frammenti di Rna che agiscono come proiettili molto precisi,
andando ad attaccarsi alle molecole di RNA cellulari da cui
verrebbe prodotto VEGF. I piccoli proiettili non
interagiscono invece con il Dna, scongiurando il rischio di
alterare il patrimonio genetico emerso con altre forme di
terapia genica. Questo meccanismo d'azione, efficace e
specifico, avviene normalmente all'interno delle nostre
cellule per regolare l'espressione dei geni e quindi la
successiva produzione delle proteine. Il tutto acquisisce un
interesse clinico in situazioni in cui ci sia un'abbondante
produzione di una proteina chiave come Vegf alla base di
una malattia: è in questi casi che piccole molecole di Rna
possono diventare veri e propri farmaci. Le molecole
vengono dunque prodotte artificialmente in modo da
raggiungere la destinazione prescelta per spegnere il segnale
desiderato. Devono quindi essere specifiche, agire per un
periodo di tempo sufficiente e non causare effetti collaterali
rilevanti: tutte caratteristiche che Bevasiranib sembra
possedere. Lo studio che ha coinvolto circa 130 persone
affette da degenerazione maculare senile avanzata, nelle
quali si è osservata una riduzione della crescita dei vasi
sanguigni nella retina con un evidente miglioramento della
capacità visiva. Alla dose più bassa gli effetti durano qualche
mese, a quella più alta perdurano più a lungo senza effetti
collaterali di rilievo fatta eccezione per una reazione
infiammatoria nel sito dell'iniezione. La somministrazione
locale del farmaco rappresenta un vantaggio per il suo
profilo di sicurezza, accanto a una maggiore praticità di
somministrazione.
Secondo i ricercatori che lo hanno sviluppato, Bevasiranib
potrebbe essere usato anche in combinazione con altri
farmaci che agiscono sempre su Vegf andando a bloccare la
proteina già prodotta. In questo modo si produrrebbe un
effetto sinergico di blocco totale della proteina patogena.
Questo e altri aspetti verranno indagati negli studi futuri:
l'azienda si è già impegnata a comunicare i risultati degli
studi di fase due entro il prossimo settembre e ad avviare
l'anno successivo quelli di fase tre.
I GENI CHE CAUSANO LA DEGENERAZIONE
MACULARE
Proviene dalla Sardegna un'importante scoperta in ambito
di genetica della degenerazione maculare, malattia che
colpisce quasi il 20% degli anziani dell'Occidente,
diventando poi la prima causa di cecità. Una scoperta che ha
ottenuto anche un prestigioso riconoscimento
internazionale. Secondo i dati più aggiornati, circa il 18-20%
degli anziani dell'Occidente soffrono di degenerazione
maculare, deterioramento della retina che diventa poi la
prima causa di cecità.
Si tratta quindi di un problema notevole, rispetto al quale
appare importante capire come si sviluppi e se vi siano dei
geni che ne determinano la predisposizione. Recentemente,
in Sardegna, le ricercatrici Annalisa Loi Zedda (Società di
Ricerca Genomica Shardna) e Maria Cristina Mallocci
(Clinica Oculistica dell'Università di Cagliari) hanno messo
in luce la scoperta che effettivamente un gruppo di geni è
interessato a questa patologia che colpisce la parte più
delicata dell'occhio.
La ricerca - premiata con il Premio Internazionale Alcon,
uno dei più prestigiosi riconoscimenti di oculistica - è il
frutto di una collaborazione tra la già citata Società Shardna
e l'Istituto di Genetica delle Popolazioni del CNR di Sassari.
Un'immagine di Talana, nell'area dell'Ogliastra, in provincia
di NuoroA fornire un ottimo campo di indagine sono state le
caratteristiche genetiche della popolazione dell'Ogliastra,
zona della Sardegna dove è presente una percentuale tra le
più alte al mondo di ultracentenari. Il relativo isolamento di
tale territorio ha in sostanza permesso il formarsi di
popolazioni con caratteristiche genetiche particolari e molto
fertili per questi tipi di studi.
Nei laboratori si indaga in particolare sulla genotipizzazione,
ovvero sull'individuazione dei polimorfismi, le variazioni
delle quattro basi del DNA. Questa viene poi incrociata con
le caratteristiche ambientali e di vita dei vari paesi. Durante
appunto una di queste ricerche - condotta nella località di
Talana - Loi Zedda e Mallocci hanno rilevato che in un
consistente numero di persone non vi era traccia né della
degenerazione maculare senile, né dei geni che alcuni
ricercatori statunitensi avevano ipotizzato essere la causa di
questa malattia. Un risultato, quindi, che la comunità
scientifica ha giudicato come un'importante dimostrazione
indiretta del collegamento di quei geni con quella patologia.
di S. B. da "Superando.it"
CELLULE STAMINALI UMANE: UTILI PER
MALATTIE DEGLI OCCHI - STUDI NEGLI USA
WASHINGTON - Le cellule staminali embrionali umane
possono parzialmente ripristinare la vista nelle cavie cieche
e possono essere una fonte di trapianto per alcune malattie
degli occhi. Lo rende noto una ricerca Usa, pubblicata sul
giornale Cloning and Stem Cells.
"Abbiamo sviluppato una tecnologia che speriamo possa
essere usata per curare le malattie oculari degenerative
come la maculopatia", ha detto il dottor Robert Lanza della
Advanced Cell Technology a Worcester, Massachusetts, che
ha condotto lo studio.
"Abbiamo dimostrato che queste cellule staminali
embrionali possono salvare le funzioni visive in animali che
altrimenti diventerebbero ciechi", ha spiegato Lanza in una
e-mail.
Le staminali sono un tipo di cellule che riescono a
riprodurre vari tipi di tessuti. Quelle prese dagli embrioni
sono particolarmente malleabili e possono produrre
qualsiasi cellula o tessuto del corpo. Il loro uso e la loro
produzione è controversa, con gli oppositori che sostengono
che non sia etico usare embrioni umani a questo scopo.
Il presidente Usa George W. Bush ha ristretto i
finanziamenti federali per la ricerca sulle staminali
embrionali a poche sequenze di cellule già esistenti
nell'agosto 2001. Le società private come Advanced Cell
Technology possono fare come vogliono, e il team di Lanza
ha usato alcune delle sequenze del 2001 e altri prodotte
usando finanziamenti privati.
di "Reuters Italia"
TELETHON FINANZIA CON 645 MILA EURO LA
RICERCA SULLE NEUROPATIE OTTICHE
EREDITARI
E' stato finanziato da Telethon con 645.000 euro il progetto
di ricerca sulla Degenerazione del nervo ottico nelle
neuropatie mitocondriali presentato dal Dott. Valerio Carelli
del Dipartimento di Scienze Neurologiche dell'Università di
Bologna, che coordina una rete di 6 laboratori italiani. Il
progetto riguarda un gruppo di malattie ereditarie - la
neuropatia ottica ereditaria di Leber (LHON) e l'atrofia
ottica dominante (DOA) - che portano a cecità.
Si tratta di malattie rare ma non rarissime (stime da
1:10.000 a 1:50.000). Il significato della ricerca sta anche
nel fatto che queste neuropatie ottiche sono malattie
mitocondriali pure, e quindi costituiscono un importante
modello di studio per il funzionamento dei mitocondri. I
mitocondri sono la "centrale energetica" di tutte le cellule, e
la ricerca può quindi fornire indicazioni sul coinvolgimento
di questi organelli in patologie neurodegenerative più
frequenti come la malattia di Alzheimer e la malattia di
Parkinson e su possibili interventi terapeutici. Spiega il dr.
Carelli: "Le neuropatie ottiche mitocondriali sono
caratterizzate da una degenerazione selettiva di uno
specifico tipo cellulare retinico, le cellule ganglionari della
retina. La maggior parte dei pazienti perde la vista in età
precoce (prima dei 10 anni nella DOA, e nell'adolescenza per
i maschi nella LHON). Le basi genetiche di entrambe le
malattie sono ora conosciute e coinvolgono proteine che
svolgono le loro funzioni nei mitocondri. Nonostante
l'origine genetica di queste malattie sia nota e le funzioni dei
mitocondri siano ben conosciute, non siamo ancora in grado
di capire perché solo le cellule ganglionari della retina sono
colpite e perché non tutti gli individui portatori del difetto
genetico si ammalino. Inoltre, al momento non esiste una
cura per questi pazienti. Il nostro progetto ha l'obiettivo di
chiarire gli enigmi sopra menzionati usando diverse
strategie che includono ulteriori studi genetici, studi
funzionali in vivo, e modelli cellulari di malattia. In
particolare, abbiamo già dei risultati preliminari che ci
indicano come le cellule siano in grado di organizzare una
strategia compensatoria al malfunzionamento mitocondriale
tale che alcuni individui non sviluppano mai la malattia.
Crediamo fermamente che la conoscenza dettagliata di
questi meccanismi sia di grande aiuto per la comprensione
della malattia e possa fornirci dei mezzi per la terapia".
Il prof. Baruzzi, direttore del Dipartimento di Scienze
Neurologiche aggiunge: "Questo finanziamento, ottenuto da
una istituzione scientifica di alto prestigio come il Telethon,
è anche un riconoscimento all'impegno del nostro
dipartimento nella ricerca scientifica sulle malattie
neurodegenerative, su cui abbiamo fatto negli ultimi 10 anni
importanti investimenti in termini di persone e strutture". Il
progetto si svolgerà in un arco di tempo di 3 anni, e oltre all'
Università di Bologna come centro principale con 3
laboratori (il coordinatore dott. Valerio Carelli, Laboratorio
di Genetica, Dipartimento di Scienze Neurologiche - la
dott.sa Anna Ghelli, Laboratorio di Biologia cellulare del
Dipartimento di Biologia - il prof. Raffaele Lodi, Diagnostica
con Spettroscopia RM del Dipartimento di Medicina Clinica
e Biotecnologia Applicata), partecipano anche l'Università
"La Sapienza" di Roma (la prof.ssa Giulia D'Amati,
Dipartimento di Medicina Sperimentale e Patologia),
l'IRCCS "E. Medea" (il dott. Andrea Martinuzzi), e
l'Università di Bari (il prof. Palmiro Cantatore, Dipartimento
di Biochimica e Biologia Molecolare).
di "Bur.it"
UN BIOCHIP CHE RESTITUISCE LA VISTA
Chip impiantati direttamente nell'occhio per sconfiggere
definitivamente le malattie degenerative dell'apparato
visivo. Fantascienza? No, anzi, alcuni progetti avanzati sono
in dirittura d'arrivo.
ROMA - Prove di fusione definitiva tra apparati organici e
processori al silicio, scienze biologiche e tecnologie
informatiche. Non siamo ancora arrivati alla macchina di
Turing biologica, ma gli innesti di microscopiche unità
computazionali all'interno di organismi viventi complessi fa
un altro passo verso la direzione indicata da anni dal
fortunato filone della fantascienza cyber-punk. Scienziati
dell'Università della Pennsylvania hanno annunciato la
realizzazione di un chip impiantabile direttamente
nell'occhio umano, capace di restituire la vista alle persone
affette da gravi forme di degenerazione cellulare
dell'apparato visivo.
Il microchip è frutto di un progetto pensato per combattere
la retinite pigmentosa, malattia degenerativa di origine
genetica che distrugge progressivamente tutte le cellule
retinali, unità biologiche fondamentali responsabili della
transcodifica dei segnali luminosi catturati dall'occhio in
impulsi nervosi inviati al cervello per l'interpretazione. Col
passare degli anni, l'intero tessuto retinico viene affetto dal
morbo fino alla perdita totale della vista da parte del
paziente.
Non è la prima volta che si registrano notizie di ricerche
volte a restituire la vista con innesti tecnologici: ricordiamo
a tal proposito il lavoro dei ricercatori dell'Università di
Stanford, e il loro occhio bionico servo-assistito da computer
e mini-telecamere esterne. In quel caso, la microcamera
montata su uno speciale paio di occhiali catturava le
informazioni visive che, una volta processate da un
computer esterno, venivano trasmesse in wireless all'innesto
tecnologico presente nell'occhio del paziente.
L'approccio del Penn è molto più sofisticato: il
microprocessore viene impiantato a diretto contatto con il
nervo ottico, e da solo si occupa di catturare la luce e
convertirla in segnali interpretabili dalla zona della corteccia
adibita alla visione, esattamente come gli impulsi nervosi
generati da un apparato visivo perfettamente funzionante.
Non solo: il bio-chip sarebbe in grado di replicare la maniera
in cui una retina sana percepisce il movimento e adatta
l'intensità della luce e il contrasto dei colori.
Un design minimale e perfettamente mimetizzato nel
tessuto vitale dell'organismo, una fusione quasi perfetta di
corpo e macchina in grado finalmente di realizzare uno dei
sogni a lungo accarezzati dai ricercatori e dagli scienziati di
mezzo mondo. O almeno è quello che promettono quelli del
Penn: prima che si passi alle sperimentazioni cliniche, e si
possa quindi verificare quanto di concreto ci possa essere
nelle speranze suscitate da annunci del genere, il prossimo
passo della ricerca consisterà nel ridurre ulteriormente le
dimensioni del chip-innesto e il consumo di energia
necessario al suo funzionamento continuato.
da "Punto Informatico"
IL RECETTORE CHIAVE DELLA MACULOPATIA
Aumentare le possibilità di cura: un obiettivo al quale punta
attivamente la ricerca nel campo della degenerazione
maculare o maculopatia senile, problema crescente nei paesi
avanzati in quanto legato all’invecchiamento (colpisce circa
un ultrasettancinquenne su tre) e prima causa di cecità
legalmente riconosciuta dopo i 55 anni. Una malattia della
quale si conoscono i fattori di rischio, soprattutto età
avanzata, familiarità, fumo, esposizione non protetta alla
luce solare, deficit di vitamine antiossidanti,
ipercolesterolemia, ipertensione, obesità, ma non sono note
le cause. Si sa che insorge per degenerazione di strutture
vicino alla macula retinica (epitelio pigmentato retinico,
membrana di Bruch e coriocapillari) che iniziano decenni
prima delle manifestazioni dei sintomi e conducono a due
situazioni: la maculopatia secca, non essudativa o non
neovascolare, più comune e graduale, e la maculopatia
umida o essudativa o neovascolare, che annovera il 10% dei
casi ma causa l’80% delle cecità, nella quale c’è una
neoformazione di vasi sanguigni che danno essudazione ed
emorragie sub-retiniche. Per quest’ultima è d’elezione la
terapia fotodinamica con iniezione endovenosa di una
sostanza fotosensibile (verteporfina) che aderisce ai neovasi
e viene attivata da un laser, un trattamento efficace ma che
in genere va ripetuto più volte e in molti casi non è
sufficiente. Da qualche anno l’attenzione è puntata sull’uso
di farmaci antiangiogenesi, che contrastano cioè la
formazione dei nuovi vasi sanguigni, da combinare con la
terapia fotodinamica.
Il caso unico dei lamantini.
L’angiogenesi è un processo da tempo individuato come
essenziale per la crescita tumorale e più di recente è emerso
che in esso svolge un ruolo importante la molecola VEGF
(fattore di crescita endoteliale vascolare), contro la quale si
potevano quindi dirigere terapie anticancro. Inibitori del
VEGF sono anche gli anticorpi monoclonali bevacizumab e
ranibizumab, il primo approvato dalla FDA statunitense per
il tumore del colon ma dimostratosi valido pure nella
maculopatia e il secondo approvato l’estate scorsa per
questa patologia. Ad ampliare le prospettive d’impiego degli
anti-VEGF nella terapie di combinazione della
degenerazione maculare senile giunge ora una ricerca
pubblicata sulla rivista Nature che ha individuato l’esistenza
di una proteina che impedisce l’angiogenesi nella cornea.
Questo tessuto trasparente di rivestimento dell’occhio
permette una visione ottimale grazie all’assenza di vasi
sanguigni, nonostante sia presente il VEGF: un’apparente
contraddizione a lungo irrisolta, che adesso si spiega con la
scoperta del recettore in forma solubile sVEGFR-1, o sflt-1,
che a differenza della forma di membrana non consente la
trasmissione del segnale per interazione con la proteina
VEGF. I ricercatori hanno osservato che in topi
geneticamente manipolati l’sflt-1 era asse e c’era
vascolarizzazione corneale spontanea; la stessa mancanza è
stata verificata nei lamantini, trichechi acquatici di zone
costiere dell’ordine dei Sirenidi (da cui la leggenda) che sono
gli unici animali noti con la cornea vascolarizzata, mentre
l’sflt-1 è presente negli stessi altri Sirenidi come i dugonghi e
in loro parenti terrestri come gli elefanti, suggerendo un
significato evolutivo.
Verso modulatori dell’angiogenesi.
Dalla scoperta del ruolo del sflt-1 nel mantenere la cornea
avascolarizzata si potrà forse arrivare alla messa a punto di
modulatori dell’angiogenesi da utilizzare nelle patologie a
base neovascolare, compresa la maculopatia senile.
Nonostante i progressi compiuti, per quest’ultima la terapia
ha ancora vari problemi da risolvere, tra i quali anche la
possibilità di trattamenti meno costosi e impegnativi, da non
ripetere più volte e con un migliore impatto sul benessere
dei pazienti. Un aspetto finora poco analizzato ma che si lega
alla mancanza di strumenti ottimali per valutare la qualità di
vita dell’anziano colpito dalla malattia (sottovalutata finché
non c’è un rapido peggioramento e non si estende all’altro
occhio). All’inizio possono infatti non essere riconosciuti
sintomi quali riduzione od oscuramento della visione
centrale e distorsione delle linee diritte, che si accertano con
il semplice test della griglia di Amsler; sono opportuni
comunque periodici controlli della vista. Per la prevenzione
si può poi agire sui fattori di rischio evitabili, dalla
protezione degli occhi esposti a forte luce solare
all’astensione dal fumo al consumo di alimenti (frutta e
verdure tra cui quelle a foglia verde) ricchi di antiossidanti
come vitamina C, E, beta-carotene e luteina.
da "Dica 33"
DEGENERAZIONE MACULARE SENILE: TROVATO
IL GENE DELLA FORMA AGGRESSIVA
È la prima causa di cecità nelle persone over 50.
Una variante di un gene aumenta il rischio di sviluppare la
forma "umida" della degenerazione maculare senile, che
rappresenta la più importante causa di cecità nella
popolazione ultracinquantenne. La scoperta è stata fatta da
ricercatori della Yale School of Medicine, che ne danno
notizia sull'ultimo numero di "Science". La degenerazione
maculare, che porta a una progressiva perdita della visione
centrale, si presenta in due forme: quella "secca", che
progredisce lentamente nel corso di molti anni, e quella
"umida" che è molto più aggressiva. Lo scorso anno lo stesso
gruppo di ricerca diretto da Josephine Hoh aveva
identificato un gene legato alla forma secca e scoperto che
entrambe le forme erano correlate a una variante nel gene
CFH (complement factor H), che esprime una molecola
coinvolta nella risposta infiammatoria dell'organismo ad
agenti patogeni. Nel nuovo studio Hoh e collaboratori hanno
scoperto che il rischio di sviluppare la malattia nella sua
forma più grave è fortemente condizionato dalla presenza di
un polimorfismo a singolo nucleotide (ossia nel
cambiamento di una sola base nucleotidica nella sequenza
del gene) a carico del gene HTRA1 presente sul cromosoma
10. In particolare, mentre la variante del gene CFH porta
alla lenta formazione a livello retinico di depositi di
materiali metabolici di scarto, la proteina espressa dal gene
HTRA1 influenza lo sviluppo dei vasi sanguigni nella retina,
il carattere distintivo della forma umida della maculopatia.
La compresenza di entrambi i meccanismi, pur alquanto
rara, porta alle forme più complesse della patologia.
da "Le Scienze
TRAPIANTO DI CELLULE DELLA RETINA E
ALCUNE CAVIE TORNANO A VEDERE
Lo studio, pubblicato su "Nature", è stato condotto sui
topolini. Apre nuove speranze per la cura delle malattie
degenerative
LONDRA - Riacquistare la vista grazie ad un trapianto di
cellule della retina. Per la prima volta, in un esperimento
condotto su topi, un gruppo di ricercatori è riuscito a
ripristinare parzialmente la facoltà visiva, trapiantando negli
animali cellule precursori della retina. Lo studio, pubblicato
sulla rivista Nature, si deve all'équipe del London Institute
of Ophthalmology ed ha utilizzato un nuovo approccio
sviluppato alla University of Michigan, presso il Kellogg Eye
Center. I risultati sono stati accolti con grande interesse
dalla comunità scientifica e potrebbero aprire la strada ad
una cura per moltissime malattie degenerative della retina,
compreso il diabete che affligge milioni di persone. Gli
scienziati hanno utilizzato cellule staminali adulte e quindi,
già con una destinazione precisa: fotorecettori, cioè cellule
della retina, "in fieri".
Nelle retinopatie si ha una degenerazione per cui i
fotorecettori che costituiscono la retina, ovvero le cellule
chiamate coni e bastoncelli, muoiono. Sono i fotorecettori a
tradurre gli impulsi visivi in un messaggio che poi,
viaggiando sul nervo ottico, arriva al cervello, che lo traduce
in immagini. Senza fotorecettori al cervello non arriva nulla
e la capacità visiva è persa.
E' da tempo che si tenta il trapianto di cellule nella retina
per ricostituire coni e bastoncelli, ma finora non c'erano
sono stati progressi significativi. Una delle coordinatrici
dello studio, la dottoressa Jane Sowden, si è detta sorpresa
dei risultati ottenuti: "Abbiamo riscontrato che la retina
matura, che finora si credeva non avesse la capacità di
rigenerarsi, è invece in grado di sopportare lo sviluppo di
nuovi fotorecettori funzionali", ha spiegato. I ricercatori
guidati da Robin Ali, dell'istituto londinese, hanno invece
pensato di trapiantare cellule "semi-adulte", cioè già
abbastanza sviluppate e avviate a diventare cellule della
retina, ma senza aver ancora raggiunto lo stadio di
maturazione completa. Ha funzionato: trapiantando cellule
della retina "in fieri", gli scienziati hanno ottenuto un
miglioramento parziale della funzione visiva nei topi.
Le cellule trapiantate hanno ultimato il loro sviluppo
trasformandosi in fotorecettori, e si sono integrate nella
retina, allacciando contatti con le terminazioni del nervo
ottico, diventando così funzionali.
"E' una ricerca sorprendente, che in futuro potrebbe portare
ad un trapianto per curare la cecità nell'uomo", ha
commentato alla Bbc il professor AndrewBrick
dell'università di Bristol. Il cammino verso un'applicazione
clinica è ancora molto lungo, ma già nel giro di cinque anni
si potrebbero avere i primi sviluppi verso un utilizzo di
questa nuova procedura anche sull'uomo.
da "Superabile.it"
Dopo questo e altri articoli sullo stesso argomento apparsi
su vari organi di stampa, crediamo opportuno ospitare un
intervento della Dottoressa Cristiana Marchese.
Ricercatori inglesi e americani hanno riportato sul numero
del 6 Novembre 2006 della rivista “Nature” i risultati
ottenuti in seguito al trapianto di precursori di fotorecettori
nella retina di topi sani e nella retina di topi con
degenerazione retinica. Molte malattie che portano a cecità
sono infatti causate dalla perdita progressiva dei
fotorecettori (bastoncelli e coni) e un trapianto di
fotorecettori sembra il modo più ovvio di curare queste
malattie. I fotorecettori sono cellule specializzate del sistema
nervoso il cui ruolo è quello di trasformare l'energia
luminosa in stimolo elettrico che viene poi trasferito tramite
altre cellule nervose retiniche e il nervo ottico alla zona del
nostro cervello che ha il compito di trasformare in immagine
lo stimolo ricevuto. Perché il trapianto sia efficace è però
necessario non solo che i fotorecettori trapiantati
sopravvivano, ma che stabiliscano le necessarie connessioni
(sinapsi) per poter trasmettere lo stimolo ricevuto.
Poiché le malattie causate dalla degenerazione dei
fotorecettori lasciano, almeno inizialmente, integro il
sistema delle altre cellule nervose retiniche, se il trapianto di
fotorecettori avviene in tempi precoci, questi hanno ancora
disponibile tutto il resto della rete di cellule retiniche
funzionanti e devono stabilire soltanto un tipo di
connessione (sinapsi). Sino ad ora i tentativi di trapianto
con cellule staminali derivate dal cervello o dalla retina non
avevano dato buoni risultati, infatti non si integravano nella
giusta parte della retina, non si differenziavano in
fotorecettori e non stabilivano adeguate connessioni con le
altre cellule.
In questo esperimento i ricercatori hanno trapiantato cellule
(precursori di fotorecettori) derivate da una retina in corso
di maturazione e giunte ad un punto di differenziazione
subito precedente la trasformazione delle cellule stesse in
fotorecettori (in particolare bastoncelli). Le cellule iniettate
nella retina sia di animali sani che di animali con
degenerazione retinica si sono inserite nella parte corretta
della retina, si sono differenziate in bastoncelli , hanno
stabilito le corrette connessioni (sinapsi) e i test
elettrofisiologici hanno dimostrato ilfunzionamento dei
fotorecettori trapiantati.
Questo è un importante passo verso la comprensione dei
meccanismi che regolano la differenziazione delle cellule in
fotorecettori. Si tratta di una ricerca eseguita su animali da
esperimento ed è necessaria una certa cautela prima di ,
poter affermare che è applicabile ai pazienti con
degenerazione retinica, anche se per la prima volta èstato
dimostrato che le cellule trapiantate sono in grado di
funzionare correttamente.
Dottoressa Cristiana Marchese
Dirigente Medico Genetista ASO Ordine Mauriziano Torino
IL MERIDIONE, AVANGUARDIA
CONTRO LA RETINITE PIGMENTOSA
ISTITUTI GENETICA E BIOFISICA
di Michele D'Urso
Dirigente di Ricerca dell'Istituto Internazionale
di Genetica e Biofisica del CNR, Napoli
LA SCHEDA
SCHEDA 1
Prima tappa contro la cecità.
Isolato uno dei geni della retinite pigmentosa.
La retinite pigmentosa è una malattia ereditaria dell'occhio
che provoca nel corso degli anni una progressiva riduzione
del campo visivo e rappresenta una delle più frequenti
cause di cecità. Si stima che nel mondo ne siano affetti più di
un milione e mezzo di individui; in Italia sono almeno
quarantacinquemila le persone colpite. Da alcuni anni i
ricercatori di tutto il mondo sono perciò impegnati ad
individuarne le cause genetiche. L'impresa è tutt'altro che
semplice, in quanto la retinite pigmentosa si può presentare
in forme diverse (si parla infatti di retiniti pigmentose), con
differenti modelli ereditari di trasmissione; non è
improbabile che le alterazioni (mutazioni) dei geni
responsabili siano addirittura alcune decine. È possibile,
quindi, che in due famiglie affette da retinite pigmentosa
apparentemente simili per epoca di insorgenza e decorso
clinico, i geni che inducono la malattia siano diversi.
Risultati incoraggianti sono stati ottenuti, grazie
all'identificazione di alcuni di questi geni implicati nella
malattia. È facile comprendere quale sia l'interesse pratico di
queste acquisizioni: conoscere infatti la causa di malattie
genetiche come questa, che spesso possono insorgere non
alla nascita (ma anche nell'infanzia, nella giovinezza e
perfino in età adulta), significa essere in grado di eseguire
una diagnosi preclinica ed addirittura prenatale, in modo da
attuare programmi di prevenzione, ed aprire la strada a
modelli di cura del tutto innovativi quali la terapia genica.
Un'importante scoperta, pubblicata nel numero di maggio
della prestigiosa rivista Nature Genetics, riguarda appunto
l'isolamento del gene RP3 che, trovandosi su uno dei
cromosomi sessuali X, colpisce in modo severo solo i
maschi, che ereditano il cromosoma con il gene sbagliato,
mentre le femmine (le madri e le figlie degli affetti, ma
anche alcune delle loro sorelle) sono soltanto portatrici e,
pertanto, a rischio a loro volta di generare altri maschi
malati. Circa il 20% dei malati di retinite pigmentosa sono
affetti da questa forma. L'RP3 è il primo gene per la retinite
pigmentosa identificato mediante la nuova strategia di
sequenziamento (sequenziamento posizionale). Le
implicazioni di questa scoperta hanno una rilevante
importanza non solo ai fini nosografici (classificazione di
malattia), ma anche per i nuovi sviluppi della ricerca
biochimica, in quanto la proteina predetta rappresenta una
nuova classe di proteine coinvolte nel processo della
fotorecezione.
Introduzione.
Con il termine retinite pigmentosa (RP) viene indicato un
insieme di retinopatie ereditarie, eterogenee dal punto di
vista clinico e genetico, e che colpiscono, primitivamente ed
in maniera progressiva, i fotorecettori e l'epitelio
pigmentato della retina. La malattia ha una frequenza nella
popolazione di 1:4000 e quindi costituisce la causa più
frequente di cecità nell'uomo. La sua trasmissione genetica
può essere distinta in tre forme: autosomica dominante
(ADRP), autosomica recessiva (ARRP) e legata al
cromosoma X (XLRP). Si può attualmente affermare che la
causa primaria della distruzione progressiva dei
fotorecettori nella malattia risieda in anomalie strutturali e
funzionali di proteine retiniche coinvolte nel ciclo visivo,
risultanti da differenti difetti genetici. Negli ultimi cinque
anni nello studio della retinite pigmentosa sono state
impiegate con successo tecniche di genetica molecolare che
hanno portato a una maggiore comprensione dei
meccanismi patogenetici di questo gruppo di malattie.
Infatti l'identificazione di molti marcatori genetici altamente
polimorfici ha facilitato la localizzazione di alcuni geni
responsabili della malattia, mentre lo sviluppo di tecniche
che permettono di individuare rapidamente ed
efficientemente mutazioni ha consentito di indagare su geni
"candidati" per la malattia. La retinite pigmentosa mostra un
elevato grado di eterogeneità genetica e sinora sono stati
clonati e/o mappati oltre 50 geni coinvolti nelle
degenerazioni retiniche. Al momento, ben 30 loci distinti
sembrano causare forme sindromiche e non per la sola
retinite pigmentosa, 9 dei quali sono stati anche identificati.
I geni finora identificati tengono però conto solo di una
piccola percentuale delle famiglie affette, lasciando perciò
presupporre l'esistenza di una complessa cascata di eventi
metabolici che regola il processo visivo con il
coinvolgimento di un numero ancora imprecisato di geni.
Isolamento e caratterizzazione del gene
RPGR (RP3).
L'eterogeneità genetica evidente nelle forme autosomiche è
riscontrabile anche nelle forme di retinite pigmentosa legate
al cromosoma X. La XLRP, che in alcune popolazioni ha
un'incidenza di circa il 20%, è forse la più devastante forma
di RP, a causa dell'esordio precoce (nelle prime due decadi
di vita nel maschio affetto) e della severità della malattia,
che porta alla cecità completa nel giro di 10-20 anni. La
tabella 1 mostra le tappe più importanti nello studio
genetico e molecolare della Retinite Pigmentosa legata al
cromosoma X. Nel 1984 S.S. Bhattacharya localizzò il primo
gene RP sul braccio corto del cromosoma X (Xp11.3),
mediante analisi di linkage ad un marcatore polimorfico di
DNA. Questo gene, presente tra dieci, forse cento altri geni
in una regione di 5 cM, non è stato ancora identificato.
Subito dopo, analisi di linkage e tecniche di citogenetica
confermarono la presenza di almeno altri due loci genetici
per la XLRP e, grazie ad un'estesa delezione cromosomica
osservata nel DNA di un individuo affetto da RP, il locus
RP3 venne mappato in una regione relativamente piccola e
compresa tra due marcatori polimorfici OTC e DXS1110 in
Xp21.1.
Tabella 1: tappe significative nello studio genetico e
molecolare dell'XLRP
1984 Mappaggio per linkage del locus RP2 in Xp11.3.
1985 Eterogeneità genetica in XLRP.
Evidenze di un altro locus in Xp21.
Lunga delezione cromosomica nel paziente BB (affetto da
DMD,
CGD, fenotipo McLeod e RP).
1988 Conferma della localizzazione RP3 in Xp21.
1990 L'esistenza del locus RP6 viene suggerita per analisi
statistica, in posizione distale a DMD in Xp21.
1995 Un altro locus RP15 viene mappato in Xp22. E' lo
stesso di RP6?
1996 Primo gene XLRP, RPGR, che viene identificato.
Più tardi l'analisi genetica rivelò la presenza di due altri loci
(RP6 e RP15), in posizione più telomerica (Xp22).
Attualmente sono stati quindi individuati ben quattro
possibili loci candidati tra le regioni Xp11.21 e Xp22.13; RP2
in posizione Xp11.4-p11.23; RP3 in Xp21.11; RP6 in
posizione Xp21.3-p21.2 e RP15 in Xp22.13-p22.11. I primi tre
seguono una trasmissione genetica recessiva mentre l'ultimo
locus è dominante. Nel corso di questi anni vari gruppi di
ricerca si erano concentrati sull'isolamento del gene RP3
impiegando le tecniche classiche del "clonaggio posizionale"
quali la "cDNA selection" e la "exons trapping" senza alcun
successo soddisfacente. L'isolamento del gene RPGR è stata
invece possibile grazie ad un'approccio che potremo
definire di "sequenziamento posizionale". Questa tecnica
richiede il sequenziamento in automatico della regione
candidata per il gene responsabile della malattia e,
successivamente, l'analisi computazionale della sequenza
mediante l'uso di programmi che consentono la predizione
di unità di trascrizione. Ciò è possibile solo dopo che
l'analisi genetica, con l'uso di marcatori polimorfici, ha
determinato la localizzazione cromosomica del locus
malattia. Naturalmente lo studio può essere ulteriormente
facilitato dalla presenza di grossi riarrangiamenti
cromosomici che possono permettere di concentrare l'analisi
molecolare su specifiche regioni cromosomiche. Infatti, una
delezione all'interno della regione Xp21, presente in un
paziente affetto da distrofia muscolare di Duchenne, da
granulomatosi cronica, da fenotipo McLeod e retinite
pigmentosa, ha portato all'isolamento dei geni responsabili
delle patologie sopra indicate.
L'individuazione del gene RP3 è risultata estremamente
complicata non solo a causa della inaspettata eterogeneità
del locus ma è stata ulteriormente complicata dallo studio di
un paziente RP, chiamato B.B., che presentava una grossa
delezione di 3500 Kb, a cui il gene sembrava strettamente
associato. L'analisi di questo paziente aveva inizialmente
portato a localizzare il gene RP3 all'interno della delezione
stessa, grazie anche all'analisi di linkage di un marcatore
genetico DXS7 che è risultato poi essere associato anche al
locus RP2 (Figura 1). Successivamente e con l'impiego di
altri marcatori polimorfici fiancheggianti la delezione, il
locus RP3 è stato, invece, individuato fuori di essa. Il gene
infatti è risultato centromerico rispetto alla regione
prossimale della delezione, ad una distanza di 400 Kb, dalla
stessa. Una mappatura più fine del locus RP3 è stata
effettuata grazie all'analisi di un paziente affetto dalla sola
RP, chiamato M.O., che presentava una delezione di 75 Kb
della regione. È stato possibile quindi costruire una mappa
fisica in cloni cosmidici della regione ed isolare un primo
gene candidato (SRPX), nel quale però non sono state
rilevate mutazioni in pazienti affetti da RP3.
Quindi sequenziando per intero due cloni cosmidici che
coprono la delezione del paziente M.O. è stato possibile
prima individuare una EST (Expressed Sequence-Tagged)
nella regione e poi predire, attraverso il programma GRAIL,
una possibile unità di trascrizione (il gene RPGR appunto)
che si estende per circa 60 kb ed è costituito da 19 esoni.
Un'analisi più accurata ha evidenziato che questo gene è
espresso in tutti i tessuti, codifica un trascritto di 2784 bp la
cui open reading frame (ORF) è di 2446 bp e che la proteina
prodotta è costituita da 815 aminoacidi. Tale proteina è stata
innanzitutto chiamata RPGR (RP GTPase Regulator) perché
nella regione ammino terminale sono presenti sei elementi
RCC1 (regolatore della condensazione cromosomica)
ripetuti in tandem e caratteristici di un fattore di scambio
della guanina altamente conservato, la cui funzione è quella
di regolare una piccola proteina nucleare GTPase-Ran (Rasrelated nuclear protein).
Tabella 2: Mutazioni in 7 famiglie con RP3
Nome Sostituzione del Mutazione
Famiglia(#DNA) nucleotide (nt)
Dex14/15 delezione esoni delezione RP16/489 14,15 inframe
G52X nt213G -> T Gly -> Stop a 52 RP07/122
H89Q nt353C -> A His -> Gln a 98 F47/29
W194X nt640G -> A Trp -> Stop a 194 F50/31
G215V nt703G -> T Gly -> Val a 215 RP49/S474
C250R nt807T -> C Cys -> Arg a 250 F75/52
DTISY1296 delezione di 16 bp delezione RP0259
nt945-959 in-frame.
Quest'ultima appartiene alla superfamiglia delle proteine
Ras (piccole GTPase) che, a differenza delle altre ha
prevalentemente una localizzazione nucleare e manca, nella
regione C-terminale, di un sito di isoprenilazione
indispensabile per il suo ancoraggio. La proteina Ran è
ubiquitaria ed è implicata in numerose funzioni cellulari
quali il ciclo cellulare, il trasporto intra- ed extra-nucleare, la
sintesi del DNA e la maturazione dell'RNA. RCC1 è un
polipeptide di 421 aminoacidi che promuove la
fosforilazione del GDP a GTP e stabilizza la conformazione
attiva di Ran-GTP. La presenza dei repeats RCC1 in RPGR
suggerisce una possibile interazione con una nuova
isoforma di Ran comportandosi come fattore di regolazione
del ciclo cellulare e del trasporto molecolare nella retina.
Analisi delle mutazioni in pazienti XLRP.
Nonostante il locus RP3 risulti strettamente associato al
gene malattia (70-90% dei casi a seconda delle popolazioni
studiate), solo in 7 pazienti su 74 sono state individuate
mutazioni in questo gene (due delezioni e cinque mutazioni
puntiformi, comprendenti due mutazioni non senso e tre
missenso) (Tabella 2). Probabilmente ciò può dipendere dal
fatto che le comuni mutazioni del gene RPGR non sono tutte
individuabili mediante la tecnica della SSCP usata per
l'analisi, oppure che le mutazioni possono essere presenti
nel promotore o in una regione del gene non ancora
identificata, oppure possono essere presenti in un trascritto
con siti di splicing alternativi. È probabile che RPGR non sia
l'unico responsabile della malattia nella maggior parte dei
casi di RP3, e che quindi restino da identificare altri geni
coinvolti in RP3. Un'analisi sistematica delle sequenze di
RPGR in pazienti geneticamente definiti RP3, e il clonaggio
dell'intero cDNA dell'RPGR dalla retina, può aiutare a
scegliere tra le possibili ipotesi. Comunque tutte le
mutazioni individuate cadono nei repeats RCC1 che,
essendo altamente conservati, costituiscono una regione
critica per la funzione della proteina. Malgrado il basso
numero di mutazioni individuate risulta evidente una
grande variabilità genetica, ed è quindi probabile che la
maggior parte delle mutazioni RP3 abbia origine
indipendente. Le recenti analisi genetiche delle famiglie
XLRP, usando markers polimorfici per la regione RP3,
mostrano che la maggior parte dei pazienti non imparentati
hanno un differente substrato genetico. Questi risultati
possono suggerire che il gene (o i geni) RP3 ha un'alta
percentuale di mutazioni e che può anche essere
responsabile di un inaspettato alto numero di casi sporadici
di RP.
Conclusioni.
RP3 è, quindi, il primo gene della XLRP identificato
mediante la tecnica del sequenziamento posizionale. La sua
identificazione ha un'importanza rilevante poiché consente
la diagnosi delle forme XLRP anche nei casi in cui non è
possibile classificare la malattia sulla base dei segni clinici e
consente l'individuazione delle femmine portatrici le quali
hanno il rischio del 50% di trasmettere la malattia. Inoltre
permetterebbe di risolvere almeno in parte quel 40% di casi
"sporadici"di RP con storia familiare negativa. Nella stessa
regione genomica sono state mappate due altre distrofie
retiniche, la distrofia retinica dei coni (COD1) e l'emeralopia
notturna congenita stazionaria (CSNB); sarà perciò
importante stabilire se mutazioni nel gene RPGR possono
causare anche queste malattie. Infine, la scoperta di una
nuova famiglia di proteine "RCC1-related" fornirà
sicuramente nuovi strumenti per il loro studio biochimico
ed il loro ruolo nella funzione retinica nello stato normale e
patologico.
Ringraziamenti.
Il presente lavoro è stato svolto nel laboratorio del Dr.
Michele D'Urso, dirigente di ricerca dell'Istituto
Internazionale di Genetica e Biofisica del CNR di Napoli,
presso l'Unità di Sequenziamento dell'Area di Ricerca di
Napoli, grazie al contributo essenziale dei Dottori Alfredo
Ciccodicola, Carmela Migliaccio, Vincenzo Cirigliano ed
allo sforzo collaborativo di prestigiosi Centri Europei di
Ricerca. Tale gruppo di studio è già da anni coinvolto nel
progetto Genoma Umano con rilevanti risultati, e dispone di
una notevole esperienza sul sequenziamento automatico del
DNA, tecnica con la quale è stata resa possibile questa
scoperta. Questi risultati sono stati ottenuti grazie anche ai
finanziamenti concessi dall' Associazione Telethon, per lo
studio delle malattie genetiche. Inoltre va sottolineato che
già da qualche anno è stato intrapreso a Napoli uno studio
interdisciplinare sulla retinite pigmentosa, che vede
impegnati il CNR di Napoli (con il gruppo del Dr. Michele
D'Urso), la Clinica Oculistica della II Università degli Studi
di Napoli (Prof. Ernesto Rinaldi, Dott. Francesca Simonelli)
e il Servizio di Genetica Medica dell'Ospedale A. Cardarelli
(Prof. Valerio Ventruto), nonché l'ORAO, Associazione
Campana di non vedenti (Prof. Giuseppe Imbucci).
FORI MACULARI, ITALIANI
INDIVIDUANO LA TECNICA
CHIRURGICA MIGLIORE
Colpiscono circa 3 soggetti su mille e sono più frequenti
nella popolazione tra i 60 e gli 80 anni, in particolare di
sesso femminile . Sono i fori maculari, veri e propri “buchi”
della macula, la parte centrale della retina, ovvero quella
che ci permette di vedere distintamente gli oggetti, di
leggere da vicino, di vedere le cose piccole da lontano, di
percepire i colori. Per fortuna, in genere, il foro non si
presenta improvvisamente, ma progredisce secondo quattro
stadi. Sull’utilità o meno delle diverse tecniche chirurgiche
per guarire i fori maculari ci sono sempre state opinioni
contrastanti. A far chiarezza sull’argomento ci ha pensato
un gruppo di ricercatori della Clinica Oculistica
dell’Università di Trieste, diretta da Giuseppe Ravalico. Lo
studio, coordinato da Daniele Tognetto, ha raccolto
l’esperienza di 28 chirurghi internazionali su più di 1500
casi di foro maculare ed è stato pubblicato su
Ophthalmology, una delle riviste più autorevoli del settore.
Una delle tecniche microchirurgiche utilizzate nella
chirurgia del foro maculare consiste nel togliere la
membrana limitante interna, una sottile pellicola presente
nella parte più interna della retina, permettendo così la
chiusura spontanea del foro ed il recupero della capacità
visiva. Lo studio ha messo a confronto un gruppo di
pazienti cui è stata rimossa la membrana e un gruppo con
cui sono state utilizzate altre tecniche. La tecnica di
rimozione della membrana ha dimostrato di garantire un
maggior successo chirurgico rispetto alla tecnica senza
rimozione. Infatti nel gruppo di pazienti in cui è stata
effettuata la rimozione si è ottenuta la chiusura del foro nel
94,1 per cento dei casi, mentre in quello in cui la membrana
è stata lasciata in sede solo l’89 per cento dei casi ha
raggiunto la chiusura del foro. E’ stato possibile dimostrare
inoltre che la probabilità della chiusura del foro è maggiore
nel caso di fori più piccoli, cioè allo stadio 2 e 3. L’alto
numero di variabili prese in considerazione, tra cui l’età del
paziente, la durata dei sintomi, lo stadio del foro maculare
ed il tipo di tecnica utilizzata, ha permesso inoltre di
stabilire quali siano i fattori più importanti per una corretta
prognosi e per il migliore approccio chirurgico secondo le
peculiari caratteristiche di ciascun paziente.
da "Yahoo Notizie"