DAL SEGNO CLINICO ALLA DIAGNOSI
IN NEONATOLOGIA: CUORE
Maria Alice Donati
Sezione Malattie Metaboliche e Muscolari
Ereditarie
Clinica di Neurologia Pediatrica
AOU “Anna Meyer” Firenze
ATP
Tessuto altamente ossidativo
>90% energia da respirazione mitocondriale
CARBURANTE MAGGIORE
PERIODO FETALE
GLUCOSIO
POSTNATALE
ACIDI GRASSI
31a SETTIMANA: ECO FETALE
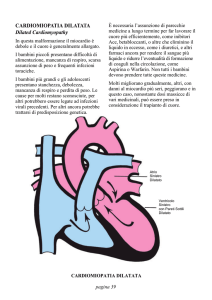
CARDIOMIOPATIA
INSUFFICIENZA CARDIACA
SCARSO ACCRESCIMENTO
→ PARTO CESAREO
P 1060g (5° perc) L 40cm (25° perc) CC 27cm (15° perc)
ECOCARDIO: CARDIOMIOPATIA DILATATIVA
→ EXITUS A 3 GIORNI DI VITA
ISTOLOGIA FEGATO: STEATOSI
CUORE (M.E.): PROLIFERAZIONE MITOCONDRIALE
SCREENING NEONATALE (2 giorni di vita):
C16OH ↑; C18:1-OH ↑; C18:2-OH ↑
→
LCHAD
OMOZIGOTE MUT gene TFP α−sub
Genitori consanguinei
DIFETTI
β–OSSIDAZIONE MITOCONDRIALE
CARDIOMIOPATIA e/o ARITMIA
IPOGLICEMIA IPOCHETOTICA
EPATOPATIA
SINDROME REYE-LIKE
MIOPATIA (CK ↑)
CONVULSIONI
COMA
M. N.
2°giorno di vita: ipotonia, ipotermia, distress respiratorio, ipoglicemia
AST 286 U/L
ALT 72 U/L
CK 14000 U/L
52 h: BRADICARDIA → RIANIMAZIONE e VENTILAZIONE ASSISTITA
-
53 h: EXITUS
Sospetto deficit
β-ossidazione
-
AUTOPSIA: CARDIOMEGALIA
“MIOCARDOSI TORBIDO”-GRASSA EPATOMEGALIA
STUDIO ISTOLOGICO STEATOSI EPATOCELLULARE
INFILTRAZIONE GRASSA DELLE FIBRE MIOCARDICHE
ACIDI GRASSI PLASMATICI A MEDIA-LUNGA CATENA: ACIDO TETRADECANOICO
BIOPSIA CUTANEA PERIMORTEM:
DEFICIT VLCAD SU FIBROBLASTI
ACILCARNITINE MS/MS in
DIFETTO di VLCAD
RECUPERATO SPOT NEONATALE → LCHAD
C16:1-OH
C16-OH
C14:1-OH
C18-OH
C14-OH
C18:1-OH
C14:1
Ricerca mutazione comune c.1528G>C gene HADHA:
Paziente è omozigote per la mutazione
DIFETTO CARNITINA PALMITOIL TRANSFERASI II
(CPTII) NEONATALE
CARDIOMIOPATIA
CARDIOMEGALIA
ALTERAZIONI RITMO
DISMORFISMI
DISPLASIA RENALE CISTICA
DIFETTO MIGRAZIONE NEURONALE
IPOGLICEMIA
CONVULSIONI
EXITUS 1°MESE
EPATOMEGALIA
ACIDOSI METABOLICA
IPERAMMONIEMIA
North K JPediatr 1995;127,414-20
Hug G NeuEngJMed 1991;325,1862-64
MALATTIE MITOCONDRIALI
CARDIOENCEFALOPATIA
MITOCHONDRIAL MEDICINE 2006
S. Di Mauro, M. Hirano, E.A.Schon
CARDIOMIOPATIA (HCM)
LEUCODISTROFIA
DIFETTO COMPLESSO I CATENA RESPIRATORIA
MUTAZIONI GENI NDUF S2; NDUF V2; NDUF S8; NDUF S4
CARDIOMIOPATIA FATALE (HCM)
ENCEFALOPATIA tipo Sindrome di Leigh
DIFETTO COMPLESSO IV CATENA RESPIRATORIA
MUTAZIONI GENE SCO2; COX 15
MALATTIE MITOCONDRIALI
CARDIOMIOPATIA (HCM)
ACIDOSI LATTICA NEONATALE
ENCEFALOPATIA
MIOPATIA
ESORDIO PRIMI GIORNI
EXITUS PRECOCE
3-METILGLUTACONICO ACIDURIA
DIFETTO COMPLESSO V CATENA RESPIRATORIA
Gene ATP 12 Ackerman SH Prog Nucl Acid Res 2005; 80:95-133
Gene TMEM70 Cizkova A Nat Gen 2008;40:1288-90
CARDIOENCEFALOPATIA
MUTAZIONI DNA MITOCONDRIALE
RARE AD ESORDIO NEONATALE-INFANTILE
MUTAZIONE
MUTAZIONE
C4320T Gene tRNA Ile
T8993G Gene ATPase 6
MITOCHONDRIAL MEDICINE 2006
S. Di Mauro, M. Hirano, E.A.Schon
6 ore di vita:
IPOTONIA, IPOTERMIA
DIFFICOLTA’ AD ALIMENTARSI
DOSAGGIO CoQ 10 in MUSCOLO
LATTICOACIDEMIA ↑
FIBROBLASTI
CONVULSIONI
LEUCOCITI
CARDIOMIOPATIA (HCM)
DISFUNZIONE TUBULARE RENALE
EXITUS ≅ 2 ANNI
BIOPSIA MUSCOLARE: ACCUMULO LIPIDICO
DIFETTO I+III e II+III CATENA RESPIRATORIA
DIFETTO CoQ 10
MUTAZIONI NUOVO GENE CoQ 9
SINDROME DI BARTH
I:1
II:1
II:2
III:1 III:2
II:3
I:2
II:4
II:5
III:4 III:5
III:3
IV: 1
IV: 2
Am J Hum Genet.
1 997, 61 : 862- 7
II:6
IV: 3
X-LINKED
II:7
III:6
IV: 4
III:7
IV: 5 IV: 6
CARDIOMIOPATIA INFANTILE con mitocondri abnormi
MIOPATIA
NEUTROPENIA
3-METILGLUTACONICO ACIDURIA
(Kelley, J Pediatr 1991, 119, 738)
mutazione gene TAZ
c.877G>A p.G197R
M.A.Donati et al. J Inherit Metab Dis.
2006;29:684.
SINDROME DI BARTH
ANAMNESI FAMILIARE
SESSO
ETA’ DI ESORDIO
REPERTI CARDIOLOGICI
3-METILGLUTACONICO
ACIDURIA
ANALISI MOLECOLARE
GENE TAZ (G 4.5) Xq28
(Bione, Nat Genet, 1996,12, 385)
(D’Adamo, Am J Hum Genet 1997, 61, 862)
GENE TAZ (G 4.5) PROTEINA TAFAZZINA
ACILTRANSFERASI coinvolta nel metabolismo della cardiolipina
MONOLISOCARDIOLIPINA ↑ CARDIOLIPINA ↓
CARDIOLIPINA alterata quantitativamente
e qualitativamente in fibroblasti e piastrine
Alterata funzione
mitocondriale
(Valianpour, Clin Chem 2002, 48, 1390)
ANALISI MS/MS SPOT SANGUE
MONOLISOCARDIOLIPINA/CARDIOLIPINA
Kulik W. Clin Chem. 2008;54:371
SINDROME DI BARTH
CARDIOMIOPATIA
ESORDIO PRIMO ANNO DI VITA
INSUFFICIENZA CARDIACA:
Spencer CT et al. Pediatrics. 2006; 118:337
D.D.
MIOCARDITE VIRALE
DILATAZIONE
RIDOTTA CONTRATTILITA’
LIEVE-MODERATA IPERTROFIA VS o VS + VD
MORTE IMPROVVISA
MUTAZIONE
GENE TAZ
(FIBRILLAZIONE VENTRICOLARE ?)
X-LINKED ISOLATED NON COMPACTION
OF LEFT VENTRICULAR MYOCARDIUM (INVM)
(Bleyl Am J Med Genet 1997, 61,868)
(Bleyl Am J Med Genet 1999, 85,190)
AUTOPSIA:
FIBROELASTOSI ENDOCARDICA
DILATAZIONE- INSPESSIMENTO MIOCARDIO
M.E.: STRUTTURA FIBRILLARE NORMALE - MITOCONDRI ALTERATI
MALATTIA DI POMPE
ESORDIO INFANTILE
DEFICIT DELL’ENZIMA LISOSOMIALE α-GLUCOSIDASI (GAA)
CUORE
Esordio primi mesi
MUSCOLO
SCHELETRICO
MARCATA CARDIOMEGALIA
CARDIOMIOPATIA
INSUFFICIENZA CARDIACA
DEBOLEZZA MUSCOLARE PROGRESSIVA
IPOTONIA/“FLOPPY BABY”
RITARDO MOTORIO
POLMONI
INFEZIONI RESPIRATORIE
INSUFFICIENZA CARDIO-RESPIRATORIA
GASTROINTESTINALE MACROGLOSSIA
DIFFICOLTÀ AD ALIMENTARSI
SCARSO ACCRESCIMENTO
EPATOMEGALIA
MALATTIE DI POMPE INFANTILE
Manifestazioni cardiache
Esordio primi mesi
• Cardiomegalia
– RX torace
• Cardiomiopatia
– Ecocardiogramma
• Insufficienza
cardiaca
progressiva
M.di POMPE INFANTILE
Cardiomiopatia ad esordio prenatale
ECG: PR corto Ecocardio fetale a 32° sett
QRS di grande ampiezza
ONDA T invertita
Hamdan MA JIMD 2008
marcata deviazione asse SX
DIAGNOSI ENZIMATICA → DEFICIT di α−GLUCOSIDASI
LINFOCITI, FIBROBLASTI, MUSCOLO
DOSAGGIO ENZIMATICO MS/MS
SU SPOT SANGUE
DIAGNOSI di MALATTIA di POMPE
ANALISI ENZIMATICA in MS/MS su SPOT di SANGUE
Anno Nascita 2008
Mantova
DISTRESS RESPIRATORIO
CARDIOMIOPATIA
DIAGNOSI A 6 GIORNI DI VITA
Anno Nascita 2009
Milano
CARDIOMIOPATIA
DIAGNOSI A 2 MESI e 1/2 DI VITA
Anno Nascita 2009 Firenze
CARDIOMIOPATIA
DIAGNOSI A 14 GIORNI DI VITA
Pediatrics 2008;122;e39-e45
F.G.
I:1
GANGLIOSIDOSI GM1 TIPO 1
I:2
I:3
I:4
(VARIANTE CON CARDIOMIOPATIA)
Ridotta crescita fetale, parto cesareo
36asettimana, PN Kg. 2,250
1a settimana di vita: edema generalizzato transitorio
epatomegalia lieve
A 4 mesi: Rx torace: CARDIOMEGALIA
Ecocardio: CARDIOMIOPATIA DILATATIVA
II:1
II:2
P263L
III:1
IN LISTA PER TRAPIANTO
III:2
W92X
/P263L
II:3
W92X+/-
III:3 III:4
T.A.
II:4
W92X+/
II:5
II:6
III:5
W92X+/
A 5 mesi: Facies dismorfica, RITARDO PSICOMOTORIO
ipotonia generalizzata ROT iperattivi
edemi arti inferiori teleangectasie
EPATOSPLENOMEGALIA
Striscio di sangue periferico:
LINFOCITI VACUOLATI
Rx scheletro: DISOSTOSI MULTIPLA
TLC oligosaccaridi urinari: ALTERATI
Exitus a 18 mesi
β-Galattosidasi leucociti
e fibroblasti: <1% vn
Lattosio
Normale
GM1 tipo 1
M.P.
a 15 gg: DIFFICOLTÀ ad ALIMENTARSI, IPOREATTIVITÀ
a 20 gg: IPOTONIA GENERALIZZATA, CUTE MAREZZATA,
TACHIPNEA, CANDIDOSI
ECO cuore: GRAVE CARDIOPATIA IPOCINETICA
METILMALONICO ACIDURIA
ECO cerebrale:
MALONICO ACIDURIA
IPERSONORITÀ dei NUCLEI della BASE
PROPIONICO ACIDURIA
IPERTROFIA CONCENTRICA VSX
ACIDOSI METABOLICA, ANEMIA,
LEUCOPENIA, PIASTRINOPENIA
METILMALONICO ACIDURIA con omocistinuria (Cbl C)
TERAPIA: IDROSSICOBALAMINA, L-CARNITINA,BETAINA
CARDIOMIOPATIA
• ANAMNESI FAMILIARE
• SESSO
• ETA’ DI ESORDIO
• CLINICA
TIPO DI ESORDIO
REPERTI CARDIOLOGICI
ALTRI ORGANI O
APPARATI COINVOLTI
• LABORATORIO
DIAGNOSI di MALATTIA METABOLICA
CARDIOMIOPATIE
ITER DIAGNOSTICO
• ESAMI EMATOCHIMICI
GLICEMIA, EMOCROMO, EMOGAS,
CK, LDH, ALT, AST
ACIDO URICO, AMMONIO
LATTATO (LATTATO/PIRUVATO)
• BIOPSIA MUSCOLARE
MORFOLOGIA (ISTOCHIMICA,
• ALTRI ESAMI
ACILCARNITINE (MS/MS)
ACIDI ORGANICI URINARI (GC/MS)
AMINOACIDI
OLIGOSACCARIDI
……………… STRISCIO
COLTURE DI FIBROBLASTI
DOSAGGI ENZIMATICI
ISTOLOGIA)
BIOCHIMICA
ANALISI MOLECOLARE
EXITUS IMPROVVISO
Protocollo raccolta campioni
PLASMA* (3 ml)
SIERO* (2 ml)
SANGUE INTERO IN EDTA (5 ml) (PER ESTRAZIONE DNA)
SPOT DI SANGUE SU CARTA DA FILTRO
URINE* (10 ml) (se necessario da catetere o da puntura sovrapubica)
LIQUOR* (1-2 ml)
BIOPSIA CUTANEA (10 X 5 mm) in MEM o S.Fis.
BIOPSIA MUSCOLARE
(20-50 mg, IN AZOTO LIQUIDO O A -80°)
BIOPSIA EPATICA e CARDIACA
(10-20 mg, IN AZOTO LIQUIDO O A -80°)
AUTOPSIA (conservare i tessuti in FORMALDEIDE 4% E KARNOFSKI)
SPOT DI BILE SU CARTA DA FILTRO
* = Conservare a -20°
A.L.
D.N. 22.03.2005
3 mesi
Cardiomiopatia
Ipotonia generalizzata
Deficit suzione
↓
Maltasi acida <1%
Linfociti e fibroblasti
MALATTIA DI POMPE INFANTILE
Anno nascita 2005
A 4 mesi:
RISPOSTA A ERT
Programma di accesso allargato
Uso compassionevole DM 8/5/2003
Inizio ERT
1 anno: Deambulazione autonoma, miglioramento quadro cardiaco
Curva di sopravvivenza (Kaplan-Meier)
1.0
Proportion of Patients Alive
0.9
Pazienti sottoposti a ERT (n=17)
0.8
0.7
Pazienti non trattati
0.6
Media di età all’ Exitus:
8.7 mesi
0.5
Sopravvivenza
Sopravvivenza
Sopravvivenza
Sopravvivenza
25.7%
14.4%
9.0%
7.1%
0.4
0.3
a
a
a
a
12 mesi:
18 mesi:
24 mesi:
36 mesi:
0.2
0.1
0.0
0
6
12
18
24
30
Age (months)
36
42
48
54
60
CURE NON RISOLUTIVE in ALCUNE MALATTIE
DIAGNOSI PRECISA
EVITARE ITER DIAGNOSTICI ERRATI-INUTILI
CONSIGLIO GENETICO
“ADEGUATO E PRECOCE”
DIAGNOSI PRENATALE
Pier della Francesca “Madonna del parto”
Pediatrics 2008;122;e39-e45
132538 neonati
0.091% RR
4 diagnosi
Eta’ 9-22 giorni
PREVALENZA 1:33333
Eta’ diagnosi
gruppo controllo
3-6 mesi
DCM: 31 ≠ geni
HCM: 20 ≠ geni
STUDIO SU MUSCOLO CARDIACO
IMPORTANTE SE STUDIO SU MUSCOLO SCHELETRICO E
ALTRE ANALISI INCONCLUDENTI
• ISTOCHIMICO
(Kennaway et al. Pediatr Res 1990,28,529)
• BIOCHIMICO
• ULTRASTRUTTURALE
IMPORTANTE SE DIFETTO TESSUTO CARDIACO SPECIFICO
Es: CARDIOMIOPATIA ISTIOCITOIDE DELL’INFANZIA
STUDIO: MORFOLOGICO: fibre muscolari ingrandite (histiocyte-like) con accumulo di
mitocondri
BIOCHIMICO: difetto di complesso III e citocromo b (normali in muscolo e fegato)
(Papadimitriu et al. Pediatr Res 1985,18,1023)
CARDIOMIOPATIA (HCM) FATALE
ACCUMULO DI GLICOGENO EXTRALISOSOMIALE in
MIOCARDIO e MUSCOLO SCHELETRICO
SINDROME di SENGERS
FORMA FATALE NEONATALE
Sengers RCA J Pediatr 1975;86:873-80
CARDIOMIOPATIA IPERTROFICA
CATARATTA CONGENITA
MIOPATIA
ACIDOSI LATTICA
In alcuni casi:
DIFETTO I CATENA RESPIRATORIA
RM ENCEFALO: IPOPLASIA CEREBELLARE e TRONCO
Morava E Eur J Pediatr 2004;163:467-71
Perry MS Ped Neurol 2008;39:113-15
1 Difetto enzimatico
COMPLESSO
MULTIPROTEICO
COMPLESSO I
45 SUBUNITA’
nDNA+7 geni mtDNA
DIAGNOSI GENETICO MOLECOLARE COMPLESSA
NECESSARIA PER IDENTIFICAZIONE
DIFETTO NUCLEARE O MITOCONDRIALE
IDENTIFICAZIONE PORTATORI
DIAGNOSI PRENATALE
Pediatrics 2008;122;e39-e45
132538 neonati
0.091% RR
4 diagnosi
Eta’ 9-22 giorni
PREVALENZA 1:33333
Eta’ diagnosi
gruppo controllo
3-6 mesi