caricato da
giorgiodamiani13
I LEGAMI, Materiali per l'ingegneria elettrica

A cura di:
Giorgio Damiani,
Orazio Pazienza,
Davide Giannoccaro,
Antonluca Romoli.
1. SCHEMI INIZIALI
1.1 Generalità degli stati di aggregazione della materia
La materia come siamo abituati a conoscerla ed interpretarla risulta presentarsi in natura secondo
tre differenti “stati di aggregazione”, disposizioni particolarmente legate di atomi, da non
interpretare come proprietà fisse di una sostanza. Nello specifico essi sono tre: stato solido, stato
liquido e stato gassoso, e possono tutti, al variare delle condizioni ambientali, essere perseguiti da
moltissime delle sostanze presenti in natura (in primis l’acqua). I materiali allo stato solido
risultano possedere forma e volume proprio, e le molecole al loro interno risultano essere
reciprocamente legate da forze interne abbastanza intense da pregiudicarne il moto libero nello
spazio. Al pari dello stato solido anche i materiali allo stato liquido risultano possedere un proprio
volume, ma assumono la forma del recipiente che li contiene, minimizzando la superficie di
contatto con il fluido immiscibile con il quale sono a contatto (un banale esempio è un bicchiere
pieno d’acqua, a contatto con l’aria). I legami che caratterizzano lo stato liquido sono inoltre meno
intensi rispetto allo stato solido permettendo uno scorrimento delle molecole l’una sull’altra.
Questi due primi stati di aggregazione, vista la particolare attitudine delle molecole ad
agglomerarsi e non disperdersi casualmente, prendono il nome di stati condensati, e possiedono
un’attitudine opposta a quello che è il
nostro ultimo stato di aggregazione: lo
stato gassoso. Nello specifico, i materiali
allo
stato
gassoso
risultano
non
possedere né forma né volume proprio e
hanno la particolare attitudine, se liberati
in un ambiente, di andare ad occupare
tutto lo spazio a loro disposizione, vista
l’assenza di legami intercorrenti tra le
particelle.
1.1.1 Passaggi di stato
Come già accennato in precedenza, ogni
sostanza può cambiare stato in relazione alle trasformazioni caratteristiche del sistema
termodinamico nel quale viene inserito. In particolare, un passaggio di stato si innesta in relazione
ad un assorbimento o ad un rilascio di energia sotto forma di calore. Il passaggio da solido a
liquido ad esempio prende il nome di fusione, e avvenendo in condizioni di innalzamento di
temperatura, costituisce un processo con assorbimento di energia. Passaggio simmetrico di stato è
la solidificazione che avviene a fronte di un rilascio di energia (calore latente) causato dal
precedente abbassamento di temperatura. Il passaggio da liquido a gas, poi, prende il nome di
evaporazione (assorbe energia) ed il suo processo inverso prende il nome di condensazione. Con il
termine sublimazione si fa poi riferimento al passaggio diretto da stato solido a stato gassoso, ed
infine con brinamento si indica il passaggio diretto da gas a solido (un interessante esempio di
sublimazione è relativo al gas che si libera dall’emissione di ghiaccio secco ad alta pressione,
costituito da nient’altro che anidride carbonica allo stato solido).
Uno strumento di particolare interesse nello studio dei passaggi di stato è il “diagramma di stato”,
una relazione grafico analitica che ci permette di osservare in relazione alla variazione delle
condizioni ambientali quello che è il caratteristico stato di aggregazione di una sostanza, e quelle
che sono le sue temperature di fusione o evaporazione. Dal diagramma di stato posto di fianco,
relativo all’acqua, si osserva come alla pressione atmosferica le temperature caratteristiche dei
passaggi di stato risultano proprio essere quelle notevoli, di cento gradi Celsius per l’evaporazione
e di zero gradi Celsius per la fusione.
1.2 Struttura della materia
Avendo, in via generale, descritto quelle che sono le proprietà distintive dei tre stati di
aggregazione, andiamo ora ad osservare più nello specifico quelle che sono le particolari strutture
distintive dei materiali in questi stati.
1.2.1 Stato solido e reticolo cristallino
La particolare struttura dei materiali solidi, aventi forma e volume propri, dipende, a livello
atomico, dall’equilibrio tra le particelle stesse e le forze intense che esercitano gli uni sugli altri.
Questo fa sì che un solido possegga una struttura atomica ordinata con atomi collocati in posizioni
fisse nello spazio, senza che essi siano liberi di muoversi proprio a causa dei legami (di molteplice
tipologia) precedentemente instaurati: una struttura solida si presenta generalmente in tre forme,
quali, “cristallina”, ovvero secondo una disposizione ordinata di atomi, “amorfa”, ovvero secondo
una distribuzione disordinata di particelle (struttura caratterizzata da minori intervalli di fusione)
ed infine “semi-amorfa”.
Con il termine cristallo si fa riferimento, allora, ad una struttura ordinata di atomi la cui ripetizione
nello spazio provoca la formazione di un reticolo: infatti, osservando un’ipotetica geometria di
atomi, osserviamo come essi, schematizzati in forma sferica, vadano ad occupare i vertici di un
solido regolare, che potrà assumere forma differente (cubo, prisma, tetraedro, ecc.) in relazione
proprio a quella che è la natura particolare del materiale. Tale geometria prende il nome di “cella
elementare” e la sua ripetizione e sovrapposizione nello spazio, provoca la formazione del reticolo
ristallino. E’ importante sottolineare come queste approssimazioni di natura microscopica, hanno
un
estrema
rilevanza
macroscopico,
poiché
individuare
breve
in
dal
punto
ci
permettono
di
proprietà
del
tempo
di
vista
materiale in analisi quali, ad esempio, la densità
(mediante
il
calcolo
del
FCA,
fattore
di
compattamento atomico, calcolato su di una cella
elementare) e la temperatura di fusione, senza necessariamente bisogno di
un’analisi empirica. Alcune celle elementari di necessaria menzione per la loro
estrema diffusione in natura sono la struttura cubica a corpo centrato (C.C.C.)
avente una geometria cubica, completata dall’inserimento di un atomo
collocato nel baricentro del cubo; la struttura cubica a facce centrate (C.F.C)
avente anch’essa una geometria cubica con inserimento questa volta di sei
ulteriori atomi collocati al centro di ogni faccia del cubo, ed infine la struttura
esagonale compatta (E.C), caratterizzata da una geometria prismatica a base esagonale, con atomi
posti, oltre che sui vertici, al centro delle due facce di base (uno per faccia) e all’interno della
struttura, con tre atomi posti internamente alla struttura prismatica a metà dell’altezza
complessiva della cella. Le tre strutture differiscono in tutti quelli che sono i parametri utili a
valutare le proprietà macroscopiche del materiale, ovvero, il numero di atomi interamente
presenti nell’unità (2 per la C.C.C, 4 per la C.F.C, 6 per la E.C) il numero di coordinazione, ovvero il
numero con cui ciascun atomo è in contatto, la costante reticolare, ovvero la dimensione uguale in
tutte e tre le direzioni per la cella (il lato del cubo per C.C.C e C.F.C, rapporto tra altezza e lato di
base nella E.C.) e proprio il fattore di compattamento, prima menzionato, pari al rapporto tra il
volume effettivamente occupato dagli atomi nella cella ed il volume della cella.
1.2.1.1 Metodo di Miller
Un' altra importante parentesi che risulta necessario aprire nel momento in cui ci si sofferma su
quelle che sono le applicazioni pratiche relative allo studio del reticolo cristallino risulta essere
legata all'individuazione di posizioni e piani specifici all'interno del reticolo cristallino. In
particolare, il metodo utilizzato per l'individuazione delle località specifiche in un cristallo, prende
il nome di "metodo di Miller", e adopera un sistema di individuazione dei punti del reticolo
mediante una successione di tre numeri interi, ognuno indicante la distanza caratteristica della
coordinata del punto in questione con quella che è l'origine del reticolo cristallino stesso: tale
distanza sarà misurata impiegando come unità di misura i lati della cella unitaria del cristallo.
L'origine, poi, viene individuata arbitrariamente mediante l'individuazione degli assi cristallografici,
ovvero un sistema di assi collocato arbitrariamente in uno dei vertici della nostra cella elementare.
Proprio da qui si parte per l'individuazione delle direzioni: considerato infatti un vettore passante
per l'origine degli assi cristallografici, andiamo a fissare su di esso le coordinate di posizione del
vettore nel suo punto conclusivo (frazionandole rispetto alla cella unitaria); andiamo poi a
considerare per le 3 l'intero minore, attraverso il semplice calcolo del minimo comune multiplo, e
riscriviamo per ognuna delle direzioni cristallografiche, [x;y;z], i rispetivi valori interi [u;v;w]. Un
insieme di vettori aventi gli stessi indici di direzione risulta costituire una famiglia di "direzioni
equivalenti", indicata con <u;v;w>. Un processo fondamentalmente analogo potrà essere seguito
per l'individuazione di un piano: in primis si procede all'individuazione dello stesso, che per ipotesi
NON dovrà essere passante per l'origine. A seguito si individuano le intercette del piano stesso con
gli assi cristallografici (sempre frazionate rispetto alla cella unitaria) per poi andare ad individuare
il reciproco dei valori in questione. A seguito si riducono i valori, per semplicità di scrittura,
all'intero minore e si completa la notazione con la scrittura degli indici come (h;k;l) . Anche in
questo caso, piani aventi i medesimi indici prendono il nome di famiglie di piani equivalenti, e si
identificano come {h;k;l}.
1.2.2 Forme allotropiche e difetti del reticolo cristallino
E’ inoltre necessario sottolineare come la struttura cristallina di un materiale solido, e quindi la
caratteristica cella elementare, possono variare a fronte di variazioni di temperatura, secondo
particolari trasformazioni denominate “trasformazioni allotropiche”: un esempio, è dato dalla
struttura cristallina del ferro, il quale presenta tre forme allotropiche, ferro alpha, stabile fino ad
una temperatura di 912°, ferro gamma, stabile per un intervallo di temperatura compreso tra i
912° ed i 1394° ed infine il ferro delta, stabile dai 1394° fino a fusione. Proprio le tre forme
allotropiche presentano due differenti celle elementari: se il ferro alpha presenta infatti una
struttura C.C.C (al pari del ferro delta), il ferro gamma risulterà possedere una struttura C.F.C,
influendo sulle proprietà macroscopiche del materiale.
Un’ultima menzione relativa alla struttura della materia allo
stato solido è relativa ai difetti del reticolo cristallino, utili a
permetterci di distinguere il grado di nobiltà di un
materiale. Si definisce difetto, ogni forma di disomogeneità
o mancanza nel reticolo cristallino, e tanto meno difetti
saranno riscontrabili, tanto più nobile sarà il materiale in questione. I difetti possono essere
catalogati in puntuali, lineari e superficiali. Ai primi appartengono difetti quali “le vacanze”
identificabili come cavità del reticolo in posizioni nelle quali la disposizione avrebbe previsto la
presenza di particelle, difetti interstiziali, caratterizzati dalla presenza di atomi, omogenei o
eterogenei al reticolo in posizioni nelle quali il reticolo non avrebbe previsto la presenza di atomi,
ed infine i difetti sostituzionali, caratterizzati dalla presenza di atomi eterogenei al reticolo in
posizioni che sarebbero dovute esser occupate da atomi omogenei al reticolo, e quindi di
differente dimensione.
All’interno dei difetti lineari rientrano poi, impurezze quali “le dislocazioni a spigolo” o “a vite”
caratterizzati rispettivamente dalla presenza in difetto di un semipiano di atomi per la prima e
dalla presenza di un semipiano aggiuntivo nel secondo caso, mentre nei difetti superficiali si fa
riferimento a quelle impurezze presenti sull’interfaccia tra differenti cristalli, aventi avuto in
precedenza direzioni di crescita differenti (nella solidificazione ad esempio) e sulle quali superfici è
frequente che si vengano a formare cricche o zone dalle minori prestazioni in termini di proprietà
macroscopiche.
1.2.3 Stato liquido
Lo stato liquido è stato precedentemente definito come quello stato condensato per il quale il
materiale risulta avere volume proprio ma una forma dettata dal recipiente che lo contiene. I
materiali di questo tipo risultano caratterizzati da molecole soggette a forze di attrazione
elettrostatica reciproca e ad interazioni intermolecolari, responsabili della disposizione per strati
sovrapposti, abbastanza forti da non consentire la separazione tra di esse, ma non così intense da
bloccare le molecole in punti fissi dello spazio come avveniva per lo stato solido. Le molecole
quindi sono in uno stato di continuo movimento reciproco, con scorrimento relativo tra le
particelle senza che esse si separino. L’attitudine degli strati di liquido a scorrere gli uni sugli altri
viene valutata attraverso la “viscosità”, utile a valutare la resistenza che incontrano i piani a
scorrere reciprocamente: essa sarà tanto più bassa tanto più elevati risulteranno essere i valori di
temperatura. Analiticamente la viscosità può essere ricavata supponendo di avere una particella di
fluido a forma di parallelepipedo, applicando su di essa uno sforzo tangenziale. La relazione che ne
risulterà, risulterà essere la relazione caratteristica di tutti i fluidi Newtoniani:
dove µ è proprio la viscosità dinamica del liquido.
1.2.3.1 Dimostrazione
Consideriamo, come detto, una particella di fluido a forma di parallelepipedo, sottoposta per
ipotesi ad uno stato di tensione tangenziale 𝜏, applicato sulla faccia superiore della geometria in
questione. In un intervallo di tempo ∆𝑡 la nostra geometria avrà necessariamente subito uno
spostamento verso destra come in figura, percorrendo con la propria faccia superiore uno spazio
pari a U*∆𝑡 (U=velocità [m/s]). Osservando la geometria della regione triangolare formatasi, ora,
possiamo mettere in correlazione la distanza percorsa con l'angolo di inclinazione "∆𝛾" che si
viene a formare rispetto alla verticale: tg(∆𝛾)= (U*∆t)/d , indicata con d la dimensione verticale
della particella. Per infinitesimi intervalli di tempo, sappiamo di poter approssimare la tg di un
angolo all'angolo stesso, da cui ricaviamo che ∆𝛾/∆t = U/d. Volendo a questo punto risalire
all'accelerazione angolare (𝜑 ) derivante dallo sforzo, che sappiamo, dalla meccanica tradizionale,
∆𝛾
essere direttamente proporzionale allo sforzo stesso, osserviamo come 𝜑= lim ( ∆𝑡 )sarà, per
∆𝑡→0
sostituzione dall'equazione precedente, pari a U/d, e per intervalli di tempo infinitesimi e per
distanze infinitesime pari a dU/dy (essendo d diretta lungo y). Sostituendo quest'ultima relazione
nell'equazione di diretta proporzionalità di tra lo sforzo tangenziale e l’accelerazione angolare,
otteniamo che 𝜏=k*𝜑=k*(U/d) = k*(dU/dy), per gli infinitesimi. Isolando ora k, otteniamo che essa
è pari a 𝜏*(d/U) e costituirà proprio la nostra "viscosità dinamica" C.V.D. Dal punto di vista
dimensionale, essa sarà misurabile in [kg/m*s], e proprio da essa possiamo risalire, dividendo per
la densità, ad un valore di "viscosità cinematica", dimensionalmente più pratica, misurabile quindi
come:
"𝑛𝑖" = 𝜇 /𝜌" [m^2/s]
L’osservazione della viscosità rende altresì evidente quella che è una delle proprietà caratteristiche
dei liquidi, ovvero l’elasticità, individuata come l’attitudine di un fluido a deformarsi in seguito a
l’azione di uno sforzo tangenziale, senza che riprendano al cessare della sollecitazione la forma
originaria. Un’altra proprietà caratteristica dei materiali in questo stato di aggregazione è poi, la
quasi totale incomprimibilità, che fa sì che in uno stato di pressione e temperatura costanti essi
non possano praticamente essere compressi, secondo un fattore di comprimibilità che varia in
funzione della natura del liquido stesso.
1.2.4 Cristalli liquidi e ferrofluidi
Un’interessante situazione intermedia tra stato solido e stato liquido, è data dai “cristalli liquidi”
ovvero particolari composti di matrice organica, che anziché possedere un passaggio di stato ben
delineato tra fase solida e fase liquida risultano stabilizzarsi, in particolari condizioni intermedie,
secondo mesofasi (fasi intermedie) che presentano caratteristiche sia dello stato solido che di
quello liquido: essi in particolare sono totalmente anisotropi dal punto di vista ottico e termico
(ovvero l’applicazione di calore o di radiazioni risulta essere indipendente dalla direzione di
applicazione degli stessi) al pari dei solidi ma risultano possedere fluidità e mobilità molecolare al
pari dei liquidi. Per quanto concerne la geometria della formazione dei cristalli liquidi, essi sono
usualmente costituiti da molecole caratterizzate da una forte asimmetria, aventi una forma
allungata caratterizzata da una direzione orientata analoga a quella dei solidi, ma una
distribuzione casuale dei centri delle loro molecole, al pari dei fluidi. La loro particolare attitudine
a variare poi la propria colorazione in relazione agli squilibri termici, li rendono di grande impiego
per quanto concerne l’ingegneria dei sensori termici.
Un'altra interessante forma di ibrido liquido-solido, di larghissima applicazione nelle moderne
tecnologie elettriche, è relativa ai "ferrofluidi". I ferrofluidi sono composti di particelle
ferromagnetiche sospese in un veicolo fluido, molto spesso un solvente organico oppure acqua. I
ferrofluidi sono composti da nanoparticelle ferromagnetiche, solitamente magnetite, ematite o
qualche altro composto contenente Fe2+ o Fe3+. Le dimensioni delle nano-particelle sono
tipicamente nell'ordine dei 10 nm; tali dimensioni sono contenute al punto da far sì che
l'agitazione termica le disperda uniformemente all'interno del solvente, e che le particelle
contribuiscano alla risposta magnetica complessiva del fluido. I veri ferrofluidi sono stabili. Ciò
significa che le particelle solide non si agglomerano o creano fasi separatamente, anche quando
immerse in campi magnetici estremamente forti. Comunque, i tensioattivi tendono a spezzarsi con
il tempo (pochi anni), e alla fine le nanoparticelle si agglomereranno, separandosi e cessando di
contribuire alla risposta magnetica del fluido. I ferrofluidi sono usati comunemente negli
altoparlanti e per formare dei sigilli liquidi attorno agli assi di rotazione degli hard disk. Possiedono
inoltre caratteristiche di riduzione dell'attrito. Se applicati sulla superficie di un magnete
sufficientemente potente, come uno fatto in neodimio, esso può planare su superfici lisce con una
resistenza minima.
1.2.5 Stato gassoso
Lo stato gassoso, infine, risulta essere, risulta essere uno stato di aggregazione non condensato,
caratterizzato da particelle svincolate le une dalle altre, in uno stato di totale indipendenza tra le
molecole in questione, le quali sono liberamente disposte a muoversi continuamente in tutto lo
spazio a loro disposizione. Proprio questo perpetuo stato di moto risulta compiersi a causa degli
urti continui tra le molecole libere in un ipotetico recipiente o tra le molecole stesse e le pareti del
recipiente in questione. Questi scontri, provocano un moto per segmenti di retta, con continui
cambi di direzione. Proprio a causa dell’assenza di legami intermolecolari, allora, un gas non avrà
ne forma ne volume proprio, ma andrà ad occupare tutto lo spazio a propria disposizione. Inoltre
l’assenza di interazioni chimiche fa si che, se per i solidi ed i liquidi come vedremo a seguito sarà
possibile individuare una corrispondenza diretta tra le proprietà fisiche del materiale in questione
ed il legame di interesse, per i gas esiste un comportamento molto uniforme tra le sostanze anche
in presenza di una variazione dei parametri fisici che caratterizzano il loro stato iniziale. Per
l’analisi dei gas sarà allora necessario provvedere ad uno studio delle variabili di stato del sistema,
quali temperatura, pressione, volume e numero di moli, la cui indicazione sarà fondamentale per
la caratterizzazione di un gas particolare, piuttosto che per un solido la cui indicazione della massa
risultava sufficiente. Proprio a questo scopo nasce la legge dei gas ideali, ovvero una teorizzazione
avente la specifica finalità di individuare una relazione tra le grandezze prima indicate, sempre
nell’ipotesi di totale assenza di vincoli tra le particelle. La legge afferma in particolare che, indicata
con P la pressione, con V il volume occupato dal gas, on T la temperatura, con n il numero di moli e
con R una costante detta “costante dei gas ideali” che:
PxV=nxRxT
Questa legge mostra quelle che sono le attitudini generali di un gas, permettendoci di osservare il
suo comportamento a fronte di una compressione (vista la natura dei gas che a differenza dei
liquidi sono comprimibili, a causa dell’assenza di vincoli intermolecolari) o a fronte di un
innalzamento di temperatura.
1.3 I legami
La materia è costituita da atomi. In natura, solo pochi elementi esistono come atomi isolati: è il
caso dei gas nobili, elementi dell’ottavo gruppo (VIIIA) della tavola periodica. Tutti gli altri elementi
tendono a legarsi formando dei composti. I legami chimici sono delle forze che tengono uniti gli
atomi e, il principio di base che spiega la loro formazione è quello della minima energia. Infatti, gli
atomi isolati hanno un’energia maggiore (b) rispetto a quando sono legati mediante legame
chimico (a). In altre parole, si ha, in generale, che il legame chimico dà luogo ad un sistema più
stabile, cioè a minore energia rispetto agli atomi isolati. L’energia coinvolta nella formazione (o
nella rottura) di un legame è detta “energia di legame”. Quest’ultima è l’energia necessaria per
rompere un dato legame quando la sostanza si trova in fase gassosa, o l’energia liberata quando il
legame si forma.
I legami chimici che un atomo può formare dipendono dagli elettroni più esterni, cioè dagli
elettroni di valenza. Un metodo utile per mettere in evidenza gli elettroni del livello più esterno di
un atomo è quello che fa uso dei simboli di Lewis, dal nome del chimico Gilbert N. Lewis che
propose di rappresentare gli elettroni valenza con puntini disposti attorno al simbolo dell’atomo.
Lewis notò che gli elementi dell’ottavo gruppo (VIIIA), i gas nobili, hanno tutti otto elettroni di
valenza (ad eccezione dell’elio, che ne ha solo 2) e sono poco reattivi. Lewis pertanto concluse che
la presenza di otto elettroni esterni rappresenta una condizione di particolare stabilità alla quale
tendono gli atomi di tutti gli elementi. La configurazione ad otto elettroni di valenza è chiamata
“ottetto”. L’elio, invece, ha due elettroni esterni e anche questa configurazione è particolarmente
stabile, perché i due elettroni completano il primo livello energetico. Secondo la teoria di Lewis un
legame chimico è il risultato di un trasferimento o di una condivisione di elettroni in seguito ai
quali i due atomi raggiungono la configurazione elettronica del gas nobile più vicino che, in genere,
ha un ottetto completo. Secondo la “regola dell’ottetto” quando un atomo forma un legame,
tende o a cedere, o ad acquistare, o a condividere elettroni in modo da raggiungere la
configurazione esterna dell’ottetto completo.
1.3.1 Classificazione dei legami
Esistono tre tipologie di legame: legame covalente, legame ionico e legame metallico.
Per comprendere che tipo di legame tiene uniti due atomi dobbiamo far riferimento
all’elettronegatività degli elementi coinvolti. Quest’ultima indica la tendenza di un atomo ad
attirare gli elettroni di legame. Avremo che:
-
Se la differenza di elettronegatività, ΔE, è compresa tra 0 e 0,4 => legame covalente puro (o
omopolare);
-
Se la differenza di elettronegatività, ΔE, è compresa tra 0,4 e 1,7 => legame covalente
polare;
-
Se la differenza di elettronegatività, ΔE, è maggiore di 1,7 => legame ionico.
1.3.2 Elettronegatività
Gli atomi manifestano una diversa capacità di attrazione nei confronti degli elettroni coinvolti nei
legami chimici. Questa capacità viene descritta da una proprietà chiamata elettronegatività. Il
concetto di elettronegatività viene espresso quantitativamente assegnando a ogni elemento un
valore numerico. Il metodo più ampiamente utilizzato per determinare l’elettronegatività è stato
quello messo a punto dal chimico statunitense Linus Pauling; i valori numerici sono stati ottenuti
mettendo in relazione i dati sperimentali relativi all’energia di ionizzazione (l’energia necessaria
per strappare un elettrone all’atomo), all’affinità elettronica (l’energia che si ottiene quando un
elettrone viene aggiunto a un atomo) e all’energia di legame (l’energia richiesta per rompere un
legame).
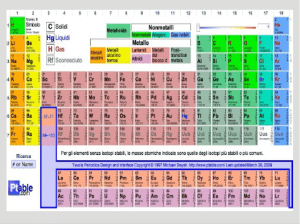
La figura che segue riproduce una parte della tavola periodica in cui i valori di elettronegatività
degli elementi sono rappresentati anche sotto forma di istogrammi.
Come si vede, l’elettronegatività aumenta in un gruppo dal basso verso l’alto e in un periodo da
sinistra verso destra: di conseguenza gli elementi più elettronegativi si trovano nell’angolo in alto a
destra mentre quelli meno elettronegativi si trovano dalla parte opposta, nell’angolo in basso a
sinistra. Si può notare che l’elettronegatività dei gas nobili non è stata determinata, dato che
normalmente questi elementi non formano legami.
1.3.3 Il legame covalente
Nel legame covalente due atomi condividono una o più coppie di elettroni di valenza. Il composto
che si forma è detto composto molecolare (o covalente).
Un legame tra due atomi in cui gli elettroni di legame sono equamente condivisi è detto “Legame
covalente puro” o “omopolare”. Ad esempio:
Nella molecola del cloro i due atomi mettono in comune una sola coppia di elettroni: si tratta di un
“legame semplice”. I due elettroni che tengono uniti gli atomi nella molecola del cloro Cl 2 sono
condivisi in egual misura da parte dei due nuclei.
Talvolta, per raggiungere l’ottetto è necessario condividere più coppie di elettroni. Il legame
covalente è pertanto definito doppio o triplo. Ad esempio:
In generale i legami doppi sono più corti e più forti dei legami singoli, e quelli tripli sono più forti di
quelli doppi.
Il “legame covalente polare” si forma mediante condivisione di elettroni tra atomi la cui differenza
di elettronegatività è compresa tra 0,4 e 1,7. In queste molecole gli elettroni di legame non sono
equamente condivisi ed esse risultano polari. Ciò significa che gli elettroni di legame sono spostati
verso l’atomo più elettronegativo, che acquisisce una parziale carica negativa (δ-), mentre l'altro
atomo acquisisce una parziale carica positiva (δ+). Si forma così un dipolo.
Un particolare legame covalente è il “legame dativo”. In esso la coppia di elettroni di legame è
fornita da uno solo dei due atomi che partecipano al legame. L’atomo che dona gli elettroni si dice
donatore, quello che li riceve prende il nome di accettore. Questo legame una volta formatosi non
è distinguibile dal normale legame covalente.
1.3.3.1 Caratteristiche dei solidi covalenti (come ad esempio il diamante)
I solidi covalenti o reticolari si formano grazie a una rete tridimensionale di legami covalenti fra gli
atomi. Il legame covalente, anziché caratterizzare una singola molecola vista come entità
microscopica a sé stante, può caratterizzare un intero reticolo cristallino. Proprietà caratteristiche
dei solidi covalenti sono:
-
Elevata durezza;
-
poco volatili;
-
temperatura di fusione elevata;
-
bassissima conducibilità elettrica;
-
non sono solubili in acqua.
1.3.4 Il legame ionico
Il legame ionico è un legame di natura elettrostatica che si forma quando si combinano fra loro gli
atomi di elementi aventi, rispettivamente, una bassa energia di ionizzazione ( la minima energia
che si deve fornire ad un atomo neutro allo stato gassoso per allontanare da esso l’elettrone del
livello energetico più esterno) ed una elevata affinità elettronica (energia che si libera quando un
atomo neutro allo stato gassoso acquista un elettrone).
Il legame ionico si forma quando la differenza di elettronegatività tra due atomi è molto alta e
indicativamente maggiore di 1,7. L’atomo più elettronegativo acquista l’elettrone e diventa uno
ione negativo, l’altro, che perde l’elettrone, diviene uno ione positivo. Tra ioni di carica opposta si
stabilisce una forza di attrazione elettrostatica che costituisce il legame ionico. Formano legami
ionici elementi spiccatamente metallici (quelli che cedono facilmente un elettrone,
elettronegatività piccola) uniti a elementi spiccatamente non-metallici (quelli che accettano
facilmente un elettrone, elettronegatività elevata). Gli ioni in un composto ionico sono disposti
secondo uno schema ben preciso e danno luogo ad un reticolo cristallino.
La formula dei composti ionici è definita “unità formula”. Essa indica il rapporto di combinazione
tra ioni positivi e negativi ma non rappresenta la molecola di un composto ( che si ha solo nei
composti caratterizzati dalla presenza di legami covalenti) perché nei cristalli non si distinguono
unità molecolari.
Caratteristiche dei composti ionici sono le seguenti:
-
Sono solidi a temperatura ambiente e nelle normali condizioni di pressione;
-
Hanno alte temperature di fusione;
-
Si presentano in forme cristalline regolari e geometricamente ben definite;
-
Conducono la corrente solo allo stato fuso o in soluzione;
-
In forma solida non conducono la corrente;
-
Quasi tutti sono solubili in acqua e in altri solventi polari;
-
Rispondono alle tensioni in modo fragile a volte rompendosi con piani di sfaldamento.
1.3.5 Il legame metallico
Il legame metallico è dovuto all’attrazione tra gli ioni metallici positivi e gli elettroni mobili che li
circondano.
In un metallo gli atomi perdono i loro elettroni di valenza trasformandosi in cationi (ioni positivi).
I cationi si dispongono in modo da impacchettarsi nel miglior modo possibile, creando strutture
geometriche ben definite. Gli elettroni di valenza sono delocalizzati su tutto il reticolo, cioè non
appartengono più ai singoli atomi ma sono liberi di muoversi tra i vari cationi e, nonostante la
mobilità della nube elettronica, la neutralità del sistema è garantita.
Le proprietà tipiche dei metalli sono dovute alla libertà di movimento degli elettroni delocalizzati.
Esse sono:
- solidi a temperatura ambiente;
- conducibilità elettrica e del calore;
- duttilità;
- malleabilità.
La conducibilità elettrica, ovvero la capacità di condurre corrente elettrica, e la conducibilità
termica, ovvero la capacità di condurre calore, sono da attribuire alla bassa energia di ionizzazione
ed alla elevata mobilità degli elettroni di legame.
La duttilità, cioè la capacità di lasciarsi forgiare in fili, e la malleabilità, cioè la capacità di lasciarsi
forgiare in lamine sottili, sono la conseguenza della flessibilità del legame metallico. Gli elettroni
mobili, infatti, consentono ai piani ionici di “scivolare” l’uno sull’altro lasciando inalterate le
interazioni di legame tra i cationi e non facendo insorgere forze repulsive.
1.3.6 Forze intermolecolari (o legami secondari)
Tali forze sono chiamate intermolecolari per distinguerle dai legami covalenti che agiscono
all’interno delle molecole o dai legami ionici che vincolano gli ioni in un cristallo.
Le forze intermolecolari sono forze di natura elettrostatica che si manifestano tra molecole
elettricamente neutre o tra ioni e molecole polari. Questi legami hanno energia di legame minore
rispetto a quella dei legami chimici primari, pertanto sono detti anche “interazioni deboli” e sono
importanti per determinare le proprietà fisiche di sostanze allo stato liquido o solido.
I legami intermolecolari possono essere classificati in diverse categorie in ragione del tipo di forza
che interviene tra esse:
-
Interazione ione-dipolo;
-
Forze di Van der Waals (interazione dipolo-dipolo, interazione dipolo permanente-dipolo
indotto e Forze di London, ovvero interazione dipolo istantaneo- dipolo indotto);
-
Legame ad idrogeno.
1.3.6.1 Interazione ione-dipolo
L’interazione o legame ione - dipolo è la forza di attrazione elettrostatica che si stabilisce tra uno
ione e una molecola polare. Si forma quando un composto ionico o molecolare, in acqua, si
dissocia in ioni di carica opposta. Gli ioni positivi orientano e attirano la parziale carica negativa
localizzata sull’atomo di ossigeno dell’ H2O, mentre ioni negativi orientano e attirano la parziale
carica positiva localizzata sui due atomi di idrogeno, con la conseguenza che ogni ione è circondato
da un numero elevato di molecole di H2O (idratazione).
1.3.6.2 Interazione dipolo-dipolo
Sono deboli forze attrattive che si esercitano tra molecole polari inorganiche allo stato liquido o
solido, oppure tra molecole organiche allo stato liquido. Le molecole polari sono dipoli elettrici che
presentano una parziale carica positiva ad una estremità (δ+) ed una parziale carica negativa (δ-)
all’altra estremità. Molecole di questo tipo, quando sono molto vicine, si orientano in modo tale
da stabilire forze attrattive tra le parziali cariche di segno opposto. Le forze dipolari in un liquido
sono più deboli che nello stato solido.
1.3.6.3 Interazione dipolo permanente-dipolo indotto
Sono deboli forze attrattive che si esercitano tra molecole polari (H2O) e molecole apolari (I2). La
molecola polare induce una separazione di cariche elettriche (polarizzazione) nella molecola
apolare (dipolo indotto) permettendo lo stabilirsi di interazioni elettrostatiche.
1.3.6.4 Forze di London (Interazione dipolo istantaneo - dipolo indotto)
Sono interazioni elettrostatiche che si stabiliscono tra molecole apolari (H2, N2, F2, CCl4) allo stato
liquido o solido. Tali interazioni sono possibili a causa del continuo cambiamento di posizione degli
elettroni in una molecola che può dar luogo, in un dato momento, a una densità elettronica
asimmetrica, ovvero a un dipolo istantaneo. Questo può determinare per induzione in una
molecola adiacente una temporanea polarizzazione (dipolo indotto) permettendo così lo stabilirsi
di forze attrattive.
1.3.6.5 Legame ad idrogeno
Tale legame si stabilisce quando in una molecola polare è presente l’atomo di idrogeno legato
covalentemente ad un atomo dal raggio atomico piccolo e molto elettronegativo (N, O, F).
Rispetto a tutti gli altri legami secondari, il legame ad idrogeno è quello caratterizzato da una forza
di legame maggiore e determina l’elevato punto di ebollizione delle sostanze che lo presentano
(come ad esempio l’acqua).