DISPENSE DEL CORSO DI PATOLOGIA GENERALE
1
DISPENSE DEL CORSO DI PATOLOGIA GENERALE
1. ALTERAZIONI POST-MORTALI
Sono le alterazioni che si verificano dopo la morte dell’animale. Se si sovrappongono a
lesioni patologiche già presenti precedentemente al decesso, possono portare ad errori di
valutazione.
In ordine cronologico le alterazioni post-mortali sono:
- Pallore o lividità cadaverica
- Raffreddamento del cadavere o algor mortis
- Ipostasi o macchie cadaveriche
- Rigidità cadaverica o rigor mortis
- Coagulazione del sangue
- Alterazioni dell’occhio
- Putrefazione
1.1 PALLORE O LIVIDITA’ CADAVERICA
Con l’arresto cardiaco e della circolazione il sangue scompare dai vasi superficiali,
inducendo un caratteristico pallore al corpo. Negli animali la lividità cadaverica si osserva
meglio a livello di mucose apparenti (gengive, occhio e mucose genitali), mentre sulla cute
è più evidente nelle zone dove il pelo è più sottile e più rado (logge ascellare ed inguinale,
regioni prepubica, ombelicale e sternale).
La cute e le mucose appaiono di colorito pallido, fino al bianco-cereo, che diventa ancor
più evidente se l’animale è morto per emorragie esterne o interne.
Nel caso invece di morte per fenomeni infettivi o tossici, si può osservare un colorito
rosso-violaceo dovuto ad alterazioni delle pareti vasali.
In alcune malattie (ittero o patologie epatiche) può comparire una colorazione giallastra o
giallo-verdastra di cute e mucose.
1.2 RAFFREDDAMENTO DEL CADAVERE
La cessazione di tutti i processi metabolici e dell’attività dei centri termoregolatori
comporta il raffreddamento del cadavere. La diminuzione della temperatura è di circa 1°C
all’ora a temperatura ambiente, valore influenzato da diversi fattori, quali la temperatura
esterna, l’umidità, l’aerazione, lo stato di nutrizione dell’animale, ecc.
In genere nei grossi animali si arriva al raffreddamento completo dopo circa 15-24 ore, nei
piccoli animali dopo 6-12 ore.
La temperatura decresce più rapidamente in caso di animali molto magri, anemici, con
epatiti o paralisi. Viceversa il raffreddamento è più lento in animali in buone condizioni di
nutrizione, in caso di morte improvvisa per malattie acute.
Nel caso di malattie infettive accompagnate da stati febbrili (tetano, carbonchio ematico,
meningiti) si può verificare un aumento della temperatura di alcuni gradi nelle prime ore
dopo la morte (nel tetano si possono raggiungere i 45°C).
1.3 IPOSTASI E MACCHIE CADAVERICHE
Dopo la morte dell’animale il sangue si sposta dai vasi superficiali e, per gravità, tende ad
accumularsi nei vasi delle regioni più declivi del corpo. Ciò provoca il pallore cadaverico
(vedi1.1) nelle porzioni più alte del cadavere, e la comparsa delle macchie ipostatiche
2
(macchie di colorito rossastro) nelle parti declivi. Tali fenomeni si verificano tra le 3 e le 6
ore dopo la morte dell’animale.
L’ipostasi ante-mortem è invece un fenomeno che si verifica quando, per compromissione
cardiaca, l’agonia dell’animale è particolarmente prolungata.
Le macchie ipostatiche si dividono in :
- macchie di I grado, prima delle 8-12 ore dalla morte; sono causate dal riempimento
dei vasi sanguigni superficiali, e tendono a “migrare” verso la parte declive se il
cadavere viene spostato o girato o se si applica una forza compressiva.
- macchie di II grado, dovute alla coagulazione del sangue nei vasi declivi; lo
spostamento del cadavere o la loro compressione, non comporta una scomparsa.
Le macchie ipostatiche possono presentare varie forme ed un colorito dal rosa al violaceo,
si riscontrano sia sulla cute sia sugli organi interni, non deformano la parte, non colorano i
tessuti e scompaiono alla compressione. Possono essere differenziate dalle emorragie
verificatesi durante la vita dell’animale, in cui la parte si presenterà deformata, il tessuto
rosso-violaceo ed alla compressione la macchia non si attenuerà.
1.4 RIGIDITA’ CADAVERICA O RIGOR MORTIS
La perdita dell’eccitabilità muscolare dopo la morte determina la rigidità cadaverica.
Nell’animale appena morto le masse muscolari sono molli e cedevoli e sono presenti
contrazioni spontanee che perdurano per circa 3 ore. In seguito, a causa della scomparsa
del glicogeno muscolare e dell’acidificazione del pH (da 7 a 5,4) dovuta all’aumento
dell’acido lattico, actina e miosina si legano andando a formare il complesso actomiosina;
le fibre perdono acqua e tendono ad accorciarsi.
La muscolatura appare quindi dura, inestensibile e contratta.
La rigidità cadaverica segue la “Legge di Nysten”: compare in ordine cronologico in cuore,
diaframma, collo, nuca, lingua, muscoli masticatori, testa, collo, tronco, arti anteriori e
infine arti posteriori. Inizia dopo 2-3 ore dalla morte e può perdurare fino a 48 ore.
Alcune patologie possono allungare la durata del rigor mortis (tetano, avvelenamenti), altre
lo accorciano (folgorazione, setticemie). Esso compare precocemente in caso di
emorragie acute, annegamento ed a temperature ambientali elevate, è più tardivo se la
temperatura ambientale è bassa.
3
1.5 COAGULAZIONE DEL SANGUE
Si verifica nei vasi di grosso calibro e nelle cavità cardiache a causa della trasformazione
del fibrinogeno in fibrina. Di norma si trovano coaguli dopo 4-5 ore dalla morte.
La coagulazione è incompleta in casi di deperimento organico, shock anafilattico,
avvelenamenti, leucemie, carbonchio ematico.
I coaguli possono essere di due tipi:
- rossi o cruorosi (massa di fibrina che imprigiona numerosi globuli rossi; appaiono
rossastri, molli, gelatinosi, lisci)
- bianchi, costituiti prevalentemente da fibrina, biancastri o giallastri.
I coaguli non aderiscono alle pareti vasali e sono facilmente asportabili; possono quindi
essere differenziati dai trombi, masse solide che si formano su lesioni della parete vasale,
costituiti da fibrina, globuli rossi e piastrine, e caratterizzati da anelasticità e friabilità.
1.6 ALTERAZIONI DELL’OCCHIO
Sono rappresentate da:
- rilasciamento dell’iride (per cessazione attività nervi))
- opacamento della cornea (6-7 ore dopo la morte, conseguenza dell’autolisi
enzimatica)
- avvizzimento del globo oculare (per evaporazione dei liquidi dell’occhio; l’occhio
rimpicciolisce e si infossa nella cavità orbitale)
- macchie nerastre sulla sclera.
1.7 PUTREFAZIONE
Si tratta della degradazione della carcassa operata dagli enzimi batterici sui tessuti morti.
E’ preceduta da fenomeni di autolisi asettica, degradazione enzimatica ad opera di
enzimi proteolitici intracellulari (catepsine), liberati dai lisosomi delle cellule morte.
L’autolisi asettica si manifesta con opacamento della cornea, risoluzione della rigidità
cadaverica, intorbidamento dell’urina e del liquido cefalo-rachidiano soprattutto ad alte
temperature e a pH acido (4-5,8).
La putrefazione avviene invece per opera dei batteri presenti in gran numero nel contenuto
intestinale. E’ favorita dalle temperature elevate ed umide e compare dopo 8-24 ore dalla
morte dell’animale. Si verifica più rapidamente in animali in buono stato di nutrizione, in
animali poco dissanguati, in casi di setticemia, tetano, diabete, uremia ed intossicazioni.
Nelle prime ore dopo la morte prevalgono i germi aerobi (Bacillus subtilis, B. mesentericus,
Proteus vulgaris), che trasformano gli zuccheri in acidi. Poi intervengono i colibacilli, che,
attraverso la produzione di ammoniaca; favoriscono la moltiplicazione dei germi anaerobi,
con attività proteolitica, che colliquano i tessuti.
Le tappe della putrefazione sono:
- macchie della putrefazione, sulla cute, di colore verdastro (emoglobina + idrogeno
solforato → solfoemoglobina)
- timpanismo o meteorismo cadaverico, a causa della produzione di gas per
fenomeni biochimici e germi nell’intestino. L’addome si gonfia e si distende; si può
avere prolasso rettale o vaginale.
- enfisema cadaverico, sviluppo di gas nel connettivo sottocutaneo; la cute diviene
crepitante alla palpazione.
4
-
odore della putrefazione, nauseabondo, dolciastro, dovuto all’idrogeno solforato e
ad altri gas.
5
2. NORME PER IL PRELIEVO, LA CONSERVAZIONE E L’INVIO DI CAMPIONI AI
LABORATORI
2.1 INTRODUZIONE
I risultati dell’esame clinico ed anatomo-patologico di molte malattie degli animali sono
così simili che per ottenere una diagnosi esatta sono necessarie indagini di laboratorio.
La validità di tali esami dipende da:
- scelta accurata del campione
- preparazione
- confezionamento
- spedizione
I campioni possono essere prelevati dal tessuto animale o dall’ambiente; gli scopi, oltre
alla diagnosi corretta, possono essere vigilanza sanitaria, ricerche epidemiologiche,
controlli di trattamenti vaccinali.
Le regole generali da seguire sono:
- i campioni ottenuti da animali vivi devono essere raccolti con cura, evitando stress
per l’animale e pericolo per l’operatore
- i campioni ottenuti da animali morti vanno prelevati subito dopo la morte
dell’animale
- adottare misure igieniche idonee al fine di evitare contaminazioni crociate tra i
campioni
- conservare in modo appropriato ed inviare al laboratorio il prima possibile
- ogni campione va accompagnato da una scheda che riporti tutte le informazioni
raccolte, dall’anamnesi ai dati anatomo-patologici.
2.2 ESAMI BATTERIOLOGICI
Il materiale per gli esami batteriologici deve essere ottenuto in modo da evitare le
contaminazioni esterne.
Gli strumenti utilizzati (bisturi, coltelli, forbici o pinzette) devono essere sterili e comunque
sterilizzati dopo ogni procedura con acqua bollente per 15 minuti oppure sostanze
chimiche o ancora vapore; in seguito vanno conservati in contenitori in metallo (pure
sterile).
Se il materiale prelevato è solido (fegato, rene, cervello), si dovrebbe evitare la superficie
esterna; meglio prelevare in profondità dopo aver sterilizzato la superficie con un ferro
arroventato.
6
Nel caso di un sospetto di setticemia si consiglia il prelievo di porzioni di fegato, rene
milza, linfonodi, polmone, cervello ed osso lungo (femore o tibia), posti in buste di plastica
ben chiuse. Se si prospetta la possibilità di fuoriuscita di liquido dalla busta, meglio
utilizzare più buste, una dentro l’altra.
I liquidi (essudati o trasudati) vanno prelevati con siringa sterile e raccolti in provette pure
sterili a chiusura ermetica.
Il contenuto intestinale va conservato in barattoli a bocca larga, chiusi ermeticamente.
Sono inoltre disponibili tamponi per batteriologia contenenti terreni di trasporto.
Ogni contenitore va poi etichettato e siglato.
Per la conservazione dei campioni è richiesta la temperatura di refrigerazione (2-8°C).
2.3 ESAMI VIROLOGICI
La maggior parte di virus che colpiscono gli animali dimostrano preferenza per determinati
tessuti o organi (tropismo virale). Si consiglia pertanto un’accurata selezione dei campioni,
evitando sempre la contaminazione batterica.
Per i prelievi valgono le stesse regole degli esami batteriologici; per la conservazione ed il
trasporto è preferibile il congelamento.
Se si sospetta la rabbia va inviata al laboratorio l’intera testa dell’animale, in contenitori a
tenuta ermetica.
2.4 ESAMI ISTOLOGICI
I tessuti prelevati dovrebbero essere fissati immediatamente, per evitare processi autolitici
e putrefattivi che altererebbero la morfologia dei tessuti stessi.
Le regole generali sono:
- utilizzare coltelli o lame molto affilati (si evitano dentellature nei tessuti)
- eseguire incisioni accurate
- preparare blocchetti di dimensioni appropriate (1-2 cm) con superfici di taglio
parallele
- comprendere nel prelievo parte di tessuto patologico e parte di tessuto sano
- porre i campioni in formalina tamponata (in un volume che sia almeno 10 volte
superiore al volume del prelievo).
- conservare a temperatura ambiente
Formalina tamponata:
formalina
acqua distillata
NaH2PO4
Na2HPO4
100 ml
900 ml
4 gr
6,5 gr
I campioni che tendono a galleggiare (polmone e midollo osseo) vengono sommersi
mediante cotone o garza.
Incidere gli organi cavi (intestino) così da facilitare l’ingresso del fissativo.
Utilizzare sempre barattoli a bocca larga (il pezzo una volta fissato non mantiene la
caratteristica “morbidezza” del fresco e tende, se voluminoso, a non uscire più dal
barattolo).
Etichettare i barattoli ed inviarli sempre accompagnati dalla scheda.
7
2.5 ESAMI EMATOLOGICI E SIEROLOGICI
Per gli esami ematologici (ricerca di batteri, virus, protozoi) il sangue va addizionato ad
anticoagulanti (eparina); nel caso invece di esami sierologici (ricerca dei soli anticorpi) il
sangue va lasciato sierare.
Prelevare i campioni per via endovenosa (in asepsi!!), dalla vena giugulare o caudale nei
grossi animali, dalla brachiale nei piccoli animali e negli uccelli (vena alare).
NB: Lavare, rasare e sterilizzare con cura la zona!
I campioni vanno poi conservati in provette a 4°C se sangue intero, congelati nel caso si
tratti di sieri.
2.6 ESAMI CHIMICI E TOSSICOLOGICI
Se si sospetta la morte per avvelenamento.
Il materiale da inviare comprende gli organi filtro (fegato e rene) oltre a sangue, contenuto
gastrico ed intestinale ed urine.
Quantità da prelevare:
- sangue
10 ml
- urina
50 ml
- fegato
100 gr
- rene
100 gr
- grasso
100 gr
- cervello
100 gr
- contenuto gastrico
500 gr
- alimenti
500 gr
Porre i materiali in contenitori separati, puliti ed ermetici. Congelare o refrigerare per
evitare i fenomeni putrefattivi.
8
3. PATOLOGIA GENERALE
3.1 INTRODUZIONE
La patologia è la scienza che studia la malattia nella sua natura, nelle sue cause, nel suo
evolvere e nelle sue conseguenze.
La malattia è un’alterazione strutturale e funzionale di una cellula, tessuto o organo che si
ripercuote sull’economia generale dell’organismo. Il concetto di malattia è diverso da
quello di “stato morboso”, alterazione senza ripercussioni sull’omeostasi dell’organismo.
Malattia quindi può essere un processo infiammatorio di un arto, che comporterà uno stato
febbrile dell’animale, mentre lo stato morboso può essere la mancanza di uno o più dita,
che non stravolge la sopravvivenza del soggetto.
Per valutare una patologia dobbiamo tenere in considerazione due criteri fondamentali:
- alterazione rispetto allo stato normale (dove esiste armonia tra le diverse funzioni
dell’organismo)
- modificazione dell’omeostasi
Ma qual è il fine ultimo della patologia? Quello di capire le cause ed i processi evolutivi di
una malattia per giungere ad una corretta diagnosi.
3.2 COMPONENTI DEL PROCESSO PATOLOGICO
3.2.1 LESIONI
Si tratta di modificazioni strutturali e funzionali che si verificano nell’organismo. Si possono
dividere in:
- macroscopiche (visibili ad un esame anatomo-patologico)
- microscopiche (visibili all’istologia)
- biochimiche (conosciute dopo specifici esami di laboratorio)
Dall’esame delle lesioni si può arrivare ad enunciare una diagnosi morfologica, basata
sulla lesione predominante in una malattia (ad esempio epatite linfocitaria o steatosi), ma
non sempre si arriva ad una diagnosi eziologica, per la quale spesso sono necessari
ulteriori esami di laboratorio.
3.2.2 PATOGENESI
Si tratta dello sviluppo dell’evento patologico, quella sequela di eventi fisiologici, patologici
e biochimici coinvolti nella risposta dell’organismo al danno. Per riuscire ad identificare la
patogenesi è necessario conoscere a fondo la causa della malattia e la risposta
dell’ospite, ma soprattutto l’interazione che si stabilisce tra questi.
3.2.3 EZIOLOGIA
E’ lo studio delle cause della malattia. Per arrivare alle cause il patologo si deve basare
sugli effetti che queste producono; se una malattia si manifesta con un effetto specifico la
causa sarà una sola (ad esempio il granuloma provocato dal germe della TBC); se invece
gli effetti manifestati sono aspecifici, alla base ci potranno essere più concause (ad
esempio la febbre, che può essere provocata da diversi agenti eziologici o stati patologici).
Inoltre le cause potranno essere:
- efficienti e sufficienti: provocano sempre l’effetto aspettato
- efficienti ed insufficienti: per provocare gli effetti necessitano di quantità maggiori.
9
In ogni caso la certezza che sia una determinata causa a determinare un effetto si realizza
con la ripetitività dell’effetto atteso.
Gli agenti eziologici più frequenti in patologia animale sono di diversi tipi:
a) agenti fisici:
- traumi meccanici (tagli, compressioni)
- elettricità (correnti ad elevate intensità)
- calore (colpi di sole, febbre, ustioni)
- freddo (shock da freddo, geloni soprattutto alle estremità)
- radiazioni (UV, raggi X)
- pressione (provoca squilibri al sistema di regolazione della pressione endogena)
b)
-
agenti chimici
tossine biologiche (presenti in funghi, batteri, ed alcuni vegetali)
pesticidi, erbicidi (Paraquat, organofosforati, responsabili di sindromi neurologiche)
tossine ambientali (inquinamento ambientale da metalli e nitriti)
c)
-
agenti biologici
acellulari (virus e prioni)
procarioti (batteri)
eucarioti (funghi, protozoi ed alghe)
parassiti (cestodi, nematodi, trematodi ed insetti)
d) agenti esterni o interni
- deficit o eccessi nutrizionali (eccessi di proteine causano danni renali, deficit di
vitamina D danni al sistema riproduttivo, eccesso di vitamina A danni alle ossa,
deficit di vitamina C è causa dello scorbuto)
- carenze ambientali (acqua, ossigeno o calore)
- età
- difetti del sistema immunitario (rendono più sensibili alle malattie)
- difetti genetici
3.2.4 CONSEGUENZE CLINICHE
Per arrivare ad una diagnosi corretta il clinico e l’anatomo-patologo devono valutare gli
effetti patologici attraverso la vista (osservare bene le lesioni), il tatto (palpare per valutare
variazioni di consistenza e presenza di tessuti “anomali”), l’udito (l’enfisema cadaverico
provoca un caratteristico crepitio del tessuto sottocutaneo) e l’odorato (caratteristico
l’odore dolciastro e nauseante dei tessuti putrefatti o quello di mandorle amare nei casi di
avvelenamento da cianuri).
Molto importante, se la diagnosi certa non è possibile, è valutare le diverse diagnosi
differenziali, vale a dire una lista delle malattie che presentano una sintomatologia simile,
così da restringere il campo delle diagnosi possibili in vista di ulteriori esami di laboratorio.
Una volta arrivati ad una diagnosi corretta è possibile stabilire una prognosi, cioè cosa ci si
aspetta che accada in futuro; la prognosi può essere fausta se la via intrapresa è quella
della guarigione; infausta, se l’animale non potrà sopravvivere alla malattia; riservata se
non è possibile prevedere gli eventi.
10
3.3 DANNO LETALE E MORTE CELLULARE
Quando un agente eziologico agisce sulle cellule di un animale, queste possono
rispondere in due diversi modi:
- adattandosi al danno subito con un aumento delle attività metaboliche e
conseguente aumento di volume (IPERTROFIA)
- subendo danni alla struttura con arresto di alcune attività metaboliche; la cellula
quindi diminuirà di volume (ATROFIA)
E’ fondamentale ricordare che vi sono alcune strutture essenziali per la sopravvivenza
cellulare, come la membrana cellulare, che protegge dall’ambiente esterno, i sistemi di
respirazione cellulare e i meccanismi di sintesi di enzimi e proteine, che permettono il
corretto nutrimento, ed il DNA. Quando un qualsiasi agente eziologico va ad agire su uno
di questi step fondamentali, si assisterà ad un danno cellulare, reversibile od irreversibile a
seconda della durata e della sede colpita.
11
3.3.1 EVENTI BIOCHIMICI COMUNI
Alla base del danno cellulare vi sono meccanismi biochimici specifici che vanno ad agire
proprio su quelle strutture vitali precedentemente elencate; essi sono:
- diminuzione dell’ossigeno nella cellula con conseguente produzione di radicali liberi
(O2-, H2O2, OHo), responsabili della perossidazione dei lipidi (costituenti essenziali
della membrana cellulare).
- aumento dei livelli di calcio per influsso dall’esterno (la membrana cellulare è
danneggiata!) e liberazione dai depositi intracellulari. Il calcio è responsabile
dell’attivazione di enzimi litici quali le fosfolipasi, le proteasi, le ATPasi e le
endonucleasi.
- diminuzione dei livelli di ATP (ATPasi dipendenti) che rendono meno attivo il
metabolismo ed il ricambio cellulare
- aumento della permeabilità della membrana per effetti diretti di enzimi (fosfolipasi
batteriche) o indiretto (enzimi intracellulari)
Ne conseguono alterazioni strutturali della cellula, che come abbiamo visto in precedenza
potranno essere reversibili o meno.
CAUSE IPOSSICHE
A) FASE REVERSIBILE
Il danno è contenuto ed il processo può evolvere verso la completa guarigione.
La diminuzione dell’ossigeno provoca alterazioni della permeabilità di membrana e della
fosforilazione ossidativa. Ne consegue una diminuzione dell’ATP che ha diverse
conseguenze:
- carenza energetica per la cellula; si attivano quindi le vie alternative (glicolisi) che
comportano consumo del glicogeno e produzione di acido lattico; quest’ultimo
provoca uno stato di acidosi (con addensamento della cromatina nucleare)
- edema cellulare, per riduzione della pompa Na/K (ATPasi sensibile) ed accumulo di
cataboliti nelle cellule. Il danno alla pompa ionica provoca squilibri osmotici con
entrata di Na+, Ca++ ed acqua ed uscita di K+.
- Distacco dei ribosomi dal reticolo endoplasmatico, disgregazione dei ribosomi liberi
(formazione di monosomi) e conseguente diminuzione della sintesi proteica (nel
caso delle lipoproteine si assisterà ad un accumulo dei lipidi non utilizzati)
12
Al microscopio si osserverà dilatazione del reticolo endoplasmatico, rigonfiamento dei
mitocondri, presenza di “myelin figures” (rottura dei fosfolipidi di membrana con
esposizione di fosfatidi che intrappolano acqua tra le strutture lamellari)
B) FASE IRREVERSIBILE
Come detto in precedenza il danno diventa irreversibile in base al tempo ed alla sede
colpita; in genere il tempo varia da 1-2 ore per gli epatociti (cellule ad alta capacità
rigenerativa) ai 4-5 minuti per i neuroni (cellule perenni).
- le lesioni mitocondriali progrediscono con separazione delle creste e
vacuolizzazione;
- si aggravano i danni alla membrana per la continua azione fosfolipasica e per il
Ca+ in ingresso; inoltre le proteasi Ca+ dipendenti danneggiano il citoscheletro
(distacco della vinculina). Radicali liberi vengono prodotti anche dall’endotelio
vascolare daneggiato e da alcune cellule del sistema immunitario (perossidazione
ulteriore dei lipidi di membrana)
- perdita di aminoacidi
Al microscopio: rottura della membrana cellulare, con ingresso di acqua e perdita di
enzimi, metaboliti ed acidi nucleici, rigonfiamento e rottura dei lisosomi, con rilascio di
enzimi nel citoplasma.
13
CAUSE OSSIDATIVE: I RADICALI LIBERI
I radicali liberi sono composti instabili e reattivi che possiedono un solo elettrone
nell’orbitale più esterno. Essi possono danneggiare direttamente le molecole organiche e,
attraverso reazioni a catena, generare altri radicali liberi.
Possono derivare da radiazioni ionizzanti (RX) o eccitanti (UV), da normali reazioni redox
intracellulari (come la superossido dismutasi SOD), oppure dal metabolismo di alcune
sostanze tossiche (es. citocromo P450).
I radicali liberi più rappresentativi sono:
- derivati del carbonio : dal metabolismo del tetracloruro di carbonio (CCL4 → CCl3o +
Cl-)
- derivati dell’azoto: ossido nitrico (NOo) e perossinitrito (ONOO-)
- derivati dell’ossigeno:
anione superossido (O2- o O2o), prodotto nel citoplasma, nei mitocondri e nel
reticolo endoplasmatico
perossido di idrogeno (H2O2), prodotto nei perossisomi o nel citoplasma
radicale ossidrile (OHo), prodotto per ionizzazione dell’acqua da parte dei
raggi X.
I radicali liberi agiscono secondo diversi meccanismi d’azione:
- perossidazione dei lipidi di membrana (si legano ai doppi legami degli acidi grassi
generando perossidi, che sono a loro volta radicali liberi – effetto a catena)
- ossidazione e frammentazione delle proteine per unione con gli atomi di zolfo,
- ossidazione del DNA per legame con la timina, rottura di singole eliche e
conseguente morte cellulare o trasformazione tumorale.
I radicali liberi possono andare incontro a decadimento spontaneo (processo abbastanza
raro e soprattutto molto lento). In genere persistono e la cellula tenta di difendersi
mettendo in gioco agenti anti-ossidanti, esogeni o endogeni:
- vitamina E
- cisteina e glutatione (composti con gruppi sulfidrici che si legano ai radicali liberi al
posto degli enzimi)
- proteine di trasporto (albumina, transferrina)
- enzimi specifici: SOD, catalasi e glutatione perossidasi (dal perossido di idrogeno
formano acqua).
14
CAUSE CHIMICHE (TOSSICHE)
Gli agenti chimici agiscono sulle cellule in modi diversi.
- Azione diretta per legame con le strutture cellulari. E’ tipica di composti idrosolubili
sulle cellule deputate alla loro eliminazione; ad esempio il Cloruro di mercurio si
lega ai gruppi sulfidrici delle proteine di membrana aumentandone la permeabilità; i
cianuri si legano alla citocromo ossidasi mitocondriale bloccando la fosforilazione
ossidativa.
- Azione indiretta: si convertono in metaboliti attivi oppure provocano la formazione di
radicali liberi (es. il tetracloruro di carbonio viene convertito in radicale libero dalla
citocromo P450 ossidasi, che agisce anche sull’acetaminofene, trasformato poi in
metabolita tossico ossidante.
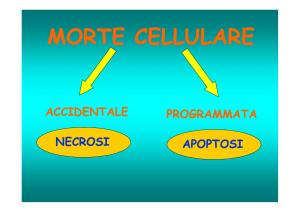
3.3.2 NECROSI
Espressione morfologica di alterazioni indotte, dopo la morte cellulare, da:
- Digestione enzimatica ad opera di enzimi lisosomiali (autolisi) o leucocitari
(eterolisi). In questo caso si arriva alla necrosi colliquativa, caratteristica di infezioni
batteriche (che richiamano polimorfonucleati).
- Denaturazione proteica e degli enzimi a causa dell’acidosi e del danno cellulare. Si
avrà quindi necrosi coagulativa, caratteristica di stati ipossici o tossici.
La necrosi coagulativa può trasformarsi in colliquativa se si ha l’intervento di leucociti.
Altri tipi particolari di necrosi sono la caseosa, un tipo di necrosi coagulativa caratterizzata
da tipici infiltrati granulomatosi (TBC) e la steatonecrosi, saponificazione degli acidi grassi
ad opera di lipasi specifiche.
Se un tessuto necrotico viene colonizzato da batteri saprofiti si ha la gangrena (o necrosi
gangrenosa), che può essere umida o secca a seconda dell’intervento o meno di batteri
piogeni.
Il tessuto necrotico si presenta macroscopicamente come una massa pallida o
emorragica, coagulata o colliquata, diversa dai tessuti adiacenti. Il danno cellulare è
evidente, mentre l’architettura tissutale si mantiene.
Le caratteristiche istopatologiche della necrosi cellulare sono una maggiore eosinofilia del
citoplasma (a causa delle proteine denaturate), che presenta aspetto vitreo (per perdita
del glicogeno) e vacuolizzato (per lisi degli organuli); morte nucleare con cariolisi (nuclei
frammentati), picnosi (nuclei piccoli e scuri) e carioressi (nuclei “disintegrati”).
3.3.3 APOPTOSI
E’ un meccanismo di morte cellulare programmata geneticamente. Ha un duplice
significato:
- fisiopatologici: in tessuti naturalmente soggetti ad involuzione (embriogenesi, atrofie
o involuzioni ormono-dipendenti ad esempio l’utero dopo il parto, ricambio di cellule
proliferanti, immunomodulazione per ristabilire l’equilibrio tra linfociti T e B).
- patologico: nel caso di tumori, morte cellulare indotta da linfociti T o virus, basse
dosi di agenti necrotizzanti.
Meccanismo d’azione: si stabilisce un equilibrio tra geni cosiddetti “inducenti” (ced-3,4; cmyc; p53) e geni “inibenti” (ced-9; bcl2) l’apoptosi. Questo implica:
- induzione delle transaminasi che vanno ad agire sulle proteine coartandole; da qui
si ha una diminuzione del volume cellulare, addensamento degli organuli, e
formazione dei “corpi apoptotici”.
15
-
Attivazione di endonucleasi calcio-dipendenti, che frammentano il DNA in multipli di
180-200 basi. La cromatina si condensa alla periferia del nucleo.
Espressione di antigeni specifici (β-integrine) sulla superficie cellulare che si legano
a recettori di cellule adiacenti. Questo favorisce la fagocitosi da parte delle cellule
vicine.
All’esame istopatologico le cellule apoptotiche appaiono intensamente eosinofiliche in
assenza di reazioni infiammatorie.
3.3.4 LESIONI SUBCELLULARI
A) LISOSOMI
I lisosomi sono coinvolti in due processi fondamentali:
- autofagia (nel caso di fenomeni involutivi o atrofici) in cui il lisosoma fagocita le
sostanze di scarto intracellulari. Con tale processo si forma il vacuolo autofagico.
- Eterofagia, per portare alla cellula i nutrimenti e le sostanze necessarie alla
sopravvivenza. Attraverso i processi di fagocitosi (solidi) e pinocitosi (liquidi), si ha
la formazione di fagosomi (o lisosomi secondari).
16
Quando il lisosoma va incontro a degenerazione dà luogo alla formazione di corpi residui
intracellulari, definiti lipofuscine.
B) RETICOLO ENDOPLASMATICO LISCIO
Può andare incontro ad iperplasia per induzione enzimatica, in caso di terapie con
barbiturici che stimolano l’enzima P450.
C) MITOCONDRI
Si possono avere:
- diminuzioni di numero, in caso di ipotrofia cellulare (la cellula riceve meno
nutrimento)
- aumento di numero, nelle ipertrofie cellulari
- aumento di volume (megamitocondri) in caso di intossicazioni da alcool, carenze
nutrizionali, oncocitomi (tumori del settore cervicale) o in caso di miopatia
mitocondriale (malattia ereditaria dell’uomo)
D) CITOSCHELETRO
Il citoscheletro è formato da filamenti di actina (6-8 µm), miosina (15 µm), filamenti
intermedi (10 µm) e microtubuli (20-25 µm). Essi possono andare incontro a:
17
-
-
patologie del movimento cellulare: sindrome di Chediak-Higashi, discinesia ciliare
(apparato respiratorio, segnalata nei cani di razza springer spaniel) e durante
somministrazione di citocalasina B (che provoca mancata polimerizzazione dei
monomeri, che impediscono la fagocitosi)
disgregazione con accumulo di materiale fibrillare nella cellula (soprattutto i
filamenti intermedi); nelle intossicazioni da alcool si formano i “corpi del Mallory”,
mentre nella malattia di Alzheimer le “neurofibrillary tangles”.
Tubuline α e β
18
3.4 MALATTIE DA ACCUMULO
Sono malattie caratterizzate dall’accumulo intracellulare di:
- normali costituenti cellulari
- sostanze animali esogene o endogene
- pigmenti
Possono essere:
- temporanee o permanenti
- costituite da materiali innocui o dannosi
- reversibili o irreversibili
- intracitoplasmatiche o intranucleari
Il meccanismo patogenetico si basa sul fatto che nella cellula si possano verificare
alterazioni del metabolismo (aumento dell’anabolismo o diminuzione del catabolismo),
oppure arresto di alcune reazioni metaboliche (carenze enzimatiche ereditarie), ancora la
presenza all’interno della cellula di sostanze che essa non è in grado di digerire (pigmenti
o inclusioni virali).
3.4.1 ACCUMULO DI PROTEINE (PROTEINOSI)
La valutazione dell’accumulo di materiale proteico nella cellula è importante perché in
alcuni casi permette di arrivare ad una diagnosi eziologica (nel caso ad esempio di
malattie virali). Le inclusioni proteiche possono essere intracitoplasmatiche o intranucleari,
eosinofiliche o basofiliche, e di solito hanno forma sferica o globulare.
Possono originare da:
- processi di assorbimento, in alcune malattie renali con un riassorbimento eccessivo
di albumina dal lume tubulare;
- eccesso di secrezione, nelle plasmacellule durante processi flogistici con accumulo
di anticorpi nel citoplasma (corpi di Russel)
- processi tossici, in caso di intossicazione da metalli che alterano la funzionalità
epatica
- malattie virali (presenza di DNA o RNA virale nel nucleo o nel citoplasma delle
cellule infette)
- alterazione nell’aggregazione delle proteine (malattia di Alzheimer, encefalopatie
spongiformi).
3.4.2 ACCUMULO DI GLICOGENO
Il glicogeno è la forma di riserva del glucosio e viene immagazzinato soprattutto nel fegato.
Un accumulo di questo materiale (glicogenosi) si verifica quindi in prevalenza nelle cellule
epatiche, ma si ritrova anche nel rene e nel muscolo.
19
La glicogenosi può derivare da:
- iperglicemia, nei casi di diabete, con accumulo negli epiteli renali, fegato, cuore e
nelle cellule β del pancreas;
- malattie metaboliche provocate da farmaci (cortisone che aumenta la glicemia)
- malattie ereditarie con deficit enzimatici
- tumori (epatocellulari e pancreatici)
All’esame istopatologico la cellula appare intensamente eosinofila, con il citoplasma
ripieno di gocciole chiare, mentre il nucleo rimane centrale.
3.4.3 ACCUMULO DI LIPIDI
E’ un processo definito lipidosi, steatosi o degenerazione grassa. I lipidi che si accumulano
nelle cellule possono avere diversa natura (fosfolipidi, trigliceridi e glicolipidi).
Si possono distinguere:
- LIPIDOSI: accumulo di lipidi diversi da colesterolo e trigliceridi; è una condizione
molto rara dovuta a difetti enzimatici ereditari.
-
COLESTERINOSI: accumulo di colesterolo per deficit enzimatici o ingestione di
segmenti di membrana cellulare. Ne fanno parte:
a) Xantomi, forme simil-neoplastiche con presenza di cellule a piccoli vacuoli
(foamy cells) subepiteliali in cute e tendini
b) Macrofagi “foamy”, in sedi di necrosi per ingestione di membrane rotte
(fosfolipidi)
c) Colesterolosi, accumulo di macrofagi contenenti colesterolo nella colecisti
d) Ateromi, molto diffusi nell’uomo e nel suino, sono placche focali nell’intima
vasale di aorta, coronarie e biforcazione di grosse arterie costituite da una
capsula fibrosa (connettivo denso, cellule muscolari lisce, e macrofagi), una
zona ricca in linfociti T alla periferia della capsula, un core necrotico
giallastro e granuloso (materiale lipidico, cristalli di colesterolo, foamy cells e
detriti) ed abbondante neovascolarizzazione in periferia. Gli ateromi possono
provocare l’occlusione di piccole arterie, con conseguenti fenomeni
ischemici; possono indebolire la parete di arterie più grosse e dare origine ad
aneurismi (sfiancamenti) o trombi. Ancora frammenti dell’ateroma possono
staccarsi e finire in circolo, andando a costituire i cosiddetti “ateroemboli”.
20
Meccanismi patogenetici: esistono fattori scatenanti primari (iperlipidemia,
ipercolesterolemia) e fattori secondari (ipertensione, fumo, diabete,
anticoncezionali, difetti enzimatici), che in sinergia provocano un aumento
concentrazione locale di LDL. Quest’ultimo tende a legarsi all’endotelio
vasale, in genere alterato (in caso di danni funzionali o di difetti genetici che
espongono molecole di adesione). Oltre ai lipidi si ha poi l’adesione
endoteliale di monociti e piastrine.
STEATOSI pd: accumulo di trigliceridi più frequentemente nel fegato (a causa del
suo ruolo centrale nel metabolismo lipidico). I lipidi alimentari una volta ingeriti
vengono trasformati in chilomicroni (micelle lipoproteiche) che passano al fegato.
Qui vengono trasformati in acidi grassi non esterificati (NEFA) che serviranno da
una parte per produrre energia, dall’altra per essere trasformati di nuovo in
trigliceridi e poi in lipoproteine come riserva (accumulo in tessuto adiposo) e come
fonte di energia per i tessuti.
Tra le steatosi si possono riscontrare (a seconda del punto metabolico implicato):
a) Steatosi da sovraccarico: in caso di diete iperlipidiche (soprattutto monogastrici),
iperlipidemie ereditarie (uomo) e nel post-partum del bovino (sindrome della
vacca grassa). Si verifica una saturazione dei sistemi di smaltimento con
accumulo intraepatocitario (steatosi macroguttulare – una sola goccia lipidica
che spinge il nucleo in periferia) che porta un’ulteriore alterazione del
metabolismo cellulare.
b) Steatosi tossiche (microguttulari) dovute o ad aumento della produzione di lipidi
a partire da precursori (etanolo → acetaldeide → acetato → lipidi) oppure per
riduzione nello smaltimento lipidico conseguente al blocco della trascrizione
dell’RNA (aflatossine o α-amanitina) o blocco dell’esocitosi delle lipoproteine
sintetizzate (falloidina, CCL4)
c) Steatosi carenziali: da carenza relativa o assoluta di acidi grassi poli-insaturi, di
colina o metionina ed aminoacidi essenziali, che rendono impossibile
l’assemblaggio delle lipoproteine.
-
21
3.4.4 ACCUMULO DI PRODOTTI NON DEGRADABILI
a) LIPOFUSCINA: pigmento bruno che deriva dal metabolismo dei lipidi insaturi.
Aumenta con la vecchiaia (aging pigment) e si ritrova frequentemente a livello del
SNC. Da ricordare nell’uomo le ceroidolipofuscinosi (malattie ereditarie)
b) FOSFOLIPIDI: accumulo intralisosomiale a seguito di alterazioni dei meccanismi di
riparazione della membrana cellulare. Si verifica in caso di disordini genetici,
eccesso di farmaci (tranquillanti) o veleni vegetali.
c) SALI DI CALCIO: provocano calcificazioni dei tessuti e sono spesso accompagnati
da sali di Mg e Fe. Le calcificazioni possono essere di due tipi:
Distrofica: in tessuti morti (necrosi, ateromi) o su valvole cardiache,
indipendentemente dalla calcemia.
22
Metastatica: si verifica in tessuti normali ed è legata a disturbi del
metabolismo del calcio (ipercalcemia da iperparatiroidismo, ipervitaminosi D,
neoplasie osee osteolitiche, insufficienza renale con ritenzione di fosfati)
La formazione di fosfato di calcio in forma di apatite (simile all’idrossiapatite ossea
avviene in due fasi:
Iniziazione: a) extracellulare (++ in forme distrofiche) in cui si ha la formazione di
vescicole con membrana propria derivante da membrane in disfacimento per
azione delle fosfolipasi. b) intracellulare (forme metastatiche), con fissazione di
calcio all’interno dei mitocondri.
Propagazione: aumento della quantità di calcio legata, sia nelle sedi intra che extracellulari con velocità diversa in funzione della quantità di calcio e fosfati e della
presenza di inibitori o stimolatori (osteoponina – proteina legante il calcio).
-
-
Le LITIASI sono accumuli di calcio ed altri minerali all’interno dei lume di organi
cavi. Le cause sono da ricercarsi nella stasi, nelle ripetute infezioni, soprattutto
batteriche, e nell’elevato contenuto in minerali. Vengono comunemente denominati
“calcoli”.
3.4.5 ACCUMULO DI PIGMENTI
Si distinguono diversi tipi di pigmenti che possono essere endogeni o esogeni. Tra i primi
ricordiamo:
- Melanina: da ossidazione della tirosina in diidrossifenilalanina, è un pigmento bruno
presente in peli, capelli, occhio, ed è contenuta in granuli all’interno di melanociti.
- Emosiderina (micelle di ferritina aggregate): derivante dal catabolismo del ferro, che
si verifica in casi di alterato metabolismo del ferro, anemie emolitiche, trasfusioni.
- Bilirubina: conseguenza dell’ittero; si accumula in fegato e rene.
Tra i pigmenti esogeni:
- Antracosi: accumulo di granuli di carbone nel tessuto polmonare, nei macrofagi
locali e nei linfonodi satellite
- Tetracicline: se in eccesso danno colorazione giallastra in osso e denti.
23
3.4.6 AMILOIDOSI
Il termine amiloidosi definisce un gruppo di malattie caratterizzate dalla deposizione di
sostanza amiloide in sede extracellulare.
La sostanza amiloide è una sostanza amorfa, di aspetto eosinofilico e ialino
accumulandosi porta ad atrofia da compressione le cellule adiacenti. Esistono diversi tipi
di amiloide, a seconda della natura chimica (2 tipi principali).
Caratteristiche fisiche (comuni ai diversi tipi):
- struttura formata da fibrille non intrecciate di lunghezza variabile e diametro 7,5-10
nm, con disposizione β (a foglietto ripiegato)
- componente P a struttura pentagonale “a bombolone” che tiene unite le fibrille.
Caratteristiche tintoriali: dipendono dalla struttura fisica
- Positività al rosso congo (con birifrangenza verdastra all’esame a luce polarizzata)
- Metacromasia al crystal violetto
- Positività alla tioflavina S (fluorescenza giallastra)
Caratteristiche chimiche:
- 5% struttura glicoproteica dovuta alla componente P (responsabile anche della PAS
positività); tale componente è sempre presente in tutte le forme di amiloide, ha una
struttura simile alla proteina C reattiva, possiede alta affinità per le strutture fibrillari
ed è indispensabile per la deposizione delle stesse.
- 95% strutture proteiche ripiegate a foglietti β, responsabili della positività al rosso
congo. Sono presenti almeno 15 diversi tipi.
AL (amyloid light chain) costituita da catene leggere anticorpali (soprattutto
λ) o da frammenti NH terminali di catene leggere. E’ associata a disturbi
nella produzione anticorpale di linfociti B e plasmacellule.
AA (amyloid associated protein), una proteina di 76 aminoacidi derivante
dalla SAA (serum amyloid associated protein), una proteina infiammatoria di
fase acuta presente nel siero e legata a lipoproteine.
TRR (transtyrethin), proteina di trasporto di tiroxina e retinolo, tipica
dell’amiloidosi senile e di alcune forme ereditarie.
β2macroglobulina: proteina di fase acuta associata al complesso maggiore di
istocompatibilità I (MHCI) che in alcune forme di amiloidosi si può depositare
in forma di fibrille.
β2 amyloid protein (Aβ) proteina di 4000 dalton tipica dei depositi di amiloide
cerebrali durante la malattia di Alzheimer. Deriva dalla APP (amyloid
precursor protein) presente sulla membrana cellulare del neurone
Procalcitonina
Cheratina
24
Classificazione delle amiloidosi:
Sono presenti diversi tipi di classificazione, su base chimica (AA,AL ecc), su base clinica
(n funzione della malattia che determinano), in base alla sede (localizzata o generalizzata)
ed in base all’evoluzione (primaria, secondaria)
Esempi:
- Discrasie degli immunociti: sono amiloidosi primarie legate a disturbi delle cellule
immunitarie. Possono essere sistemiche e sono di tipo AL. A volte associate a
mielomi multipli (nel 5-15% dei casi), con la produzione di un solo tipo di anticorpo o
di anticorpi “incompleti”; nel rimanente 85-95% dei casi sono associate a discrasie
dei linfociti B che producono anticorpi anomali.
- Amiloidosi reattiva sistemica, tipo AA, secondaria, solitamente localizzata a
parenchimi ed organi linfoidi, associata ad infezioni, flogosi, neoplasie con protratto
danno cellulare, TBC, osteomieliti, artriti.
- Amiloidosi associata all’emodialisi: sistemica, con localizzazione a tendini e sinovie,
tipo Aβ2m, secondaria, dovuta a ritenzione in circolo della proteina di fase acuta
non filtrata dagli apparecchi per la dialisi.
- Amiloidosi eredo-familiare: ad esempio la febbre mediterranea ereditaria, tipo AA,
secondaria con febbri ed infezioni ricorrenti e la polineuropatia amiloidotica
ereditaria, primaria, tipo ATTR, che colpisce diverse strutture nervose.
- Amiloidosi localizzate nodulari, forme tumor-like, nodulari su diversi organi
(polmone, laringe, cute, lingua, occhio) caratterizzata da noduli di amiloide con
presenza di istiociti e linfociti.
25
-
Amiloidosi endocrina, localizzata, con accumulo di tipo variabile (ormoni, IAPP =
islet amyloid peptide), secondaria, in presenza di tumori endocrini (tumori tiroidei,
delle isole pancreatiche, feocromocitomi) e diabete.
Amiloidosi senili: cardiaca, con accumulo di TTR nei ventricoli, oppure cerebrale,
localizzata (tipo Aβ2), secondaria all’Alzheimer.
Patogenesi:
a) Forma AA: a causa di stimoli flogistici e distruzione tissutale protratta, vengono
prodotti alcuni mediatori chimici (IL1 e IL6), che determinano un aumento della
sintesi epatica di SAA. L’accumulo di SAA può essere dovuto a deficit enzimatico
(ereditario??) che ne arresta la normale degradazione, oppure ad un’anomalia
genetica della stessa SAA che resiste alla degradazione enzimatica.
b) Forma AL: a causa di uno stimolo neoplastico o flogistico si ha un’iperproduzione di
anticorpi da parte di linfociti B e plasmacellule. L’amiloide si forma per deposizione
di frammenti di anticorpi (dopo elaborazione macrofagica), per incompleta proteolisi
delle catene leggere o per anomalie genetiche degli stessi che diventano resistenti
alla lisi.
3.5 INVECCHIAMENTO CELLULARE
Nel processo di invecchiamento cerebrale sono implicate sollecitazioni esogene e stimoli
endogeni.
Tra le prime ricordiamo:
- ripetuti insulti da parte dei radicali liberi (con conseguente accumulo di lipofuscine)
- modificazioni post-traslazionali delle proteine (glicosilazioni non enzimatiche)
La cellula risponde con stimoli endogeni:
- accorciamento dei telomeri (stabilizzatori dei cromosomi) con conseguente perdita
di DNA
- attivazione di geni specifici della senescenza (nell’uomo cromosomi 1 e 4), perdita
di geni regolatori ed induzione di geni inibitori della crescita.
Tutto questo comporta il rallentamento di tutti i processi fondamentali per il metabolismo
cellulare (ciclo di Krebs, sintesi di DNA, RNA, enzimi e proteine), la vacuolizzazione dei
mitocondri, la diminuzione dell’attività degli organuli e l’accumulo di pigmenti.
3.6 CRESCITA CELLULARE
La crescita cellulare è un meccanismo fondamentale per la riparazione dei tessuti
danneggiati. Come risposta ad un danno tissutale le cellule sopravvissute iniziano a
proliferare, si differenziano nel tipo cellulare finale e danno origine ad un nuovo tessuto del
tutto simile a quello presente prima della lesione. In questo processo è molto importante
l’interazione tra le cellule e la matrice extracellulare, che guida le cellule nella crescita,
nella migrazione e nella differenziazione.
26
CICLO CELLULARE:
Cellule in rapida divisione (labili)
Epiteli, midollo
G2
MITOSI
S
G1
Cellule permanenti (perenni)
cuore, muscolo e SNC
Cellule quiescenti (stabili)
Parenchimi, endoteli, mesenchima
G0
Meccanismi della crescita cellulare:
Intervento di fattori solubili (fattori di crescita) con attività paracrina (su cellule adiacenti) ed
autocrina (sulla stessa cellula) e fattori insolubili (matrice extracellulare ECM, che guida la
crescita cellulare).
FATTORI DI CRESCITA:
a) EGF family: ne fanno parte l’EGF (epidermal growth factor) e il TGFα (transforming
growth factor α), con caratteristiche simili. In vitro agiscono come mitogeni per gli
epiteli ed i fibroblasti, in vivo mitogeni per gli epatociti. Hanno un’azione mediata da
un recettore specifico (c-erb B1) e sono presenti in saliva, urine ed intestino.
27
b) Patelet growth factor (PGF): formato da due catene (A e B) che induce
proliferazione di fibroblasti, cellule muscolari lisce e monociti. La sua azione è
mediata da recettori specifici (α e β). Si trova in piastrine, macrofagi attivati endoteli,
cellule muscolari lisce e cellule tumorali.
c) Fibroblast growth factor (FGF) basico (β) ed acido (α), che induce angiogenesi
(formazione di nuovi vasi) ed è prodotto in molti tessuti dai macrofagi attivati.
d) Transforming growth factor β family (TGFβ)che inibisce in vitro la crescita epiteliale,
mentre in vivo agisce su cellule muscolari lisce e fibroblasti. A basse dosi è un
mitogeno, a dosi elevate inibitore. E’ prodotto da macrofagi attivati, piastrine e
cellule endoteliali.
MATRICE EXTRACELLULARE
E’ formata da :
a) Proteine strutturali fibrose come il collagene: formato da 3 eliche di catene α con
sequenza ripetitiva. Esistono 15 tipi diversi di collagene a seconda della struttura
fibrillare.
b) Glicoproteine adesive, prodotte da fibroblasti, macrofagi attivati ed endoteli, si
legano da un lato alle proteine strutturali fibrose, dall’altra alle cellule. Ne fanno
parte fibronectina, coinvolta nella motilità cellulare; laminina che influenza crescita,
sopravvivenza e motilità di molte cellule; tenascina e trombospondina.
c) Matrice di glicosaminoglicani (acido ialuronico) e proteoglicani (dermatan solfato ed
eparan solfato) che interagiscono con le classi precedenti regolando la struttura e la
permeabilità del connettivo.
3.6.1 ATTIVAZIONE CELLULARE
Quando uno stimolo agisce su una cellula l’interazione segue degli step predeterminati:
a) Legame ligando-recettore: in cui il ligando è generalmente un fattore di crescita,
mentre il recettore è una molecola localizzata sulla membrana cellulare.
b) Attivazione del recettore (che di solito ha attività tirosina-kinasica)
c) Trasduzione del segnale: si verificano una serie di reazioni a cascata provocate
dall’interazione tra la tirosina-kinasi attivata ed alcune proteine di segnale. Al
termine di tali reazioni si attiva un enzima definito MAPK (mitogen activated protein
kinase)
d) Trascrizione del DNA: il MAPK dà un segnale al nucleo che inizia la trascrizione di
geni regolatori precoci (protooncogeni) e tardivi, che stimolano la sintesi di DNA.
e) Attivazione delle cicline: proteine che si legano alle kinasi citoplasmatiche e
fosforilano proteine coinvolte nella sintesi di DNA.
3.6.2 ADATTAMENTI CELLULARI
Possono essere fisiologici (come risposta a stimoli endogeni “normali” ad esempio
ormonali) e patologici (risposta a stimoli dannosi – tentativo di sopravvivere in condizioni
sfavorevoli).
Il meccanismo fondamentale si basa su una up-down regulation di recettori che attivano
determinati processi cellulari, un aumento delle sintesi proteiche ed una variazione
dell’indirizzo produttivo (metaplasia)
28
a) IPERPLASIA: aumento del numero di cellule dovuto ad aumento delle divisioni
cellulari. Può essere:
- Fisiologica: ormonale (ad esempio l’utero gravido) oppure compensatoria (fegato
dopo epatectomia parziale). Il numero di cellule aumenta più di 100 volte,
probabilmente in risposta a TGFα e HGF (hepatic growth factor) prodotti dalle
cellule mesenchimali. Al ristabilimento del volume dell’organo la crescita è inibita
dal TGFβ.
- Patologica: conseguente ad eccessi di stimoli ormonali (endometrio per squilibrio
tra estrogeni e progesterone → forme pre-tumorali)
b) IPERTROFIA: aumento delle dimensioni delle cellule per aumento della produzione
delle componenti strutturali (tipica di cellule che non sono in grado di replicare, ad
esempio il muscolo). Anche in questo caso può essere:
- Fisiologica: ormonale (cellule muscolari dell’utero gravido) o compensatoria
(muscolo sottoposto a iperlavoro). Spesso associata a modificazioni fenotipiche.
- Patologica: stessi stimoli e meccanismi della precedente con l’impossibilità del
sistema vascolare a sostenere l’ipertrofia. Il danno sarà un’ischemia della parte.
c) ATROFIA: diminuzione del volume cellulare per perdita di componenti cellulari.
Fisiologica o patologica, deriva da un minor apporto di sangue alla zona oppure da
una diminuzione del lavoro cellulare in assenza di stimoli endocrini o di senescenza
cellulare. In questo caso il catabolismo cellulare supera l’anabolismo su azione
delle proteasi citoplasmatiche (sia lisosomiali – formazione di vacuoli autofagici, che
non lisosomiali).
d) METAPLASIA: trasformazione di una cellula in un tipo cellulare più resistente.
Forme più comuni comprendono la trasformazione dell’epitelio laringeo da
colonnare in squamoso (nei fumatori e in casi di ipervitaminosi A), la metaplasia
squamosa dell’epitelio urinario durante le calcolosi; la metaplasia ossea del tessuto
connettivo in alcuni tumori. In ogni caso la cellula metaplastica è caratterizzata dalla
perdita delle funzioni originarie. Le cause sono da ricercarsi in stimoli esterni, fattori
di crescita (TGF) e sostanze che agiscono sul DNA.
29
30
3.7 PROCESSO INFIAMMATORIO
E’ una reazione tissutale protettiva necessaria per ridurre i danni provocati da eventi
esterni e per ricostruire i tessuti.
3.7.1 PROCESSO INFIAMMATORIO ACUTO
E’ la reazione immediata del tessuto al danno ed ha lo scopo di portare costituenti difensivi
del plasma. Si articola in 4 fasi:
a) Aumento del flusso di sangue:
subito dopo l’insulto si verifica uno stato di vasocostrizione transitoria; in seguito,
attraverso l’intervento di istamina e prostaglandine, le arteriole vasodilatano. Si ha così
un aumento del letto capillare, con fuoriuscita di liquidi ed aumento della viscosità del
sangue. La conseguenza è un rallentamento del circolo (stasi) che permette la
emarginazione (avvicinamento all’endotelio) dei leucociti circolanti.
Dopo uno stim olo infiamm atorio alcuni m ediatori (IST, ser..)
e riflessi neurogeni provocano dilatazione arteriolare
b) Permeabilizzazione dei vasi:
La pressione osmotica ed oncotica superano quella idrostatica provocando quindi la
fuoriuscita di liquidi dai vasi. Inoltre si verifica l’apertura dei gap intercellulari
dell’endotelio per azione di istamina, bradichinina e leucotrieni; TNF e IL1 agiscono sul
citoscheletro provocando retrazione endoteliale; batteri ed ustioni provocano un danno
endoteliale diretto; i leucociti rilasciano prodotti tossici che agiscono direttamente
sull’endotelio
31
G li elem enti cellulari si ridistribuiscono con stasi eritrocitaria
.
c) Marginazione dei leucociti:
nel sangue esiste un pool di leucociti circolante ed uno marginale. In seguito allo
stimolo flogogeno, con l’aumento della viscosità del sangue, tutti i leucociti tendono ad
addossarsi alla parete del vaso. Grazie a molecole di adesione particolari le cellule si
attaccano alla parete, la attraversano servendosi dei gap intercellulari aperti ed
arrivano al tessuto danneggiato
I leucociti migrano negli spazi extravascolari e iniziano a muoversi (chemiotassi)
Verso lo stimolo infiammatorio.
32
I leucociti (prevalentemente neutrofili nella flogosi acuta), si accumulano nel focus
infiammatorio
d) Attivazione dei leucociti (fagocitosi):
E’ un processo simile a quello visto per le altre cellule. Gli attivatori possono essere
sostanze esogene (lipopolisaccaridi batterici, peptidi formilati) o endogeni (componenti
del complemento, leucotrieni, chemokine – citochine ad azione chemiotattica). I
recettori a cui si legano gli attivatori sono molecole di membrana che variano a
seconda della specie e dello stimolo.
Quando avviene il legame tra attivatore e recettore si attiva la proteina G che stimola la
fosfolipasi C. Questa fa rilasciare dai depositi intracellulari il calcio che va ad agire su
actina e miosina responsabili dei meccanismi di chemiotassi e fagocitosi; inoltre attiva
la proteinkinasi C che attraverso alcune fosforilazioni innesca i meccanismi di
degranulazione.
- La chemiotassi è un movimento direzionale della cellula e per questo si distingue
dalla chemiocinesi, un movimento casuale. Essa si compie attraverso la
polimerizzazione di actina nel citosol (calcio dipendente), che legandosi alla
miosina è responsabile dell’emissione di pseudopodi.
- La fagocitosi p.d. è rappresentata dalla ricognizione dell’antigene circolante e dalla
sua ingestione. Si basa sul legame tra i recettori cellulari e l’anticorpo legato
all’antigene. Questo fa sì che si attivi all’interno della cellula un sistema di
microtubuli che permettono l’emissione di pseudopodi e la formazione del
fagolisosoma.
- Una volta formato il fagolisosoma si ha l’uccisione del batterio e la degradazione,
meccanismi ossigeno-dipendenti (respiratory burst). Dalla reazione tra NADPH e
ossigeno si formano NADH e 2O2o, ed in seguito H2O2. Questa si lega al Cl- e va ad
agire sulla membrana batterica (alogenazione e lipoperossidazione).
33
Opsonizzazione delle particelle infiammatorie con stimolo degli specifici
recettori sui leucociti.
Le particelle attaccate sono inglobate nel fagosoma(A); uccise dopo fusione del
fago-lisosoma (B); digerite(C); ed espulse (D).
34
A. Attacco ad un microorganismo
B. Gli intermedi dell’O2 sono incorporati
nei fagosomi.
sulla membrana cellulare
Formazione di prodotti intermedi dell’O2
.
C. Formazione di un fagolisosoma con
Distruzione del microrganismo da parte dei
radicali dell’O2
−
−
−
−
MEDIATORI CHIMICI DELL’INFIAMMAZIONE
Sono sostanze che intervengono durante il processo infiammatorio; possono essere
plasmatici (ad esempio precursori del complemento) o cellulari (istamina) o ancora
neoformati (prostanoidi). Hanno un’azione mediata da recettori diversa in funzione del
bersaglio e un’emivita breve.
A. Amine vasoattive:
- Istamina: presente nei granuli di mastociti, basofili e piastrine. Reagisce a stimoli
fisici, immunologici (ipersensibilità di I tipo), neuropeptidi e citochine. E’
responsabile della dilatazione delle arteriole e della permeabilizzazione delle venule
35
B.
-
-
C.
-
D.
E.
F.
G.
-
H.
I.
-
Serotonina: in mastociti (topo), piastrine, cellule enterocromaffini. Reagisce
all’aggregazione piastrinica ed al contatto con la matrice. Ha azione simile
all’istamina.
Proteasi plasmatiche:
Complemento: composto da 20 componenti plasmatiche che si attivano in
successione. Reagisce a stimoli anticorpali attivando la via classica ed a fattori
tissutali attivando una via alternativa. E’ responsabile di vasodilatazione se risponde
a fenomeni vascolari; chemiotassi ed aderenza se reagisce all’attivazione dei
leucociti.
Chinine: sono attivate da alcuni fattori della coagulazione. Hanno azione
vasopermeabilizzante, di contrazione della muscolatura liscia e dolorifica.
Fattori della coagulazione: dall’attivazione del fattore XII si arriva alla formazione di
trombina; questa è responsabile dell’aderenza dei leucociti e della proliferazione dei
fibroblasti che a loro volta trasformano il fibrinogeno in fibrina (chemiotassi e
vasodilatazione). Inoltre la fibrinolisi genera plasmino, che attiva il complemento.
Derivati dell’acido arachidonico (prostanoidi o eicosanoidi):
Acido arachidonico: acido poliinsaturo derivato dall’acido linoleico, presente nei
fosfolipidi di membrana. Reagisce a stimoli che attivano le fosfolipasi. Dalle
cicloossigenasi attraverso enzimi specifici (presenti in piastrine, endoteli e
mastociti) si arriva alla produzione di prostaglandine (vasodilatazione ed edema) e
prostacicline (vasodilatazione ed inibizione dell’aggregazione piastrinica). Dalle
lipoossigenasi invece derivano i leucotrieni (chemiotassi, vasocostrizione,
broncospasmo).
Patelet activating factor (PAF):
prodotto da mastociti, neutrofili, endotelio e piastrine per attivazione delle
fosfolipasi; è responsabile di vasocostrizione/vasodilatazione (in funzione della
dose), broncocostrizione, aggregazione piastrinica ed attivazione dei leucociti.
Citochine:
IL1, TNFα (presente in macrofagi) e TNFβ (linfociti B); reagiscono alla presenza di
endotossine ed immunocomplessi. Agiscono su endoteli (attivazione di molecole di
adesione, produzione citochine) su neutrofili e fibroblasti.
Ossido nitrico (NO):
gas libero solubile sintetizzato dall’endotelio a partire da L-arginina O2 e NADPH
per mezzo della NO-sintetasi . Ha un’azione transitoria e rapida che porta a
vasodilatazione, aggregazione piastrinica e citotossicità.
Enzimi lisosomiali:
lattoferrina, lisozima, fosfatasi alcalina e collagenasi; presenti nei granuli specifici
(secondari) dei neutrofili
mieloperossidasi, defensine, lisozima idrolasi acide e proteasi neutre; nei granuli
primari dei neutrofili.
Catepsina e proteine basiche; nei granuli del terzo tipo (ruminanti).
Radicali liberi dell’ossigeno:
provocano danni all’endotelio, attivazione di metalloproteasi ed inattivazione di
antiproteasi.
Altri mediatori:
neuropeptidi (sostanza P) che provoca permeabilizzazione diretta o mediata
dall’istamina, fattori di crescita (TGFβ) che stimola la chemiotassi.
36
Vasoconstriction
Vasodilation
Chemotaxis/Leuk activation
↑ Vasc permeability
TXA2
LTC4, LTD4, LTE4
Endothelin
PAF
PGI2, NO
Histamine,Serotonin
Bradykinin
PGD2, PGE2, PGF2α
C3a,C5a – release histamine
LXA4
PAF (low doses)
C5a, LTB4, HETE
IL-8, IL-1, TNFα
Bact products: LPS, formyl peptides
PAF
fMLP
Monos: C5a, MCP1, TGFβ, PDGF
Histamine, Serotonin
Bradykinin*
C5a, C3a*
PAF*
Substance P*
LTC4, LTD4, LTE4 (venules)
Factor Xa
Reactive O2 species
In inflam sites, ‘end target-derived’
products formyl peptides and C5a are dominant
over ‘regulatory cell-derived’ products LTB4 or
IL-8
N∅ migrate by haptotaxis
(chemoattractants are immobilized to ECM,
insoluble, up the gradient)
dolore: PGD2, PGE2, PGF2α, Bradykinin
febbre: IL-1, IL-6, TNF, PGD2, PGE2, PGF2α
danno tissutale: Lysosomal enzymes from N∅, m∅s, Oxygen metabolites, NO
37
* via ↑ release of histamine from
mast cells
EVOLUZIONE DEL PROCESSO INFIAMMATORIO ACUTO:
L’essudato presente in fase acuta può subire modificazioni in base al tipo di sostanze
presenti:
- liquidi e proteine a basso PM: infiammazione sierosa
- proteine ad elevato PM (fibrina): infiammazione fibrinosa.
Quando si ha organizzazione della fibrina: fibrosi
- se oltre ai precedenti sono presenti cellule a motilità attiva (PMNn): infiammazione
purulenta
- Se sono presenti cellule a motilità passiva (globuli rossi): infiammazione emorragica
- Se vi sono lesioni tissutali: infiammazione necrotica
Gli effetti sistemici dell’infiammazione acuta sono: sonno, anoressia, ipotensione,
catabolismo proteico e febbre, causati da:
- sintesi epatica di proteine di fase acuta: proteina C reattiva, SAA, fattori del
complemento e della coagulazione.
- Sintesi di citochine (leucociti): IL1 ed IL6 che agiscono su recettori del centro
ipotalamico di termoregolazione.
A livello ematico si avrà leucocitosi con neutrofilia dovuta a :
- rilascio del pool marginale (indotto dai corticosteroidi) con aumento dei leucociti
maturi.
- Rilascio del pool midollare (IL1 e TNF) con aumento dei leucociti immaturi.
- Alcune forme particolari rilasciano linfociti (mononucleosi e morbillo), o eosinofili
(asma, parassitosi) o ancora inducono leucopenia (virus).
A) Risoluzione completa:
In caso di lesioni limitate, in tessuti rigeneranti (epiteli), in presenza di agenti eziologici a
bassa virulenza ed emivita breve.
In questo caso si attivano sistemi di difesa cellulari e tissutali:
- antiossidanti (mieloperossidasi)
- antiproteasi tissutali
- metalloproteinasi
- Chaperonine (garantiscono il mantenimento conformazionale delle proteine) ed
ubiquitina (degradano completamente molecole già denaturate in parte.
Si ha l’arresto degli stimoli chemiotattici e della diapedesi leucocitaria, il riassorbimento
l’edema (attraverso i vasi linfatici) e la rimozione dei detriti cellulari e dei PMN per
intervento dei macrofagi.
La situazione finale un tessuto uguale a quello presente prima del danno (restitutio ad
integrum)
B) Ascessualizzazione
Per azione di germi piogeni (streptococchi e stafilococchi); questi producono esotossine
(emolisine) ed endotossine (LPS) che agiscono sui tessuti. Prevalgono i fenomeni di
diapedesi leucocitaria e di necrosi tissutale con grave azione litica dovuta sia ai prodotti
batterici che agli enzimi leucocitari.
La conseguenza è la formazione di un ascesso, zona di necrosi circondata da una capsula
connettivale e da una “membrana piogena” (formata dai PMN).
38
C) Fibrosi
In caso di estese lesioni tissutali ed in tessuti non rigeneranti. Si ha un’estesa produzione
di tessuto connettivo a causa dello stimolo di fattori di crescita e citochine (fibrogenesi ed
angiogenesi)
D) Infiammazione cronica
A causa di stimoli infiammatori persistenti o problemi nella riparazione dei tessuti. Dovuta
ad una prevalenza dei fenomeni tissutali e cellulari della flogosi.
3.7.2 PROCESSO INFIAMMATORIO CRONICO
Infiammazione di lunga durata in cui coesistono fenomeni infiammatori, distruzione di
tessuto e tentativi di riparazione.
Può essere conseguenza di forme acute oppure forma primaria di fenomeni a “lenta
evoluzione”(infiammazioni cosiddette specifiche) ad esempio:
- infezioni da germi a “bassa patogenicità” (TBC, lebbra)
- prolungata espositione a tossici endogeni o esogeni (silicosi, carbonio)
- reazioni autoimmuni
Patogenesi:
a) Infiltrazione cellulare (mononucleati): nel tessuto vengono richiamati monociti
ematici e macrofagi tissutali differenziati (cellule di Kupfer o glia). I monociti una volta
nei tessuti si trasformano in macrofagi e si attivano grazie all’intervento di linfokine,
endotossine e per contatto con la matrice. Vengono prodotti metaboliti tossici (radicali
liberi, NO, proteasi, collagenasi) ed amplificatori della flogosi (derivati dell’acido
arachidonico, complemento); i linfociti T, B, e killer agiscono in cooperazione con i
macrofagi, producendo interferone (che a sua volta attiva i macrofagi).
In alcune situazioni (malattie parassitarie) vengono richiamati eosinofili, che producono
MBP (major basic protein) ad azione litica sulle cuticole parassitarie ma anche sugli
endoteli.
Nell’infiammazione cronica le cellule meno rappresentate sono i PMN e le
plasmacellule.
b) Distruzione tissutale cellulo-mediata per produzione dei metaboliti tossici sopra
citati.
c) Rigenerazione connettivale (fibrosi): grazie a vari fattori di crescita si verificano
fenomeni di angiogenesi, migrazione e proliferazione dei fibroblasti (con sintesi di
collagene e deposizione della matrice extracellulare) e rimodellamento connettivale
(per le metallo-proteasi)
FLOGOSI GRANULOMATOSE:
Particolari forme di flogosi cronica caratterizzate dalla presenza di macrofagi attivati con
aspetto simil-epiteliale (cellule epitelioidi) e da cellule giganti, circondate da linfociti e da
un’eventuale capsula fibrosa.
Sono causate in genere da germi persistenti (micobatteri), parassiti e funghi che
provocano la formazione di “granulomi immunologici” in cui prevalgono linfociti e materiale
inorganico (granuloma da corpo estraneo in cui le cellule immunologiche sono molto rare e
prevalgono i macrofagi).
39
3.7.3 RIPARAZIONE DELLE FERITE
Riparazione per prima intenzione:
si verifica quando si ha morte di poche cellule e modico danno connettivale (es. sutura
chirurgica). Nei primi momenti si ha afflusso di sangue nella ferita e formazione di fibrina;
durante il 1° giorno arrivano i neutrofili, le cellule basali cominciano a proliferare formando
“getti” epiteliali e si forma la membrana basale. Dopo 3 giorni compare il tessuto di
granulazione ed i macrofagi; cominciano a riconoscersi alcune fibre collagene. Il 5° giorno
il tessuto di granulazione, la vascolarizzazione e l’epitelio di rivestimento sono completi.
Entro 1 mese vengono rimossi l’edema ed i leucociti e si ha la formazione di una cicatrice
costituita da connettivo ricoperto da epitelio.
Riparazione per seconda intenzione:
Rispetto alla precedente si ha:
- intensa necrosi e reazione infiammatoria più marcata;
- maggiore quantità di tessuto di granulazione;
- la fibrosi terminale determina un fenomeno definito “retrazione cicatriziale” a causa
della presenza di miofibroblasti con attività contrattile simile alle cellule muscolari
lisce.
Meccanismo della cicatrizzazione:
Entrano in gioco 14 tipi diversi di collagene, stimolati da PDGF, FGF, TGFβ, IL1 ed IL4. La
sintesi del collagene avviene in diverse fasi:
- fase intracellulare: dai ribosomi vengono prodotte le cateneα in seguito idrossilate
dalla vitamina C a generare il procollagene.
- fase intermedia: con taglio del polipeptide con formazione di fibrille di collagene
- fase extracellulare: con ossidazione delle lisine e legame tra le cateneα per
stabilizzare la struttura.
La degradazione del collagene ed il suo rimodellamento avviene ad opera di diversi enzimi
quali la serina-proteasi e le metallo-proteinasi (collagenasi interstiziali, gelatinasi), prodotte
da fibroblasti, macrofagi, neutrofili e cellule sinoviali su azione di fattori di crescita, IL1 e
TNFα. Questi enzimi tagliano le catene fibrillari in frammenti uguali digeribili anche dalle
proteasi tissutali.
Patologie della cicatrizzazione:
- cheloide: cicatrice estesa e deturpante
- desmoide: o fibrogenesi aggressiva con eccessiva proliferazione di fibroblasti.
40
RIPARAZIONE PER PIRMA INTENZIONE
A. La ferita si riempie di sangue e det
detriti coagulati
B. L’epitelio prolifera
Ponti di tessuto connettivo
si formano tra I lembi epiteliali
C. I lembi di epitelio si avvicinano
Il tex sottoepiteliale è normale
D.
Il tessuto torna normale
RIPARAZIONE PER SECONDA INTENZIONE
A.
Ampia ferita che distrugge il tex
detriti e sangue riempiono
la cavità
B.
L’epitelio prolifera
sviluppo tex di granulazione
che causa contrazione
della ferita
C.
I fibroblasti producono collagene
per riparare il tessuto
danneggiato
.
D.
Guarigione completa
Rimane tessuto connettivo
(cicatrice)
41
3.8
DISTURBI DI CIRCOLO
3.8.1 EDEMA
Accumulo di fluidi (trasudato = poche proteine, basso peso specifico) negli spazi
intercellulari o nelle cavità preformate.
Può essere:
- localizzato se intracavitario: idrotorace, idropericardio, ascite (cavo addominale),
idrarto.
- generalizzato: anasarca.
Le conseguenze sono diverse a seconda dell’organo colpito. Risultano più gravi gli edemi
intracranici o intracavitari perchè compressivi.
Si divide in edema infiammatorio (se provocato da stimoli flogistici) ed edema non
infiammatorio o emodinamico (se derivante da squilibri nel sistema vascolare).
Patogenesi: alterazione degli equilibri pressori a livello di arteriole e venule:
linfatici
normale
PI > PO
PI < PO
PI = PO
A) Aumento della pressione idrostatica (edema da stasi):
- Locale: quando vi è un blocco del deflusso venoso (trombi, neoplasie o linfonodi
aumentati di volume)
- Generalizzato (cardiogeno): legato ad aumento della pressione venosa e riduzione
della gittata cardiaca (che attiva nel rene il sistema renina-angiotensina
responsabile della ritenzione di Na e H2O e quindi di un aumento di volume del
plasma → trasudazione)
B) Riduzione della pressione oncotica:
Per perdita di albumine (danni renali, enteropatie proteino-disperdenti, ascite), o
diminuzione della produzione delle stesse (epatopatie, malnutrizione e carenza di
aminoacidi essenziali)
C) Ostruzione al deflusso linfatico:
Flogosi, neoplasie, linfoadenopatie, parassitosi nei vasi)
D) Aumento del volume plasmatico:
ritenzione di Na e H2O per eccessivo apporto alimentare (ruminanti e selvatici) o per
iperattività del sistema renina-angiotensina.
42
Cause:
↑ pressione
idrostatica
CHF
Ascite
Trombi
Compressioni
dall’esterno
Dilatazione
arteriolare: calore
o neurogena
↓ pressione oncotica
ostruzione linfatica
Ritenzione di Na
Infiammazione
Enteropatie proteinodisperdenti
Cirrosi epatica
Malnutritione
Nefropatie proteinodisperdenti
Infiammazione
Neoplasia
Post-chirurgia
Post-irradiazione
Ipoperfusione
renale
Aumento del
riassorbimento di
Na
Insufficienza
renale
Acuta
Cronica
Angiogenesi
3.8.2 IPEREMIA/CONGESTIONE
Aumento del volume di sangue in un organo o in una sua parte.
- Iperemia (iperemia attiva): se aumenta l’apporto arterioso. Si parla di “rubor”
dell’organo dovuto all’azione neurogena di sostanze vasoattive (cfr infiammazione).
- Congestione (iperemia passiva): se è ridotto il deflusso venoso. Si parla di “cianosi”
dell’organo.
Può evolvere in edema.
Nel fegato in seguito a congestione si ha un quadro particolare, definito fegato a “noce
moscata”, con dilatazione della vena centrolobulare, atrofia degli epatociti adiacenti e
steatosi periferica. Evolve in necrosi.
3.8.3 EMORRAGIA
Fuoriuscita di sangue dai vasi per rottura della parete. Le cause sono da ricercarsi in
traumi, arteriosclerosi, erosioni della parete.
Si possono distinguere emorragie interne (ematomi se nel sottocute, emopericardio,
emoperitoneo, emartro se in cavità articolare) o esterne.
Conseguenze:
- Emorragie iperacute (perdita di più del 20% del volume): shock ipovolemico e morte
- Emorragie acute e croniche: anemia di diversa gravità.
3.8.4 TROMBOSI
Formazione di coaguli intravascolari in presenza di lesioni endoteliali.
Fasi della coagulazione:
- Vasocostrizione momentanea dovuta a fattori neurogenici (endotelina)
- Emostasi primaria: esposizione di collagene trombogenico subendoteliale, adesione
ed attivazione piastrinica e formazione del tappo piastrinico.
- Emostasi secondaria: fattori tissutali e piastrinici attivano la cascata della
coagulazione che porta alla formazione di trombina che trasforma il fibrinogeno in
fibrina.
- Formazione del tappo permanente per interazione della fibrina con il tappo
piastrinico primario.
43
Coagulazione del sangue:
Fattori antitrombotici
Antiplatelet
Intact Endothelial cells
PGI2
NO
Adenosina difosfato
Fattori Protrombotici
Cellule endoteliali:
ECM vWF ed endothelial derived vWF
Tissue Factor
(indotti da LPS, IL-1, TNF)
PAIs: depress fibrinolytic system
Platelets
Anticoagulanti
Antithrombin III
Thrombomodulin
Heparin-like molecules
VWF & matrice EC
Fibrinolitici
t-PA
Elementi coinvolti:
a) Endotelio: possiede proprietà antitrombotiche
- antipiastriniche con liberazione di NO e PGI2 (vasodilatatori ed antiaggreganti)
- anticoagulanti attraverso molecole heparin-like di membrana che attivano
l’antitrombina III tissutale, e la trombomodulina che lega la trombina e le proteine C
ed S endoteliali.
- Procoagulanti: l’endotelio attivato produce il fattore di von Willebrand (vWF) ed
alcuni recettori per i fattori della coagulazione.
44
b) Piastrine: sono provviste di recettori per le integrine e per le P-selectine; possiedono
granuliα che contengono fibrinogeno, fibronectina ed alcuni fattori della coagulazione e
granuli elettrondensi contenenti ATP, istamina, serotonina ed epinefrina.
Le piastrine vengono attivate attraverso il legame di proteine endoteliali e la
glicoproteine piastrinica. Quindi iniziano a secernere Ca++ (cofattore della
coagulazione) e ADP (aggregante). In seguito attivano la fosfolipasi C, espongono
sulla superficie un “complesso fosfolipidico” sul quale si svilupperà la cascata
coagulativa e cominciano a secernere trombossano A. Quest’ultimo insieme all’ADP è
responsabile dell’aggregazione delle piastrine e della formazione del tappo primario.
Una volta formato il tappo primario si attiva l’actomiosina piastrinica che provoca la
retrazione del coagulo (metamorfosi viscosa) e la formazione del tappo secondario.
c) Cascata della coagulazione: si instaura su di un substrato lipidico (complesso
fosfolipidico (vedi sopra) . Si basa sull’attivazione di proenzimi in enzimi in presenza di
cofattori (es. fattore Va trasforma il fibrinogeno in fibrina). E’ stimolata da fattori tissutali
come lipoproteine cellulari liberate su stimolo di IL1 e TNF dai tessuti danneggiati,
endoteli e macrofagi.
d) Fattori anticoagulanti:
- antitrombina III: tissutale, attivata dal legame con molecole heparin-like endoteliali
- Proteine C ed S: inattivano i cofattori Va e VIIa
- Plasminogeno e plasmino: scindono la fibrina
- Attivatori del plasminogeno (PA): di cui si conoscono l’uPA (urokinasi) che agisce
sul plasminogeno circolante ed il tPA che agisce sul plasminogeno tissutale.
PATOGENESI DEL TROMBO
Il trombo si forma per la coesistenza di diversi fattori:
- Danni endoteliali: frequenti in cuore (infarti, endocarditi valvolari) e arterie (ateromi,
ipertensione, deposizione di immunocomplessi).
- Alterazioni del flusso ematico: diverse a seconda del distretto colpito. Si possono
avere turbolenze arteriose (in corrispondenza di ateromi ed aneurismi) o stasi
venose (vene varicose, iperviscosità del sangue) che possono provocare: lesioni
endoteliali dirette, interruzione del flusso laminare assiale (con contatto tra piastrine
ed endotelio), ritardo dell’arrivo degli inibitori della coagulazione.
- Ipercoagulabilità: che riconosce cause genetiche o acquisite (danno renale, traumi,
ustioni, neoplasie, età, obesità e fenomeni immunomediati).
45
EVOLUZIONE DEL TROMBO
-
Ostruzione: con effetti diversi a seconda del distretto colpito (nell’uomo trombosi
superficiali agli arti provocano stasi ed edema, mentre quelli profondi embolizzano
al polmone).
Propagazione: lenta e progressiva ostruzione per apposizione di nuovi trombi
Embolismo
Dissoluzione: per attivazione della fibrinolisi entro i primi 2 giorni (terapie
farmacologiche)
Organizzazione e ricanalizzazione: all’interno del trombo si ha la formazione di
tessuto di granulazione di cellule muscolari lisce e mesenchimali, nuova
angiogenesi con formazione di una rete di anastomosi collaterali
Rammollimento per azione di enzimi autolitici (rischio di contaminazione batterica)
CID (Coagulazione Intravasale disseminata):Formazione di molti microtrombi che
consegue all’attivazione dei sistemi coagulativi. Le conseguenze possono essere
una coagulopatia da consumo oppure una fibrinolisi attiva (produzione di sostanze
che degradano il fibrinogeno con effetti anticoagulanti).
3.8.5 EMBOLIA
L’embolo è una massa solida, liquida o gassosa circolante all’interno dei vasi sanguigni. In
patologia generale si possono osservare diverse situazioni:
- (Trombo) embolismo polmonare: origina da emboli sviluppatisi da trombi nei vasi
profondi degli arti posteriori. Ha effetti diversi a seconda delle dimensioni: se
occlude vasi grandi come l’arteria polmonare causa infarti polmonari e morte, se
occlude arterie terminali del microcircolo provoca emorragie polmonari o
ipertensione. Da ricordare il fenomeno degli emboli paradossi, piccoli emboli che
dal polmone, dopo superamento del cuore, arrivano al grande circolo.
- (Trombo) embolismo sistemico: origina nell’uomo da trombi arteriosi o cardiaci, nel
gatto da trombi alle arterie iliache. Gli emboli possono arrivare a diverse sedi: SNC,
visceri, arti.
- Embolismo (infusione) amniotica: nella donna, durante il parto, per passaggio in
circolo di liquido amniotico o materiale fetale.
- Emboli gassosi: per “barotraumi” durante il parto o pneumotorace, oppure a
decompressione nei sub (morbo di Caisson), dove, a causa dell’alta pressione, si
solubilizzano i gas presenti nel sangue (ossigeno, azoto ed elio).
- Emboli grassi: liberazione di midollo giallo in seguito a fratture delle ossa spugnose.
E’ una forma rara.
46
3.8.6 INFARTO
E’ un’area di necrosi ischemica prodotta da un’occlusione arteriosa o venosa. Le cause
possono essere svariate, ma soprattutto trombo-embolismi arteriosi. I trombo-embolismi
venosi sono invece più rari e coinvolgono prevalentemente testicolo e ovaio, in cui vi è
un’unica vena efferente.
Si possono trovare infarti:
- bianchi: dovuti ad occlusione arteriosa di tessuti solidi (cuore, rene, milza)
- rossi: dovuti ad occlusione venosa in tessuti lassi con conseguenti fenomeni
congestizi.
- Settici o asettici a seconda che vi sia o meno contaminazione batterica.
Le conseguenze possono essere diverse; in genere la funzionalità dell’organo è
preservata se si sviluppa un circolo collaterale efficace.
3.8.7 SHOCK
Lo shock è uno stato di ipoperfusione tissutale dovuto ad una diminuzione del sangue o
della gittata cardiaca con conseguente riduzione del volume ematico efficace; le sue
cause possono essere molteplici: emorragie, ustioni, traumi, infarti, emboli, sepsi.
47
Esistono tipi diversi di shock:
- Cardiogeno: per problemi cardiaci (infarti, aritmie), o extracardiaci (tamponamento
cardiaco, trombo-embolismo polmonare).
- Ipovolemico o emorragico: da traumi.
- Neurogeno: in seguito a stimoli nervosi che provocano vasodilatazione imponente
di tutto il distretto circolatorio. Di solito è conseguente ad anestesie.
- Anafilattico: da ipersensibilità di tipo I.
- Settico o endotossico: da batteri Gram negativi (Coli, Klebsiella) che producono
endotossine (lipopolisaccaridi-LPS) ad azione vasodilatatoria. La sua patogenesi si
sviluppa in diversi step:
a) i LPS si legano a diversi recettori di leucociti ed endoteli
b) si sviluppa un danno diretto per lesioni di membrana
c) segue un danno indiretto per:
stimolo macrofagi e monociti
↓
produzione di IL1 e TNFα
↓
danno al microcircolo con attivazione dei leucociti di endoteli e tessuti
↓
↓
↓
produzione di IL6,PG,leucotrieni
prod. NO e PAF
Attivazione chinine
⇓
Vasodilatazione
Lo shock riconosce vari stadi:
- non progressiva: in cui si ha l’attivazione di sistemi di compensazione
(catecolamine, endorfine, ADH) che provocano tachicardia, vasocostrizione e
ritenzione urinaria.
- Progressiva: in cui, superati i suddetti sistemi di compensazione, cominciano a
verificarsi disturbi metabolici (acidosi per glicolisi anaerobia e vasodilatazione per
paralisi da acidemia).
- Irreversibile: con danni cellulari permanenti (necrosi tubulare acuta nel rene) e
produzione di MDF (fattore depressante il miocardio).
48
3.9
MALATTIE GENETICHE
Sono malattie che interessano i cromosomi ed il codice genetico.
Possono essere:
- malattie geneticamente determinate – dovute alla presenza di geni “anomali” che
inducono o la predispongono alla patologia in presenza di agenti ambientali (ad
esempio un individuo in cui mancano i geni “immunologici” sarà sensibile alle
infezioni)
- malattie ereditarie – malattie geneticamente determinate trasmesse dai genitori
- malattie familiari – trasmissione di un carattere patologico nelle generazioni (ad
esempio l’emofilia)
- malattie congenite – patologie acquisite durante la vita uterina, ma non
necessariamente genetiche (ad esempio le malformazioni o la TBC)
Lo studio delle malattie genetiche si serve della biologia molecolare, che utilizza:
a) metodi classici: da un’anomalia in una proteina si risale al gene alterato
b) “reverse genetic” o “positional cloning”: non si conosce la proteina alterata ma si
cerca di analizzare frammenti genetici vicino a quello che si sospetta alterato.
c) Animali transgenici: topi, ratti, o altri animali in cui è stato inserito DNA anomalo per
studiarne gli effetti.
d) Animali “knock out” in cui è stato tolto un gene per evidenziare le conseguenze
dell’assenza di tele gene sulla vita.
e) Terapia genica: inserzione di geni normali al posto di quelli alterati.
MUTAZIONI
Sono modificazioni permanenti del DNA spontanee o indotte (raggi X, virus, sostanze
chimiche).
Se colpiscono cellule somatiche provocano malformazioni congenite o tumori, mentre se
avvengono in cellule germinali danno origine a malattie trasmissibili geneticamente.
Le mutazioni si dividono in:
- genomiche: si ha la variazione numerica dei cromosomi (monosomia se uno solo o
trisomia se tre)
49
cromosomiche: si verifica lo spostamento di geni (riarrangiamento genetico)
all’interno di un cromosoma con conseguente modificazione della sua forma.
Solitamente è incompatibile con la vita
- geniche: modificazione di un gene o di poche basi del gene. Possono essere
a) puntiformi (si modifica una base sola)
b) “frameshift” con inserzione o delezione di più basi.
Le conseguenze possono essere proteine con aminoacidi diversi dal normale oppure
proteine a cui mancano determinati aminoacidi.
-
3.9.1 DISORDINI MENDELIANI
Sono malattie in cui i geni mutati hanno effetti sistemici. Possono essere:
- recessive: evidenti solo in omozigoti
- dominanti: evidenti in omozigoti ma anche in eterozigoti con espressione parziale
- codominanti: ad esempio nei gruppi sanguigni
Tipi di trasmissione:
- Autosomica dominante:
- Indipendente dal sesso
- spesso ereditaria
- a volte compare in età avanzata
- bassa % di individui manifesta la patologia (bassa penetranza)
- coinvolge proteine di vie metaboliche complesse (enzimi, prot. di trasporto)
- Autosomica recessiva:
- Indipendente dal sesso
- esclusivamente ereditaria
- comparsa precoce
- penetranza completa (colpiti tutti i portatori)
- espressione uniforme
Esempi: malattie da accumulo lisosomiale, glicogenosi
- X – LINKED
- Legata al sesso (cromosoma X),
- spesso recessiva
- Patogenesi: X e Y non sono omologhi: geni su X non hanno alleli su Y
- malattia solo nei maschi
- trasmissione alla figlie femmine di maschi malati
- malattia nelle femmine solo se inattivo X sano
- femmina eterozigote trasmette al 50% della prole
Esempi: emofilia, distrofia muscolare di Duchenne
50
3.9.2 DISORDINI MULTIFATTORIALI O POLIGENICI
• Azione combinata di due o più mutazioni con effetto additivo in presenza di fattori
ambientali (ad esempio colore dei capelli, peso, intelligenza, diabete)
• Rischio condizionato dal numero di geni mutanti
• Forte influenza ambientale (es. alimentazione)
• Tasso incidenza = in tutti i parenti di I grado
• Probabilità di trasmettere la malattia aumenta con il numero di gravidanze
3.9.3 DISORDINI CROMOSOMICI (CITOGENETICI)
Alterazioni di numero
- Euploidia (multiplo del numero normale o aploide)
- Aneuploidia (non multiplo per errori di mitosi/meiosi)Esempi: trisomia 21 (sindrome
di Down), Sindromi di Turner / Klinefelter
51
4. TUMORI
Un tumore è una massa di tessuto anomalo, la cui crescita eccede e non è coordinata a
quella del tessuto normale e persiste nello stesso modo anche dopo la fine dello stimolo
che ha evocato il cambiamento.Willis, 1942Un tumore o neoplasia è la crescita nuova ed
anomala di un tessuto
4.1 TUMORI BENIGNI
Sono caratterizzati da:
a) differenziazione cellulare (le cellule somigliano molto al tipo originario)
b) Espansione molto lenta (le mitosi si susseguono ma lasciano un certo tempo di
adattamento al tessuto)
c) Non invadono la membrana basale (ad esempio nei tumori ghiandolari le cellule
neoplastiche si sviluppano all’interno del lume ghiandolare, senza infiltrare il tessuto
adiacente)
d) Generalmente possiedono una parete di tessuto connettivo
e) Per la loro denominazione si utilizza il suffisso OMA dopo il nome del tessuto di
origine (angioma, fibroma)
f) Eventuali problemi: compressione dei parenchimi e delle strutture vicine
4.2. TUMORI MALIGNI
Sono caratterizzati da:
a) mancata differenziazione cellulare (anaplasia)
b) Espansione rapida
c) Invasione della membrana basale
d) Non ben delimitate da eventuali capsule
e) Il loro nome dipende dalla derivazione: se epiteliale→carcinoma; se
connettivale→sarcoma, se ghiandolare→adenocarcinomaMitosi incontrollate
- Perdita di coesione con cellule vicine (essenziale per le metastasi)
52
- Citoscheletro non funzionaleAlterazioni metabolicheLo stroma è caratterizzato da
numerosissimi vasi neoformati (neo-angiogenesi) e da una grande quantità di tessuto
connettivo (desmoplasia)
Le metastasi, colonizzazione delle cellule tumorali di tessuti diversi da quello di origine
possono avvenire in diversi modi:
- per contatto diretto (ad esempio dal carcinoma intestinale si arriva al rene)
- attraverso i vasi linfatici (che drenano il distretto) → linfonodi regionali → altri organi
- attraverso i vasi ematici → vari organi
SISTEMI DI DIFESA CELLULARI:
53
Le cellule tumorali esprimono sulla superficie antigeni “non self”, quindi verranno riconosciute dalle cellule
immunitarie come cellule “anormali”.
54