“SAPIENZA”
Università di Roma
Dipartimento di Biotecnologie Cellulari ed Ematologia
Sezione di Ematologia
DOTTORATO DI RICERCA IN SCIENZE EMATOLOGICHE - XXII CICLO
Coordinatore Prof. Robin Foà
TESI DI DOTTORATO
“Caratterizzazione di Funzioni Cellulari nelle Leucemie”
Relatore:
Dottoranda:
Prof.ssa Anna Guarini
Dott.ssa Nadia Peragine
Anno Accademico 2008/2009
1
INDICE
PREFAZIONE .......................................................................................................................... 6
La Leucemia Linfatica Cronica (LLC) ................................................................................... 6
PARTE I - Studio delle conseguenze funzionali della stimolazione in vitro delle cellule B
neoplastiche di pazienti affetti da Leucemia Linfatica Cronica (LLC) ................................. 10
1. INTRODUZIONE .............................................................................................................. 11
1.1 Il complesso recettoriale per l’antigene dei linfociti B (BCR) ...................................... 11
1.1.1 Composizione e ruolo del BCR nel processo di ontogenesi B cellulare ....................... 11
1.2 Ruolo del BCR nella patogenesi della LLC .................................................................... 14
1.2.1 Caratteristiche strutturali del BCR delle cellule B di LLC ........................................... 14
1.2.2 Stato maturativo e d’attivazione delle cellule B di LLC ............................................... 19
1.2.3 Natura degli antigeni implicati nella patogenesi ed evoluzione della LLC .................. 25
1.2.4 Segnali mediati dal BCR nelle cellule B di LLC ........................................................... 27
1.3 Scopo del progetto di ricerca ........................................................................................... 33
2. MATERIALI E METODI ................................................................................................. 35
2.1 Pazienti .............................................................................................................................. 35
2.2 Isolamento e coltura delle cellule mononucleate dei pazienti affetti da LLC ............. 35
2.3 Separazione immunomagnetica delle cellule B leucemiche CD19+ dei pazienti affetti
da LLC ..................................................................................................................................... 35
2.4 Stimolazione delle cellule B leucemiche CD19+ ............................................................. 36
2.5 Estrazione dell’RNA e analisi del profilo di espressione genica................................... 36
2.6 Analisi del ciclo cellulare.................................................................................................. 37
2.7 Saggio di proliferazione ................................................................................................... 37
2.8 Analisi del tasso apoptotico.............................................................................................. 37
2.9 Analisi statistica per gli studi di proliferazione e apoptosi ........................................... 38
2.10 Visualizzazione dell’actina intracitoplasmatica ........................................................... 38
3. RISULTATI ........................................................................................................................ 40
2
3.1 Analisi del profilo di espressione genica delle cellule B neoplastiche di pazienti affetti
da LLC dopo stimolazione del BCR ..................................................................................... 40
3.2 Effetti della stimolazione del BCR delle cellule B neoplastiche di LLC sulla
modulazione del ciclo cellulare .............................................................................................. 42
3.3 Studio dell’attività proliferativa delle cellule B neoplastiche di LLC dopo
stimolazione del BCR ............................................................................................................. 45
3.4 Valutazione degli effetti della stimolazione del BCR delle cellule B neoplastiche di
LLC sulla modulazione del processo apoptotico ................................................................. 50
3.5 Analisi dei cambiamenti citoscheletrici delle cellule B neoplastiche di LLC dopo
stimolazione del BCR ............................................................................................................. 56
4. DISCUSSIONE ................................................................................................................... 60
5. REFERENZE ...................................................................................................................... 73
PARTE II - Studio della funzionalità della proteina p53 in campioni primari di cellule
neoplastiche di pazienti affetti da Leucemia Linfatica Cronica (LLC) ................................. 83
1. INTRODUZIONE .............................................................................................................. 84
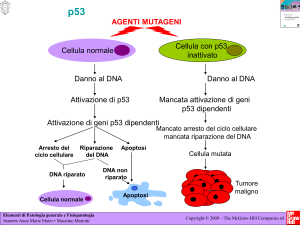
1.1 La proteina p53 ................................................................................................................. 84
1.1.1 Cenni introduttivi .......................................................................................................... 84
1.1.2 Struttura della proteina p53 .......................................................................................... 85
1.1.3 Regolazione dei meccanismi di attivazione della proteina p53 .................................... 88
1.1.4 Ruolo della proteina p53 nella soppressione tumorale ................................................ 96
1.1.5 Funzioni citoplasmatiche della proteina p53 ................................................................ 99
1.1.6 Ruolo dei microRNA nel controllo dell’attività della proteina p53 ............................ 102
1.2 Meccanismi di deregolazione a carico del gene TP53 e carcinogenesi ....................... 104
1.2.1 Mutazioni del gene TP53 ............................................................................................ 104
1.2.2 Impatto dello stato del gene TP53 sulla prognosi tumorale ....................................... 109
1.2.3 Ripristino della funzionalità della proteina p53 nei tumori umani............................. 111
1.3 Meccanismi di deregolazione della via di p53 in pazienti affetti da LLC ................. 114
1.4 Scopo del progetto di ricerca ......................................................................................... 120
3
2. MATERIALI E METODI ............................................................................................... 121
2.1 Pazienti e Controlli ......................................................................................................... 121
2.2 Induzione dell’attivazione della proteina oncosoppressoria p53 ............................... 121
2.3 Valutazione dell’attivazione della proteina oncosoppressoria p53 ............................ 122
2.4 Analisi del tasso apoptotico............................................................................................ 123
2.5 Analisi statistica per gli studi di apoptosi ..................................................................... 124
3. RISULTATI ...................................................................................................................... 125
3.1 Analisi della funzionalità della proteina p53 in campioni primari di cellule
neoplastiche di pazienti affetti da LLC a differente stadio di malattia ........................... 125
3.2 Correlazione della funzionalità della proteina p53 con lo stato del gene TP53 ........ 135
3.3 Correlazione della funzionalità della proteina p53 con i principali fattori prognostici
caratteristici della LLC ........................................................................................................ 138
3.4 Correlazione della funzionalità della proteina p53 con la capacità di induzione del
processo apoptotico .............................................................................................................. 141
4. DISCUSSIONE ................................................................................................................. 146
5. REFERENZE .................................................................................................................... 158
PARTE III - Caratterizzazione fenotipica e funzionale delle cellule T regolatorie di pazienti
affetti da neoplasie ematologiche sottoposti a trapianto allogenico di cellule staminali
emopoietiche .......................................................................................................................... 167
1. INTRODUZIONE ............................................................................................................ 168
1.1 Il Trapianto di Cellule Staminali Emopoietiche (TCSE) ............................................ 168
1.1.1 Tipologie di TCSE e principi relativi all’impiego del TCSE in pazienti affetti da
neoplasie ematologiche ........................................................................................................ 168
1.1.2 Scelta della fonte di CSE ............................................................................................. 171
1.1.3 Il Condizionamento pre-TSCE .................................................................................... 175
1.1.4 Attecchimento e ricostituzione del sistema emopoietico post-TCSE........................... 175
1.1.5 Alloreattività post-TCSE: il rigetto, la GvHD e la GvL.............................................. 181
1.2. Le cellule T regolatorie “naturali” ............................................................................... 181
4
1.2.1 Ontogenesi e caratterizzazione delle cellule T regolatorie “naturali” ...................... 181
1.2.2 Peculiarità intrinseche delle cellule T regolatorie “naturali” in vitro e in vivo ........ 188
1.2.3 Meccanismi di soppressione della risposta immune mediati dalle cellule T regolatorie
“naturali” ............................................................................................................................. 190
1.2.4 Ruolo delle cellule T regolatorie “naturali” nel TCSE .............................................. 196
1.3 Scopo del progetto di ricerca ......................................................................................... 200
2. MATERIALI E METODI ............................................................................................... 201
2.1 Pazienti e Controlli ......................................................................................................... 201
2.2 Purificazione dei linfociti T CD4+CD25+ ...................................................................... 201
2.3 Espansione dei linfociti T CD4+CD25+ isolati .............................................................. 202
2.4 Analisi Immunofenotipica delle cellule T CD4+CD25+ espanse .................................. 202
2.5 Valutazione delle capacità soppressorie delle cellule T CD4+CD25+ espanse ........... 203
2.6 Valutazione della produzione di IL-10 da parte delle cellule T CD4+CD25+
espanse ................................................................................................................................... 203
3. RISULTATI ...................................................................................................................... 205
3.1 Capacità di espansione e caratteristiche immunofenotipiche delle cellule T
CD4+CD25+ isolate ................................................................................................................ 205
3.2 Capacità soppressoria delle cellule T regolatorie CD4+CD25+ espanse ..................... 209
3.3 Produzione di citochine immunosoppressorie da parte delle cellule T regolatorie
CD4+CD25+ espanse ............................................................................................................. 212
4. DISCUSSIONE ................................................................................................................. 214
5. REFERENZE .................................................................................................................... 222
5
PREFAZIONE
La Leucemia Linfatica Cronica (LLC)
La Leucemia Linfatica Cronica (LLC) è un disordine ematologico, di natura monoclonale,
caratterizzato dall‟espansione e accumulo di piccoli linfociti B nel sangue periferico, nel
midollo osseo e negli organi linfatici secondari, quali linfonodi e milza [1]. E‟ la forma di
leucemia di più frequente riscontro nell‟emisfero occidentale [2]. Viene tipicamente
considerata una malattia dell‟adulto, in quanto la maggior parte dei pazienti ha più di 55 anni
e l‟età media alla diagnosi è intorno ai 65 anni, mentre solo il 15% dei pazienti ha un‟età
inferiore a 50 anni. E‟, inoltre, più frequente tra gli individui di sesso maschile essendo il
rapporto maschio/femmina affetti di 2:1. È una delle poche neoplasie ematologiche a non
essere stata mai associata all‟esposizione a radiazioni ionizzanti o ad altri fattori eziologici
ambientali, occupazionali, infettivi [3-4]. Una predisposizione genetica appare invece
rilevante nella patogenesi della LLC, come sembra indicare l‟evidenza epidemiologica
secondo cui nel 5-10% dei casi esiste una suscettibilità alla LLC di tipo familiare, per cui si
ammalano due o più componenti di una stessa famiglia [5].
Nella maggior parte dei casi la LLC è asintomatica, mentre nei pazienti sintomatici si presenta
principalmente
attraverso
l‟aumento
delle
dimensioni
delle
ghiandole
linfatiche
(linfoadenomegalia), della milza (splenomegalia) e del fegato (epatomegalia) [6]. La diagnosi
di LLC è un processo a più stadi che richiede: un‟analisi emocromocitometrica, morfologica,
immunofenotipica, citogenetica e molecolare. L‟esame dell‟emocromo normalmente rivela la
presenza nel sangue periferico di una linfocitosi, vale a dire un numero di linfociti superiore a
5000/ l ed è utile per diagnosticare la malattia nel caso di pazienti asintomatici. La
valutazione dell‟aspirato midollare e della biopsia osteomidollare non sono generalmente
richiesti per la diagnosi. Tuttavia possono essere utili, qualora la conta linfocitaria risulti
inferiore a 5000/ l, per una conferma della diagnosi di LLC e per una valutazione diagnostica
differenziale nei confronti di altri disordini linfoproliferativi [7]. Secondo quanto riportato dal
French-American-British (FAB) Cooperative Group è possibile distinguere due principali
varianti morfologiche di LLC: la forma tipica e atipica. Nella forma tipica, l‟analisi
morfologica di uno striscio di sangue periferico mostra un prevalente accumulo di piccoli
linfociti maturi con scarso citoplasma e cromatina nucleare parzialmente aggregata [8].
Quadri citomorfologici rappresentati da più del 10% di linfociti con aspetto diverso dal
piccolo linfocita, definiscono invece la forma atipica della malattia, frequentemente associata
ad un decorso clinico più aggressivo e una prognosi più sfavorevole.
6
Da un punto di vista immunofenotipico la cellula B di LLC esprime sulla sua superficie
antigeni di membrana propri della linea differenziativa B come il CD19, il CD20 e il CD23.
Inoltre, i linfociti B leucemici esprimono caratteristicamente sulla loro membrana un antigene
comunemente espresso dai linfociti T, il CD5 [9]. Un altro aspetto importante che
contraddistingue le cellule neoplastiche è la bassa densità di espressione delle
Immunoglobuline (Ig) di superficie, componenti del recettore della cellula B (BCR), e la
restrizione kappa o lambda delle catene leggere delle Ig, indice della clonalità della
popolazione neoplastica. L‟analisi immunofenotipica mostra anche negatività o debole
espressione degli antigeni CD22, FMC7 e CD79b [10].
Lo studio citogenetico e l‟analisi molecolare hanno essenzialmente una finalità prognostica
[11]. Attraverso “fluorescence in situ hybridization” (FISH) è possibile determinare anomalie
del cariotipo in più dell‟80% dei casi di LLC, anche se nessuna di queste anomalie può essere
considerata specifica della patologia. A differenza degli altri disordini linfoproliferativi, le
traslocazioni sono eventi meno frequenti; al contrario, prevalgono le aberrazioni
cromosomiche caratterizzate da acquisto o perdita di materiale genico. Le principali sono: la
delezione del braccio lungo del cromosoma 13 (del13q14, in più del 50% dei casi), la trisomia
del cromosoma 12 (nel 10-20% dei casi), la delezione del braccio lungo del cromosoma 11
(del11q23, nel 10-20% dei casi) e la delezione del braccio corto del cromosoma 17 (del17p13,
nel 5-10% dei casi) [12]. Inoltre le cellule di LLC possono presentare, al momento della
diagnosi, anche alterazioni di natura genica, sebbene si tratti di un evento raro. Tra queste le
più consuete sono rappresentate da mutazioni puntiformi e microdelezioni dei geni TP53 e
ATM (Ataxia Telangiectasia Mutated) che mappano rispettivamente sui cromosomi 17 e 11.
A prescindere dalla presentazione clinica iniziale, l‟evoluzione della LLC è molto eterogenea;
la sopravvivenza varia da mesi a decine di anni, con una mediana di circa 7,5 anni. Diventa
quindi di fondamentale importanza essere a conoscenza già alla diagnosi di fattori prognostici
che possano predire l‟evoluzione nel tempo della malattia. I fattori prognostici tradizionali, di
natura clinica, sono rappresentati dalla stadiazione secondo Rai o Binet; dal tempo di
raddoppiamento della conta linfocitaria (LDT) e dal dosaggio di una serie di parametri
sierologici quali: la beta2microglobulina (β2M), la timidina chinasi (TK), la lattico
deidrogenasi (LDH) e l‟antigene CD23 in forma solubile (sCD23). I livelli di TK correlano
con la massa tumorale e l‟attività proliferativa delle cellule di LLC e predicono la
progressione di malattia; livelli elevati di β2M e sCD23 sono direttamente correlati a una
prognosi negativa [13]. I suddetti criteri però, nonostante siano utili a definire l‟estensione
della malattia e a determinarne la progressione durante il follow-up clinico, non risultano
7
sufficienti a predire l‟evoluzione della malattia nella fase iniziale di diagnosi. Al fine di
stabilire meglio la prognosi, sono stati quindi identificati nuovi parametri prognostici di
natura biologica, indipendenti da quelli clinici convenzionali. Attualmente lo “stato
mutazionale” ovvero la frequenza delle mutazioni nei geni che codificano per la regione
variabile delle catene pesanti delle Immunoglobuline (IgHV) è considerato il parametro più
affidabile per definire la prognosi dei pazienti di LLC, data la sua stabilità durante il corso
della malattia [14]. In relazione allo stato mutazionale della regione IgHV sono stati
caratterizzati 2 gruppi di pazienti: uno a configurazione IgHV non mutata e uno a
configurazione IgHV mutata, ciascuno caratterizzato da un andamento clinico diverso [1517]. Recentemente, è stata inoltre segnalata l‟importanza prognostica dell‟espressione di due
proteine, quali il CD38, glicoproteina di membrana mediatrice dell‟interazione cellula-cellula,
e ZAP-70, proteina tirosin-chinasica comunemente espressa dalle cellule T e cellule NK, ma
non dai linfociti B normali circolanti. Pazienti che mostrano, allo studio immunofenotipico,
una positività per entrambe le molecole, considerando un cut-off del 30% per il CD38 e del
20% per ZAP-70, appaiono avere una prognosi meno favorevole [18-21]. Entrambe le
molecole sembrerebbero agire potenziando il segnale del BCR nelle cellule B di LLC. Il
CD38, quando legato al suo ligando, il CD31, induce una cascata del segnale che porta la
cellula B di LLC a proliferare e ad incrementare la propria sopravvivenza [22-24].
L‟espressione di ZAP-70 è invece associata non solo ad un aumento del segnale attraverso il
complesso del BCR ma esercita anche un ruolo nel modulare la lunghezza di tale segnale. Le
cellule ZAP-70 positive (ZAP-70+) sembrerebbero più responsive rispetto alle cellule ZAP-70
negative (ZAP-70-), perché in grado di ricevere maggiori stimoli proliferativi e segnali di
sopravvivenza [25-26]. Tali eventi potrebbero contribuire ad una progressione clinica rapida
verso gli stadi più avanzati della malattia con tempi di sopravvivenza minori.
La LLC rimane ancora oggi una malattia difficilmente curabile. Nella decisione terapeutica
assume sempre più importanza la presenza o assenza di alcuni fattori di natura biologica come
lo stato mutazionale dei geni delle Immunoglobuline, l‟espressione di ZAP-70, del CD38 e la
presenza di specifiche alterazioni citogenetiche [27]. Per diversi decenni le terapie a base di
agenti alchilanti hanno costituito il trattamento standard di prima linea nei pazienti affetti da
LLC, come agenti singoli o in combinazione. Dalla metà degli anni „80 si è passati all‟utilizzo
di analoghi delle purine (es: Fludarabina) da soli o in combinazione con anticorpi monoclonali
quali l‟Alemtuzumab, una proteina diretta contro l‟antigene CD52, e il Rituximab, un
anticorpo monoclonale diretto contro l‟antigene CD20 [28].
8
I progressi compiuti negli ultimi anni nell‟ambito della ricerca scientifica hanno permesso di
modificare la gestione terapeutica della LLC. La tendenza attuale va verso una terapia
biologicamente-orientata, in cui la scelta del momento in cui iniziare la terapia e la strategia
terapeutica sono basati sull'integrazione delle caratteristiche cliniche e biologiche della
malattia. Gli studi clinici in corso sono, infatti, volti proprio alla stratificazione dei pazienti e
quindi alla valutazione dell'efficacia di una strategia terapeutica basata sulle caratteristiche
individuali di rischio.
9
PARTE I
Studio delle conseguenze funzionali della
stimolazione in vitro delle cellule B neoplastiche di
pazienti affetti da Leucemia Linfatica Cronica
(LLC)
10
1. INTRODUZIONE
1.1 Il complesso recettoriale per l’antigene dei linfociti B (BCR)
1.1.1 Composizione e ruolo del BCR nel processo di ontogenesi B cellulare
Il BCR è un complesso multimerico prodotto dall‟assemblaggio non covalente di una
molecola immunoglobulinica di membrana (sIg), coinvolta nel riconoscimento antigenico, e
un eterodimero Igα/Igβ (CD79a/CD79b), la cui funzione è di avviare la cascata di trasduzione
del segnale in seguito alla stimolazione antigenica [Figura 1] [29]. Ciascuna cellula B matura
esprime sulla sua superficie un recettore unico, selezionato da un ampio repertorio. La
diversità del repertorio B recettoriale è il risultato di un processo di riarrangiamento, regolato
dal prodotto dei geni attivanti la ricombinazione RAG1 e RAG2, a carico di multipli segmenti
genici presenti all‟interno dei loci delle Immunoglobuline. Nell'uomo, tali loci sono distinti in
un locus codificante per tutti i differenti isotipi di catena pesante (locus H) e due loci
codificanti per le catene leggere (loci κ e λ), ciascuno situato su un cromosoma diverso. Il
locus della catena pesante consiste dei tratti VH, D, JH e della porzione costante C. Quello
della catena leggera è organizzato in modo simile ma manca del tratto D [2]. Il
riarrangiamento dei suddetti segmenti genici si verifica durante il processo di ontogenesi B
cellulare all‟interno del midollo osseo e comporta la sintesi di una cellula B matura attraverso
il differenziamento del progenitore staminale ematopoietico pluripotente, prima in cellula proB quindi pre-B. Una volta completato il processo di riarrangiamento del locus H, il gene
riarrangiato viene trascritto e il relativo RNA messaggero (mRNA) primario va incontro ad un
processo di splicing alternativo, consentito grazie alla presenza di sequenze ripetute di switch
(S) situate a monte di ciascun esone, in seguito al quale il segmento VHDJH è collegato al
primo
dominio
della
regione
C,
che
determina
l‟isotipo
anticorpale.
Esistono
complessivamente 5 isotipi anticorpali, M, G, A, D ed E, codificati ciascuno da uno dei
rispettivi domini della regione CH, quali μ, γ, α, δ e ε [30]. Il completamento della transizione
da cellula pro-B a pre-B richiede, a questo punto, che l‟mRNA maturo neosintetizzato sia
tradotto nella catena pesante delle Immunoglobuline di cui viene, in un primo momento,
verificata l‟idoneità in base alla capacità di associarsi, all‟interno del reticolo endoplasmatico
(RE), ad un surrogato della catena leggera (SCL) composto da due polipeptidi omologhi ai
domini VL e CL, denominati rispettivamente VpreB e λ5. L‟eterodimero Igα/Igβ rappresenta
quindi la componente finale nell‟assemblaggio del recettore della cellula pre-B (pre-BCR),
consentendone inoltre il trasporto attraverso l‟apparato di Golgi verso la superficie cellulare
[31].
11
Figura 1. Struttura del complesso recettoriale per l’antigene dei linfociti B (BCR). Il BCR è formato
dall‟assemblaggio di Immunoglobuline di membrana (sIg) e l‟eterodimero Igα/Igβ provvisto, nella coda
intracitoplasmatica, di alcuni motivi ricchi in residui di tirosina chiamati domini ITAM, indispensabili per la
trasduzione del segnale.
Lo stadio pre-B costituisce la fase più precoce dello sviluppo B linfoide durante cui è richiesta
la segnalazione attraverso il BCR [32]. Mutazioni target a carico dei geni codificanti i
componenti del pre-BCR comportano, infatti, l‟arresto del processo di differenziamento
durante la transizione pre-B/pro-B. Si ritiene che la segnalazione attraverso il pre-BCR
permetta il verificarsi del fenomeno dell‟esclusione allelica, un meccanismo grazie al quale il
linfocita esprime i geni riarrangiati della catena pesante e leggera appartenenti a uno solo dei
due cromosomi omologhi, preservando in questo modo la specificità della risposta
immunitaria. Conseguentemente alla formazione del pre-BCR la cellula blocca, inoltre, la
sintesi delle proteine implicate nella ricombinazione somatica e le proteine surrogato della
catena leggera e, dopo una serie di cicli proliferativi, perde la sua responsività all‟IL-7 [33].
Non è chiaro quale sia il meccanismo alla base della trasduzione dei primi segnali attraverso il
pre-BCR, sebbene sia stato supposto che lo stesso assemblaggio di questa forma recettoriale
12
sarebbe sufficiente a tale scopo. In realtà, probabilmente, la presenza stessa del pre-BCR sulla
superficie cellulare potrebbe favorire dei processi di aggregazione recettoriale spontanei,
come accade nel caso del BCR maturo, o mediati dalla localizzazione in microdomini ricchi
in colesterolo e sfingolipidi noti come “zattere lipidiche”. Alcuni autori hanno invece
postulato l‟esistenza di un ligando midollare stromale, come ad esempio la galectina,
potenzialmente coinvolto nel mediare l‟aggregazione dei recettori della cellula pre-B, sebbene
il contributo di tale ligando rimanga tuttora controverso [34]. Ad ogni modo, il pre-BCR è
espresso solo transientemente sulla superficie della cellula pre-B, e la sua downregolazione
richiede il riarrangiamento dei segmenti genici VL e JL del locus della catena leggera delle
Immunoglobuline, evento che si verifica, attraverso un meccanismo simile a quello impiegato
per la produzione della catena pesante, durante la transizione del linfocita B dallo stadio pre-B
a quello di cellula B immatura. Tale riarrangiamento viene in realtà indotto grazie ad un
meccanismo di feedback negativo, mediato dallo stesso pre-BCR, basato sull‟inibizione della
sintesi delle proteine surrogato della catena leggera, che impedisce l‟ulteriore espressione del
recettore della cellula pre-B. Una volta sintetizzata, la catena leggera si sostituisce al
surrogato nell‟appaiamento con la catena pesante e, a questo punto, viene espresso sulla
superficie del linfocita B immaturo, un complesso recettoriale maturo antigene-specifico
unico [35]. Tale unicità è, in primo luogo, frutto del vasto numero di eventi combinatoriali tra
i segmenti genici V, D e J di ciascuna catena immunoglobulinica, che rappresentano il primo
stadio nella generazione della diversità anticorpale caratteristica del sistema immune. In
secondo luogo, a garantire ulteriormente l‟unicità del BCR, il processo di diversificazione
giunzionale, contraddistinto da eventi di aggiunta o rimozione casuale di sequenze
nucleotidiche in corrispondenza dei punti di giunzione dei tratti V, D e J. La generazione della
diversità del repertorio B recettoriale risulta indispensabile per la difesa dell‟organismo dalla
molteplicità di agenti patogeni cui è continuamente esposto. Di conseguenza è stato calcolato
che, considerando unicamente il numero di ciascuno dei segmenti genici H e L, vi sarebbero
potenzialmente oltre 1.6 milioni di combinazioni possibili, una cifra che rende
comprensibilmente insignificante la probabilità di osservare due complessi B recettoriali
identici, non solo nello stesso soggetto ma anche tra soggetti diversi.
Questo presupposto diventa interessante quando si esaminano le caratteristiche del BCR delle
cellule neoplastiche di pazienti affetti da LLC, il cui studio, come vedremo, ha condotto
all‟ipotesi della stimolazione antigenica come meccanismo patogenetico.
13
1.2 Ruolo del BCR nella patogenesi della LLC
1.2.1 Caratteristiche strutturali del BCR delle cellule B di LLC
La LLC è stata a lungo considerata una patologia derivante dalla trasformazione neoplastica
di linfociti B immaturi, immunologicamente incompetenti e scarsamente proliferanti,
caratterizzati da resistenza all‟apoptosi e conseguente tendenza ad accumularsi nel paziente. I
progressi nello studio dei geni IgV, del loro assemblaggio e delle modifiche che subiscono
durante il differenziamento e la stimolazione antigenica B cellulare hanno permesso di
analizzare il livello di competenza, esperienza e stato di maturazione delle cellule B,
rivelandosi utili a migliorare la comprensione della LLC e a ridefinire il concetto della sua
patogenesi. Diversamente dal passato, oggi la LLC viene, infatti, considerata una neoplasia
derivante dall‟espansione clonale di cellule B mature, antigenicamente esperte, la cui
tendenza ad accumularsi rappresenta, in realtà, il risultato di una condizione dinamica
caratterizzata da uno squilibrio tra morte e proliferazione [36].
Le prime evidenze ad aver favorito un avanzamento nella comprensione della patogenesi della
LLC sono derivate dallo studio delle caratteristiche dei geni IgHV e LV delle cellule B
neoplastiche. Tali studi sono stati incentivati dall‟osservazione dell‟eterogeneità del decorso
clinico dei pazienti affetti da LLC, una peculiarità caratteristica di questa malattia nonostante
la presenza di un profilo morfologico, immunofenotipico e genomico pressoché omogeneo, ed
hanno evidenziato la presenza di una correlazione tra la suddetta variabilità e le caratteristiche
strutturali e funzionali del BCR delle cellule B patologiche. In primo luogo, il
sequenziamento genico ha consentito di identificare lo stato mutazionale del locus IgHV
come il parametro più affidabile per definire la prognosi dei pazienti di LLC. I pazienti
caratterizzati da una configurazione IgHV non mutata sono, infatti, associati a un andamento
aggressivo della malattia e, quindi, a una prognosi più sfavorevole; mentre i pazienti
caratterizzati da una configurazione IgHV mutata presentano solitamente decorso indolente,
lenta progressione della malattia e lungo tempo di sopravvivenza. Questo dato ha fornito le
basi per sostenere l‟ipotesi che le due sottoclassi prognostiche potessero differire
primariamente in relazione alla reattività del BCR dei linfociti B neoplastici; ipotesi
successivamente avvalorata dagli studi relativi alle caratteristiche del repertorio di geni IgHV
e LV impiegato da tali cellule [37]. All‟interno del locus IgH sono presenti 51 geni HV
funzionali, raggruppati in 7 famiglie sulla base di una omologia di sequenza pari almeno
all‟80%. Nella LLC, un impiego non casuale dei componenti delle suddette famiglie geniche,
è stato innanzitutto riportato in seguito all‟osservazione di un riscontro più frequente di alcuni
14
geni, di quanto atteso probabilisticamente. Le cellule B neoplastiche utilizzano, infatti,
preferenzialmente i geni delle famiglie IgHV1, HV3 e HV4 secondo una distribuzione
differente rispetto alla controparte B normale, rappresentata convenzionalmente dalle cellule
B CD5+, sebbene, considerando la possibilità che il CD5 venga acquisito dopo la stimolazione
antigenica, qualsiasi popolazione B linfoide potrebbe costituire il precursore della LLC. Le
cellule B neoplastiche mostrano, inoltre, anche all‟interno di queste famiglie, una preferenza
per alcuni geni quali IgHV1-69, HV3-07, HV3-21, HV3-23 e HV4-34 [38]. Di questi, il gene
IgHV1-69, come dimostrato da una serie di studi condotti a partire dalla fine degli anni ‟80,
risulta nel complesso il gene IgHV maggiormente impiegato dalle cellule B di LLC; a seguire
il gene IgHV3-21. E‟ possibile che le percentuali relative all‟uso ristretto di specifici geni
IgHV subiscano leggere variazioni all‟interno delle diverse coorti di pazienti esaminate, ad
ogni modo, questo comportamento può essere spiegato considerando l‟impatto della
distribuzione geografica e, quindi, il possibile effetto di un agente ambientale di origine
regionale nella selezione del clone leucemico dotato di una peculiare specificità recettoriale.
Nei pazienti affetti da LLC è stata anche rilevata un‟associazione specifica di alcuni tratti HV
al riarrangiamento preferenziale di determinati tratti HJ e HD. Ad esempio l‟impiego del gene
IgHV1-69 è risultato caratteristicamente associato a quello dei segmenti genici J6 e D3-3 o
D2-2. Analogamente la maggior parte dei casi esprimenti il gene IgHV3-07 utilizza i
segmenti genici delle famiglie J4 (90% dei casi) e D3 [39]. Sorprendentemente, anche la
presenza o assenza di ipermutazioni somatiche appare associata all‟uso di specifici e ricorrenti
geni IgHV e segue una precisa gerarchia basata sia sulla famiglia che sul singolo segmento
genico selezionato. Sono, infatti, maggiormente inclini all‟acquisto di mutazioni somatiche i
geni della famiglia IgHV3, a seguire IgHV4 e IgHV1. Inoltre, all‟interno di tali famiglie, i
geni IgHV4-34 sono quelli preferibilmente espressi nelle forme mutate di LLC, mentre i geni
IgHV1-69 e HV4-39 nelle forme non mutate [40]. Fa eccezione il gene IgHV3-21, distribuito
più equamente tra riarrangiamenti mutati e non mutati, e caratterizzato dalla peculiarità di
essere associato a cattiva prognosi indipendentemente dallo stato mutazionale [41]. Le
mutazioni osservate all‟interno dei tratti genici HV sono di natura missenso, cambiano quindi
la composizione aminoacidica della molecola immunoglobulinica, e manifestano una
localizzazione preferenziale all‟interno di sei domini noti con il termine di “Regioni
Determinati Complementarità” (CDR). Le regioni CDR, tre in ciascun riarrangiamento
immunoglobulinico, sono regioni ipervariabili coinvolte nella formazione del sito di
riconoscimento antigenico del BCR. La più variabile delle suddette regioni è la CDR3 della
catena pesante (HCDR3) che si estende a livello del sito di giunzione dei segmenti genici VDJ
15
e, di conseguenza, rappresenta il principale determinante della specificità del sito di legame
per l‟antigene [42]. A tale proposito è importante sottolineare come anche le regioni HCDR3
mostrino delle caratteristiche peculiari nei pazienti affetti da LLC. E‟ stato osservato che
l‟impiego di specifici geni IGHV condiziona sia la lunghezza sia la composizione
aminoacidica all‟interno della regione HCDR3. Ad esempio, i casi caratterizzati dal
riarrangiamento HV1-69 presentano una regione HCDR3 molto lunga; al contrario, i casi
caratterizzati dal riarrangiamento HV3-07 hanno una regione HCDR3 piuttosto corta. Vi sono
poi tratti genici associati a molteplici tipologie di regioni HCDR3. In base alla lunghezza di
tali regioni, i pazienti IgHV4-34+ segregano, ad esempio, in 2 sottogruppi: i casi HV4-34+ non
mutati hanno HCDR3 più lunghi, contenenti solitamente i segmenti J6 o J5, mentre quelli
HV4-34+ mutati hanno HCDR3 più corti in associazione al segmento J4 [43]. Ad ogni modo,
in tutti i casi la lunghezza delle regioni HCDR3 appare significativamente differente nelle
cellule B neoplastiche di LLC rispetto alle cellule B normali. Una caratteristica peculiare delle
cellule di LLC che utilizzano la famiglia genica IgHV1 è inoltre la presenza di code
tirosiniche a livello della regione 3‟, codificate parzialmente dal segmento genico J6. Infine,
la carica del segmento HCDR3 definita in termini di punto isoelettrico (pI), rappresenta
un‟altra distinzione che caratterizza le cellule di LLC relativamente all‟uso dei geni IgHV. Le
cellule leucemiche che esprimono i geni della famiglia IgHV1 hanno, ad esempio, il pI
minore mentre le cellule IgHV3+ hanno il pI molto più alto. I casi IgHV4+ mostrano valori
intermedi. Complessivamente, quindi, la lunghezza, la composizione aminoacidica e la carica
della regione HCDR3 delle cellule leucemiche tendono a variare dipendentemente dal gene
IgHV espresso [44].
Al di là delle differenze nelle regioni HCDR3 proprie delle cellule di LLC rispetto alla
controparte B normale, è di grande interesse l‟osservazione che cloni B leucemici
appartenenti a differenti pazienti, possano presentare un utilizzo di regioni HCDR3 molto
simili tra loro. Tale considerazione ha condotto al concetto del cosiddetto “recettore
stereotipato”. La somiglianza nella composizione del BCR rilevata in pazienti affetti da LLC
appartenenti anche ad aree geograficamente lontane, costituisce una peculiarità unica di
questa malattia, con un tasso medio del 20% circa. Si ritiene comunque che tale percentuale
sia sottostimata in quanto, in molti casi, l‟ipermutazione somatica comporta la sostituzione di
un aminoacido presente nella regione HCDR3 del recettore con un altro, dotato di
caratteristiche fisiche e chimiche analoghe, che conseguentemente dà origine a un sito di
legame antigenico molto simile in termini di specificità. Questa osservazione supporterebbe
l‟ipotesi relativa all‟esistenza di un insieme di antigeni o classi di epitopi strutturalmente
16
simili con un ruolo primario nella selezione del clone B leucemico [45]. L‟espressione di
BCR stereotipati è più frequente in pazienti di LLC con geni IgHV non mutati, dove può
raggiungere valori intorno al 40%, rispetto ai casi IgHV mutati, dove si attesta a valori pari al
10% [46]. Inoltre, i pazienti con stato IgHV non mutato contraddistinti da BCR stereotipati
presentano un pattern distintivo di mutazioni somatiche rispetto ai pazienti con
riarrangiamenti casuali del BCR. Alcuni geni IgHV sono poi maggiormente associati a
regioni HCDR3 stereotipate. E‟ il caso dei geni IgHV3-21; HV1-69, HV1-2, HV1-3 e HV439; al contrario i geni IgHV3-7, HV3-74 e HV2-5 hanno una bassa probabilità di mostrare
HCDR3 stereotipati. In alcuni casi, l‟assenza di HCDR3 omologhi si correla alla comparsa di
omologie strutturali a livello delle cosiddette “regioni cornice” (FWR). Le regioni FWR
separano le regioni CDR e sono altamente conservate tra i vari membri delle famiglie geniche.
In passato si riteneva che la loro funzione fosse il mantenimento della struttura terziaria delle
Immunoglobuline, ma negli ultimi anni sono state identificate alcune proteine di natura
microbica ed endogena in grado di interagire direttamente, in qualità di superantigeni, con tali
regioni, rivelando l‟esistenza di ipotetici siti di legame ad esse associati. Di conseguenza, la
bassa frequenza di HCDR3 stereotipati potrebbe indicare una forma di riconoscimento
antigenico, da parte del BCR, di natura non convenzionale, ovvero al di fuori dei siti
caratteristici del legame classico, rappresentati appunto dalle regioni HCDR3 [47]. La
presenza di un‟omologia strutturale a carico del BCR influenza solitamente in modo negativo
l‟andamento prognostico della malattia, associandosi a caratteristiche fenotipiche, molecolari
e cliniche aggressive. Il primo studio finalizzato alla valutazione dell‟impatto clinico della
stereotipia risale al 2004 e mostra in una serie di 5 pazienti con composizione similare dei
rispettivi BCR, un decorso aggressivo della malattia, complicato da ricorrenti infezioni,
trasformazione in sindrome di Richter e formazione di tumori solidi [48]. In seguito, è stato
anche dimostrato come l‟influenza prognostica della stereotipia possa avere importanti
implicazioni prognostiche addizionali e/o indipendenti dallo stato mutazionale. E‟ il caso dei
pazienti IgHV3-21+ [45].
Diversamente dalle catene pesanti, meno note sono le peculiarità del repertorio dei geni LV e
LJ caratteristico delle cellule B di LLC. Sembrerebbe che tale repertorio non differisca
sostanzialmente da quello impiegato dalla controparte B normale, sebbene alcune differenze
nei riarrangiamenti IgLV-J siano state osservate, maggiormente a carico dei geni del locus k.
Ad ogni modo, i dati disponibili indicano, nel complesso, non solo un impiego peculiare di
specifici tratti IgLV nella LLC, ma anche la presenza di un appaiamento non stocastico delle
catene leggere alle catene pesanti del BCR, a sostegno del ruolo complementare delle catene
17
leggere nel riconoscimento antigenico del recettore clonotipico. Per quanto riguarda l‟uso di
un ristretto repertorio di geni IgLV nella LLC, è stato osservato, in un recente studio
effettuato su un totale di 276 pazienti, l‟impiego preferenziale nei casi k+ dei geni IgkV3-20,
IgkV1-39, IgkV1-5, IgkV4-1 e IgkV2-30 e nei casi λ+ dei geni IgλV3-21, IgλV2-8 e IgλV214. La restrizione del repertorio IgLV è stata primariamente osservata nei casi di BCR
stereotipati, dove anche i motivi LCDR3 appaiono omologhi laddove viene impiegato lo
stesso gene k o λ. La presenza di mutazioni somatiche è risultata invece caratteristica del
50.3% dei pazienti k+ e del 45.4% dei pazienti λ+, con l‟osservazione che, similmente a quello
che accade nel caso delle catene pesanti, l‟accumulo delle mutazioni avviene in modo
differenziale all‟interno delle famiglie e dei segmenti genici e che alcuni cambiamenti
aminoacidici sono preferiti e quindi ricorrono rispetto ad altri [49]. In questo studio, come in
una serie di studi paralleli, è stata inoltre rivelata un‟associazione specifica tra domini IgkVJ/IgλV-J e domini IgHV-D-J impiegati all‟interno delle cellule B neoplastiche. L‟utilizzo del
gene IgkV1-39 è risultato ad esempio frequente nei casi IgHV1-69+, mentre quello dei geni
Igk2-30 e Igλ3-21 è apparso rispettivamente in associazione ai riarrangiamenti IgHV4-34 e
IgHV3-21 [40]. Recentemente il concetto dell‟implicazione delle catene leggere nel
riconoscimento antigenico delle cellule B di LLC, è stato anche rafforzato grazie alla
considerazione che una proporzione di pazienti con espressione monotipica del locus IgL
risulta caratterizzata da riarrangiamenti IgL funzionali multipli, alludendo alla possibilità che
riarrangiamenti secondari del locus IgL possano verificarsi nel contesto del processo di
editing recettoriale indotto dall‟esposizione ad un (auto)antigene [50]. In conformità a tali
dati, è possibile sostenere l‟ipotesi secondo cui la reattività nei confronti dell‟antigene
coinvolto nella selezione del clone B patologico non dipenda esclusivamente dalle
caratteristiche delle componenti IgHV e HCDR3, ma dalla struttura complessiva del BCR
stereotipato. Di conseguenza, la formazione di un distintivo sito di riconoscimento antigenico
richiederebbe sia il contributo delle catene pesanti che di quelle leggere e il riarrangiamento
dei loci IgH e IgL potrebbe riflettere l‟ancestore della LLC e la storia dell‟esposizione
antigenica di ciascun paziente.
Accanto alle peculiarità dei riarrangiamenti a carico dei loci IgH e IgL, sono spesso osservate
nelle cellule B di LLC, alterazioni strutturali della componente immunoglobulinica del
recettore così come delle componenti Igα/Igβ, coinvolte nella trasduzione del segnale. Queste
alterazioni sono in parte responsabili della bassa espressione superficiale del BCR, sebbene il
meccanismo attraverso cui vengano indotte non sia stato ancora chiarito [51]. E‟ stato
ipotizzato che alla base vi sia un‟alterazione dei processi di folding dei singoli elementi che
18
costituiscono il complesso recettoriale. Recentemente è stato, infatti, dimostrato che la catena
pesante μ delle IgM di superficie e la catena Igα sono trattenute nel reticolo endoplasmatico
delle cellule B di LLC. Questo fenomeno è associato a un difetto nella glicosilazione di
entrambe le molecole e determina una loro ridotta traslocazione sulla membrana cellulare, cui
consegue una diminuzione della densità di superficie del BCR [52]. In molti casi di LLC è
stata anche identificata una variante tronca della catena Igβ, prodotta come conseguenza della
delezione dell‟esone 3, codificante per il dominio extracellulare di tale molecola [53]. Questo
dato suggerisce come, oltre al processo di folding proteico, anche il meccanismo di splicing
alternativo possa essere implicato nel causare ridotti livelli di espressione del BCR sulla
superficie delle cellule neoplastiche, una caratteristica solitamente presente in cellule B
linfoidi anergizzate, che non necessariamente si associa ad una difettiva o assente
segnalazione attraverso il BCR.
Il BCR espresso dalle cellule B di LLC presenta, quindi, una serie di caratteristiche strutturali
peculiari. L‟insieme di tali caratteristiche supporta la tesi del ruolo fondamentale
dell‟interazione con l‟antigene nella patogenesi della leucemia e nella selezione del clone
leucemico. Conseguentemente, la comprensione del loro impatto sulla funzionalità del BCR
diventa di grande importanza per la comprensione della modalità mediante la quale il BCR
possa influenzare il destino della popolazione B neoplastica.
1.2.2 Stato maturativo e d’attivazione delle cellule B di LLC
Le osservazioni relative alle caratteristiche del BCR delle cellule B leucemiche hanno
condotto ad avvalorare la tesi secondo la quale la stimolazione antigenica abbia un ruolo
fondamentale nella patogenesi della LLC. In realtà, considerando che il processo di
diversificazione del repertorio immunoglobulinico richiede la stimolazione del BCR mediata
dalla presenza dell‟antigene, si potrebbe erroneamente dedurre che i casi di LLC caratterizzati
da una configurazione IgHV mutata derivino da cellule antigenicamente esperte e, al
contrario, che i casi di LLC con configurazione IgHV non mutata originino da cellule B naive.
Questa apparente contraddizione può essere risolta tenendo presente che l‟assenza di
mutazioni somatiche a carico delle regioni variabili delle catene pesanti delle
Immunoglobuline non necessariamente equivale all‟assenza di una precedente stimolazione
antigenica e che tali casi potrebbero derivare da cellule B che, pur avendo incontrato
l‟antigene, non hanno accumulato mutazioni. A sua volta, il mancato accumulo di mutazioni
potrebbe dipendere dal tipo di stimolazione antigenica che la cellula ha ricevuto o essere il
risultato del momento in cui si è verificato l‟evento trasformante. Sebbene il meccanismo alla
19
base di tale diversificazione non sia tuttora chiaro, oltre agli studi relativi alla struttura del
BCR, anche i dati ottenuti analizzando lo stato maturativo e d‟attivazione delle cellule B
leucemiche, supportano la tesi dell‟origine comune di entrambi i sottotipi di LLC dalla
trasformazione neoplastica di una cellula B esposta alla stimolazione antigenica, definendo la
presenza dell‟antigene il prerequisito per l‟evoluzione clonale della malattia, anche in quei
casi che non esibiscono mutazioni nella regione IgHV [Figura 2].
In primo luogo, diversi studi immunofenotipici hanno evidenziato nelle cellule di LLC, non
solo l‟espressione di una serie marcatori caratteristici di cellule B attivate a seguito della
stimolazione antigenica, ma anche uno stato d‟attivazione più avanzato rispetto a quello di
cellule B normali. Quando confrontate con linfociti B normali CD5+, le cellule B neoplastiche
mostrano, infatti, un‟iperespressione dei marcatori di attivazione CD23, CD25, CD39, CD40,
CD69 e CD71, affiancata ad un‟elevata e omogenea densità di espressione del CD27, un
marcatore tipico delle cellule B memoria. Allo stesso tempo, tali cellule sono caratterizzate da
una riduzione dell‟espressione del CD22, del CD79b, del CD32 (FCγBIIb) e dell‟isotipo D
delle Immunoglobuline, evento consistente con la tesi dell‟attivazione in vivo dei linfociti B
patologici, in quanto le suddette molecole sono normalmente sottoposte ad un controllo
negativo in seguito alla stimolazione antigenica [54]. E‟ stato, infatti, osservato che le cellule
B stimolate attraverso il BCR sono in grado di downmodulare l‟espressione del CD79b,
attraverso la modulazione sia della trascrizione che del processo di splicing dell‟mRNA [55].
Allo stesso modo, il ridotto rapporto tra le IgM e le IgD di superficie, riportato a carico delle
cellule B di LLC, potrebbe essere il risultato di una modulazione indotta dall‟incontro con
l‟antigene, dal momento che le cellule B mature che co-esprimono molecole
immunoglobuliniche di entrambi gli isotipi, downregolano l‟espressione delle sIgD come
conseguenza della segnalazione attraverso il BCR [56]. Questo spiegherebbe il motivo della
ridotta densità superficiale delle Immunoglobuline, caratteristico delle cellule neoplastiche
della maggior parte dei pazienti affetti da LLC [57]. Anche la bassa espressione del CD22 e
del CD32, molecole entrambe coinvolte nella regolazione negativa delle risposte B cellulari
indotte sia attraverso il BCR che il CD38, rafforza il concetto secondo cui le cellule B di LLC
siano in grado di rispondere all‟antigene andando incontro, di conseguenza, alla perdita di
fattori di natura regolatoria [58]. Le suddette caratteristiche fenotipiche sono le stesse sia nei
casi di LLC IgHV mutata che non mutata. I due sottogruppi differiscono unicamente nella
percentuale di cellule neoplastiche positive per l‟espressione degli specifici marcatori di
superficie.
20
Figura 2. La LLC come un disordine della cellula antigenicamente esperta. I due principali sottotipi di LLC
sono distinti in base alla presenza o assenza di mutazioni nella regione IgHV. Poiché lo sviluppo di mutazioni
nella regione IgHV richiede la stimolazione dell‟antigene e quindi l‟attivazione del BCR, molto probabilmente i
casi di LLC mutata derivano da cellule B stimolate che passano attraverso il centro germinativo (CG) e
subiscono il processo di ipermutazione somatica. Per questa stessa ragione i casi di LLC non-mutata dovrebbero
derivare da cellule B naive, ma poiché la mancanza di mutazioni nella regione IgHV non necessariamente
significa assenza di stimolazione antigenica, questi casi potrebbero trarre origine da cellule B stimolate
dall‟antigene che non hanno ricevuto stimoli sufficienti per formare il CG e accumulare mutazioni. Di
conseguenza, in entrambi i casi di LLC la stimolazione antigenica sembra essere un prerequisito essenziale per
l‟evoluzione della leucemia e la continua e prolungata esposizione all‟antigene può fornire stimoli proliferativi,
resistenza all‟apoptosi o anergia, a seconda delle caratteristiche delle cellule B neoplastiche.
Infatti, i casi non mutati presentano sovente, una percentuale maggiore di cellule CD38+,
CD40+ e CD69+, mentre i casi mutati mostrano una maggiore espressione dei marcatori CD39
e CD71. L‟intervallo temporale tra l‟attivazione cellulare e la modulazione di questi marcatori
varia notevolmente all‟interno della popolazione B normale; ad esempio, l‟upregolazione del
CD69 si verifica anticipatamente rispetto a quella del CD71 [59]. Di conseguenza,
considerando la relazione inversa tra l‟espressione del CD69 e del CD71 nell‟ambito dei due
sottogruppi di LLC, si potrebbe ipotizzare che i casi non mutati ricordino delle cellule B
21
temporalmente più prossime allo stimolo antigenico e quindi ad uno stadio più precoce di
attivazione rispetto ai casi mutati.
La presenza di un fenotipo attivato indipendentemente dallo stato mutazionale, consente di
confermare la tesi secondo cui entrambe le forme di LLC abbiano origine da una cellula B
antigenicamente esperta. Sono state proposte diverse ipotesi per spiegare l‟assenza di
mutazioni somatiche a carico dei geni IgHV, caratteristica di una percentuale di pazienti
affetti da LLC. E‟ stato ad esempio supposto che le cellule B neoplastiche possano non aver
accumulato mutazioni come conseguenza della mancata attivazione del macchinario
mutazionale. In alternativa, si potrebbe pensare che tali cellule siano state esposte a un
antigene non in grado di indurre il meccanismo di ipermutazione somatica e/o le reazioni del
centro germinativo, come nel caso di antigeni T-indipendenti, o che siano state attivate da un
antigene T-dipendente, ma trasformate prima dell‟ingresso nel centro germinativo. Ancora,
dopo la trasformazione maligna, si potrebbe essere verificata un‟esclusione dei linfociti
patologici dal centro germinativo stesso o semplicemente le cellule IgHV non mutate
potrebbero aver avuto origine da linfociti specifici per antigeni maggiormente reattivi in
presenza di geni IgHV non mutati [60]. Ciononostante, non è possibile escludere la possibilità
che i dati relativi al fenotipo attivato delle cellule B di LLC siano una conseguenza degli
effetti della trasformazione neoplastica sulla deregolazione dell‟espressione dei geni
codificanti molecole attivatorie. Ad ogni modo, la tesi dell‟attivazione in vivo delle cellule B
di
LLC,
viene
anche
supportata
dall‟osservazione,
all‟interno
di
tali
cellule,
dell‟overespressione della ciclina D2, coinvolta nella progressione del ciclo cellulare; della
traslocazione costitutiva del fattore nucleare delle cellule attivate (NF-ATp); della
fosforilazione dei fattori STAT-1 e STAT-3, mediatori della trasduzione del segnale e
dell‟attivazione trascrizionale a valle del BCR, e della sintesi di numerose citochine
immunomodulatorie tra cui l‟interleuchina (IL)-1α, IL-1β, IL-6, IL-7, IL-8, IL-10, IL-13,
l‟interferone (IFN)-γ, il fattore di necrosi tumorale (TNF), il fattore stimolante la formazione
di colonie di granulociti e macrofagi (GM-CSF) e il fattore di crescita tumorale (TGF)-β1 [6162]. Tali citochine, normalmente prodotte da cellule B attivate, sono non solo sintetizzate ma
anche secrete dalle cellule B neoplastiche. Il loro ruolo nello sviluppo e nella progressione
della LLC non è del tutto chiaro; è stato ipotizzato che alcune citochine come il TNF-α e l‟IL8, possano promuovere la crescita cellulare in modo autocrino, altre in modo paracrino.
La valutazione, all‟interno delle cellule B neoplastiche, della lunghezza dei telomeri fornisce
informazioni addizionali per la comprensione della storia proliferativa della LLC. I telomeri
sono strutture nucleotidiche esameriche ripetute (TTAGGG) presenti all‟estremità di ciascun
22
cromosoma, la cui lunghezza si riduce ad ogni divisione cellulare come conseguenza di un
normale processo di senescenza. Durante la reazione del centro germinativo, le cellule B
normali dimostrano un elevato livello di attività della telomerasi, enzima deputato al processo
di allungamento dei telomeri, che non si osserva in nessun altro tipo cellulare. E‟ stato
dimostrato che le cellule B dei pazienti di LLC sono caratterizzate da telomeri di lunghezza
ridotta rispetto a quelli di cellule B normali isolate da soggetti aventi la stessa età. Questo
dimostra, contrariamente a quanto si pensava decenni fa, non solo che le cellule di LLC si
dividono, ma che lo fanno con frequenza maggiore rispetto alla controparte normale. Inoltre,
quando si confrontano i due sottogruppi di LLC distinti in base allo stato mutazionale dei geni
IGHV, si osserva che le cellule B a configurazione non mutata hanno telomeri più corti
rispetto alle cellule B a configurazione mutata, un dato indicativo di una storia replicativa più
lunga, in linea con la prognosi più sfavorevole di tale gruppo [63]. Ciononostante, il grado di
accorciamento telomerico è lo stesso in entrambi i casi per cui, con molta probabilità, i
maggiori cambiamenti si verificano prima della trasformazione neoplastica. In apparente
contraddizione con quanto riportato, l‟attività enzimatica della telomerasi è più elevata nelle
cellule non mutate, ovvero con telomeri più corti. Considerando che la telomerasi viene
attivata quando i telomeri raggiungono una lunghezza tale da minacciare la sopravvivenza
cellulare, è stato ipotizzato, nel caso della LLC IgHV non mutata, che l‟induzione della
telomerasi sia utile a compensare l‟eccessiva perdita di lunghezza dei telomeri che si verifica
durante la fase preleucemica e leucemica e che risulta proporzionale al numero di divisioni
cellulari [64].
Un‟idea più precisa del tasso proliferativo delle cellule B di LLC e, quindi, del loro turnover
di crescita è stata fornita attraverso una misurazione in vivo basata sull‟impiego del deuterio,
un isotopo non radioattivo dell‟idrogeno. La somministrazione di D2O a soggetti affetti da
LLC, allo scopo di marcare il DNA cellulare, ha consentito di valutare non solo la quota di
cellule di nuova generazione, ma anche la crescita in termini esponenziali. Attraverso
l‟impiego di questo approccio, è stato possibile rilevare un tasso di nuova generazione di un
intero clone di LLC pari allo 0.1-1.75% al giorno, equivalente a 109-1012 cellule, e un tasso di
crescita tra –1.052% e +0.712% al giorno [2]. Ovviamente i livelli di cellule B neoplastiche
circolanti in ciascun paziente di LLC sono proporzionali alle dinamiche di nascita e morte
cellulare, quest‟ultima calcolata sottraendo al tasso di crescita quello di nuova generazione. In
base a tale presupposto è stato osservato che nelle forme più aggressive di LLC la nascita
supera la morte cellulare, contrariamente a quanto si verifica nei casi più stabili, dove i due
valori sono pressoché uguali. Inoltre, la rilevazione dell‟espansione del clone B neoplastico
23
richiede almeno 2 settimane, un dato interessante che supporta la tesi secondo la quale le
cellule B leucemiche siano prodotte all‟interno di compartimenti solidi come il midollo osseo,
la milza, i linfonodi, e in seguito rilasciate a livello ematico. Complessivamente, i dati
riportati confermano la tesi secondo la quale la LLC sarebbe una patologia dinamica in cui
l‟accumulo delle cellule B tumorali rappresenta il risultato dello squilibrio tra eventi
proliferativi e apoptotici che, a sua volta, appare strettamente correlato alle interazioni tra il
BCR neoplastico e lo specifico stimolo antigenico.
Anche gli studi di espressione genica dei linfociti B neoplastici supportano la tesi della
stimolazione antigenica nella patogenesi della LLC. E‟ stato, infatti, dimostrato che tali
cellule manifestano, indipendentemente dallo stato mutazionale, un pattern di espressione
uniforme, tipico di cellule B memoria e, allo stesso tempo, distinto da quello caratteristico di
altre neoplasie di natura B cellulare. Delle migliaia di geni caratteristicamente espressi dalle
cellule di LLC, solo un numero esiguo che può variare, a seconda degli studi, tra 20 e 200,
mostra un‟espressione differenziale tra casi IgHV mutati e non [65-66]. Quest‟osservazione
suggerisce che entrambi i gruppi possano derivare da un precursore comune attraverso lo
stesso meccanismo patogenetico, che implica l‟attivazione funzionale della cellula B
leucemica e si verifica probabilmente allo stadio di cellula B memoria. I risultati ottenuti dagli
studi di espressione genica sono supportati dal fatto che la LLC è l‟unica tra le malattie
neoplastiche linfoidi a non presentare le traslocazioni reciproche bilanciate. Questo tipo di
aberrazioni si verifica durante la ricombinazione VDJ nelle prime fasi di sviluppo delle cellule
B, o all‟interno dei centri germinativi durante il processo di ipermutazione somatica e lo
scambio di classe. Quindi, se il processo neoplastico ha inizio nelle cellule B memoria non
può implicare le traslocazioni cromosomiche, visto che tale processo nelle cellule B memoria
è stato già inattivato. Tale dato è consistente con la predominante presenza, nelle cellule di
LLC, di alterazioni genetiche come le delezioni o le amplificazioni, peculiari di neoplasie che
colpiscono tessuti non esposti ai processi di ipermutazione somatica. Inoltre, l‟acquisizione di
tali anomalie citogenetiche supporta ulteriormente il concetto dell‟evoluzione clonale della
LLC dal momento che la maggior parte di queste alterazioni si manifesta in fase tardiva di
malattia, non essendo presente all‟inizio del processo di tumorigenesi. Molto probabilmente,
la continua e ripetuta stimolazione delle cellule neoplastiche da parte dell‟antigene,
contribuisce all‟accumulo delle aberrazioni cromosomiche, con conseguente influenza sulla
progressione tumorale.
24
1.2.3 Natura degli antigeni implicati nella patogenesi ed evoluzione della LLC
Data la moltitudine di dati a favore dell‟implicazione della stimolazione antigenica nello
sviluppo della LLC, uno degli aspetti di maggior interesse è rappresentato dal tentativo di
comprendere quale sia la natura dell‟antigene coinvolto nella selezione tumorale.
Quest‟ultima, non può essere definitivamente dedotta dalle sequenze dei geni delle
Immunoglobuline espressi dalle cellule B neoplastiche ma, allo stesso tempo, può fornire
alcune importanti informazioni avvalendosi della conoscenza di anticorpi con specificità nota.
Le sequenze codificanti dei geni IgHV e IgLV espressi nella LLC sono state usate per
generare dei “trasfettomi” producenti anticorpi allo scopo di studiarne l‟attività di legame.
Attraverso questo approccio, è stata osservata una reattività delle cellule B leucemiche nei
confronti di autoantigeni, antigeni microbici e apteni. In particolare, una forte omologia di
sequenza tra le componenti immunoglobuliniche del BCR delle cellule B neoplastiche e
anticorpi diretti contro antigeni self, come ssDNA, tireoglobulina, actina, mioglobina e
cardiolipina è risultata peculiare della maggior parte dei casi di LLC in cui il clone leucemico
presenta regioni CDR3 stereotipate [67]. Recenti studi basati sull‟impiego di anticorpi
ricombinanti espressi da pazienti di LLC, hanno poi rivelato che l‟80% e il 15% degli
anticorpi di LLC rispettivamente IgHV non mutata e mutata, reagisce con antigeni self e non
secondo una modalità polireattiva. Sebbene in modelli murini transgenici l‟espressione di
recettori polireattivi non porta di per se allo sviluppo della leucemia, il basso livello di
stimolazione continua di tali recettori solitamente a bassa affinità, potrebbe potenzialmente
aumentare il rischio di LLC. E‟ stato inoltre osservato che anticorpi ottenuti da casi di LLC
IgHV mutata non polireattivi, possono acquisire una polirettività dopo reversione della loro
sequenza genica da mutata a germline. Questo suggerisce che entrambi i sottogruppi di LLC
possano derivare da cellule B poli/self-reattive e che le caratteristiche del BCR potrebbero
essere rilevanti nel condurre all‟anergia cellulare in un caso e a una prominente espansione
nell‟altro [68]. L‟omologia della catena pesante e leggera del BCR neoplastico ad
autoanticorpi “naturali” polireattivi ha condotto a paragonare la LLC a un disordine di natura
autoimmune. La considerazione della LLC come un modello di patologia a cavallo tra
neoplasia e autoimmunità è stata rafforzata anche dall‟osservazione della positività dei
linfociti patologici per il CD5, un antigene spesso caratteristico di popolazioni B cellulari
implicate nella reattività al self, e dal riscontro di manifestazioni autoimmuni nella maggior
parte dei pazienti. Le cellule CD5+, note anche come B1, sono deputate alla produzione di
anticorpi, prevalentemente di isotipo IgM, a bassa affinità per componenti batteriche.
Ciononostante, in alcune circostanze, tali anticorpi naturali sono in grado di reagire in modo
25
polireattivo con antigeni self con tendenza a mostrare crossreattività nei confronti di antigeni
batterici, un dato che ha condotto all‟ipotesi della derivazione della LLC dalla trasformazione
neoplastica di cellule B CD5+ presenti, nell‟uomo, all‟interno della zona marginale
extrafollicolare [69]. In realtà la possibilità che il CD5 sia un marcatore d‟attivazione
piuttosto che di linea, ha ridirezionato l‟opinione scientifica nel considerare l‟origine della
LLC in un precursore B ancora non identificato, seppur attivato dall‟antigene. Per quanto
riguarda la presenza di fenomeni di natura autoimmune, nei pazienti di LLC è stata riportata
una reattività virtualmente esclusiva nei confronti delle componenti del sistema
ematopoietico; al contrario, non è stata rilevata alcuna associazione con disordini autoimmuni
sistemici o a livello di altri organi. L‟anemia emolitica autoimmune si verifica nel 10-25% dei
casi, la trombocitopenia immune nel 2%, mentre più rare sono la neutropenia autoimmune e
l‟aplasia eritroide [70]. Non ci sono tuttora spiegazioni conclusive riguardo le forme di
autoreattività policlonale osservate nella LLC, è stato comunque ipotizzato che possano
derivare da difetti nel compartimento B e T normale, indotti dallo stesso clone neoplastico
[71]. La reattività delle cellule B di LLC è comunque indirizzata verso strutture cellulari di
natura self intracitoplasmatiche piuttosto che nucleari, diversamente da quanto accade nei
disordini autoimmuni come la SLE, dove gli antigeni riconosciuti sono quasi esclusivamente
di natura nucleare [72]. Alcuni linfociti patologici reagiscono con autoantigeni esposti sulla
membrana citoplasmatica o strutture chimiche prodotte in seguito all‟apoptosi o ad altri
processi catabolici. Si tratta di proteine o lipoproteine non riconosciute nel loro stato nativo
ma solamente dopo modifiche, ad esempio, di natura ossidativa [73]. Dall‟altro lato, però, le
stesse cellule autoreattive possono crossreagire con antigeni batterici, di conseguenza, come
anticipato, non è possibile escludere il contributo di antigeni esogeni nella patogenesi della
LLC. Essi sembrano, infatti, essere implicati nella selezione del clone leucemico soprattutto
nei casi di LLC in cui raramente si identificano recettori stereotipati. Questo potrebbe
verificarsi sia attraverso l‟azione di superantigeni, come la proteina A dello Stafilococco,
riconosciuti da siti antigenici esterni a quelli convenzionali, sia attraverso l‟azione di antigeni
esogeni riconosciuti secondo la modalità classica CDR3-dipendente. A tale proposito, è
interessante notare la correlazione inversa tra lo sviluppo della LLC e la malattia cardiaca
cronica reumatica. I soggetti affetti da questa patologia sono protetti dalla leucemogenesi
probabilmente come conseguenza dell‟uso a lungo termine di antibiotici, un dato a sostegno
della tesi degli antigeni batterici nella patogenesi della LLC [74].
Secondo quanto riportato, si potrebbe ipotizzare che la LLC abbia inizio come patologia
monoclonale delle cellule B poli/auto-reattive. L‟antigene self sarebbe necessario per
26
selezionare le cellule B suscettibili alla trasformazione neoplastica dal repertorio B linfoide
normale e la sua azione potrebbe essere facilitata e rafforzata dall‟intermittente, seppur meno
frequente, stimolazione esogena di natura microbica. Nella maggior parte dei cloni IgHV non
mutati, la persistenza di ripetute interazioni tra il BCR polireattivo e l‟autoantigene, potrebbe
impedire l‟acquisto di mutazioni somatiche, permettendo a tali cellule di mantenere la loro
poli/auto-reattività [75]. Al contrario, nei cloni IgHV mutati, l‟acquisto di mutazioni
somatiche comporterebbe la perdita della polireattività a favore o di una maggiore specificità
per l‟autoantigene o di una perdita di affinità nei suoi confronti. In quest‟ultimo caso, le
mutazioni, privando il BCR del suo potenziale autoreattivo potrebbero indurre uno stato di
anergia cellulare. In quei casi che mantengono la poli/auto-reattività, preferenzialmente IgHV
non mutati ma talvolta, seppur in misura minore, anche IgHV mutati, la stimolazione del BCR
continuerebbe invece a fornire segnali più o meno positivi a seconda delle caratteristiche di
affinità e avidità del BCR e della valenza, forma e concentrazione dell‟antigene. I segnali
positivi di sopravvivenza o espansione cellulare potrebbero essere maggiormente caratteristici
delle forme IgHV non mutate, quelli negativi delle forme IgHV mutate, in linea con la
prognosi più infausta del primo sottogruppo di LLC [76]. L‟identificazione della natura
dell‟antigene coinvolto nella patogenesi della LLC è di grande interesse anche per la
comprensione del sito anatomico all‟interno del quale si verifica l‟incontro con il precursore
leucemico. Risulta infatti più plausibile che l‟incontro con un autoantigene polivalente ed
immobilizzato abbia luogo nei tessuti linfoidi solidi piuttosto che nel sangue periferico, dove
la maggior parte degli antigeni è monovalente e solubile.
1.2.4 Segnali mediati dal BCR nelle cellule B di LLC
I segnali mediati dal complesso del BCR possono avere svariate conseguenze sulla cellula B
linfoide in base al suo stato maturativo e alla sua attivazione. Uno stesso segnale può ad
esempio indurre il processo apoptotico in una cellula caratterizzata da uno stadio maturativo
precoce, viceversa stimolare la proliferazione se la cellula è in uno stadio maturativo avanzato
[Figura 3]. I dati relativi alle caratteristiche del BCR delle cellule B di LLC hanno avvalorato
nel tempo l‟ipotesi che l‟eterogeneità del decorso clinico peculiare di tale malattia, possa
dipendere primariamente dalla reattività del suddetto complesso recettoriale. Questo
presupposto ha condotto a numerosi studi finalizzati alla valutazione della funzionalità del
BCR patologico in seguito alla stimolazione in vitro delle cellule B leucemiche con anticorpi
anti-IgM e/o anti-IgD, in grado di mimare ciò che accade in vivo in presenza dell‟antigene.
L‟obiettivo è rappresentato dalla possibilità di comprendere i meccanismi patogenetici della
27
LLC legati alla stimolazione antigenica e, quindi, le basi molecolari della prevalenza dei
segnali di sopravvivenza, proliferazione o anergia cellulare, con il fine ultimo di correlare tale
risposta all‟andamento prognostico della malattia. Una delle caratteristiche prominenti delle
cellule B leucemiche è rappresentata dalla bassa espressione superficiale delle componenti
immunoglobuliniche del BCR. A causa di questa osservazione, è stato per lungo tempo
ritenuto che i linfociti B neoplastici dei pazienti di LLC fossero indistintamente caratterizzati
da una bassa responsività alla stimolazione del BCR [77]. Tale proprietà risultava anche
causata dall‟espressione di una variante tronca o mutata della componente Igβ del recettore o,
in alternativa, da disfunzioni o inadeguati livelli della tirosin-chinasi SYK [51]. Studi
successivi hanno poi dimostrato che, indipendentemente da questi fattori, i livelli di
fosforilazione della tirosin-chinasi SYK così come il rilascio intracellulare di Ca2+, variano
sostanzialmente in rapporto alla stimolazione recettoriale. In primo luogo, pur esprimendo
livelli normali di SYK, le cellule B leucemiche di circa la metà dei pazienti affetti da LLC
mancano dell‟abilità di indurre la sua fosforilazione dopo crosslinking del BCR [78].
H Niiro. Nat Immunol 2, 2002
Figura 3. Vie di trasduzione del segnale indotte nelle cellule B linfoidi dal legame dell‟antigene al BCR.
28
In secondo luogo, pazienti caratterizzati da un basso rilascio di Ca2+ presentano un pattern
complessivo di fosforilazione notevolmente ridotto dopo stimolazione dell‟isotipo IgM [79].
Dall‟altro lato, nonostante la relativa bassa espressione delle Immunoglobuline di superficie, è
possibile osservare in una buona percentuale di casi una risposta alla stimolazione recettoriale,
talvolta simile a quella di linfociti B normali. Questi dati hanno rafforzato la tesi
dell‟eterogeneità della risposta dei linfociti B di LLC, almeno in termini di eventi di
segnalazione prossimali, alla stimolazione del BCR neoplastico, incoraggiando la valutazione
dell‟associazione di tali differenze molecolari e funzionali alla presenza di fattori indicatori di
prognosi, come lo stato mutazionale della regione IgHV, l‟espressione di ZAP-70, del CD38 e
la presenza di anomalie citogenetiche [Figura 4]. I primi studi in tal senso risalgono alla fine
degli anni ‟90. Il CD38 è stato il primo fattore prognostico ad essere associato alla tipologia di
risposta del BCR delle cellule B leucemiche. E‟ stato dimostrato che l‟espressione del CD38
sulla superficie dei linfociti B di LLC correla con una rapida mobilizzazione di Ca2+
intracellulare, seguita dall‟attivazione del processo apoptotico, in seguito alla stimolazione
con anticorpi anti-IgM. Al contrario, tale risposta non si osserva nei casi di LLC CD38-,
nonostante una densità delle sIgM paragonabile in entrambi i sottogruppi, indicando una
risposta difettiva al crosslinking del BCR in termini di segnalazione intracellulare [80]. La
mobilizzazione di Ca2+ non è indotta direttamente dal CD38, dal momento che l‟esposizione
ad anticorpi anti-CD38 non influenza la concentrazione intracellulare di Ca2+, piuttosto
l‟espressione del CD38 sembra identificare un sottogruppo di cellule B di LLC con peculiari
proprietà funzionali del BCR. E‟ stato in seguito dimostrato come anche il crosslinking del
BCR mediante anticorpi anti-IgD susciti una risposta esclusivamente in cellule B CD38+,
sebbene secondo una cinetica differente rispetto all‟isotipo IgM. In questo caso però i segnali
trasdotti sono essenzialmente di sopravvivenza e pro-differenziativi [81]. Questi dati hanno
rappresentato le prime evidenze non solo della presenza di un‟eterogeneità comportamentale
delle cellule B di LLC, probabilmente legata ad uno stadio maturativo diverso, ma anche della
possibilità che isotipi immunoglobulinici differenti possano trasdurre segnali opposti e
contribuire alla fine regolazione della risposta cellulare. La risposta delle cellule B
neoplastiche ad anticorpi anti-IgM, si è in realtà rivelata caratterizzata da una dicotomia.
Alcuni casi di LLC mostrano a seguito della stimolazione induzione del processo apoptotico,
altri della proliferazione cellulare. La spiegazione della suddetta dicotomia risiede
nell‟attivazione differenziale di una serie di chinasi intracellulari; la MAPK p38 sembra
essere un prerequisito per lo stimolo apoptotico mentre la MAPK ERK per la proliferazione.
29
T Zenz. Nat Rev Cancer 10, 2010
Figura 4. Le cellule di LLC manifestano comportamenti biologici e clinici marcatamente differenti in
relazione alla configurazione mutata o non mutata delle regioni IgHV. a) Le cellule IgHV non mutate sono
caratterizzate da livelli più elevati della tirosin-chinasi ZAP-70 e dell‟antigene CD38; inducono, inoltre, vie di
trasduzione del segnale chiave in risposta all‟attivazione del BCR, coinvolgenti LYN, SYK, ERK e AKT; b) Le
cellule IgHV non mutate hanno una maggiore probabilità di acquisire lesioni genetiche (es: del17p13 e
del11q23) rispetto alle forme IgHV mutate; c) Le suddette differenze biologiche potrebbero essere alla base del
differente decorso clinico delle due sottoforme di LLC.
Una spiegazione alternativa presuppone invece che la presenza di risposte contraddittorie sia
il risultato della tipologia di stimolazione effettuata in vitro. In alcuni studi si è, infatti, ricorso
all‟impiego di anticorpi anti-IgM solubili, in altri casi sono stati impiegati anticorpi anti-IgM
immobilizzati. E‟ possibile che l‟uso di anticorpi solubili non rappresenti accuratamente
quello che accade in vivo in relazione alla stimolazione recettoriale, in quanto tali anticorpi
sono rapidamente internalizzati come conseguenza dell‟endocitosi recettoriale; al contrario le
30
forme immobilizzate sono trattenute sulla superficie cellulare per oltre 24 h [82]. Per di più, la
stimolazione con anticorpi immobilizzati induce un‟efficiente degradazione di IkB e una
prolungata attivazione di AKT ed ERK, con aumento dell‟attività metabolica delle cellule B
leucemiche, induzione dell‟ingresso nel ciclo cellulare e acquisizione di una resistenza
all‟apoptosi indotta dai chemioterapici [83]. Queste osservazioni aiuterebbero a superare
l‟apparente contraddizione insita nell‟associazione tra la risposta apoptotica che si verifica
dopo crosslinking delle IgM e i fattori prognostici sfavorevoli caratteristici delle cellule B di
LLC responsive alla stimolazione.
Studi più recenti hanno inoltre dimostrato come i casi responsivi allo stimolo del BCR
appaiono prevalentemente associati a fattori di cattiva prognosi, rappresentati non solo dal
CD38 ma anche dalla positività per l‟espressione della proteina ZAP-70 e dalla
configurazione IgHV non mutata, un dato che implica una correlazione tra l‟aggressività della
LLC e la funzionalità del BCR neoplastico [84]. ZAP-70 è una molecola segnale caratteristica
dei linfociti T ma espressa anche in una porzione di casi di LLC. E‟ strutturalmente omologa a
SYK e si associa al BCR delle cellule B leucemiche stimolate dall‟antigene, indicando una
sua possibile funzione nella modulazione dei segnali a valle [85]. A supporto di questa tesi, è
stato osservato che cellule B di LLC ZAP-70+, stimolate con anticorpi anti-IgM, sono
caratterizzate da una massiva induzione di eventi di fosforilazione a carico di proteine
citosoliche tra cui SYK, BLNK e PLCγ, con conseguente mobilizzazione di Ca2+
intracellulare. La stessa ZAP-70 viene fosforilata e si associa alla componente Igβ del
recettore, un evento indicativo del suo contributo alla segnalazione del BCR [86]. In realtà
tale contributo sembrerebbe indipendente dalla fosforilazione di ZAP-70 e quindi dalla sua
attività chinasica, dal momento che esperimenti di trasfezione di linfociti B leucemici con
vettori codificanti una forma normale della proteina e una priva del dominio chinasico, hanno
mostrato risultati sovrapponibili in termini di modulazione dell‟attività dei target intracellulari
[87]. E‟ stato anche provato che la cellula B neoplastica utilizzi prevalentemente la tirosinchinasi SYK, in quanto, la sua espressione aumenta nei casi responsivi alla stimolazione del
BCR, contrariamente a quanto accade a ZAP-70, i cui livelli si mantengono pressoché costanti
[22]. Questo dato ha condotto a ipotizzare che il ruolo di ZAP-70 sia principalmente
riconducibile a quello di adattatore molecolare di proteine segnale richieste per i successivi
eventi citoplasmatici legati al crosslinking del BCR. La sua funzione consisterebbe quindi
nell‟accelerazione dell‟attivazione e nell‟aumento della durata dello stimolo. Per quanto
riguarda la correlazione tra lo stato mutazionale della regione IgHV e la risposta allo stimolo
del BCR neoplastico, è stato dimostrato che i casi IgHV non mutati non solo rispondono al
31
crosslinking delle IgM con l‟aumento intracellulare di Ca2+ ma anche con la traslocazione del
recettore a livello delle zattere lipidiche presenti nella membrana cellulare grazie ad una serie
di interazioni SRC-mediate tra l‟actina citoscheletrica e il BCR stesso [88]. Questi eventi si
traducono nella proliferazione dei linfociti patologici, contribuendo alla cattiva prognosi dei
pazienti IgHV non mutati. Al contrario, l‟iporesponsività dei cloni IgHV mutati presuppone
che si siano probabilmente sviluppati a uno stadio in cui l‟esposizione cronica all‟antigene li
ha resi relativamente tolleranti alla stimolazione, di conseguenza, la mancata traslocazione del
BCR a livello delle zattere lipidiche, un evento normalmente tipico di cellule B anergiche,
rappresenterebbe un meccanismo utile al controllo negativo della segnalazione a valle del
recettore. Recentemente, è emerso che l‟anergia funzionale di tale porzione di cellule B
leucemiche presenta un pattern molecolare uniforme, contraddistinto dall‟attivazione
costitutiva delle MAPK ERK1/2 e MEK1/2 e dall‟incrementata transattivazione di NF-AT, in
assenza dell‟induzione di AKT [89]. La tesi dell‟anergizzazione dei linfociti B di LLC
irresponsivi alla stimolazione recettoriale viene supportata dal fatto che nei sistemi murini lo
stato anergico è caratterizzato dalla non responsività alla stimolazione mediante anticorpi antiIgM mentre la segnalazione via Igα/Igβ rimane intatta ed è, inoltre, in accordo con
l‟osservazione che questo stato di basso vigore metabolico può essere revertito dopo coltura in
vitro, evento che suggerisce la possibilità in vivo di una persistente interazione antigene-BCR
[84]. Alcuni recenti lavori hanno inoltre evidenziato come l‟anergia sia isotipo-dipendente
nelle cellule B di LLC, in quanto tali cellule appaiono, nella quasi totalità dei casi, responsive
alla stimolazione dell‟isotipo IgD [37]. In realtà, a causa del numero tuttora limitato di studi
relativi al crosslinking dell‟isotipo IgD, non è ancora chiaro se i segnali specifici trasdotti da
ciascuna forma recettoriale siano indipendenti tra loro o, almeno in vivo, sia presente una
forma di “crosstalk”. Infine, oltre alla trasduzione di segnali mediati dalla presenza
dell‟antigene, il BCR delle cellule B di LLC risulta anche in grado di fornire stimoli di
sopravvivenza attraverso il cosiddetto segnale tonico antigene-indipendente, secondo un
meccanismo paragonabile a quello che si verifica nelle cellule B mature normali. Questo
spiegherebbe l‟assenza di varianti isotipo-negative di LLC così come il motivo per il quale le
traslocazioni a carico dei loci delle Immunoglobuline coinvolgano quasi sempre l‟allele non
produttivo [90].
In relazione a quanto riportato, il perfezionamento delle conoscenze relative ai meccanismi
molecolari e funzionali alla base della stimolazione del BCR delle cellule B leucemiche, oggi
ancora preliminari, diventa di grande importanza per la comprensione del comportamento dei
processi alla base della patogenesi ed evoluzione della malattia.
32
1.3 Scopo del progetto di ricerca
Diversamente da altri disordini linfoproliferativi, la patogenesi e la logica di alcune
caratteristiche biologiche peculiari della LLC hanno eluso per anni una definizione precisa.
Questo è dipeso non solo dall‟assenza, in tale leucemia, di un‟alterazione cromosomica
patogenetica, ma anche dalla relativa incertezza riguardo le sue origini cellulari e dal
differente grado di immunocompetenza delle cellule B leucemiche e dei loro progenitori. Le
conoscenze acquisite nel tempo relativamente alle caratteristiche del BCR neoplastico hanno
fornito informazioni utili a migliorare la comprensione sia dell‟origine cellulare della LLC
che dei meccanismi di natura patogenetica alla base dell‟insorgenza di tale malattia. Dal
momento che l‟eterogeneità del decorso clinico della LLC appare altamente dipendente
dall‟acquisizione di mutazioni somatiche a carico dei geni IgV delle componenti
immunoglobuliniche recettoriali, una delle ipotesi più accreditate identifica la stimolazione
antigenica come il presupposto imprescindibile per lo sviluppo e l‟evoluzione clonale della
LLC. Ciò ha reso lo studio delle proprietà funzionali del BCR neoplastico di grande interesse
per la comprensione del modo attraverso il quale le differenze nella composizione e struttura
del recettore possano influenzare il comportamento delle cellule B leucemiche a seguito
dell‟incontro con l‟antigene e, di conseguenza, la variabilità dell‟andamento prognostico della
malattia.
In tale contesto si inserisce il presente progetto di ricerca incentrato sullo studio degli effetti
della stimolazione in vitro del BCR delle cellule B di LLC, nel tentativo sia di rilevare la
presenza di differenze funzionali a carico del recettore, che di correlare tali differenze alle
caratteristiche biologiche e cliniche di natura prognostica dei pazienti in esame. Sono stati
inclusi nello studio pazienti affetti da LLC a diversa prognosi, non precedentemente esposti
ad alcuna terapia antitumorale. La funzionalità del BCR è stata esaminata mediante
stimolazione delle cellule B leucemiche con anticorpi anti-IgM e anti-IgD, diretti contro la
porzione costante della catena pesante delle molecole immunoglobuliniche a isotipo μ e δ,
componenti del complesso recettoriale B linfoide. Quindi, i campioni stimolati e non, sono
stati analizzati e confrontati in termini di modulazione del profilo di espressione genica,
variazione dell‟attività proliferativa e apoptotica e, infine, alterazione delle proprietà
citoscheletriche.
L‟obiettivo finale del suddetto studio è rappresentato dall‟opportunità di esaminare l‟influenza
della reattività del BCR neoplastico sull‟andamento clinico eterogeneo della malattia e
identificare quei pazienti affetti da LLC che potrebbero beneficiare di un approccio
33
terapeutico innovativo basato sull‟inibizione farmacologica della segnalazione del BCR. Si
potrebbe così ottenere un doppio beneficio garantendo l‟interruzione sia delle vie antigenedipendenti che indipendenti indispensabili per la sopravvivenza e crescita del clone maligno.
34
2. MATERIALI E METODI
2.1 Pazienti
Nel nostro studio sono stati inclusi 48 pazienti affetti da LLC, seguiti presso la Sezione di
Ematologia del Dipartimento di Biotecnologie Cellulari ed Ematologia dell‟Università degli
studi di Roma “Sapienza”. I pazienti in esame non sono stati sottoposti ad alcuna terapia
antileucemica al momento e durante il corso dello studio e il prelievo dei campioni di sangue
periferico e le analisi biologiche sono state eseguite previo consenso informato fornito da
ciascuno di loro in accordo con la Dichiarazione di Helsinki. La diagnosi di LLC è stata
effettuata attraverso l‟identificazione, nel sangue venoso periferico di ciascun paziente, di una
quantità di linfociti superiore a 5x109/L con una morfologia e immunofenotipo caratteristici di
LLC (CD5+CD20+CD23+CD22+sIg±CD10-). Sono state eseguite nei pazienti studiati analisi di
routine quali: la valutazione dello stato mutazionale dei geni IgHV [16]; l‟analisi
citofluorimetrica dell‟espressione della proteina ZAP-70 [26] e dell‟antigene CD38 [24];
l‟analisi FISH (ibridazione in situ fluorescente) per l‟identificazione delle aberrazioni
citogenetiche coinvolgenti le regioni 11q22-23, 13q14, 6q21, 17p13 e la trisomia 12, e il
sequenziamento del gene TP53 [16].
2.2 Isolamento e coltura delle cellule mononucleate dei pazienti affetti da LLC
Le cellule mononucleate del sangue periferico (PBMC) dei pazienti affetti da LLC sono state
ottenute mediante separazione su gradiente di densità (Lymphoprep; Nycomed Pharma, Oslo,
Norway) e coltivate a 37 C in atmosfera umidificata con un tasso di CO2 pari al 5%, in
terreno colturale RPMI 1640 (Cambrex Bio Science Verviers, Belgium) addizionato con 10%
di siero fetale bovino (FBS, HyClone, South Logan, UT), 1% di L-glutammina (EuroClone,
Europe) e 1% di Pen-Strepto (EuroClone, Europe). La vitalità cellulare di ciascun campione è
stata determinata attraverso l‟impiego del Trypan Blue (Sigma-Aldrich CO. St. Louis, USA)
ed è risultata essere superiore al 95% in tutti i casi esaminati.
2.3 Separazione magnetica delle cellule B leucemiche CD19+ dei pazienti affetti da LLC
Le cellule B leucemiche dei pazienti affetti da LLC sono state isolate dal resto delle PBMC
mediante Separazione Cellulare Magnetica (MACS; Miltenyi Biotec), una metodica basata
sull‟impiego di un kit di Isolamento Immunomagnetico che permette di separare cellule
35
altamente purificate sfruttando l‟espressione di marcatori antigenici specifici, presenti sulla
superficie cellulare. Nel caso dei linfociti B, la separazione al MACS è stata effettuata
attraverso una selezione positiva delle cellule B neoplastiche CD19+ mediante l‟uso di
specifici anticorpi anti-CD19 coniugati a biglie magnetiche (Miltenyi Biotec, Auburn, CA,
USA). Successivamente all‟isolamento immunomagnetico, la purezza delle cellule ottenute è
stata valutata mediante analisi citofluorimetrica. Le cellule CD19+ isolate sono state marcate
per 20 min a 4°C in PBS privo di ioni Ca2+ e Mg2+ (DPBS, Cambrex Bio Science Verviers,
Belgium), con i seguenti mAbs: CD20FITC, CD5PE, CD3PerCP (Becton Dickinson,
Mountain View, CA, USA), al fine di distinguere la popolazione d‟interesse,
CD20+CD5+CD3-, e identificare l‟eventuale presenza di linfociti T contaminanti CD20CD3+CD5+/-. In tutti gli esperimenti sono stati utilizzati isotipi aspecifici di Immunoglobuline
(IgG1, IgG2), coniugati con FITC, PE e PerCP, come controllo negativo. L‟acquisizione dei
dati è avvenuta mediante l‟utilizzo di un citofluorimetro FACScan e la loro analisi è stata
realizzata utilizzando il programma CellQuest (Becton Dickinson).
2.4 Stimolazione delle cellule B leucemiche CD19+
Per la stimolazione, le cellule B leucemiche purificate sono state coltivate in piastre da 96
pozzetti a fondo ad U. Prima di allestire la coltura, le piastre impiegate per la stimolazione
sono state preparate in modo da formare sul fondo di ciascun pozzetto, un monostrato di
anticorpi anti-IgG su cui far successivamente adsorbire le cellule. A tale scopo una soluzione
di PBS e di anticorpi anti-IgG (Sigma-Aldrich CO, St Louis, MS) ad una concentrazione
finale pari a 50 μg/ml è stata aggiunta ad ogni pozzetto e la piastra è stata incubata a +4°C per
tutta la notte. Il giorno successivo, il PBS è stato rimosso accuratamente, evitando di intaccare
il monostrato di anticorpi anti-IgG e le cellule B CD19+ sono state piastrate ad una
concentrazione di 5×105 per pozzetto in RPMI completo. La stimolazione del BCR è stata
indotta aggiungendo a ciascun pozzetto della piastra anticorpi anti-IgM o anti-IgD (SigmaAldrich), diretti rispettivamente contro la porzione costante della catena pesante delle sIgM e
delle sIgD, entrambi ad una concentrazione finale di 10 μg/ml.
2.5 Estrazione dell’RNA e analisi del profilo di espressione genica
Dopo 24 e 48 h di incubazione, le cellule stimolate (S) e non stimolate (NS) sono state lisate e
l‟RNA totale è stato estratto utilizzando il reagente Trizol (Life Technologies, Grand Island,
36
NY). Per determinare il profilo di espressione genica sono stati impiegati i chips HGU133
Plus 2.0 (Affymetrix, Santa Clara, CA). Il protocollo seguito è disponibile su
“http://www.affymetrix.com/support/technical/manual/expressionmanual.affx”.
2.6 Analisi del ciclo cellulare
I cambiamenti nella distribuzione del ciclo cellulare delle cellule B leucemiche S e NS sono
stati valutati, a 24 e 48 h di coltura, mediante l‟impiego della tecnica dell‟Arancio di Acridina
(AO). La percentuale di cellule in fase G0, G1, S, G2M e il contenuto medio di RNA delle
cellule in fase G0/1 sono stati determinati misurando simultaneamente il contenuto cellulare
totale di DNA e RNA [91]. L‟analisi citofluorimetrica è stata effettuata utilizzando un
citofluorimetro FACSCan (Becton Dickinson) a 488 nm, allo scopo di rilevare la fluorescenza
verde (F530-DNA) e rossa (F>620-RNA). L‟acquisizione e l‟analisi dei dati sono state
eseguite mediante il software CellQuest (Becton Dickinson). L‟analisi della distribuzione del
ciclo cellulare è stata effettuata mediante il software ModFit LT (Verity Software House,
Topsham, ME).
2.7 Saggio di proliferazione
Per valutare l‟attività proliferativa delle cellule B neoplastiche S e NS, per ciascun paziente,
5×105 cellule B CD19+ sono state piastrate in triplicato in piastre da 96 pozzetti a fondo ad U
e coltivate a 37° C in atmosfera umidificata al 5% di CO2, per 24 e 48 h, in presenza e assenza
di anticorpi anti-IgM o anti-IgD. Allo scadere della coltura, le cellule sono state incubate con
1 μCi di timidina triziata ([3H]timidina) (Amersham, Arlington Heights, IL) per 18 h e la
lettura della radioattività è stata effettuata mediante l‟utilizzo di un beta-counter (Packard
Bioscience, Groningen, The Netherlands). I risultati sono espressi come media dei colpi per
minuti (Cpm) ± deviazione standard.
2.8 Analisi del tasso apoptotico
Lo studio dell‟apoptosi delle cellule B leucemiche S e NS è stato effettuato attraverso
l‟impiego di due differenti metodiche. Nel primo caso, dopo 24 h di coltura in presenza e
assenza di stimolazione con anticorpi anti-IgM e anti-IgD, le cellule B leucemiche sono state
marcate per 10 min al buio e a temperatura ambiente con Annexina-V FITC e Ioduro di
37
Propidio (Immunotech Research, Quebec, Canada), entrambi alla concentrazione finale di 1
g/ml, in Binding buffer 1x (Binding buffer 10x: 100mM HEPES/NaOH pH 7.5; 1.4M NaCl,
25mM CaCl2). Dopo la marcatura, le cellule apoptotiche sono state determinate mediante
citometria a flusso e i dati analizzati utilizzando il software CellQuest (Becton Dickinson).
Nel secondo caso, l‟apoptosi è stata misurata sia a 24 che 48 h di coltura valutando il sub
picco G0/1 sugli istogrammi delle frequenze di DNA ottenuti mediante la tecnica dell‟AO,
che misura il tasso di cellule apoptotiche tenendo conto della minore colorabilità degli
elementi apoptotici in fluorescenza verde (F530-DNA) associata ad una aumentata
colorabilità in fluorescenza rossa (F>620-RNA), evento conseguente al fenomeno della
condensazione cromatinica.
2.9 Analisi statistica per gli studi di proliferazione e apoptosi
I dati sono analizzati statisticamente mediante l‟utilizzo del metodo “Student‟s t test”. I
risultati sono espressi come media ± deviazione standard.
2.10 Visualizzazione dell’actina intracitoplasmatica
Per valutare la presenza di eventuali cambiamenti citoscheletrici nelle cellule B CD19+ di
LLC, dopo stimolazione con anticorpi anti-IgM e anti-IgD, è stata impiegata la Falloidina, un
veleno che lega ad alta affinità l‟actina filamentosa (F-actina). Le cellule sono state raccolte ai
diversi tempi di studio, quali 24 e 48 h, ed è stato effettuato un test di marcatura
intracitoplasmatica basato sull‟utilizzo del kit di permeabilizzazione “Fix & Perm” (Caltag
Laboratories Burlingame, USA). Per la marcatura i campioni d‟interesse sono stati incubati in
presenza di 100 μl di reagente A (medium di fissazione) per 15 min al buio a temperatura
ambiente. Terminata l‟incubazione, dopo opportuno lavaggio in PBS, ai campioni sono stati
aggiunti 100 μl di reagente B (medium di permeabilizzazione) e 50 μg/ml di Falloidina e
ciascun campione è stato incubato per 25 min, in agitazione continua, a temperatura ambiente.
A questo punto, un‟aliquota delle cellule B CD19+, raccolta ai diversi tempi di studio, è stata
analizzata mediante citometria a flusso. La percentuale di espressione dell‟actina all‟interno
dei diversi campioni S e NS è stata, in tal caso, riportata come Intensità Media di
Fluorescenza (MIF), espressa come rapporto tra la fluorescenza del campione e quella
dell‟isotipo usato per la marcatura. L‟aliquota cellulare residua è stata invece ulteriormente
marcata per 5 min al buio, con 4‟,6Diamidino-2-fenilindolo cloridrato (DAPI), un colorante
38
fluorescente impiegato per la colorazione nucleare. Al termine dei 5 min di incubazione con il
DAPI, la localizzazione intracellulare dell‟actina e gli eventuali cambiamenti citoscheletrici
delle cellule B leucemiche S e NS dei pazienti affetti da LLC sono stati visualizzati attraverso
l‟utilizzo del microscopio a fluorescenza ad un ingrandimento 100X.
39
3. RISULTATI
3.1 Analisi del profilo di espressione genica delle cellule B neoplastiche di pazienti affetti
da LLC dopo stimolazione del BCR
Allo scopo di analizzare la funzionalità del BCR, sono stati in primo luogo valutati dal gruppo
di studio del Profilo di Espressione Genica del nostro laboratorio, gli effetti della stimolazione
recettoriale, effettuata per 24 e 48 h mediante l‟impiego di anticorpi anti-IgM e anti-IgD, sulla
modulazione dell‟espressione genica dei campioni primari di cellule B leucemiche CD19+,
isolate da pazienti affetti da LLC. I profili di espressione genica dei campioni stimolati (S) e
non stimolati (NS) sono stati poi confrontati in relazione allo stato dei geni IgHV e
all‟andamento clinico della malattia, al fine di valutare eventuali differenze nei diversi
sottogruppi, e correlare la reattività del BCR leucemico alle caratteristiche prognostiche dei
pazienti in esame.
Nel caso della stimolazione dell‟isotipo recettoriale M, gli studi di espressione genica sono
stati eseguiti su un totale di 10 pazienti: 5 caratterizzati da una configurazione IgHV non
mutata e una malattia in progressione (C.C., B.A.M., S.P., P.A., P.G.) e 5 da una
configurazione IgHV mutata e una malattia stabile (D.M.A., F.A., U.R., P.G., N.C.). L‟analisi
del profilo di espressione genica effettuata mediante un approccio “supervised” ha
evidenziato, esclusivamente nei casi di LLC non mutata e in progressione la presenza di 197
geni differenzialmente espressi tra il controllo NS e il campione S, a seguito del trattamento
con anticorpi anti-IgM. Sono stati osservati deregolati principalmente geni per proteine
implicate nell‟attivazione e nella trasmissione del segnale tramite il complesso del BCR, tra i
quali: geni coinvolti nella trasduzione del segnale (DUSP-2/4/6, SYK, CXCR4); geni per
fattori di trascrizione (EGR-1/2/3, CHES1, NEDD9); geni per regolatori del metabolismo
cellulare (LDHB, HSP90AB1, ATP2A3), del ciclo cellulare (CCND2, CDK4, PTPN6) e
dell‟organizzazione del citoscheletro (ACTB, ACTG1, K-ALPHA1). L‟espressione delle IgM
di superficie, positiva in tutti i campioni esaminati, è apparsa invariata in seguito alla
stimolazione del BCR, mostrando, indipendentemente dalle caratteristiche prognostiche,
valori di espressione più elevati nei casi maggiormente responsivi, a sostegno della stretta
correlazione tra la risposta cellulare e la densità di superficie del suddetto isotipo recettoriale.
Diversamente da quanto osservato nei casi di LLC caratterizzati da fattori biologici e clinici di
prognosi infausta, il confronto del profilo di espressione genica delle cellule S e NS
appartenenti a pazienti con LLC a configurazione IgHV mutata e andamento clinico stabile,
non ha evidenziato alcuna differenza di espressione realmente significativa, evidenziando un
40
comportamento anergico, almeno in termini di modulazione genica, alla stimolazione
dell‟isotipo M del BCR neoplastico. Risultati paragonabili sono stati osservati nelle 2 diverse
sottocategorie esaminate a 48 h dalla stimolazione con anticorpi anti-IgM.
Nel caso della stimolazione dell‟isotipo recettoriale D è stato incluso un numero complessivo
di 11 pazienti: 4 caratterizzati da una configurazione IgHV non mutata e una malattia in
progressione (G.V., A.G., G.P., P.G.); 3 da una configurazione IgHV mutata e una malattia
progressiva (F.A., V.C.; S.D.) e 4 da una configurazione IgHV mutata e una malattia stabile
(F.F., M.C., M.M.L., M.M.). L‟analisi “supervised” a 24 h ha rivelato, in tale ambito, la
presenza di 287 geni differenzialmente espressi tra il controllo NS e il campione S. Tra le
categorie funzionali più rappresentate all‟interno delle classi geniche, modulate in massima
parte negativamente, è stata riscontrata, anche in tal caso, la risposta alla stimolazione del
BCR in termini di attivazione e trasmissione del segnale a valle. In particolare è risulta
deregolata l‟espressione di geni codificanti fattori implicati nella trasduzione del segnale
(ANXA6, RGS14, ANXA6) e nel processo di trascrizione genica (CEBPB, POU2F2,
FOXP1). Sono apparsi downregolati anche geni coinvolti nel differenziamento (SYK,
CD79A/B, CD27, CD24) e nella regolazione della morte cellulare programmata (CEBPβ/δ,
TP53INP1, CD24, CD27, CD74). Risultati paragonabili in termini di modulazione genica
globale, indipendente dalla sottocategoria di pazienti esaminata, sono stati osservati anche a
48 h dalla stimolazione con anticorpi anti-IgD, nonostante, in tal caso, sia stato rivelato un
cambiamento nella tipologia di modulazione genica e geni interessati. Infatti, a 48 h dalla
stimolazione recettoriale, dei 229 geni differenzialmente espressi tra campioni NS e S, la
maggior parte dei geni è risultata upregolata (172/229, 75.1%) piuttosto che downregolata
(58/229; 25.0%). I geni maggiormente interessati dalla stimolazione con anticorpi anti-IgD a
48 h sono risultati coinvolti soprattutto nel controllo dell‟espressione genica e nel
metabolismo degli RNA ma anche nel ciclo cellulare e nella sua regolazione, nella biosintesi
delle macromolecole, nel metabolismo degli aminoacidi e acidi carbossilici e, infine, nel
processo di traduzione.
I dati ottenuti complessivamente dall‟analisi del profilo di espressione genica sottolineano che
le cellule neoplastiche isolate da pazienti affetti da LLC, possono esibire un comportamento
diverso, in seguito alla stimolazione in vitro del BCR, sia in relazione all‟isotipo recettoriale
M o D interessato, che ai fattori prognostici. In particolare, la stimolazione dell‟isotipo M si
associa alla modulazione, prevalentemente positiva, di geni correlati all‟induzione di stimoli
attivatori di tipo proliferativo, mentre quella dell‟isotipo D, alla modulazione, sia negativa che
positiva, di
geni
coinvolti
nel
controllo della proliferazione, dell‟apoptosi,
41
del
differenziamento e della sintesi proteica. Nel caso della stimolazione con anticorpi anti-IgM
solo i casi di LLC caratterizzati da fattori di prognosi infausta appaiono responsivi, un dato a
sostegno del ruolo del BCR nell‟espansione e progressione della malattia. Al contrario, tutti i
campioni di LLC indipendentemente dai fattori prognostici, manifestano una responsività, in
termini di modulazione genica, alla stimolazione con anticorpi anti-IgD, a supporto di un
ruolo più marginale di tale isotipo recettoriale nell‟influenzare l‟andamento della malattia.
3.2 Effetti della stimolazione del BCR delle cellule B neoplastiche di LLC sulla
modulazione del ciclo cellulare
Allo scopo di confermare a livello funzionale le indicazioni ottenute dallo studio del profilo di
espressione genica, sono stati in primo luogo esaminati gli effetti della stimolazione del BCR
sui cambiamenti nella distribuzione delle cellule B CD19+ di LLC, all‟interno delle varie fasi
del ciclo cellulare. I campioni leucemici primari sono stati stimolati per 24 e 48 h con
anticorpi anti-IgM e anti-IgD e l‟analisi del ciclo cellulare è stata effettuata mediante
l‟impiego della tecnica dell‟Arancio di Acridina (AO). Come mostrato nelle Figure 5 e 6, la
stimolazione recettoriale dell‟isotipo M esaminata nei diversi sottogruppi di LLC distinti in
base allo stato mutazionale e all‟andamento clinico della malattia, si associa all‟induzione di
una considerevole attività proliferativa esclusivamente nei casi caratterizzati da una
configurazione IgHV non mutata e una malattia in progressione. Nei casi IgHV non mutati
(11 pazienti) è stato, infatti, rilevato un incremento medio significativo della percentuale di
cellule in fase G1 del ciclo cellulare dallo 0.9% ± 1.1% nei campioni NS al 7.5% ± 3.9%,
dopo 24 h di stimolazione (P=0.037), e dal 2.7% ± 2.4% nei campioni NS al 15.3% ± 6.7%,
dopo 48 h di stimolazione (P=0.010). Allo stesso modo, nei casi di LLC in progressione (13
pazienti), la percentuale di cellule in fase G1 del ciclo cellulare pre- e post-stimolazione è
risultata pari a 0.8% ± 1.1 vs 6.3% ± 4.5% a 24h (P=0.040) e a 2.2% ± 1.0% vs 12.1% ± 6.1%
a 48 h (P=0.014). Al contrario, nei casi di LLC caratterizzati da una configurazione IgHV
mutata (8 pazienti) e una malattia stabile (6 pazienti) è stata osservata una resistenza alla
progressione del ciclo cellulare indotta dalla stimolazione con anticorpi anti-IgM, evidenziata
dalla presenza di percentuali pressoché identiche di cellule B leucemiche in fase G1 del ciclo
cellulare nei campioni NS e S, quali: 0.7% ± 1.1% vs 1.2% ± 1.7% a 24 h e 0.5% ± 0.8% vs
0.7% ± 0.8% a 48 h, nel primo caso e 0.9% ± 1.2% vs 1.6% ± 1.8% a 24 h e 0.6% ± 0.9% vs
0.8% ± 1.1% a 48 h, nel secondo caso.
42
a)
CLL IgHV non mutate
% Cellule in fase G1
25%
15%
20%
15%
CTR
8%
10%
IgM
3%
5%
1%
P=0.037
0%
P=0.010
24h
48h
b)
CLL in progressione
% Cellule in fase G1
25%
20%
12%
CTR
15%
6%
10%
IgM
2%
5%
1%
0%
P=0.040
P=0.014
24h
48h
Figura 5. Analisi del ciclo cellulare pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgM nei campioni di
LLC a configurazione IgHV non mutata (a) e in progressione di malattia (b).
43
a)
CLL IgHV mutate
% Cellule in fase G1
25%
20%
15%
CTR
10%
IgM
5%
1.2%
0.7%
0.7%
0.5%
0%
24h
48h
b)
CLL stabili
% Cellule in fase G1
25%
20%
15%
CTR
10%
IgM
5%
1.6%
0.9%
0.8%
0.6%
0%
24h
48h
Figura 6. Analisi del ciclo cellulare pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgM nei campioni di
LLC a configurazione IgHV mutata (a) e malattia stabile (b).
44
A differenza della stimolazione con anticorpi anti-IgM, l‟impiego di anticorpi anti-IgD non si
associa ad alcuna significativa progressione in fase G1 del ciclo cellulare, indipendentemente
dallo stato mutazionale dei geni IgHV e dallo stato clinico della malattia. A 24 h la
percentuale di cellule in fase G1 del ciclo cellulare, nei campioni NS e S, è risultata infatti
essere pari a 0.9% ± 1.1% vs 0.8% ± 0.9% nei campioni a configurazione IgHV non mutata
(11 pazienti); a 0.8% ± 1.1% vs 0.7% ± 0.9% nei campioni di LLC in progressione (13
pazienti); a 0.7% ± 1.1% vs 0.7% ± 0.8% nei campioni a configurazione IgHV mutata (8
pazienti) e infine a 0.9% ± 1.2% vs 1.0% ± 0.9% nei campioni di LLC stabile (6 pazienti)
[Figura 7]. Valori pressoché identici sono stati osservati dopo 48 h di stimolazione con
anticorpi anti-IgD (dati non mostrati).
Complessivamente, i risultati ottenuti dimostrano, in linea con quelli relativi al profilo di
espressione genica, come la stimolazione del BCR delle cellule B di LLC possa avere effetti
differenti, in termini di avanzamento nel ciclo cellulare, a seconda dell‟isotipo recettoriale
interessato, suggerendo l‟esistenza di funzioni cellulari isotipo-specifiche all‟interno dei
linfociti B leucemici. In secondo luogo, i dati emersi in merito alla stimolazione dell‟isotipo
recettoriale M hanno permesso di rilevare l‟esistenza di un‟associazione tra le capacità
proliferative delle cellule B di LLC e i fattori biologici e clinici di prognosi infausta quali: lo
stato non mutato dei geni IgHV e l‟andamento progressivo della LLC, rafforzando la tesi del
ruolo della stimolazione recettoriale nel sostegno delle forme più aggressive della malattia.
3.3 Studio dell’attività proliferativa delle cellule B neoplastiche di LLC dopo
stimolazione del BCR
In base ai risultati ottenuti dall‟analisi del profilo di espressione genica e dall‟esame dei
cambiamenti nella distribuzione del ciclo cellulare, il passo successivo dello studio è stato
quello di verificare gli effetti della stimolazione con anticorpi anti-IgM e anti-IgD, sulle
capacità proliferative delle cellule B CD19+ di LLC. A tal proposito i campioni primari di
LLC sono stati stimolati per 24 e 48 h ed è stato eseguito un saggio di proliferazione. Tale
saggio consente di determinare indirettamente il tasso proliferativo cellulare, attraverso la
valutazione del grado di incorporazione della [3H]timidina, un precursore nucleotidico
radioattivo che viene normalmente introdotto esclusivamente nel DNA di cellule proliferanti.
Il confronto tra i due sottogruppi di LLC distinti in base allo stato mutazionale dei geni IgHV
ha rivelato un incremento dei livelli di incorporazione della [3H]timidina esclusivamente nei
casi di LLC caratterizzati da una configurazione IgHV non mutata (18 pazienti).
45
a)
% Cellule in fase G1
25%
20%
15%
CTR 24h
10%
IgD 24h
5%
0.9%
0.8%
0.8%
0.7%
0%
CLL IgHV non mutate (n=11)
CLL in progressione (n=13)
b)
% Cellule in fase G1
25%
20%
15%
CTR 24h
10%
IgD 24h
5%
0.7%
0.9%
0.7%
1%
0%
CLL IgHV mutate (n=8)
CLL stabili (n=6)
Figura 7. Analisi del ciclo cellulare pre- e post- 24 h di stimolazione con anticorpi anti-IgD nei campioni di LLC
a configurazione IgHV non mutata e in progressione di malattia (a) e nei campioni di LLC a configurazione
IgHV mutata e malattia stabile (b).
46
In tali campioni l‟incorporazione di [3H]timidina, a 24 e 48 h dalla stimolazione, è risultata
aumentata rispettivamente di 1.6 e 1.8 volte rispetto al controllo NS. A 24 h è stato, infatti,
rilevato un valore medio di Cpm pari a 174.0 ± 112.8 nei campioni IgHV non mutati NS e
285.1 ± 168.4 in quelli S, mentre a 48 h il valore medio di Cpm è risultato equivalente a 303.0
± 136.9 in assenza di stimolazione e 552 ± 115.9 dopo stimolazione [Figura 8a]. Al contrario,
nessuna variazione significativa dell‟attività proliferativa è stata osservata, sia 24 che a 48 h
dalla stimolazione, nelle cellule NS e S dei pazienti IgHV mutati analizzati (10 pazienti). Tali
casi si comportano quindi manifestando una risposta anergica alla stimolazione dell‟isotipo M
recettoriale. Nelle forme di LLC IgHV mutata il valore medio dei Cpm è risultato infatti pari a
130.9 ± 49.2 nei campioni NS e 156.5 ± 86.2 in quelli S (Fold di incremento=1.2) a 24 h, e a
211.3 ± 104.5 nel controllo NS e 239.6 ± 133.7 nei campioni S (Fold di incremento=1.1) a 48
h [Figura 8b]. In accordo con la stretta associazione tra lo stato mutazionale dei geni IgHV e
la prognosi della LLC, risultati simili sono stati ottenuti confrontando gli effetti della
stimolazione dell‟isotipo M in rapporto all‟andamento clinico della malattia. Nei campioni di
LLC in progressione (19 pazienti; 2 IgHV mutati e 17 IgHV non mutati) è stato infatti
rilevato, a 24 e 48 h dalla stimolazione, un incremento del tasso proliferativo pari a 1.5
(Valori medi Cpm: 161.8 ± 122.8 vs 236.6 ± 179.4) e 1.7 (Valori medi Cpm: 309.0 ± 167.4 vs
535.0 ± 115.3) volte rispetto al controllo [Figura 9a]. Al contrario, nei campioni di LLC
stabile (7 pazienti; 7/7 IgHV mutati), è stata evidenziata la presenza di un tasso proliferativo
pressoché identico pre- e post-stimolazione (Valori medi Cpm: 129.1 ± 18.1 vs 144.8 ± 71.2 a
24 h, Fold di incremento=1.1 e 199.8 ± 106.7 vs 240.6 ± 106.9, Fold di incremento=1.2 a 48
h) [Figura 9b].
Diversamente da quanto osservato in merito all‟impiego di anticorpi anti-IgM, ma in accordo
con i dati relativi all‟analisi del ciclo cellulare, la stimolazione dell‟isotipo recettoriale D è
apparsa associata a una riduzione piuttosto che a un aumento del tasso proliferativo delle
cellule B di LLC. Tale effetto è risultato inoltre indipendente sia dallo stato mutazionale dei
geni IgHV che dall‟andamento clinico della malattia. I risultati del saggio di proliferazione
hanno, infatti, evidenziato, a 24 e 48 h dalla stimolazione, un abbassamento dei Cpm pari a
1.6 e 1.4 volte rispetto al controllo NS, nei campioni IgHV non mutati (11 pazienti). In
particolare, sono stati osservati in tale gruppo valori medi rispettivi pre- e post-stimolazione di
215.4 ± 122.8 vs 134.9 ± 59.3 e 354.1 ± 153.4 vs 251.7 ± 174.9 [Figura 10a].
47
a)
CLL IgHV non mutate
552
700
600
285
Cpm
500
303
CTR
400
IgM
174
300
200
100
0
24h
48h
b)
CLL IgHV mutate
700
600
Cpm
500
CTR
240
400
300
200
IgM
211
157
131
100
0
24h
48h
Figura 8. Analisi dell‟attività proliferativa dei campioni di LLC a configurazione IgHV non mutata (a) e IgHV
mutata (b), pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgM.
48
a)
CLL in progressione
700
535
Cpm
600
237
400
300
CTR
309
500
IgM
162
200
100
0
24h
48h
b)
CLL stabili
700
Cpm
600
CTR
500
400
199
300
200
240
IgM
145
129
100
0
24h
48h
Figura 9. Analisi dell‟attività proliferativa dei campioni di LLC caratterizzati da una malattia progressiva (a) e
stabile (b), pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgM.
49
Allo stesso modo, nei campioni IgHV mutati (7 pazienti), il valore medio di Cpm dopo 24 e
48 h di stimolazione, è risultato diminuito in ambo i casi di 1.4 volte rispetto al controllo
(Valori medi Cpm 24 h; 158.7 ± 28.1 vs 113.6 ± 63.9; Valori medi Cpm 48 h; 278.4 ± 91.8 vs
191.6 ± 100.6) [Figura 10b]. Infine, nei campioni di LLC isolati da pazienti con malattia in
progressione (13 pazienti; 3 IgHV mutati e 10 IgHV non mutati) è stata osservata una
riduzione del tasso proliferativo di 1.4 volte (187.5 ± 112.1 vs 137.9 ± 77.4) a 24 h e 1.4 volte
(319.6 ± 167.5 vs 232.7 ± 172.9) a 48 h rispetto al controllo NS e in quelli isolati da pazienti
con malattia stabile (4 pazienti; 4/4 IgHV mutati) di 1.4 volte (145.5 ± 16.6 vs 104.2 ± 28.2) e
1.3 volte (236.6 ± 102.9 vs 180.1 ± 106.8), rispettivamente [Figura 11].
I dati ottenuti in tale ambito, in linea con quanto osservato in merito all‟avanzamento nel ciclo
cellulare, confermano la possibilità di modulare la risposta delle cellule B di LLC alla
stimolazione del BCR attraverso la scelta dell‟isotipo recettoriale coinvolto nella trasmissione
della segnalazione a valle, evidenziando l‟abilità dei due isotipi M e D di mediare effetti
diametralmente opposti. Inoltre rafforzano l‟ipotesi dell‟esistenza di un‟associazione tra la
risposta alla stimolazione dell‟isotipo recettoriale M e i parametri di prognosi sfavorevole,
avvalorando la tesi secondo la quale le differenze nella composizione e struttura del recettore
possano influenzare il comportamento delle cellule B leucemiche a seguito dell‟incontro con
l‟antigene e, di conseguenza, la variabilità dell‟andamento della malattia.
3.4 Valutazione degli effetti della stimolazione del BCR delle cellule B neoplastiche di
LLC sulla modulazione del processo apoptotico
Per comprendere se gli effetti proliferativi indotti dalla stimolazione con anticorpi anti-IgM
nei campioni di LLC IgHV non mutata e di LLC progressiva potessero essere associati ad una
attività citoprotettiva e se, al contrario, la riduzione del tasso proliferativo nei campioni
stimolati con anticorpi anti-IgD potesse dipendere dall‟attivazione del processo di morte
cellulare, le cellule B leucemiche sono state stimolate per 24 e 48 h con anticorpi anti-IgM e
anti-IgD e l‟analisi del ciclo cellulare è stata effettuata mediante l‟impiego delle tecniche
dell‟Annexina V e dell‟Arancio di Acridina (AO). A sostegno della prima ipotesi, l‟analisi dei
risultati relativi alla stimolazione dell‟isotipo recettoriale M ha evidenziato nei campioni di
LLC caratterizzati da uno stato IgHV non mutato e una malattia in progressione, un
incremento non significativo o, in alcuni casi un decremento, del tasso apoptotico dopo
esposizione ad anticorpi anti-IgM.
50
a)
CLL IgHV non mutate
700
Cpm
600
354
500
400
CTR
251
IgD
215
300
135
200
100
0
24h
48h
b)
CLL IgHV mutate
700
Cpm
600
CTR
500
191
300
200
IgD
278
400
158
114
100
0
24h
48h
Figura 10. Analisi dell‟attività proliferativa dei campioni di LLC a configurazione IgHV non mutata (a) e IgHV
mutata (b), pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgD.
51
a)
CLL in progressione
700
Cpm
600
319
500
CTR
232
400
300
IgD
187
137
200
100
0
24h
48h
b)
CLL stabili
700
600
Cpm
500
CTR
400
180
300
200
IgD
237
145
104
100
0
24h
48h
Figura 11. Analisi dell‟attività proliferativa dei campioni di LLC caratterizzati da una malattia progressiva (a) e
stabile (b), pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgD.
52
A 24 h, nei casi IgHV non mutati (10 pazienti) sono stati, infatti, osservati livelli medi di
apoptosi pari a 25.2% ± 14.7% nelle cellule NS e a 28.7% ± 11.6% in quelle S (Incremento
apoptosi: 1.1 volte). Allo stesso modo, i campioni di LLC in progressione (14 pazienti: 4
IgHV mutati e 10 IgHV non mutati) presentavano livelli di apoptosi del 30.5% ± 15.5% pre- e
del 31.7% ± 12.3% post-stimolazione (Incremento apoptosi: 1.04 volte). Questi dati sono stati
ulteriormente corroborati dall‟analisi dei livelli apoptotici effettuata a 48 h dalla stimolazione
recettoriale, che ha evidenziato la persistenza di valori pressoché identici di cellule
apoptotiche in entrambe le sottoclassi (Campioni IgHV non mutati: 24.4% ± 14.5% vs 31.7%
± 22.6%, Incremento apoptosi: 1.2 volte; Campioni di LLC in progressione: 25.6% ± 13.4%
vs 34.2% ± 21.1%, Incremento apoptosi: 1.3) [Figura 12a]. Al contrario, la stimolazione
dell‟isotipo M induce una sensibilizzazione nei confronti del processo apoptotico nei casi di
LLC a configurazione IgHV mutata e malattia stabile, evidenziabile già a 24 h. Nei campioni
IgHV mutati (12 pazienti) è stata, infatti, rilevata una percentuale media di cellule Annexina
V+, dopo 24 e 48 h di stimolazione, rispettivamente pari a 41.8% ± 8.5% e 54.1% ± 12.2%
contro un tasso apoptotico basale del 26.3% ± 8.6% e 39.4% ± 7.9% (Incremento apoptosi di
1.6 volte a 24 h e 1.4 a 48 h; P=0.004), rispettivamente. Ugualmente, i livelli apoptotici
caratteristici delle cellule B di pazienti affetti da LLC stabile (8 pazienti: 8/8 IgHV mutati)
sono apparsi incrementati di 1.6 (25.0% ± 7.4% vs 40.8% ± 9.5%) e 1.5 (41.1% ± 9.5% vs
59.9 % ± 13.2%) volte ai due tempi di stimolazione (P=0.006) [Figura 12b]. I risultati
complessivi ottenuti mediante la tecnica dell‟Annexina V, nelle 4 sottoclassi di pazienti
distinti in base allo stato mutazionale dei geni IgHV e all‟andamento clinico della malattia,
sono stati ulteriormente confermati da quelli conseguiti con la tecnica dell‟AO. Il crosslinking
delle sIgM è risultato in grado di indurre un incremento non significativo o un decremento
della percentuale di cellule in fase subG0/1 nei casi IgHV non mutati e/o progressivi,
contrariamente ai casi IgHV mutati stabili, in cui è stato osservato un incremento della
percentuale di cellule in fase subG0/1, proporzionalmente all‟aumento del tasso apoptotico
(dati non mostrati).
Anche in termini di induzione del processo apoptotico il comportamento delle cellule B
leucemiche in risposta alla stimolazione dell‟isotipo recettoriale D è apparso differente
rispetto a quello osservato in seguito alla stimolazione dell‟isotipo recettoriale M.
53
a)
% Cellule AnnV+
100%
80%
CTR 24h
40%
34%
32%
60%
25% 28%
30% 31%
24%
26%
IgM 24h
CTR 48h
IgM 48h
20%
0%
CLL IgHV non mutate (n=10)
CLL in progressione (n=14)
b)
100%
CTR 24h
% Cellule AnnV+
80%
60%
60%
54%
IgM 24h
42%
39%
26%
40%
41%
41%
CTR 48h
25%
IgM 48h
20%
0%
P=0.004
P=0.004
P=0.006
CLL IgHV mutate (n=12)
P=0.006
CLL stabili (n=8)
Figura 12. Analisi dei livelli apoptotici pre- e post- 24 e 48 h di stimolazione con anticorpi anti-IgM nei
campioni di LLC a configurazione IgHV non mutata e in progressione di malattia (a) e nei campioni di LLC a
configurazione IgHV mutata e malattia stabile (b).
54
L‟incubazione dei linfociti B di LLC con anticorpi anti-IgD è apparsa associata a un aumento
significativo dei livelli apoptotici in tutte le sottocategorie esaminate, indipendentemente sia
dallo stato mutazionale dei geni IgHV che dall‟andamento clinico della malattia. A 24 h è
stato, infatti, rilevato un incremento medio del tasso apoptotico di 1.59 volte nei casi IgHV
non mutati (11 pazienti; 26.4% ± 13.8% vs 41.9% ± 24.2%, P=0.006); di 1.43 nei casi di LLC
in progressione (13 pazienti; 29.0% ± 15.6% vs 41.6% ± 21.6%, P=0.002); di 1.49 volte nei
casi IgHV mutati (8 pazienti; 32.7% ± 13.9% vs 49.0% ± 11.9%, P=0.002) e infine di 1.78
volte nei casi di LLC stabile (7 pazienti; 29.2% ± 10.4% vs 52.1% ± 14.6%, P<0.0001)
[Figura 13]. Risultati sovrapponibili sono stati ottenuti a 48 h, in cui il tasso di incremento del
processo apoptotico è risultato, nelle 4 sottoclassi, rispettivamente pari a 2.07 (P=0.0014),
1.98 (P=0.00015), 2.10 (P<0.0001) e 2.22 (P=0.0004) volte.
In conclusione, i dati rilevati in tale ambito dimostrano come la stimolazione dell‟isotipo
recettoriale M abbia un impatto differenziale sull‟induzione del processo di morte cellulare
dei linfociti B patologici a seconda delle caratteristiche biologico-cliniche della malattia. Al
contrario, la stimolazione dell‟isotipo recettoriale D risulta indipendente dalle suddette
proprietà, rivelando una risposta funzionale comune che suggerisce una dominanza degli
effetti isotipo-specifici sui fattori prognostici predittivi del comportamento della malattia.
% Cellule AnnV+
100%
80%
42%
42%
60%
40%
26%
52%
49%
CTR 24h
33%
29%
29%
IgD 24h
20%
0%
P=0.006
P=0.002
P=0.002
P<0.0001
CLL IgHV NM (n=11) CLLprog (n=13) CLL IgHV M (n=8) CLL stab (n=7)
Figura 13. Analisi dei livelli apoptotici pre- e post- 24 h di stimolazione con anticorpi anti-IgD della totalità dei
casi di LLC esaminati, distinti in base allo stato mutazionale della regione IgHV (non mutato o mutato) e
all‟andamento clinico della malattia (progressivo o stabile).
55
3.5 Analisi dei cambiamenti citoscheletrici delle cellule B neoplastiche di LLC dopo
stimolazione del BCR
I risultati ottenuti dall‟analisi del profilo di espressione genica hanno rivelato, principalmente
a seguito della stimolazione del BCR neoplastico con anticorpi anti-IgM, una deregolazione
dei geni coinvolti nell‟organizzazione dell‟actina citoscheletrica e nel controllo della motilità,
nei campioni di cellule B di LLC a configurazione IgHV non mutata. Per valutare se i
cambiamenti a livello genico osservati in tale sottogruppo, fossero tradotti anche a livello
proteico e se, nel caso della stimolazione dell‟isotipo recettoriale D, l‟attivazione delle vie di
segnalazione a valle del BCR potesse implicare una riorganizzazione del citoscheletro
indipendente dalla modulazione del pattern di espressione genica, è stata inclusa nel nostro
studio un‟analisi dei livelli e della localizzazione intracitoplasmatica dell‟actina in forma
polimerizzata (F-actina). A tale scopo, le cellule B leucemiche isolate da pazienti di LLC con
differenti caratteristiche prognostiche, sono state stimolate per 24 e 48 h con anticorpi antiIgM e anti-IgD quindi, l‟espressione intracellulare dell‟actina è stata valutata attraverso
analisi citofluorimetrica e espressa come Intensità Media di Fluorescenza (MIF); al contrario,
la sua visualizzazione intracellulare è stata eseguita mediante l‟utilizzo di un microscopio a
fluorescenza, dopo marcatura con Falloidina fluoresceinata.
Nel caso della stimolazione dell‟isotipo recettoriale M, i risultati dell‟analisi citofluorimetrica
hanno evidenziato a 48 h di trattamento con anticorpi anti-IgM, un incremento dei livelli
intracitoplasmatici di actina esclusivamente nelle cellule B di LLC a configurazione IgHV
non mutata (10 pazienti) e in progressione di malattia (12 pazienti: 2 IgHV mutati e 10 IgHV
non mutati), con valori di MIF pari a 51.50 ± 10.2 nei campioni NS e 81.5 ± 24.2 nei
campioni S nel primo caso, e a 56.3 ± 21.8 pre- e 66.1 ± 24.9 post-stimolazione, nel secondo
caso. Viceversa, a 24 h sono stati osservati valori della MIF pressoché costanti nel controllo e
nel campione S, rispettivamente equivalenti a 92.6 ± 46.0 vs 93.8 ± 43.3 e a 89.7 ± 43.1 vs
90.4 ± 41.5 [Figura 14]. Nei campioni di LLC a configurazione IgHV mutata e malattia
stabile (7 pazienti) non è stato invece possibile rilevare, come atteso in relazione ai dati
ottenuti dallo studio del profilo di espressione genica, nessuna variazione significativa
nell‟espressione dell‟actina intracitoplasmatica, ad entrambi i tempi di studio. Infatti, in tali
casi, il valore della MIF a 24 h è risultato pari a 103.7 ± 26.9 nei campioni NS e a 100.6 ±
29.6 in quelli S, mentre a 48 h a 73.6 ± 13.7 nel controllo e a 69.0 ± 21.9 dopo stimolazione
[Figura 15].
56
CLL IgHV non mutate (n=10)
a)
94
IgM 24h
92
CTR 24h
IgM 48h
82
CTR 48h
51
0
20
40
60
80
100
120
MIF Actina intracitoplasmatica
b)
CLL progressive (n=12)
90
IgM 24h
89
CTR 24h
IgM 48h
66
CTR 48h
56
0
20
40
60
80
100
120
MIF Actina intracitoplasmatica
Figura 14. Analisi dei livelli intracitoplasmatici di actina pre- e post- 24 e 48 h di stimolazione con anticorpi
anti-IgM nei campioni di LLC a configurazione IgHV non mutata (a) e in progressione di malattia (b). I risultati
dell‟espressione dei livelli di actina sono riportati come MIF.
57
Tali risultati sono stati corroborati dalla visualizzazione al microscopio a fluorescenza,
dell‟actina intracitoplasmatica, che ha evidenziato non solo una riorganizzazione della
proteina ma anche la formazione di regioni più dense di polarizzazione, solamente in quei
campioni S di LLC a configurazione IgHV non mutata e in progressione di malattia, in cui era
stato in precedenza rilevato un cambiamento quantitativo nei livelli di actina [Figura 16].
Diversamente da quanto osservato con l‟impiego di anticorpi anti-IgM, ma in linea con
l‟assente modulazione genica di molecole citoscheletriche, la stimolazione con anticorpi antiIgD non si traduce nella modulazione dell‟actina, né in termini quantitativi che qualitativi, in
nessuna delle sottocategorie esaminate. Più precisamente i dati ottenuti dallo studio
citofluorimetrico rivelano a 24 h, valori della MIF pre- e post-stimolazione di 99.7 ± 60.9 vs
97.2 ± 56.0 nei casi di LLC IgHV non mutata (5 pazienti); di 93.5 ± 53.2 vs 92.4 ± 50.0 nei
casi di LLC in progressione (7 pazienti: 2 IgHV mutati e 5 IgHV non mutati) e infine di 78.0 ±
27.0 vs 80.5 ± 26.5 nei casi di LLC IgHV mutata stabile (2 pazienti). Valori equivalenti sono
stati osservati a 48 h dalla stimolazione (dati non mostrati). Allo stesso modo, non è stato
possibile rilevare alcuna tipologia di riorganizzazione citoscheletrica in seguito all‟analisi
CLL IgHV mutate stabili (n=8)
delle cellule B leucemiche al microscopio a fluorescenza.
100
IgM 24h
103
CTR 24h
IgM 48h
69
CTR 48h
74
0
20
40
60
80
100
120
MIF Actina intracitoplasmatica
Figura 15. Analisi dei livelli intracitoplasmatici di actina pre- e post- 24 e 48 h di stimolazione con anticorpi
anti-IgM nei campioni di LLC a configurazione IgHV mutata e malattia stabile. I risultati dell‟espressione dei
livelli di actina sono riportati come MIF.
58
Questi risultati dimostrano come cellule di LLC con differenti caratteristiche prognostiche
rispondano in maniera differenziale alla stimolazione dell‟isotipo recettoriale M non solo in
termini di espressione genica, ciclo cellulare, proliferazione e apoptosi, ma anche in termini di
cambiamenti morfologici. Al contrario, la stimolazione dell‟isotipo recettoriale D risulta
ancora una volta incapace di indurre una risposta differenziale sia in relazione allo stato
mutazionale delle cellule neoplastiche che all‟andamento clinico della malattia, confermando
l‟ipotesi di una dominanza degli effetti isotipo-specifici sui principali parametri indicatori di
prognosi.
a)
b)
Figura 16. Cambiamenti citoscheletrici delle cellule B di LLC IgHV non-mutata dopo stimolazione con
anticorpi anti-IgM. E‟ riportata, a titolo esemplificativo la visualizzazione al microscopio a fluorescenza, a un
ingrandimento 100X, dell‟actina intracitoplasmatica delle cellule B di LLC in progressione a configurazione
IgHV non-mutata, NS (a) e S (b), di uno dei pazienti studiati. E‟ possibile osservare, dopo 48 h di stimolazione,
una riorganizzazione dell‟actina intracitoplasmatica e la presenza di regioni più dense di polarizzazione, assenti
nelle cellule NS.
59
4. DISCUSSIONE
L‟eterogeneità del decorso clinico della LLC rappresenta un affascinante quesito di natura
biologica, peculiare di tale malattia nell‟ampio spettro delle neoplasie ematologiche. Per
questa ragione, numerose speculazioni sono state formulate nel tentativo di comprendere i
fattori predisponenti la progressione o, in alternativa, la stabilità del suo decorso clinico. In
realtà, nonostante l‟identificazione di parametri clinici e biologici di significato prognostico
abbia permesso di classificare i pazienti in sottocategorie di rischio, contribuendo a fornire
indicazioni sull‟evoluzione della malattia nel tempo, le conoscenze acquisite risultano tuttora
incomplete e non sempre adeguate a predire il comportamento delle cellule B leucemiche e la
risposta farmacologica. La LLC continua, infatti, ad essere una patologia incurabile,
caratterizzata, nella maggior parte dei casi, dall‟acquisizione di resistenza alla terapia
citotossica convenzionale. I dati relativi alle caratteristiche del BCR delle cellule B di LLC
hanno avvalorato nel tempo l‟ipotesi che l‟eterogeneità del decorso clinico peculiare di tale
malattia, possa dipendere primariamente dalla reattività del suddetto complesso recettoriale.
La segnalazione attraverso il BCR potrebbe influenzare, in combinazione con le altre
caratteristiche biologiche delle cellule B leucemiche, il comportamento più o meno aggressivo
della patologia. Questo presupposto ha condotto a esplorare la possibilità che la stimolazione
antigenica possa rivestire un ruolo chiave non solo nel processo di espansione clonale ma
anche nella patogenesi della LLC. Sulla base di tali premesse, il presente progetto di ricerca si
è incentrato sullo studio della funzionalità del BCR dei linfociti B leucemici, con l‟obiettivo
finale di comprendere i meccanismi alla base della patogenesi e progressione della LLC legati
alla stimolazione antigenica, quindi, le basi molecolari della prevalenza dei segnali di
sopravvivenza, proliferazione o anergia cellulare, e correlare tale risposta all‟andamento
prognostico della malattia. A tale scopo è stato impiegato un modello sperimentale, basato
sulla stimolazione in vitro del BCR delle cellule B leucemiche con anticorpi anti-IgM o antiIgD immobilizzati, in grado di mimare la fisiopatologia della LLC, ovvero gli eventi che si
verificano in vivo in presenza dell‟antigene. La risposta alla stimolazione in termini di
variazione del profilo di espressione genica, dell‟attività proliferativa ed apoptotica e della
struttura citoscheletrica dei linfociti B patologici, è stata esaminata sia in relazione allo stato
mutazionale dei geni IgHV che all‟andamento, stabile o progressivo, della malattia.
I risultati ottenuti in merito all‟impiego di anticorpi anti-IgM hanno rivelato l‟esistenza di una
differenza nella responsività al suddetto isotipo recettoriale, in relazione all‟andamento
prognostico della malattia. E‟ stato, infatti, possibile osservare una sensibilità alla
60
stimolazione, relativamente alla modulazione del profilo di espressione genica, solo nei casi
di LLC caratterizzati da una configurazione IgHV non mutata e una malattia in progressione.
Al contrario, nessun cambiamento significativo del profilo di espressione genica è stato
osservato nei casi a configurazione IgHV mutata e malattia stabile. Complessivamente, i dati
ottenuti in merito allo studio del profilo di espressione genica sono risultati in linea con
quanto riportato nel lavoro di Vallat LD et al. [92] in cui la stimolazione dell‟isotipo
recettoriale M rivela un pattern trascrizionale BCR-specifico, con prevalenza di geni coinvolti
nel ciclo cellulare e nella proliferazione come MYC, NF-kB e YWHAQ, nei casi di LLC a
prognosi infausta, ovvero caratterizzati da uno stato IgHV non mutato, dalla positività per la
chinasi ZAP-70 e dalla presenza di aberrazioni citogenetiche. La responsività al crosslinking
delle sIgM dei casi di LLC caratterizzati da fattori di prognosi infausta, rilevata nel nostro
studio, potrebbe quindi supportare in modo ulteriore l‟ipotesi del ruolo centrale del BCR
nell‟espansione e progressione della malattia; al contrario l‟iporesponsività alla stimolazione
dei casi di LLC stabile IgHV mutata potrebbe evidenziare uno stato anergico potenzialmente
correlabile alla stimolazione (auto)antigenica cronica che si verifica in vivo. In un recente
studio, è stato, infatti, dimostrato che l‟iporesponsività agli anticorpi anti-IgM non solo
correla con i fattori prognostici favorevoli, ma risulta anche reversibile dopo coltura in vitro o
induzione del processo di endocitosi recettoriale e ri-espressione dell‟isotipo M sulla
membrana cellulare [93]. Queste osservazioni potrebbero confermare l‟esistenza in vivo di un
legame anergizzante antigene-BCR patologico, la cui reversibilità rende marginale la presenza
di difetti intrinseci nella biosintesi del BCR per giustificare le diverse competenze di
segnalazione delle cellule B di LLC.
Contrariamente a quanto osservato nel caso dell‟impiego di anticorpi anti-IgM, la
stimolazione dell‟isotipo recettoriale D ha mostrato una risposta più omogenea in termini di
modulazione del pattern di espressione genica, nei diversi sottogruppi di pazienti distinti in
base allo stato mutazionale e all‟andamento della malattia, evidenziando la presenza di un
ruolo più marginale di tale isotipo recettoriale nell‟influenzare la prognosi dei pazienti affetti
da LLC. La stimolazione con anticorpi anti-IgD è inoltre risultata associata ad una inversione
nella tipologia di modulazione genica in relazione ai due tempi di stimolazione, mostrando
una preferenziale downregolazione dei geni implicati nel controllo della trasduzione del
segnale, della trascrizione genica, del differenziamento e della morte cellulare programmata a
24 h, e una marcata upregolazione dei geni coinvolti nel controllo dei processi metabolici
dell‟RNA, degli acidi carbossilici e della sintesi proteica a 48 h. Questi risultati ci hanno di
conseguenza condotto a ipotizzare l‟esistenza di una risposta attivatoria più tardiva nel caso
61
della stimolazione delle sIgD. Ad ogni modo, non vi sono in letteratura studi relativi alla
modulazione genica post-stimolazione con anticorpi anti-IgD, di conseguenza tali dati
andrebbero confermati in una casistica più ampia di pazienti.
Allo scopo di validare a livello funzionale le osservazioni emerse dall‟analisi del profilo di
espressione genica, sono stati quindi esaminati gli effetti della stimolazione del BCR sui
cambiamenti nella distribuzione delle cellule B di LLC, all‟interno delle varie fasi del ciclo
cellulare. In linea con l‟upregolazione dei geni coinvolti nel controllo del ciclo cellulare,
rilevata nei casi di LLC progressiva IgHV non mutata, la stimolazione con anticorpi anti-IgM
è apparsa associata a un incremento significativo della quota di cellule in fase G1,
esclusivamente nei suddetti casi. Non è stata invece osservata alcuna modulazione del ciclo
cellulare nei campioni di LLC caratterizzati da fattori di prognosi favorevole, un dato atteso
vista l‟iporesponsività di tali campioni all‟acquisizione di cambiamenti nel pattern di
espressione genica dopo stimolazione. Gli effetti della stimolazione dell‟isotipo recettoriale M
sul ciclo cellulare sono stati poi ulteriormente confermati dall‟esame dell‟attività proliferativa
dei linfociti B di LLC. Anche in tal caso, è stato, infatti, possibile osservare una responsività,
identificabile nell‟incremento del tasso di proliferazione in vitro, solamente nei campioni
cellulari con caratteristiche prognostiche infauste. Questi risultati hanno permesso di supporre
che l‟esposizione all‟antigene, promuovendo l‟espansione dei cloni non mutati, contribuisca
alla cattiva prognosi di questo sottogruppo. Al contrario i cloni iporeponsivi in vitro,
potrebbero essersi sviluppati in vivo come conseguenza di una transizione verso uno stadio
tollerogenico da esposizione antigenica continua dovuto alla selezione di un clone
inizialmente responsivo poi condotto a uno stato di desensibilizzazione recettoriale; di
conseguenza l‟anergia agli stimoli proliferativi giustificherebbe il miglior decorso clinico
della malattia. La capacità delle cellule di LLC caratterizzate da fattori prognostici
sfavorevoli, di rispondere a stimoli di natura proliferativa è stata anche dimostrata in vitro
dopo incubazione con oligonucleotidi immunostimolatori. Cellule di LLC IgHV non mutate
isolate da pazienti con malattia in progressione sono indotte in tali condizioni a entrare nel
ciclo cellulare, proliferare e manifestare un incremento della sopravvivenza [94].
L‟associazione tra l‟entrata in fase G1 del ciclo cellulare e le caratteristiche prognostiche
infauste delle cellule B leucemiche, osservata nel nostro studio, è inoltre apparsa in linea con
quanto riportato nel lavoro di Deglesne PA et al., in cui l‟incremento della percentuale di
cellule in fase G1, osservato in seguito alla stimolazione dell‟isotipo recettoriale M con
anticorpi immobilizzati, è apparso associato preferenzialmente con lo stato non mutato dei
geni IgHV, la positività per la chinasi ZAP-70, la stadiazione Binet, il tempo di
62
raddoppiamento dei linfociti e i livelli sierici della timidina chinasi [95]. In realtà, in questo
studio, né lo stato mutazionale né ZAP-70 risultano sufficienti ad identificare la popolazione
cellulare progressiva, in quanto anche casi IgHV mutati e ZAP-70- manifestano, sebbene con
una minor frequenza, una responsività alla stimolazione in vitro, un‟osservazione a sostegno
dell‟importanza dell‟analisi della reattività del BCR patologico per l‟identificazione di quei
casi, apparentemente a prognosi favorevole ma a rischio di progressione. Anche nel nostro
studio, a differenza di quanto osservato in merito allo stato mutazionale, non è stata osservata
alcuna associazione tra l‟espressione della chinasi ZAP-70 e la risposta alla stimolazione
recettoriale. I livelli di tale chinasi, diversamente da quelli della chinasi SYK, non cambiano
dopo incubazione con anticorpi anti-IgM, confermando dati precedenti in merito al ruolo
predominante di SYK nella trasduzione del segnale anche nei linfociti B patologici [77-96].
Infatti, sebbene inizialmente ZAP-70 sia stata identificata come un fattore prognostico
negativo associato alla capacità di segnalazione a valle del BCR delle cellule B leucemiche, è
stato postulato che il suo ruolo nella trasduzione dei segnali mediati dalle sIgM potrebbe
essere quello di prolungare la persistenza dello stimolo agendo da adattatore molecolare, dal
momento che è la positività per tale chinasi, indipendentemente dal suo grado di
fosforilazione, a favorire gli eventi di fosforilazione tirosinica a carico di SYK, BLNK e
PLC e l‟aumento del flusso intracellulare di Ca2+ [87]. Nel lavoro di Deglesne PA et al. gli
autori ipotizzano l‟esistenza di un‟associazione tra l‟entrata in fase G1 del ciclo cellulare e
una maggiore capacità proliferativa in vivo, supportata dall‟induzione, nei linfociti B
leucemici responsivi, della ciclina D2 e di CDK4. Ciononostante, in accordo con i nostri
risultati, la frazione di cellule in fase S del ciclo rimane invariata, un dato giustificato, nello
studio di Deglesne PA et al., dalla persistente espressione dell‟inibitore del ciclo cellulare
p27kip-1, che suggerisce la necessaria presenza di segnali costimolatori addizionali affinché
la transizione G1-S possa verificarsi.
In realtà, piuttosto che sul ciclo cellulare e l‟attività proliferativa, la maggior parte degli studi
precedenti relativi alla stimolazione recettoriale si è focalizzata sull‟analisi del ruolo delle
sIgM sulla modulazione del processo apoptotico, evidenziando, pur all‟interno dello stesso
sottogruppo prognostico, una dicotomia nel comportamento dei linfociti B leucemici,
rappresentata, in una percentuale di casi, dall‟induzione della sopravvivenza e nell‟altra,
dall‟accelerazione della morte cellulare. Nel nostro sistema sperimentale, come atteso da
quanto osservato in merito agli studi funzionali relativi alle capacità di espansione del clone
leucemico, l‟esame dei livelli apoptotici ha rilevato la presenza di un effetto citoprotettivo nei
campioni caratterizzati da LLC in progressione IGHV non mutata e, al contrario, una
63
sensibilizzazione nei confronti del processo apoptotico nei casi di LLC a configurazione
IgHV mutata e malattia stabile, a sostegno del ruolo dell‟antigene nell‟attivazione di un
programma di sopravvivenza esclusivamente nelle forme più aggressive della malattia. In
particolare, l‟effetto citoprotettivo potrebbe garantire alle cellule IgHV non mutate, almeno
nelle prime fasi di malattia, il tempo necessario a ricevere il secondo segnale utile nel
processo di attivazione antigene-dipendente. Un‟inibizione del processo apoptotico nella
maggior parte dei campioni di LLC, ma senza correlazioni con specifici sottogruppi
prognostici, è stata rilevata nello studio di Bernal A et al., in seguito all‟incubazione delle
cellule B neoplastiche con anticorpi anti-IgM immobilizzati. La risposta cellulare è stata
osservata, in tal caso, in associazione non solo a una riduzione significativa dose-dipendente
dei livelli di apoptosi spontanea, ma anche all‟inibizione dell‟attività delle caspasi,
all‟induzione di NF-kB e all‟espressione di MCL-1, Bcl-2 e BFL-1. Le differenze di
responsività sono inoltre apparse correlate alla via della chinasi AKT, in quanto l‟inibizione
della chinasi PI3-K sarebbe in grado di annullare quasi del tutto gli effetti della stimolazione
mentre la presenza di un‟agonista del CD40 come segnale costimolatorio, di potenziarli [97].
L‟assenza di una correlazione con i principali fattori prognostici rilevata nel suddetto studio,
viene giustificata in quanto consistente con la biologia di una forma tumorale indolente in cui
le cellule maligne generano sIgM con la capacità di riconoscere antigeni autologhi, tuttavia,
questa spiegazione elude le basi dell‟eterogeneità del decorso clinico della malattia, e
meriterebbe un maggior approfondimento. La variabilità della risposta alla stimolazione
dell‟isotipo recettoriale M è anche evidente nello studio di Allsup DJ et al. in cui solo le
forme di LLC IgHV non mutata rispondono incrementando il flusso intracellulare di Ca2+, la
fosforilazione del CD79a e la traslocazione del BCR a livello delle zattere lipidiche. Questo si
traduce nell‟aumento del tasso di sopravvivenza, nonostante tale risposta risulti indipendente
sia dallo stadio di malattia, che, a differenza di quanto riportato nel nostro studio,
dall‟espressione delle sIgM [98]. Nel nostro caso, la densità delle sIgM si è rivelata, infatti,
essere il miglior parametro per discriminare la responsività alla stimolazione del BCR, dal
momento che i casi IgHV non mutati sono risultati caratterizzati dalla maggiore espressione di
tale isotipo recettoriale e la presenza di una riduzione nei livelli di espressione in un caso di
LLC progressiva IgHV non mutata ne ha determinato la clusterizzazione con i casi
iporesponsivi di LLC stabile IgHV mutata.
I risultati relativi alla funzione anti-apoptotica delle sIgM riportati nel nostro studio, sono in
realtà in contrasto con una serie di lavori in cui l‟isotipo M appare invece associato
all‟induzione
di
stimoli
pro-apoptotici.
Molteplici
64
possibili
spiegazioni,
correlate
primariamente
sia
alla
modalità
eterogenea
di
stimolazione
recettoriale
(natura
monoclonale/policlonale degli anticorpi e/o dosaggio) che ai differenti fattori indicatori di
prognosi presi in esame, possono essere addotte per interpretare le differenze rilevate in
merito al comportamento delle cellule B leucemiche. In uno dei primi studi, risalenti agli anni
‟90, è stato dimostrato che l‟impiego di anticorpi anti-IgM è in grado di indurre, nelle cellule
di LLC, la frammentazione del DNA con conseguente apoptosi, similmente a quanto accade
in presenza di glucorticoidi [99]. E‟ possibile in tal caso che le differenze tra gli anticorpi antiIgM impiegati, e fattori come la concentrazione di cellule B leucemiche coltivate, possano
aver contribuito a quanto rilevato in vitro, essendo tali variabili difficili da controllare.
Successivamente, anche in una serie di lavori di Zupo S et al., il cross-linking delle sIgM è
stato osservato prevalentemente in associazione alla trasduzione di segnali pro-apoptotici,
sebbene solo in cellule CD38+ [80-81]. Tale effetto potrebbe essere in parte originato
dall‟impiego di anticorpi solubili piuttosto che immobilizzati. Esiste, infatti, una pletora di
evidenze a supporto della diversa responsività alla stimolazione del BCR patologico in
relazione alla natura solubile o immobilizzata degli anticorpi anti-IgM impiegati, che
influenza sia la durata sia la segnalazione a valle del recettore. In particolare, l‟impiego, nel
nostro sistema sperimentale, della modalità di stimolazione con anticorpi immobilizzati è
stato scelto perché presumibilmente in grado di riprodurre più fedelmente quanto accade in
vivo in relazione alla presentazione antigenica a livello tissutale, partendo dal presupposto che
l‟incontro con l‟antigene abbia luogo all‟interno dei tessuti linfoidi secondari. Un‟altra
importante differenza rispetto al nostro studio è poi rappresentata dalla scelta di analizzare la
risposta alla stimolazione del BCR in relazione ai livelli di espressione dell‟antigene CD38
sulle cellule B leucemiche, e di ricorrere alla selezione dei linfociti B CD38 + come
popolazione da utilizzare negli esperimenti di stimolazione. Sebbene, infatti, la responsività
agli anticorpi anti-IgM appaia correlata esclusivamente alla positività per l‟antigene CD38, la
selezione della popolazione leucemica impone un notevole bias, in quanto in primo luogo
l‟espressione del CD38 potrebbe non rappresentare un fattore invariabile nel tempo, in
secondo luogo, il comportamento delle cellule CD38+ potrebbe essere diverso rispetto a
quello della popolazione CD19+ impiegata nel nostro sistema, a causa del presumibile minor
grado di differenziamento. In alternativa, la contraddizione insita nell‟associazione tra un
marcatore a prognosi sfavorevole e l‟induzione del processo apoptotico dopo stimolazione del
BCR patologico, potrebbe dipendere dall‟assenza nel sistema in vitro, dei segnali antiapoptotici determinati in vivo dal supporto dei componenti del microambiente. Nel nostro ma
anche in altri lavori, nonostante il numero ridotto di casi CD38+, non è stata, infatti, rilevata
65
alcuna influenza della positività per tale antigene sulla capacità di segnalazione a valle del
BCR dopo crosslinking delle sIgM, un dato che supporta ulteriormente la probabilità che la
selezione magnetica delle cellule CD38+ possa alterare la risposta alla stimolazione, che il
CD38 possa intervenire solo negli eventi secondari alla stimolazione o sia semplicemente un
marcatore di attivazione [93]. Nel lavoro di Nédellec S et al. è stata invece osservata non solo
una distinzione tra i casi di LLC responsivi al crosslinking delle sIgM e quelli non responsivi,
ma anche una dicotomia nella tipologia di risposta. I casi di LLC iporesponsivi agli anticorpi
anti-IgM sono, come nel nostro studio, caratterizzati da una prognosi favorevole, al contrario i
casi responsivi si distinguono in due classi. Alla prima classe appartengono quelli con regioni
IgHV non-mutate e livelli più elevati di positività per ZAP-70 e CD38, che vanno incontro ad
apoptosi dopo stimolazione; alla seconda classe quelli con prognosi intermedia, che
rispondono alla stimolazione proliferando [100]. Ad ogni modo, gli autori non specificano gli
effetti della stimolazione distinguendo in modo assoluto le due classi prognostiche, di
conseguenza, la contemporanea presenza di casi IgHV mutati e non mutati, ZAP-70+ e ZAP70- così come CD38+ e CD38- nelle due classi rispondenti, introducendo una notevole
eterogeneità nella popolazione in esame, rende tali risultati meno facilmente interpretabili.
L‟analisi degli effetti funzionali della stimolazione dell‟isotipo recettoriale D ha invece
rivelato degli aspetti inattesi. In primo luogo, diversamente da quanto osservato nel caso della
stimolazione con anticorpi anti-IgM, l‟impiego di anticorpi anti-IgD, a parità di
concentrazione, non è risultato associato ad alcuna significativa progressione in fase G1 del
ciclo cellulare, indipendentemente sia dallo stato mutazionale dei geni IgHV che
dall‟andamento clinico della malattia. In accordo con tali dati, tutti i campioni cellulari di
LLC si sono mostrati indistintamente caratterizzati da una riduzione del tasso proliferativo
piuttosto che da un aumento. L‟osservazione di una diminuzione del tasso proliferativo
potrebbe essere un‟indicazione indiretta dell‟attivazione di un pathway apoptotico
indipendente dalle caspasi. Alternativamente, la modulazione di espressione del gene CHOP
rilevata dall‟analisi del profilo di espressione genica, potrebbe supportare l‟attivazione, dopo
incubazione con anticorpi anti-IgD, di una risposta allo stress cellulare (UPR) che solitamente
si verifica nelle cellule B dopo la stimolazione del BCR e precedentemente al
differenziamento in plasmacellule. La mancanza di espressione differenziale di altri geni
implicati in questo processo potrebbe essere una conseguenza della ridondanza del pathway o
della durata insufficiente della stimolazione. A sostegno della correlazione tra la riduzione del
tasso proliferativo e l‟attivazione del processo di morte cellulare, e in linea con l‟osservazione
della modulazione dei geni coinvolti nel suddetto processo emersa dall‟analisi del profilo di
66
espressione genica, l‟incubazione dei linfociti B di LLC con anticorpi anti-IgD è infatti
apparsa contraddistinta dall‟aumento significativo dei livelli apoptotici in tutte le
sottocategorie esaminate, senza differenze in relazione alla densità superficiale delle sIgD.
Quest‟ultimo dato potrebbe implicare che tale isotipo recettoriale è in grado di trasdurre
segnali intracellulari anche a seguito di un basso livello di crosslinking superficiale oppure
che l‟induzione dell‟apoptosi è indipendente dei livelli di espressione delle sIgD.
Nel complesso, i risultati ottenuti hanno permesso in primo luogo di confermare l‟esistenza di
funzioni recettoriali isotipo-specifiche nei linfociti B leucemici, fornendo il presupposto per la
modulazione della risposta delle cellule B di LLC, attraverso la scelta dell‟isotipo recettoriale
coinvolto nella trasmissione della segnalazione a valle. L‟esistenza di una specializzazione
funzionale dei due isotipi è stata a lungo postulata in base all‟osservazione della presenza di
numerose
differenze
molecolari
e
strutturali
a
carico
delle
due
componenti
immunoglobuliniche. Tali differenze sono emerse da una serie di studi finalizzati alla
comprensione del significato, tuttora enigmatico, della loro coespressione sulla membrana
delle cellule B mature. Gli isotipi immunoglobulinici M e D differiscono primariamente in
relazione alla composizione delle loro regioni costanti e trasmembrana e alla loro espressione
differenziale durante lo sviluppo B linfoide. E‟ stata inoltre osservata non solo una maggiore
stabilità di espressione superficiale delle sIgD rispetto alle sIgM, correlabile al suo elevato
grado di glicosilazione, ma anche un ridotto tasso di endocitosi costitutiva antigeneindipendente, un processo molto efficiente nell‟ambito dei meccanismi di controllo della
densità di superficie delle sIgM [101]. Le IgD, a differenza delle IgM, sono scarsamente
secrete in quanto presenti quasi esclusivamente sulla membrana cellulare e non appaiono
inoltre interessate dal processo di downregolazione recettoriale che si verifica in vivo in
seguito all‟induzione dello stato anergico, evento che presumibilmente interessa le forme
IgHV mutate di LLC. Un‟altra caratteristica distintiva delle sIgD è rappresentata dalla
possibilità di essere espresse attraverso una duplice modalità: quella classica, in associazione
alle molecole Igα/Igβ, e quella alternativa, in associazione ai lipidi di membrana attraverso la
mediazione del glicositolo, correlata alla peculiare induzione della cascata dell‟AMP ciclico
[102]. Oltre alla presenza di diversità strutturali, la differenza nei livelli di crosslinking,
l‟identificazione di proteine intracellulari specificamente associate all‟uno o all‟altro isotipo e
la trasmissione di segnali a valle differenti in termini di cinetica e intensità hanno
ulteriormente incentivato la tesi di un loro ruolo differenziale nella trasduzione dei segnali
intracellulari. In particolare è stato osservato che la fosforilazione indotta dopo stimolazione
dell‟isotipo M è caratterizzata da un picco a 1 min con riduzione già a 60 min, mentre quella
67
indotta dalla stimolazione dell‟isotipo D cresce più lentamente e si mantiene per un tempo
superiore ai 240 min, un dato indicativo dell‟assenza in quest‟ultimo caso di un controllo di
feedback negativo che potrebbe consentire la persistenza degli eventi di fosforilazione
intracellulari [103]. Ad ogni modo, sebbene i segnali trasmessi dai due isotipi differiscano, la
mancanza di un isotipo sembra essere compensata dalla presenza dell‟altro e viceversa, come
dimostrato dal fenotipo mite di topi deficitari di sIgM o sIgD [101]. Questo dato potrebbe
mostrare una capacità di compensazione funzionale delle sIgM e sIgD, evidenziabile solo in
specifiche condizioni. Sulla base delle suddette osservazioni, la presenza di una risposta alla
stimolazione delle sIgD differenziale rispetto a quella delle sIgM, e l‟uniformità di tale
risposta, osservata nel nostro studio in tutte le sottocategorie prognostiche esaminate,
potrebbe dipendere in primo luogo dalle caratteristiche intrinseche dell‟isotipo recettoriale in
esame. A causa della parzialità delle conoscenze relative al ruolo delle sIgD nella fisiologia
dei linfociti B, sia normali che patologici, la maggior parte degli studi in vitro relativi agli
effetti della stimolazione del BCR sulle cellule B di LLC si sono concentrati sull‟isotipo
recettoriale M, rendendo le conoscenze relative al ruolo delle sIgD piuttosto limitate. Ad ogni
modo, i risultati riportati in letteratura, sebbene esigui, appaiono, anche relativamente agli
effetti della stimolazione dell‟isotipo D del BCR, contraddittori. In linea con la competenza di
segnalazione e quindi la responsività indistinta dei campioni di LLC in relazione ai fattori
prognostici osservata nel nostro studio, un recente lavoro ha evidenziato l‟associazione tra la
segnalazione attraverso le sIgD e la presenza di una risposta universalmente positiva, in
termini di incremento intracellulare di Ca2+, nei casi di LLC IgHV mutati e non [93]. Queste
osservazioni hanno permesso di supporre che l‟isotipo D non sia anergizzato da meccanismi
operanti in vivo, e che l‟anergia possa rivelarsi isotipo-dipendente nel caso della LLC.
Evidenze non solo dell‟esistenza di segnali isotipo-specifici ma anche del ruolo proapoptotico delle sIgD derivano anche da una serie di studi effettuati su cellule spleniche
murine. In tali cellule, l‟incubazione con anticorpi anti-IgD sia mono- che poli-clonali risulta
infatti nell‟accelerazione del processo apoptotico in misura più efficace che in presenza di
anticorpi anti-IgM, a meno che non si ricorra all‟incubazione con IL-4 o un agonista del
CD40L, utili a fornire il secondo segnale per l‟attivazione B linfoide. E‟ interessante
sottolineare che questo effetto è più evidente nel range di concentrazioni tra 0.1 e 10 g/ml,
dal momento che 10 g/ml è la concentrazione anticorpale impiegata nel nostro studio. Allo
stesso tempo, le sIgD possono causare l‟entrata delle cellule B spleniche murine in fase G1
del ciclo cellulare. Questo però si verifica solo se gli anticorpi anti-IgD sono impiegati in
soluzione e a concentrazioni superiori a 10
68
g/ml, di conseguenza la scelta di utilizzare
anticorpi immobilizzati a concentrazioni pari a 10
g/ml potrebbe aver impedito
l‟osservazione, nei nostri esperimenti, dell‟attività pro-sopravvivenza delle sIgD [104-105].
Il concetto che le sIgD trasducano segnali apoptotici è al tempo stesso contrario a gran parte
dei dati riportati in letteratura. E‟ noto che l‟espressione delle sIgD compare solo quando i
linfociti B sono maturi da migrare in periferia. L‟osservazione che la co-espressione
sIgM/sIgD su tali linfociti si associa, in presenza degli opportuni segnali antigenici,
all‟attivazione cellulare mentre la mancanza delle sIgD sui linfociti B neonatali causa
l‟induzione della tolleranza in risposta agli stessi stimoli antigenici, ha condotto a ipotizzare il
ruolo negativo delle sIgD nel controllo del suddetto processo. In realtà l‟attivazione precoce
dell‟apoptosi in condizioni di limitato crosslinking, osservata nei modelli murini di cui sopra,
favorisce la tesi di un ruolo positivo di tale isotipo recettoriale nella tolleranza immunologica.
Probabilmente, quando la concentrazione dell‟antigene è elevata, come accade nei follicoli
linfoidi, i linfociti dipendono in misura minore dai segnali costimolatori, e possono entrare in
ciclo anche solo a seguito della stimolazione recettoriale. Al contrario, lontano dai follicoli
linfoidi, quando la concentrazione antigenica è bassa, le cellule B si attivano o vanno incontro
all‟apoptosi a seconda che il secondo segnale sia o meno presente. Di conseguenza l‟effetto
apoptotico della stimolazione dell‟isotipo D del BCR patologico osservata nei nostri
esperimenti, potrebbe semplicemente riprodurre quanto accade in vivo in assenza di
costimolazione, probabilmente a causa di un crosslinking sub-ottimale per l‟induzione
dell‟attivazione; in alternativa, potrebbe derivare dalla presenza di alterazioni della
segnalazione prossimale a valle delle sIgD, peculiari delle cellule B di LLC.
Più recentemente, l‟effetto pro-sopravvivenza delle sIgD è stato in realtà osservato anche in
cellule di LLC CD38+ ed è risultato caratterizzato dall‟induzione di un aumento del flusso di
Ca2+ e della fosforilazione intracellulare simile, in termini di cinetica e intensità, a quello che
si verifica nel caso della stimolazione dell‟isotipo M. In realtà, la discrepanza tra il nostro e il
suddetto studio, potrebbe essere solo apparente, in quanto l‟incremento della sopravvivenza è
risultato rilevabile nello studio di Zupo S et al. solo al 4°-5° giorno di coltura, diventando 2
volte superiore al controllo non stimolato al 10° giorno. Nel nostro caso, invece, l‟analisi
degli effetti funzionali della stimolazione con anticorpi anti-IgD è stata effettuata entro le 48 h
di stimolazione, di conseguenza la scelta di tempi più precoci potrebbe aver impedito
l‟identificazione di un programma di sopravvivenza con attivazione più tardiva. Questo dato
è, infatti, supportato dal cambiamento nella tipologia e nella modalità di regolazione, da
negativa a positiva, dei geni differenzialmente espressi a 48 h in risposta allo stimolo
recettoriale. Potrebbe quindi essere opportuno prolungare il tempo della stimolazione con
69
anticorpi anti-IgD, per verificarne gli effetti funzionali a tempi di esposizione più lunghi.
Sempre nello stesso studio, gli autori evidenziano inoltre la capacità delle sIgD di trasdurre
stimoli di natura differenziativa. Questo risultato non è in realtà una conseguenza genuina
degli effetti della stimolazione in vitro in quanto è indotto dall‟aggiunta di IL-2 ricombinante
e non è infatti rilevabile in presenza esclusiva di anticorpi anti-IgD. In linea, infatti, con i
nostri risultati, l‟isotipo D non è in grado di indurre il differenziamento dei linfociti B
leucemici in plasmacellule, come evidente dall‟assenza di variazioni nelle caratteristiche di
SSC e FSC della popolazione in esame e dalla persistenza di elevati livelli di CD19 (dati non
mostrati). Risultati più eterogenei in merito all‟analisi degli effetti della stimolazione delle
sIgD sono stati invece osservati nello studio di una popolazione multicentrica di pazienti
affetti da LLC [106]. In tal caso, le cellule B leucemiche sono risultate distinte in 3 gruppi in
relazione alla tipologia di risposta dopo stimolazione, rappresentata da inibizione, induzione o
mancata influenza sul processo apoptotico. Dall‟analisi dei casi rispondenti è emerso un
ridotto tempo al trattamento dei pazienti interessati, che ha condotto gli autori a postulare
l‟importanza della responsività alle sIgD, indipendentemente dal tipo di risposta, nella
prognosi della LLC. Nel lavoro però non si fa riferimento ai parametri prognostici presi in
considerazione nel nostro studio, quali lo stato mutazionale e l‟andamento clinico della
malattia; inoltre la scelta arbitraria di un cut-off di riduzione o incremento dei livelli
apoptotici del 20% potrebbe aver condotto a un‟esasperazione dei dati ottenuti e, allo stesso
tempo, alla perdita di informazioni presumibilmente importanti correlate ai campioni cellulari
con valori di variazione dei livelli apoptotici più deboli.
Un dato interessante emerge dal nostro studio integrando le differenze di comportamento
delle cellule B di LLC, distinte in relazione ai fattori prognostici, in rapporto agli effetti della
stimolazione delle sIgM o sIgD. Le cellule di LLC stabile e/o IgHV mutate manifestano,
infatti, un comportamento univoco nei confronti della stimolazione recettoriale. Vanno,
infatti, incontro ad apoptosi sia in presenza di anticorpi anti-IgM che anti-IgD evidenziando
non solo una probabile indipendenza dalla tipologia isotipica ma anche una risposta peculiare
di cellule tollerogeniche, un dato indicativo di una stimolazione antigenica cronica in vivo. Al
contrario, le cellule di LLC progressiva e/o IgHV non mutate sono caratterizzate da una
risposta differenziale in relazione all‟isotipo considerato, traducibile nella proliferazione o
nell‟apoptosi. Questo dato implica una dipendenza comportamentale di tali cellule dall‟isotipo
stimolato ed evidenzia l‟esistenza di un probabile stato maturativo e d‟attivazione differente
rispetto alle forme di LLC a prognosi favorevole, che ne preserva la capacità di risposta
differenziale alla stimolazione, indispensabile, soprattutto considerando l‟integrità dei segnali
70
proliferativi, alla progressione di malattia. Infatti, molto probabilmente in vivo è
l‟integrazione tra gli effetti specifici di ciascun isotipo, mediati dall‟(auto)antigene, a
determinare il comportamento tumorale. Di conseguenza sarebbe di grande interesse
verificare, attraverso l‟impiego del nostro sistema sperimentale, gli effetti in vitro della coincubazione delle cellule B di LLC con anticorpi anti-IgM e anti-IgD.
Partendo dal presupposto che la funzionalità del BCR implica dei cambiamenti nella struttura
citoscheletrica intracellulare è stata inclusa, nell‟ultima parte del nostro studio, la valutazione
dell‟espressione e della distribuzione intracitoplamatica dell‟actina in forma polimerizzata (Factina) a 24 e 48 h di stimolazione con anticorpi anti-IgM e anti-IgD. Nel caso della
stimolazione dell‟isotipo recettoriale M, l‟analisi eseguita mediante citometria a flusso, ha
rivelato un incremento tardivo nei livelli d‟espressione della proteina in questione,
esclusivamente nei campioni di LLC progressiva IgHV non mutata, un dato confermato anche
dalla visualizzazione al microscopio a fluorescenza di una riorganizzazione della proteina
all‟interno del citoscheletro, con formazione di aree più dense di polarizzazione alla periferia
cellulare. Quest‟osservazione è risultata in accordo con quanto suggerito dall‟analisi del
profilo di espressione genica in termini di deregolazione dei geni coinvolti nel controllo della
motilità cellulare (ALPHA1, H2-ALPHA e PSMA7) e nell‟organizzazione dei filamenti
dell‟actina citoscheletrica (ACTG1 e ACTB), all‟interno dei campioni a prognosi infausta,
dopo stimolazione con anticorpi anti-IgM. Questi risultati implicano che il BCR possa
modulare il comportamento delle cellule B leucemiche non solo in termini di sopravvivenza o
apoptosi, ma anche di traffico e homing negli organi linfoidi secondari, partecipando nella
formazione e nel mantenimento dell‟architettura del microambiente tissutale tumorale.
Diventa quindi di grande interesse valutare se i cambiamenti nell‟espressione dell‟actina e
nella riorganizzazione del citoscheletro osservati dopo stimolazione nei campioni di LLC a
prognosi sfavorevole possano tradursi in un maggiore grado di invasività tissutale e quindi
aggressività della malattia. E‟ stato, infatti, dimostrato che la capacità di rispondere alla
chemochina SDF1α secreta dalle cellule stromali midollari da parte delle cellule B leucemiche
ZAP-70+/CD38+, correla con un incremento delle potenzialità migratorie, e quindi un
comportamento neoplastico più aggressivo e una prognosi infausta [107-108]. In questo
contesto risultati paragonabili a quelli del nostro studio sono stati ottenuti in due recenti lavori
in cui la stimolazione delle cellule B di LLC con anticorpi anti-IgM si associa,
preferenzialmente nei casi IgHV non mutati, ZAP-70+ e CD38+, non solo alla trasduzione di
segnali pro-sopravvivenza ma anche alla secrezione delle chemochine chemotattiche CCL3 e
CCL4, implicate nella localizzazione delle cellule B a livello dei pseudofollicoli, e alla
71
modulazione dell‟espressione dei recettori per chemochine e delle molecole di adesione
implicate nel traffico delle cellule B nel midollo osseo e all‟interno dei tessuti linfatici [109110]. Nessuna modulazione quantitativa o qualitativa dell‟actina citoscheletrica è stata invece
osservata in alcuna delle sottocategorie di LLC dopo stimolazione dell‟isotipo recettoriale D.
Anche in questo caso i risultati hanno confermato quanto atteso dallo studio del profilo di
espressione genica pre- e post-stimolazione, scarsamente rappresentato dalla modulazione di
geni coinvolti nel riarrangiamento del citoscheletro intracellulare. Non vi sono dati in
letteratura relativi al possibile coinvolgimento delle sIgD nella modulazione dell‟actina
citoscheletrica, ad ogni modo, il numero dei campioni di LLC valutati nel nostro studio, in
citometria a flusso e microscopia confocale, è piuttosto limitato, per cui i risultati ottenuti
sono da considerarsi preliminari. Per approfondire il ruolo della stimolazione dell‟isotipo
recettoriale D nella riorganizzazione citoscheletrica, sarebbe opportuno aumentare la casistica
ed eseguire la stimolazione a intervalli di tempo più precoci e tardivi rispetto alle 24 e 48 h.
L‟impiego del nostro sistema sperimentale ci ha quindi permesso di ottenere interessanti
informazioni sulle peculiarità biologiche comportamentali delle cellule B leucemiche,
rivelando non solo la presenza di funzioni recettoriali isotipo-specifiche nelle cellule B di
LLC ma anche una diversità, negli effetti della stimolazione attraverso il BCR, in relazione
alle caratteristiche prognostiche dei campioni leucemici, avvalorando l‟ipotesi del contributo
dell‟antigene nella storia naturale della malattia. Sebbene nel nostro studio le cellule B
leucemiche siano invariabilmente destinate all‟apoptosi in seguito alla stimolazione delle
sIgD, mostrando invece un comportamento duplice, a seconda delle caratteristiche
prognostiche, in seguito alla stimolazione delle sIgM, questo potrebbe non essere
necessariamente vero in vivo dopo interazione con il putativo antigene. In tali condizioni la
quota relativa dei due isotipi, la loro capacità di legame antigenico e di modulazione
dell‟attività dei linfociti T potrebbero avere un ruolo nel determinare il risultato della risposta.
Sulla base di tali premesse, un affascinante quesito è rappresentato dalla comprensione delle
basi molecolari della modalità di trasmissione e degli effetti della stimolazione degli isotipi
recettoriali M e D e delle loro correlazioni con i diversi fattori prognostici della LLC. Queste
informazioni potrebbero rivelarsi utili al fine di identificare nuovi target terapeutici per la cura
della LLC e disegnare nuovi farmaci, quali agonisti o antagonisti del BCR, da adottare nel
trattamento della leucemia per l‟innalzamento del tasso di remissioni complete e quindi
dell‟aspettativa di sopravvivenza.
72
5. REFERENZE
1. Boelens J, Lust S, Vanhoecke B, Offner F. Chronic lymphocytic leukaemia. Anticancer
Res 2009; 29:605-615
2. Chiorazzi N. Cell proliferation and death: forgotten features of chronic lymphocytic
leukaemia B cells. Best Pract Res Clin Haematol 2007; 20:399-413
3. Montserrat E, Moreno C. Chronic lymphocytic leukaemia: a short overview. Ann Oncol
2008; 19:320-325
4. Eichhorst B, Hallek M. Revision of the guidelines for diagnosis and therapy of chronic
lymphocytic leukemia (CLL). Best Pract Res Clin Haematol 2007; 20:469-477
5. Mauro FR, Giammartini E, Gentile M, Sperduti I, Valle V, Pizzuti A, Guarini A,
Giannarelli D, Foà R. Clinical features and outcome of familial chronic lymphocytic
leukemia. Haematologica 2006; 91:1117-1120
6. Caligaris-Cappio F. Biology of chronic lymphocytic leukemia. Rev Clin Exp Hematol
2000; 4:5-21
7. Foà R, Hoffbrand AV. Biology of chronic lymphocytic leucemia. Rev Clin Exp Hematol
2000; 4:1-100
8. Matutes E, Polliack A. Morphological and immunophenotypic features of chronic
lymphocytic leukemia. Rev Clin Exp Hematol 2000; 4:22-47
9. Matutes E, Owusu-Ankomah K, Morilla R. The immunological profile of B-cell disorders
and proposal of a scoring system for the diagnosis of CLL. Leukemia 1994; 8:1640-1645
10.
Zomas AP, Matutes E, Morilla R. Expression of the immunoglobulin associated protein
B29 in B-cell disorders with the monoclonal antibody SN8 (CD79b). Leukemia 1996;
10:1966-1970
11.
Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S, Castellano L,
Magrelli A, Citarella F, Messina M, Maggio R, Peragine N, Santangelo S, Mauro FR,
Landgraf P, Tuschl T, Weir DB, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M,
Guarini A, Foà R, Macino G. Quantitative technologies establish a novel microRNA profile
of chronic lymphocytic leukemia. Blood 2007; 109:4944-4951
12.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, Dohner K,
Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N
Engl J Med 2000; 343:1910-1916
73
13.
Kharfan-Dabaja MA, Chavez JC, Khorfan KA, Pinilla-Ibarz J. Clinical and therapeutic
implications of the mutational status of IgVH in patients with chronic lymphocytic leukemia.
Cancer 2008; 113:897-906
14.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated
immunoglobulin VH genes are associated with a more aggressive form of chronic lymphocytic
leukemia. Blood 1999; 94:1848-1854
15.
Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005;
24:804-815
16.
Guarini A, Gaidano G, Mauro FR, Capello D, Mancini F, De Propris MS, Mancini M,
Orsini E, Gentile M, Breccia M, Cuneo A, Castoldi G, Foa R. Chronic lymphocytic leukemia
patients with highly stable and indolent disease show distinctive phenotypic and genotypic
features. Blood 2003; 102:1035-1041
17.
Hallek M; German CLL Study Group. Prognostic factors in chronic lymphocytic
leukemia. Ann Oncol 2008; 19:51-53
18.
Ibrahim S, Keating M, Do KA, O'Brien S, Huh YO, Jilani I, Lerner S, Kantarjian HM,
Albitar M. CD38 expression as an important prognostic factor in B-cell chronic lymphocytic
leukemia. Blood 2001; 98:181-186
19.
Del Poeta G, Maurillo L, Venditti A, Buccisano F, Epiceno AM, Capelli G, Tamburini
A, Suppo G, Battaglia A, Del Principe MI, Del Moro B, Masi M, Amadori S. Clinical
significance of CD38 expression in chronic lymphocytic leukemia. Blood 2001; 98:26332639
20.
Au-Yeung BB, Deindl S, Hsu LY, Palacios EH, Levin SE, Kuriyan J, Weiss A. The
structure, regulation, and function of ZAP-70. Immunol Rev 2009; 228:41-57
21.
Orchard JA, Ibbotson RE, Davis Z, Wiestner A, Rosenwald A, Thomas PW, Hamblin
TJ, Staudt LM, Oscier DG. ZAP-70 expression and prognosis in chronic lymphocytic
leukaemia. Lancet 2004; 10:105-111
22.
Guarini A, Chiaretti S, Tavolaro S, Maggio R, Peragine N, Citarella F, Ricciardi MR,
Santangelo S, Marinelli M, De Propris MS, Messina M, Mauro FR, Del Giudice I, Foà R.
BCR ligation induced by IgM stimulation results in gene expression and functional changes
only in IgV H unmutated chronic lymphocytic leukemia (CLL) cells. Blood 2008; 112:782792
23.
Del Giudice I, Chiaretti S, Tavolaro S, De Propris MS, Maggio R, Mancini F, Peragine
N, Santangelo S, Marinelli M, Mauro FR, Guarini A, Foà R. Spontaneous regression of
74
chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood 2009; 114:638646
24.
Gentile M, Mauro FR, Calabrese E, De Propris MS, Giammartini E, Mancini F, Milani
ML, Guarini A, Foà R. The prognostic value of CD38 expression in chronic lymphocytic
leukaemia patients studied prospectively at diagnosis: a single institute experience. Br J
Haematol 2005; 130:549-557
25.
Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, Marce S, Lopez-
Guillermo A, Campo E, Montserrat E. ZAP-70 expression as a surrogate for immunoglobulinvariable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003; 348:17641775
26.
Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, Neuberg DS, Flinn
IW, Rai KR, Byrd JC, Kay NE, Greaves A, Weiss A, Kipps TJ. ZAP-70 compared with
immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in
chronic lymphocytic leukemia. N Engl J Med 2004; 35:893-901
27.
Hallek M. Chronic Lymphocytic Leukemia (CLL): first-line treatment. Haematology
2005; 285-291
28.
Montserrat E. CLL therapy: progress at last! Blood 2005; 105:2-3
29.
Matsuchi L, Gold MR. New views of BCR structure and organization. Curr Opin
Immunol 2001; 13:270-277
30.
Zarrin AA, Tian M, Wang J. Influence of switch region length on immunoglobulin class
switch recombination. Proc Natl Acad Sci 2005; 102:2466-2470
31.
Deaglio S, Vaisitti T, Bergui L. CD38 and CD100 lead a network of surface receptors
relaying positive signals for B-CLL growth and survival. Blood 2005; 105:3042-3050
32.
Zhang M, Srivastava G, Lu L. The pre-B cell receptor and its function during B cell
development. Cell Mol Immunol 2004; 1:89-94
33.
Mårtensson I, Keenan R., Licence S. The pre-B-cell receptor. Curr Opin Immunol 2007;
19:137-142
34.
Gauthier L, Rossi B, Roux F, Termine E, Schiff C. Galectin-1 is a stromal cell ligand of
the pre-B cell receptor (BCR) implicated in synapse formation between pre-B and stromal
cells and in pre-BCR triggering. Proc Natl Acad Sci 2002, 99:13014-13019
35.
Melchers F, ten Boekel E, Rollink AG. Repertoire selection by pre-B-cell receptors and
B-cell receptors, and genetic control of B-cell development from immature to mature B cells.
Immunol Rev 2000; 175:33-46
75
36.
Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from
studies of the B cell antigen receptor. Ann Rev Immunol 2003; 21:841-894
37.
Efremov DG, Gobessi S, Longo PG. Signaling pathways activated by antigen-receptor
engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev 2007; 7:102-108
38.
Tobin G, Rosén A, Rosenquist R. What is the current evidence for antigen involvement
in the development of chronic lymphocytic leukemia? Hematol Oncol 2006; 24:7-13
39.
Fais F, Ghiotto F, Hashimoto S. Chronic lymphocytic leukemia B cells express
restricted sets of mutated and unmutated antigen receptors. J Clin Invest 1998; 102:1515-1525
40.
Tobin G, Thunberg U, Karlsson K. Subsets with restricted immunoglobulin gene
rearrangement features indicate a role for antigen selection in the development of chronic
lymphocytic leukemia. Blood 2004; 104:2879-2885
41.
Tobin G, Thunberg U, Johnson A, Eriksson I, Söderberg O, Karlsson K, Merup M,
Juliusson G, Vilpo J, Enblad G, Sundström C, Roos G, Rosenquist R. Chronic lymphocytic
leukemias utilizing the VH3-21 gene display highly restricted Vlambda2-14 gene use and
homologous CDR3s: implicating recognition of a common antigen epitope. Blood 2003;
101:4952-4957
42.
Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody
specificities. Immunity 2000; 13:37-45
43.
Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, Foa R, Damle
RN, Fais F, Messmer D, Rai KR, Ferrarini M, Chiorazzi N. Multiple distinct sets of
stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic
leukemia. J Exp Med 2004; 200:519-525
44.
Rosenquist R, Lindström A, Holmberg D, Lindh J, Roos G. V(H) gene family
utilization in different B-cell lymphoma subgroups. Eur J Haematol 1999; 62:123-128
45.
Ghia P, Stamatopoulos K, Belessi C, Moreno C, Stella S, Guida G, Michel A, Crespo
M, Laoutaris N, Montserrat E, Anagnostopoulos A, Dighiero G, Fassas A, Caligaris-Cappio
F, Davi F. Geographic patterns and pathogenetic implications of IGHV gene usage in chronic
lymphocytic leukemia: the lesson of the IGHV3-21 gene. Blood 2005; 105:1678-1685
46.
Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, Belhoul
L, Stella S, Stavroyianni N, Crespo M, Hadzidimitriou A, Sutton L, Bosch F, Laoutaris N,
Anagnostopoulos A, Montserrat E, Fassas A, Dighiero G, Caligaris-Cappio F, Merle-Béral H,
Ghia P, Davi F. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped
receptors: pathogenetic implications and clinical correlations. Blood 2007; 109:259-270
76
47.
Ghia P, Chiorazzi N, Stamatopoulos K. Microenvironmental influences in chronic
lymphocytic leukaemia: the role of antigen stimulation. J Intern Med 2008; 264:549-562
48.
Ghiotto F, Fais F, Valetto A, Albesiano E, Hashimoto S, Dono M, Ikematsu H, Allen
SL, Kolitz J, Rai KR, Nardini M, Tramontano A, Ferrarini M, Chiorazzi N. Remarkably
similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J
Clin Invest 2004; 113:1008-1016
49.
Stamatopoulos K, Belessi C, Hadzidimitriou A, Smilevska T, Kalagiakou E, Hatzi K,
Stavroyianni N, Athanasiadou A, Tsompanakou A, Papadaki T, Kokkini G, Paterakis G,
Saloum R, Laoutaris N, Anagnostopoulos A, Fassas A. Immunoglobulin light chain repertoire
in chronic lymphocytic leukemia Blood 2005; 106:3575-3583
50.
Hadzidimitriou A, Darzentas N, Murray F, Smilevska T, Arvaniti E, Tresoldi C,
Tsaftaris A, Laoutaris N, Anagnostopoulos A, Davi F, Ghia P, Rosenquist R, Stamatopoulos
K, Belessi C. Evidence for the significant role of immunoglobulin light chains in antigen
recognition and selection in chronic lymphocytic leukemia. Blood 2009; 113:403-411
51.
Lankester AC, van Schijndel GMW, van der Schoot CE, van Oers MHJ, van Noesel
CJM, van Lier RAW. Antigen receptor nonresponsiveness in chronic lymphocytic leukemia B
cells. Blood 1995; 86:1090-1097
52.
Dighiero G. CLL biology and prognosis. Haematology 2005; 278-284
53.
Alfarano A, Indraccolo S, Circosta P, Minuzzo S, Vallario A, Zamarchi R, Fregonese
A, Calderazzo F, Faldella A, Aragno M, Camaschella C, Amadori A, Caligaris-Cappio F. An
alternatively spliced form of CD79b gene may account for altered B-cell receptor expression
in B-chronic lymphocytic leukemia. Blood 1999; 93:2327-2335
54.
Damle RN, Ghiotto F, Valetto A, Albesiano E, Fais F, Yan X, Sison CP, Allen SL,
Kolitz J, Schulman P, Vinciguerra VP, Budde P, Frey J, Rai KR, Ferrarini M, Chiorazzi N. Bcell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated,
antigen-experienced B lymphocytes. Blood 2002; 11:4087-4093
55.
Hashimoto S, Chiorazzi N, Gregersen P. Alternative splicing of CD79a and CD79b
RNA transcripts in human B cells. Mol Immunol 1995; 32:651-659
56.
Monroe JG, Havran WL, Cambier JC. B lymphocyte activation: entry into cell cycle is
accompanied by decreased expression of IgD but not IgM. Eur J Immunol 1983; 13:208-213
57.
Ternynck T, Dighiero G, Follezou J, Binet JL. Comparison of normal and CLL
lymphocyte surface Ig determinants using peroxidase-labeled antibodies, I: detection and
quantitation of light chain determinants. Blood 1974; 43:789-795
77
58.
Dorken B, Moldenhauer G, Pezzutto A. HD39 (B3), a B lineage-restricted antigen
whosecell surface expression is limited to resting and activated human B lymphocytes. J
Immunol 1986; 136:4470-4479
59.
Simms PE, Ellis TM. Utility of flow cytometric detection of CD69 expression as a rapid
method for determining poly- and oligoclonal lymphocyte activation. Clin Diagn Lab
Immunol 1996; 3:301-304
60.
Damle RN, Ghiotto F, Valetto A, Albesiano E, Fais F, Yan XJ, Sison CP, Allen SL,
Kolitz J, Schulman P, Vinciguerra VP, Budde P, Frey J, Rai KR, Ferrarini M, Chiorazzi N. Bcell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated,
antigen-experienced B lymphocytes. Blood 2002; 99:4087-4093
61.
Schuh K, Avots A, Tony HP, Serfling E, Kneitz C. Nuclear NF-ATp is a hallmark of
unstimulated B cells from B-CLL patients. Leuk Lymphoma 1996; 23:583-592
62.
Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic
leukemia containsignal transducer and activator of transcription (STAT) 1 and STAT3
constitutively phosphorylatedon serine residues. J Clin Invest 1997; 100:3140-3148
63.
Chiorazzi N, Ferrarini M. Evolving view of the in-vivo kinetics of chronic lymphocytic
leukemia B cell. Haematology 2006; 273-278
64.
Grabowski P, Hultdin M, Karlsson K. Telomere length as a prognostic parameter in
chronic lymphocytic leukemia with special reference to VH gene mutation status. Blood
2005; 105:4807-4812
65.
Keating MJ, Chiarazzi N, Messmer B, Damle RN, Allen SL, Rai KR, Ferrarini M,
Kipps TJ. Biology and treatment of chronic lymphocytic leukemia. Haematology 2003; 153175
66.
Rodríguez A, Villuendas R, Yáñez L, Gómez ME, Díaz R, Pollán M, Hernández N, de
la Cueva P, Marín MC, Swat A, Ruix E, Cuadrado MA, Conde E, Lombardía L, Cifuentes F,
Gonzalez M, García-Marco JA, Piris MA. Molecular heterogeneity in chronic lymphocytic
leukaemia is dependent on BCR signalling: clinical correlation. Leukemia 2007; 21:1-8
67.
Ghia P, Scielzo C, Frenquelli M, Muzio M, Caligaris-Cappio F. From normal to clonal
B cells: Chronic lymphocytic leukemia (CLL) at the crossroad between neoplasia and
autoimmunity. Autoimmun Rev 2007; 7:127-131
68.
Besmer E, Gourzi P, Papavasiliou FN. The regulation of somatic hypermutation. Curr
Opin Immunol 2004; 16:241-245
69.
Dighiero G. Natural autoantibodies, tolerance, and autoimmunity. Ann NY Acad Sci
1997; 815:182-192
78
70.
Landgren O, Engels EA, Caporaso NE, Gridley G, Mellemkjaer L, Hemminki K.
Patterns of autoimmunity and subsequent chronic lymphocytic leukemia in Nordic countries.
Blood 2006; 108:292-296
71.
Myint H, Copplestone JA, Orchard J, Craig V, Curtis D, Prentice AG, Hamon MD,
Oscier DG, Hamblin TJ. Fludarabine-related autoimmune haemolytic anaemia in patients
with chronic lymphocytic leukaemia. Br J Haematol 1995; 91:341-344
72.
Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, Chiorazzi N,
Meffre E. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B
cell precursors despite expressing different antibody reactivity. J Clin Invest 2005; 115:16361643
73.
Lanemo Myhrinder A, Hellqvist E, Sidorova E. A new perspective: molecular motifs on
oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia
antibodies. Blood 2008; 111:3838-3848
74.
Landgren O, Engels EA, Caporaso NE, Gridley G, Mellemkjaer L, Hemminki K.
Patterns of autoimmunity and subsequent chronic lymphocytic leukemia in Nordic countries.
Blood 2006; 108:292-296
75.
Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the
B-cell receptor. Blood 2004; 103:4389-4395
76.
Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005;
352:804-815
77.
Kipps TJ. The B-cell receptor and ZAP-70 in chronic lymphocytic leukemia. Best Pract
Res Clin Haematol 2007; 20:415-424
78.
Semichon M, Merle-Beral H, Lang V, Bismuth G. Normal Syk protein level but
abnormal tyrosine phosphorylation in B-CLL cells. Leukemia 1997; 11:1921-1928
79.
Michel F, Merle-Beral H, Legac E, Michel A, Debré P, Bismuth G. Defective calcium
response in B-chronic lymphocytic leukemia cells. Alteration of early protein tyrosine
phosphorylation and of the mechanism responsible for cell calcium influx. J Immunol 1993;
150:3624-3633
80.
Zupo S, Isnardi L, Megna M, Massara R, Malavasi F, Dono M, Cosulich E, Ferrarini M.
CD38 expression distinguishes two groups of B-cell chronic lymphocytic leukemias with
different responses to antiIgM antibodies and propensity to apoptosis. Blood 1996; 88:13651374
79
81.
Zupo S, Massara R, Dono M, Rossi E, Malavasi F, Cosulich ME, Ferrarini M.
Apoptosis or plasma cell differentiation of CD38-positive B-chronic lymphocytic leukemia
cells induced by cross-linking of surface IgM or IgD. Blood 2000; 95:1199-1206
82.
Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S. Sustained signaling
through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic
leukemia B-cells. Blood 2005; 105:4820-4827
83.
Deglesne PA, Chevallier N, Letestu R, Baran-Marszak F, Beitar T, Salanoubat C,
Sanhes L, Nataf J, Roger C, Varin-Blank N, Ajchenbaum-Cymbalista F. AjchenbaumCymbalista F. Survival response to B-cell receptor ligation is restricted to progressive chronic
lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res 2006; 15:7158-7166
84.
Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F, Packham G. Differential
signaling via surface IgM is associated with VH gene mutational status and CD38 expression
in chronic lymphocytic leukemia. Blood 2003; 101:1087-1093
85.
Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A. Expression of ZAP-70 is
associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood
2002; 100:4609-4614
86.
Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, Weiss A, Kipps
TJ. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood 2005;
105:2036-2041
87.
Gobessi S, Laurenti L, Longo PG, Sica S, Leone G, Efremov DG. ZAP-70 enhances B-
cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic
lymphocytic leukemia and lymphoma B cells. Blood 2007; 109:2032-2039
88.
Allsup DJ, Kamiguti AS, Lin K, Sherrington P, Matrai Z, Slupsky JR, Cawley J, Zuzel
M. B cell receptor translocation to lipid rafts and associated signaling differ between
prognostically important subgroups of chronic lymphocytic leukemia. Cancer Res 2005;
65:7328-7377
89.
Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, Caligaris-
Cappio F, Ghia P. Constitutive activation of distinct BCR-signaling pathways in a subset of
CLL patients: a molecular signature of anergy. Blood 2008; 112:188-195
90.
Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer 2005;
5:251-262
91.
Ricciardi MR, Petrucci MT, Gregorj C, Ariola C, Lemoli RM, Fogli M, Mauro FR,
Cerretti R, Foà R, Mandelli F, Tafuri A. Reduced susceptibility to apoptosis correlates with
80
kinetic quiescence in disease progression of chronic lymphocytic leukaemia. Br J Haematol
2001; 113:391-399
92.
Vallat LD, Park Y, Li C, Gribben JG. Temporal genetic program following B-cell
receptor cross-linking: altered balance between proliferation and death in healthy and
malignant B cells. Blood 2007; 109:3989-3997
93.
Mockridge CI, Potter KN, Wheatley I, Louise AL, Packham G, Stevenson FK.
Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene
mutational status. Blood 2007; 109:4424-4431
94.
Longo PG, Laurenti L, Gobessi S, Petlickovski A, Pelosi M, Chiusolo P, Sica S, Leone
G, Efremov DG. The Akt signaling pathway determines the different proliferative capacity of
chronic lymphocytic leukemia B-cells from patients with progressive and stable disease.
Leukemia 2006; 21:110-120
95.
Deglesne PA, Chevallier N, Letestu R, Baran-Marszak F, Beitar T, Salanoubat C,
Sanhes L, Nataf J, Roger C, Varin-Blank N, Ajchenbaum-Cymbalista F. Survival response to
B-cell receptor ligation restricted to progressive chronic lymphocytic leukemia cells
irrespective of ZAP-70 expresssion. Cancer Res 2006; 66:7158-7166
96.
Tavolaro S, Chiaretti S, Messina M, Peragine N, Del Giudice I, Marinelli M, Santangelo
S, Mauro FR, Guarini A, Foà R. Gene expression profile of protein kinases reveals a
distinctive signature in chronic lymphocytic leukemia and in vitro experiments support a role
of second generation protein kinase inhibitors. Leuk Res 2010; 34:733-741.
97.
Bernal A, Pastore RD, Asgary Z, Keller SA, Cesarman E, Liou H, Schattner EJ.
Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood 2001;
98:3050-3057
98.
Allsup DJ, Kamiguti AS, Sherrington PD, Matrai Z, Slupsky JR, Cawley JC, Zuzel M.
B-cell receptor translocation to lipid rafts and associated signalling differ between
prognostically important subgroups of chronic lymphoid leukemia. Canc Res 2005; 65:73287337
99.
McConkey DJ, Aguilar-Santelises M, Hartzell P, Eriksson I, Mellstedt H, Orrenius S,
Jondal M. Induction of DNA fragmentation in chronic B-lymphocytic leukemia cells. J
Immunol 1991; 146:1072-1076
100. Nédellec S, Renaudineau Y, Bordron A, Berthou C, Porakishvili N, Lydyard PM, Pers
J, Youinou P. B cell response to surface IgM cross-linking identifies different prognostic
groups of B-chronic lymphocytic leukemia patients. J Immunol 2005; 174:3749-3756
81
101. Geisberger R, Lamers M, Achatz G. The riddle of the dual expression of IgM and IgD.
Immunology 2006; 118:429-437
102. Geisberger R, Königsberger S, Achatz G. Membrane IgM influences membrane IgD
mediated antigen internalization in the B cell line Bcl1. Immunol Lett 2006; 102:169-176
103. Kim KM, Reth M. Signaling difference between class IgM and IgD antigen receptors.
Ann NY Acad Sci 1995; 766:81-88
104. Peckham D, Andersen-Nissen E, Finkelman FD, Stunz LL, Ashman RF. Difference in
apoptosis induction between surface IgD and IgM. Int Immunol 2001; 13:285-295
105. Sieckmann DG, Finkelman FD, Scher I. IgD as a receptor in signaling the proliferation
of mouse B-lymphocytes. Ann NY Acad Sci 1982; 399:277-289
106. Morabito F, Cutrona G, Gentile M, Fabbi M, Matis S, Colombo M, Reverberi D, Megna
M, Spriano M, Callea V, Vigna E, Rossi E, Lucia E, Festini G, Zupo S, Molica S, Neri A,
Ferrarini M. Prognostic relevance of in vitro response to cell stimulation via surface IgD in
binet stage a CLL. Br J Haematol 2010; 149:160-163
107. Batista DF, Harwood NE. The who, how and where of antigen presentation to B cells.
Nat Immunol 2009; 9:15-27
108. Niiro H, Clark EA. Regulation of B cell fate by antigen-receptor signals. Nat Immunol
2002; 2:945-956
109. Burger JA, Quiroga MP, Hartmann E, Bürkle A, Wierda WG, Keating MJ, Rosenwald
A. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic
leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood 2009;
113:3050-3058
110. Vlad A, Deglesne PA, Letestu R, Saint-Georges S, Chevallier N, Baran-Marszak F,
Varin-Blank N, Ajchenbaum-Cymbalista F, Ledoux D. Down-regulation of CXCR4 and
CD62L in chronic lymphocytic leukemia cells is triggered by B-cell receptor ligation and
associated with progressive disease. Cancer Res 2009; 69:6387-6395
82
PARTE II
Studio della funzionalità della proteina p53 in
campioni primari di cellule neoplastiche di pazienti
affetti da Leucemia Linfatica Cronica (LLC) a
differente stadio di malattia
83
1. INTRODUZIONE
1.1 La proteina p53
1.1.1 Cenni introduttivi
Le cellule sono continuamente soggette a condizioni e stimoli esterni, inclusi il metabolismo
ossidativo e l‟esposizione a radiazioni, che risultano nello stress genotossico. Di conseguenza,
la capacità di rilevare e riparare un conseguente danno a carico del DNA rappresenta una
funzione critica per le cellule normali. L‟osservazione secondo cui l‟inefficiente riparazione
del danno al DNA cellulare possa condurre alla trasformazione neoplastica, ha consentito
l‟identificazione delle basi funzionali della soppressione tumorale nel riparo o
nell‟eliminazione delle cellule danneggiate e quindi potenzialmente deleterie. In tale ambito,
un ruolo di primaria importanza riveste la proteina ad attività oncossoppressoria p53. Tale
proteina fu identificata per la prima volta nel 1979 all‟interno di cellule infettate dal virus
SV40 (simian virus 40) come una fosfoproteina che co-immunoprecipitava con l‟antigene
“large T”. Poiché sembrava non condividere determinanti antigenici con l‟antigene “large T”
di SV40 e dal momento che venne in seguito dimostrata la sua iperespressione anche in
cellule di carcinoma embrionale murino non infettate da SV40, si concluse che la proteina p53
fosse codificata da un gene cellulare e non da un gene virale [1]. La successsiva
dimostrazione che la sequenza aminoacidica di tale proteina risultasse altamente conservata
nelle diverse specie di vertebrati, fece quindi supporre un suo ruolo chiave nel mantenimento
dell‟omeostasi tissutale e della fisiologia cellulare.
I primi studi condussero a definire erroneamente la proteina p53 come il prodotto di un
oncogene. Tale proteina risultava, infatti, presente a basse concentrazioni all‟interno delle
cellule normali, mentre la sua concentrazione appariva significativamente più elevata nelle
linee cellulari tumorali coltivate in vitro e nei tessuti tumorali in vivo. La base molecolare di
questo aumento dei livelli di p53 nelle cellule tumorali era in realtà da attribuire
probabilmente ad un fenomeno di stabilizzazione post-trascrizionale, dovuta a cambiamenti
nell‟ambiente cellulare, o alla formazione di complessi con altre proteine sia cellulari che
virali. Il risultato era, in ogni caso, un aumento del tempo di emivita di p53, con conseguente
accumulo della proteina stessa e, dunque, aumento della sua concentrazione [2].
L‟osservazione che le cellule trasformate potessero contenere una quantità maggiore della
proteina p53 rispetto alle corrispondenti cellule normali fece pensare che potesse giocare un
ruolo importante nel controllo del ciclo cellulare e che l‟aumentata espressione di p53
portasse ad un aumento della proliferazione cellulare. Osservazioni successive relative alla
84
capacità del prodotto del gene TP53 di cooperare con l‟oncogene Ras nella trasformazione dei
fibroblasti di embrione di ratto, continuarono a sostenere l‟ipotesi che p53 fosse un oncogene
dominante e che contribuisse, quindi, alla tumorigenesi, fornendo uno stimolo per la crescita
cellulare quando iperespresso [3]. Solo nel 1989 Hinds PW et al. dimostrarono che in realtà
molti degli studi in cui il gene TP53 era stato cotrasfettato con il gene Ras erano stati
realizzati utilizzando la forma mutata del gene TP53. Quando questi esperimenti di
cotrasfezione furono ripetuti con il cDNA del gene TP53 “wild type” (non mutato), non si
verificò la trasformazione dei fibroblasti. Questi risultati permisero di stabilire, quindi, la vera
natura di TP53, un gene oncosoppressore che normalmente inibisce la crescita cellulare. In
realtà, alcune mutazioni che colpiscono questo gene possono, in taluni casi, dare origine a
proteine p53 mutanti che non solo perdono la loro normale funzione di inibizione sulla
proliferazione cellulare, ma che acquistano anche una nuova attività di tipo oncogenico, ossia
stimolatoria nei confronti della crescita della cellula [4]. Questa particolare caratteristica di
alcuni mutanti del gene TP53 ha reso più difficoltosa e più confusa la sua classificazione
come gene oncosoppressore.
1.1.2 Struttura della proteina p53
La proteina umana p53 è codificata dal gene oncosoppressore TP53, localizzato sul
cromosoma 17p13.1. Questo gene è composto da undici esoni (il primo dei quali non
codificante), per una lunghezza complessiva di circa 20 Kb [5]. Il gene TP53 è espresso
praticamente in tutti i tessuti ed è altamente conservato in tutte le specie di vertebrati. Il
prodotto proteico del gene TP53 è una fosfoproteina nucleare composta da 393 aminoacidi
(Peso Molecolare: 53 KDa), primariamente coinvolta nella regolazione della risposta cellulare
allo stress genotossico, attraverso il controllo del ciclo cellulare, della riparazione del DNA,
dell‟apoptosi e della senescenza. L‟attività multifunzionale della proteina p53 è insita nella
intrinseca flessibilità della sua organizzazione quaternaria, resa possibile grazie alle peculiari
caratteristiche dei suoi domini. Gli studi di cristallografia e spettroscopia NMR hanno
permesso di isolare e caratterizzare tali domini distinguendoli in: una regione N-terminale
detta di transattivazione (TAD, transactivation domain; residui 1-98), una regione ad alta
affinità di legame per il DNA (DBD, DNA-binding domain; residui 98-303), una regione di
localizzazione nucleare (resisui 303-323), una regione di oligomerizzazione (residui 323-363)
e una regione C-terminale (CTD, C-terminal domain; residui 363-393) [6-7]. La regione Nterminale consiste di due sub-domini di attivazione trascrizionale contigui (TADI e TADII) e
una regione ricca in residui di prolina (PRD, prolin-rich-domain) [Figura 1a]. L‟attività
85
trascrizionale di p53 è modulata grazie al legame delle molecole target a livello della regione
TAD. L‟analisi dei frammenti N-terminali di p53 complessati con altre proteine rivela la
formazione di α-eliche, mentre studi su frammenti isolati non rivelano alcuna struttura
terziaria definita [8]. Il dominio centrale (core domain; residui 102-292) all‟interno del DBD,
implicato nel legame con il DNA, è stato studiato tramite cristallografia a raggi X e la sua
struttura è stata risolta sia in forma isolata, che complessata con DNA e altre proteine. Questi
studi strutturali hanno dimostrato che il dominio si organizza in un β-sandwich antiparallelo
che coordina 2 loop che a loro volta prendono contatto con il DNA. Questi loop sono
stabilizzati dallo ione zinco, a formare un motivo “loop-sheet-helix” [Figura 1b] [9].
Mutazioni all‟interno del dominio di legame al DNA hanno effetti deleteri sull‟attività di p53,
comportando la perdita della sua attività trascrizionale e/o alterando l‟organizzazione
strutturale di tale dominio. Il dominio di oligomerizzazione media la formazione di complessi
omotetramerici della proteina p53, evento indispensabile per il corretto funzionamento della
stessa. Tale dominio forma un tetrametro simmetrico di 20 kDa con una topologia costituita
da un dimero di dimeri [Figura 1c]. Ognuno dei due dimeri principali è costituito da due
eliche antiparallele collegate ad un β-foglietto antiparallelo. Un β-foglietto e un‟elica sono
forniti da ogni monomero. L‟interfaccia tra i due dimeri che formano il tetrametro è mediata
solamente dai contatti elica-elica. Il risultato finale è un insieme di quattro eliche simmetriche,
con le eliche adiacenti orientate in senso antiparallelo le une con le altre e con i due β-foglietti
antiparalleli collocati sulla faccia opposta della molecola [10]. Indicazioni relative
all‟importanza
del
processo
di
oligomerizzazione
per
l‟esplicazione
dell‟attività
oncosoppressoria di p53, derivano principalmente dallo studio dei siti di legame della proteina
al DNA cellulare. La caratterizzazione di tali siti all‟interno del genoma, ha evidenziato come
ognuno di essi contenga due copie del decanucleotide 5‟Pu-Pu-Pu-C-(A/T)-(A/T)-G-Py-PyPy-3‟ separate da 0-13 coppie di basi con sequenza casuale. I siti che legano p53 hanno,
quindi, un‟ovvia simmetria (quattro copie dell‟emisito 5‟- (A/T)-G-Py-Py-Py-3‟ sono
orientate, a due a due, in direzione opposta a formare una struttura palindromica) [11]. Ciò
suggerisce come la proteina p53 possa legarsi a questi siti sotto forma di tetramero; risultato,
questo, confermato anche da studi biofisici che indicano che la proteina p53 in soluzione è
tetramerica. Diversamente da altri fattori di trascrizione, p53 possiede, inoltre, un secondo
dominio di legame al DNA che mappa nella regione C-terminale, caratterizzata
prevalentemente da α-eliche [Figura 1d].
86
AL Okorokov. Curr Opin Struct Biol 19, 2009
Figura 1. Struttura dei domini della proteina p53. (a) Dominio N-terminale di transattivazione (TAD,
transactivation domain; residui 1-98); (b) Dominio centrale localizzato all‟interno del DBD (core domain;
residui 102-292); (c) Dominio di oligomerizzazione (residui 323-363); (d) Dominio C-terminale (CTD, Cterminal domain; residui 363-393).
Tale dominio conferisce alla proteina p53 la capacità di formare complessi con sequenze non
specifiche di DNA, stabilizzando probabilmente l‟interazione del dominio centrale della
proteina con il DNA stesso e modulandone l‟attività di fattore trascrizionale.
La conformazione della proteina p53 è strettamente correlata alla sua funzione: un‟alterazione
di tale conformazione a causa, ad esempio, di eventi mutageni, può infatti comportare uno
switch nella funzione di p53 da repressore della crescita cellulare a promotore della
duplicazione cellulare. La struttura terziaria della proteina p53 sembra essere stabilizzata dalla
chelazione di residui conservati di cisteina con ioni metallici, probabilmente con ioni zinco
[12]. Due probabili regioni di legame per lo ione zinco (Cys135-X5-Cys141-X34-Cys176X2-His179 e Cys238-X3-Cys242-X32-Cys275-X1-Cys277), identificate nella proteina p53,
87
potrebbero formare un ponte tra i diversi domini e questo spiegherebbe come mutazioni
lontane anche più di 150 aminoacidi risultino avere un effetto simile sulla struttura terziaria.
Numerosi studi in vitro hanno, inoltre, contribuito a dimostrare come un altro fattore in grado
di influenzare lo stato conformazionale di p53 e quindi la sua funzione biologica, sia
rappresentato dalle modifiche post-traduzionali a cui viene esposta tale proteina, una volta
indotta [13]. Lo stato di fosforilazione di p53 viene classicamente considerato come lo step
cruciale per la sua stabilizzazione e attivazione. Una vasta gamma di differenti chinasi, tra cui
ATM/ATR/CDK2/PKC, media l‟aggiunta di gruppi fosfato ai residui di serina di p53 [14]. E‟
stato dimostrato che i tetrametri della proteina p53 possono essere presenti in forma latente o
attivata e che gli eventi di fosforilazione aumentano la capacità dei tetrameri in forma latente
di legarsi al DNA, convertendoli nella forma attivata. In conclusione, la proteina p53 è un
tetramero regolato in modo allosterico, normalmente presente in forma latente ma attivato
reversibilmente in seguito a condizioni di stress cellulare [15].
1.1.3 Regolazione dei meccanismi di attivazione della proteina p53
La proteina p53 è stata denominata “guardiano del genoma” a causa del suo ruolo centrale nel
coordinare le risposte cellulari alle varie tipologie e livelli di stress, in termini di arresto del
ciclo cellulare, riparo del DNA, apoptosi e senescenza. Il meccanismo primario attraverso cui
p53 media tali risposte è rappresentato dall‟attivazione di una serie di geni target. La
transattivazione p53-dipendente rappresenta quindi una caratteristica essenziale della risposta
cellulare allo stress, sebbene alcuni effetti di p53 possano essere indotti indipendentemente
dalla trascrizione genica. Recentemente sono stati annoverati 129 geni come target diretti di
p53 benché, con molta probabilità, questo numero sia destinato a crescere; inoltre, il numero
di geni la cui espressione risulta alterata indirettamente dopo induzione di p53 è dell‟ordine di
migliaia. Quindi, considerando che, come fattore trascrizionale, p53 è in grado di modulare
positivamente o negativamente un vasto numero di geni, il controllo della sua attività rende
necessario un complesso network di meccanismi di regolazione. Tali meccanismi sono
rappresentati principalmente da modifiche post-traduzionali cui va incontro la proteina sia in
condizioni di omeostasi che di stress cellulare. E‟ stato infatti dimostrato che più di 36
aminoacidi all‟interno della sequenza di p53 rappresentano potenziali target di modifiche
biochimiche [16]. Il modello “classico” di induzione della proteina p53 consiste in tre fasi
attivatorie sequenziali: 1) Stabilizzazione stress-dipendente di p53; 2) Legame sequenza
specifico di p53 al DNA; 3) Attivazione dei geni target mediante interazione con il
macchinario di trascrizione genica [Figura 2].
88
JP Kruse. Cell 137, 2009
Figura 2. Modello “cclassico” di induzione della proteina p53. Il modello “classico” di induzione della proteina p53 consiste
in tre fasi attivatorie sequenziali: 1) Stabilizzazione stress-dipendente di p53; 2) Legame sequenza specifico di p53 al DNA;
3) Attivazione dei geni target mediante interazione con il macchinario di trascrizione genica.
La prima fase è mediata da eventi di fosforilazione ATM/ATR-dipendenti a carico dei residui
di serina 15 e 28 presenti all‟interno del dominio N-terminale di transattivazione della
proteina p53. La fosforilazione di tali serine ha rappresentato una delle prime modifiche posttraduzionali identificate a carico di p53. Tale evento comporta la stabilizzazione della proteina
impedendone il legame con il suo principale inibitore, rappresentato da MDM2. In condizioni
di omeostasi, infatti, p53 è caratterizzata da una ridotta emivita ed è presente all‟interno della
cellula a livelli negligibili. Tali livelli sono mantenuti non solo grazie al controllo negativo
della traduzione dell‟RNA messaggero (mRNA) di p53, evento reso possibile grazie al
legame della proteina ribosomale L26 alla regione 5‟ non tradotta di tale mRNA, ma
principalmente grazie all‟attività della proteina MDM2 [17]. Tale proteina lega p53 a livello
del dominio N-terminale di transattivazione e inattiva p53 funzionando come ubiquitina-ligasi
E3, promuovendone, quindi, la degradazione attraverso la via del proteasoma, dopo l‟aggiunta
di molecole di ubiquitina [Figura 3]. MDM2 non ha in realtà un‟attività enzimatica intrinseca,
ma coopera con gli enzimi UbcH5B e UbcH5C per l‟ubiquitinazione di p53, che si verifica
prevalentemente a carico di residui di lisina presenti a livello dei domini di oligomerizzazione
e C-terminale della proteina. MDM2 è in grado di indurre sia la monoubiquitinazione che la
poliubiquitinazione di p53. Quale di questi eventi si verifichi dipende dai livelli di attivazione
di MDM2. Bassi livelli di attivazione inducono la monoubiquitinazione di p53 e il suo esporto
89
nucleare, con successiva degradazione a livello dei proteasomi citosolici o deubiquitinazione
e induzione della forma citosolica apoptoticamente attiva, mentre alti livelli ne promuovono la
poliubiquitinazione e la degradazione sia a livello dei proteasomi citosolici che nucleari [18].
In realtà, sebbene i livelli della proteina p53 siano elevati in assenza di MDM2, p53 continua
ad essere degradata in cellule murine MDM2-/-, suggerendo l‟esistenza di una via alternativa
di degradazione MDM2-indipendente [19]. A tale proposito, è stato recentemente dimostrato,
sia in colture tissutali che in esperimenti biochimici in vitro, il coinvolgimento di una serie di
altre ligasi E3, tra cui COP1, Pirh2 e Arf-BP1, nel controllo dei livelli intracellulari di p53.
Nonostante ciò, il ruolo dei meccanismi di degradazione MDM2-indipendente di p53, nella
risposta allo stress cellulare, deve essere ancora delucidato.
AN Bullock. Nat Rev Cancer 1, 2001
Figura 3. Meccanismo di degradazione della proteina p53 MDM2-dipendente. L‟oncosoppressore MDM2 lega
la proteina p53 a livello del dominio N-terminale di transattivazione e la inattiva funzionando come ubiquitinaligasi E3, promuovendone la degradazione attraverso la via del proteasoma, dopo l‟aggiunta di molecole di
ubiquitina.
90
Sono diversi i livelli di regolazione che correlano la funzione di MDM2 alla stabilizzazione
della proteina p53 durante la risposta allo stress. Il principale regolatore di MDM2 è
rappresentato dall‟oncosoppressore ARF [20]. ARF è una proteina nucleolare in grado di
stabilizzare p53 sia interferendo con il legame di p53 ad MDM2, sia inibendo direttamente
l‟attività di ubiquitina-ligasi di MDM2. I ridotti livelli di ARF in condizioni di omeostasi sono
infatti drammaticamente incrementati in condizioni di stress oncogenico, garantendo così il
controllo della proliferazione cellulare attraverso il blocco del ciclo e l‟apoptosi p53dipendenti. Un altro regolatore di MDM2 è rappresentato dalla proteina MdmX (anche nota
come MDM4) che, oltre alla sua capacità di interagire direttamente con p53, è in grado di
stabilizzare MDM2 e promuovere la sua attività di ubiquitina-ligasi. Inoltre, lo stesso MDM2
è sottoposto a regolazione mediante fosforilazione e acetilazione, entrambe modifiche in
grado di favorire, a seconda del sito interessato e dell‟enzima coinvolto, sia l‟inibizione che
l‟attivazione di MDM2.
Una volta ridotta l‟efficacia del ruolo inibitorio di MDM2 nei confronti di p53 grazie agli
eventi iniziali di fosforilazione a carico delle serine 15 e 28, un‟ulteriore fosforilazione della
treonina in posizione 18 consente il legame della proteina PIN1 (una prolil-isomerasi) a p53
con conseguente polimerizzazione dei domini PRD di p53, evento che riduce ulteriormente
l‟affinità di legame con MDM2 e, nel contempo, determina l‟aumento dell‟affinità di legame
con l‟acetil-trasferasi CBP/p300. Il legame covalente di un gruppo acetile ad un residuo di
lisina rappresenta un evento primariamente identificato a carico degli istoni e l‟acetilazione
istonica è nota avere un impatto importante sulla regolazione trascrizionale. p53 è stata
identificata come prima proteina non istonica in grado di essere regolata funzionalmente dalle
modifiche di acetilazione e deacetilazione. Tali eventi sono importanti per un efficiente
reclutamento dei cofattori trascrizionali e per l‟attivazione dei geni target di p53 in vivo.
L‟acetil-trasferasi CBP/p300 rappresenta il principale enzima acetilante di p53, sebbene
recentemente sia stato dimostrato il coinvolgimento di differenti acetil-trasferasi, quali Tip60
e hMof, nella regolazione dell‟attività di p53. I siti target di acetilazione sono rappresentati da
una sequenza di 6 lisine presente nella regione C-terminale e dalle lisine in posizione 120 e
164 presenti all‟interno del DBD. Una volta localizzate a livello dei promotori genici grazie al
legame con la proteina p53, le acetil-trasferasi facilitano la trascrizione non solo agendo
direttamente su p53 ma anche acetilando gli istoni presenti nelle vicinanze dei geni target,
stabilendo in questo modo una conformazione cromatinica più flessibile. Le conseguenze
funzionali dell‟acetilazione di p53 suggeriscono che il “timing” degli eventi di acetilazione
delle differenti regioni della proteina risulta importante per una sua accurata regolazione e per
91
la determinazione del destino cellulare. In realtà, a complicare ulteriormente il modello
relativo all‟attivazione di p53, bisogna ricordare che tale proteina rappresenta il target di
numerose altre modifiche post-traduzionali, tra le quali: metilazione, sumonilazione e
neddilazione, tutte coinvolte nel controllo della sua attività trascrizionale. Ad esempio, sono
state identificate tre differenti metil-transferasi in grado di metilare i residui di lisina Cterminali della proteina p53. La monometilazione mediata dalla metil-trasferasi Set7/9 a
livello della lisina K372 promuove l‟attivazione di p53, in particolare favorendo la
transattivazione di p21, mentre la monometilazione delle lisine K370 e K382 Smyd2- e
Set8/PR-Set7-dipendenti reprime l‟attività di p53 [21]. Inoltre, a sostegno dell‟importanza
degli eventi di metilazione nella regolazione di p53, vi è l‟osservazione che sia p53 sia gli
istoni che circondano la proteina una volta legata al DNA, sono metilati a livello dei residui di
arginina e che tale evento garantisce la specificità di legame di p53 ai promotori dei geni
target attraverso una modifica dell‟affinità.
Per ciascuna tipologia di risposta allo stress, almeno tre classi di geni sono attivate da p53. La
prima classe di geni target protegge le cellule dai potenziali effetti deleteri, sulla vitalità
cellulare, di un‟eccessiva attivazione di p53. Tali geni includono molecole come MDM2 o
MdmX e la loro induzione è indipendente dall‟acetilazione di p53. La regolazione positiva
dell‟espressione di MDM2 comporta un aumento della sua concentrazione intracellulare in
relazione alla presenza di p53, con induzione, quindi, di un meccanismo di feedback negativo
che permette una fine regolazione dell‟attività di p53 all‟interno della cellula [22]. La seconda
classe di geni comprende molecole implicate nell‟induzione dell‟arresto della crescita
cellulare, la cui attivazione rappresenta un evento fondamentale per garantire l‟inizio dei
processi di riparazione del DNA cellulare danneggiato in seguito all‟esposizione ai fattori di
stress. Tale evento richiede una parziale acetilazione della proteina p53. Il target principale di
tale classe di geni è rappresentato da p21, una molecola di 165 aminoacidi appartenente alla
famiglia degli inibitori delle cicline chinasi-dipendenti (CDK) che, attraverso il legame con il
complesso CycE/CDK2, induce il blocco del passaggio dalla fase G1 alla fase S del ciclo
cllulare [23]. Altri geni appartenenti alla seconda classe e regolati positivamente da p53 sono:
il gene GADD45, la cui espressione indotta da danni al DNA dovuti a radiazioni ionizzanti è
associata al blocco del ciclo cellulare in fase G1 mediato dall‟inibizione della formazione del
complesso CycB/Cdc2, e il gene TP53 stesso. Infine, la terza classe di geni indotti è costituita
dai regolatori del processo apoptotico, rappresentati principalmente da PUMA, Bax, NOXA e
APAF-1. Dal momento che l‟induzione del processo apoptotico, che si verifica qualora la
cellula non sia stata in grado di riparare efficacemente il danno a carico del DNA, rappresenta
92
un evento irreversibile, la sua piena attivazione presuppone che: tutti i coattivatori chiave
siano stati reclutati; p53 sia completamente acetilato e che i corepressori (MDM2 e MdmX)
siano stati completamente allontanati dai promotori target. In realta, come vedremo in seguito,
l‟induzione dell‟apoptosi non rappresenta l‟unico meccanismo attraverso cui p53 è in grado di
indurre l‟eliminazione di cellule caratterizzate da lesioni genetiche irreparabili. Un altro
importante meccanismo è infatti rappresentato dalla senescenza cellulare, un processo meno
noto da un punto di vista molecolare, ma altrettanto efficace nella soppressione della crescita
di cellule pre-cancerose.
E‟ stato recentemente dimostrato come la proteina p53 sia in grado non solo di attivare ma
anche di reprimere la trascrizione genica, sebbene i meccanismi di transrepressione così come
i target transrepressi non siano ancora del tutto compresi. Sembrerebbe che p53 regoli
negativamente quei geni la cui espressione risulta controllata da promotori che contengono
sequenze TATA-box o CAAT-box, probabilmente interagendo con i fattori trascrizionali che
si legano a tali sequenze e/o reclutando enzimi deacetilanti (DACs) a livello di tali promotori
[24]. E‟ stato ad esempio osservato come la soppressione della crescita indotta da p53 sia
accompagnata dalla diminuzione dell‟espressione del gene PCNA, codificante per l‟Antigene
Nucleare della Proliferazione Cellulare, una proteina nucleare che agisce come cofattore della
DNA polimerasi δ. Anche l‟espressione dell‟oncogene B-myb e del gene per la DNA
polimerasi α diminuiscono in relazione al blocco del ciclo cellulare mediato da p53. Si ritiene
che p53 controlli l‟espressione del gene B-myb direttamente e che la proteina prodotta da tale
oncogene regoli a sua volta la trascrizione del gene PCNA e del gene per la DNA polimerasi
α [25]. Inoltre, p53 potrebbe reprimere l‟espressione genica indirettamente, ovvero attraverso
la transattivazione di geni codificanti microRNA, regolatori negativi della trascrizione.
A complicare ulteriormente il modello relativo ai meccanismi di transattivazione e
transrepressione mediati dalla proteina p53 vi è poi la difficoltà di predire e identificare la
totalità dei geni da essa modulati considerando che: 1) non tutte le sequenze consenso
riconosciute da p53 a livello dei promotori genici sono occupate da tale proteina durante la
risposta allo stress cellulare; 2) non tutte le regioni del DNA cui p53 si associa una volta
indotto sono conformi alle sequenze consenso per p53; 3) non tutti i geni nelle vicinanze dei
siti di legame per p53 sono modulati positivamente come conseguenza di tale legame; 4) la
presenza di repressori della trascrizione genica potrebbe contrastare l‟attività di
transattivazione di p53 [26].
Una volta mediata la sua azione, la proteina p53 deve essere naturalmente sottoposta ad un
controllo negativo. A tale scopo, uno dei principali eventi di modulazione negativa è
93
rappresentato dalla sua deacetilazione, catalizzata da una serie di complessi di istonedeacetilasi (HDACs) contenenti HDAC1 o Sir2α /Sirt1 [27]. Le reazioni di deacetilazione si
verificano piuttosto rapidamente, una volta che l‟attivazione trascrizionale p53-dipendente
non è più richiesta, e il ripristino dei livelli normali di p53 all‟interno della cellula, dopo il
completamento del riparo del DNA, è cruciale per il ritorno alla normale omeostasi. Una delle
principali conseguenze della deacetilazione di p53 è quella di rendere la proteina suscettibile
all‟ubiquitinazione MDM2-dipendente, dal momento che l‟acetilazione di p53 inibisce tale
evento, come dimostrato sia in vitro che in vivo [28].
Nel complesso, il modello “classico” di attivazione di p53 consiste quindi nella
stabilizzazione stress-indotta della proteina, seguita da un intricato network regolatorio
finalizzato all‟induzione del legame al DNA e al controllo dell‟espressione dei geni target. In
realtà, recenti studi genetici effettuati mediante l‟introduzione di mutazioni puntiformi nella
sequenza di TP53, hanno rivelato aspetti relativi alla regolazione della proteina, che non
possono essere spiegati sufficientemente secondo il modello “classico”. E‟ stata di
conseguenza proposta l‟esistenza di una ridondanza regolatoria tra le modifiche posttrasduzionali di p53 che assicurerebbe il controllo appropriato della proteina in risposta ai
diversi stimoli di stress e alle loro differenti intensità, e che quindi assicurerebbe l‟appropriata
specificità tissutale. Tali osservazioni hanno inoltre condotto a ridefinire il modello “classico”
di attivazione di p53 mediante l‟introduzione di un‟ulteriore fase rappresentata dagli eventi di
antirepressione a carico della proteina, eventi che avverrebbero a seguito della sua
stabilizzazione e prima dell‟attivazione trascrizionale dei geni target e contribuirebbero
all‟induzione della proteina p53 in relazione all‟insieme di tutte le modifiche post-traduzionali
a suo carico [Figura 4]. L‟introduzione di tale fase deriva dall‟osservazione che gli inibitori
MDM2 e MdmX sono in grado di legare p53 anche a livello dei promotori dei geni target,
nascondendo
il
suo
dominio
di
transattivazione
al
complesso
di
trascrizione.
Conseguentemente, MDM2 e MdmX non solo mediano la degradazione di p53 attraverso la
via del proteasoma, ma agiscono anche reprimendone direttamente l‟attività di fattore
trascrizionale [29]. Tale osservazione ha indotto a supporre che p53 sia probabilmente
intrinsecamente attivo all‟interno della cellula e che necessiti di una continua repressione. Il
legame di p53 ai suoi repressori a livello del DNA viene principalmente regolato da eventi di
fosforilazione e acetilazione a carico di residui aminoacidici chiave. Naturalmente, gli eventi
di antirepressione, da soli, garantiscono solo l‟attivazione di geni altamente responsivi come
quelli coinvolti nel blocco del ciclo cellulare, ma risultano insufficienti per l‟induzione dei
geni mediatori dell‟apoptosi, i cui promotori, infatti, sono legati da p53 a bassa affinità, per
94
cui tale evento richiederebbe modifiche post-traduzionali addizionali di p53. Di conseguenza,
un segnale di stress dovrebbe indurre un arresto reversibile del ciclo cellulare mentre la
risposta apoptotica irreversibile richiederebbe eventi stimolatori addizionali.
1. Stabilization
2. Antirepression
3. Promoter-specific activation
JP Kruse. Cell 137, 2009
Figura 4. Modello “rrifinito” di attivazione della proteina p53. L‟osservazione dell‟esistenza di aspetti relativi
alla regolazione della proteina p53 che non possono essere spiegati sufficientemente secondo il modello
“classico” di attivazione ha condotto a ridefinire tale modello mediante l‟introduzione di una ulteriore fase
rappresentata dagli eventi di antirepressione a carico della proteina p53. Questi eventi avverrebbero a seguito
della stabilizzazione di p53 e prima dell‟attivazione trascrizionale dei geni target e contribuirebbero
all‟induzione della proteina in rapporto all‟insieme di tutte le modifiche post-traduzionali a suo carico.
95
1.1.4 Ruolo della proteina p53 nella soppressione tumorale
La proteina p53, grazie alla sua capacità di avvertire la presenza di un danno a carico del
DNA con conseguente induzione dell‟arresto del ciclo cellulare e della riparazione del DNA
o, nel caso in cui il danno sia esteso, promozione dell‟apoptosi o della senescenza, svolge un
ruolo cruciale nella soppressione tumorale, prevenendo l‟espansione di cellule anormali
potenzialmente cancerose. A sostegno dell‟importanza di p53 nel controllo della tumorigenesi
è stato infatti dimostrato che oltre il 50% di tutti i tumori umani presentano mutazioni del
gene TP53 e che alterazioni genetiche a carico di proteine che regolano o mediano l‟attività di
p53 si riscontrano in buona parte della restante metà dei tumori; il gene MDM2, ad esempio, è
amplificato in almeno il 7% dei tumori umani p53 negativi [30]. Inoltre, i pazienti affetti dalla
sindrome di Li-Fraumeni, una rara patologia ereditaria a carattere autosomico dominante il cui
difetto genetico è rappresentato, nella maggior parte dei casi, dalla presenza di una mutazione
somatica a carico di uno dei due alleli del gene TP53, sono caratterizzati da un‟elevata
predisposizione allo sviluppo di tumori multipli in età precoce.
I principali meccanismi antitumorali mediati dalla proteina p53 sono rappresentati
dall‟apoptosi
e dalla
senescenza
cellulare.
L‟apoptosi
è un
processo
coinvolto
nell‟eliminazione di cellule ad alto rischio di trasformazione neoplastica mentre la senescenza
cellulare opera da barriera della tumorigenesi mediante l‟induzione di un arresto irreversibile
del ciclo cellulare. p53 agisce direttamente o indirettamente e a multipli livelli nell‟induzione
dell‟uno o dell‟altro processo. L‟apoptosi è un fenomeno universale negli organismi
multicellulari ed è alla base di processi fondamentali come l‟organogenesi, l‟omeostasi
tissutale e la selezione negativa dei cloni linfocitari autoreattivi. Viene normalmente indotta
dai processi di controllo dello sviluppo ma anche insulti tossici per la cellula sono alla base
della sua induzione. La proteina p53 è in grado di attivare la via apoptotica sia in modo
trascrizione-dipendente che indipendente. Quest‟ultimo meccanismo sarà esaminato nel
sottoparagrafo successivo relativo alle funzioni citoplasmatiche della proteina p53, mentre per
quanto riguarda l‟induzione dell‟apoptosi in modo trascrizione dipendente, tale processo
consiste essenzialmente nella transattivazione di target pro-apoptotici mediatori sia della via
intrinseca che estrinseca del processo di morte cellulare. Nell‟ambito dei target coinvolti nella
via intrinseca possono essere annoverati i geni codificanti le molecole appartenenti alla
famiglia Bcl-2, quali Bax e le proteine BH3-only (Bid, PUMA, NOXA) che possono fungere
da attivatori o derepressori dei fattori pro-apoptotici e OKL38, un oncosoppressore in grado di
localizzarsi a livello mitocondriale e favorire il rilascio del Citocromo c. Per quanto riguarda i
target coinvolti nella via estrinseca, p53 induce l‟espressione dei recettori e dei ligandi di
96
morte cellulare quali DR5 e CD95, TRAIL e FasL, rispettivamente. p53 è inoltre in grado di
attivare la trascrizione dei geni codificanti per i componenti del macchinario effettore
dell‟apoptosi, rappresentati da APAF-1 e le Caspasi 6 e 8, e per fattori meno noti come la
proteina PIG-3, che induce stress ossidativo e causa un aumento dei radicali reattivi
dell‟ossigeno, e IGF-BD (Insulin Growth Factor-Binding Protein), che a sua volta compete
per il legame con il recettore del fattore di crescita insulinico 1 (IGF1R), bloccando la via di
trasduzione dei segnali di sopravvivenza trasmessi da questo recettore in presenza di IGF1.
Contemporaneamente, p53 reprime la trascrizione di proteine anti-apoptotiche come Bcl-2,
Bcl-XL, la proteina ARC che contrasta le funzioni apoptotiche di PUMA e Bad, la Survivina
e la Galectina-3, un inibitore del rilascio di Citocromo c mitocondriale [31]. E‟ importante
sottolineare che l‟apoptosi mediata da p53 agisce non solo come meccanismo di difesa per
proteggere l‟organismo dalla propagazione di cloni cellulari con mutazioni permanenti, ma è
anche coinvolto nella modulazione dell‟attività citotossica di diversi farmaci antitumorali.
Studiando la relazione esistente tra l‟attività di p53 e la sensibilità o resistenza a vari agenti
antitumorali è stato dimostrato che tale proteina è necessaria per la morte cellulare
programmata indotta indirettamente da questi farmaci. Le cellule tumorali con mutazioni del
gene TP53 potrebbero, di conseguenza, diventare resistenti agli agenti antitumorali con
peggioramento della prognosi del paziente.
In realtà, diverse evidenze indicano che funzioni mediate da p53 e diverse dall‟apoptosi sono
ugualmente importanti nella soppressione tumorale. E‟ stato ad esempio osservato che topi
PUMA-/- sono protetti dalla tumorigenesi sebbene caratterizzati dalla compromissione di uno
dei principali mediatori apoptotici di p53. Inoltre, topi esprimenti proteine p53 mutanti
contraddistinte da perdita della capacità di induzione della morte cellulare ma da preservata
capacità di induzione dell‟arresto della crescita, non sviluppano, nella maggior parte dei casi,
tumori. Tale osservazione ha consentito di identificare il blocco del ciclo come ulteriore
meccanismo mediato dalla proteina p53, efficace nella soppressione della crescita di cellule
pre-cancerose. Comunque, un arresto transiente della crescita potrebbe essere rischioso, se il
potenziale oncogenico cellulare non fosse contrastato e la cellula riprendesse a proliferare; di
conseguenza, la risposta più appropriata consiste nell‟arresto irreversibile della crescita, noto
con il termine di “Senescenza cellulare” [32]. L‟accorciamento dei telomeri è un meccanismo
universale utile a eliminare il potenziale proliferativo delle cellule normali a seguito di
estensivi cicli di divisioni cellulari. Tale processo è noto come “Senescenza Replicativa” ed è
stato ipotizzato che l‟erosione telomerica oltre un limite definito, possa comportare
l‟attivazione di una risposta al danno a carico del DNA associata all‟induzione della via
97
ATM/ATR-p53 e all‟arresto del ciclo cellulare. In realtà, è stata recentemente dimostrata
anche l‟esistenza di una forma di senescenza nota con il termine di “Senescenza Accelerata” o
“Senescenza Prematura” che risulta indipendente dall‟accorciamento telomerico ma viene
indotta dalla risposta delle cellule a condizioni di stress [33]. In ogni caso, le cellule che non
sono in grado di andare incontro a senescenza e continuano a proliferare nonostante siano
caratterizzate da telomeri disfunzionali o siano sottoposte a condizioni di stress, acquistano, di
conseguenza, aberrazioni cromosomiche che possono comportare la loro trasformazione
maligna. La senescenza rappresenta quindi un meccanismo evolutivamente conservato di
soppressione tumorale, che agisce come barriera naturale all‟immortalizzazione cellulare. La
proteina p53 è in grado di controllare la senescenza cellulare sia dipendente che indipendente
dall‟accorciamento telomerico. I principali segnali induttori di senescenza attraverso la via di
p53 indipendente dall‟accorciamento telomerico, sono rappresentati da: danno al DNA;
attivazione inappropriata di oncogeni (es: Ras, RUNX1 e RUNX1-ETO); stress ossidativo,
deplezione di chaperoni molecolari (es: Hsp70) e attività di farmaci chemioterapici (es:
Ciclofosfamide; Etoposide) [34]. I meccanismi responsabili dell‟attivazione di p53 nelle
cellule senescenti sono solo parzialmente compresi, sebbene le evidenze molecolari stiano
crescendo. Tra le cause di attivazione identificate sinora, l‟incremento dell‟espressione
dell‟oncosoppressore ARF, che agisce inibendo la regolazione negativa MDM2-dipendente di
p53, e l‟attivazione dell‟oncosoppressore PML che interagisce con l‟acetiltrasferasi CBP/p300
stabilizzando indirettamente p53 mediante la sua acetilazione [35-36]. Anche i meccanismi
attraverso cui p53 induce la senescenza sono attualmente incompresi. L‟induzione di p21
risulta importante per la senescenza indotta dal danno al DNA tanto quanto lo è per
l‟induzione dell‟arresto transiente della crescita, sebbene quale meccanismo sia alla base della
decisione tra l‟arresto definitivo o transiente della crescita, attraverso l‟attivazione di p21, non
sia stato ancora del tutto delucidato. E‟ stato suggerito che un efficiente riparo del DNA
comporti l‟inibizione della via p53-p21, consentendo la progressione nel ciclo cellulare,
mentre lesioni irreparabili del DNA sostengano l‟attivazione della via ATM/ATR-p53-p21
con acquisizione del fenotipo di senescenza. p53 sembra inoltre indurre la senescenza
cellulare anche attraverso la regolazione dell‟espressione dell‟attivatore del plasminogeno
(PAI-1), un noto marcatore di senescenza, e del gene MIC-1, codificante una citochina della
famiglia del TGF-β [37]. Una volta attivata, p53 può indurre, quindi, a seconda delle
circostanze, l‟apoptosi o l‟arresto irreversibile del ciclo cellulare. Tuttavia, non è ancora del
tutto chiaro quali meccanismi siano alla base della scelta di un destino cellulare di morte o
senescenza, sebbene la loro conoscenza potrebbe essere importante per modulare la risposta di
98
p53 nelle cellule tumorali. Per la comprensione di tali meccanismi è necessario considerare la
tipologia di stress cellulare, la sua durata e intensità, quindi lo stato quantitativo e qualitativo
della proteina p53 in termini di conformazione, localizzazione, stabilità e attività,
caratteristiche che a loro volta dipendono sia da modifiche post-traduzionali a carico di p53
sia da interazioni dirette proteina-proteina. E‟ stato ad esempio dimostrato che l‟acetilazione
della lisina 120, presente all‟interno del DBD di p53, da parte delle acetil-trasferasi Tip60 e
hMOF, favorisce l‟induzione dell‟apoptosi e che la prevenzione di tale evento si associa ad
una selettiva riduzione della transattivazione dei geni pro-apoptotici Bax e PUMA [38]. Lo
stesso effetto ha la fosforilazione della serina 46 di p53 mediata dalle chinasi p38, DYRK2 o
HIPK2, grazie all‟induzione di un cambio conformazionale che consente a p53 di dissociarsi
dall‟inibitore dell‟apoptosi iASPP e allo stesso tempo altera la specificità di legame di p53 ai
promotori e co-attivatori specifici per l‟apoptosi quali p63, p73 e STAT1 [39]. Inoltre, in un
recente studio è stato dimostrato come l‟acetilazione della lisina 320 PCAF-mediata causi
l‟ipofosforilazione dei residui N-terminali di p53 consentendo l‟induzione di p21, mentre
l‟acetilazione della lisina 373 induca la fosforilazione dei residui N-terminali della proteina
con attivazione dei geni pro-apoptotici [40]. Sembrerebbe poi che l‟attivazione di alcuni
oncogeni, come MYC, sia coinvolta nell‟induzione preferenziale dell‟apoptosi ARF/p53dipendente attraverso la soppressione specifica del promotore di p21; mentre l‟oncogene Ras
rappresenta, ad esempio, un tipico mediatore di senescenza nei fibroblasti primari [41-42].
Infine, l‟attivazione dell‟uno o dell‟altro meccanismo di soppressione tumorale sarebbe
influenzata dal tipo di cellula interessata. Ad esempio, le cellule linfoidi sono intrinsecamente
predisposte ad andare incontro all‟apoptosi, contrariamente ai fibroblasti e alle cellule
epiteliali che vanno normalmente incontro a senescenza.
Considerando la complessità dei meccanismi alla base dell‟induzione dei diversi processi di
soppressione tumorale all‟interno delle cellule, acquisire una maggiore conoscenza dei
determinanti molecolari alla loro base diviene quindi particolarmente importante per il
disegno di strategie terapeutiche antineoplastiche.
1.1.5 Funzioni citoplasmatiche della proteina p53
In aggiunta alle sue funzioni nucleari di attivatore trascrizionale, p53 possiede, in realtà,
anche attività biologiche citosoliche trascrizione-indipendenti. E‟ stato osservato che
l‟overespressione di una forma mutata del gene TP53, caratterizzata dalla mancanza della
maggior parte del DBD e completamente inefficiente nella funzione di transattivazione, può
ancora essere associata all‟induzione del processo apoptotico. Similmente, l‟attivazione di
99
p53 risulta nell‟apoptosi anche in cellule caratterizzate dall‟assenza del nucleo o in condizioni
di parziale o totale blocco trascrizionale [43]. L‟abilità di p53 di indurre la via apoptotica in
maniera indipendente dalla trascrizione genica è una conseguenza degli effetti
dell‟associazione di tale proteina alla membrana mitocondriale. Una serie di differenti
condizioni di stress cellulare, tra cui l‟esposizione a radiazioni ionizzanti, stimolano, infatti, la
rapida traslocazione di p53 sulla superficie esterna della membrana mitocondriale (MOM), a
livello della quale p53 induce una variazione di permeabilità, causando il rilascio di fattori
pro-apoptotici dallo spazio trasmembrana mitocondriale [44]. La permeabilizzazione della
MOM (MOMP) è solitamente inibita dal legame di molecole anti-apoptotiche appartenenti
alla famiglia Bcl-2 (quali: Bcl-2, Bcl-XL e Mcl1) e, al contrario, attivata da molecole
appartenenti alla stessa famiglia, ma con funzione pro-apoptotica (Bim, Bak e Bax). In
relazione alla loro affinità ai componenti della famiglia Bcl-2, una serie di proteine proapoptotiche “BH3-only” (come PUMA e NOXA) è in grado di interagire direttamente con
Bax o Bak inducendone l‟omo-oligomerizzazione, favorendo in questo modo la MOMP e/o
contrastando l‟attività dei fattori anti-apoptotici. Una serie crescente di evidenze ha
dimostrato come in condizioni pro-apoptotiche p53 si comporti, a livello citosolico, come una
proteina “BH3-only”, funzionando da attivatore diretto di Bax e Bak o da derepressore. Sono
stati proposti due diversi modelli relativi all‟azione di p53 a livello mitocondriale. Secondo il
primo modello, p53 in condizioni di omeostasi è sequestrata, a livello della MOM, dalle
proteine anti-apoptotiche Bcl-2 e Bcl-XL. I segnali di stress cellulare inducono il rilascio del
legame di p53 dai fattori anti-apoptotici grazie all‟azione di spiazzamento esercitata dalla
proteina PUMA. In questo modo p53 può essere attivata e può associarsi alle proteine Bax e
Bak, inducendo la loro oligomerizzazione, con conseguente aumento della permeabilità
mitocondriale e rilascio del Citocromo c [45]. In base al secondo modello, invece, p53 è in
grado di legare esclusivamente Bcl-2 e Bcl-XL; questo legame permette a Bak e Bax di
sganciarsi da Bcl-2 e Bcl-XL, dalle quali sono inattivate all‟interno delle cellule, in condizioni
normali. L‟azione di p53 diventa in tal caso quella di inattivare Bcl-2 e Bcl-XL, in modo tale
che Bak e Bax possano a loro volta essere attivate dalla forma tronca di Bid (tBid), un fattore
pro-apoptotico che induce la loro oligomerizzazione, con conseguente formazione dei pori
sulla membrana mitocondriale esterna [46]. Quindi, secondo il primo modello p53 si
comporterebbe, al pari di Bid, come un attivatore dell‟apoptosi, in quanto in grado di legare
direttamente Bax e Bak; in base al secondo modello, invece, p53 si comporterebbe, al pari di
Bad, come un sensibilizzatore dell‟apoptosi, in quanto in grado di legare direttamente solo
Bcl-2 e Bcl-XL e, quindi, di attivare solo indirettamente Bak e Bax. In realtà, gli effetti
100
citoplasmatici pro-apoptotici di p53 sono trascrizione-indipendenti solo in parte, in quanto il
controllo della trascrizione mediato da p53 contribuisce decisivamente alle sue funzioni
citoplasmatiche. Infatti, senza la regolazione dell‟espressione di MDM2 da parte di p53, il
sistema non sarebbe in grado di rispondere ai fattori di stress cellulare in quanto p53 non
potrebbe stabilizzarsi e accumularsi a livello sia citosolico che nucleare. Inoltre, anche la
proteina PUMA rappresenta un target dell‟attività di transattivazione di p53, senza il quale
p53 non potrebbe contrastare il sequestro citoplasmatico mediato da Bcl-2 e Bcl-XL. In
conclusione, le due vie di induzione dell‟apoptosi trascrizione-dipendente e trascrizioneindipendente sono strettamente correlate, e quindi, mutazioni del gene TP53 in cellule
neoplastiche hanno la potenzialità di abolire contemporaneamente entrambe le attività.
Un‟ulteriore funzione della proteina p53 citoplasmatica è rappresentata dalla capacità di
controllare il fenomeno dell‟autofagia, ovvero il sequestro e la successiva digestione, da parte
delle cellule, di porzioni del citoplasma, finalizzata all‟adattamento a condizioni di stress così
come alla rimozione di strutture citosoliche danneggiate e quindi potenzialmente dannose. Dal
momento che l‟autofagia ha un ruolo essenziale nel mantenimento della stabilità genomica,
l‟inibizione dell‟autofagia rappresenta un evento oncogenico. Sebbene p53 sia in grado di
transattivare geni induttori di autofagia come DRAM e le Sestrine 1 e 2, livelli normali di p53
mediano invece un segnale tonico inibitorio nei confronti dell‟autofagia. La soppressione
dell‟autofagia è mediata dalla proteina p53 citoplasmatica attraverso il blocco dell‟attività
della chinasi AMP-dipendente, un regolatore positivo dell‟autofagia, e l‟induzione di mTOR,
un target della Rapamicina con funzione di regolatore negativo dell‟autofagia. E‟ stato
proposto un modello secondo cui l‟inibizione dell‟autofagia e l‟induzione della MOMP
costituiscono una risposta coordinata finalizzata a facilitare l‟attivazione della morte cellulare.
L‟autofagia, infatti, attraverso la rimozione dei mitocondri danneggiati o permeabilizzati,
contrasta gli effetti letali della MOMP, per cui, la sua inibizione mediata da p53 potrebbe
facilitare l‟esecuzione della morte cellulare tramite MOMP. Non sono tuttavia ancora chiari i
meccanismi attraverso cui p53 possa passare dalla sua funzione basale di inibitore
dell‟autofagia a quella di induttore della MOMP a livello citoplasmatico [47]. Potrebbe
apparire paradossale che livelli citoplasmatici normali di p53 inibiscano l‟autofagia,
considerando che tale evento è essenzialmente oncogenico. In realtà, tale paradosso può
essere risolto prendendo in considerazione l‟osservazione che la maggior parte delle forme
mutate di p53, che ha quindi perso la sua attività di transattivazione genica, è ancora in grado
di inibire l‟autofagia. Di conseguenza, nei casi in cui p53 non è più in grado di esercitare la
sua azione di fattore pro-apoptotico, la soppressione dell‟autofagia può diventare
101
sostanzialmente dannosa, promuovendo la progressione tumorale. Questo potrebbe
contribuire alla potente azione oncogenica di alcuni mutanti, difficilmente spiegabile
considerando esclusivamente la mera inibizione delle capacità oncosoppressive di p53.
Gli effetti della proteina p53 citoplasmatica e di quella nucleare sono determinati da una serie
di modificazioni post-traduzionali, che stabiliscono il destino della proteina modulandone
l‟interazione con altri fattori e/o lo spostamento da un compartimento cellulare all‟altro. E‟
stato dimostrato che la poli(ADP)ribosilazione di p53 comporta un accumulo nucleare della
proteina; al contrario, la sua monoubiquitinazione da parte di MDM2 induce un esporto
nucleare della proteina che, una volta giunta a livello dei mitocondri, può essere
deubiquitinata dall‟enzima mitocondriale HAUSP trasformandosi nella forma citosolica
apoptoticamente attiva. Altre modificazioni post-traduzionali di p53, come la fosforilazione
delle serine C-terminali, possono stimolare l‟esporto nucleare e/o l‟associazione alla MOM.
Inoltre, è stato osservato che il fattore trascrizionale Foxo3a promuove l‟accumulo
citoplasmatico di p53, aumentando il suo esporto nucleare. Infine, la proteina p53 nucleare
risulta acetilata, mentre la forma citoplasmatica è normalmente deacetilata. Ulteriori studi
sarebbero comunque utili al fine di chiarire meglio quali modifiche post-traduzionali di p53
determinano la sua attività pro-MOMP o anti-autofagia.
1.1.6 Ruolo dei microRNA nel controllo dell’attività della proteina p53
Fino a poco tempo fa, si riteneva che i componenti del network di p53 fossero rappresentati
esclusivamente da geni codificanti prodotti proteici, inclusi quelli coinvolti nella regolazione
dell‟attività di p53, sia a monte che a valle, e nella sua mediazione. Recenti studi hanno
invece dimostrato che l‟oncosoppressore p53 è in grado di modulare anche l‟espressione di
una serie di microRNA (miR), tra i quali un ruolo primario è stato attribuito alla famiglia dei
miR34. L‟identificazione di tali miR come componenti della via di p53 ha rappresentato la
prima rivelazione dell‟esistenza di una correlazione tra proteine e RNA non codificanti,
nell‟ambito della soppressione tumorale. I miR sono una classe di piccoli RNA ad attività
regolatoria coinvolti nel silenziamento post-trascrizionale di specifici mRNA target. La
famiglia dei miR34 comprende un insieme di microRNA (mir34-a, mir34-b e mir34-c)
altamente conservati da un punto di vista evolutivo, direttamente indotti da p53 in risposta
alla presenza di un danno a carico del DNA o in condizioni di stress oncogenico [48]. Sia
stimoli di natura esogena che endogena sono infatti in grado di indurre l‟espressione dei
miR34, secondo una modalità p53-dipendente, in sistemi cellulari in vitro e in tessuti animali.
Tale induzione non dipende da sintesi proteica de-novo, ma dalla presenza di siti di legame
102
intatti per la proteina p53 a livello dei promotori genici dei miR34 [49]. La cinetica e
l‟intensità di induzione dell‟espressione di tali miR sono paragonabili a quelle di altri target
canonici di p53, come p21. E‟ stato dimostrato che l‟espressione ectopica di tali miR
ricapitola gli effetti di p53, con induzione dell‟arresto della crescita o dell‟apoptosi, attraverso
l‟abilità di reprimere l‟espressione di geni proliferativi e anti-apoptotici. In particolare,
l‟overespressione del miR34-a in fibroblasti primari e in alcune linee tumorali, causa un
significativo arresto del ciclo cellulare, evidente dall‟incremento di cellule in fase G1 e
diminuzione in fase S. Inoltre, a seconda del tipo cellulare, l‟overespressione ectopica del
miR34-a è in grado di indurre senescenza, come avviene in fibroblasti primari di polmone
umano, o apoptosi, come nel caso della maggior parte delle linee tumorali [50]. Ad ogni
modo, data la potenzialità dei miR di riconoscere molteplici target a causa della capacità di
appaiamento imperfetto di basi, gli effetti pleiotropici del miR34-a potrebbero semplicemente
riflettere il vasto spettro di mRNA disponibili come target all‟interno di un dato sistema
cellulare. Approcci bioinformatici e sperimentali sono stati impiegati per identificare i target
attraverso cui i miR34 contribuiscono all‟attività di p53: gli studi di espressione genica hanno
rivelato che l‟attivazione di tali miR è in grado di causare la downmodulazione di centinaia di
mRNA, rappresentati soprattutto da quelli codificanti i regolatori del ciclo cellulare quali
CDK4, CDK6, la ciclina E2, i fattori trascrizionali E2F1/3 e Notch1. A differenza dei geni
proliferativi, i geni anti-apoptotici non sono particolarmente rappresentati nell‟insieme dei
target dei miR34; tuttavia Bcl-2, un regolatore negativo dell‟apoptosi, risulta downregolato
dai miR34 in svariati tipi cellulari, confermando il ruolo di tali miR nell‟apoptosi indotta da
p53 [51].
La scoperta del contributo dei miR34 alla regolazione dell‟attività di p53 ha condotto ad
intraprendere numerosi studi finalizzati all‟identificazione di eventuali ulteriori miR target di
p53. E‟ stata in questo modo individuata una dozzina di miR caratterizzati da pattern di
espressione indicativi di una regolazione mediata da p53, sebbene solo un numero esiguo di
essi è stato confermato quale target della proteina in studi multipli. Tra i suddetti miR vanno
inclusi: i miR15 e 16, coinvolti nella regolazione post-trascrizionale di Bcl-2; let7, modulatore
negativo di Ras e del miR221 che a sua volta downmodula l‟inibitore delle CDK p27 e i
miR372 e 373, che contrastano la senescenza p53-dipendente indotta dall‟oncogene Ras [52].
In accordo con il loro ruolo nel controllo dell‟attività di p53, i miR34 rappresentano frequenti
target di eventi di mutagenesi nell‟ambito delle patologie tumorali. Ridotti livelli di tali miR
sono stati riscontrati sia in tumori umani che in linee tumorali in vitro. Sebbene infatti la
pressione selettiva per l‟acquisizione di delezioni a carico dei geni codificanti i miR34 sia
103
alleviata dall‟elevata frequenza di mutazioni a carico del gene TP53, tale evento è stato
riportato in diversi forme tumorali umane TP53 wild-type. Ad esempio, la regione 1p36 su cui
mappa il gene codificante il miR34-a, rappresenta un sito di frequente delezione eterozigote.
Recentemente un altro gene presente all‟interno di tale regione, codificante la proteina
cromodominio CHD5, è stato implicato nell‟attività oncossoppressiva del suddetto locus. E‟
stato proposto che CHD5 agisca a monte di p53 regolandone l‟espressione, in risposta a
diverse condizione di stress, attraverso l‟attivazione di ARF. In questo modo, la delezione del
locus 1p36 influenzerebbe l‟integrità della via di p53 sia a monte e a valle [53].
Considerando il ruolo dell‟alterazione del funzionamento dei miR nella tumorigenesi, come
accade nel caso del contributo alla perdita di integrità della via di p53, diviene quindi
importante capire se essi hanno uno specifico ruolo di oncosoppressori nella biologia cellulare
o se tale osservazione riflette semplicemente la loro funzione di regolatori flessibili dei
programmi di espressione genica.
1.2 Meccanismi di deregolazione a carico del gene TP53 e carcinogenesi
1.2.1 Mutazioni del gene TP53
Noto il ruolo primario della proteina p53 nella soppressione tumorale, mutazioni a carico del
gene TP53 rappresentano le più comuni alterazioni genetiche associate a tumori umani. Esse
avvengono con differente frequenza in quasi tutte le neoplasie ereditarie e sporadiche finora
studiate, compresi il carcinoma del colon, il carcinoma della mammella, il carcinoma
epatocellulare, i tumori del polmone e dell‟esofago, i tumori cerebrali, le leucemie e i linfomi,
il tumore della vescica e quello dell‟ovaio. Mutazioni germinali in uno dei due alleli del gene
TP53 determinano, inoltre, un‟alta predisposizione allo sviluppo di tumori multipli; è questa
una condizione familiare nota come sindrome di Li-Fraumeni [54]. Sia mutazioni somatiche
che acquisite di TP53 possono poi essere accompagnate dal fenomeno della “perdita di
eterozigosità” nel corso della progressione tumorale; un evento causato da una forza selettiva
di inattivazione del rimanente allele wild-type. In questi ultimi anni si è iniziato a chiarire
come l‟inattivazione della funzione oncosoppressiva p53-dipendente, rappresenti una tappa
quasi universale nello sviluppo dei tumori umani, in quanto la funzione fisiologica della
proteina p53 risulta essenziale per il mantenimento del fenotipo normale, non neoplastico,
delle cellule [55]. La perdita della funzione oncosoppressiva della proteina p53 è in molti casi
un evento tardivo associato alla progressione del tumore da neoplasia benigna a neoplasia
maligna. Nei carcinomi mammari, polmonari, prostatici, renali, nel neuroblastoma, nei
104
carcinomi della testa e del collo, nell‟epatocarcinoma e nel melanoma, le mutazioni del gene
TP53 e l‟accumulo della proteina anomala sono particolarmente rilevanti nei casi
clinicamente più aggressivi e meno differenziati.
La localizzazione e le caratteristiche delle mutazioni di TP53 rappresentano importanti
indicatori dell‟eziologia e della patogenesi molecolare dei tumori. In generale, la maggior
parte delle mutazioni di TP53 possono essere classificate, in relazione al loro effetto sulla
stabilità termodinamica della proteina, in due categorie, note con il termine di mutazioni di
“contatto” e “conformazionali” [56]. Alla prima classe appartengono mutazioni a carico di
residui direttamente coinvolti nel legame della proteina p53 al DNA; la seconda classe
comprende invece mutazioni che causano distorsioni nella conformazione di p53 di natura
locale o globale. A differenza della maggior parte degli altri geni oncosoppressori, che vanno
tipicamente incontro a inattivazione biallelica mediante l‟acquisto di delezioni o mutazioni
troncanti nel corso della tumorigenesi, TP53 viene inattivato, in circa il 75% dei casi, da
singole mutazioni monoalleliche di natura missenso, che causano la produzione di una
proteina alterata altamente stabile e facilmente rivelabile all‟interno delle cellule tumorali
[57]. Tali mutazioni colpiscono solitamente le porzioni di sequenza codificanti quattro regioni
della proteina p53, altamente conservate nell‟evoluzione. Queste regioni comprendono i
codoni 117-142, 171-181, 236-258 e 270-286, che mappano all‟interno del DBD di p53; per
cui mutazioni a carico di tali siti comportano solitamente l‟abrogazione della capacità di
legame di p53 al DNA [Figura 5]. Inoltre, almeno 1/3 di tutte le mutazioni missenso interessa
6 residui definiti “punti caldi” di mutazione e rappresentati dai codoni R248 e R273, che
rientrano nella classe dei mutanti di “contatto”; G245 e R249 che rappresentano mutanti
“conformazionali” con influenza locale sulla distorsione delle proteina p53; e infine R175 e
R282, mutanti “conformazionali” con effetto globale sulla distorsione strutturale di p53 [58].
La frequenza e la distribuzione dei punti caldi di mutagenesi varia a seconda della diversa
tipologia tumorale, probabilmente in relazione al tipo e alla quantità di mutageno che può
raggiungere il tessuto e anche in relazione alle diverse pressioni selettive sulla crescita
cellulare alle quali il tessuto è sottoposto [59]. Oltre alle mutazioni puntiformi, che sono
quelle che intervengono più frequentemente nell‟inattivazione del gene TP53, sono state
riscontrate anche piccole delezioni o inserzioni nel gene, nonché alterazioni della proteina
dovute al legame con oncoproteine virali o cellulari, che inibiscono la capacità della forma
“wild type” di legarsi al DNA e attivare la trascrizione genica.
La tipologia delle mutazioni di TP53 è influenzata non solo da processi di selezione, ma
anche da fattori intrinseci che interessano, differenzialmente, specifici nucleotidi e regioni del
105
gene, e rivelano un coinvolgimento di meccanismi mutagenici endogeni nella formazione del
tumore. Le transizioni a livello dei siti CpG sono infatti più frequenti a causa dell‟elevata
mutagenicità di tali regioni e rappresentano una forza chiave nell‟influenzare la distribuzione
delle mutazioni nel gene TP53. Ad esempio, l‟alta frequenza di transizioni da C a T in siti
dinucleotidici CpG nel carcinoma del colon è molto probabilmente dovuta a meccanismi di
deaminazione endogena [60]. Lo spettro di mutazioni del gene TP53 può inoltre dipendere e,
quindi, dare informazioni sui particolari carcinogeni e meccanismi biologici che intervengono
nella formazione di uno specifico tumore [61]. Infatti, è stato dimostrato che differenti
carcinogeni causano mutazioni caratteristiche specifiche. Ad esempio, le radiazioni UV
determinano preferenzialmente transizioni a livello di siti dipirimidinici sul DNA;
l‟esposizione all‟aflatossina B1 è correlata a trasversioni da G:C a T:A che portano ad una
sostituzione aminoacidica del residuo 249 della proteina p53 nel carcinoma epatocellulare;
l‟esposizione al fumo di sigaretta provoca trasversioni da G:C a T:A nei carcinomi del
polmone, della testa e del collo.
CJ Brown. Nat Rev Cancer 9, 2009
Figura 5. Siti della sequenza del gene TP53 preferenzialmente interessati dalle mutazioni. La maggior parte
delle mutazioni inattivanti ricade all‟interno del dominio di legame con il DNA (DBD), mentre il dominio di
transattivazione è relativamente libero da mutazioni. Le mutazioni rappresentate in giallo (R248; R273)
interessano i punti di contatto con il DNA; quelle in azzurro (G245; R249) causano distorsioni locali della
struttura proteica di p53 e quelle in blu (R175; R282) eventi di denaturazione globali.
Gli effetti fenotipici delle mutazioni del gene TP53 possono sostanzialmente essere
classificati in tre gruppi non mutualmente esclusivi. Al primo gruppo appartiene la maggior
parte delle mutazioni di TP53 osservate nei tumori umani; tali mutazioni sono caratterizzate
dalla capacità di abrogare o attenuare l‟efficienza di legame di p53 al DNA e, di conseguenza,
106
impediscono la regolazione dei promotori genici attivati dalla proteina p53. In genetica, tali
mutazioni sono definite “ipomorfiche” o “amorfiche”, a seconda che causino una parziale o
totale perdita di funzione. La perdita di funzione è frequente tra le mutazioni missenso,
sebbene sia particolarmente rilevante nel caso di mutazioni nonsenso, troncanti o di splicing,
così come nel caso delle delezioni. Il secondo gruppo comprende quelle mutazioni che
causano la produzione di una proteina con effetto dominante-negativo (DN), ovvero in grado
di inibire, a svariati livelli, la funzione della proteina normale prodotta dall‟allele “wild type”.
Questo effetto DN “antimorfico” può essere ottenuto grazie alla capacità della proteina mutata
di formare con il prodotto “wild type” etero-oligomeri inattivi o difettivi funzionalmente,
incapaci cioè di attivare la trascrizione dei geni bersaglio di p53 [62]. La fosfoproteina
nucleare p53 agisce, infatti, solo dopo avere formato complessi oligomerici (omotetrameri).
Se tali complessi sono costituiti, anche parzialmente, da una proteina p53 alterata in siti critici
della molecola, la loro capacità di interagire con il DNA risulta compromessa e le specifiche
funzioni soppresse. E' quindi sufficiente che una sola copia del gene TP53 subisca una
mutazione perché il suo prodotto, interagendo con quello dell'allele normale, formi
omotetrameri a scarsa affinità per il DNA. L‟osservazione, tuttavia, che nella maggior parte
dei tipi cellulari tumorali con mutazioni nel gene TP53 venga perso anche l‟altro allele “wild
type”, sembra indicare che le mutazioni dominanti negative siano in realtà piuttosto rare.
Infine, al terzo gruppo appartengono quelle mutazioni di TP53, solitamente di natura
missenso, caratterizzate dall‟abilità di conferire alla proteina mutata, non solo la perdita della
funzione soppressiva sulla crescita cellulare propria della proteina “wild type”, ma anche
l‟acquisto di nuove funzioni tumorigeniche, indipendenti dalla proteina “wild type”. Tali
proprietà “neomorfiche” di guadagno di funzione (GOF) sono di natura oncogenica
dominante e possono essere dimostrate sperimentalmente in assenza della proteina p53
funzionale, overesprimendo, ad esempio, in cellule TP53-/-, una forma mutata di p53 con
presunte proprietà GOF [63-65]. Si ritiene che la maggior parte di tali proprietà dipenda
dall‟abilità della forma mutata di p53 di interagire con proteine cellulari, come i fattori
trascrizionali, alterandone la normale attività. Sebbene quindi la maggior parte delle
mutazioni di TP53 causi una perdita della capacità di transattivazione dei geni target, la
modulazione della trascrizione genica rappresenta uno dei principali meccanismi GOF della
proteina mutante. Ad esempio, le mutazioni di TP53 sono spesso associate a resistenza ai
farmaci in svariate neoplasie. Tale fenomeno sembrerebbe dipendere in parte dall‟attivazione
trascrizionale, da parte della proteina p53 mutata, del gene MDR1, codificante un fattore
coinvolto nella resistenza multifarmaco [66]. A partire dalla dimostrazione del controllo della
107
trascrizione genica di MDR1, numerosi altri geni sono stati identificati quali target della forma
mutata di p53. La maggior parte di tali geni manca della sequenza consenso di legame al
DNA per p53, per cui è stato ipotizzato che i mutanti di p53, sebbene difettivi nel legame
sequenza-specifico al DNA, mantengano l‟abilità di legare ad alta affinità specifiche strutture
genomiche ricche in elementi ripetuti [67]. Tra le altre proprietà oncogeniche della forma
mutata di p53, coinvolte nel controllo della trascrizione genica, va menzionata anche l‟abilità
di interagire con fattori trascrizionali sequenza-specifici. Alcuni mutanti di p53 sono, ad
esempio, in grado di legare e inattivare gli altri membri della sua famiglia proteica, p63 e p73,
con inibizione della loro capacità di indurre il blocco del ciclo cellulare [68]. Infine, dati
recenti implicano la forma mutata di p53 nel controllo dell‟espressione dei geni coinvolti nei
fenomeni infiammatori, come quelli codificanti citochine, chemochine e modulatori della
matrice extracellulare. In realtà, esistono anche forme di GOF indipendenti dalla modulazione
della trascrizione genica. E‟ stato dimostrato che i mutanti p53R248W e p53R273H sono in grado
di legare MRE11, un componente a monte della via di risposta al danno a carico del DNA
ATM-dipendente, con conseguente inibizione del riparo di rotture a doppio filamento del
DNA [69]. Tale evento non solo giustificherebbe l‟associazione tra la forma mutata di p53 e
l‟instabilità cromosomica dei tumori umani, ma avrebbe importanti implicazioni cliniche,
probabilmente favorendo la produzione di cloni neoplastici resistenti a quegli agenti
terapeutici che agiscono inducendo rotture a doppio filamento a carico del DNA [70].
Le forme mutate di p53 presentano, ad ogni modo, alcune caratteristiche generali che le
contraddistinguono dalla proteina “wild type”. In particolare, hanno un tempo di emivita
molto maggiore e pertanto una concentrazione intracellulare molto più elevata a livello basale,
di conseguenza, la rilevazione immunoistochimica di p53 nelle cellule tumorali indica
solitamente la presenza di mutazioni missenso e può garantire informazioni prognostiche
predittive; hanno, inoltre, una conformazione diversa e quindi espongono epitopi non presenti
nella conformazione “wild type”. La capacità di accumulo intracellulare della forma mutata di
p53 sembra dipendere dalla sua impossibilità di transattivare il gene codificante MDM2 che,
normalmente, controlla i livelli di p53 inducendone la degradazione attraverso la via del
proteasoma [71]. In realtà, è stato recentemente dimostrato che le mutazioni missenso
inattivanti sono insufficienti, da sole, a causare l‟accumulo della proteina mutata e che, con
molta probabilità, eventi addizionali sono richiesti, durante la tumorigenesi, per garantire tale
effetto. Questo spiegherebbe il perché la proteina p53 mutata non si accumula in cellule sane
di pazienti affetti dalla sindrome di Li-Fraumeni, ma solo in cellule tumorali dello stesso
soggetto [72]. Esistono inoltre anche meccanismi di accumulo MDM2-indipendenti; alcune
108
forme mutate, ad esempio, pur mantenendo la proprietà di legare MDM2 caratteristica della
proteina “wild-type”, sono resistenti all‟ubiquitinazione mediata da MDM2; altre forme
mutate possono essere invece stabilizzate dall‟interazione con fattori quali PTEN o le “heat
shock proteins” Hsp90 e 70, che ne prevengono la degradazione dopo ubiquitinazione [73].
L‟accumulo della proteina mutata diventa, quindi, il presupposto per favorire la sua attività
DN e/o GOF, come dimostrato in diversi modelli sperimentali sia in vitro che in vivo. In
cellule embrionali murine derivate da un topo knock-in per una forma mutata di p53,
all‟accumulo della proteina consegue infatti un incremento nel tasso di crescita; una ridotta
inibizione da contatto della crescita e un‟aumentata abilità di cooperazione con l‟oncogene
Ras [74]. E‟ ancora oggetto di dibattito quale proprietà della forma mutata di p53 sia
preferenzialmente selezionata durante lo sviluppo tumorale: la perdita di funzione, l‟acquisto
di proprietà dominanti negative, il guadagno di nuove funzioni oncogeniche o addirittura una
combinazione di tutte e tre le caratteristiche sopraelencate. Ad ogni modo, la natura della
mutazione, la sua estensione all‟interno della popolazione neoplastica, la capacità di formare
oligomeri e la localizzazione cellulare della proteina, sono tutti fattori che influenzano la
capacità trasformante delle forme mutanti di p53. La conoscenza quindi del tipo e numero di
mutazioni che riguardano il gene TP53, del loro effetto e della specifica relazione tra
mutazione ed esposizione ad un particolare carcinogeno assumono un grande interesse, perché
permettono di identificare tra i soggetti sani le persone ad alto rischio per lo sviluppo di una
neoplasia, e possono permettere di ottenere, in questo modo, una diagnosi precoce che
dovrebbe consentire un miglioramento dell‟approccio terapeutico alla malattia. Inoltre
mutazioni di questo genere potrebbero avere diverse e specifiche connotazioni prognostiche e
terapeutiche nelle varie forme neoplastiche in campo clinico.
1.2.2 Impatto dello stato del gene TP53 sulla prognosi tumorale
Dati iniziali piuttosto inconsistenti, relativi all‟associazione tra la presenza di mutazioni a
carico del gene TP53 e la sopravvivenza o la risposta alla terapia, hanno condotto ad un
dibattito circa il ruolo prognostico e predittivo dello stato di TP53 nelle diverse forme
tumorali, ritardando, di conseguenza, l‟introduzione della valutazione dello stato di tale gene
in ambito clinico [75]. Una delle ragioni principali di tale inconsistenza è rappresentata
dall‟impiego, nella maggior parte degli studi effettuati sino a poco tempo fa, della rilevazione
immunocitochimica (ICC) dell‟accumulo intracellulare della proteina p53 mutata. Infatti,
l‟assegnazione indiretta dello stato del gene TP53 mediante il solo impiego dell‟ICC risulta
spesso inaccurata, dal momento che in molti tumori caratterizzati dalla presenza di mutazioni
109
di TP53 non si osserva un accumulo intracellulare della proteina; questo risulta
particolarmente vero nel caso di mutazioni frameshift, nonsenso e di splicing. Inoltre, non
tutti i tumori con mutazioni missenso di TP53 appaiono ICC positivi; alcuni tumori
accumulano la forma “wild-type” di p53 a causa della persistenza di segnali di stress e infine,
altre forme tumorali inattivano la funzione della proteina normale attraverso meccanismi
indipendenti da eventi mutazionali come l‟amplificazione di MDM2 o la deregolazione di altri
componenti a monte o a valle della via di p53 [76]. A complicare ulteriormente il quadro, la
mancanza di protocolli standard e di cut-off univoci di rilevamento dell‟accumulo della
proteina mutata. Più recentemente, grazie all‟introduzione di metodiche di sequenziamento
genico, è stato possibile ottenere una valutazione più accurata dello stato di TP53 e, di
conseguenza, un‟associazione maggiormente affidabile tra mutazioni di TP53 e caratteristiche
cliniche della malattia tumorale, con la tendenza generale ad associare tali alterazioni a ridotta
sopravvivenza, ridotto intervallo di malattia libero dal trattamento e scarsa risposta
terapeutica. In realtà, in neoplasie coinvolgenti il seno, il collo, l‟intestino ed il sistema
emopoietico, il 65-90% degli studi riscontra un‟associazione tra mutazioni del gene TP53 e
prognosi avversa, mentre nei tumori polmonari o ovarici tale correlazione risulta meno chiara.
Ad ogni modo, una combinazione di ICC e sequenziamento genico potrebbe rappresentare un
migliore strumento diagnostico; è stato infatti dimostrato che mutanti del gene TP53 ICC+
hanno una prognosi peggiore dei mutanti di TP53 ICC- [77]. Inoltre, per una fine correlazione
delle disfunzioni di p53 alla prognosi tumorale, diviene necessario non solo il rilevamento di
mutazioni a carico del gene TP53, ma anche un attento esame degli effetti delle singole
mutazioni sulla struttura e/o funzione della proteina, che a loro volta dipendono dall‟esone o
dal dominio funzionale interessato e dallo specifico residuo mutato, quindi un‟analisi dello
stato di addizionali oncosoppressori ed oncogeni regolatori o mediatori della via di p53.
Nell‟ambito di uno studio su larga scala comprendente 1749 pazienti affetti da tumore al seno
è stato, ad esempio, dimostrato non solo che mutazioni missenso del gene TP53 a livello della
regione del DBD sono associate a prognosi peggiore rispetto a mutazioni missenso che
ricadono all‟esterno del DBD, ma anche che nell‟insieme delle mutazioni missenso, alcune
risultano più aggressive di altre e che, infine, pazienti caratterizzati da mutazioni troncanti o
nonsenso presentano solitamente una prognosi più infausta [78].
Lo stato del gene TP53 deve essere quindi considerato non come una variabile binaria, ma un
insieme multi-parametrico di dati, affinchè la sua analisi possa acquisire un valore reale nella
pratica clinica tanto per la diagnosi tumorale precoce, il rilevamento della recidiva, la
valuatazione della prognosi e la predizione della resistenza alla terapia antineoplastica quanto
110
per il disegno di innovative strategie terapeutiche finalizzate al ripristino dell‟attività della
proteina mutata disfunzionale.
1.2.3 Ripristino della funzionalità della proteina p53 nei tumori umani
Considerando l‟elevata frequenza di alterazioni molecolari e funzionali a carico
dell‟oncosoppressore p53 nei tumori umani, il ripristino della sua normale attività continua a
rappresentare una delle ambizioni primarie nell‟ambito delle terapia antineoplastica.
Naturalmente, il potenziale di tale approccio è strettamente correlato alla natura della proteina
p53 endogena. Infatti, a causa della ridondanza funzionale dei componenti a monte e a valle
della via di p53, mentre una metà delle forme tumorali si caratterizza per l‟inattivazione
funzionale diretta della proteina, a causa della presenza di mutazioni a carico del gene TP53,
nella restante metà l‟inattivazione della proteina è una conseguenza indiretta dell‟alterazione
di molecole coinvolte nel controllo o nella mediazione della sua attività. E‟ stato ipotizzato
che le proprietà pro-sopravvivenza di p53, facilitando il recupero di cellule danneggiate o in
condizioni transienti di stress, possano favorire il mantenimento della sua integrità e ridirigere
la pressione selettiva nei confronti di altri target della via di p53. Questo potrebbe significare
che le cellule tumorali sono, almeno all‟inizio, maggiormente dipendenti dalle funzioni di p53
rispetto alla loro controparte normale, almeno fino a che mutazioni oncogeniche addizionali
non fanno perdere qualsiasi tipo di competenza omeostatica. Alternativamente, p53 potrebbe
essere selezionata per una perdita parziale di attività, con mantenimento delle funzioni
vantaggiose per la crescita tumorale, ed acquisire nuove proprietà oncogeniche quando, ad
esempio, indotta da segnali di stress. Tutti questi elementi che dipendono, ovviamente, dal
tessuto e dalla linea cellulare interessati e riflettono la vulnerabilità degli elementi genetici in
relazione al loro grado di espressione o stato cromatinico, influenzano la diversità dei
meccanismi di inattivazione della proteina p53 nelle patologie tumorali [79].
Nell‟ambito delle forme tumorali caratterizzate dalla presenza della proteina p53 “wild-type”,
numerosi studi si sono focalizzati sull‟identificazione di piccole molecole o peptidi in grado di
proteggere p53 dall‟attività del suo inibitore cellulare, MDM2. Infatti, l‟inattivazione di p53
ottenuta attraverso l‟overespressione di MDM2, evento conseguente, nella maggior parte dei
casi, all‟amplificazione del segmento cromosomico su cui mappa tale gene, è molto frequente
nei tumori umani. Molteplici studi hanno permesso di validare MDM2 quale target
terapeutico; è stato ad esempio dimostrato, in sistemi murini caratterizzati da bassi livelli di
espressione di MDM2 a causa della presenza di un allele ipomorfico del suo gene, che
minime riduzioni nei livelli di tale proteina bastano a garantire un incremento di attività della
111
forma “wild-type” di p53 [80]. I primi antagonisti di MDM2 con attività in vivo identificati
sono rappresentati dalle “Nutline”, una serie di molecole derivate da composti di cisimidazolina. Le “Nutline” agiscono legando il sito di interazione per p53 presente all‟interno
di MDM2 attraverso una modalità che mima le interazioni molecolari tra MDM2 ed una serie
di residui aminoacidici cruciali della proteina p53 [81]. In questo modo, le “Nutline”
impediscono il riconoscimento di p53 da parte del suo inibitore per cui la proteina non viene
ubiquitinata e degradata attraverso la via del proteasoma ma si accumula con conseguente
aumento dei livelli intracellulari ed attivazione dei target trascrizionali. Le “Nutline”, di cui
“Nutlin-3” rappresenta il prototipo, agiscono in modo altamente specifico ed indipendente
dalla stabilizzazione di p53 associata all‟induzione di un danno a carico del DNA e la loro
efficacia è stata dimostrata in diversi modelli sperimentali. Cellule cancerose proliferanti che
esprimono la proteina p53 “wild-type” sono efficacemente arrestate in fase G1 e G2 del ciclo
o vanno incontro ad apoptosi se trattate con concentrazioni micromolari di “Nutline”; questo
implica che la tipologia cellulare influenza l‟induzione di una risposta differenziale sebbene i
meccanismi molecolari alla base di tale differenza siano ancora oggetto di studio [82].
In realtà, nonostante vi siano risultati promettenti relativamente all‟uso degli inibitori di
MDM2, bisogna ricordare le potenziali problematiche correlate alla scelta dell‟interazione
MDM2-p53 come target terapeutico. In primo luogo, MDM2 viene indotto dall‟attivazione di
p53 come parte di un meccanismo di feedback negativo di regolazione, di conseguenza
farmaci come “Nutlin-3” causerebbero un‟induzione del loro stesso target, con limitazione
dell‟efficacia medica; in secondo luogo le molecole sinora prodotte appaiono inefficaci
nell‟inibizione di MdmX che ha un‟attività sovrapponibile a quella di MDM2 e risulta allo
stesso modo overespresso in una vasta quantità di forme tumorali.
Approcci differenti sono stati invece identificati per la riattivazione della funzione di p53
nelle cellule tumorali caratterizzate dalla presenza di forme mutate della proteina. Tali
approcci sono resi possibili grazie all‟osservazione che le mutazioni a carico di TP53 cadono
nella maggior parte dei casi nei siti “hot spots” all‟interno del DBD, il loro effetto è
reversibile e tali mutazioni causano solitamente la stabilizzazione e l‟accumulo di p53 nelle
cellule tumorali, al contrario di quanto accade per altri oncosoppressori (pRB e p16) la cui
inattivazione si realizza tramite una riduzione dell‟espressione. La ricostituzione della corretta
funzionalità di p53 permetterebbe l‟eliminazione delle cellule tumorali, andando a ristabilire i
programmi di riparo e/o garantendo l‟eliminazione delle cellule il cui DNA risulta
danneggiato [83]. Il presupposto di tali approcci è rappresentato dal ripristino della
conformazione normale di p53 e quindi della sua capacità di legame al DNA, mediante
112
“correzione chimica”. Infatti, sebbene le mutazioni a carico del gene TP53 siano da sole
insufficienti ad indurre lo sviluppo tumorale, la conversione della forma mutata di p53 in
“wild-type” può rivelarsi costrittiva nei confronti della crescita, forse a causa dei livelli
anormalmente elevati di tale proteina e dei continui segnali attivatori che permeano le cellule
neoplastiche. E‟ noto che il dominio centrale di p53 è piuttosto instabile, con un‟emivita
media di 9 minuti a temperatura corporea. Le mutazioni del gene TP53 causano solitamente
un‟ulteriore destabilizzazione termica della proteina a temperatura corporea quindi, sulla base
della dimostrazione che i processi di unfolding e/o misfolding del dominio centrale delle
forme mutanti di p53 non sono irreversibili e che la loro gravità varia tra le diverse forme
mutanti per cui una sostanza capace di legarsi alla frazione correttamente foldata può spostare
l‟equilibrio verso il folding nativo, una serie di studi sono stati condotti per l‟identificazione
di molecole capaci di stabilizzare la forma mutata di p53 nella sua conformazione biologica
[84]. A tale proposito, le estremità C- ed N-terminale rappresentano dei target idonei, nota la
loro capacità di influenzare e regolare il legame del DBD di p53 al DNA. I primi risultati
hanno portato alla realizzazione di anticorpi, come pAb421, in grado di legare la porzione Cterminale di p53 ed aumentare la resistenza del dominio centrale alla denaturazione termica. È
stato quindi dimostrato che un peptide sintetico derivato dal dominio C-terminale di p53, il
peptide 46, può ripristinare il corretto legame al DNA e la funzione di transattivazione
trascrizionale di un certo numero di proteine p53 mutanti e che il recupero della funzione
delle proteine p53 endogene mutate risulta nell‟inibizione della crescita cellulare e
nell‟induzione dell‟apoptosi. È stato ipotizzato che i peptidi derivanti dall‟estremità Cterminale di p53 siano in grado di riattivare le forme di p53 mutate attraverso la
stabilizzazione del folding del dominio core e/o stabilendo nuovi contatti col DNA [85]. La
stabilizzazione del dominio centrale di p53 può essere altresì mediata dal legame a chaperoni
cellulari. La proteina p53 può legarsi a Hsp40, Hsp70 e Hsp90. Le forme mutate di p53 si
legano a Hsp70 con un‟affinità maggiore della forma “wild-type”. Le proteine Hsp
stabilizzano p53 nella sua conformazione “unfolded”; la distruzione del loro legame alla
forma mutata di p53 consente il recupero della conformazione di p53 [86]. Sono state inoltre
identificate, grazie allo screening di librerie di piccole molecole, sostanze di natura non
peptidica in grado di ripristinare l‟attività dei mutanti di p53. Tra queste, CP-31398 ha
dimostrato la capacità di indurre arresto del ciclo cellulare ed apoptosi p53-dipendente sia in
vitro che in vivo in modelli murini atimici e modelli murini di cancro [87]. Il suo meccanismo
di azione rimane tuttora ignoto. Gli studi tramite MNR non hanno rilevato nessun legame tra
la molecola e il dominio centrale di p53. E‟ stato supposto che CP-31398 agisca aumentando i
113
livelli dalla proteina p53 “wild-type” nelle cellule, prevenendone l‟ubiquitinazione. Mediante
l‟utilizzo di approcci basati sull‟impiego di sistemi cellulari è stato invece identificato un
composto, noto con il termine di “PRIMA-1” (p53 reactivation and initiation of massive
apoptosis), efficace nella riattivazione della transattivazione dei geni target in cellule
caratterizzate dalla forma mutata di p53, secondo un meccanismo dipendente dall‟induzione
dell‟espressione della proteina Hsp90 e dall‟attivazione della via della chinasi JNK (Jun-NH2kinase) [88]. Infine, sono state proposte anche strategie di terapia genica finalizzata
all‟introduzione di copie extra del gene TP53; all‟induzione dell‟espressione selettiva di geni
killer nelle cellule contenenti copie di p53 non funzionali o all‟utilizzo di virus replicazionedifettivi che possano propagarsi nelle cellule p53-deficienti. Tuttavia, l‟assenza di un
efficiente “sistema di consegna e distribuzione” ha precluso, almeno finora, gli approcci
mirati ad una somministrazione sistemica.
La pletora di composti attivi identificati si dimostra tuttavia inutilizzabile nella pratica clinica,
per problemi legati sia alla tossicità che alla reale efficacia. L‟ulteriore accumulo di
informazioni riguardo la struttura di p53 e il miglioramento delle tecniche di screening e
sviluppo di nuovi farmaci diventano quindi necessari per la sperimentazione di molecole
innovative sempre più efficaci e meno tossiche.
1.3 Meccanismi di deregolazione della via di p53 in pazienti affetti da LLC
L‟importanza dello studio dei meccanismi di deregolazione nei componenti della via di p53
nell‟ambito della LLC è emersa sostanzialmente grazie alla dimostrazione della differenza
fondamentale del decorso clinico dei pazienti caratterizzati dalla delezione del braccio corto
del cromosoma 17 (del17p13). La del17p13 viene riscontrata nel 5-7% dei pazienti in fase
precoce di malattia e nel 25-40% dei pazienti affetti da LLC in stadio avanzato. Tali pazienti
presentano, al contrario della maggior parte dei soggetti affetti da LLC, una prognosi
altamente infausta, contraddistinta da rapida progressione di malattia, resistenza o refrattarietà
al trattamento chemioterapico e sopravvivenza media da 2 a 3 anni dalla diagnosi [89]. La
cattiva prognosi associata alla del17p13 è legata all‟evidenza che il gene TP53 mappa sul
braccio corto del cromosoma 17 e che delezioni di questo cromosoma interessano nella quasi
totalità dei casi il suddetto gene [90]. Inoltre, nella maggior parte dei pazienti caratterizzati
dalla del17p13 (del17p13+), l‟inattivazione del rimanente allele del gene TP53 attraverso
meccanismi mutageni, contribuisce in modo fondamentale all‟avversità della prognosi. E‟
importante poi sottolineare come la del17p13 non si presenti quasi mai come unica
114
aberrazione cromosomica all‟interno delle cellule tumorali. Uno studio del profilo genomico
dei pazienti affetti da LLC del17p13+ ha, infatti, rivelato che la presenza della suddetta
delezione si associa solitamente a un cariotipo complesso, caratterizzato dalla comparsa di
anomalie citogenetiche addizionali come la del13q14, che interessa il locus codificante i
miR15 e 16; la trisomia 12 e, sebbene in minor percentuale, le alterazioni a carico delle
regioni cromosomiche 8p, 9q, 2p e 3q. In particolare, la del8p21-p23, interessando il locus
dell‟oncosoppressore TRAIL1/2, sembrerebbe essere associata a resistenza al Nutlin-3, un
farmaco impiegato nelle patologie neoplastiche con alterazioni della via di p53 causate da
aberrante attività di MDM2, la cui azione apoptotica nelle cellule di LLC è TRAIL2dipendente [91]. La del17p13 rappresenterebbe, quindi, un marcatore di cellule caratterizzate
da un genoma instabile, incline all‟acquisizione di lesioni genetiche e all‟evoluzione clonale.
E‟ stato dimostrato, in due diversi studi prospettici, che la comparsa della del17p13 in pazienti
affetti da LLC è in grado di predire una riduzione del tasso di risposta alla terapia
convenzionale a base di analoghi purinici ed una riduzione della sopravvivenza media [92].
Tali effetti risultano, inoltre, nell‟ambito di tali studi, strettamente correlati al dosaggio della
del17p13, in quanto pazienti caratterizzati dalla presenza del 5-20% di cellule nucleate delete
mostrano una prognosi simile a pazienti non deleti (del17p13-), mentre pazienti con una
percentuale di delezione superiore al 20 presentano una prognosi infausta, con tasso di
risposta terapeutica del 13% e sopravvivenza media di soli 11 mesi [93].
In realtà, la maggior parte degli studi relativi al decorso clinico dei pazienti del17p13+ affetti
da LLC è stata condotta esaminando soggetti con malattia progressiva destinati ad essere
inclusi in protocolli terapeutici di prima linea o precedentemente sottoposti a terapia
antitumorale. A tal proposito le osservazioni riportate, nonostante la loro importanza sia ormai
consolidata, non garantiscono la comprensione degli effetti relativi alla presenza della
delezione al momento della diagnosi piuttosto che durante il follow-up del paziente. La
scarsità di dati in tale ambito deriva essenzialmente dalla difficoltà di osservare pazienti affetti
da LLC del17p13+ asintomatici e in stadi precoci di malattia. E‟ stato, infatti, osservato che le
anomalie citogenetiche sono normalmente acquisite durante il decorso della malattia
neoplastica, per cui la del17p13 costituisce frequentemente un evento secondario.
Un recente studio è stato condotto al fine di comprendere la valenza prognostica della
presenza della del17p13 in pazienti affetti da LLC non progressiva, non esposti a terapia. Dai
risultati del suddetto studio è emerso che circa il 50% dei soggetti con del17p13 “de novo”
sviluppa una malattia progressiva che richiede terapia entro 12-18 mesi dalla diagnosi, mentre
la restante metà è caratterizzata da una malattia relativamente stabile che non necessita di
115
terapia ad un follow-up di 70 mesi [94]. Queste osservazioni sottolineano l‟importanza che
assume il momento in cui la delezione viene acquisita dal paziente nel determinare le
implicazioni cliniche ad essa correlate e sostengono la possibilità di una associazione non
assoluta tra presenza della del17p13 e avversità della prognosi. Di conseguenza, data
l‟eterogeneità del decorso clinico dei pazienti con del17p13 “de novo”, risulta indispensabile
evitare di sottoporre i suddetti pazienti a un approccio terapeutico differente, esclusivamente
sulla base della presenza della delezione. La sopravvivenza dovrebbe, in tal caso, essere
predetta attraverso la valutazione di ulteriori parametri sia clinici che biologici. Diventa ad
esempio di grande interesse, dal momento che la prognosi avversa della del17p13 è
solitamente correlata ai suoi effetti sulla via di p53, valutare se i pazienti con LLC del17p13 +
apparentemente benigna sono caratterizzati dall‟allele non deleto del gene TP53 “wild-type” o
da una normale funzione della proteina, entrambi fattori che permetterebbero di giustificare
l‟andamento favorevole della malattia in tale sottogruppo. Infatti, nonostante sia stata
osservata una frequente associazione tra la presenza della del17p13 e la comparsa di
mutazioni a carico del gene TP53 sull‟allele non deleto, è stato tuttavia riscontrato un numero
minore di casi di LLC in cui la delezione non risulta associata a tali mutazioni (0.8% dei casi)
o viceversa, l‟acquisizione delle mutazioni di TP53 risulta indipendente dalla del17p13 (4.5%
dei casi) [95].
Sebbene la presenza di mutazioni a carico del gene TP53 nei pazienti affetti da LLC sia nota
sin dalla fine degli anni ‟90, il suo preciso significato prognostico, in particolare nei casi in
cui non si associa alla del17p13, è meno chiaro. La sua comprensione diventa di grande
rilevanza clinica se si considera che il gene TP53 risulta mutato nel 5-40% dei pazienti affetti
da LLC, con una incidenza del 5-10% alla diagnosi, in soggetti non trattati, e del 25-40% in
fase di progressione di malattia o nei casi di LLC resistenti alla chemioterapia a base di
analoghi purinici o agenti alchilanti [96]. Inoltre, da un punto di vista clinico, identificare la
presenza di mutazioni a carico del gene TP53 come fattore prognostico indipendente,
permetterebbe di chiarire, come nel caso della del17p13 e indipendentemente da essa, il suo
ruolo nella predizione dell‟aggressività della malattia e nella resistenza o refrattarietà ai
regimi terapeutici convenzionali. Diversi studi sono stati intrapresi per comprendere se le
mutazioni del gene TP53 in assenza della del17p13 conferiscono lo stesso profilo biologico,
le stesse caratteristiche cliniche e la stessa prognosi delle mutazioni di TP53 associate a
delezione e i primi risultati sembrano confermare tale ipotesi [Figura 6] [97].
116
T Zenz. Nat Rev Cancer 10, 2009
Figura 6. Meccanismi di alterazione a carico del gene TP53 nella LLC. a) Confronto della sopravvivenza dei
pazienti affetti da LLC stratificati in base all‟assenza o presenza della del17p13 e delle mutazioni del gene TP53;
b) La del17p13 è invariabilmente associata alla perdita del gene TP53, come confermato dall‟analisi FISH; c)
Multiple aberrazioni genomiche interessano la via di p53 nella LLC: la figura mostra uno schema
esemplificativo di tale via e della risposta al danno al DNA.
Un recente studio italiano condotto su 308 pazienti affetti da LLC ha, infatti, dimostrato che le
mutazioni del gene TP53 alla diagnosi sono associate a breve sopravvivenza e resistenza alle
chemioterapie convenzionali, indipendentemente dalla del17p13 [98]. Le mutazioni di TP53
in pazienti del17p13- mostrano un profilo prognostico infausto sovrapponibile a quello dei
pazienti del17p13+, che include l‟associazione con lo stato non mutato dei geni IgHV; la
frequente presentazione in fase avanzata di malattia e la correlazione con marcatori
proliferativi. Inoltre tali mutazioni predispongono il paziente al rischio di progressione della
malattia verso la fase sintomatica, che richiede trattamento terapeutico, a chemioresistenza e a
una riduzione della sopravvivenza media. Da questo studio è inoltre emerso che, come accade
117
anche in altre forme di neoplasie umane, il 95% delle mutazioni di TP53 identificate in
pazienti affetti da LLC cade nel DBD della proteina. Sono, infatti, principalmente interessati
gli esoni 5-8. Si tratta, nella maggior parte dei casi, di mutazioni missenso sebbene siano state
identificate anche mutazioni nonsenso, mutazioni di splicing e microdelezioni o
microinserzioni che comportano uno slittamento della cornice di lettura del codice genetico.
Sono inoltre maggiormente frequenti le transizioni piuttosto che le trasversioni. Tali risultati
sono stati confermati anche da altri studi paralleli. Un gruppo di ricerca tedesco ha dimostrato
che l‟inattivazione monoallelica di TP53 nella LLC si associa a prognosi infausta e, in
particolare, all‟evoluzione clonale della malattia osservata in termini di aumento della taglia
della popolazione tumorale mutata. Inoltre, l‟analisi dei pazienti refrattari alla Fludarabina,
che appaiono caratterizzati da un‟elevata incidenza di mutazioni a carico di TP53 in assenza
di delezione (18% pazienti refrattari vs 4.5% pazienti non trattati), suggerisce l‟impatto
determinante della terapia nella selezione dei subcloni mutati, sottolineando la correlazione
tra mutazioni e chemioresistenza [99]. Infine, è stata recentemente osservata una correlazione
tra l‟incidenza delle mutazioni del gene TP53 in pazienti affetti da LLC e lo sviluppo di un
cariotipo complesso e aberrante (≥ 3 aberrazioni cromosomiche), che potrebbe contribuire a
spiegare l‟impatto il significato prognostico negativo di tali mutazioni.
Nonostante la dimostrazione della correlazione tra la presenza di mutazioni del gene TP53 e
la prognosi infausta nei pazienti affetti da LLC, è necessario comunque considerare
l‟esistenza di una considerevole variabilità nella sopravvivenza nei pazienti. Tale variabilità
riflette sostanzialmente la taglia del clone TP53 mutato, il sito e il tipo di mutazione e la
funzione della proteina mutata. Risulta inoltre indispensabile la valutazione della correlazione
delle mutazioni agli altri fattori prognostici, sia clinici che biologici. Le correnti guide linea
diagnostiche suggeriscono tuttavia di studiare i pazienti affetti da LLC per la presenza della
del17p13 ma non delle mutazioni di TP53 [100]. Di conseguenza, i soggetti caratterizzati da
mutazioni di TP53 indipendenti dalla del17p13 non possono essere riconosciuti al momento
della diagnosi e rischiano di essere sottoposti a una terapia non idonea. Noto quindi il
significato prognostico indipendente delle mutazioni del gene TP53, l‟introduzione dello
screening di tutte le diverse anomalie genetiche a suo carico nei pazienti con diagnosi di LLC,
assume un‟importanza primaria per il miglioramento della pratica clinica.
In realtà, è necessario ricordare che la via di p53 può risultare alterata a molteplici livelli a
causa di potenziali eventi addizionali che escludono la del17p13 o le mutazioni a carico del
gene TP53. In primo luogo sono stati identificati alcuni polimorfismi nella sequenza di TP53
con impatto sull‟attività biologica della proteina. In particolare, due forme polimorfe del gene
118
TP53 sono ampiamente distribuite nella popolazione umana. Esse codificano per un‟arginina
o una prolina in posizione 72 all‟interno della regione codificante il dominio ricco in proline
della sequenza genica di TP53 [101]. E‟ stato dimostrato che la presenza dell‟una o dell‟altra
forma influenza la struttura primaria della proteina p53 e, di conseguenza, la sua attività
trascrizionale e la sua capacità di induzione del processo apoptotico. In particolare, la forma
Arg72 di p53 è caratterizzata da un‟inferiore capacità di transattivazione genica ma da una
maggiore attività pro-apoptotica. Questo sembra dipendere dalla sua superiore abilità di
localizzazione mitocondriale, conseguenza della maggiore suscettibilità al legame e
all‟ubiquitinazione da parte di MDM2, evento che ne comporta l‟esporto nucleare [102]. E‟
stato ipotizzato che le differenze biologiche correlate a tale polimorfismo possano avere
importanti implicazioni nel rischio allo sviluppo del cancro o nell‟influenzare la prognosi nei
casi di perdita mono- o bi-allelica del gene TP53. Inoltre, il polimorfismo del codone 72
potrebbe alterare la sensibilità delle neoplasie agli agenti chemioterapici; si potrebbe ad
esempio predire che l‟omozigosi dell‟allele codificante la forma Arg72 di p53 risponda in
misura maggiore alle radiazioni o alla chemioterapia. Sono state inoltre osservate alterazioni a
carico di componenti sia a monte che a valle della via di p53 con effetti paragonabili a quelli
conseguenti le mutazioni del gene TP53. In particolare, in pazienti affetti da LLC, le
alterazioni a carico del gene ATM rappresentano una potenziale causa di disfunzioni della
proteina p53. Ridotti livelli di ATM sono stati riscontrati nel 30-40% dei pazienti e mutazioni
del suo gene sono state osservate in una sostanziale proporzione di casi [103]. Inoltre, circa il
20% dei soggetti affetti da LLC presenta la delezione della regione 11q22-23 su cui mappa il
gene ATM, che, in una buona percentuale dei casi, è associata alla mutazione del rimanente
allele. Altri meccanismi di deregolazione della via di p53 nella LLC sono rappresentati dai
difetti a carico del gene MDM2, correlati a eventi di amplificazione genica o alla presenza di
polimorfismi; difetti funzionali nell‟induzione di p21 ed alterazioni dell‟espressione del
miR34-a. Il miglioramento della prognosi dei pazienti di LLC con mutazioni a carico del gene
TP53 o disfunzioni nella via di p53 rimane quindi strettamente correlato alla corretta
identificazione di coloro che, allo stesso modo dei pazienti del17p13+, non beneficerebbero di
un‟intensa chemioterapia a base di farmaci citotossici standard che agiscono attraverso p53,
come gli agenti alchilanti e gli analoghi purinici. Tali pazienti dovrebbero essere candidati a
terapie alternative non convenzionali, come l‟inclusione in protocolli clinici che studiano
l‟attività di farmaci con meccanismi d‟azione indipendenti dalla via apoptotica p53dipendente, di cui esempi sono il Flavopiridolo e la Lenalidomide; terapie a base di anticorpi
monoclonali come l‟Alemtuzumab e trapianto di cellule staminali emopoietiche.
119
1.4 Scopo del progetto di ricerca
La LLC è una patologia caratterizzata da un decorso clinico altamente variabile. Parte di
questa variabilità può essere attribuita alla proteina oncosoppressoria p53. I difetti del gene
TP53 nella LLC sono fortemente associati a progressione di malattia, resistenza alla
chemioterapia ed accorciamento dell‟aspettativa di sopravvivenza. Nonostante l„osservazione
di tale correlazione, esiste, tuttavia, anche all‟interno del sottogruppo di pazienti affetti da
LLC caratterizzati da mutazioni nel gene TP53, una considerevole variabilità prognostica che
incide sull‟indice di sopravvivenza. Tale variabilità riflette sostanzialmente la taglia del clone
TP53 mutato, il profilo mutazionale in termini di sito e tipologia di mutazione e la funzione
della proteina mutata. A complicare poi ulteriormente la comprensione della variabilità del
decorso clinico dei pazienti affetti da LLC correlata all‟oncosoppressore p53, vi è la
possibilità di osservare anomalie funzionali della proteina indipendenti dalla presenza di
mutazioni a carico del gene TP53.
In tale contesto si inserisce il presente progetto di ricerca incentrato sullo studio della
funzionalità di p53 in pazienti affetti da LLC, nel tentativo sia di identificare l‟eventuale
presenza di una correlazione tra l‟attività della proteina p53 e le caratteristiche molecolari del
gene TP53, che di predire la risposta ai chemioterapici convenzionali in pazienti non trattati o
associare la risposta terapeutica alle caratteristiche funzionali di p53 in pazienti
precedentemente esposti a terapia antitumorale. Sono stati inclusi nello studio pazienti affetti
da LLC a diversi stadi di malattia, quali: esordio, progressione e resistenza a una o più linee di
terapia convenzionale a base di farmaci citotossici. La funzionalità di p53 è stata studiata
valutando, mediante Western blot, l‟induzione della proteina in risposta all‟esposizione dei
campioni primari di cellule neoplastiche di LLC a 5 Gray di radiazioni ionizzanti (IR). Una
volta esaminata la funzionalità della proteina e definite le differenti tipologie di disfunzioni, è
stato effettuato un confronto tra i dati funzionali ottenuti, lo stato del gene TP53 e le
caratteristiche prognostiche dei pazienti disfunzionali. E‟ stata infine eseguita una valutazione
della sensibilità, delle forme funzionali e disfunzionali della proteina p53 identificate, al
processo apoptotico indotto dal danno subletale causato dopo esposizione dei campioni
cellulari primari di LLC a IR. Il fine ultimo del suddetto studio è rappresentato
dall‟opportunità di esaminare il ruolo della proteina p53 nel decorso della malattia e
identificare quei pazienti affetti da LLC caratterizzati da disfunzioni a carico di p53 che non
potrebbero, quindi, beneficiare di un approccio terapeutico convenzionale ma dovrebbero
essere indirizzati verso approcci terapeutici innovativi.
120
2. MATERIALI E METODI
2.1 Pazienti e Controlli
Nel nostro studio sono stati inclusi 87 pazienti affetti da LLC a differente stadio di malattia,
seguiti presso la Sezione di Ematologia del Dipartimento di Biotecnologie Cellulari ed
Ematologia dell‟Università degli studi di Roma “Sapienza”. All‟interno di tale coorte, 13
pazienti presentavano una malattia all‟esordio, 54 pazienti risultavano caratterizzati da una
malattia in progressione e 20 pazienti da una malattia resistente a una o più linee di terapia
convenzionale. Il prelievo dei campioni di sangue periferico e le analisi biologiche sono stati
eseguiti previo consenso informato fornito da ciascuno di loro in accordo con la Dichiarazione
di Helsinki. La diagnosi di LLC è stata effettuata attraverso l‟identificazione, nel sangue
venoso periferico (SVP) di ciascun paziente, di una quantità di linfociti superiore a 5x10 9/L,
con
una
morfologia
ed
un
profilo
immunofenotipico
caratteristici
di
LLC
(CD5+CD20+CD23+CD22+sIg±CD10-). Sono state effettuate nei pazienti studiati le seguenti
analisi di routine: la valutazione dello stato mutazionale dei geni IgVH [104]; l‟analisi
citofluorimetrica dell‟espressione della proteina ZAP-70 [105-106] e dell‟antigene CD38
[107]; l‟analisi FISH (ibridazione in situ fluorescente) per l‟identificazione delle aberrazioni
citogenetiche coinvolgenti le regioni 11q22-23, 13q14, 17p13 e la trisomia 12, ed il
sequenziamento del gene TP53 [104]. Al tempo dello studio, i pazienti all‟esordio di malattia
presentavano un‟età media di 52 anni e una conta media di globuli bianchi (WBC) pari a
31,704/µLx106 (range 5,790-69,870/μLx106); i pazienti caratterizzati da una malattia in
progressione, un‟età media di 64 anni e un valore medio di WBC pari a 93,566/µLx106 (range
6,200-511,400/μLx106); infine, i pazienti caratterizzati da una malattia resistente, un‟età
media di 68 anni e un valore medio di WBC pari a 97,080/µLx106 (range 12,250610,500/μLx106).
In qualità di controllo sono stati utilizzati, previo consenso informato, campioni di SVP
prelevati da donatori sani adulti ottenuti dalla Banca del Centro Trasfusionale della stessa
Università.
2.2 Induzione dell’attivazione della proteina oncosoppressoria p53
Le cellule mononucleate del sangue periferico (PBMC) dei pazienti affetti da LLC a
differente stadio di malattia, sono state ottenute mediante separazione su gradiente di densità
(Lymphoprep; Nycomed Pharma, Oslo, Norway) e coltivate overnight a 37 C in atmosfera
121
umidificata con un tasso di CO2 pari al 5%, in terreno colturale RPMI 1640 (Cambrex Bio
Science Verviers, Belgium) addizionato con 10% di siero fetale bovino (FBS, HyClone,
South Logan, UT), 1% di L-glutammina (EuroClone, Europe) e 1% di Pen-Strepto
(EuroClone, Europe). Quindi, al fine di indurre l‟attivazione della proteina p53, un‟aliquota
delle cellule mononucleate è stata esposta a 5 Gray (Gy) di radiazioni ionizzanti (IR) e,
successivamente, l‟aliquota cellulare irradiata e le PBMC non esposte a IR sono state
mantenute in coltura per ulteriori 8 e 24 h dal tempo dell‟esposizione. Allo scadere della
coltura, i campioni cellulari sono stati criopreservati come pellet secchi per il successivo
utilizzo.
2.3 Valutazione dell’attivazione della proteina oncosoppressoria p53
Allo scopo di esaminare l‟induzione dell‟attivazione della proteina p53 mediante Western
blot, le PBMC dei pazienti in esame, conservate in pellet secchi dopo 8 e 24 h di coltura
dall‟esposizione a IR, sono state sottoposte a lisi diretta mediante incubazione per 30 minuti a
4°C in una soluzione contenente: Tris-HCl 2M (pH 8); EDTA 0.5M (pH 8); NaCl 5M; NaF
0.05M (pH 7.2); Sodio Ortovanadato 0.001M; 10% Triton X-100 ed 1 pasticca composta da
una miscela di Inibitori delle Proteasi (Complete Protease Inhibitor Cocktail Tablets; Roche
Diagnostics, Mannheim, Germany). Al termine dei 30 min di incubazione, i lisati cellulari
sono stati centrifugati a 14,000 rpm per 15 min e i sovranatanti, contenenti la frazione proteica
purificata, sono stati raccolti. La concentrazione proteica dei sovranatanti è stata quindi
determinata utilizzando il Bio-Rad protein assay kit (Bio-Rad Laboratories, Munich,
Germany).
A questo punto, quantità equivalenti di campioni proteici (40 g) sono state miscelate con
Sample Buffer (una soluzione di Laemmli sample al 5% di 2-mercaptoetanolo; Bio-Rad
Laboratories) e incubate 5 min a 95°C per la denaturazione al calore; quindi separate
mediante elettroforesi su gel di Sodio Dodecil Solfato (SDS)-Poliacrilamide all‟8% (10%
SDS; 30% Soluzione di Poliacrilammide; Tris-HCl 1.5M pH 8.8; 10% Ammonio Persolfato,
6% TEMED). Al termine della corsa elettroforetica è stato effettuato a +4°C il trasferimento
overnight dei campioni proteici su membrane di nylon PVDF (Immobilon-P; Millipore,
Bedford, MA). Dopo il recupero, le membrane sono state lavate in una soluzione di TBS 1x
(1M Tris-HCl pH8 e 5M NaCl) allo 0.05% di Tween 20 (TBS-T) ed è stato effettuato il
passaggio di “blocking”, al fine di saturare i siti idrofobici liberi sulla membrana e prevenire il
legame dell‟anticorpo primario alla membrana stessa. Per il blocco è stata impiegata una
122
soluzione di TBS-T al 5% di latte scremato. Terminato il blocco di 1 h a temperatura
ambiente (TA), le membrane sono state lavate in TBS-T per poi procedere all‟incubazione
con l‟anticorpo primario. È stato utilizzato, a tale scopo, un anticorpo monoclonale di topo
diretto contro l‟isoforma umana della proteina p53 (Calbiochem-Novabiochem, La Jolla, CA)
a una diluizione 1:500 in TBS-T al 5% di latte. Le membrane sono state incubate in tale
soluzione per 1 h a TA, lavate in TBS-T e incubate nuovamente per 45 min a TA con un
anticorpo secondario policlonale di capra anti-topo coniugato con l‟enzima perossidasi (Pierce
Biotecnology, Rockford, IL), diluito 1:150 in TBS-T. Al termine dell‟incubazione le bande
proteiche specifiche sono state visualizzate utilizzando il sistema ECL (GE Healthcare,
Buckinghmshire, UK). Al fine di verificare se il caricamento di tutti i campioni fosse
avvenuto in modo quantitativamente corretto, le membrane sono state incubate
addizionalmente con un anticorpo primario monoclonale di topo diretto contro la
Gliceraldeide-3-Fosfato Deidrogenasi (GAPDH; Chemicon, Millipore) e un anticorpo
secondario policlonale di pecora anti-topo coniugato all‟enzima perossidasi (GE Healthcare),
entrambi diluiti 1:2.000.
2.4 Analisi del tasso apoptotico
Il tasso apoptotico è stato misurato sulle PBMC dei pazienti affetti da LLC, esposte e non a 5
Gy di IR, dopo 24 h di coltura. A tale scopo è stata impiegata la tecnica dell‟Annexina-V, che
prende il nome da una proteina in grado di legare la fosfatidilserina, un marcatore di apoptosi
precoce che, all‟inizio della cascata apoptotica, trasloca dalla parte interna della membrana a
quella esterna.
Per la marcatura, i campioni cellulari sono stati lavati 2 volte in PBS, quindi risospesi in
Binding buffer 1x (Binding buffer 10x: 100mM HEPES/NaOH pH 7.5; 1.4M NaCl, 25mM
CaCl2) in presenza di Annexina-V FITC e Ioduro di Propidio (IP; Immunotech Research,
Quebec, Canada) entrambi alla concentrazione finale di 1 g/ml. Lo IP è utile a marcare il
DNA delle cellule necrotiche la cui membrana risulta degradata. La miscela è stata quindi
incubata 10 min al buio a TA e, dopo la marcatura, la percentuale di cellule apoptotiche è
stata determinata mediante citometria a flusso e i dati analizzati utilizzando il software
CellQuest (Becton Dickinson). La combinazione Annexina V-FITC/IP (Immunotech
Research, Quebec, Canada) ha permesso la differenziazione tra: cellule vitali (Annexina-, IP-),
cellule in fase precoce di Apoptosi (Annexina+, IP-), cellule in fase tardiva di Apoptosi
(Annexina+, IP+), cellule necrotiche (Annexina-, IP+).
123
2.5 Analisi statistica per gli studi di apoptosi
I dati sono stati analizzati statisticamente mediante l‟utilizzo del metodo “Student‟s t test”. I
risultati sono espressi come media ± deviazione standard.
124
3. RISULTATI
3.1 Analisi della funzionalità della proteina p53 in campioni primari di cellule
neoplastiche di pazienti affetti da LLC a differente stadio di malattia
Allo scopo di esaminare la funzionalità di p53, nel nostro studio è stato valutato l‟effetto,
sull‟espressione della proteina, dell‟esposizione delle PBMC di pazienti affetti da LLC a IR. I
livelli della proteina p53, normalmente non rilevabili nelle cellule quiescenti, sono, infatti,
soggetti a un incremento in risposta a differenti tipologie di stress cellulare, in quei casi
caratterizzati da una normale funzionalità di p53. Al contrario, la presenza di alterazioni
molecolari a carico del gene TP53 o anomalie funzionali della proteina p53 indipendenti dalla
presenza di tali mutazioni, comporta solitamente un prolungamento della sua emivita, con la
possibilità di rilevare la proteina anche in condizioni basali, ovvero in assenza di qualunque
tipologia di stimolo induttore.
Nel nostro studio sono stati inclusi complessivamente 87 campioni cellulari di pazienti affetti
da LLC. All‟interno di tale coorte, 13 pazienti (14.9%) presentavano una malattia all‟esordio,
54 pazienti (62.0%) risultavano caratterizzati da una malattia in progressione e 20 pazienti
(23.1%) da una malattia resistente ad una o più linee di terapia convenzionale a base di
farmaci citotossici [Figura 7]. L‟espressione della proteina p53 è stata valutata, mediante
Western blot, sia sui campioni irradiati che sulla controparte non esposta a IR, dopo una
coltura di 8 e 24 h dal tempo dell‟esposizione. Cellule linfoidi normali, prive di alterazioni a
carico di p53, sono state impiegate come controllo negativo.
15%
23%
Pazienti alla diagnosi
Pazienti progressivi
Pazienti chemioresistenti
62%
Figura 7. Distribuzione nelle diverse fasi di malattia dei pazienti affetti da LLC inclusi nello studio.
125
Una normale funzionalità di p53, caratterizzata dall‟assenza della proteina nella controparte
non esposta IR e dal suo massivo incremento dopo irradiazione, è stata rilevata in 62/87
pazienti esaminati (71.3%) e, più precisamente, in 12/13 pazienti (92.3%) all‟esordio di
malattia; 40/54 pazienti (74.0%) in progressione di malattia e 10/20 pazienti (50.0%)
resistenti alla terapia. Al contrario, 25/87 pazienti (28.7%), di cui 1/13 pazienti (7.7%)
all‟esordio di malattia, 14/54 pazienti (26.0%) in progressione di malattia e 10/20 pazienti
(50.0%) resistenti alla terapia presentavano un‟alterata funzionalità di p53 [Tabella 1; Figura
8]. Nei pazienti caratterizzati da un‟alterata funzionalità di p53 è stato possibile distinguere 3
differenti tipologie di disfunzioni. In 13/25 pazienti disfunzionali (52.0%) i livelli di p53
risultavano costitutivamente incrementati nella controparte non irradiata ma subivano un
ulteriore accumulo dopo esposizione del campione a IR (Disfunzione di tipo I). In 11 di loro
(44.0%), nonostante la proteina p53 fosse rilevabile nella controparte non irradiata, i suoi
livelli rimanevano immutati dopo irradiazione (Disfunzione di tipo II).
Tabella 1. Analisi della funzionalità della proteina p53 nei pazienti affetti da LLC inclusi nello studio.
Pazienti inclusi
nello studio
(n=87)
Pazienti caratterizzati da
normale funzionalità di p53
(n=62; 71.3%)
Pazienti caratterizzati da
alterata funzionalità di p53
(n=25; 28.7%)
Esordio (n=13)
12/13; 92.3%
1/13; 7.7%
Progressione (n=54)
40/54; 74.0%
14/54; 26.0%
Resistenza (n=20)
10/20; 50.0%
10/20; 50.0%
126
a)
Valori percentuali
100
80
71.3%
60
40
28.7%
20
0
Normale funzionalità p53
Alterata funzionalità p53
b)
100
92.3%
74%
Valori percentuali
80
60
50%
50%
40
26%
20
7.7%
0
Pz alla diagnosi
Pz progressivi
Normale funzionalità p53
Pz chemioresistenti
Alterata funzionalità p53
Figura 8. Funzionalità della proteina p53 nella totalità dei pazienti affetti da LLC inclusi nello studio (a) e
nei pazienti affetti da LLC in relazione allo stadio di malattia (b). I campioni B cellulari leucemici sono stati
esposti a IR e l‟espressione della proteina p53 è stata valutata mediante Western blot, sia sui campioni irradiati
che sulla controparte non esposta a IR, dopo una coltura di 8 h dal tempo dell‟esposizione.
127
Infine in 1/25 pazienti (4.0%) la proteina p53 risultava assente sia nelle cellule non irradiate
che dopo esposizione a IR (Disfunzione di tipo III) [Figura 9].
L‟unico paziente (CLL I.A.) all‟esordio di malattia caratterizzato da anomalie funzionali a
carico della proteina p53, presentava una disfunzione di tipo II. Dei 14 pazienti in
progressione di malattia, 8 risultavano caratterizzati da una disfunzione di tipo I (57.1%; CLL
F.M.R.; G.M.; L.P.; S.C.; C.G.; B.V.; G.C.; D.G.), 5 da una disfunzione di tipo II (35.7%;
CLL M.M.; B.A.M.; I.A.; N.L.; S.A.) e un solo paziente da una disfunzione di tipo III (7.2%;
CLL D.C.G.). Infine, dei 10 pazienti disfunzionali con LLC chemioresistente, in 5 è stata
riscontrata una disfunzione di tipo I (50.0%; CLL P.B.; M.F.; F.C.; G.L.; D.A.A.) e nei
restanti 5 una disfunzione di tipo II (50.0%; CLL A.R.; N.E.; L.P.; D.V.A.; S.A.). La
distribuzione delle differenti tipologie di disfunzioni nell‟intera coorte di pazienti affetti da
LLC e in ciascuna delle tre classi in cui i pazienti sono stati suddivisi in relazione allo stadio
di malattia, è mostrata nella Figura 10.
Nel complesso i risultati ottenuti rivelano una maggiore incidenza delle disfunzioni a carico di
p53 nelle forme di LLC progressiva e chemioresistente piuttosto che all‟esordio di malattia.
E‟ stato possibile osservare una prevalenza delle disfunzioni di tipo I non solo nella
popolazione complessiva di pazienti affetti da LLC ma anche in 1 delle 3 classi di pazienti
suddivisi in relazione allo stadio di malattia, rappresentata dai casi progressivi. Al contrario, i
casi di LLC resistente sono apparsi ugualmente associati a disfunzioni di tipo I e II, mentre
non è stato possibile valutare la correlazione tra la tipologia di disfunzione e i pazienti con
LLC all‟esordio a causa della scarsità di campioni disfunzionali osservati nella suddetta fase
di malattia.
128
a)
0 8h
0 8h
0 8h
0 8h
p53
GAPDH 35 kDa
0 8h
0 8h
0 8h
b)
p53
GAPDH 35 kDa
0 8h
0 8h
c)
0 8h
p53
GAPDH 35 kDa
Figura 9. Rappresentazione al Western blot delle bande relative all’espressione della proteina p53 al tempo 0
e a 8 h dall’esposizione dei campioni cellulari di LLC a IR. Sono rappresentate a titolo esemplificativo le bande
relative all‟espressione della proteina in: a) 3 campioni di LLC con normale funzionalità di p53 (assenza della
proteina in condizioni basali e forte upregolazione dopo irradiazione); b) 3 campioni di LLC con disfunzione di
tipo I (costitutivo incremento della proteina nelle cellule non irradiate e suo ulteriore accumulo dopo
irradiazione); c) 2 campioni di LLC con disfunzione di tipo II (costitutivo incremento della proteina in
condizioni basali in assenza di un suo ulteriore accumulo dopo irradiazione) e 1 campione di LLC con
disfunzione di tipo III di p53 (D.C.G.; assenza della proteina sia pre- sia post-esposizione a IR). E‟ stato
impiegato in qualità di controllo negativo un campione di linfociti normali (PBL). Per evidenziare le differenze
potenzialmente attribuibili al caricamento del gel, le membrane sono state incubate addizionalmente con un
anticorpo primario monoclonale anti-GAPDH.
129
a)
Valori percentuali
100
80
52%
44%
60
40
4%
20
0
Disfunzione I
Disfunzione II
Disfunzione III
b)
100%
100
Valori percentuali
80
57.1%
50% 50%
60
35.7%
40
20
0%
7.2%
0%
0%
0
Pz alla diagnosi
Disfunzione I
Pz progressivi
Disfunzione II
Pz chemioresistenti
Disfunzione III
Figura 10. a) Distribuzione delle tipologie di disfunzioni a carico della proteina p53 nella totalità dei pazienti
(Pz) affetti da LLC inclusi nello studio. b) Distribuzione delle tipologie di disfunzioni a carico della proteina
p53 nei pazienti affetti da LLC in relazione allo stadio di malattia. I campioni cellulari leucemici sono stati
esposti a IR e l‟espressione della proteina p53 è stata valutata mediante Western blot, sia sui campioni irradiati
che sulla controparte non esposta a IR, dopo una coltura di 8 h dal tempo dell‟esposizione.
130
3.2 Correlazione della funzionalità della proteina p53 con lo stato del gene TP53
Il passo successivo dello studio è stato quello di valutare la correlazione tra la risposta
funzionale di p53 alle IR e lo stato del gene TP53, analizzato dal gruppo di studio del
Sequenziamento Genico del nostro laboratorio. Dei 62 pazienti caratterizzati da una normale
funzionalità della proteina, la totalità di essi (100%) è risultata wild type per la sequenza del
gene TP53, come atteso. In tale gruppo è stata quindi rilevata una concordanza del 100% tra
dati proteici e dati genomici. Al contrario, dei 25 pazienti con anomalie funzionali della
proteina p53, 20 hanno evidenziato mutazioni nella sequenza del gene TP53, mentre i
rimanenti 5 (CLL P.B.; M.F.; F.C.; A.R.; D.V.A./3 disfunzioni di tipo I e 2 di tipo II), tutti
contraddistinti da una malattia resistente, sono risultati wild-type per il locus TP53,
suggerendo la presenza di un difetto in un differente target della via di p53.
Conseguentemente, la concordanza tra dati proteici e dati genomici è risultata, nell‟ambito dei
casi disfunzionali, pari all‟80.0% [Tabella 2].
In particolare, all‟interno del sottogruppo di pazienti con anomalie funzionali e molecolari a
carico di p53, 19/20 (10 disfunzioni di tipo I, 8 disfunzioni di tipo II e 1 disfunzione di tipo
III) hanno evidenziato la presenza di una singola mutazione del gene TP53, mentre 1/20 (CLL
N.E., disfunzione di tipo II) la presenza di una doppia mutazione del gene TP53, ossia di due
diverse mutazioni in due differenti posizioni della sequenza genica.
Tabella 2. Correlazione tra la funzionalità della proteina p53 e lo stato del gene TP53: Valori di predittività
negativa (NPV) e positiva (PPV).
Pazienti inclusi nello
studio (n=87)
Pazienti caratterizzati da
normale funzionalità di p53
(n=62; 71.3%)
Pazienti caratterizzati da
alterata funzionalità di
p53 (n=25; 28.7%)
Pazienti TP53 wild-type
62/62; 100.0% (NPV)
5/25; 20.0%
0/62; 0.0%
20/25; 80.0% (PPV)
Pazienti TP53 mutati
131
Nell‟ambito dei 20 pazienti disfunzionali caratterizzati da mutazioni del gene TP53, sono state
complessivamente identificate mediante sequenziamento genico, 18 mutazioni missenso, 2
nonsenso ed 1 microdelezione; di queste, 15 mutazioni risultavano in omozigosi mentre 6 in
eterozigosi. Sulla base di tali dati, per una migliore comprensione dei meccanismi alla base
della comparsa di disfunzioni a carico dell‟oncosoppressore p53, è stata valutata la
correlazione tra le differenti tipologie di disfunzioni di p53 e il profilo mutazionale del gene
TP53 in termini di natura e stato allelico della mutazione [Tabella 3]. Dall‟analisi delle
mutazioni in ciascuna categoria di disfunzioni è emerso, in primo luogo, come la disfunzione
di tipo I sia associata principalmente a mutazioni eterozigoti di natura missenso a carico del
gene TP53. Infatti, 9/10 pazienti di LLC con disfunzioni di tipo I (90.0%) presentavano
mutazioni con le suddette caratteristiche. Le mutazioni di natura missenso sono risultate
peculiari anche della maggior parte dei casi disfunzionali di tipo II (7/9 pazienti; 77.8%)
mentre in tale sottogruppo è stata riscontrata una maggiore associazione con lo stato allelico
omozigote del gene TP53. Infatti, sebbene il 55.6% dei casi disfunzionali di tipo II (5/9
pazienti), appariva caratterizzato da mutazioni in eterozigosi del gene TP53, il rimanente
44.4% (4/9 pazienti) presentava mutazioni in omozigosi contrariamente all‟10.0% dei casi
disfunzionali di tipo I [Figura 11]. E‟ stata dunque analizzata la correlazione tra le differenti
classi disfunzionali di p53 e lo stato del gene TP53 in relazione al sito della mutazione,
ovvero all‟esone e codone interessato e al cambio di natura nucleotidica. La disfunzione di
tipo I è stata osservata in associazione a mutazioni del gene TP53 localizzate esclusivamente
negli esoni 5-8 della regione codificante il DBD di p53. Tali esoni, codificando per la regione
più significativa della proteina p53 dal punto di vista funzionale e filogeneticamente più
conservata, in quanto coinvolta nell‟interazione con il DNA, rappresentano, infatti, i
cosiddetti punti caldi di mutagenesi, ovvero i siti in cui si riscontra, normalmente, il maggior
numero di mutazioni. In particolare, nel 60.0% dei casi di LLC con disfunzione di tipo I (6/10
pazienti) sono state riscontrate mutazioni all‟interno dell‟esone 8, nel 20.0% mutazioni
all‟interno dell‟esone 6 (2/10 pazienti), mentre il rimanente 20% risultava equamente
caratterizzato da mutazioni all‟interno dell‟esone 7 (10.0%; 1/10 pazienti) e dell‟esone 5
(10.0%; 1/10 pazienti). Al contrario, sebbene le mutazioni del gene TP53 a livello degli esoni
8 e 6 presentavano la più alta incidenza nei casi disfunzionali di tipo II (33.4%, 3/9 pazienti e
22.2%, 2/9 pazienti rispettivamente), seguite dalle mutazioni negli esoni 7 e 6 (22.2%, 2/9
pazienti in entrambi i casi), è stata osservata, in tale sottogruppo, anche la presenza di
mutazioni a livello dell‟esone 4 (11.1%; 1/9 pazienti), in posizione 110 del DBD, e 9 (11.1%;
1/9 pazienti), in posizione 321 della regione segnale di localizzazione nucleare [Figura 12].
132
Tabella 3. Correlazione tra le disfunzioni a carico di p53 e il profilo mutazionale del gene TP53 nei pazienti affetti da LLC distinti in base allo stadio di malattia.
Fasi della
malattia
Esordio (n=1)
Progressione
(n=14)
Resistenza
(n=5)
Cambio
nucleotidico
Transizione (Ts)
vs
Trasversione (Tv)
Cambio
aminoacidico
Stato
allelico
della
mutazione
Paziente
Disfunzione
proteina p53
I.A.
II
6
193
CAT-CGT
Ts
His-Arg
Missenso
Eterozigote
B.A.M.
II
8
282
CGG-GGG
Tv
Arg-Gly
Missenso
Omozigote
F.M.R.
I
8
283
CGC-TGC
Ts
Arg-Cys
Missenso
Omozigote
G.M.
I
6
191
CCT-CAG
-
In Frame
Delezione
Eterozigote
L.P.
I
8
273
CGT-CAT
Ts
Arg-His
Missenso
Eterozigote
S.C.
I
8
275
TGT-TTT
Tv
Cys-Phe
Missenso
Eterozigote
S.A.
II
7
241
TCC-TTC
Ts
Ser-Phe
Missenso
Omozigote
D.C.G.
III
6
213
CGA-TGA
Ts
Arg-Stop
Nonsenso
Omozigote
C.G.
I
7
248
CGG-TGG
Ts
Arg-Trp
Missenso
Eterozigote
G.C.
I
5
175
CGC-CAC
Ts
Arg-His
Missenso
Eterozigote
I.A.
II
6
193
CAT-CGT
Ts
His-Arg
Missenso
Eterozigote
D.G.
I
6
195
ATC-ACC
Ts
Ile-Thr
Missenso
Eterozigote
M.M.
II
8
281
GAC-AAC
Ts
Asp-Asn
Missenso
Omozigote
N.L.
II
4
110
CGT-CTT
Tv
Arg-Leu
Missenso
Eterozigote
B.V.
I
8
275
TGT-TAT
Ts
Cys-Tyr
Missenso
Eterozigote
G.L.
I
8
273
CGT-CAT
Ts
Arg-His
Missenso
Eterozigote
L.P.
D.A.A.
II
II
II
I
9
8
7
8
321
272
235
278
AAA-TAA
GTG-ATG
AAC-GAC
CCT-CGT
Tv
Ts
Ts
Tv
Lys-Stop
Val-Met
Asn-Asp
Pro-Arg
Nonsenso
Missenso
Missenso
Missenso
Omozigote
Eterozigote
Eterozigote
Eterozigote
P.B.
II
5
126
TAC-TAG
133
Tv
Tyr- Stop
Nonsenso
Eterozigote
N.E.
Codone
TP53
mutato
Tipologia
di
mutazione
Esone
TP53
mutato
a)
100%
100
90%
78%
Valori percentuali
80
60
40
22%
10%
20
0%
0%
0%
0%
0
Disfunzione I
Disfunzione II
n=10
Disfunzione III
n=9
Mutazioni Missenso
n=1
Mutazioni Nonsenso
Microdelezioni
b)
100%
Valori percentuali
100
90%
80
55.6%
44.4%
60
40
10%
20
0%
0
Disfunzione I
Disfunzione II
Disfunzione III
n=10
n=9
n=1
Mutazioni Eterozigoti
Mutazioni Omozigoti
Figura 11. a) Correlazione tra le disfunzioni a carico di p53 e la tipologia delle mutazioni del gene TP53; b)
Correlazione tra le disfunzioni a carico di p53 e lo stato allelico delle mutazioni del gene TP53.
134
Nell‟ambito delle classi disfunzionali I e II è stato anche possibile rilevare una distribuzione
del tutto distinta dei residui TP53 interessati dalla mutazione. Infatti, mentre mutazioni del
gene TP53 a carico dei residui 175-190-191-195-248-273-275-278-283 sono state osservate
esclusivamente in associazione a disfunzioni di tipo I, mutazioni a livello dei residui 110-126193-235-241-272-281-282-321 sono risultate unicamente caratteristiche delle disfunzioni di
tipo II [Figura 13]. Inoltre, dei 10 casi disfunzionali di tipo I, 8/10 (80.0%) mostravano una
correlazione con la classe dei mutanti di “Contatto” (residui 112-141; 236-251; 271-286)
mentre 2/10 (20.0%) con la classe dei mutanti “Conformazionali” (residui 163-195). Al
contrario, nell‟ambito dei 9 casi disfunzionali di tipo II, sono stati identificati 5 mutanti di
“contatto” (55.6%); 2 mutanti “conformazionali” (22.2%) e 2 mutanti a livello di residui non
compresi nelle regioni corrispondenti alle suddette classi mutazionali (22.2%) [Figura 14a].
Infine, le disfunzioni di tipo I hanno rivelato una prevalente correlazione con le transizioni
(77.8%; 7/9 pazienti) mentre le disfunzioni di tipo II sono risultate più equamente suddivise
tra transizioni (55.6%; 5/9 pazienti) e trasversioni (44.4%; 4/9 pazienti) [Figura 14b].
100%
Valori percentuali
100
80
60%
60
33.4%
40
20
20%
10%
10%
0%
22.2% 22.2%
11.1% 11%
11.1%
0% 0%
0%
0% 0% 0%
0
Disfunzione I
n=10
Esone 4
Esone 5
Disfunzione II
n=9
Esone 6
Esone 7
Disfunzione III
n=1
Esone 8
Esone 9
Figura 12. Correlazione tra le disfunzioni a carico di p53 e gli esoni interessati dalle mutazioni del gene TP53.
135
Non è stato invece possibile identificare un pattern di associazioni peculiari o ricorrenti tra la
disfunzione di tipo III e il profilo mutazionale del gene TP53, dal momento che tale tipologia
disfunzionale è stata osservata solamente in uno dei 25 pazienti affetti da LLC e caratterizzati
da anomalie della proteina p53. Ad ogni modo, tale paziente risultava portatore di una
mutazione omozigote di natura nonsenso del gene TP53 a livello del residuo 213 dell‟esone 6.
I dati ottenuti rivelano, dunque, l‟esistenza di un‟associazione tra le differenti tipologie di
disfunzioni e le caratteristiche delle mutazioni a carico del gene TP53, identificando, in
particolar modo nel sottogruppo dei pazienti caratterizzati da disfunzioni di tipo II, un più
complesso e sfavorevole profilo di correlazione con lo stato del suddetto gene.
E‟ stato, inoltre, possibile costatare l‟esistenza di anomalie della proteina p53 indipendenti
dalla presenza di mutazioni nella sequenza codificante del gene TP53 ma fenotipicamente
sovrapponibili a quelle riscontrate in presenza di tali mutazioni.
Valori percentuali
100
80
60
40
20
0
110 126 175 191 193 195 231 235 241 248 272 273 275 278 281 282 283 321
Codoni interessati dalle mutazioni del gene TP53
Disfunzione I
Disfunzione II
Disfunzione III
n=10
n=9
n=1
Figura 13. Correlazione tra le disfunzioni a carico di p53 e i codoni interessati dalle mutazioni del gene TP53.
136
a)
100%
Valori percentuali
100
80%
80
55.6%
60
40
22.2% 22.2%
20%
20
0%
0% 0%
0
Disfunzione I
Disfunzione II
Disfunzione III
n=10
n=9
n=1
Mutanti di Contatto
Mutanti Conformazionali
Mutanti non classificabili
b)
100%
Valori percentuali
100
77.8%
80
55.6%
44.4%
60
22.2%
40
20
0%
0
Disfunzione I
Disfunzione II
n=10
n=9
Transizioni
Disfunzione III
n=1
Trasversioni
Figura 14. a) Correlazione tra le disfunzioni a carico di p53 e le classi mutazionali del gene TP53; b)
Correlazione tra le disfunzioni a carico di p53 e la natura del cambio nucleotidico nella sequenza mutata del gene
TP53.
137
3.3 Correlazione della funzionalità della proteina p53 con i principali fattori prognostici
caratteristici della LLC
Allo scopo di comprendere il possibile significato prognostico delle disfunzioni a carico della
proteina p53, è stata valutata la presenza di un‟eventuale correlazione tra le suddette tipologie
di disfunzioni e i principali fattori indicatori di prognosi nei pazienti affetti da LLC, quali: lo
stato mutazionale della regione IgHV; l‟espressione della proteina ZAP-70 e dell‟antigene
CD38; la presenza della delezione del braccio corto del cromosoma 17.
In primo luogo, dei 25 pazienti disfunzionali (13 disfunzioni di tipo I, 11 di tipo II ed 1 di tipo
III), 20 (80.0%) sono risultati caratterizzati dalla presenza di una sequenza non mutata della
regione IgHV, di cui: 9/13 (69.2%) con disfunzione di tipo I; 10/11 (90.9%) con disfunzione
di tipo II e 1/1 (100.0%) con disfunzione di tipo III. Al contrario, dei restanti 5 pazienti,
caratterizzati da una sequenza IgHV mutata, 4/13 (30.8%) presentavano una disfunzione di
tipo I ed 1/11 (9.1%) di tipo II. L‟analisi relativa alla correlazione tra le anomalie della
proteina p53 e l‟espressione della proteina ZAP-70, effettuata su un totale di 20/25 pazienti
disfunzionali (11 disfunzioni di tipo I, 8 di tipo II ed 1 di tipo III), ha rivelato la presenza, di
13/20 (60.0%) casi ZAP-70+, 7/11 (63.6%) con disfunzioni di tipo I e 6/8 (75.0%) con
disfunzioni di tipo II; al contrario, 7/20 casi (40.0%) risultavano ZAP-70-, 4/11 (36.4%) con
disfunzioni di tipo I, 2/8 (25.0%) con disfunzione di tipo II ed 1/1 (100.0%) di tipo III. I casi
disfunzionali CD38+ sono invece risultati pari al 52.2% (12/23 casi disfunzionali esaminati;
12 disfunzioni di tipo I, 10 di tipo II ed 1 di tipo III), con la seguente distribuzione all‟interno
delle differenti tipologie di disfunzioni: 33.3% (4/12 pazienti) e 80.0% (8/10 pazienti)
rispettivamente nei casi disfunzionali di tipo I e II. Nell‟ambito dei casi disfunzionali CD38-,
il 66.7% (8/12 pazienti) risultava bensì associato a disfunzioni di tipo I, il 20.0% (2/10
pazienti) a disfunzioni di tipo II e il 100.0% (1/1 pazienti) di tipo III [Tabella 4].
Infine, un cariotipo alterato è stato osservato in tutti i 20 pazienti disfunzionali in cui è stato
possibile eseguire uno studio citogenetico (10 disfunzioni di tipo I, 9 di tipo II ed 1 di tipo
III). La prevalenza della trisomia 12 e delle anomalie strutturali a carico delle regioni
cromosomiche 13q14 e 11q22-, valutata mediante FISH in 14 dei suddetti pazienti (7
disfunzioni di tipo I e 7 di tipo II) è risultata rispettivamente pari a 7.1%, 42.8% e 7.1%. La
trisomia 12 è stata osservata in 1/14 pazienti (CLL progressiva F.C.; 14.3%, 1/7 disfunzioni di
tipo I); la del13q14 in 6/14 pazienti (57.2%, 4/7 disfunzioni di tipo I e 28.6%, 2/7 disfunzioni
di tipo II) e la del11q22-23 in 1/14 pazienti (CLL progressiva S.A.; 14.3%, 1/7 disfunzioni di
tipo II) [Tabella 4].
138
Sedici dei 20 pazienti p53 disfunzionali analizzati per la presenza di anomalie strutturali a
carico della regione cromosomica 17p sono risultati caratterizzati dalla del17p13 (16/20;
80.0%). All‟interno di tale coorte, 6/16 (37.5%) pazienti presentavano una disfunzione di tipo
I (60.0%, 6/10 disfunzioni di tipo I), 9/16 (56.3%) di tipo II (100.0%, 9/9 disfunzioni di tipo
II) e 1/16 (6.2%) di tipo III (100.0%, 1/1 disfunzioni di tipo III). Prendendo in considerazione
la totalità dei pazienti disfunzionali (n=20) inclusi nell‟analisi citogenetica della regione 17p è
stato possibile rilevare valori di cellule del17p13+ compresi tra il 5% e il 20% nel 10.0%
(1/10) delle disfunzioni di tipo I e nel 33.3% (3/9) delle disfunzioni di tipo II, mentre valori
superiori al 20% sono stati osservati nel 50.0% (5/10) delle disfunzioni di tipo I, nel 66.7%
(6/9) delle disfunzioni di tipo II e nel 100% (1/1) delle disfunzioni di tipo I [Tabella 4].
Tabella 4. Caratteristiche biologiche dei pazienti affetti da LLC contraddistinti dalla presenza di difetti
funzionali a carico della proteina p53.
Caratteristiche
Biologiche
Disfunzioni
di Tipo I
Disfunzioni
di Tipo II
Disfunzioni
di Tipo III
Omologia IgHV ≥98%
9/13 (69.2%)
10/11 (90.9%)
1/1 (100.0%)
ZAP-70 ≥20
7/11 (63.6%)
6/8 (75.0%)
0/1 (0.0%)
CD38 ≥7%
4/12 (33.3%)
8/10 (80.0%)
0/1 (0.0%)
del 17p13 5≤ <20%
1/10 (10.0%)
2/9 (33.3%)
0/1 (0.0%)
del 17p13 ≥20%
5/10 (50.0%)
6/9 (66.7%)
1/1 (100.0%)
del 11q22-23 >10%
0/7 (0.0%)
1/7 (14.3%)
-
del 13q14 >5%
4/7 (57.2%)
2/7 (28.6%)
-
+12>5%
1/7 (14.3%)
0/7 (0.0%)
(-, non eseguito)
139
Quindici (75.0%) dei 20 pazienti disfunzionali analizzati, 6/10 (60.0%) disfunzioni di tipo I,
8/9 (88.9%) disfunzioni di tipo II ed 1/1 (100%) disfunzioni di tipo III, risultavano
caratterizzati sia da mutazioni a carico del gene TP53 che dalla del17p13. La sola presenza di
mutazioni del gene TP53 è stata osservata in 3/20 (15.0%) pazienti, tutti contraddistinti da una
disfunzione di tipo I (30.0%, 3/10 disfunzioni di tipo I), mentre 1/20 pazienti (5.0%; CLL
resistente A.R.; 1/9, 11.1 disfunzioni di tipo II) presentava unicamente la del17p13. Infine, in
1/20 pazienti (5.0%, CLL resistente F.C.; 10%, 1/10 disfunzioni di tipo I) non è stata rilevata
la presenza né di mutazioni del gene TP53 né della del17p13 [Figura 15]. Nei pazienti con
disfunzioni a carico della proteina p53 è stata inoltre osservata la stessa distribuzione dei
fattori prognostici indipendentemente dalla presenza di mutazioni del gene TP53.
Nel complesso, i dati ottenuti, sebbene relativi ad una piccola coorte di pazienti, hanno
rivelato una prevalente associazione tra le disfunzioni a carico della proteina p53 e i principali
fattori di prognosi infausta caratteristici della LLC. Inoltre, nell‟ambito delle differenti classi
disfunzionali è stato possibile identificare una maggiore tendenza a tale associazione nel
sottogruppo dei pazienti caratterizzati da disfunzioni di tipo II.
100%
89%
Valori percentuali
100
80
60%
60
30%
40
20
11%
10%
0% 0%
0% 0%
0%
0%
0
TP53
WT/del17p13-
TP53
mut/del17p13-
Disfunzione I
n=10
TP53
mut/del17p13+
Disfunzione II
n=9
TP53
WT/del17p13+
Disfunzione III
n=1
Figura 15. Correlazione tra la tipologia di disfunzione a carico della proteina p53 e la presenza di anomalie
mono- o bi-alleliche del gene TP53.
140
3.4 Correlazione della funzionalità della proteina p53 con la capacità di induzione del
processo apoptotico
Il passo conclusivo di questo studio è stato quello di stabilire se le disfunzioni della proteina
p53, associate o meno ad alterazioni del gene TP53, identificate all‟interno delle cellule
neoplastiche dei pazienti affetti da LLC, potessero causare un‟alterazione dei meccanismi
cellulari p53-dipendenti. Dal momento che le cellule di LLC sono prevalentemente non
ciclanti e refrattarie agli stimoli mitogeni, la valutazione della sensibilità, delle forme
funzionali e disfunzionali della proteina p53, al processo apoptotico indotto dopo esposizione
dei campioni di LLC a IR, è stato preferito rispetto all‟analisi della capacità di arresto del
ciclo cellulare. Le radiazioni ionizzanti, infatti, danneggiano il DNA cellulare inducendo la
via apoptotica p53-dipendente. Di conseguenza, le cellule caratterizzate da assenza di
alterazioni nella via di p53 vanno normalmente incontro a una progressiva morte cellulare
dopo esposizione a IR.
Il tasso apoptotico delle cellule di LLC in condizioni basali e dopo esposizione a 5 Gy di IR, è
stato valutato dopo 24 h di coltura dall‟induzione del danno al DNA, mediante doppia
marcatura con Annexina V e Ioduro di Propidio e successiva analisi citometrica a flusso.
L‟analisi è stata effettuata su un numero complessivo di 26 pazienti, di cui: 16 caratterizzati
da normale funzionalità della proteina p53 e 10 caratterizzati da una proteina disfunzionale.
Come atteso, in tutti i casi di LLC con normale funzionalità di p53 e quindi sequenza “wildtype” del gene TP53, è stato possibile osservare una preservata capacità di induzione del
processo apoptotico dopo esposizione a IR. In particolare, a 24 h è stata stimata una
percentuale media di cellule apoptotiche pari, in tali casi, a 52.4% ± 15% nelle cellule non
irradiate e 74.5% ± 14% dopo irradiazione, con un incremento medio di 1.4 volte. Tale
differenza è risultata statisticamente significativa (P=0.007). La Figura 16 mostra, a scopo
esemplificativo, la visualizzazione al citofluorimetro della percentuale di cellule apoptotiche
osservata in 1 paziente caratterizzato da normale funzionalità di p53.
In contrasto con quanto osservato nei campioni contraddistinti dalla presenza di una proteina
p53 funzionale, le cellule isolate da 9/10 pazienti studiati caratterizzati da disfunzioni di p53
hanno manifestato una resistenza al processo apoptotico indotto dall‟esposizione a IR.
Complessivamente in tali casi è stata osservata una percentuale media di apoptosi pari a
49.7% ± 25% in condizioni basali e 57.6% ± 24% dopo esposizione a IR, con un incremento
medio di 1.2 volte, confermando che la morte cellulare indotta dalle IR è, nelle cellule di
LLC, p53-dipendente [Figura 17].
141
Nell‟ambito, dei suddetti pazienti, è stato inoltre possibile distinguere, in relazione alla
tipologia di disfunzione riscontrata, due differenti livelli di resistenza al processo apoptotico
indotto da IR. Infatti, nei 4/9 pazienti resistenti caratterizzati da disfunzioni di tipo I studiati,
la percentuale media di apoptosi è risultata pari a 35.3% ± 14.9% in condizioni basali e 47.3%
± 15.7% dopo irradiazione, con un incremento medio di 1.3 volte, evidenziando una
condizione di parziale resistenza alle IR.
Figura 16. Visualizzazione al citofluorimetro della percentuale di cellule apoptotiche prima e dopo esposizione a
IR osservata in 1 paziente caratterizzato da normale funzionalità di p53 (Paziente Z.G., LLC alla diagnosi).
Al contrario, nei restanti 5/9 pazienti, di cui 4 con disfunzioni di tipo II ed 1 con disfunzione
di tipo III, è stata stimata una percentuale media di cellule apoptotiche pressoché identica
prima e dopo irradiazione, con valori rispettivamente pari a 61.2% ± 25.6% e 65.8% ± 25.9%
ed incremento medio di 1.07, indicando una condizione di completa resistenza alle IR [Figura
18]. Otto di tali pazienti risultavano caratterizzati anche da mutazioni a carico del gene TP53,
mentre in 1 paziente, affetto da LLC resistente, era stata rilevata una normale sequenza del
suddetto gene, evidenziando un‟alterazione della funzionalità di p53 indipendente dalla
142
presenza di mutazioni del gene TP53. La Figura 19 mostra la visualizzazione al
citofluorimetro della percentuale di cellule apoptotiche osservata in 3 pazienti di LLC
ciascuno caratterizzato da una delle differenti tipologie di disfunzione di p53.
Infine, in 1 dei 9 pazienti (CLL progressiva N.L., disfunzione di tipo II) studiati caratterizzati
da disfunzioni di p53, è stata riportata una normale risposta alle IR (dati non mostrati),
consistente o con la possibilità che l‟anomalia funzionale interessi un piccolo subclone della
popolazione neoplastica o con l‟attivazione di un processo apoptotico p53-indipendente.
In conclusione, i dati osservati evidenziano l‟associazione tra la presenza di disfunzioni a
carico della proteina p53 e la comparsa di alterazioni nel meccanismo di induzione del
processo apoptotico indotto dalle IR. Le differenti forme di resistenza al processo apoptotico
osservate in tale studio nei pazienti disfunzionali, mostrano, inoltre, come ciascuna tipologia
di disfunzione possa avere un impatto più o meno forte sull‟alterazione dei meccanismi
cellulari p53-dipendenti.
143
Valori percentuali
100
80
74.5%
57.6%
52.4%
CTR
49.7%
60
IRR 24h
40
20
0
LLC p53 funzionali
n=16
LLC p53 disfunzionali totali
n=9
Figura 17. Confronto delle percentuali medie di cellule apoptotiche prima e dopo esposizione a IR osservate
nella totalità dei pazienti caratterizzati da normale o alterata funzionalità di p53.
100
Valori percentuali
70.9%
76.8%
80
47.4%
60
CTR
35.3%
40
IRR 24h
22.4%
22%
20
0
Disfunzioni I
n=4
Disfunzioni II
n=4
Disfunzioni III
n=1
Figura 18. Correlazione tra le percentuali medie di cellule apoptotiche prima e dopo esposizione a IR e la
tipologia di disfunzione a carico di p53.
144
a)
b)
c)
Figura 19. Visualizzazione al citofluorimetro della percentuale di cellule apoptotiche osservata in 3 pazienti di
LLC ciascuno caratterizzato da una delle differenti tipologie di disfunzione di p53: a) Paziente L.P., disfunzione
I, LLC in progressione; b) Paziente A.R., disfunzione II, LLC resistente; c) Paziente D.C.G., disfunzione III,
LLC in progressione.
145
4. DISCUSSIONE
La comprensione dei meccanismi molecolari alla base della suscettibilità al cancro e della
resistenza a terapie farmacologiche specifiche è essenziale per migliorarne l‟esito terapeutico.
La LLC è una patologia a decorso clinico altamente variabile e sebbene l‟identificazione di
parametri prognostici, sia di natura clinica che biologica, abbia consentito la classificazione
dei pazienti in categorie di rischio, tale suddivisione non sempre si rivela adeguata alla
predizione del comportamento clinico e della risposta farmacologica. Le principali opzioni
terapeutiche destinate a pazienti affetti da LLC sono tuttora rappresentate dall‟impiego di
farmaci citotossici o di antimetaboliti, quali agenti alchilanti e analoghi purinici, che mediano
l‟eliminazione delle cellule neoplastiche attivando il processo di morte cellulare programmata
dopo induzione di un danno diretto o indiretto a carico del DNA. Di conseguenza, lo studio
delle proprietà che determinano la suscettibilità delle cellule di LLC alla terapia antitumorale
e la loro eventuale correlazione con i marcatori prognostici caratteristici della patologia,
potrebbe diventare utile per ottenere informazioni predittive addizionali. In tale ambito, un
ruolo di primaria importanza riveste la proteina oncosoppressoria p53 che, attraverso
l‟induzione dell‟arresto del ciclo cellulare, dell‟apoptosi o della senescenza in risposta a
differenti condizioni di stress, tra cui la presenza di un danno a carico del DNA, non solo
protegge il genoma dagli insulti mutageni, ma contribuisce anche all‟azione citotossica della
maggior parte dei chemioterapici. Non sorprende quindi l‟osservazione che i difetti del gene
TP53 nella LLC siano fortemente associati a progressione di malattia, resistenza alla
chemioterapia e accorciamento dell‟aspettativa di sopravvivenza. Tali dati sono emersi in
primo luogo dall‟analisi del decorso della LLC in pazienti caratterizzati dalla delezione del
braccio corto del cromosoma 17, regione su cui mappa il gene TP53, ma sono stati
successivamente consolidati dall‟analisi di pazienti contraddistinti dalla comparsa di
mutazioni a carico del suddetto gene, in associazione o meno alla del17p13. In realtà,
nonostante la dimostrazione dell‟esistenza di una correlazione tra le alterazioni molecolari a
carico del gene TP53 e il peggioramento della prognosi, esiste, anche all‟interno del
sottogruppo di pazienti affetti da LLC caratterizzati da tali alterazioni, una considerevole
variabilità prognostica. Parte di questa variabilità può essere attribuita alle proprietà
funzionali della proteina p53 mutata. A differenza, infatti, di altri geni oncosoppressori, che
normalmente sono inattivati da mutazioni associate a perdita di sintesi del prodotto genico, la
maggior parte delle mutazioni del gene TP53 è contraddistinta dalla produzione di una
proteina mutata che, a seconda dei casi, può esercitare un effetto dominante negativo sulla
146
proteina “wild-type” e/o essere caratterizzata da nuove proprietà oncogeniche. La
combinazione tra le suddette caratteristiche e l‟estensione della mutazione all‟interno della
popolazione neoplastica, che influenzano rispettivamente l‟attività residua della proteina
normale e la quantità di proteina mutata prodotta, può quindi esercitare un‟influenza più o
meno negativa sui meccanismi cellulari alla base della progressione tumorale e della
resistenza alle terapie farmacologiche antineoplastiche. Tuttora, però, gli effetti di tale
combinazione sono poco chiari e quindi oggetto di intenso dibattito.
Sulla base di tali premesse, il presente progetto di ricerca si è incentrato sullo studio della
funzionalità della proteina p53 in pazienti affetti da LLC, nel tentativo di correlare l‟attività di
tale proteina al decorso della malattia e, quindi, alle caratteristiche molecolari del gene TP53,
con lo scopo di implementare la comprensione della variabilità dell‟andamento clinico in
pazienti contraddistinti da alterazioni a carico di tale oncosoppressore e identificare,
all‟interno di tale gruppo, quei pazienti che potrebbero beneficiare di un approccio terapeutico
innovativo. Sono stati inclusi nello studio pazienti affetti da LLC a diversi stadi di malattia,
quali: esordio, progressione e resistenza a una o più linee di terapia citotossica convenzionale.
La funzionalità di p53 è stata studiata valutando l‟induzione della proteina in risposta
all‟esposizione dei campioni primari di cellule neoplastiche di LLC a radiazioni ionizzanti. Da
tale analisi è emerso, prendendo in considerazione la totalità dei pazienti inclusi nello studio,
che la proteina p53 risulta caratterizzata da una normale funzionalità nella maggior parte dei
casi e più precisamente in una quota pari al 71.3%, presentando, al contrario, anomalie
funzionali nel restante 28.7%. Questo primo dato suggerisce come, diversamente da altre
forme di neoplasie in cui la percentuale di alterazioni a carico dell‟oncosoppressore p53 si
estende sino all‟80.0% dei pazienti, nella LLC tale proteina rivesta probabilmente un ruolo
più marginale nel processo tumorale o, in alternativa, sfrutti la ridondanza della sua via di
segnalazione intracellulare per influenzare tale processo.
All‟interno dei casi disfunzionali è stato quindi possibile osservare una peculiare distribuzione
in relazione alle differenti classi di pazienti affetti da LLC. Infatti, sebbene le disfunzioni
della proteina p53 siano state rilevate anche in pazienti al momento della diagnosi, esse sono
apparse concentrate maggiormente in pazienti in progressione di malattia e in pazienti con
malattia resistente o refrattaria alle terapie citotossiche convenzionali. In seguito a tale
osservazione, è stato possibile supporre che le anomalie a carico di p53 siano rilevanti, nella
LLC, non tanto nel condizionare la patogenesi della malattia, quanto nell‟influenzare e
governare la progressione, quindi l‟espansione del clone leucemico all‟interno della
popolazione cellulare, e la suscettibilità alla terapia antitumorale, con implicazioni nello
147
sviluppo di resistenze farmacologiche. Non vi sono in letteratura studi di funzione della
proteina p53 in pazienti affetti da LLC a differente stadio di malattia, ma i risultati ottenuti in
tale ambito sono paragonabili e riflettono quanto osservato relativamente alla presenza di
alterazioni molecolari del gene TP53 nei suddetti pazienti. Infatti, tali alterazioni sono
normalmente riscontrate dal 4 al 37% dei pazienti di LLC, con la più bassa incidenza, pari al
4-10%, al momento della diagnosi e la più alta, del 25-37%, al momento della comparsa della
chemioresistenza [97]. A giustificare la bassa incidenza di alterazioni a carico della proteina
p53 al momento della diagnosi, vi è la considerazione che tra le ipotesi relative alla
patogenesi della LLC, una delle più accreditate riguarda la stimolazione antigenica del
recettore B cellulare delle cellule neoplastiche. Di conseguenza, la comparsa di tali alterazioni
potrebbe inserirsi in questo contesto ed esserne conseguenza piuttosto che causa scatenante,
considerando che un comportamento biologico più aggressivo delle cellule B neoplastiche, in
termini di rapida proliferazione e avanzato danno al DNA, potrebbe comportare una pressione
mutazionale selettiva nei confronti di p53. Al contrario, a conferma del possibile ruolo delle
alterazioni di p53 nella progressione di malattia, è emerso, da un recente studio, come tale
proteina sia in grado di controllare la polarità delle divisioni cellulari in cellule staminali
cancerose (CSC) [108]. Attraverso l‟impiego di un modello di cellule tumorali mammarie, è
stato osservato come tali cellule siano caratterizzate da una maggiore tendenza a divisioni
simmetriche e illimitate e come queste proprietà possano essere trasferite in cellule staminali
normali (CSN), mediante l‟introduzione di mutazioni a carico del gene TP53. Al contrario, la
riattivazione farmacologica di p53 si associa al ripristino dell‟asimmetria delle divisioni
cellulari sia nelle CSN che nelle CSC. Di conseguenza, l‟acquisizione di anomalie a carico di
p53 da parte delle cellule tumorali, può rappresentare un meccanismo utile a favorire,
influenzando la simmetria delle divisioni cellulari, la crescita tumorale e quindi la
progressione di malattia. La relazione tra anomalie della proteina p53 e acquisizione di
resistenze alla terapia antitumorale risulta invece più facilmente comprensibile, dal momento
che i farmaci citotossici impiegati nella LLC agiscono attivando la via apoptotica p53dipendente, per cui la loro somministrazione potrebbe favorire un meccanismo di “escape”
tumorale consistente con la comparsa o la selezione di cloni caratterizzati da alterazioni
molecolari e/o funzionali di p53 all‟interno di nicchie ematiche rese disponibili dall‟attività
dei farmaci.
Dall‟analisi dei campioni dei pazienti affetti da LLC in cui la proteina p53 è risultata
disfunzionale, è stato possibile individuare 3 tipologie di disfunzioni, classificate come
disfunzioni di tipo I, II e III. La disfunzione di tipo I si associa a un costitutivo incremento
148
della proteina p53 nelle cellule di LLC non irradiate e un suo ulteriore accumulo dopo
irradiazione. Nella disfunzione di tipo II, pur essendo presente il costitutivo incremento della
proteina in condizioni basali, non si verifica alcun ulteriore accumulo dopo irradiazione.
Infine, nella disfunzione di tipo III, la proteina p53 non risulta rilevabile né prima né dopo
esposizione delle cellule di LLC a radiazioni ionizzanti. Le disfunzioni di tipo I sono state
riscontrate con maggiore frequenza sia all‟interno della totalità dei pazienti inclusi nello
studio che in 1 delle 3 classi di pazienti suddivisi in relazione allo stadio di malattia,
rappresentata dai casi progressivi. Non essendo state riscontrate disfunzioni di tipo I nei
pazienti all‟esordio di malattia, non è stato invece possibile valutare la correlazione di tali
alterazioni all‟interno della suddetta classe.
Il dato relativo alla prevalenza delle disfunzioni di tipo I nei pazienti affetti da LLC risulta
interessante se si considera la definizione di p53 come oncosoppressore. Infatti, essendo le
disfunzioni di tipo I caratterizzate dalla co-presenza di una quota di proteina mutata e una
quota di proteina normale, giustificata dall‟aumento dell‟accumulo di p53 dopo irradiazione,
si potrebbe supporre che nella maggior parte dei casi siano selezionate, diversamente da
quanto accade per altri oncosoppressori, alterazioni monoalleliche del gene TP53, associate
alla produzione di una proteina p53 con effetto dominante negativo nei confronti della
proteina normale o con nuove proprietà oncogeniche. In ambo i casi, infatti, gli effetti deleteri
delle anomalie di p53 potrebbero essere sufficientemente esercitati anche in presenza di una
porzione normale della proteina stessa, senza dover essere necessariamente accompagnate da
inattivazione biallelica. In alternativa, si potrebbe pensare alla presenza di anomalie
addizionali di altri componenti della via di p53 che agirebbero in maniera sinergica con le
alterazioni monoalleliche a carico del gene TP53 o alla presenza di alterazioni bialleliche
estese solamente ad un subclone della popolazione neoplastica.
Per quanto riguarda le disfunzioni di tipo II, è importante sottolineare come i pazienti
chemioresistenti siano caratterizzati dalla più alta percentuale di tali alterazioni. Dal momento
che nel caso delle disfunzioni di tipo II non è stata rilevata alcuna quota normale di proteina
p53, si potrebbe supporre, in tal caso, un‟inattivazione biallelica del gene TP53 estesa
all‟intera popolazione tumorale. Come riportato da uno studio recente, l‟analisi sequenziale di
pazienti affetti da LLC TP53 “wild-type” e sottoposti a terapia citotossica, ha rivelato che la
simultanea inattivazione di ambo gli alleli del gene TP53 appare peculiare in tali casi poiché,
probabilmente, viene accelerata dalla terapia stessa e segue meccanismi diversi rispetto ai
processi di mutagenesi spontanea. L‟inattivazione biallelica appare inoltre caratterizzata, in
tale studio, da un primo evento di mutagenesi cui può associarsi, sull‟altro allele, un secondo
149
evento mutagenico o l‟acquisizione della del17p13 [97]. Queste osservazioni potrebbero
quindi giustificare la concentrazione delle disfunzioni di tipo II all‟interno della categoria dei
pazienti chemioresistenti. Infine, essendo stata osservata la disfunzione di tipo III in un unico
paziente affetto da LLC in progressione di malattia, non è stato possibile valutare, in tal caso,
una reale associazione all‟una o l‟altra classe di malattia.
Allo scopo di verificare le ipotesi relative alla correlazione tra le differenti tipologie di
disfunzioni dell‟oncosoppressore p53 e lo stato del gene TP53 è stato quindi effettuato un
confronto tra il dato funzionale di natura proteica e il dato genomico relativo all‟analisi di
sequenza del suddetto gene. Tale confronto ha rivelato, in primo luogo, una concordanza
assoluta tra la normale funzionalità della proteina p53 e lo stato “wild-type” del gene TP53.
Al contrario, è stata osservata una concordanza elevata, seppur non assoluta, tra la presenza di
disfunzioni della proteina p53 e lo stato mutato del gene TP53. L‟analisi dei casi disfunzionali
concordanti, precisamente pari all‟80.0%, ha identificato nella maggior parte mutazioni
missenso, osservate per lo più in eterozigosi. Tale dato riflette le peculiarità del profilo
mutazionale del gene TP53 che, svariati studi di pubblicazione più o meno recente, riportano
sia caratterizzato, in circa il 75% dei casi, proprio da alterazioni missenso [96-99].
Il confronto tra i dati di funzionalità e quelli genomici ha inoltre mostrato la tendenza delle
varie tipologie di disfunzioni ad associarsi a un profilo mutazionale specifico sia in termini di
natura che posizione della mutazione genica. In particolare, la disfunzione di tipo I è stata
osservata in associazione prevalente sia a mutazioni missenso che a mutazioni in eterozigosi,
osservate entrambe in circa il 90.0% dei casi, a supporto dell‟ipotesi relativa alla correlazione
tra tali disfunzioni e le forme di inattivazione monoallelica del gene TP53. Anche le
disfunzioni di tipo II sono apparse associate a mutazioni missenso del gene TP53, sebbene,
rispetto alle disfunzioni di tipo I, si sono rivelate caratterizzate da una più stretta correlazione
a mutazioni omozigoti. Quest‟ultimo dato favorisce, in tal caso, la tesi dell‟associazione delle
disfunzioni di tipo II alle forme di inattivazione biallelica del gene TP53. L‟analisi dei 3
pazienti con disfunzione di tipo II e mutazioni eterozigoti di TP53 ha inoltre permesso di
supportare ulteriormente questa tesi. Tali pazienti sono, infatti, apparsi caratterizzati da un
assetto genomico più complesso. Uno di loro presentava due diverse mutazioni del gene TP53
in eterozigosi. Se tali mutazioni fossero presenti sullo stesso allele, una mutazione con effetto
dominante negativo sul prodotto genico normale potrebbe giustificare il fenotipo di una
disfunzione di tipo II; in alternativa, se le due mutazioni fossero presenti su due alleli diversi,
tale condizione potrebbe mimare quello che si verifica nel caso di una mutazione in
omozigosi e, anche in questo caso, correlare con il fenotipo di una disfunzione di tipo II. Il
150
secondo paziente, studiato orizzontalmente al momento della diagnosi e in progressione di
malattia, ha manifestato, all‟analisi effettuata mediante FISH, la presenza addizionale di una
delezione del braccio corto del cromosoma 17 in una percentuale iniziale di cellule B
neoplastiche del 14.0%, con successiva estensione sino all‟84.0% in progressione di malattia.
In aggiunta, in tale paziente è stata riscontrata una singola mutazione missenso in eterozigosi
nel gene ATM, concordante con la mancata induzione di p53 dopo irradiazione delle cellule
tumorali. La presenza di cellule del17p13+ è stata rivelata anche nel terzo paziente, in una
percentuale pari al 75.0%. In quest‟ultimo paziente, la presenza di un fenotipo di tipo II
nonostante una mutazione eterozigote del gene TP53 e una del17p13 non estesa a tutta la
popolazione neoplastica, risulta consistente con l‟esercizio di un effetto dominante negativo
da parte della proteina p53 mutata sulla forma normale. Anche la disfunzione di tipo III,
sebbene identificata in un solo paziente, è risultata associata ad una forma di inattivazione
biallelica del gene TP53, rappresentata da una mutazione in omozigosi con introduzione di un
codone di stop. In questo caso, si potrebbe supporre che la mancata osservazione di p53 sia in
condizioni basali che dopo irradiazione, dipenda dall‟impossibilità dell‟anticorpo impiegato
nello studio di riconoscere la forma tronca della proteina o dalla tendenza della proteina
tronca ad essere riconosciuta e degradata.
E‟ stato possibile distinguere le disfunzioni di tipo I e II anche in relazione alla differente
corrispondenza con gli esoni e i codoni del gene TP53 interessati dalla mutazione. Mentre nel
caso delle disfunzioni di tipo I è stata, infatti, osservata la presenza esclusiva di mutazioni
nelle regioni codificanti il DBD, con prevalenza di mutanti di “contatto”, le disfunzioni di tipo
II sono risultate associate non solo a mutazioni nel DBD ma anche in regioni esterne al DBD,
con una correlazione più stretta con la classe dei mutanti “conformazionali”, nota per essere
associata ad un effetto prognostico peggiore. In tali casi è stata osservata anche
un‟associazione con mutanti a livello di residui non compresi nelle regioni corrispondenti alle
suddette classi mutazionali. Di conseguenza, si potrebbe supporre che le disfunzioni di tipo I
siano la manifestazione degli eventi classici di mutagenesi a carico del gene TP53 e, al
contrario, che le disfunzioni di tipo II, siano la manifestazione di eventi mutageni più
complessi, nati da una pressione selettiva in parte diversa e con un probabile maggiore
impatto prognostico sfavorevole. E‟ stato ad esempio dimostrato, attraverso la trasfezione di
differenti mutanti di TP53 in cellule di lievito, che circa il 30% delle mutazioni ha un effetto
dominante negativo sull‟allele TP53 “wild-type” e che tali mutazioni ricadono
preferenzialmente in codoni codificanti aminoacidi altamente conservati localizzati all‟interno
del DBD [109]. La correlazione delle disfunzioni di tipo I a mutazioni prevalentemente
151
localizzate nel DBD avvalora l‟ipotesi che quest‟ultime possano esercitare un meccanismo
dominante negativo sulla proteina normale e quindi, come supposto, manifestare il loro effetto
anche in assenza di meccanismi di inattivazione biallelica del gene TP53. Per quanto riguarda
la disfunzione di tipo III, essa è stata osservata in associazione a una mutazione interna al
DBD ma a livello di residui non compresi nelle regioni corrispondenti alle classi dei mutanti
di “contatto” o “conformazionali”.
I risultati ottenuti relativamente alle varie tipologie di disfunzioni e alle loro correlazioni con
lo stato del gene TP53, concordano solo in parte con i dati pubblicati in letteratura. Questo
deriva principalmente dalla scarsità di studi di funzione della proteina p53 nella LLC. Solo
due recenti lavori riportano l‟identificazione di altrettante tipologie di disfunzioni a carico di
p53 nella LLC, ma in termini non totalmente sovrapponibili a quanto rilevato in questo studio.
Sebbene, infatti, nel lavoro pubblicato dal gruppo di ricerca di Stankovic T et al. [103] sia
stata identificata una tipologia di disfunzione, definita di tipo A, sovrapponibile, sia in termini
di definizione che di relazione con lo stato di inattivazione monoallelica del gene TP53, alla
disfunzione di tipo I osservata in questo studio, non è stato possibile trovare alcuna
corrispondenza relativamente alle disfunzioni di tipo II e III. Al contrario, nel suddetto lavoro,
viene descritta l‟esistenza di una seconda tipologia di disfunzione, definita di tipo B, per
definizione ma non per correlazione con lo stato del gene TP53, paragonabile alla disfunzione
di tipo III di questo studio. L‟assenza di una corrispondenza può essere giustificata
considerando i differenti parametri presi in esame in ciascuno degli studi, sulla base dei quali
ruota la definizione della tipologia di disfunzione. In questo lavoro, è stato possibile
identificare 3 classi di disfunzioni prendendo in considerazione la sola espressione dalla
proteina p53. La classificazione suggerita dal gruppo di ricerca di Stankovic T et al., nasce
invece dall‟analisi simultanea dei livelli di espressione delle proteine p53 e p21. Di
conseguenza, la disfunzione di tipo B, che si associa ad assenza di accumulo basale di p53 e
deficit contemporaneo di induzione di entrambe le proteine dopo irradiazione, risulta
associata a mutazioni del gene ATM, confermando il ruolo di tale proteina nell‟attivazione
della via di p53 a seguito dell‟induzione di un danno al DNA. Analogamente, nel lavoro
pubblicato da Pettitt AR et al., ritroviamo un‟ulteriore classificazione delle tipologie di
disfunzioni, basata sull‟espressione di p53 e p21 [110]. In questo lavoro, viene inoltre
identificata una terza tipologia di disfunzione, definita di tipo C, caratterizzata da incapacità
della proteina p21 di accumularsi in seguito ad un danno a carico del DNA cellulare,
nonostante una normale risposta di p53. Tale disfunzione è riportata in circa il 10% dei
pazienti affetti da LLC e sembra essere causata dalla presenza di un polimorfismo nel codone
152
31 del gene p21. Questo difetto è infine associato a fenomeni di resistenza alla citotossicità
indotta dalle radiazioni ionizzanti [110]. Ad ogni modo, quello che risulta evidente dai lavori
pubblicati in letteratura e viene avvalorato da questo studio è che ad ogni tipologia di
disfunzione della proteina p53 rilevata, si associa una diversa alterazione di natura genica, che
può coinvolgere la proteina p53 stessa o diverse componenti a monte o a valle della sua via di
segnalazione.
Una quota pari al 20.0% dei pazienti disfunzionali è risultata caratterizzata dall‟assenza di
alterazioni molecolari del gene TP53. In questi casi si potrebbe supporre che la mutazione non
sia del tutto assente, ma che l‟alterazione genica sia presente in esoni non routinariamente
sequenziati, ovvero esoni esterni al DBD. Questo implica che la restrizione dell‟analisi di
sequenza agli esoni 5-8 del gene TP53 potrebbe dare origine a dei falsi negativi,
sottostimando la presenza di mutazioni non presenti nei comuni hot-spots. In realtà, in tutti i
casi discordanti, nonostante sia stato eseguito il sequenziamento dell‟intera regione
codificante del gene TP53 (esoni 2-11), non è stata rivelata presenza alcuna di mutazioni.
Questo dato conferma quanto osservato recentemente in letteratura riguardo la possibilità di
osservare anomalie funzionali e, quindi, meccanismi di stabilizzazione della proteina p53,
indipendenti dalla presenza di mutazioni a carico del gene TP53. In realtà, si potrebbe
ipotizzare che la mutazione sia presente in un subclone di ridotta estensione all‟interno della
popolazione neoplastica, tanto da risultare non rilevabile, oppure che i casi discordanti siano
contraddistinti da alterazioni di altre componenti della via di p53. E‟ stato, infatti, dimostrato
che il 15%-35% dei pazienti affetti da LLC possiede una copia soprannumeraria del
cromosoma 12, che codifica per MDM2, proteina inibitoria di p53. L‟overespressione di
MDM2 è stata, inoltre, riportata nella LLC sia come anomalia singola che in associazione alla
trisomia del cromosoma 12. Anche l‟inattivazione del gene ATM, come descritto nel lavoro
del gruppo di ricerca di Stankovic T, rappresenta un‟ulteriore potenziale causa di disfunzioni
di p53 in pazienti affetti da LLC TP53 “wild-type”. Ridotti livelli della proteina ATM sono
stati rilevati complessivamente nel 30%-40% dei pazienti di LLC e nel 20% dei casi è stata
dimostrata la presenza della del11q22-23, regione su cui mappa il gene ATM. Sia
l‟overespressione di MDM2 che l‟inattivazione di ATM comporterebbero tuttavia una
alterazione nella capacità della proteina p53 di accumularsi dopo irradiazione, in assenza di
quantità rilevabili della stessa in condizioni basali. In realtà, il fenotipo osservato nei pazienti
disfunzionali TP53 “wild-type”, di cui 3 caratterizzati da disfunzioni di tipo I e 2 di tipo II,
non mostra una correlazione con quanto atteso in presenza di anomalie di MDM2 o ATM,
essendo caratterizzato, sia nel caso delle disfunzioni di tipo I che II, da un aumento costitutivo
153
basale della proteina p53. Di conseguenza, in tali casi si potrebbe pensare ad anomalie dei
meccanismi di degradazione di p53 a carico di molecole diverse da MDM2, come
l‟oncosoppressore ARF o i regolatori del processo di ubiquitinazione. Nei pazienti
disfunzionali TP53 “wild-type” è quindi in corso uno studio volto all‟identificazione di cause
alternative di disfunzione dell‟oncosoppressore p53, che comprende comunque la valutazione
dello stato dei geni ATM e MDM2. Ad ogni modo, indipendentemente dalle cause di
stabilizzazione della proteina p53 in condizioni basali, è necessario ricordare come la
presenza di alterazioni funzionali di p53 possa predisporre il paziente a un‟eventuale ridotta o
mancata risposta terapeutica in quei casi in cui è previsto l‟impiego di farmaci citotossici che
agiscono, come le IR, inducendo, attraverso il danno al DNA, la via apoptotica p53dipendente. La rilevazione di tali disfunzioni a prescindere dal sequenziamento del gene
TP53, data la loro incidenza, pari nel nostro studio al 20.0%, diviene quindi di grande
importanza al fine di identificare quei pazienti che, nonostante siano caratterizzati da assenza
di alterazioni molecolari a carico di TP53, potrebbero presentare una scarsa risposta ai
chemioterapici convenzionali e dovrebbero quindi essere indirizzati verso approcci terapeutici
differenti. Appare inoltre interessante sottolineare come i casi disfunzionali/TP53 “wild-type”
siano tutti caratterizzati da una malattia resistente. L‟associazione tra questa fase di malattia e
la comparsa di anomalie di p53 indipendenti da eventi mutazionali supporta, infatti,
ulteriormente la tesi che i farmaci citotossici possano sfruttare la ridondanza della via di p53
per indurre chemiorefrattarietà.
Per identificare il possibile significato prognostico delle disfunzioni a carico della proteina
p53, è stata in seguito valutata la presenza di un‟eventuale correlazione tra le suddette
tipologie di disfunzioni e i principali fattori indicatori di prognosi nei pazienti affetti da LLC.
Come atteso, la maggior parte dei pazienti disfunzionali ha manifestato una corrispondenza,
più evidente nel caso delle disfunzioni di tipo II, con fattori prognostici infausti, rappresentati
dalla presenza di una sequenza non mutata dei geni IgHV, dalla positività per l‟espressione
della proteina ZAP70 e dalla presenza della del17p13. Un‟associazione meno forte è stata
invece osservata nei confronti della positività per l‟antigene CD38. Quest‟ultimo dato è in
accordo con quanto riportato nel lavoro di Lin K et al., nel quale, nonostante la conferma del
valore predittivo indipendente dello stato mutazionale, dell‟espressione del CD38 e delle
disfunzioni della proteina p53 nei pazienti affetti da LLC, è stata osservata l‟esistenza di una
correlazione di tali disfunzioni con lo stato IgHV non mutato ma non con la positività per
l‟antigene CD38 [111]. In particolare, l‟associazione delle disfunzioni di p53 con stato IgHV
non mutato potrebbe essere giustificata formulando una serie di ipotesi. Si potrebbe supporre
154
che tali disfunzioni alterino la capacità delle cellule B neoplastiche di andare incontro al
processo di ipermutazione somatica; alternativamente, le cellule B di LLC IgHV non mutate
potrebbero essere propense all‟acquisizione di lesioni genetiche che risultano nella comparsa
di disfunzioni a carico di p53 o le stesse disfunzioni della proteina p53 potrebbero facilitare
l‟espansione clonale delle cellule B tumorali con stato IgHV non mutato. Quale di queste
ipotesi sia la più corretta non è ancora noto, di conseguenza, la comprensione di questi
meccanismi diventa fondamentale per la predizione della sopravvivenza e della risposta
terapeutica in pazienti affetti da LLC con difetti a carico di p53. Anche il dato relativo
all‟associazione delle disfunzioni della proteina p53 alla del17p13 rispecchia le evidenze
riportate in letteratura, secondo le quali, nonostante la presenza di mutazioni del gene TP53 si
sia rivelata essere un fattore prognostico indipendente, la loro natura monoallelica rimane
ristretta ad una piccola percentuale di casi, essendo, tali mutazioni, spesso presenti in
concomitanza alla suddette delezioni, che conferiscono aggressività, chemioresistenza e
prognosi infausta alla patologia. Inoltre, quanto osservato in questo studio si accompagna
all‟osservazione che le delezioni del braccio corto del cromosoma 17 non si presentano quasi
mai come unica aberrazione cromosomica nel genoma dei pazienti affetti da LLC [91]. La
positività per la del17p13 anche in pazienti con disfunzioni di tipo I, delle quali era stata
supposta, in questo studio, una correlazione a forme di inattivazione monoallelica del gene
TP53, non deve essere considerata una discrepanza in quanto, in tali casi, la presenza di una
quota normale di proteina p53 può essere giustificata ipotizzando che l‟alterazione biallelica
sia estesa solo all‟interno di un subclone della popolazione neoplastica e non nella totalità di
essa.
Il passo conclusivo di questo studio è stato quello di valutare se le disfunzioni della proteina
p53 rilevate nei pazienti di LLC, causassero un difetto nell‟induzione del processo apoptotico,
dopo esposizione dei campioni a radiazioni ionizzanti. Queste, infatti, danneggiano il DNA
cellulare inducendo la via apoptotica p53-dipendente, per cui le cellule caratterizzate da
assenza di alterazioni nella via di p53 vanno normalmente incontro a una progressiva morte
cellulare dopo irradiazione. Come atteso, le cellule isolate da pazienti con normale
funzionalità di p53 hanno presentato una preservata capacità di andare incontro al processo
apoptotico a seguito dell‟insulto genotossico. Al contrario, le cellule isolate da pazienti
caratterizzati da disfunzioni di p53, eccetto che in un singolo caso, hanno mostrato,
indipendentemente dalla presenza di mutazioni a carico del gene TP53, difetti nell‟induzione
del processo apoptotico, confermando che la morte cellulare indotta dalle radiazioni ionizzanti
è p53-dipendente. La normale risposta alle radiazioni ionizzanti riportata in uno dei pazienti
155
disfunzionali studiati, potrebbe essere invece consistente con l‟induzione di una via apoptotica
p53-indipendente. Un dato interessante, emerso dall‟analisi dei risultati ottenuti in tale
ambito, è rappresentato dall‟identificazione di due diversi livelli di resistenza all‟induzione
del processo apoptotico, ciascuno strettamente correlato alla tipologia di disfunzione. Infatti,
le disfunzioni di tipo I, sono risultate caratterizzate da una resistenza “parziale” mentre sia le
disfunzioni di tipo II che III da una resistenza “totale” all‟apoptosi indotta dopo esposizione a
radiazioni ionizzanti. Questo dato correla perfettamente con quanto atteso in relazione alla
descrizione del fenotipo delle differenti disfunzioni di p53. Infatti, la resistenza parziale al
processo apoptotico è stata osservata laddove presente una quota normale di proteina p53; al
contrario, in assenza di una quota normale di proteina p53 è stata rilevata una resistenza
totale. La resistenza al processo apoptotico indotto dalle radiazioni ionizzanti, osservata in
questo studio nei pazienti p53 disfunzionali, trova conferma non solo nel lavoro di Stankovic
T e coll. ma anche in un lavoro di recente pubblicazione in cui si dimostra l‟esistenza di una
correlazione tra la suddetta forma di resistenza e la presenza di un‟elevata complessità
genomica nelle cellule B di LLC, caratterizzata non solo da mutazioni del gene TP53 ma
anche dalle delezioni delle regioni 17p13 e 13q14 e dipendente da errori nel processo di riparo
del DNA piuttosto che dall‟accorciamento telomerico [112]. Nei casi caratterizzati da
resistenza parziale, la proporzione di cellule apopototiche è risultata differente a seconda del
campione in esame. Questo dato riflette, ovviamente, la quota residua di proteina p53
funzionale specifica di ciascun paziente, la peculiarità delle caratteristiche della proteina p53
disfunzionale in termini di effetto dominante negativo o acquisto di nuove proprietà
oncogeniche e l‟eventuale contributo di vie apoptotiche p53-indipendenti. Ad ogni modo, a
prescindere dalla maggiore o minore parzialità della resistenza al processo apoptotico,
caratteristica delle disfunzioni di tipo I, in tali casi la popolazione neoplastica disfunzionale
potrebbe essere comunque selezionata positivamente in seguito all‟esposizione a farmaci
citotossici convenzionali, i quali potrebbero, a loro volta, favorire l‟acquisizione di ulteriori
anomalie a livello della via apoptotica p53-dipendente, con peggioramento della prognosi del
paziente. Tali effetti potrebbero verificarsi, in misura ancor più evidente, nei pazienti
caratterizzati da disfunzioni di tipo II o III e quindi da resistenza totale al processo apoptotico.
All‟interno di tale gruppo, nei pazienti non trattati all‟esordio o in progressione di malattia, si
potrebbe predire la possibilità di una mancata risposta ai chemioterapici citotossici
convenzionali; al contrario, nei pazienti già esposti a una o più linee di terapia si potrebbe
correlare l‟acquisizione della resistenza farmacologica alla rilevazione della specifica
alterazione molecolare e funzionale della proteina p53. Di conseguenza, in entrambi i casi
156
sarebbe necessario indirizzare i pazienti a terapie alternative, come l‟impiego di anticorpi
monoclonali o il trapianto di cellule staminali emopoietiche, che non comportino l‟ulteriore
selezione di subcloni con anomalie di p53.
Lo studio della risposta di p53 alle radiazioni ionizzanti si è rivelata, in questo studio, una
modalità efficace sia per la valutazione di anomalie funzionali a carico di tale proteina che per
l‟indicazione di anomalie molecolari a carico del gene TP53. Il fatto che i difetti funzionali
della proteina p53 siano stati osservati in tutti i casi con mutazioni del gene TP53 rivela un
alto livello di specificità della metodica, indicando una probabilità quasi irrisoria di falsi
negativi. Inoltre, la rilevazione di disfunzioni di p53 anche in pazienti TP53 “wild-type”,
evidenzia l‟importanza dello studio funzionale nell‟evitare di sottostimare le anomalie a
carico di tale oncosoppressore che potrebbero avere un impatto prognostico paragonabile a
quello osservato in presenza di mutazioni del gene TP53 e/o delezioni del braccio corto del
cromosoma 17. I pazienti p53 disfunzionali ma TP53 “wild-type” diventano oltretutto
interessanti per uno studio finalizzato all‟individuazione di meccanismi alternativi di
stabilizzazione di p53, che potrebbe comportare la scoperta di nuovi target terapeutici
destinati a tale sottogruppo. Infine, l‟osservazione dell‟assenza di un‟equivalenza funzionale
tra le differenti forme anomale della proteina p53, correlate o meno a eventi mutazionali a
carico del gene TP53, rappresenta un punto chiave nella comprensione dell‟eterogeneità
prognostica dei pazienti affetti da LLC e caratterizzati da alterazioni del suddetto
oncosoppressore.
Dal momento che le anomalie a carico di p53 si sono dimostrate rilevanti non tanto nella
patogenesi, quanto nella progressione della LLC e nello sviluppo di resistenze
farmacologiche, dimostrando come una errata linea terapeutica in questi pazienti possa
comportare lo sviluppo di forme più aggressive della malattia, sarebbe fondamentale proporre
dei nuovi algoritmi terapeutici che definiscano un “percorso individuale” per ciascun
paziente, finalizzato ad una selezione razionale della terapia medica sulla base dello stato di
p53. A tale scopo l‟analisi simultanea dello stato mutazionale del gene TP53, non ancora
incluso nelle attuali linee guida diagnostiche della LLC, e della funzionalità del suo prodotto
proteico sarebbe di grande importanza per un‟opportuna gestione del paziente che abbia,
come fine ultimo, il miglioramento della prognosi e l‟allungamento delle aspettative di
sopravvivenza.
157
5. REFERENZE
1. Lane D, Benchimol S. p53: oncogene or anti-oncogene? Genes Dev 1990; 4:1-8
2. Levine A, Momand J, Finlay C. The p53 tumor suppressor gene. Nature 1991; 351:453456
3. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of
transformation. Cell 1989; 57:1083-1093
4. Hinds PW, Finlay CA, Levine AJ. Mutation is require to activate the p53 gene for
cooperation with the ras oncogene and transformation. J Virol 1989; 63:739-746
5. Roemer K, Friedmann T. Mechanisms of action of the p53 tumor suppressor and prospects
for cancer gene therapy by reconstitution of p53 function. Ann NY Acad Sci 1994; 716:265282
6. Ho WC, Fitzgerald MX, Marmorstein R. Structure of the p53 Core Domain Dimer Bound
to DNA. J Biol Chem 2006; 281:20494-20502
7. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev
Biochem 2008; 77:557-582
8. Okorokov AL, Orlava EV. Structural biology of the p53 tumour suppressor. Curr Opin
Struct Biol 2009; 19:197-202
9. Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI,
Valle M, Fersht AR. Quaternary structures of tumor suppressor p53 and a specific p53 DNA
complex. Proc Natl Acad Sci 2007; 104:12324-12329
10.
Okorokov AL, Sherman MB, Plisson C, Grinkevich V, Sigmundsson K, Selivanova G,
Milner J, Orlova EV. The structure of p53 tumour suppressor protein reveals the basis for its
functional plasticity. EMBO J 2006; 25:5191-5200
11.
El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a
consensus binding site for p53. Nat Genet 1992; 1:45-49
12.
Hainaut P, Milner J. A Structural Role for Metal Ions in the Wild- Type Conformation
of the Tumor Suppressor Protein-p53. Cancer Res 1993; 53:1739-1742
13.
Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609-622
14.
Appella E, Anderson CW. Post-translational modifications and activation of p53 by
genotoxic stresses. Eur J Biochem 2001; 268:2764-2772
15.
Hupp TR, Lane DP. Allosteric activation of latent p53 tetramers. Curr Biol 1994; 4:865-
875
158
16.
Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell 2008; 133:930-
930
17.
Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and
induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005; 123:49-63
18.
Boehme KA. Regulation of p53-insights in a complex process. Crit Rev Biochem Mol
Biol 2009; 44:367-392
19.
Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and
continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10:501-514
20.
Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin
Genet Dev 2003; 13:77-83
21.
Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst
P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine
methylation. Nature 2004; 432:353-360
22.
Boehme KA, Blattner C. Regulation of p53-insights into a complex process. Crit Rev
Biochem Mol Biol 2009; 44:367-392
23.
Xiong Y and Beach D. p21 is a universal inhibitor of cyclin kinases. Nature 1993;
366:701-710
24.
Frebourg T, Friend SH. The Importance of p53 Gene Alterations in Human Cancer - Is
There More Than Circumstantial Evidence. J Nat Cancer Inst 1993; 85:1554-1557
25.
Stephens T. Closing in on p53‟s normal function. J NIH Res 1991; 3:32-36
26.
Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009;
137:413-431
27.
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative
control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107:137-148
28.
Kruse JP, Gu W. p53 aerobics: the major tumor suppressor fuels your workout. Cell
Metab 2006; 4:1-3
29.
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53
activation. Cell 2008; 133:612-626
30.
Harris CC, Hollstein M. Clinical Implications of the p53 Tumor-Suppressor Gene. N
Engl J Med 1993; 329:1318-1327
31.
Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death.
Oncogene 1999; 18:6145-6157
32.
Vousden KH. p53: Death Star. Cell 2000; 103:691-694
159
33.
Drayton S, Peters G. Immortalisation and transformation revisited. Curr Opin Genet
Dev 2002; 12:98-104
34.
Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by
p53: the importance of apoptosis and cellular senescence. J Pathol 2009; 219:3-15
35.
Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is
induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:20152027
36.
Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y,
Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature
senescence induced by oncogenic Ras. Nature 2000; 406:207-210
37.
Shetty S, Shetty P, Idell S, Velusamy T, Bhandary YP, Shetty RS. Regulation of
plasminogen activator inhibitor-1 expression by tumor suppressor protein p53. J Biol Chem
2008; 283:19570-19580
38.
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB.
Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006;
24:841-851
39.
Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, Lu X, Crook T, Del Sal G. The
prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis
inhibitor iASPP. Nat Struct Mol Biol 2007; 14:912-920
40.
Le Cam L, Linares LK, Paul C, Julien E, Lacroix M, Hatchi E, Triboulet R, Bossis G,
Shmueli A, Rodriguez MS, Coux O, Sardet C. E4F1 is an atypical ubiquitin ligase that
modulates p53 effector functions independently of degradation. Cell 2006; 127:775-788
41.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes
premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;
88:593-602
42.
Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF.
Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and
immortalization. Genes Dev 1998; 12:2424-2433
43.
Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacological activation of p53 elicits
Bax-dependent apoptosis in the absence of transcription. Cancer Cell 2003; 4:371-381
44.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has
a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577-590
45.
Chipuk JE, Green DR. p53's believe it or not: lessons on transcription-independent
death. J Clin Immunol 2003; 23:355-361
160
46.
Polster BM, Fiskum G. Mithocondrial mechanism of neural cell apoptosis. J
Neurochem 2004; 90:1281-1289
47.
Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature
2009; 458:1127-1130
48.
Lie H. miR join the p53 network-another piece in the tumor-suppression puzzle. Natu
Rev Canc 2007, 7:819-822
49.
Bommer G. p53-mediated activation of miRNA34 candidate tumor-suppression genes.
Curr Biol 2007; 17:1298-1307
50.
Chang TC. Transactivation of miR-34a by p53 broadly influences gene expression and
promotes apoptosis. Mol Cell 2007; 26:745-752
51.
Raver Shapira N. Trascriptional activation of miR-34a contributes to p53-mediated
apoptosis. Mol Cell 2007; 26:731-743
52.
Tarasov V. Differential regulation of microRNAs by p53 revealed by massively parallel
sequencing: miR34a is a p53 target that induces apoptosis and G1 arrest. Cell Cycle 2007;
6:1586-1593
53.
Lin He. A microRNA component of the p53 tumor suppressor network. Nature 2007;
447:1130-1135
54.
Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, Brugières L,
Chompret A, de Paillerets BB, Stoppa-Lyonnet D, Bonaïti-Pellié C, Frébourg T. Molecular
basis of the Li-Fraumeni sindrome: an update from the French LFS families. J Med Genet
2008; 45:307-356
55.
Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of
different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999;
18:477-485
56.
Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect
by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004;
23:2330-2338
57.
Monti P, Campomenosi P, Ciribilli Y, Iannone R, Inga A, Abbondandolo A, Resnick
MA, Fronza G. Tumour p53 mutations exhibit promoter selective dominance over wild type
p53. Oncogene 2002; 21:1641-1648
58.
JV Gannon, R Greavesl, Riggo and DP Lane. Activating mutations in p53 produce a
common conformational effect. A monoclonal antibody specific for the mutant form. EMBO
J 1990; 9:1595-1602
161
59.
Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers.
Science 1991; 253:49-53
60.
Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field.
Nat Rev Cancer 2009; 9:701-713
61.
Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in
human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene
2007; 26:2157-2165
62.
Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding
the function-structure and function-mutation relationships of p53 tumor suppressor protein by
high-resolution missense mutation analysis. Proc Natl Acad Sci 2003; 100:8424-8429
63.
Milner J, Medcalf EA, Cook AC. Tumor suppressor p53: analysis of wild-type and
mutant p53 complexes. Mol Cell Biol 1991; 11:12-19
64.
Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the
wild-type p53 protein into the mutant conformation. Cell 1991; 65:765-774
65.
Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the
guardian of the genome. Cancer Res 2000; 60:6788-6793
66.
Chin KV, Ueda K, Pastan I, Gottesman MM. Modulation of activity of the promoter of
the human MDR1 gene by Ras and p53. Science 1992; 255:459-462
67.
Koga H, Deppert W. Identification of genomic DNA sequences bound by mutant p53
protein (Gly245-->Ser.) in vivo. Oncogene 2000; 19:4178-4183
68.
Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53
mutants in mammalian cells. Mol Cell Biol 1999, 19:1438-1449
69.
Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic
instability by inactivating ATM. Nature Cell Biol 2001; 9:573-580
70.
Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity.
Apoptosis 2009; 14:597-606
71.
Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the
guardian of the genome. Cancer Res 2000; 60:6788-6793
72.
Soussi T. in 25 Years of p53 Research (eds P. Hainaut & K. G. Wiman) 255-292
(Springer, 2005)
73.
Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem
2006; 6:1205-1214
162
74.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA,
Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot
spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119:861-872
75.
Olivier M, Hainaut P, Borresen AL. in 25 Years of p53 Research (eds P. Hainaut & K.
G. Wiman) 321-338 (Springer, 2005)
76.
Shi H, Le Calvez F, Olivier M, Hainaut P. in 25 Years of p53 Research (eds P. Hainaut
& K. G. Wiman) 293-319 (Springer, 2005)
77.
Bartel F, Jung J, Böhnke A, Gradhand E, Zeng K, Thomssen C, Hauptmann S. Both
germ line and somatic genetics of the p53 pathway affect ovarian cancer incidence and
survival. Clin Cancer Res 2008; 14:89-96
78.
Olivier M, Langerød A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C,
Lidereau R, Bièche I, Varley J, Bignon Y, Uhrhammer N, Winqvist R, Jukkola-Vuorinen A,
Niederacher D, Kato S, Ishioka C, Hainaut P, Børresen-Dale AL. The clinical value of
somatic TP53 gene mutations in 1,794 patients with breast cancer. A large-scale analysis of
the prognostic association of TP53 mutations in breast cancer, including comparison of the
clinical effects of different mutation categories. Clin Cancer Res 2006; 12:1157-1167
79.
Junttila MR. p53-a Jack of all trades but master of none. Nat Rev Canc 2009; 9:821-829
80.
Mendrysa SM, O'Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA,
Perry ME. Tumor suppression and normal aging in mice with constitutively high p53 activity.
Genes Dev 2006; 20:16-21
81.
Gatza C, Moore L, Dumble M, Donehower L. Tumor suppressor dosage regulates stem
cell dynamics during aging. Cell Cycle 2007; 6:52-55
82.
Vijg J, Hasty P. Aging and p53: getting it straight. A commentary on a recent paper by
Gentry and Venkatachalam. Aging Cell 2005; 4:331-333
83.
Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer 2001;
1:68-76
84.
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman
J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 leads to tumour regression in vivo.
Nature 2007; 445:661-665
85.
Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and
therapeutic potential. Oncogene 2007; 26:2243-2254
86.
Martins CP, Brown-Swigart L, Evan GI. Modeling the Therapeutic Efficacy of p53
Restoration in Tumors. Cell 2006; 127:1323-1334
163
87.
Tang X, Zhu Y, Han L, Kim AL, Kopelovich L, Bickers DR, Athar M. CP-31398
restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis
in mice. J Clin Invest 2007; 117:3753-3764
88.
Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, Wiman KG.
PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005;
24:3484-3491
89.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, Döhner K,
Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N
Engl J Med 2000; 343:1910-1916
90.
Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, Diehl D, Schlenk R,
Coy J, Stilgenbauer S, Volkmann M, Galle PR, Poustka A, Hunstein W, Lichteret P. p53 gene
deletion predicts for poor survival and non-response to therapy with purine analogs in
chronicB-cell leukemias. Blood 1995; 85:1580-1589
91.
Forconi F, Rinaldi A, Kwee I, Sozzi E, Raspadori D, Rancoita PMV, Scandurra M,
Rossi D, Deambrogi C, Capello D, Zucca E, Marconi D, Bomben R, Gattei V, Lauria F,
Gaidano G, Berton F. Genome-wide DNA analysis identifies recurrent imbalances predicting
outcome in chronic lymphocytic leukaemia with 17p deletion. Brit J Haematol 2008;
143:532-536
92.
Stilgenbauer S, Krober A, Busch R. 17p Deletion predicts for inferior overall survival
after fludarabine-based first line therapy in chronic lymphocytic leukemia: first analysis of
genetics in the CLL4 Trial of the GCLLSG. Blood 2005; 106:Abstract 715
93.
Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ, Bezares RF, Pettitt AR,
Hamblin T, Milligan DW, Child JA, Hamilton MS, Dearden CE, Smith AG, Bosanquet AG,
Davis Z, Brito-Babapulle V, Else M, Wade R, Hillmen P; UK National Cancer Research
Institute (NCRI) Haematological Oncology Clinical Studies Group; NCRI Chronic
Lymphocytic Leukaemia Working Group. Assessment of fludarabine plus cyclophosphamide
for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised
controlled trial. Lancet 2007; 370:230-239
94.
Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, Van Dyke DL, O'Brien S, Ferrajoli
A, Lerner SA, Lynn A, Kay NE, Keating MJ.. De novo deletion 17p13.1 chronic lymphocytic
leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic
experience. Blood 2009; 114:957-964
95.
Oscier DG, Gardiner AC, Mould SJ, Glide S, Davis ZA, Ibbotson RE, Corcoran MM,
Chapman RM, Thomas PW, Copplestone JA, Orchard JA, Hamblin TJ. Multivariate analysis
164
of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or
mutation of the p53 gene are independent prognostic factors. Blood 2002; 100:1177-1184
96.
Zenz T, Benner A, Döhner H, Stilgenbauer S. Chronic lymphocytic leukemia and
treatment resistance in cancer. The role of the p53 pathway. Cell Cycle 2008; 24:3810-3814
97.
Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, Benner A, Denzel T, Winkler D,
Edelmann J, Schwänen C, Döhner H, Stilgenbauer S. Monoallelic TP53 inactivation is
associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed
genetic characterization with long-term follow-up. Blood 2008; 112:3322-3329
98.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, De Paoli L, Spina V, Gattei
V, Capello D, Forconi F, Lauria F, Gaidano G. The Prognostic Value of TP53 Mutations in
Chronic Lymphocytic Leukemia Is Independent of Del17p13: Implications for Overall
Survival and Chemorefractoriness. Clin Cancer Res 2009; 15:995-1004
99.
Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, Sarno A, Groner S, Mertens
D, Busch R, Hallek M, Döhner H, Stilgenbauer S. Detailed analysis of p53 pathway defects in
fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of
17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial.
Blood 2009; 114:2589-2597
100. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H,
Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ; International Workshop on Chronic
Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic
leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia
(IWCLL) updating the National Cancer Institute-Working Group (NCI-WG) 1996 guidelines.
Blood 2008; 111:5446-5456
101. Murphy ME. Polymorphic variants in the p53 pathway. Cell Death Differ 2006; 13:916920
102. Dumont P, Leu JI, Della Pietra AC 3rd, George DL, Murphy M. The codon 72
polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003;
33:357-365
103. Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53
dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to
TP53 mutation. Blood 2001; 98:814-822
104. Guarini A, Gaidano G, Mauro FR, Capello D, Mancini F, De Propris MS, Mancini M,
Orsini E, Gentile M, Breccia M, Cuneo A, Castoldi G, Foa R. Chronic lymphocytic leukemia
165
patients with highly stable and indolent disease show distinctive phenotypic and genotypic
features. Blood 2003; 102:1035-1041
105. Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, Marce S, LopezGuillermo A, Campo E, Montserrat E. ZAP-70 expression as a surrogate for immunoglobulinvariable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003; 348:17641775
106. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, Neuberg DS, Flinn
IW, Rai KR, Byrd JC, Kay NE, Greaves A, Weiss A, Kipps TJ. ZAP-70 compared with
immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in
chronic lymphocytic leukemia. N Engl J Med 2004; 35:893-901
107. Gentile M, Mauro FR, Calabrese E, De Propris MS, Giammartini E, Mancini F, Milani
ML, Guarini A, Foà R. The prognostic value of CD38 expression in chronic lymphocytic
leukaemia patients studied prospectively at diagnosis: a single institute experience. Br J
Haematol 2005; 130:549-557
108. Cicalese A, Ionizzi G, Pasi EC, Faretta M, Ronzoni S, Giulini B, Brisken C, Minacci S,
Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing
division in mammary stem cells. Cell 2009; 138:1083-1095
109. Monti P. Tumor p53 mutations exhibit promoter selective dominance over wild type
p53. Oncogene 2002; 21:1641-1648
110. Johnson GG, Sherrington PD, Carter A, Lin K, Liloglou T, Field JK, Pettitt AR. A
Novel Type of p53 Pathway Dysfunction in Chronic Lymphocytic Leukemia Resulting from
Two Interacting Single Nucleotide Polymorphisms within the p21 Gene. Cancer Res 2009;
69:5210-5217
111. Lin K, Sherrington PD, Dennis M, Matrai Z, Cawley JC, Pettitt AR. Relationship
between p53 dysfunction, CD38 expression, and IGVH mutation in chronic lymphocytic
leucemia. Blood 2002; 100:1404-1409
112. Sali H. Increased genomic alteration complexity and telomere shortening in B-CLL
cells resistant to radiation-induced apoptosis. Cytogenet Genome Res 2008; 122:343-349
166
PARTE III
Caratterizzazione fenotipica e funzionale delle cellule
T regolatorie di pazienti affetti da neoplasie
ematologiche sottoposti a trapianto allogenico di
cellule staminali emopoietiche
167
1. INTRODUZIONE
1.1 Il Trapianto di Cellule Staminali Emopoietiche (TCSE)
1.1.1 Tipologie di TCSE e principi relativi all’impiego del TCSE in pazienti affetti da
neoplasie ematologiche
Il Trapianto di Cellule Staminali Emopoietiche (TCSE) consiste nell‟infusione di CSE
normali al fine di sostituire un sistema emopoietico anormale. Viene comunemente impiegato
in ambito clinico per il controllo a lungo termine di patologie maligne, principalmente di
natura ematologica, e disordini ereditari o acquisiti, sia di origine emopoietica che nonemopoietica, come le immunodeficienze primarie, i disturbi metabolici, le emoglobinopatie e
le forme severe di anemia e/o infezioni sviluppate a seguito di aplasia midollare [1-2]. Le
prime evidenze relative alle potenzialità delle CSE nella sfera trapiantologica risalgono alla
fine della II Guerra Mondiale e derivano dalle numerose ricerche atte ad affrontare il
problema delle conseguenze dannose dell‟esposizione alle radiazioni nucleari. Nel 1952 è
stato dimostrato che la trasfusione di cellule di origine midollare garantisce la sopravvivenza
di topi esposti a un dosaggio sovramassimale letale di radiazioni ionizzanti (IR), grazie al
recupero della funzione emolinfopoietica e quindi dell‟aplasia midollare irreversibilmente
compromessa [3]. Sempre nello stesso anno è stato osservato che l‟inoculo di cellule midollari
successivo all‟esposizione a dosi mieloablative di IR permette anche di curare modelli
tumorali animali sperimentali con prolungamento della sopravvivenza libera da malattia, e
che questo evento, così come il recupero della cellularità del midollo osseo, si verifica sia nel
caso in cui si ricorre all‟impiego di un donatore di cellule midollari geneticamente identico
che non-identico [4]. Questi studi hanno rappresentato la base per l‟evoluzione di uno schema
terapeutico basato sull‟impiego del TCSE in pazienti affetti da patologie midollari. In realtà la
dizione propria di TCSE è stata introdotta solo in seguito rispetto alle prime evidenze
sperimentali. Infatti, l‟impiego iniziale di cellule di origine midollare ha comportato per
svariati decenni l‟utilizzo della terminologia di “trapianto di midollo osseo”, fino alla
dimostrazione che non solo il midollo osseo, ma anche altre sedi quali il sangue venoso
periferico e il sangue placentare, sono caratterizzate da componenti cellulari a carattere
staminale, rappresentate appunto dalle CSE, capaci di mediare la funzione emolinfopoietica.
Il principio del TSCE è quindi rappresentato dall‟impiego di elevate dosi di terapia
mieloaplastizzante, finalizzato all‟eliminazione della patologia con coinvolgimento midollare,
seguito dall‟infusione di CSE con lo scopo di ripristinare la funzione midollare emopoietica
compromessa dalla terapia stessa. Nell‟ambito delle patologie ematologiche neoplastiche,
168
l‟impiego terapeutico del TCSE è stato favorito dall‟osservazione che il soggetto può
beneficiare di un incremento della posologia farmacologica radio/chemioterapica, a cui la
risposta antitumorale risulta direttamente proporzionale, senza limitazioni correlate alla
tossicità midollare.
Esistono due principali tipologie di TCSE: autologo e allogenico. Nel primo caso, il donatore
di CSE è rappresentato dal paziente stesso, nel secondo da un soggetto diverso. Il TCSE
autologo viene impiegato principalmente in pazienti affetti da neoplasie ematologiche
radio/chemiosensibili ad alto rischio di recidiva o resistenti ai dosaggi standard di
radio/chemioterapia. Di conseguenza, il proposito di questa forma di TCSE è quello di
garantire un incremento della terapia antileucemica finalizzato al raggiungimento della
massima uccisione delle componenti tumorali, con il limite esclusivo della tossicità extramidollare sistemica [5]. Un concetto diverso è alla base del TCSE allogenico. Esso mira alla
completa sostituzione del patrimonio staminale emopoietico del ricevente mediante
l‟infusione di CSE, ottenute da un idoneo donatore compatibile, con conseguente
ricostituzione sia di un nuovo sistema ematopoietico che di un nuovo sistema immunitario [67]. Quindi, l‟obiettivo del TCSE allogenico è quello di eradicare e sostituire le cellule
ematopoietiche malate con quelle del donatore, affinché queste possano dare origine a un
sistema ematopoietico e immunitario sano, con la realizzazione di un “chimerismo” completo.
Con il termine di chimerismo si vuole intendere la coesistenza in uno stesso individuo di cloni
cellulari con patrimonio genetico diverso. Nei trapianti allogenici si instaura, infatti, un
interscambio di cloni cellulari portatori di corredi genetici diversi fra le cellule emopoietiche
del donatore e il sistema immune del ricevente, che si traduce nella realizzazione di un
chimerismo sistemico misto [8-9]. Il trapianto allogenico costituisce una procedura
complessa, ma particolarmente efficace non solo nel trattamento di varie forme di Leucemie
Acute e Croniche, Linfomi e Mieloma Multiplo, ma anche per la correzione di difetti
quantitativi o congeniti (es: Talassemia major) del sistema emopoietico. Nell‟ambito della
terapia antitumorale, esso consente al paziente di beneficiare sia dell‟incremento della
posologia antineoplastica che, grazie alla derivazione del prodotto di infusione da un donatore
compatibile, della possibilità di induzione di una risposta immune antileucemica, mediata,
come vedremo in seguito, dalle componenti alloreattive del donatore stesso. Il primo
problema da affrontare nel caso di un trapianto allogenico è quindi rappresentato dalla
disponibilità di un donatore compatibile [10]. I geni del “Complesso Maggiore di
Istocompatibilità” o HLA (Human Leukocyte Antigen), sono i loci primariamente coinvolti
nel controllo della compatibilità tra individui. Mappano sul cromosoma 6p con i geni di classe
169
II, III e I organizzati dalla regione centromerica verso la regione telomerica e sono altamente
polimorfi [11]. Tale caratteristica riflette la loro funzione primaria, che è quella di codificare
per molecole coinvolte nel legame e nella presentazione dei peptidi antigenici alle cellule T
linfoidi. I geni HLA sono codominanti; presentano una modalità di trasmissione mendeliana
aplotipica, per cui gli alleli presenti sullo stesso cromosoma omologo vengono ereditati
insieme e sono, infine, caratterizzati dal fenomeno del “Linkage Disequilibrium” per cui
alcuni alleli risultano più frequentemente associati ad altri [12]. Quest‟ultima peculiarità è
fondamentale per la sfera trapiantologica, in quanto è stato calcolato che se ciascun allele
fosse ereditato indipendentemente, vi sarebbero 1023 combinazioni HLA-A, -B, -C, -DR, -DQ
uniche, il che comporterebbe una probabilità quasi irrilevante di identificare un donatore
istocompatibile [13]. L‟estremo polimorfismo del sistema HLA comporta che l‟identità HLA
sia più probabile tra consanguinei; di conseguenza, qualora un soggetto sia candidato a un
TCSE allogenico, la ricerca del donatore ha inizio con la tipizzazione del sistema HLA
all‟interno dei membri della famiglia. La probabilità di identificare un donatore HLA-identico
familiare si verifica con una frequenza del 25% nei fratelli, ovvero in tale percentuale di casi
due fratelli ereditano gli stessi aplotipi materni e paterni [14]. Il caso di gemelli omozigoti,
quindi geneticamente identici è invece una situazione molto rara, nota con il termine di
trapianto singenico. I notevoli progressi nel campo della genetica dell‟istocompatibilità, con
conseguente definizione delle complessità del sistema HLA, hanno recentemente permesso di
estendere la ricerca di donatori compatibili anche all‟esterno dell‟ambito familiare,
garantendo così la possibilità di ricorso alla procedura trapiantologica a un maggior numero di
pazienti. A tale scopo sono stati creati dei registri internazionali mediante i quali è possibile
identificare un donatore volontario non correlato (Matched Unrelated Donor, MUD) HLAidentico (probabilità=40%) o solo parzialmente identico (HLA-mismatched), ovvero identico
per 4 o 5 dei 6 alleli dei 3 loci HLA (A, B e DR) comunemente tipizzati [15-16]. Allo stesso
modo, quasi tutti i pazienti che potrebbero beneficiare di un trapianto, ma che non hanno un
donatore compatibile, dispongono almeno di un potenziale donatore non perfettamente
compatibile tra i familiari HLA aploidentici, ovvero identici solo per uno dei due aplotipi
HLA presenti nel loro genoma (3 alleli su 6) [17-18]. Si potrebbe ricorrere a quest‟ultima
opzione nel caso in cui il paziente richieda un trapianto immediato e non possa attendere la
ricerca di un donatore non familiare, che solitamente richiede non meno di 4-5 mesi.
In realtà, anche qualora sia disponibile un donatore HLA-compatibile, esistono altre molecole
con il ruolo di barriere di compatibilità donatore-ricevente, rappresentate dagli “Antigeni del
complesso Minore di Istocompatibilità” (miHA). I miHA sono codificati sia da geni
170
autosomali che da geni presenti sui cromosomi del sesso ereditati indipendentemente dai geni
HLA e sono peptidi di derivazione self contenenti varianti polimorfiche all‟interno della
popolazione [19]. Nell‟uomo sono stati inizialmente identificati durante gli anni ‟70 come
antigeni responsabili del rigetto del prodotto di infusione allogenico nel contesto di un
trapianto da donatore HLA-identico, sebbene rivestano un ruolo chiave anche nello sviluppo
della reattività donatore vs ricevente [20]. La risposta immune indotta dagli antigeni miHA
codificati dai geni che mappano sul cromosoma Y è poi anche associata al rischio o gli effetti
positivi della combinazione donatore-ricevente di sesso diverso, il che rende indispensabile, a
seconda dei casi, la valutazione della scelta di un donatore dello stesso sesso o meno.
Recentemente è stato dimostrato come, nella definizione di un donatore compatibile, rivestano
un ruolo importante anche fattori genetici non HLA-correlati diversi dai miHA, e
rappresentati dai geni codificanti citochine di natura infiammatoria caratterizzati da
polimorfismi nella regione regolatoria al 5‟ o al 3‟ della sequenza. Ne sono esempi i geni per
il TNF-α, l‟IFN-γ, l‟IL-1, l‟IL-8 e l‟IL-10 [21]. Le forme di reattività osservate in soggetti
sottoposti a un allotrapianto, dipendono quindi dall‟impossibilità di raggiungere una totale
istocompatibilità donatore/ricevente, anche nelle condizioni più ottimali. Questo si traduce
nella coesistenza delle cellule immunocompetenti residue del ricevente, mediatrici del rigetto,
e delle cellule immunocompetenti del donatore, mediatrici della GvHD (Graft versus Host
Disease) e della GvL (Graft versus Leukemia) [22-26]. L‟attività di entrambe le popolazioni
cellulari, come vedremo in seguito, influenza ampiamente, a seconda della tipologia di
antigeni non compatibili, la prognosi del paziente post-trapianto; di conseguenza, la scelta del
donatore e la tipizzazione sia dei loci HLA-correlati che non-correlati, assume una primaria
importanza nella fase pre-allotrapianto.
1.1.2 Scelta della fonte di CSE
Indipendentemente dalla tipologia di trapianto, autologo o allogenico, la prima fase della
procedura prevede l‟isolamento delle CSE. Le CSE sono una popolazione morfologicamente
e immunologicamente eterogenea il cui elemento unificante è rappresentato dalla capacità di
generare in vitro aggregati clonali derivati sia da progenitori ontogenicamente primitivi che
orientati, e in vivo dalla capacità di ricostituire l‟emopoiesi dopo terapia mielo-linfoablativa.
Sono cellule multipotenti, linea-specifiche e funzionalmente caratterizzate dalle capacità di
autoreplicazione illimitata e differenziamento in tutti i componenti ematici sia di natura
mieloide che linfoide [27]. Le CSE sono caratterizzate dall‟espressione di superficie
dell‟antigene CD34 e di recettori di adesione che ne permettono l‟insediamento spontaneo
171
(homing) all‟interno del midollo osseo del ricevente dopo infusione, con ricostituzione di un
nuovo tessuto ematopoietico. IL CD34 rappresenta ad oggi il marcatore universalmente
riconosciuto delle CSE. E‟ una molecola coinvolta nei processi di “homing” e adesione
cellulare, caratterizzata da un pattern di espressione intraspecie altamente conservato [28].
Viene espresso, sia sulle CSE sia sui progenitori midollari precoci, mentre i suoi livelli di
espressione diminuiscono proporzionalmente al differenziamento emopoietico [29]. In realtà,
anche cellule di origine non emopoietica, come quelle dell‟endotelio vascolare, appaiono
caratteristicamente CD34+, suggerendo una funzione di tale antigene anche al di fuori
dell‟emopoiesi. La positività per il CD34 nelle CSE si associa solitamente alla presenza di un
pattern di espressione di molecole di superficie peculiare, quale Lin-, HLAlow, Thy.1low,
CD133+, CD45RAlow, CD38-. Sono state però identificate anche CSE caratterizzate dalla
negatività per il marcatore CD34 associata a: Lin-, HLA-, Thy.1-, CD133+, CD45RA-, CD38-.
Tali osservazioni hanno condotto a due differenti ipotesi. Le CSE CD34- potrebbero
rappresentare un compartimento cellulare più primitivo rispetto a quello delle CSE CD34+; in
alternativa, il CD34 potrebbe essere un marcatore d‟attivazione la cui espressione viene
modulata negativamente durante il ripristino dello stato di quiescenza cellulare [30]. In ambo i
casi, il differenziamento dei diversi componenti ematici risulta strettamente correlato alla
combinazione di segnali di natura intrinseca, caratteristici delle CSE, ed estrinseca, derivanti
dai componenti cellulari ed extra-cellulari della nicchia midollare.
Le CSE sono isolabili da 3 differenti fonti quali: il midollo osseo (MO); il sangue venoso
periferico (SVP) e il sangue placentare (SCO) [31]. Ciascuna fonte differisce in termini di
modalità di raccolta, composizione dell‟inoculo ed effetti post-trapiantologici. Il MO ha
rappresentato per molti anni l‟unica fonte di CSE. Le CSE rappresentano l‟1-3% delle cellule
mononucleate (CM) midollari totali. La raccolta delle CSE dal MO viene effettuata mediante
espianto e richiede fino a 100-200 punture delle creste iliache posteriori e, se necessario, dello
sterno, eseguite in anestesia generale [32]. La quantità totale di sangue midollare che viene
prelevata a fini trapiantologici è pari a circa 10-15 ml/kg del peso corporeo del ricevente. In
alternativa, le CSE possono essere prelevate dal SVP, mediante “Leucoaferesi”, dopo
opportuna mobilizzazione delle CSE dal MO [33-35]. La mobilizzazione avviene attraverso la
somministrazione di fattori di crescita per i 4-5 giorni antecedenti il prelievo, ed è necessaria a
incrementare la quota di CSE periferiche, poiché normalmente essa risulta pari solo allo 0.10.2% delle CM totali [36]. Il G-CSF è il fattore di crescita maggiormente impiegato a tale
scopo. Agisce inducendo la proliferazione delle CSE midollari e la loro mobilizzazione nel
SVP grazie alla downregolazione del fattore SDF-1 (stroma-derived factor 1) presente sulla
172
superficie delle cellule stromali midollari (CSM) e all‟induzione della proliferazione e del
rilascio da parte dei neutrofili midollari di enzimi proteolitici (metalloproteasi, catepsina G,
elastasi) che degradano CXCR4, recettore di SDF-1, esposto sulla superficie delle CSE [3738]. Negli ultimi dieci anni, il numero dei trapianti di CSE isolate dal SVP è rapidamente
incrementato, sostituendo il MO come principale fonte di CSE utilizzata nelle procedure
trapiantologiche. L‟impiego del SVP si associa infatti ad un ampio numero di vantaggi tra cui:
la minore invasività del prelievo, la possibilità di determinare il numero assoluto di cellule
CD34+ infuse per Kg di peso corporeo del ricevente e l‟induzione di una ricostituzione
emopoietica completa e significativamente più rapida rispetto a quella ottenuta con CSE da
MO. Il principale svantaggio deriva invece dagli effetti collaterali dovuti all‟impiego dei
fattori di crescita nella fase di mobilizzazione delle CSE. L‟impiego del G-CFS si associa ad
esempio, sebbene in un numero limitato di casi, a ingrossamento della milza, complicanze
cardiocircolatorie e aumento della probabilità di sviluppare neoplasie ematologiche (es:
leucemie) [39]. La fonte di CSE di più recente identificazione è invece rappresentata dal
sangue placentare (SCO). La raccolta delle CSE avviene in tal caso subito dopo il parto, una
volta reciso il cordone ombelicale. Tra i vantaggi relativi all‟utilizzo del SCO: l‟immediata
disponibilità delle unità di SCO (al contrario, il 20-30% dei donatori iscritti nei registri non è
infatti disponibile al momento del trapianto); la maggior probabilità di reperire donatori; il
minor rischio di trasmissione di alcuni agenti infettivi (CMV, EBV) e, infine, le caratteristiche
biologiche uniche delle CSE isolate [40]. E‟ stato, infatti, dimostrato che tali cellule,
rappresentanti lo 0.8-1.2% delle CM totali del SCO, sono caratterizzate da un elevato grado di
immaturità immunologica che si traduce sia in una capacità di replicazione e differenziazione
verso le diverse linee cellulari emopoietiche, superiore a quella delle corrispondenti cellule
midollari e periferiche, che, nel contesto di un allotrapianto, in una ridotta capacità delle
stesse di esercitare un‟aggressione immunologica nei confronti dei tessuti del ricevente e
pertanto in una ridotta incidenza di GvHD acuta e cronica. Questa forma di allotolleranza
riduce di conseguenza l‟importanza del grado di compatibilità HLA donatore/ricevente,
consentendo un TCSE anche in situazioni di non consanguineità e/o di parziale compatibilità
HLA donatore/ricevente [41-43]. La raccolta del SCO è inoltre un‟operazione semplice e
rapida, che non comporta alcun rischio legato a procedure invasive di prelievo per la madre o
per il neonato. Gli svantaggi sono, invece, essenzialmente rappresentati dall‟impossibilità di
riutilizzo del prodotto di infusione in caso di rigetto, mancato attecchimento o recidiva, e dal
numero limitato di cellule CD34+ infuse, legato anche alla quantità di SCO che può essere
raccolta da una placenta al momento del parto. Esso influenza la ricostituzione dell‟intero
173
sistema immunitario del ricevente, che manifesta una cinetica più lenta, con conseguente
effetto negativo sulla sopravvivenza del paziente, dovuto a una maggiore possibilità di
insorgenza delle infezioni [44].
La scelta di una piuttosto che di un‟altra fonte di CSE dipende essenzialmente dalla tipologia
di trapianto. Nel caso di un TCSE autologo, il principale fattore influenzante l‟impatto clinico
dell‟inoculo, dipende principalmente dalla quantità di CSE e dei loro effetti sulle cinetiche di
recupero del sistema emolinfopoietico. Di conseguenza il SVP, distinto, grazie al processo di
mobilizzazione, dal maggior numero di progenitori emopoietici, rappresenta la fonte di
impiego preferenziale in tale forma di TCSE. Al contrario, più complessa risulta la scelta
della fonte di CSE nel TCSE allogenico. In tal caso, infatti, il decorso post-trapianto dipende
non solo dalla quantità di progenitori emopoietici e dalla loro influenza sulle cinetiche di
ricostituzione emopoietica, ma anche dalla quantità di cellule accessorie sempre di origine
ematica, quali linfociti T, cellule NK e cellule dendritiche (DC), contaminanti l‟inoculo di
CSE [45]. Tali componenti influenzano sia le cinetiche di recupero del sistema
emolinfopoietico che le reazioni immunologiche donatore-ricevente e viceversa. Si ritiene che
il MO possa costituire la fonte preferenziale del TCSE allogenico in pazienti affetti da
patologie midollari non maligne o neoplasie ematologiche in stadio precoce; e, al contrario,
che il SVP possa essere impiegato nelle neoplasie ematologiche in stadio tardivo o nelle
forme di
TCSE associate ad un regime
preparativo radio/chemioterapico non-
mieloaplastizzate. In questi ultimi casi, il ricevente potrebbe beneficiare degli effetti di
accelerazione delle cinetiche correlate alla produzione di un nuovo sistema emopoietico e del
controllo della patologia mediato dalla potenziale reattività delle cellule immunocompetenti
del donatore nei confronti delle cellule patologiche del ricevente. L‟impiego delle CSE isolate
da SCO è infine previsto, a causa del numero modesto di CSE isolabili da tale fonte, solo in
ambito allogenico in pazienti pediatrici [46-47]. Per superare le barriere del dosaggio cellulare
nel trapianto da SCO, sono stati però intrapresi numerosi studi, per valutare alcune soluzioni
possibili, come ad esempio l‟infusione di due unità di SCO o la stimolazione dell‟espansione,
in vitro, dei progenitori isolati dal cordone ombelicale [48-53]. In base a quanto riportato
appare quindi evidente l‟importanza della scelta della fonte più appropriata di CSE in ambito
trapiantologico. Tale processo dovrebbe essere adattato al singolo paziente per garantire i
maggiori vantaggi correlati a ciascuna delle tre fonti.
174
1.1.3 Il Condizionamento pre-TSCE
Il Condizionamento è un regime terapeutico preparativo cui è sottoposto il ricevente di un
TCSE, sia autologo che allogenico, indispensabile per il successo del TCSE. Consiste in un
trattamento
intensivo
chemio/radioterapico
caratterizzato
dall‟impiego
di
dosi
“sovramassimali” in grado di causare un danno irreversibile a carico dei componenti
emopoietici midollari tale da non consentire un recupero ematologico spontaneo. Lo scopo del
Condizionamento e quello di eradicare la patologia da cui il ricevente è affetto; favorire
l‟homing e l‟attecchimento delle CSE trapiantate e prevenire e/o sopprimere l‟attività del
sistema immune del ricevente per evitare il rigetto e la GvHD nel caso del TCSE allogenico
[54]. Esistono 2 tipologie di Condizionamento: Mieloablativo ad alte dosi e ad Intensità
Ridotta (RIC). Il primo include una combinazione di agenti citotossici e terapia radiante “total
body” a elevato dosaggio e garantisce la necessità di una minor stringenza nell‟identità HLA
donatore-ricevente. Il suo impiego si associa a una maggior mortalità trapianto-correlata ma
anche a più rapide cinetiche di ricostituzione emopoietica e a una minor probabilità di
recidiva. Al contrario, il RIC si basa sull‟impiego di una combinazione di agenti
immunosoppressivi (analoghi delle Purine), terapia radiante linfoide e/o uso di anticorpi per
T-deplezione [55-58]. Mediante l‟impiego del RIC è possibile candidare al TCSE anche
pazienti anziani, pazienti per cui il condizionamento mieloablativo è controindicato e pazienti
affetti da co-morbidità. Naturalmente la ridotta intensità della terapia antitumorale comporta
una maggior probabilità di recidiva post-TCSE, e un rallentamento delle cinetiche di recupero
dell‟emopoiesi, pur riducendo la mortalità. Per tali ragioni, l‟impiego del RIC è indicato nei
casi di neoplasie a più lenta crescita o per offrire un TCSE allogenico a pazienti che hanno
fallito un TCSE autologo.
Il Condizionamento si sta attualmente evolvendo da una radio/chemioterapia standard ad un
trattamento personalizzato. In base alla tipologia di malattia, all‟età, allo stato di salute
complessivo e alle caratteristiche della fonte di CSE e del tipo di donatore, può, infatti, variare
considerevolmente, con lo scopo di garantire al paziente il maggior beneficio e migliorarne il
decorso post-trapianto.
1.1.4 Attecchimento e ricostituzione del sistema emopoietico post-TCSE
Una volta che il paziente candidato a un TCSE è stato esposto al regime di Condizionamento
preparativo, avviene l‟infusione delle CSE attraverso un catetere venoso centrale, mediante
una procedura del tutto non invasiva. L‟infusione deve essere seguita, affinché un TCSE
possa essere considerato riuscito, dal processo di attecchimento, attraverso il quale le CSE
175
circolanti in modo transiente nel SVP sono convertite in cellule residenti midollari, e dalla
Ricostituzione Emopoietica (RE) e Immunologica (RI). L‟attecchimento prevede il
reclutamento delle CSE dalla microvascolatura midollare, la migrazione transendoteliale delle
CSE a livello dei cordoni emopoietici extravascolari del MO e infine la migrazione delle CSE
all‟interno del MO verso le “nicchie” midollari; eventi che si verificano grazie all‟attività di
molecole di adesione e citochine prodotte da tutte le componenti cellulari in esame. Una volta
nelle nicchie midollari, le CSE si sviluppano in stretto contatto con i componenti dello stroma
midollare sia di natura cellulare (Cellule Reticolari, Adipociti, Cellule Osteogeniche, Cellule
Endoteliali,
Macrofagi)
che
della
matrice
extracellulare
(Collagene,
Laminina,
Trombospondina, Proteoglicani) che ne controllano l‟automantenimento e il differenziamento.
La RE e la RI rappresentano invece il recupero numerico e funzionale di tutte le componenti
ematiche. In alcuni casi possono non coincidere temporalmente e questo si traduce
solitamente nell‟incremento del rischio infettivo [59]. E‟ noto che i componenti dell‟Immunità
Adattativa sono caratterizzati da cinetiche di RE/RI più lente rispetto ai componenti
dell‟Immunità Innata perché richiedono durante il loro sviluppo il supporto di uno specifico
microambiente (es: il timo per i linfociti T); sono coinvolti nei processi alloreattivi e sono
suscettibili alla profilassi/terapia anti-GvHD [60]. Tra i fattori pre/peri-trapiantologici
influenzanti la RE e RI: il tipo di malattia, la precedente chemioterapia, il grado di identità
HLA, la fonte di CSE, la mobilizzazione delle CSE, la manipolazione dell‟inoculo, la
tipologia di TCSE e il Condizionamento. Tra i fattori pre/post-trapiantologici: la profilassi e/o
terapia anti-GvHD e la profilassi e/o terapia delle infezioni. Per fronteggiare lo stato di
immunodepressione che si instaura nel paziente, a causa del tempo necessario al ripristino
delle componenti emopoietiche, si ricorre solitamente ad una profilassi a base di farmaci antibatterici, anti-fungini e anti-virali ad ampio spettro o, in alternativa, all‟accelerazione delle
cinetiche di recupero. Nel caso dei linfociti T, ad esempio, si utilizza la somministrazione di
fattori solubili stimolanti la funzionalità timica o il meccanismo timo-indipendente di
“Espansione Omeostatica Periferica”, oppure all‟infusione di cellule T linfoidi del donatore
(Donor Limphocyte Infusion; DLI) isolate mediante singola aferesi [61].
Negli ultimi anni sono stati intrapresi numerosi studi al fine di identificare approcci
innovativi, come la vaccinazione peptidica o la manipolazione del prodotto di DLI, finalizzati
sia alla riduzione del rischio infettivo post-TCSE che all‟aumento della specificità della
terapia anti-microbica, con il fine ultimo di garantire un miglior decorso del paziente in
seguito all‟infusione di CSE, con aumento della probabilità di sopravvivenza.
176
1.1.5 Alloreattività post-TCSE: il rigetto, la GvHD e la GvL
Nel TCSE allogenico esiste una “doppia barriera immunologica” dovuta alla contemporanea
presenza delle cellule immunocompetenti del ricevente sopravvissute al regime di
Condizionamento e delle cellule immunocompetenti del donatore contenute nell‟inoculo e/o
prodotte dopo attecchimento delle CSE infuse. Questa doppia barriera comporta il potenziale
sviluppo di due differenti forme di alloreattività. La prima è rappresentata dall‟insieme delle
reazioni mediate dalle cellule immunocompetenti del ricevente nei confronti dei componenti
emopoietici dell‟inoculo (HVG) e si traduce nel rigetto del TCSE. La seconda è costituita
dalle reazioni mediate dalle cellule immunocompetenti del donatore (GVH) nei confronti sia
dei tessuti sani che patologici del ricevente e comporta nel primo caso lo sviluppo di GvHD,
nel secondo lo sviluppo di GvL [62]. La GvHD e la GvL rappresenterebbero quindi due
aspetti contrapposti dello stesso processo. Gli antigeni causa di alloreattività possono essere, a
seconda del grado di compatibilità donatore-ricevente: di natura ubiquitaria (HLA e miHA);
tessuto-specifici o espressi in modo aberrante dalle cellule patologiche (miHA e proteine self
normali); specifici delle cellule patologiche (Prodotti di traslocazioni cromosomiche: t(9;22)
nella Leucemia Mieloide Cronica) o di derivazione virale (CMV: pp65; EBV: LMP-1/2).
Il rigetto è solitamente tipico di un TCSE da donatore familiare HLA parzialmente
compatibile o aploidentico o da donatore non-familiare HLA compatibile (MUD). E‟ stato
dimostrato che i principali mediatori del rigetto sono rappresentati dalle cellule NK, dalle
cellule T CD4+ e CD8+, dalle cellule T
+
e dalle cellule B del ricevente [63]. I meccanismi
effettori del rigetto differiscono in base alla precedente sensibilizzazione del ricevente agli
alloantigeni target. Infatti, nel caso di un ricevente non-sensibilizzato il rigetto delle molecole
non condivise, viene mediato principalmente da linfociti T naive CD8+ attraverso i
meccanismi classici di Perforina/Granzima e Fas/FasL mentre, nel caso di un ricevente
sensibilizzato in seguito ad esempio a trasfusioni o gravidanze, il rigetto delle molecole non
condivise dipende dall‟attività di linfociti T memoria CD8+, attraverso meccanismi non noti
[64]. Il rigetto può essere valutato esaminando la conta leucocitaria, la cellularità midollare,
l‟ematocrito o il grado di chimerismo donatore/ricevente, e il rischio che si verifichi postallotrapianto è influenzato principalmente dal regime di Condizionamento. La probabilità di
rigetto risulta, infatti, maggiore nel caso di un TCSE preceduto da RIC piuttosto che da
Condizionamento mieloablativo, un dato che dimostra l‟importanza dell‟eliminazione preTCSE delle cellule alloreattive del ricevente.
La GvHD è invece una Patologia Infiammatoria, mediata principalmente dalle cellule
accessorie T linfoidi alloreattive del donatore contaminanti l‟inoculo di CSE. Deriva dal
177
riconoscimento, da parte delle cellule T del donatore, dei componenti tissutali sani del
ricevente e rappresenta, di conseguenza, una delle principali complicanze del TCSE
allogenico. Il suo sviluppo richiede naturalmente che le cellule del ricevente siano incapaci di
rigettare adeguatamente il TCSE; che l‟inoculo di CSE contenga cellule immunocompetenti e
che vi siano incompatibilità antigeniche donatore/ricevente [65]. La GvHD è una patologia
complessa multifase [Figura 1]. La prima fase è contraddistinta dal “priming” della risposta
immune indotto dal danneggiamento tissutale causato dal regime di Condizionamento
preparativo, che comporta il rilascio di numerose citochine e fattori pro-infiammatori
coinvolti nell‟attivazione e nella maturazione delle cellule presentanti l‟antigene (APC). Il
ruolo del Condizionamento nella GvHD è stato ampiamente dimostrato grazie
all‟osservazione che la GvHD ha una comparsa ritardata in pazienti esposti a una terapia non
mieloaplastizzante, e che l‟aumento scalare del dosaggio di radiazioni ionizzanti, in modelli
murini, comporta una forma di GvHD più severa [66]. La seconda fase della GvHD prevede,
invece, l‟induzione e la rapida amplificazione della risposta delle cellule T linfoidi del
donatore, che ha inizio con il riconoscimento dei peptidi antigenici associati alle molecole
HLA sulla superficie delle APC all‟interno dei tessuti linfoidi secondari [67]. Sia le APC del
donatore che quelle del ricevente partecipano all‟induzione di GvHD. E‟ stato dimostrato che
mentre nel TCSE HLA parzialmente compatibile o aploidentico, le APC del ricevente sono
indispensabili per la presentazione degli antigeni target ai linfociti del donatore, nel TCSE
HLA identico sono indispensabili sia le APC del ricevente sia quelle del donatore per la
presentazione degli alloantigeni target [68]. Nella terza fase della GvHD, i linfociti alloreattivi
del donatore si espandono e differenziano nelle sottopopolazioni Th1 e Th2, diversamente
associate alle manifestazioni della suddetta patologia infiammatoria, e nella quarta, migrano
verso i tessuti target [69]. Questo evento è seguito dal reclutamento al loro interno di altre
popolazioni leucocitarie effettrici, mediata da citochine infiammatorie e chemochine. Lo
stadio finale effettore della GvHD prevede quindi la distruzione dei tessuti target attraverso
meccanismi di contatto cellula-cellula o mediati da fattori solubili quali FasL, TNF-α e IFNγ che, comportando il rilascio ulteriore di fattori pro-infiammatori, non fanno altro che
esacerbare il quadro clinico [70]. I mediatori della GvHD sono principalmente rappresentati
dai linfociti T αβ+ del donatore; la loro deplezione dall‟inoculo previene infatti la GvHD sia in
modelli murini che nell‟uomo. In particolare, nel TCSE HLA parzialmente compatibile o
aploidentico sia le cellule T naive CD4+ che CD8+ mediano la GvHD mentre nel TCSE HLA
identico sono le cellule T naive CD8+ le principali mediatrici.
178
LA Welniak. Annu Rev Immunol 25, 2007
Figura 1. Patofisiologia multifasica della GvHD nel trapianto allogenico di cellule staminali emopoietiche.
a) “Priming” della risposta immune indotto dal danneggiamento tissutale causato dal regime di Condizionamento
preparativo; b) Induzione e c) Amplificazione della risposta delle cellule T linfoidi del donatore; d) Migrazione
delle cellule T linfoidi del donatore verso i tessuti target e reclutamento di altre popolazioni leucocitarie
effettrici; e) distruzione dei tessuti target attraverso meccanismi di contatto cellula-cellula o mediati da citochine
infiammatorie e chemochine.
Le cellule T memoria sono invece implicate nel mantenimento della GvHD, piuttosto che
nella sua patogenesi, a causa della debole espressione del CD62L e del CCR7, entrambi
fondamentali per il ricircolo a livello degli organi linfoidi secondari; del repertorio HLA
179
ristretto, associato a una ridotta frequenza di cellule T alloreattive, e della ridotta capacità di
espansione clonale [71].
La GvHD può essere classificata, in base alle caratteristiche cliniche e istologiche, in cronica
e acuta. La GvHD acuta compare entro i primi 100 giorni dal trapianto, si presenta con necrosi
delle cellule epiteliali a carico di cute, fegato, intestino e polmoni, ed è caratterizzata da una
risposta cellulare di tipo Th1. La GvHD cronica compare dopo i 100 giorni dal trapianto,
colpisce non solo cute e mucose ma anche ghiandole esocrine e membrane sierose ed è
caratterizzata da una risposta cellulare di tipo Th2. Origina da meccanismi patogenetici più
complessi, e provoca sostanzialmente quadri clinici molto simili a quelli delle malattie
autoimmuni per alterazione della selezione timica negativa [72-73]. La prevenzione della
GvHD si basa sull‟impiego di agenti immunosoppressori (Ciclosporina, Metotrexate,
Corticosteroidi) e di inibitori dell‟attivazione T-cellulare (Rapamicina), in grado di mantenere
“quiescenti” i linfociti del donatore, o sul ricorso alla manipolazione dell‟inoculo (Tdeplezione). I limiti di tali approcci sono però rappresentati dall‟aumento della probabilità di
mancato attecchimento, recidiva e infezioni. Si stanno di conseguenza affiancando approcci
più innovativi basati sull‟uso di agenti bloccanti le citochine (ETANERCEPT), sulla
modulazione dell‟attività delle APC mediante fotoferesi extracorporea e sull‟impiego di
terapie cellulari, basate sul trasferimento cellulare adottivo, finalizzato a modulare le risposte
immunitarie allogeniche attraverso l‟induzione di una tolleranza immunitaria “periferica [7476]. In questo contesto, sta emergendo il ruolo di una sottopopolazione di cellule T ad attività
regolatoria, responsabili dell‟induzione e del mantenimento della tolleranza e omeostasi
immunologica, nella modulazione e soppressione della risposta immune non solo fisiologica
ma anche patologica, come nel caso della GvHD.
La GvL, al contrario delle GvHD, rappresenta una “componente vantaggiosa” delle reazioni
allogeniche mediate dalle cellule immunocompetenti del donatore, nel caso di un TCSE in un
soggetto affetto da una neoplasia ematologica. Consiste nell‟eliminazione delle cellule
tumorali residue del ricevente mediata dai linfociti T CD4+ e T CD8+ e dalle cellule NK
alloreattive del donatore. Rappresenta, insieme al Condizionamento, uno dei principali motivi
di successo del TCSE allogenico, in quanto contribuisce alle sue proprietà antileucemiche, ed
è il motivo per cui il TCSE allogenico viene tuttora considerato una forma di “Immunoterapia
Adottiva”. La GvL richiede per il suo sviluppo gli stessi presupposti della GvHD e i suoi
target sono sia molecole espresse selettivamente dalle cellule neoplastiche (BCR/ABL nella
Leucemia Mieloide Cronica) sia miHA non condivisi tra donatore e ricevente espressi dalle
cellule neoplastiche. Un ruolo fondamentale rivestono le cellule NK in questa forma di
180
alloreattività. La parziale istocompatibilità donatore-ricevente si traduce, infatti, nel
potenziale mismatch tra i KIR (Killer cell Ig-like Receptors) sulle cellule NK del donatore e le
molecole HLA di classe I presenti sulle cellule del ricevente e viceversa. I KIR modulano
negativamente l‟attività litica delle cellule NK, attraverso il riconoscimento di specifici
epitopi condivisi dagli HLA self di classe I [77]. Nell‟ambito di un TCSE allogenico
parzialmente identico/aploidentico o da MUD tale reattività si manifesta nei confronti delle
cellule target allogeniche del ricevente che non esprimono gli HLA complementari ai KIR
espressi dalle cellule NK del donatore (“Missing self” recognition). Questo si traduce nella
lisi mediata dalle cellule NK del donatore diretta prevalentemente verso le cellule del sistema
emopoietico del ricevente, piuttosto che degli altri tessuti, con induzione dell‟effetto GvL. Lo
studio dell‟incidenza della GvL e GvHD in TCSE aploidentici KIR-incompatibili ha suggerito
che le cellule NK incrementano lo sviluppo delle reazioni antitumorali senza influenzare la
comparsa di GvHD [78]. Il trasferimento adottivo di cellule NK dopo TCSE inibisce la GvHD
e promuove la GvL in svariati modelli murini, sebbene attraverso un meccanismo d‟azione
ancora non noto, ma che potrebbe implicare l‟accelerazione dell‟eliminazione delle APC del
ricevente o la secrezione del TGF-β 79 .
Sostanziali progressi sono stati raggiunti nella pratica clinica del TCSE allogenico e nella
comprensione della biologia sottostante questa complessa procedura. L‟ampliamento delle
conoscenze nelle tecniche diagnostiche, il perfezionamento delle cure supportive e del
controllo delle componenti immunoterapiche, diviene quindi importante per il miglioramento
della prognosi post-TCSE, con possibile indirizzamento verso l‟individualizzazione della
procedura trapiantologica.
1.2. Le cellule T regolatorie “naturali”
1.2.1 Ontogenesi e caratterizzazione delle cellule T regolatorie “naturali”
L‟abilità del sistema immune di discriminare le componenti self da quelle non self si traduce
nel fenomeno della “Tolleranza Immunologica”, che può essere definita come la mancanza di
responsività nei confronti di specifiche molecole, acquisita attraverso meccanismi centrali o
periferici. La tolleranza di natura “Centrale” si verifica durante l‟ontogenesi delle cellule T
linfoidi e comporta l‟eliminazione dei linfociti potenzialmente autoreattivi, generati come
risultato dei processi di riarrangiamento casuale delle sequenze geniche codificanti il recettore
T cellulare (TCR), principalmente attraverso un processo di delezione clonale timica. Al
contrario, la tolleranza di natura “Periferica” ha luogo durante il corso della vita ed è
181
finalizzata al controllo delle risposte immuni mediate da cellule T linfoidi autoreattive
sfuggite alla selezione negativa timica perché dirette contro antigeni a bassa affinità/avidità o
non sufficientemente presenti negli organi linfatici primari perché prodotti tardivamente o in
modo transiente, o perché sequestrati all‟interno dei tessuti periferici [80]. In realtà, in alcuni
casi, l‟organismo può anche sviluppare, in seguito ad esposizione cronica, una forma di
tolleranza dannosa, diretta ad esempio nei confronti di antigeni estranei come quelli virali o
tumorali. I meccanismi di tolleranza “Periferica” includono l‟induzione dell‟anergia cellulare
e la soppressione dell‟attività delle componenti autoreattive, realizzata principalmente
attraverso il contributo di specifiche sottopopolazioni T linfoidi di natura regolatoria (Treg)
[81]. Le cellule Treg rappresentano quindi una componente essenziale per il mantenimento
della tolleranza al self e la prevenzione di fenomeni autoimmuni, grazie alla loro capacità di
controllare negativamente le cellule T autoreattive in vivo [82]. Sono comunemente distinte in
“naturali” e “indotte”, ciascuno dei due sottotipi caratterizzato da pattern differenziativi e
funzioni specializzate distinte. Si definiscono “naturali” le cellule Treg che originano nel
timo, mentre vengono definite “indotte” o “adattative” quelle generate in periferia, in seguito
a particolari condizioni di presentazione antigenica e in presenza di specifiche citochine
[Figura 2]. Queste ultime comprendono diverse popolazioni linfoidi con funzione regolatoria,
tra cui le cellule T CD4+ di tipo 1 (o Tr1) secernenti Interleuchina (IL)-10 e di tipo 3 (Th3)
secernenti TGF-β, le cellule T CD8+CD28-, le cellule T CD8+CD122+, le cellule T
e le
cellule NK-T [83]. Diversamente dalle cellule Treg “indotte” (iTreg), quelle “naturali”
(nTreg) si identificano in una singola popolazione cellulare CD4+ caratterizzata
dall‟espressione costitutiva di elevati livelli della catena α del recettore per l‟IL-2 (CD25), un
marcatore normalmente utilizzato per l‟identificazione di linfociti T attivati in seguito
all‟incontro con l‟antigene, la cui positività sulle nTreg denota il ruolo centrale dell‟IL-2 nella
biologia di tali cellule. Le nTreg corrispondono nel topo all‟8-12%, nell‟uomo al 5-10% del
compartimento periferico dei linfociti T CD4+. Sono contraddistinte, come già anticipato, da
un‟ontogenesi timica, vengono quindi prodotte durante i normali processi di maturazione
delle popolazioni T linfoidi che si verificano nel timo, e il loro repertorio di TCR è in
massima parte self-reattivo [84]. E‟ stato suggerito che i timociti regolatori CD4+CD25+
vengano prodotti all‟interno del timo a partire da timociti doppio positivi CD4+CD28+
selezionati attraverso il riconoscimento di peptidi autologhi in associazione a molecole HLA
di classe II, speculando che l‟interazione sia ad avidità sufficientemente elevata da consentire
il processo di selezione positiva ma allo stesso tempo sfuggire quello di selezione negativa,
normalmente deputato alla delezione delle cellule autoreattive. Questa particolare forma di
182
selezione è stata definita con il termine di “selezione alterata” [85]. I timociti sono selezionati
positivamente all‟interno dell‟epitelio corticale timico, mentre la selezione negativa ha luogo
a livello della giunzione cortico-midollare e midollare timica, dove appaiono le cellule T
mature singolo-positive [86]. Diversi dati hanno in passato supportato l‟ipotesi secondo la
quale la regione midollare timica potesse costituire il sito della selezione positiva delle nTreg.
Tali cellule sono, infatti, localizzate nel topo, così come nell‟uomo, a livello della suddetta
regione; inoltre, il fenotipo regolatorio non compare sino al momento in cui i linfociti T hanno
raggiunto lo stadio maturativo di cellule singolo-positive, che accade abbastanza tardivamente
nel processo di selezione timica [87].
SM Kang. Am J Trasplant 7, 2007
Figura 2. Vie differenziative delle cellule T regolatorie naturali e adattative. pDC=cellule dendritiche plasmacitoidi.
In realtà, sebbene sia stato dimostrato, in diversi modelli murini, che le componenti stromali
epiteliali timiche siano necessarie e sufficienti per la selezione delle nTreg, è stato
recentemente osservato che l‟induzione dell‟espressione delle molecole MHC di classe II
unicamente a livello dell‟epitelio corticale timico, consente il normale sviluppo di cellule T
regolatorie CD4+CD25+ funzionali sia in vitro che in vivo [88].
183
Queste osservazioni hanno portato a supporre che le nTreg, siano selezionate positivamente
nell‟epitelio corticale timico attraverso le interazioni tra il loro TCR e i peptidi self presentati
in associazione alle molecole HLA di classe II. Alcune di queste cellule vengono poi
sottoposte a selezione negativa nella regione midollare timica, e delete secondo un processo
molto simile a quello che si verifica nel caso delle sottopopolazioni T ad attività effettrice,
con la sola differenza nell‟avidità delle interazioni antigene-recettore. Si potrebbe immaginare
che il grado di avidità comporti sia la selezione delle nTreg che la trasduzione al loro interno
di segnali che le rendono anergiche alla stimolazione antigenica e ne inducono la produzione
di molecole che a loro volta le proteggono dai segnali apoptotici che ricevono nella regione
midollare timica, a causa dell‟alta affinità per il self [89]. Naturalmente, il repertorio di
specificità antigenica delle nTreg è influenzato dalla tipologia di antigeni self presentati
all‟interno del timo. Molto probabilmente, anche molecole differenti rispetto al TCR e ai
complessi peptide self-HLA e dotate di attività costimolatoria o adesiva, intervengono nella
selezione e nel differenziamento delle nTreg. Ne sono esempi il CD28, B7, il CTLA-4, LFA-1
e ICAM-1 [90]. In realtà, è stato suggerito che in particolari circostanze le cellule T
regolatorie CD4+CD25+ possano essere generate anche in periferia. Quando ottenute da
cellule mononucleate di sangue periferico umano appaiono, infatti, caratterizzate da telomeri
più corti; questo dato, combinato con la bassa espressione dell‟antigene CD45RB, potrebbe
indicare che si tratti cellule altamente differenziate stimolate ripetutamente dall‟incontro con
l‟antigene. Quest‟osservazione ha portato a supporre che le nTreg periferiche derivino da
cellule memoria attraverso l‟interazione con antigeni presentati da cellule APC nonprofessionali che, mancando dei sufficienti segnali costimolatori indispensabili per
l‟attivazione T cellulare, risulterebbero in grado di indurre anergia piuttosto che attivazione
della risposta immune [91].
Dopo la loro produzione timica, le nTreg migrano in periferia. Qui, diversamente da quanto
accade nel caso dei linfociti T effettori che, per la loro sopravvivenza, richiedono un contatto
continuo con gli stessi complessi peptide-HLA coinvolti nella loro selezione, le nTreg
sopravvivono grazie all‟attività di citochine e molecole costimolatorie. E‟ stato, infatti,
dimostrato che topi IL-2-/- e CD28-/- non sviluppano nTreg e che il loro numero risulta ridotto
nel caso di un deficit delle molecole B7-1/2 [92]. Tutti questi fattori sono coinvolti
essenzialmente nella produzione di IL-2 o nella co-stimolazione della produzione di IL-2, un
dato che evidenzia il ruolo centrale di questa citochina nel mantenimento delle nTreg così
come nel loro differenziamento. Questo spiegherebbe il motivo per il quale topi con un deficit
di IL-2 sviluppano patologie autoimmuni organo-specifiche. Dal momento che le nTreg non
184
producono IL-2, molto probabilmente l‟IL-2 prodotta da altre componenti cellulari riveste una
importanza cruciale a tale proposito. Ad ogni modo, anche altre citochine come l‟IL-4
sembrerebbero avere un ruolo nel mantenimento delle nTreg in periferia [93].
Le nTreg sono state inizialmente definite utilizzando la sola espressione delle molecole di
superficie CD4 e CD25. Questa modalità di classificazione risulta in realtà insufficiente per
l‟identificazione di tale sottotipo linfocitario, in quanto in condizioni di infiammazione o
attivazione del sistema immune, il CD25 va incontro ad una rapida upregolazione,
mantenendosi stabile sulla superficie delle cellule convenzionali T CD4+ e CD8+ effettrici per
diversi giorni. Va inoltre ricordato che nell‟uomo i linfociti convenzionali possono esprimere
il CD25 anche in assenza di uno stimolo attivatorio [94]. Conseguentemente, è stata condotta
un‟estensiva ricerca nel tentativo di identificare marcatori fenotipici addizionali che potessero
essere utilizzati per distinguere in modo più accurato le nTreg dalle altre popolazioni T
linfoidi. Diverse molecole sono apparse caratteristicamente presenti sulla superficie delle
nTreg ma tutte si sono rivelate potenzialmente upregolabili in cellule T effettrici in condizioni
attivatorie o espresse in altre componenti del sistema immune. Ad ogni modo le nTreg, oltre
al CD25, esprimono numerose molecole di superficie come il CD45RB; le selectine (CD62
ligando o CD62L) e le integrine (CD103), indispensabili per il processo di homing
linfonodale e tissutale; le chemochine (CCR5); il recettore del TNF indotto dai
glucocorticoidi (GITR); l‟antigene-4 associato ai linfociti T citotossici (CTLA-4 o CD152); il
TGF-β e diversi membri della famiglia dei recettori Toll-like (inclusi TLR-4, TLR-5, TLR-7 e
TLR-8). GITR è coinvolto nella soppressione dell‟attività delle nTreg attraverso un
meccanismo non del tutto noto. La somministrazione di anticorpi anti-GITR in vivo induce la
comparsa di patologie autoimmuni; mentre in altri modelli la stimolazione di GITR rende i
linfociti T effettori resistenti alla soppressione mediata dalle nTreg [95]. Il CTLA-4 è espresso
costitutivamente dalle nTreg e la sua inibizione causa un‟abrogazione dell‟attività di tali
cellule, suggerendo un suo ruolo nella costimolazione delle nTreg. In realtà, il CTLA-4 è
anche in grado di mediare direttamente l‟attivazione delle nTreg grazie all‟induzione
dell‟enzima indoleamina 2, 3-diossigenasi (IDO) in seguito all‟interazione con le molecole
CD80 e CD86 presenti sulla superficie delle DC, evento che conduce al catabolismo del
triptofano e quindi all‟immunosoppressione localizzata [96]. L‟espressione dell‟enzima IDO è
anche utilizzata da un gran numero di tumori umani per evadere l‟attacco da parte del sistema
immunitario, in quanto la sua espressione correla con il mancato accumulo di linfociti T
antigene-specifici nel sito tumorale [97]. I TLR rappresentano, invece, un sistema di
riconoscimento dei patogeni con funzione fondamentale nella risposta immune di natura
185
innata. E‟ stato dimostrato che la stimolazione di nTreg con il lipopolisaccaride attraverso il
TLR-4 si traduce in una risposta proliferativa e nell‟incremento delle funzioni soppressorie
[98]. In realtà, questo dato appare contraddittorio se si pensa che normalmente i TLR sono
coinvolti nell‟attivazione piuttosto che nella soppressione della risposta immune. Inoltre, la
stimolazione delle DC attraverso i TLR induce la produzione di citochine, come l‟IL-6, che
rendono le cellule T convenzionali resistenti all‟attività soppressoria delle nTreg [99]; di
conseguenza molto probabilmente i TLR potrebbero essere implicati nel controllo negativo
delle nTreg. Oltre ai suddetti marcatori, gli studi volti a migliorare l‟identificazione delle
nTreg, hanno di recente permesso di individuare anche altri antigeni di superficie caratteristici
di tali cellule, come ad esempio la catena α del recettore dell‟interleuchina 7 (CD127),
espressa a bassa intensità, e gli antigeni CD39 e CD73, che sembrerebbero regolare la
capacità inibitoria delle nTreg [100].
Ad ogni modo, al di là delle molecole di superficie, il marcatore più specifico e di maggior
contributo per l‟identificazione delle nTreg è attualmente rappresentato dal fattore di
trascrizionale nucleare, Forkhead Transcription Factor Family 3, FOXP3. E‟ stato identificato
agli inizi degli anni 2000 da tre differenti gruppi di ricerca e la sua espressione costituisce
oggi il “gold standard” per la distinzione delle nTreg dalle cellule T linfoidi effettrici. La sua
identificazione è stata resa possibile grazie allo studio della patologia “scurfy” sviluppatasi
spontaneamente in topi del “Oak Ridge National Laboratory” nel 1949 [101]. La mutazione
genica causale la malattia, interessa un gene X-linked inizialmente chiamato “scurfin”. Gli
animali affetti da tale mutazione solitamente non sopravvivono oltre la terza settimana di vita
e presentano lesioni interne simili a quelle osservate in corso di GvHD, infiltrate da cellule T
linfoidi proliferanti e secernenti citochine infiammatorie [102]. Il clonaggio del gene “scurfin”
nel 2001 ha permesso di identificare il prodotto del suddetto gene in un fattore della famiglia
dei fattori di trascrizione Forkhead, da cui la sigla di FOXP3. Topi affetti dalla patologia
“scurfy” possono essere curati attraverso la trasfezione del gene esprimente FOXP3 o, in
alternativa mediante ricostituzione del compartimento di cellule T CD4+CD25+ [103].
Attraverso l‟impiego di un sistema di delezione condizionale, è stato in seguito dimostrato
come il silenziamento del gene FOXP3 possa comportare l‟immediata comparsa di patologie
autoimmuni persino in topi adulti, fornendo una prova del ruolo di FOXP3 e delle nTreg nel
controllo dell‟autoimmunità [104]. Nel timo l‟espressione di FOXP3 è rilevabile allo stadio
maturativo T doppio-positivo e cellule CD4+FOXP3+ sono osservabili nel topo già a 3 giorni
dalla nascita, con una buona correlazione con il dato che la timectomia effettuata dopo il terzo
giorno dalla nascita induce patologie autoimmuni organo-specifiche che possono essere
186
prevenute attraverso l‟inoculo di nTreg [105]. Ulteriori studi su modelli murini hanno
dimostrato come i linfociti CD4+CD25+ rappresentino il sottotipo cellulare FOXP3+
predominante e come sia possibile riprogrammare le cellule T effettrici CD4+ o CD8+, in
cellule con funzioni soppressorie, mediante trasferimento del gene codificante FOXP3 [106].
La trasfezione retrovirale di linfociti T naive con l‟mRNA di FOXP3 li rende anergici e
soppressori in vitro , con acquisizione di caratteristiche fenotipiche sovrapponibili a quelle
delle nTreg, sia in termini di molecole di superficie che di citochine secrete. FOXP3 ha quindi
un ruolo fondamentale sia nei processi di sviluppo che nell‟esplicazione delle funzioni
regolatorie delle nTreg e non rappresenta, come il CD25 o GITR, un mero marcatore di
attivazione, in quanto la stimolazione delle cellule T linfoidi con anticorpi anti-TCR o antiCD28 non si traduce nell‟upregolazione del suo mRNA [107].
In realtà, quando si prende in considerazione l‟uomo, esistono svariate differenze rispetto a
quanto rilevato nei modelli murini. In primo luogo, l‟espressione di FOXP3 nell‟uomo,
diversamente da quanto accade nel topo, risulta caratteristica anche di cellule T effettrici,
sebbene a livelli ridotti. Nell‟uomo esistono, inoltre, 2 isoforme del gene FOXP3, una
codificante la forma “full length” della proteina, l‟altra codificante per un‟isoforma più corta,
mancante degli aminoacidi 71-105, e l‟overespressione di una o entrambe le isoforme umane
di FOXP3 in cellule T linfoidi naive non risulta efficace nella generazione di nTreg. E‟ stato
supposto che tali differenze dipendano principalmente dalla stretta correlazione presente nelle
cellule T CD4+, tra l‟espressione di FOXP3 e l‟attivazione del TCR. La regione promotrice
del gene codificante FOXP3 contiene, infatti, diversi siti di legame per i fattori AP-1 e NFAT, entrambi coinvolti nella trasduzione dei segnali a valle del TCR, e la formazione del
complesso NF-AT/FOXP3 è indispensabile per l‟attività soppressoria delle nTreg [108].
Comunque, anche nell‟uomo è stata identificata una patologia, nota con il termine di sindrome
“IPEX”, correlata, nella maggior parte dei casi, alla presenza di mutazioni del gene
codificante FOXP3. I soggetti che ne sono affetti mostrano, come accade nei topi “scurfy”,
disordini autoimmuni quali diabete mellito insulino-dipendente, anemia emolitica e lesioni
cutanee infiammatorie [109]. Di conseguenza, sia il modello murino che umano rendono
ampiamente chiaro come i difetti a carico di FOXP3 si traducano nella compromissione delle
funzioni delle nTreg e nell‟autoimmunità multisistemica, rendendo il suo studio un approccio
indispensabile per la comprensione dei meccanismi alla base della soppressione mediata dalle
nTreg e per l‟identificazione di nuovi marcatori specifici di questa popolazione cellulare.
187
1.2.2 Peculiarità intrinseche delle cellule T regolatorie “naturali” in vitro e in vivo
I primi studi in vitro hanno evidenziato una fondamentale caratteristica distintiva delle nTreg
rispetto ai linfociti T convenzionali. Tali cellule, infatti, non proliferano in risposta alla
stimolazione del TCR in coltura, ma possono essere indotte a dividersi in presenza di
anticorpi anti-CD3 e IL-2 esogena [110]. Lo stato anergico delle nTreg osservato in vitro può
essere in parte spiegato sulla base della tipologia di APC impiegata nei differenti studi.
Generalmente, le cellule spleniche sono impiegate come fonte di APC, e tale popolazione è
relativamente debole nell‟induzione dell‟espansione di nTreg antigene-specifiche anche in
presenza di IL-2, molto probabilmente a causa di uno stato funzionale immaturo [111]. Al
contrario, le nTreg vanno incontro a estensivi cicli di proliferazione in vitro se stimolate da
DC mature, specialmente in presenza di IL-2. Tale effetto non va inoltre a scapito della loro
attività, in quanto le nTreg espanse mostrano un‟abilità di soppressione uguale o addirittura
superiore a quella delle cellule pre-espansione [112]. Naturalmente, la stimolazione della
proliferazione delle nTreg così come della loro funzione è, almeno nella prima fase, antigene
specifica. Infatti, in una co-coltura di cellule T effettrici e nTreg dotate di una differente
specificità, la soppressione non si verifica sino al momento in cui l‟antigene per cui le nTreg
sono specifiche viene aggiunto, sebbene una volta attivate, le nTreg, come vedremo in
seguito, possono sopprimere anche risposte immuni dirette contro antigeni non correlati. Allo
stesso tempo, le nTreg policlonali sono in grado di sopprimere l‟attività di linfociti T
convenzionali anch‟essi policlonali quando entrambe le popolazioni sono stimolate con
anticorpi anti-CD3, nonostante si tratti di una forma di soppressione meno efficace rispetto a
quella antigene-specifica [113].
Le nTreg proliferano in risposta all‟IL-2 anche in vivo, come dimostrato in topi trattati con
anticorpi neutralizzanti anti-IL-2, che manifestano una riduzione del numero di nTreg, o in
topi con un deficit genetico di IL-2 o del suo recettore, che sviluppano patologie autoimmuni
[114]. La riduzione quantitativa delle nTreg in assenza di IL-2 evidenzia il ruolo di tale
citochina non solo nell‟induzione dell‟espansione delle nTreg durante il corso della risposta
immune, ma anche nel mantenimento di tali cellule in condizioni omeostatiche. Inoltre, anche
in vivo così come accade in vitro, lo stato maturativo delle DC influenza in misura
differenziale la capacità di espansione e la funzionalità delle nTreg. L‟attività proliferativa
delle nTreg è stata seguita in vivo iniettando nTreg CD62Lhi singeniche in topi esprimenti un
diverso allele thy1. In tali condizioni sperimentali, un subset di nTreg appare quiescente,
mentre un altro, contraddistinto dalla forte positività per l‟antigene CD44, va incontro a
espansione, con acquisizione di un fenotipo attivato, per poi diminuire numericamente [115].
188
Inoltre, quando nTreg HA-specifiche sono iniettate in topi esprimenti l‟antigene HA sotto il
controllo del promotore dell‟insulina, tali cellule si dividono esclusivamente nei linfonodi
pancreatici, noti per contenere le DC presentanti gli antigeni di derivazione dalle cellule delle
isole β pancreatiche [116]. L‟espansione delle nTreg osservata in vivo viene quindi molto
probabilmente indotta da DC mature presentanti l‟antigene. La correlazione tra lo stadio
maturativo delle DC e l‟effetto proliferativo indotto nelle nTreg può essere attribuito al fatto
che la risposta di quest‟ultime è maggiormente dipendente dalle molecole costimolatorie
CD80 e CD86 rispetto a quella dei linfociti T effettori. Infatti, l‟espansione delle nTreg
indotta da DC isolate da topi CD80-/- e CD86-/- risulta inibita in misura superiore quando le
cellule rispondenti sono rappresentate in vitro dalle nTreg piuttosto che dai linfociti T
CD4+CD25- [117]. Il ruolo delle DC mature in vivo è stato inoltre chiarito da un recente
modello murino basato sul trasferimento adottivo di cellule T CD4+ transgeniche per il TCR e
sull‟internalizzazione guidata dell‟antigene corrispondente, attraverso l‟impiego di anticorpi
specifici per DEC205, un recettore endocitico espresso ad alti livelli sulle DC [118]. Il
direzionamento dell‟antigene a livello delle DC causa, infatti, un incremento di tutta la
popolazione T transgenica, ma la frazione CD4+CD25+ aumenta preferenzialmente. È stato
ipotizzato che la proliferazione indotta nelle nTreg da parte delle DC mature sia
indispensabile a bloccare l‟autoimmunità e lo sviluppo di uno stato infiammatorio cronico,
durante le rispose immuni anti-microbiche. Infatti, durante le infezioni, le DC sia di natura
mieloide che plasmacitoide, attivate e indotte a maturare in presenza degli stimoli
infiammatori, presentano un insieme di peptidi antigenici sia di derivazione esogena,
potenzialmente cross-reagenti con molecole self, che endogena. Il repertorio delle nTreg è
specifico per i suddetti antigeni endogeni e si espande in concomitanza con l‟espansione dei
linfociti T effettori microbo-specifici per garantire un fine controllo della risposta immune.
Un ruolo diverso è attribuibile invece alle DC immature o semi-mature, caratterizzate da
livelli intermedi di molecole costimolatorie ma prive di altre funzioni specifiche di DC
mature, che in condizioni omeostatiche trasportano gli antigeni self periferici a livello dei
linfonodi drenanti. E‟ stato ipotizzato che tali cellule, principalmente rappresentate dal
sottotipo mieloide, favoriscano il mantenimento della tolleranza al self attraverso il
reclutamento preferenziale delle nTreg mediato dall‟elevata produzione delle chemochine
CCL17 e CCL22, garantendone l‟espansione in condizioni di omeostasi [119].
Nel complesso le osservazioni effettuate suggeriscono che le nTreg vanno incontro a un
costante rinnovamento in vivo sia in condizioni di omeostasi, in risposta all‟IL-2 e alle DC
immature, sia in presenza di uno stato infiammatorio, attraverso il riconoscimento di antigeni
189
self endogeni presentati da DC mature. Quindi, le nTreg antigene-specifiche non sono
anergiche in vivo, ma possono proliferare e tale processo si verifica preferenzialmente negli
organi linfoidi che drenano il sito di accumulo antigenico.
1.2.3 Meccanismi di soppressione della risposta immune mediati dalle cellule T regolatorie
“naturali”
Studi effettuati principalmente in vitro hanno rivelato che le nTreg sono in grado di esercitare
effetti soppressivi nei confronti di multiple popolazioni cellulari coinvolte nell‟immunità e nei
processi infiammatori. In principio è stata osservata la capacità delle nTreg di inibire
l‟attivazione e l‟espansione delle cellule T CD4+ attraverso esperimenti in cui la deplezione
delle nTreg risultava associata allo sviluppo di manifestazioni autoimmuni, mentre la loro
inoculazione comportava l‟inibizione di tali manifestazioni [120]. In realtà è stato in seguito
dimostrato che l‟azione soppressoria delle nTreg interessa l‟induzione così come le funzioni
effettrici dei linfociti T sia CD4+ che CD8+ naive e memoria e si estende all‟inibizione della
proliferazione, della produzione di Immunoglobuline e dello scambio isotipico dei linfociti B,
all‟inibizione dell‟attività citotossica delle cellule NK e NK-T, al controllo della funzione e
maturazione delle DC e dell‟attività, quindi della sopravvivenza, dei neutrofili. L‟attività
soppressoria delle nTreg manifesta una peculiarità unica, identificabile con il termine di
“linked suppression”. Le nTreg possono essere infatti attivate attraverso una modalità
antigene-specifica e successivamente sopprimere le risposte immuni dirette contro antigeni
non correlati [121]. Tale forma di soppressione può essere osservata in vitro così come in vivo
solo in presenza di particolari condizioni e, dal momento che l‟attivazione delle nTreg in vivo
è sotto il controllo stringente dell‟antigene per cui sono specifiche, potrebbe verificarsi solo in
circostanze altamente specializzate come nel caso di infezioni virali organo-specifiche. In
condizioni normali, la specificità della soppressione potrebbe quindi essere ottenuta grazie
alla prossimità spazio-temporale delle cellule T effettrici, delle nTreg e delle APC [122].
Diversi studi hanno dimostrato che il pool delle nTreg è spazialmente organizzato, con cellule
reclutate o trattenute nei siti di incontro del loro antigene self target. I linfociti B e le APC
professionali esprimono diverse citochine in condizioni fisiologiche di attivazione antigenemediata; tra di esse, il CCL4 sembra mediare il reclutamento preferenziale delle nTreg nel sito
interessato, di conseguenza la sua deplezione sistemica causa un‟accelerazione della risposta
immune e la comparsa di fenomeni autoimmuni generalizzati.
A seconda del sistema sperimentale utilizzato, sono stati proposti vari modelli in vitro per
spiegare il meccanismo attraverso cui le nTreg regolano le risposte immuni. Questi vedono
190
coinvolti il contatto cellula-cellula e la secrezione di citochine, come l‟IL-10 e il TGF-β o di
molecole ad attività citotossica, come la perforina e il granzima A [Figura 3]. Tra le molecole
espresse dalle nTreg con un ruolo chiave nel meccanismo di inibizione da contatto, una delle
principali è rappresentata dal CTLA-4, che possiede un‟elevata affinità per le molecole
costimolatorie CD80 e CD86 espresse sulle DC. Le nTreg esprimono il CTLA-4
costitutivamente, mentre le cellule T effettrici lo esprimono solamente in seguito ad
attivazione, grazie anche all‟interazione con le nTreg [123]. L‟inibizione dell‟attività del
CTLA-4 in topi sani causa lo sviluppo di patologie autoimmuni grazie all‟abrogazione della
soppressione mediata dalle nTreg sui linfociti T effettori. Inoltre, l‟induzione dell‟espressione
di FOXP3 all‟interno di cellule T naive si traduce nell‟upregolazione del CTLA-4 attraverso
una modalità FOXP3-dipendente [124]. E‟ stato dimostrato che il CTLA-4, media sia
indirettamente che direttamente l‟attività soppressoria delle nTreg. Interagendo con i suoi
ligandi sulle DC, trasduce segnali costimolatori che a loro volta inducono, in concomitanza
con i segnali trasdotti dal TCR, l‟attivazione delle nTreg grazie alla competizione del legame
CD28-CD80/86 e all‟upregolazione dell‟antigene associato alla funzione linfoide (LFA)-1,
che favorisce l‟interazione fisica tra le nTreg e le APC. Una volta attivate, le nTreg agiscono
anche reprimendo l‟espressione del CD80 e del CD86 sulle DC, che vengono quindi rese
meno efficienti in termini di presentazione antigenica, e inducendo l‟espressione dell‟enzima
IDO con conversione del triptofano in metaboliti, come la kinurenina, dotati di potenti effetti
immunosoppressivi nel microambiente locale delle DC. In realtà, il CTLA-4 presente sulla
superficie delle nTreg è altresì in grado di interagire con le molecole CD80 e CD86 espresse
dai linfociti T effettori target della loro attività soppressoria, con trasmissione diretta di
segnali inibitori che convergono nel controllo negativo della trascrizione del gene per l‟IL-2,
il principale fattore proliferativo delle cellule T linfoidi [125]. Un altro fattore coinvolto nella
soppressione contatto-dipedente è rappresentato da LAG3, una molecola di adesione CD4associata espressa sulle nTreg in seguito ad attivazione, che lega le molecole MHC di classe II
presenti sia sulle cellule APC che sui linfociti T effettori. Il suo meccanismo d‟azione è poco
noto, ma anticorpi anti-LAG3 sopprimono l‟attività delle nTreg in vivo e in vitro, sebbene topi
LAG3-/- non esibiscano la comparsa di alcuna manifestazione autoimmune, diversamente da
quanto accade nel caso del deficit di CTLA-4 [126]. Le nTreg sono in realtà in grado di
svolgere la loro funzione anche in assenza di contatto con i rispettivi target cellulari, come
dimostrato in vitro dall‟integrità delle loro funzioni quando separate dai linfociti T effettori da
una membrana semipermeabile. Per quanto riguarda i meccanismi di soppressione contattoindipendente, numerose citochine sono coinvolte nel mediare l‟attività in vivo delle nTreg.
191
M Miyara. Trends Mol Med 13, 2007
Figura 3. Meccanismi di soppressione mediati dalle nTreg. Vari eventi molecolari e cellulari sono stati descritti
per spiegare come le nTreg possano sopprimere la risposta immune. Essi includono l‟inibizione dell‟espressione
del gene per l‟IL-2, la modulazione delle molecole costimolatorie sulla superficie delle APC e l‟interazione di
LAG-3 con le molecole MHC di classe II (a); la secrezione di citochine immunosoppressorie (b); l‟induzione del
catabolismo del triptofano attraverso il CTLA-4 (c) e la citotossicità (d). E‟ anche possibile che vi siano
meccanismi chiave non ancora noti (e).
L‟IL-10 viene descritta come una citochina inibitoria, con un ruolo fondamentale nel
mantenimento dell‟omeostasi immunologica. E‟ richiesta per il mantenimento del numero
delle cellule T linfoidi da parte delle nTreg; infatti, nTreg isolate da topi knock-out per il gene
dell‟IL-10 perdono la suddetta funzione. Allo stesso modo, il trasferimento adottivo di nTreg
192
in topi sottoposti a trapianto allogenico cutaneo induce allotolleranza, ma l‟iniezione di
bloccanti del recettore per tale citochina accelera il rigetto, confermando il ruolo
fondamentale dell‟IL-10 nel contesto dell‟allotrapianto [127]. Esperimenti condotti in vitro
hanno anche dimostrato un‟azione inibitoria, mediata dall‟IL-10 prodotta dalle nTreg, nei
confronti dell‟espressione delle molecole MHC di classe II e delle molecole costimolatorie
presenti sulle DC. Allo stesso modo, il TGF-β gioca un ruolo chiave nella funzione
soppressoria e nell‟omeostasi delle nTreg periferiche, principalmente attraverso l‟induzione e
il mantenimento dell‟espressione di FOXP3. Topi TGF-β-/- mostrano numeri ridotti di nTreg e
l‟espressione di FOXP3 può essere indotta nelle cellule T CD4+ naive quando coltivate in
presenza di TGF-β. Le cellule T FOXP3 indotte risultano inoltre efficaci nella soppressione
della proliferazione T come le nTreg [128]. Esistono in realtà diversi dati sull‟espressione di
membrana del TGF-β sulle cellule nTreg sia murine che umane. Questo renderebbe possibile
un suo coinvolgimento anche nella modalità di soppressione contatto-dipendente. Secondo
una delle ipotesi più accreditate, l‟espressione di membrana del TGF-β sarebbe indispensabile
per la sintesi di NOTCH1, una molecola utile al commissionamento della linea T, il cui
legame con il rispettivo ligando HES1 sui linfociti effettori favorisce la trasmissione di
segnali inibitori [129]. Come già anticipato, anche l‟IL-2 è importante per il mantenimento e
la funzione delle nTreg. Oltre al fatto che tali cellule esprimono costitutivamente il recettore
per l‟IL-2, il loro numero risulta sostanzialmente ridotto in topi IL-2-/- [130]. Lo stesso si
verifica con la neutralizzazione dell‟IL-2 circolante. Nella soppressione mediata dalle nTreg è
stato supposto che il recettore ad alta affinità per l‟IL-2 presente sulla loro membrana competa
per l‟IL-2 con i linfociti T effettori, riducendo l‟effetto di tale citochina sulla loro
proliferazione. Inoltre nell‟uomo l‟iniezione di IL-2 in pazienti linfopenici incrementa
l‟espansione delle nTreg e l‟IL-2 è in grado di indurre l‟espressione di FOXP3 nelle nTreg
umane attraverso l‟azione di STAT3 e STAT5 [131]. Le nTreg possono infine esercitare la
loro funzione attraverso il rilascio di Perforina e Granzima A, che inducono morte cellulare
non solo nei linfociti T effettori ma anche nei monociti e nelle DC. Anticorpi diretti contro il
CD18 interferiscono con tale meccanismo di uccisione, suggerendo il suo coinvolgimento
nell‟interazione delle nTreg con i rispettivi target [132].
Nello studio del meccanismo d‟azione delle nTreg in vivo è importante considerare l‟abilità di
localizzazione di tali cellule nei siti di induzione della risposta immune, in modo tale che vi
sia uno stretto contatto con i target da inibire. Di conseguenza, le molecole mediatrici dei
processi di migrazione e homing linfoide diventano di grande importanza per garantire il
funzionamento delle nTreg. Esse, infatti, ricircolano dal sangue ai tessuti linfoidi secondari in
193
quanto, come accade per i linfociti T effettori, le nTreg, in seguito alla loro stimolazione, si
localizzano, grazie all‟espressione del CD62L, a livello dei linfonodi drenanti l‟antigene dove
vanno incontro a molteplici cicli di proliferazione [133]. Anche i recettori per chemochine
quali CCR4, CCR5 e CCR8 sono importanti per il traffico delle nTreg. I loro ligandi
CCL17/22, CCL4 e CCL1, sono prodotti principalmente dalle DC, dai monociti e dai
macrofagi e consentono di direzionare le nTreg verso i loro target. Al tempo stesso, durante il
corso della stimolazione antigenica le nTreg cambiano la loro attività migratoria e
abbandonano i tessuti infiammati, un processo cruciale nel controllo di quelle popolazioni T
linfoidi che non ricircolano più dal sangue ai tessuti linfoidi secondari ma si localizzano nei
tessuti non-linfoidi. Nel topo, le nTreg acquistano ad esempio la positività per l‟integrina αEβ
perdendo quella per il CD62L, che ne permette l‟allontanamento dai siti di infiammazione
[134].
Indipendentemente da quali meccanismi siano utilizzati dalle nTreg per svolgere la loro
funzione, il controllo della magnitudine della risposta soppressoria mediata da tali cellule
risulta indispensabile, in quanto un eccesso di soppressione renderebbe l‟ospite suscettibile
non solo allo sviluppo di infezioni e tumori ma anche alla comparsa di manifestazioni
autoimmuni e allergie. Sono diversi i meccanismi di controllo dell‟attività delle nTreg [Figura
4]. In primo luogo, la modulazione della forza del segnale attraverso il TCR permette di
modulare la suscettibilità dei linfociti T effettori alla soppressione nTreg-mediata. Una debole
stimolazione policlonale del TCR con anticorpi anti-CD3 a basso dosaggio rende i linfociti T
effettori resistenti alle nTreg, mentre il contrario si verifica in presenza di anticorpi anti-CD3
ad elevato dosaggio. Lo stesso effetto hanno una forte costimolazione attraverso il CD80 e il
CD86, espressi sulle cellule T convenzionali, e la secrezione di IL6 [135]. Altri due fattori
implicati nel controllo negativo delle nTreg sono rappresentati da GITR e i TLR. L‟inibizione
di GITR favorisce la risposta immune attenuando sia in vitro che in vivo la soppressione
mediata dalle nTreg. Inoltre, l‟iniezione di agonisti di GITR in topi affetti da tumore si associa
a un incremento della magnitudine della risposta immune, con eradicazione del tumore. Il
ligando di GITR è espresso su tutte le ACP nel topo e sulle DC plasmacitoidi nell‟uomo ma
anche sui linfociti T effettori attivati [136]. La proliferazione di cellule T convenzionali con
un deficit a carico di GITR può essere inibita dalle nTreg anche in presenza di agonisti di
GITR, mentre nTregs GITR-/- non riescono a sopprimere la proliferazione di linfociti T
effettori normali in presenza dei suddetti anticorpi, a conferma del ruolo della stimolazione
attraverso GITR nell‟induzione di resistenza alla soppressione mediata dalle nTreg, nelle
cellule T convenzionali [137].
194
M Miyara. Trends Mol Med 13, 2007
Figura 4. Inibizione della soppressione mediata dalle nTreg. Il numero e la funzione delle nTreg deve essere
necessariamente controllato per garantire l‟appropriata magnitudine della risposta immune. Una forte
costimolazione attraverso il TCR, l‟IL-6, alte dosi di IL-2 e l‟attivazione di GITR sulle cellule T effettrici le
rende resistenti alla soppressione nTreg-dipendente. Altri meccanismi inibitori dell‟attività delle nTreg sono
rappresentati dall‟attivazione di TLR-4 e TLR-9 sulle APC, che si traduce nell‟induzione della secrezione di
citochine pro-infiammatorie come l‟IL-6 e il TNF-α.
Per quanto riguarda i TLR, è stato osservato che le nTreg ne esprimono alcuni sottotipi, come
il TLR2 e il TLR8, in maniera costitutiva. Il ligando del TLR-8 (CpG-A) è in grado di inibire
le nTreg, incrementando la risposta immune; mentre l‟attivazione delle nTreg attraverso il
TLR2 induce la proliferazione di tali cellule con concomitante sospensione transiente
dell‟attività soppressoria [138]. Di conseguenza, durante la fase acuta del processo infettivo,
195
le nTreg potrebbero essere rese silenti attraverso la segnalazione del TLR2 per poi riacquisire
la loro abilità soppressiva con la riduzione ma persistenza del patogeno nel sito interessato.
Questo spiegherebbe come le nTreg siano in grado di prevenire la totale eliminazione del
patogeno per garantire il mantenimento di un‟efficace memoria immunologica [139].
Nonostante numerosi progressi nella conoscenza dei meccanismi d‟azione delle nTreg, rimane
ancora controverso la modalità attraverso la quale tali cellule sopprimono la risposta immune
a livello molecolare. Molto probabilmente, più di un meccanismo di soppressione risulta
operativo nel controllo della risposta immune nTreg-mediata e vari fattori, tra cui la forza e la
natura dello stimolo antigenico, contribuiscono a determinare la magnitudine della
soppressione.
1.2.4 Ruolo delle cellule T regolatorie “naturali” nel TCSE
Evidenze relative al ruolo dell‟immunoregolazione CD4+-mediata, specialmente nella sfera
trapiantologica, risalgono a più di due decadi fa. Nel 1985, è stata dimostrata l‟induzione di
attività soppressoria in cellule T CD4+ di ratti riceventi un trapianto di cuore in associazione a
una terapia a base di ciclosporine e nel 1989 è stata riportata, sempre in modelli murini, la
comparsa di attività soppressoria in seguito a trasfusioni donatore-specifiche (TDS) [140].
Qualche anno più tardi è stato invece provato che l‟induzione di tolleranza allospecifica
attraverso l‟impiego di anticorpi anti-CD4 e anti-CD8, dipende dall‟attività di linfociti CD4+
soppressori e che sono tali cellule a garantire il prolungamento della sopravvivenza in seguito
a TDS [141]. Malgrado la pletora di indicazioni sperimentali, l‟importanza critica delle nTreg
in tale ambito è però emersa solo quando, grazie all‟impiego di un modello di trasferimento
adottivo di autoimmunità, è stato possibile identificare nel sottotipo T linfoide CD4+CD25+, la
proprietà di soppressione della risposta immune. Queste osservazioni hanno quindi
rappresentato il presupposto per lo sviluppo dell‟ipotesi secondo cui l‟induzione di
allotolleranza nel contesto di un TCSE potesse dipendere dagli stessi meccanismi tipici della
tolleranza “dominante”, diretta verso gli antigeni self. I primi esperimenti a conferma della
suddetta ipotesi sono stati basati sull‟impiego della procedura di xenotrapianto all‟interno del
sistema pollo-quaglia e hanno condotto a dimostrare l‟induzione di tolleranza dopo infusione
di xeno-trapianti di cellule epiteliali allo stadio embrionale. Lo stesso è stato dimostrato in
modelli murini [142].
Nell‟ambito delle emopatie maligne, l‟alloreattività rappresenta, in termini di GvL, un cardine
essenziale della procedura trapiantologia, in quanto, attraverso i sofisticati meccanismi
cellulari della competenza immunologica, rende possibile l‟aggressione e la distruzione di
196
eventuali cellule neoplastiche sfuggite ai trattamenti chemioterapici e ai farmaci utilizzati nel
regime di condizionamento pre-trapianto [143]. Tuttavia, tale alloreattività si traduce anche
nello sviluppo di GvHD, che, di fatto, rappresenta una delle principali complicanze del
decorso post-trapiantologico, in quanto si caratterizza per l‟induzione di una risposta immune
contro i tessuti sani del ricevente. Di conseguenza, lo stato ideale di tolleranza nel contesto di
un allotrapianto dovrebbe favorire la GvL inibendo al contempo la GvHD. A tal proposito,
diversi studi sperimentali, condotti su modelli animali e sull‟uomo, hanno confermato
l‟importanza delle nTreg nel controllo dell‟alloimmunità e nella prevenzione della GvHD,
dopo TCSE allogenico, apparentemente senza alcuna influenza sulla GvL [144].
L‟iniezione di colture linfocitarie miste C57BL/6-anti-B6.CH2bm12 in topi bm12 irradiati
subletalmente causa induzione di una forma letale di GvHD. Al contrario, l‟iniezione di
colture in presenza di anticorpi anti-CD154 o anti-CD80/86 non induce la patologia. Quando
le nTreg CD4+CD25+ sono depletate da tali colture prima dell‟iniezione, lo sviluppo della
GvHD continua ad aver luogo [145]. Questa ha rappresentato la prima reale dimostrazione
che le nTreg sono in grado di inibire la GvHD. E‟ stato in seguito dimostrato che l‟infusione
di linfociti allogenici in topi irradiati letalmente e sottoposti ad un TCSE T-depleto, non si
associa all‟induzione di GvHD. Questo fenomeno può essere spiegato ipotizzando lo sviluppo
di nTreg del donatore all‟interno del timo dell‟ospite, con conseguente protezione nei
confronti della GvHD mediata dai linfociti T infusi del donatore. Il fenotipo delle cellule ad
attività protettiva nei confronti della GvHD è, infatti, CD4+, in quanto lo stesso TCSE depleto
dei linfociti CD4+ è associato a GvHD letale dopo DLI [146]. In un modello murino simile, la
deplezione di cellule CD25+ comporta l‟accelerazione della GvHD letale, mentre l‟infusione
di linfociti CD4+CD25+ del donatore ritarda la GvHD, specialmente nel caso in cui il rapporto
nTreg/T effettori risulta elevato [147]. Nell‟uomo, è stato dimostrato che la scarsa
ricostituzione delle nTreg comporta uno sbilanciamento immunitario persistente, che
favorisce l‟insorgenza della GvHD. E‟ stata osservata una correlazione inversa tra
l‟espressione del gene codificante FOXP3 e il grado di GvHD in pazienti con GvHD acuta;
l‟espressione di FOXP3 risulta infatti consistentemente ridotta quando la malattia è attiva,
mentre normali livelli di FOXP3 garantiscono la risoluzione della GvHD [148]. Questi dati
sono stati ulteriormente confermati dall‟osservazione dell‟esistenza di un‟associazione tra la
presenza di bassi livelli di nTreg e la fase iniziale della GvHD acuta, suggerendo un
potenziale coinvolgimento di tali cellule nella patogenesi di questa forma patogenica di
alloreattività [149]. Dati conflittuali sono stati invece ottenuti in relazione al ruolo delle nTreg
nello sviluppo della GvHD cronica. In due recenti studi è stato riportato che pazienti con
197
GvHD cronica presentano un significativo aumento delle cellule T con fenotipo e funzionalità
regolatoria, rispetto a pazienti senza segni di GvHD cronica, mentre in un altro lavoro
condotto sempre su pazienti affetti da GvHD cronica, è stata segnalata una ridotta frequenza
di cellule Treg CD4+CD25+FOXP3+ [150]. Questi dati suggeriscono che una difettosa
normalizzazione dei livelli di nTreg durante la ricostituzione immunitaria successiva al
trapianto, potrebbe contribuire allo sviluppo di GvHD cronica; o, in alternativa, che la GvHD
cronica potrebbe causare un danno alla funzionalità del timo con possibile diminuzione della
produzione di nTreg.
Ma quali sono i meccanismi effettori attraverso cui le nTreg garantiscono la protezione dalla
GvHD? L‟osservazione che le nTreg esprimenti alti livelli dell‟antigene CD62L inibiscono la
letalità della GvHD indica che tali cellule agiscono inizialmente all‟interno degli organi
linfoidi secondari [151]. Allo stesso tempo, è stato dimostrato che le nTreg sono presenti
anche all‟interno dei tessuti target di GvHD come la cute, il fegato, l‟intestino e i polmoni.
Questo è reso possibile grazie all‟attività del recettore per chemochine CCR5, coinvolto nella
localizzazione delle nTreg nei tessuti infiammati [152]. In modelli sperimentali di colite, è
stato invece evidenziato il coinvolgimento di fattori solubili come l‟IL-10 e il TGFβ L'infusione di nTreg IL-10-/- garantisce un ritardo nella comparsa di GvHD, nonostante ciò,
solo il 40% degli animali sopravvive a 100 giorni dal trapianto e quelli che sopravvivono
mostrano segni di GvHD. Il ritardo nella comparsa di GvHD può essere giustificato
dall‟attività ridondante del TGF-β e del CTLA-4, che indica come le nTreg utilizzino
meccanismi effettori multipli. Alcuni studi riportano che l‟effetto di prevenzione della GvHD
ottenuto dall‟infusione delle nTreg, si realizza senza influenzare l‟effetto benefico della GvL,
per cui le nTreg sarebbero in grado di discriminare le due componenti dei meccanismi
alloreattivi [153]. La spiegazione dei meccanismi alla base di tale capacità risulta tuttora poco
nota. Una delle più recenti ipotesi nasce dall‟osservazione che la GvHD dipendente da
molecole MHC di classe II e quindi mediata dai linfociti T CD4+, risulta più severa rispetto
quella indotta da molecole MHC di classe I e quindi mediata dai linfociti T CD8+, e che le
nTreg hanno la capacità intrinseca di inibire maggiormente linfociti T CD4+. Su tali basi, le
nTreg, potrebbero garantire la protezione dalla GvHD, direzionando la loro attività
soppressoria sulle cellule T CD4+ piuttosto che CD8+, che rappresentano i linfociti
preferenzialmente deputati all'eliminazione dei blasti leucemici residui e quindi responsabili
della GvL [154]. Come anticipato, un rapporto relativamente elevato nTreg/T effettori è
richiesto per la protezione dalla GvHD. Questo potrebbe imporre un serio limite sull‟impiego
clinico delle nTreg. Per tale motivo, è stata valutata la possibilità di espandere nTreg ex-vivo
198
preservandone l‟attività regolatoria. Sono stati proposti diversi protocolli di espansione, basati
sull‟impiego di APC o anticorpi anti-CD3 e/o anti-CD28 immobilizzati, sempre in presenza di
elevate concentrazioni di IL-2. Il loro impiego consente di ottenere un fold di espansione fino
a 104 in colture di 6 settimane, e l‟infusione delle nTreg espanse in vitro, protegge
dall‟insorgenza della GvHD in diversi modelli murini [155-159]. In realtà, la capacità di
espansione così come di soppressione delle nTreg, risulta maggiore in vivo, caso in cui la
stimolazione ex-vivo viene effettuata mediante l‟impiego delle APC del ricevente del TCSE,
piuttosto che degli anticorpi anti-CD3 e/o anti-CD28, molto probabilmente a causa della
specificità delle nTreg espanse infuse. Tuttavia, nell‟uomo, la bassa frequenza delle nTreg e la
presenza di contaminanti CD4+CD25low nel sangue periferico umano pongono dei problemi
tecnici per l‟isolamento e la successiva espansione in vitro della popolazione cellulare di
interesse. Una valida alternativa al sangue periferico come fonte di nTreg per l‟espansione exvivo delle nTreg è rappresentata dal sangue di cordone ombelicale. Diversi studi hanno
indicato la possibilità di generare in vitro delle linee cellulari di nTreg umane da sangue
placentare dopo stimolo con anticorpi monoclonali anti-CD3, anti-CD28 e IL-2. La
popolazione nTreg predominante nel SCO è rappresentata da cellule con fenotipo immaturo,
CD45RA+CD45RO- e tale caratteristica è confermata anche da una minore espressione di
FOXP3 [160-162]. A questo proposito, alcuni studi hanno dimostrato una potente capacità
immunosoppressiva delle nTreg isolate dal SCO, in grado di persistere per diverse settimane e
probabilmente strettamente correlata al loro fenotipo immaturo [163-164]. Ciò potrebbe
spiegare la più bassa incidenza di GvHD che si osserva dopo trapianto di CSE da SCO,
piuttosto che da MO o SVP. Sulla base di questi risultati preliminari si apre quindi la
prospettiva per l‟utilizzo delle banche di cordone ombelicale come fonte di pronta
disponibilità di nTreg a scopo immunoterapico.
Per il ruolo cruciale che svolgono nella regolazione della risposta immune, nel mantenimento
della tolleranza immunitaria e nel controllo dei fenomeni autoimmuni, le nTreg rappresentano
quindi una potenziale terapia alternativa nell‟ambito di un TCSE allogenico che mira alla
prevenzione o al controllo della GvHD e alla promozione dell‟attecchimento e del
chimerismo, attraverso l‟induzione di allotolleranza. Per questo motivo, tali cellule,
costituendo una popolazione di linfociti T fenotipicamente e molecolarmente distinta, sono
diventate, negli ultimi anni, oggetto di studio di numerosissime ricerche applicate alla clinica.
199
1.3 Scopo del progetto di ricerca
Le nTreg rappresentano una sottopopolazione T linfoide CD4+ di derivazione timica con un
ruolo cruciale nel mantenimento della tolleranza immunologica al self e nel controllo della
risposta immune fisiologica e patologica. Nell‟ambito dell‟immunoterapia, una delle
applicazioni più promettenti delle nTreg è rappresentato dal tentativo di ridurre la tossicità
associata al trapianto allogenico di CSE, impiegato principalmente per la cura delle neoplasie
ematologiche. Le recenti scoperte sulla biologia delle nTreg e gli studi effettuati su modelli
murini, suggeriscono, infatti, un loro ruolo nella modulazione della prognosi post-trapianto, in
particolar modo attraverso la possibilità di controllare la GvHD senza interferire con l‟effetto
della GvL. In tale ambito si inserisce il nostro studio, volto ad indagare il ruolo delle nTreg
nella patogenesi e nel controllo della GvHD acuta e cronica post trapianto allogenico, per il
successivo disegno di un protocollo di Immunoterapia basato sull‟utilizzo delle nTreg e
destinato a pazienti allotrapiantati con complicanze di GvHD.
A tale scopo è stata analizzata dapprima la possibilità di isolare le nTreg da pazienti sottoposti
a TCSE allogenico, in assenza o presenza di GvHD, ed espanderle in vitro in presenza di
anticorpi anti-CD3 e anti-CD28 e IL-2. Sono state quindi esaminate le caratteristiche
fenotipiche delle nTreg espanse e la loro funzionalità in termini di attività imunosoppressoria
e produzione di citochine immunoregolatorie. La valutazione della soppressione nTregmediata è stata effettuata attraverso una co-coltura di cellule T effettrici CD4+CD25autologhe e DC allogeniche, in presenza o assenza di nTreg, finalizzata a mimare la risposta
allogenica che si verifica in vivo nel corso della GvHD. Nell‟ambito di tale studio è stata
anche avviata un‟analisi comparativa tra le nTreg isolate da pazienti sottoposti a TCSE
allogenico e quelle isolate da SVP e SCO, con lo scopo di evidenziare la possibile presenza di
differenze nelle peculiarità di tali cellule, in relazione alla fonte di origine.
Il fine ultimo del suddetto studio è rappresentato dalla possibilità di migliorare la
comprensione della biologia delle nTreg per lo sviluppo di un progetto immunoterapeutico,
basato sull‟utilizzo di tali cellule, che possa tradursi in un beneficio clinico attraverso la
riduzione del fabbisogno della terapia convenzionale immunosoppressiva nel TCSE
allogenico. Inoltre, le implicazioni funzionali di tale terapia potrebbero rappresentare il
razionale per un ulteriore approfondimento della comprensione della biologia del trapianto,
per la prevenzione e il trattamento della GvHD e per la ricostituzione immunologica.
200
2. MATERIALI E METODI
2.1 Pazienti e Controlli
Nel nostro studio sono stati inclusi 18 pazienti affetti da neoplasie ematologiche di differente
natura, sottoposti, presso il nostro Istituto, a TCSE allogenico. Il prelievo dei campioni di
SVP di tali pazienti è stato effettuato a circa un anno dal momento del trapianto. Quattordici
pazienti erano di sesso maschile e 4 di sesso femminile, con un‟età mediana di 36 anni
(intervallo: 10-61 anni). Cinque pazienti risultavano affetti da Leucemia Linfoide Acuta
(LLA), 3 da Leucemia Mieloide Acuta (LMA), 3 da Leucemia Mieloide Cronica (LMC), 3 da
Leucemia Linfatica Cronica (LLC), 2 da Mieloma Multiplo (MM), 1 da Linfoma di Hodgkin
(LH) ed 1 da Trombocitemia Essenziale (TE). Quindici pazienti sono stati sottoposti a
trapianto allogenico da donatore familiare HLA-compatibile, 3 pazienti a trapianto allogenico
da donatore non correlato HLA-compatibile. Il SVP è stato impiegato come fonte di cellule
staminali emopoietiche in 11 casi, il MO in 6 casi e il SCO in un caso. In 13 pazienti è stato
eseguito un regime di condizionamento di tipo mieloablativo e in 5 pazienti di tipo non
mieloablativo.
Le 18 unità di SCO incluse nello studio sono state ottenute dalla Banca del Cordone
Ombelicale dell‟Università degli studi di Roma “ Sapienza”. Tali unità comprendono quelle
sacche di SCO che non soddisfano i criteri di sicurezza che ne permettono l‟immissione nella
rete della banca. In qualità di controllo sono stati utilizzati, previo consenso informato, 13
campioni di SVP, prelevati da donatori sani adulti, ottenuti dal Centro Trasfusionale della
stessa Università.
Le cellule mononucleate di SCO (CBMC) e di SVP (PBMC) sono state ottenute mediante
separazione su gradiente di densità e coltivate a 37 C in atmosfera umidificata con un tasso di
CO2 pari al 5%, in terreno colturale RPMI 1640 (Cambrex Bio Science Verviers, Belgium)
addizionato con 10% di siero fetale bovino (FBS, HyClone, South Logan, UT), 1% di Lglutammina (EuroClone, Europe) e 1% di Pen-Strepto (EuroClone, Europe).
2.2 Purificazione dei linfociti T CD4+CD25+
Le cellule T CD4+CD25+ sono state isolate dal resto delle CBMC e delle PBMC sia dei
pazienti sottoposti a TCSE allogenico che dei donatori sani adulti mediante separazione
cellulare magnetica, attraverso l‟impiego di un kit di isolamento specifico per cellule Treg
CD4+CD25+ (Miltenyi Biotec Auburn, CA, USA). La procedura di isolamento è stata eseguita
201
in due passaggi. In primo luogo, allo scopo di arricchire i campioni in cellule T CD4+, è stata
effettuata una marcatura indiretta delle cellule T CD4- con una miscela di anticorpi biotinilati
diretti contro gli antigeni CD8, CD14, CD16, CD19, CD36, CD56, CD123, TCR /
Glicoforina A, e biglie magnetiche anti-biotina. A questo punto, i campioni arricchiti in
cellule T CD4+, sono stati sottoposti a una selezione positiva delle cellule T CD4+CD25+
mediante l‟uso di specifici anticorpi anti-CD25 coniugati a biglie magnetiche.
2.3 Espansione dei linfociti T CD4+CD25+ isolati
Le cellule T CD4+CD25+ ottenute dopo isolamento immunomagnetico, sono state espanse per
6 giorni in piastre da 96 pozzetti con fondo ad U, coniugate ad anticorpi monoclonali antiCD3 (5 g/ml) e anti-CD28 (5 g/ml), in presenza di IL-2 (100 U/ml), aggiunta al 1° e 3°
giorno di coltura. La frazione negativa ottenuta dopo purificazione, ovvero i linfociti T
CD4+CD25-, è stata risospesa in una soluzione di FBS al 10% di DMSO e criopreservata in
azoto liquido per il successivo impiego come popolazione cellulare T effettrice autologa.
2.4 Analisi Immunofenotipica delle cellule T CD4+CD25+ espanse
Le cellule T CD4+CD25+ espanse sono state caratterizzate immunofenotipicamente mediante
la valutazione dell‟espressione di superficie degli antigeni CD4, CD25, CD62L e
dell‟espressione intracellulare delle molecole CTLA-4 e FOXP3. Per la determinazione
dell‟espressione degli antigeni di superficie, le cellule sono state marcate con anticorpi
monoclonali (mAbs) di topo coniugati con i fluorocromi FITC, PE e PerCP e incubate per 20
minuti al buio a 4°C in PBS, privo di ioni Ca2+ e Mg2+ (DPBS, Cambrex Bio Science
Verviers, Belgium). In particolare, nel nostro sistema sperimentale sono stati impiegati i
seguenti mAbs di superficie: CD4FITC, CD25PE, CD3PERCP, (Becton Dickinson Bioscience,
San Jose, California), CD62LFITC (EBioscience). In tutti gli esperimenti, oltre agli mAbs
diretti contro antigeni di superficie caratteristicamente espressi dalle cellule Treg, sono stati
utilizzati isotipi aspecifici di Ig (IgG1, IgG2) coniugati con i fluorocromi FITC, PE e PerCP,
come controllo negativo. Per la valutazione dell‟espressione degli antigeni intracellulari
CTLA4 e FOXP3 è stato, invece, effettuato un test di marcatura intracitoplasmatica, basato
sull‟utilizzo del kit di permeabilizzazione cellulare “Fix&Perm” (Caltag Laboratories,
Hamburg, Germany). L‟acquisizione dei dati è avvenuta, in entrambi i casi, mediante
202
l‟utilizzo di un citofluorimetro “FACScan” e la loro analisi è stata realizzata utilizzando il
programma “CellQuest” (Becton Dickinson).
2.5 Valutazione delle capacità soppressorie delle cellule T CD4+CD25+ espanse
Per valutare le capacità soppressorie delle cellule T CD4+CD25+ espanse nei confronti
dell‟attività proliferativa dei linfociti T autologhi è stato allestito un test di proliferazione. A
tale scopo, le cellule T CD4+CD25+ espanse isolate da SCO e da SVP di pazienti trapiantati o
donatori sani sono state coltivate, in piastre da 96 pozzetti a fondo a U, secondo un rapporto
1:1 con cellule T effettrici CD4+CD25- autologhe a loro volta stimolate con DC generate da
monociti di donatori sani e caricate con corpi apoptotici originati da blasti leucemici
allogenici, come descritto in precedenza [165]. Allo scadere del 6° giorno di coltura, è stata
introdotto all‟interno di ciascun pozzetto 1 μCi di Timidina triziata (3H-TdR; Amersham,
Arlington Heights, IL) e, dopo un‟incubazione di circa 18 h a 37 C in atmosfera al 5% di
CO2, la capacità soppressoria delle cellule T CD4+CD25+ è stata valuta mediante l‟analisi
dell‟incorporazione di 3H-TdR da parte dei linfociti T effettori, in presenza o assenza di
cellule T CD4+CD25+ espanse, effettuata mediante l‟ausilio di un Beta-counter (Packard
Bioscience, Groningen, The Netherlands). I risultati sono stati riportati come media dei colpi
per minuto (Cpm) ± deviazione standard.
2.6 Valutazione della produzione di IL-10 da parte delle cellule T CD4+CD25+ espanse
La valutazione della produzione di IL-10 da parte delle cellule T CD4+CD25+ è stata
effettuata, mediante ELISA (Saggio di Immunoassorbimento con Enzima Coniugato), su
campioni di surnatanti prelevati al 6° giorno della coltura allestita per l‟espansione delle
cellule T CD4+CD25+ di SCO e di SVP di pazienti trapiantati o donatori sani, dopo
isolamento immunomagnetico.
In particolare, il test ELISA è stato realizzato attraverso l‟impiego di un kit per la
determinazione della concentrazione dell‟IL-10 (Quantikine®, R&D Systems) nei surnatanti
della coltura delle cellule T CD4+CD25+ espanse da 3 unità di SCO, 3 campioni di SVP di
pazienti senza segni di GvHD, 3 con segni di GvHD e infine 3 campioni di SVP di donatori
sani. Sono state impiegate come controllo diluizioni seriali di IL-10 umana ricombinante a
concentrazione nota, utili per la realizzazione di una curva standard necessaria a risalire alla
quantità di IL-10 presente nei campioni. Come standard più elevato è stata utilizzata una
203
diluizione di IL-10 pari a 1000 pg/ml, mentre il diluente fornito all‟interno del kit è stato
scelto come controllo negativo. L‟analisi dei dati è stata effettuata rilevando l‟assorbanza di
ciascun campione a 450 nm mediante l‟impiego di uno spettrofotometro.
204
3. RISULTATI
3.1 Capacità di espansione e caratteristiche immunofenotipiche delle cellule T
CD4+CD25+ isolate
Allo scopo di studiare le caratteristiche molecolari e funzionali delle cellule T regolatorie, è
stata in primo luogo esaminata la possibilità di purificarle ed espanderle in vitro a partire da
cellule mononucleate. Lo studio è stato condotto su 18 campioni di SVP prelevato da pazienti
affetti da neoplasie ematologiche ad almeno un anno dal trapianto allogenico di CSE, in
assenza (9 pazienti) o in presenza di GvHD (9 pazienti: 3 con GvHD acuta, 3 con GvHD
cronica e 3 con entrambi i tipi di GvHD); su 13 campioni di SVP prelevato da donatori sani
adulti e 18 unità di SCO.
Dai risultati ottenuti in questa prima fase dello studio, è emersa la possibilità di isolare, da
tutte le fonti esaminate, cellule T CD4+CD25+, con valori di purezza medi complessivamente
pari al 99.0% ± 0.1% (intervallo: 98-100%), mediante una primaria delezione dei componenti
CD4-, seguita da 2 cicli di selezione positiva per CD25. La valutazione della quantità di
cellule T CD4+CD25+ ottenute ha evidenziato percentuali medie di resa pressoché simili sia
nel caso del SVP dei pazienti allotrapiantati e dei donatori sani che nel caso delle unità di
SCO, con valori rispettivamente pari a 0.3% ± 0.4%, 0.6% ± 0.3% e 0.3% ± 0.3%.
Una volta isolate, le cellule T CD4+CD25+ sono state incubate in presenza di fattori stimolanti
la crescita e proliferazione delle cellule T linfoidi attraverso una modalità sia TCR-dipendente
che indipendente, rappresentati dagli anticorpi anti-CD3 e anti-CD28 e dall‟IL-2, aggiunta al
1° e 3° giorno di coltura. Al termine della 1a settimana, sebbene sia stato possibile osservare
una responsività alla stimolazione da parte delle cellule T CD4+CD25+ ottenute da tutte le tre
diverse fonti, è stata rilevata, in tal caso, una risposta quantitativamente diversa in relazione
alla tipologia di fonte. Infatti, le cellule T CD4+CD25+ isolate dalle unità di SCO hanno
mostrato una capacità di espansione superiore sia rispetto a quelle isolate dal SVP dei pazienti
allotrapiantati che dei donatori sani. In particolare, è stato calcolato un valore medio di
espansione di 11.0 ± 6.9 volte (631.9x103 ± 446.2x103 Treg pre- vs 6959.4x103 ± 655.4x103
Treg post-espansione) nel caso delle unità di SCO, di 6.7 ± 5.4 volte (355.8x103 ± 291.4x103
Treg pre- vs 2336.9x103 ± 274.5x103 Treg post-espansione) nel caso dei pazienti
allotrapiantati e di 4.4 ± 2.4 volte (680.6x103 ± 505.2x103 Treg pre- vs 2991.8x103 ±
175.1x103 Treg post-espansione) nel caso dei donatori sani [Tabella 1; Figura 5].
205
La differenza nei livelli di espansione è risultata significativa confrontando i valori medi di
espansione delle cellule T CD4+CD25+ isolate dalle unità di SCO e dal SVP dei donatori sani
(P=0.001); al contrario, nessuna differenza significativa è stata osservata paragonando la
capacità di espansione delle cellule T CD4+CD25+ isolate dai pazienti sottoposti a trapianto, a
quella caratteristica della controparte normale. Le cellule T regolatorie CD4+CD25+ isolate
sono inoltre apparse iporesponsive alla stimolazione sia in presenza di alte dosi di IL-2 in
assenza di anticorpi anti-CD3 e anti-CD28, sia in presenza dei suddetti anticorpi da soli o in
combinazione, mostrando invece una normale risposta alla stimolazione mediata da stimoli
TCR-indipendenti, rappresentati primariamente dalla fitoemoagglutinina (PHA) (dati non
mostrati).
Le cellule T CD4+CD25+ espanse sono state quindi analizzate mediante citometria a flusso
per
la
valutazione
delle
caratteristiche
fenotipiche,
finalizzata
all‟identificazione
dell‟espressione di specifici marcatori di superficie e intracellulari comunemente associati
all‟attività regolatoria. In tutti i casi esaminati, indipendentemente dalla fonte di isolamento, la
totalità delle cellule T CD4+CD25+ isolate espanse è risultata positiva per l‟espressione dei
marcatori CD4 e CD25, con valori medi complessivamente pari al 99.0% ± 0.4% (intervallo:
97-100%).
Tabella 1. Incremento medio di espansione delle cellule T regolatorie CD4 +CD25+ isolate mediante separazione
immunomagnetica dal SVP dei pazienti allotrapiantati e dei donatori sani e dalle unità di SCO e coltivate per 6
giorni in vitro in presenza di anticorpi anti-CD3 (5 g/ml) e anti-CD28 (5 g/ml) e IL-2 (100 U/ml).
Fonte di isolamento
delle cellule T CD4+CD25+
Incremento medio di espansione
delle cellule T CD4+CD25+
Unità di SCO (n=18)
11.0 volte ± 6.9 (intervallo: 1.8-24.0)
SVP dei pazienti allotrapiantati (n=18)
6.7 volte ± 5.4 (intervallo: 1.5-23.3)
SVP dei donatori sani (n=13)
4.4 volte ± 2.4 (intervallo: 1.5-13.6)
206
Quantità di cellule TCD4+CD25+ x 1000
6959.4
8000
7000
Cellule T
CD4+CD25+
pre-espansione
6000
5000
4000
2000
1000
2991.8
2336.9
3000
680.6
631.9
Cellule T
CD4+CD25+
post-espansione
355.8
0
Pz allotrapiantati
Donatori sani
Unità di SCO
Figura 5. Confronto tra le quantità medie pre- e post-espansione di cellule T regolatorie CD4+CD25+ isolate dal
SVP dei pazienti (Pz) allotrapiantati e dei donatori sani e dalle unità di SCO.
La quasi totalità delle cellule T CD4+CD25+ ottenute, è apparsa inoltre caratterizzata dalla
positività per l‟espressione del CD62L, una selectina coinvolta nella migrazione delle cellule
linfoidi a livello dei linfonodi o dei tessuti infiammatori interessati dalla GvHD, con valori
medi di intensità del 76.0% ± 28.0% per le cellule T CD4+CD25+ dei pazienti allotrapiantati,
dell'89.0% ± 1.0% per le cellule T CD4+CD25+ dei donatori sani e del 96.0% ± 9.0% per
quelle isolate dalle unità di SCO. In ciascuna popolazione di cellule T CD4+CD25+ è stata
quindi evidenziata l‟espressione dell‟antigene CTLA-4, direttamente coinvolto nel controllo
dell‟attività regolatoria, con valori medi anche in tal caso paragonabili e complessivamente
pari all‟85.0% ± 15.0% (intervallo: 81-88%). L‟espressione della molecola CTLA-4 è stata
valutata a livello intracitoplasmatico, a causa di una debole presenza in superficie, dovuta
probabilmente a un processo di rapida endocitosi strettamente correlato all‟impiego del
CTLA-4 durante la fase attivatoria delle cellule T regolatorie. Sempre all‟interno del
compartimento intracitoplasmatico, è stata valutata l‟espressione di FOXP3, data la sua
importanza nell‟identificazione della popolazione T CD4+CD25+ ad attività regolatoria. In
tutti i casi studiati, è stata osservata un‟elevata espressione di FOXP3, caratterizzata dalla
presenza di livelli equivalenti nelle cellule T CD4+CD25+ espanse ottenute dai pazienti, dai
donatori adulti e dalle unità di SCO (SVP dei pazienti: 86.0% ± 15.0%; SVP dei donatori:
80.0% ± 11.0%; Unità di SCO: 85.0% ± 14.0%). E‟ stata infine confermata, mediante analisi
207
immunofenotipica, la totale assenza di cellule T CD8+ contaminanti in tutte le popolazioni
studiate, un dato a sostegno dell‟esclusiva espansione, nel nostro sistema sperimentale, di
cellule T a fenotipo CD4+CD25+ [Tabella 2; Figura 6].
Nel complesso, i dati ottenuti hanno rivelato la possibilità di isolare ed espandere cellule T
CD4+CD25+ ad attività regolatoria a partire sia dal SVP di pazienti sottoposti a trapianto e
donatori adulti, che da unità di SCO, senza alcuna peculiarità o differenza di espressione degli
antigeni di superficie e citoplasmatici, in relazione alla fonte di derivazione. Le cellule T
CD4+CD25+ isolate dalle unità di SCO hanno inoltre mostrato una maggiore responsività agli
stimoli proliferativi forniti in vitro, un dato in accordo con le caratteristiche di immaturità
tipiche dei componenti cellulari emopoietici presenti all‟interno di tale fonte.
Tabella 2. Valori percentuali medi di espressione dei marcatori di superficie e intracellulari esaminati per la
caratterizzazione immunofenotipica delle cellule T regolatorie CD4 +CD25+ espanse isolate dal SVP dei pazienti
allotrapiantati e dei donatori sani e dalle unità di SCO.
Marcatori
esaminati
SVP dei pazienti
allotrapiantati
(n=18)
SVP dei donatori
sani (n=13)
Unità di SCO
(n=18)
CD4/CD25
98.5% ± 0.3%
99.0% ± 0.2%
98.0% ± 0.5%
CD62L
76.0% ± 28.0%
89.0% ± 1.0%
96.0% ± 9.0%
CTLA-4
87.0% ± 16.0%
84.0% ± 13.9%
86.0% ± 14.0%
FOXP-3
86.0% ± 15.0%
80.0% ± 11.0%
85.0% ± 14.0%
208
Donatori Sani
Unità di SCO
98%
99%
93%
99%
80%
85%
sCD25
99%
Pz allotrapiantati
sCD4
cCTLA-4
99%
sCD62L
sCD25
89%
cFOXP3
Figura 6. Confronto della caratterizzazione immunofenotipica delle cellule T regolatorie CD4 +CD25+ espanse
isolate dal SVP dei pazienti (Pz) allotrapiantati e dei donatori sani e dalle unità di SCO. A titolo esemplificativo
sono stati riportati i diagrammi relativi a un singolo campione di cellule T regolatorie per ciascuna fonte di
isolamento.
3.2 Capacità soppressoria delle cellule T regolatorie CD4+CD25+ espanse
Per la caratterizzazione delle proprietà funzionali, le cellule Treg CD4+CD25+ espanse, sono
state coltivate in presenza di cellule T effettrici autologhe CD4+CD25-, stimolate da DC
allogeniche caricate con corpi apoptotici leucemici, anch‟essi di natura allogenica. E‟ stata
quindi esaminata, al termine del 6° giorno di coltura linfocitaria mista, la capacità delle cellule
T regolatorie CD4+CD25+ di sopprimere l‟attività proliferativa dei linfociti T effettori
autologhi, attraverso la quantificazione del grado di inibizione dell‟incorporazione di 3HTdR. Le cellule Treg CD4+CD25+ espanse isolate da tutte le fonti in esame, hanno
manifestato, negli esperimenti di co-coltura, un‟efficiente abilità soppressoria nei confronti
dei linfociti T effettori autologhi. L‟inibizione della proliferazione è risultata dose-dipendente,
209
mostrando valori massimi ad un rapporto finale cellule T regolatorie/T effettori di 1:1. In tali
condizioni, la più elevata capacità soppressoria è stata osservata da parte delle cellule Treg
CD4+CD25+ dei donatori sani, a confronto con le altre fonti di derivazione. E‟ stata, infatti,
misurata in tal caso una riduzione media del tasso proliferativo pari a 12.0 ± 9.1 volte
(8416.0Cpm ± 3387.2Cpm in assenza di Treg vs 701.3Cpm ± 387.2Cpm in presenza di Treg).
Al contrario, l‟analisi delle cellule Treg CD4+CD25+ originate dal SVP dei pazienti
allotrapiantati e dalle unità di SCO, ha rivelato una riduzione media del tasso proliferativo
rispettivamente pari a 7.5 ± 5.6 volte (13948.7Cpm ± 833.6Cpm in assenza di Treg vs
1859.8Cpm ± 356.9Cpm in presenza di Treg) e 7.1 ± 4.5 volte (22365.1Cpm ± 2500.9Cpm in
assenza di Treg vs 3150.0Cpm ± 2151.1Cpm in presenza di Treg) [Figura 7]. Risultati
paragonabili sono stati ottenuti sia quando è stata utilizzata la PHA come fattore di
stimolazione negli esperimenti di co-coltura sia quando la popolazione complessiva di cellule
T linfoidi autologhe CD4+CD25- e CD8+CD25- è stata impiegata come popolazione T
effettrice nel test di soppressione della risposta immune alloreattiva (dati non mostrati). Non è
stata invece osservata alcuna soppressione della proliferazione dei linfociti T effettori attivati
quando le cellule Treg CD4+CD25+ espanse sono state co-coltivate in assenza di DC,
impiegate come APC. Lo studio della funzionalità delle cellule Treg CD4+CD25+ isolate dal
SVP dei pazienti allotrapiantati ha, inoltre, rivelato la presenza di una correlazione tra la
capacità soppressoria e l‟occorrenza di GvHD nei suddetti pazienti, al momento
dell‟isolamento di tali cellule. In particolare, le cellule Treg CD4+CD25+ ottenute da pazienti
privi di segni clinici di GvHD (n=5) hanno manifestato una capacità di inibizione della
proliferazione dei linfociti T effettori autologhi CD4+CD25-, significativamente maggiore
rispetto a quella osservata nei campioni di cellule Treg CD4+CD25+ ottenute da pazienti con
manifestazioni di GvHD sia acuta che cronica (n=5) (P=0.04). In media la riduzione del tasso
proliferativo indotta nel primo caso è risultata pari a 10.9 ± 2.7 volte (intervallo: 5.3-21.8;
19722.2Cpm ± 6340.2Cpm in assenza di Treg vs 1809.4Cpm ± 140.7Cpm in presenza di
Treg) mentre, nel secondo caso, a 4.0 ± 0.8 volte (intervallo: 1.5-6.3; 10943.4Cpm ±
4967.8Cpm in assenza di Treg vs 2735.8Cpm ± 1012.6Cpm in presenza di Treg) [Figura 8].
I risultati ottenuti mostrano la presenza di una competenza funzionale, in termini di capacità
di soppressione della risposta allogenica, delle cellule Treg CD4+CD25+ isolate ed espanse exvivo, esplicata indipendentemente dalla fonte di derivazione. Evidenziano, inoltre,
un‟alterazione dell‟efficienza di tali cellule quando isolate da pazienti con esperienza di
GvHD, un dato a sostegno del ruolo primario della scarsa regolazione dei meccanismi
alloreattivi nella comparsa e/o nell‟esacerbazione di questa immunopatologia infiammatoria.
210
Tasso proliferativo (cpm)
30000
22365.1
25000
mDC/Apo +
T4+25-
20000
13948.7
15000
8416
mDC/Apo +
T4+25- + Treg
10000
3150
1858.8
5000
701.3
0
Pz allotrapiantati
Donatori sani
Unità di SCO
Figura 7. Capacità soppressoria media delle cellule T regolatorie CD4 +CD25+ espanse isolate dal SVP dei
pazienti (Pz) allotrapiantati e dei donatori sani e dalle unità di SCO. Le cellule T regolatorie sono state coltivate
per 6 giorni in presenza di cellule T effettrici autologhe CD4 +CD25-, stimolate da DC allogeniche caricate con
corpi apoptotici leucemici.
4
Pz con segni di
GvHD (n=5)
10.9
0
3
6
9
12
Pz privi di segni di
GvHD (n=5)
15
Indice di soppressione
Figura 8. Confronto della capacità soppressoria media delle cellule T regolatorie CD4 +CD25+ espanse isolate
dal SVP di pazienti (Pz) allotrapiantati in assenza o presenza di segni clinici di GvHD.
211
3.3 Produzione di citochine immunosoppressorie da parte delle cellule T regolatorie
CD4+CD25+ espanse
Tra i fattori solubili coinvolti nel controllo dei meccanismi di tolleranza periferica, l‟IL-10
svolge un ruolo determinante attraverso l‟induzione, nelle cellule T linfoidi, di una forma di
anergia antigene-specifica. Tale funzione è resa possibile sia grazie ad un‟azione diretta sui
linfociti T, che si traduce nella diminuzione della produzione di IL-2 e nella riduzione della
proliferazione cellulare, sia grazie ad un‟azione indiretta, identificabile nella diminuzione di
espressione delle molecole costimolatorie e nell‟inibizione della produzione di citochine
proinfiammatorie da parte delle APC professionali. La produzione e secrezione di IL-10
sembra rappresentare uno dei meccanismi effettori contatto-indipendenti mediante i quali le
cellule Treg esplicano la loro funzione soppressoria. Allo scopo di verificare tale possibilità, il
rilascio di IL-10 da parte delle cellule Treg CD4+CD25+ isolate, è stato quantizzato in vitro,
mediante un saggio ELISA, nei surnatanti raccolti al termine dei 6 giorni di coltura in
presenza di anticorpi anti-CD3, anti-CD28 e IL-2.
E‟ stato possibile osservare una produzione quantitativamente simile di IL-10 da parte delle
cellule Treg CD4+CD25+ espanse isolate da tutte le fonti in esame. In particolare, sono stati
misurati livelli medi di IL-10 secreta pari a 471.5pg/ml ± 99.7pg/ml nel caso del SVP dei
pazienti allotrapiantati, a 382.0pg/ml ± 60.8pg/ml nel caso del SVP dei donatori sani e a
326.5pg/ml ± 72.5pg/ml nel caso delle unità di SCO [Figura 9]. Anche relativamente alla
produzione di IL-10 è stata riscontrata una differenza nelle cellule Treg CD4+CD25+ isolate
dal SVP dei pazienti allotrapiantati, in relazione alla presenza o meno di GvHD nei suddetti
pazienti al momento dello studio. Nei surnatanti delle colture di cellule Treg CD4+CD25+
ottenute dai pazienti senza segni di GvHD (n=3), il dosaggio di IL-10 è infatti risultato
lievemente maggiore rispetto alle colture di cellule Treg CD4+CD25+ ottenute dai pazienti con
segni di GvHD (n=3: 1 paziente con GvHD acuta; 1 con GvHD cronica ed 1 con entrambe le
forme) (pazienti senza GvHD vs pazienti con GvHD: 563.0pg/ml ± 102.1pg/ml vs 380.0pg/ml
± 76.0pg/ml) [Figura 10].
Questi risultati confermano la normale funzionalità delle cellule Treg CD4+CD25+ isolate ed
espanse ex-vivo, anche in termini di produzione di citochine immunomodulatorie, a supporto
dell‟esistenza di meccanismi di soppressione contatto-indipendenti Treg-mediati. Inoltre,
mentre la fonte di derivazione non influenza la capacità di secrezione dell‟IL-10, questa, come
accade per la funzione soppressoria della risposta alloreattiva, risente invece degli effetti
negativi che la GvHD esercita sulle componenti del compartimento immunitario.
212
471.6
600
IL-10 (pg/ml)
500
Pz allotrapiantati
382
326.5
400
Donatori sani
300
SCO
200
100
0
Figura 9. Valutazione della produzione di citochine immunosoppressorie da parte delle cellule T regolatorie
CD4+CD25+ espanse isolate dal SVP dei pazienti (Pz) allotrapiantati e dei donatori sani e dalle unità di SCO. Il
rilascio di IL-10 è stato quantizzato in vitro, mediante un saggio ELISA, nei surnatanti raccolti al termine dei 6
giorni di coltura.
Pz con segni di
GvHD (n=3)
380
563
0
100
200
300
400
500
600
Pz privi di segni
di GvHD (n=3)
700
Produzione di IL-10 (pg/ml)
Figura 10. Confronto della produzione di citochine immunosoppressorie da parte delle cellule T regolatorie
CD4+CD25+ espanse isolate dal SVP di pazienti (Pz) allotrapiantati in assenza o presenza di segni clinici di
GvHD.
213
4. DISCUSSIONE
Il TCSE allogenico rappresenta una terapia altamente efficace per la cura delle malattie oncoematologiche, principalmente grazie agli effetti benefici correlati all‟abilità dei componenti T
linfoidi presenti nell‟inoculo, di riconoscere ed eradicare la quota cellulare neoplastica
residua. Tuttavia, queste stesse componenti alloreattive contribuiscono all‟insorgenza della
GvHD, che rappresenta attualmente una delle maggiori cause di morbilità e mortalità posttrapianto. Di conseguenza, la riduzione dell‟incidenza e della severità della GvHD, senza
perdita dell‟effetto GvL, costituisce una delle prerogative più importanti nella sfera
trapiantologica, in quanto consentirebbe da un lato il miglioramento della prognosi del
paziente, dall‟altro l‟aumento del numero di pazienti eleggibili al TCSE, come conseguenza di
una riduzione dell‟importanza del grado di compatibilità HLA donatore/ricevente. La
prevenzione e la terapia della GvHD è stata in passato limitata alla delezione e/o
all‟inattivazione funzionale dei linfociti T alloreattivi mediante l‟impiego di farmaci
immunosoppressivi [166]. Nonostante l‟individuazione di strategie immunosoppressive
innovative, il loro impiego risulta comunque limitato dall‟incremento del rischio di recidiva
tumorale, dall‟aumento di incidenza delle infezioni opportunistiche e dalla significativa
tossicità correlata agli agenti in uso. Lo sfruttamento delle conoscenze relative ai meccanismi
immunoregolatori intrinseci del sistema immune potrebbe evitare alcuni di questi
inconvenienti. Le cellule T regolatorie CD4+CD25+ rappresentano una delle sottopopolazioni
T con un ruolo chiave nel mantenimento della tolleranza al self e nel controllo dell‟omeostasi
del compartimento T linfoide periferico. E‟ stato recentemente documentato il loro
coinvolgimento non solo nell‟induzione dell‟allotolleranza nel contesto di un trapianto solido,
ma anche nella protezione dalla letalità della GvHD in diversi modelli murini di TCSE [167169]. Quando infuse in presenza di elevate concentrazioni di linfociti T effettori CD25-, le
cellule T regolatorie CD4+CD25+ non inducono di per se la GvHD ma ne impediscono la
comparsa. In base alle combinazioni donatore/ricevente, il grado di protezione dalla GvHD
può inoltre variare dalla riduzione della severità alla completa inibizione delle manifestazioni
cliniche e della letalità GvHD-correlate. Allo stesso modo, la deplezione ex vivo delle cellule
T regolatorie CD4+CD25+ dall‟inoculo comporta un‟accelerazione delle cinetiche di comparsa
della GvHD post-TCSE, mentre la loro infusione ritardata fornisce il miglioramento della
suddetta patologia infiammatoria, suggerendo un loro potenziale impiego non solo per la
prevenzione ma anche per la terapia della GvHD, almeno nel caso in cui il decorso non sia
esageratamente aggressivo. Inoltre, nello stesso modello sperimentale, l‟attività di protezione
214
dalla GvHD mediata dalle cellule T CD4+CD25+ non interferisce, almeno apparentemente,
con l‟attecchimento delle CSE e la capacità dei linfociti T effettori CD8 + di riconoscere e
aggredire le cellule tumorali, un dato indicativo della loro incapacità a mediare una completa
paralisi del sistema immune, proprietà indispensabile per preservare la GvL [170]. In base a
tali premesse, le cellule T regolatorie CD4+CD25+, grazie alla loro capacità di induzione del
fenomeno
dell‟allotolleranza,
potrebbero
rappresentare
una
promettente
alternativa
terapeutica di natura immunomodulatoria all‟uso di farmaci immunosoppressivi per il
controllo della GvHD. La bassa percentuale, nell‟uomo, di tali cellule all‟interno della
popolazione di cellule mononucleate, ne ha in realtà inizialmente limitato l‟impiego nella
modulazione della tolleranza post-trapianto, incentivando una serie di ricerche finalizzate al
tentativo di superare i suddetti limiti quantitativi. In tale contesto si inserisce il nostro studio,
volto da un lato ad indagare il ruolo delle cellule Treg nella patogenesi e nel controllo della
GvHD post trapianto, dall‟altro ad identificare un protocollo sperimentale ottimale per
l‟isolamento ed espansione in vitro di cellule T regolatorie CD4+CD25+ funzionali,
potenzialmente utilizzabili nel contesto di un TCSE allogenico per la modulazione della
risposta alloreattiva. E‟ stata in primo luogo esaminata la possibilità di isolare cellule T
CD4+CD25+ mediante l‟impiego di una metodica di separazione magnetica basata sulla
costitutiva espressione del CD25 sulla superficie delle cellule Treg, per l‟arricchimento
cellulare. Il presupposto per l‟impiego di tale procedura è stato quello di sviluppare una
strategia che assicurasse un efficiente e riproducibile sistema di isolamento delle cellule T
CD4+CD25+ umane da prodotti di leucoaferesi o placentari, con il minimo numero di reagenti
e passaggi di selezione. E‟ stato impiegato un protocollo finale contraddistinto da un primo
ciclo di deplezione dei componenti CD4-, indispensabile per evitare la presenza di
contaminanti nel prodotto finale, rappresentati principalmente dai linfociti B e T effettori,
caratteristicamente CD25+ dopo attivazione antigenica, seguito da 2 cicli di selezione positiva
per CD25. Attraverso i suddetti passaggi, è stato possibile isolare cellule T CD4+CD25+
umane sia dal sangue periferico di pazienti allotrapiantati e donatori sani che dal sangue
placentare. Il prodotto finale è risultato caratterizzato da una purezza assoluta in tutti i casi
esaminati; inoltre, la ripetuta selezione positiva per CD25 ha consentito di isolare
preferenzialmente quelle cellule ad intensità elevata piuttosto che intermedia del suddetto
antigene, risultando nella completa eliminazione dei componenti CD25-. Questo è stato
realizzato senza sottoporre i campioni ad un ulteriore lavaggio o step di incubazione in
presenza di biglie magnetiche, importante per evitare sia il prolungamento della procedura che
la perdita di cellule. Le percentuali di resa sono apparse, partendo da campioni
215
quantitativamente equivalenti per la procedura di isolamento, assolutamente paragonabili in
relazione alla fonte di derivazione cellulare.
Indipendentemente dalla tipologia di protocollo sperimentale, il problema principale correlato
alle procedure di isolamento è in tal caso rappresentato dal numero esiguo di cellule T
CD4+CD25+ isolabili, conseguenza della bassa percentuale di tali componenti a livello
ematico. Nel nostro studio, sono state ottenute post-isolamento dalle 0.24x106 alle 24x106
cellule T CD4+CD25+, una quantità del tutto insufficiente non solo per l‟analisi delle loro
proprietà funzionali in vitro ma soprattutto per il loro potenziale impiego nel contesto di una
terapia adottiva finalizzata al controllo della GvHD. Per questo motivo, il passo successivo è
stato quello di identificare un protocollo utile per l‟espansione ex-vivo delle cellule isolate,
tale da garantire quantità clinicamente rilevanti. Nel nostro sistema, è stato calcolato il più
elevato tasso settimanale di espansione dopo esposizione delle cellule T CD4+CD25+ ad
anticorpi anti-CD3 e anti-CD28 immobilizzati, in presenza di alte dosi di IL-2 (100U); al
contrario, tali cellule sono apparse iporesponsive alla stimolazione in assenza di IL-2, o dei
suddetti anticorpi da soli o in combinazione, mostrando invece una normale risposta alla
stimolazione mediata da stimoli TCR-indipendenti, come la PHA. Questi risultati concordano
non solo con quanto noto in letteratura relativamente alle caratteristiche di anergia della
popolazione delle cellule Treg in vitro, ma anche con i dati riportati da studi precedenti o
paralleli [171-172] che dimostrano la mancata responsività di tali cellule ai segnali mediati dal
TCR, indipendentemente dalla presenza di costimolazione, e la forte dipendenza nei confronti
dell‟IL-2 per la loro crescita e sopravvivenza. La possibilità, infatti, di superare la condizione
di iporesponsività attraverso l‟aggiunta di alte dosi di IL-2, evidenzia come tale citochina sia
necessaria a rompere lo stato anergico della popolazione T regolatoria dovuto probabilmente a
un difetto nel segnale del TCR, conseguenza di una diminuzione nella mobilizzazione del
calcio, uno ione indispensabile per l‟attivazione dei fattori di trascrizione che inducono la
risposta cellulare [173]. Le cellule T CD4+CD25+ isolate dal sangue placentare hanno
manifestato, nel nostro sistema sperimentale, la più elevata responsività alla stimolazione in
vitro. Questo dato, giustificato dall‟elevato grado di immaturità delle cellule di cordone
ombelicale, che le rende capaci di rispondere più rapidamente a fattori di natura proliferativa,
diventa rilevante nella scelta della fonte di cellule Treg da impiegare per lo sviluppo di una
terapia immunomodulatoria [174]. La maggiore responsività alla stimolazione garantisce,
infatti, la possibilità di ottenere un numero quantitativamente più elevato di cellule nel minor
tempo possibile, estendendo l‟impiego dei componenti di origine placentare, dopo opportuna
216
manipolazione ex-vivo, anche a pazienti adulti, senza i limiti correlati alla quantità di cellule
da infondere per kilogrammo di peso corporeo.
Una volta espanse, le cellule T CD4+CD25+ policlonali ottenute sono state caratterizzate
fenotipicamente e funzionalmente allo scopo di validare la loro natura regolatoria. L‟analisi
immunofenotipica
ha
confermato
la
presenza,
sia
a
livello
superficiale
che
intracitoplasmatico, di una serie di marcatori caratteristicamente associati al sottotipo T
linfoide delle cellule Treg naturali (nTreg), senza differenze in rapporto alla fonte di
derivazione. Le cellule T regolatorie espanse in vitro sono inoltre risultate assolutamente
paragonabili da un punto di vista fenotipico a quelle appena isolate, evidenziando la mancata
influenza degli agenti stimolanti e della coltura ex-vivo, nella modulazione dell‟espressione
dei marcatori peculiari di questa popolazione. Sono, infatti, apparse uniformemente
caratterizzate dall‟espressione di elevati livelli di CD4, CD25, FOXP-3, CTLA-4 e CD62L,
un dato indicativo non solo della loro potenziale attività regolatoria ma anche della mancata
espansione nel nostro sistema sperimentale di contaminanti T linfoidi di natura effettrice.
Diverse evidenze sperimentali hanno dimostrato l‟importanza di FOXP-3 e del CTLA-4 nella
regolazione dell‟attività delle cellule nTreg. In particolare, il ruolo di FOXP-3 risulta evidente
dall‟esame delle conseguenze della presenza di mutazioni a carico del suo gene codificante in
soggetti affetti dalla sindrome IPEX, caratterizzata dallo sviluppo di fenomeni autoimmuni e
infiammatori cronici, causati appunto dalla mancanza di cellule T regolatorie CD4+CD25+.
Allo stesso modo, è stato dimostrato in vivo che l‟induzione del blocco del CTLA-4 esacerba
la risposta immunologica, attraverso l‟inibizione della soppressione mediata dalle nTreg,
indispensabile per il normale ripristino dell‟omeostasi. Tuttavia, in aggiunta alle molecole
direttamente coinvolte nella mediazione delle funzioni delle nTreg, è risultata di grande
interesse l‟elevata positività per il CD62L osservata nella popolazione espansa. Il CD62L è
una selectina indispensabile alla migrazione dei linfociti verso gli organi linfoidi secondari e i
siti infiammatori, di conseguenza la sua presenza sulla superficie delle cellule T regolatorie
espanse potrebbe garantirne la co-localizzazione con i componenti autoreattivi o alloreattivi in
corso di GvHD. A tal proposito, è stata recentemente osservata, in un modello murino di
allotrapianto, una correlazione tra l‟elevata espressione del CD62L sulla superficie delle
cellule T regolatorie, e una maggiore capacità di interferenza nei confronti dell‟attivazione ed
espansione degli effettori T cellulari coinvolti nel fenomeno della GvHD acuta [175]. Una
spiegazione plausibile di tale fenomeno è rappresentata dalla necessità che il “priming” dei
linfociti T convenzionali così come la loro inibizione avvenga in un sito che richiede
l‟interazione tra il CD62L e i suoi ligandi endoteliali. Le cellule nTreg CD62L+ sarebbero
217
quindi in grado di spostarsi più efficientemente a livello degli organi linfoidi secondari,
giungendo in prossimità dei linfociti alloreattivi e inibendone l‟espansione in misura
maggiore rispetto alla controparte CD62L-, dotata solo in vitro, dove il fenomeno dell‟homing
non è riproducibile, delle stesse capacità regolatorie della sottopopolazione CD62L+. La
presenza di elevati livelli dei marcatori esaminati ci permette quindi di affermare non solo che
le cellule T CD4+CD25+ espanse nel nostro sistema sperimentale presentano un fenotipo ad
attività regolatoria, ma soprattutto che la loro espressione potrebbe associarsi a un‟inibizione
ottimizzata degli effetti della GvHD.
A questo punto, è stata esaminata la capacità delle cellule T CD4+CD25+ espanse di inibire la
risposta dei linfociti effettori autologhi stimolati da DC caricate con alloantigeni, generate in
vitro, prima della co-coltura, da monociti in presenza di GM-CSF e IL-4. I risultati degli
esperimenti di soppressione hanno rivelato una normale funzionalità, identificabile nell‟abilità
di inibizione della proliferazione dei linfociti T autologhi alloreattivi, delle cellule T
CD4+CD25+ derivate da tutte le fonti in esame. Sono stati calcolati valori medi di inibizione
del tasso proliferativo dalle 7 alle 12 volte, con valori massimi superiori alle 20 volte,
traducibili in un‟inibizione media superiore all‟85% in tutti i casi esaminati. Questi dati,
suffragati da una vasta serie di studi paralleli, implicano in primo luogo che la coltura e la
stimolazione delle cellule nTreg in vitro non alterano il potenziale immunomodulatorio di tale
popolazione, preservandone piuttosto l‟attività, e che la soppressione può verificarsi
attraverso una modalità, probabilmente non esclusiva, contatto-dipendente [176-178]. Un dato
interessante è inoltre rappresentato dalla dimostrazione che le cellule nTreg presenti nel
sangue placentare possono diventare, a dispetto della loro immaturità immunologica, dopo
isolamento e opportuna stimolazione in vitro, una potente popolazione ad attività
soppressoria. Tale caratteristica è in accordo con quanto osservato nel lavoro di Wayne R et
al., dove il sangue placentare si dimostra essere una fonte di cellule T regolatorie CD4+CD25+
non solo più facilmente isolabili rispetto al sangue venoso periferico ma anche in grado di
manifestare una soppressione della risposta alloreattiva nel corso di una reazione linfocitaria
mista, superiore al 95% [179].
Diversi aspetti nel modello di soppressione impiegato nel nostro studio, appaiono utili a
offrire alcune riflessioni sulla funzione delle cellule nTreg in vivo. La regolazione negativa
dell‟attività dei linfociti T effettori sembra richiedere, infatti, che la popolazione T regolatoria
mediatrice sia esposta e presumibilmente attivata attraverso il TCR e, in particolare, secondo
una modalità antigene specifica. Questo dato è confermato dall‟impossibilità di osservare un
effetto soppressorio quando le cellule T regolatorie CD4+CD25+ sono coltivate in presenza
218
dei target attivati ma in assenza delle APC, nonostante la precedente stimolazione, ai fini
dell‟espansione, con anticorpi anti-CD3 e anti-CD28. Al contrario, la risposta della stessa cocoltura alla stimolazione effettuata mediante PHA, risulta efficacemente soppressa. Sebbene
nel nostro sistema non sia stato possibile identificare la specificità delle cellule T regolatorie
CD4+CD25+, a causa dell‟impossibilità di separare le richieste per l‟attivazione della
popolazione T effettrice e quella T regolatoria, i dati sono consistenti con un modello secondo
il quale le due popolazioni competono sulla superficie delle APC per l‟antigene e i segnali
costimolatori. Quest‟osservazione è stata anche confermata dal lavoro di Lombardi G et al.
[180] in cui la competizione sulla superficie delle APC rappresenta un meccanismo mediante
il quale i cloni T anergici antigene-specifici inibiscono l‟espansione dei cloni T normali.
L‟importanza delle DC nel controllo dell‟attività soppressoria delle cellule nTreg è stata
anche recentemente dimostrata grazie all‟osservazione che il blocco delle molecole
costimolatorie presenti sulla loro superficie riduce il loro numero e favorisce lo sviluppo di
reazioni autoimmuni in topi diabetici non obesi [181]. Le DC sono inoltre in grado di
produrre IL-2, un fattore indispensabile per la sopravvivenza ed espansione delle cellule
nTreg.
Un dato rilevante osservato nel nostro sistema sperimentale è emerso dalla possibilità di
correlare la capacità funzionale delle cellule T CD4+CD25+ isolate dai pazienti allotrapiantati,
alla presenza di GvHD al momento del loro isolamento. Le cellule T regolatorie derivate da
pazienti con segni di GvHD hanno, infatti, manifestato, in vitro, una scarsa efficienza nella
soppressione della proliferazione dei linfociti T convenzionali alloreattivi. Tale risultato
potrebbe essere giustificato da due differenti osservazioni. La scarsa funzionalità di
immunomodulazione negativa della risposta immune mediata dalle cellule nTreg prodotte nel
ricevente di un allotrapianto in seguito all‟infusione delle CSE, dovuta a un‟inefficiente
ricostituzione ematologica e immunologica post-trapianto o a un difettivo priming in vivo
delle nTreg, potrebbe essere una concausa dei fenomeni associati alla GvHD rilevati nei
pazienti studiati. Quindi, quanto osservato in vitro non sarebbe altro che una conseguenza dei
difetti intrinseci della popolazione T regolatoria presente in vivo. In alternativa, l‟esacerbarsi
della GvHD nel decorso post-TCSE potrebbe aver indotto un‟alterazione o un‟inibizione
irreversibile dei meccanismi di soppressione delle cellule T regolatorie CD4+CD25+ in origine
perfettamente funzionali; in tal caso, il difetto funzionale registrato in vitro sarebbe il risultato
degli effetti che la GvHD esercita in vivo sulle componenti del sistema immunitario.
Naturalmente non è da escludere il verificarsi di una combinazione delle due situazioni
sopradescritte. In ambo i casi, il paziente potrebbe trarre beneficio dall‟infusione di cellule T
219
regolatorie isolate dal donatore delle CSE e manipolate ex-vivo prima dell‟inoculo;
quest‟ultimo passaggio garantirebbe, infatti, il trasferimento adottivo di una popolazione
funzionale potenzialmente in grado di sopperire non solo al deficit di attività ma anche al
numero limitato di precursori antigene-specifici dei componenti T regolatori presenti in vivo,
ipotizzando che l‟espansione policlonale ex-vivo delle cellule nTreg aumenti anche la
frequenza dei componenti alloreattivi. A tal proposito, un recente studio murino ha dimostrato
l‟efficacia delle cellule T CD4+CD25+ espanse policlonalmente ex-vivo, di sopprimere la
GvHD e favorire la sopravvivenza in un contesto completamente allogenico [144].
Per verificare la potenziale esistenza di meccanismi di soppressione contatto-indipendenti
mediati dalle cellule nTreg, è stata valutata, nell‟ultima fase del nostro studio, la capacità delle
cellule T regolatorie CD4+CD25+ isolate ed espanse ex-vivo, di produrre e secernere citochine
immunomodulatorie. La scelta è stata incentrata sull‟IL-10, come conseguenza del suo
coinvolgimento nei meccanismi di protezione dalla letalità della GvHD. Attraverso il nostro
sistema sperimentale è stato possibile confermare, indipendentemente dalla fonte di
derivazione, la normale funzionalità delle cellule T regolatorie CD4+CD25+ isolate ed espanse
ex-vivo, anche in termini di produzione di IL-10. Inoltre, come osservato in precedenza
relativamente alla funzione soppressoria della risposta alloreattiva, anche la secrezione di IL10, è apparsa inversamente correlata alla presenza di GvHD nei pazienti sottoposti a TCSE
allogenico. L‟importanza di quest‟ultimo dato, emerge non solo da un recente studio
preclinico nel quale i livelli ematici di IL-10 in pazienti sottoposti ad allotrapianto, sono
risultati correlati allo sviluppo della GvHD acuta, ma anche dalla recente dimostrazione
dell‟influenza, sul decorso della GvHD, della presenza di polimorfismi a singolo nucleotide
(SNP) a carico dei geni codificanti citochine infiammatorie tra cui l‟IL-10, l‟IL-6 e il TNF-α
[182-183]. Dal momento che il rilascio di IL-10 influenza e al tempo stesso è influenzato
dalla risposta alloreattiva, il trasferimento adottivo di una popolazione di cellule T regolatorie
manipolate ex-vivo e funzionali nella produzione di IL-10, potrebbe quindi favorire una
protezione contatto-indipendente nei confronti della GvHD.
In conclusione, l‟abilità di espandere cellule T regolatorie CD4+CD25+ in vitro presenta
molteplici implicazioni. In primo luogo, la popolazione espansa rappresenta una fonte
cellulare numericamente ampia e manipolabile, potenzialmente indagabile molecolarmente,
biochimicamente e funzionalmente. In tal senso, la possibilità di identificare nuovi marcatori
o meccanismi di soppressione, potrebbe ad esempio migliorare la nostra comprensione delle
basi dell‟immunoregolazione, sia in vitro che in vivo, garantendo un utilizzo razionale delle
potenzialità di tali cellule. In secondo luogo, le cellule T regolatorie CD4+CD25+ espanse
220
potrebbero attualmente essere considerate un approccio terapeutico realizzabile per una
varietà di indicazioni cliniche, come l‟immunoterapia adottiva per il controllo della severità
della GvHD nel contesto dell‟allotrapianto. In particolare, la possibilità di utilizzare il sangue
placentare come fonte di una popolazione espandibile e funzionale di nTreg, data la pronta
disponibilità alla richiesta, offrirebbe numerosi vantaggi per lo sviluppo di una terapia di
soppressione del riconoscimento allogenico, suggerendo una possibile applicazione
indipendentemente dai limiti imposti dalla scarsa percentuale dei suddetti componenti postisolamento.
La scoperta di nuovi agenti in grado di prevenire o modulare le risposte allo- e auto-reattive è
considerata un passaggio obbligato per lo sviluppo di nuovi protocolli tollerogenici. Di
conseguenza, un progetto immunoterapico basato sull‟utilizzo delle cellule nTreg espanse exvivo in condizioni di Good Manufacturing Practice (GMP), potrebbe tradursi in un rilevante
beneficio clinico, riducendo il fabbisogno della terapia convenzionale immunosoppressiva nel
TCSE allogenico.
221
5. REFERENZE
1. Holland AM, Stanley EG. Stems cells and the price of immortality. Stem Cell Research
2009; 2:26-28
2. Bonnet D. Haematopoietic stem cells. J Pathol 2002; 197:430-440
3. Lorenz E, Congdon C, Uphoff D. Modification of acute irradiation injury in mice and
guinea-pigs by bone marrow injections. Radiology 1952; 58:863-877
4. Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogenic hematopoietic stem
cell trasplantation. Ann Rev Immunol 2007; 25:139-158
5. Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA,
Weissman IL. Biology of haematopoietic stem cells and progenitors: Implications for clinical
application. Ann Rev Immunol 2003; 21:759-806
6. Maury S, Mary JY, Rabian C, Schwarzinger M, Toubert A, Scieux C, Carmagnat M,
Esperou H, Ribaud P, Devergie A, Guardiola P, Vexiau P, Charron D, Gluckman E, Socié G.
Prolonged immune deficiency following allogeneic stem cells transplantation: risk factors and
complications in adulte patients. Br J Haematol 2001; 115:630-641
7. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet 2009;
373:1550-1561
8. Yunis EJ, Zuniga J, Romero V. Chimerism and tetragametic chimerism in humans:
implications in autoimmunity, allorecognition and tolerance. Immunol Research 2007;
38:213-236
9. Engelhardt M, Lubbert M, Guo Y. CD34+ or CD34–: which is the more primitive?
Leukemia 2002; 16:1603-1608
10.
Lisbeth A, Welniak, Blazar BR, Murphy WJ. Immunobiology of allogenic
hematopoietic stem cell transplantation. Ann Rev Immunol 2007; 25:139-170
11.
Petersdorf EW. Risk assessment in haematopoietic stem cell trasplantation:
Histocompatibility. Best Pract Res Clin Haematol 2007; 20:155-170
12.
Begovich AB, McClure GR, Suraj VC, Helmuth RC, Fildes N, Bugawan TL, Erlich
HA, Klitz W. Polymorphism, recombination, and linkage disequilibrium within the HLA
class II region. J Immunol 1992; 148:249-258
13.
Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S,
Christiansen F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8,
DR3) with multiple immunopathological diseases. Immunol Rev 1999; 167:257-274
222
14.
Schreuder GM, Hurley CK, Marsh SG, Lau M, Maiers M, Kollman C, Noreen HJ. The
HLA Dictionary 2001: a summary of HLA-A, -B, -C, -DRB1/3/4/5 and -DQB1 alleles and
their association with serologically defined HLA-A, -B, -C, -DR and -DQ antigens, Eur J
Immunogenet 2001; 28:565-596
15.
Nowak J. Role of HLA in hematopoietic SCT. Bone Marrow Transplant 2008; 42 Suppl
2:S71-76
16.
Hahn T, McCarthy PL Jr, Zhang MJ, Wang D, Arora M, Frangoul H, Gale RP, Hale
GA, Horan J, Isola L, Maziarz RT, van Rood JJ, Gupta V, Halter J, Reddy V, Tiberghien P,
Litzow M, Anasetti C, Pavletic S, Ringdén O. Risk factors for acute graft-versus-host disease
after human leukocyte antigen-identical sibling transplants for adults with leukemia. J Clin
Oncol 2008; 26:5728-5734
17.
Kang Y, Chao NJ, Aversa F. Unmanipulated or CD34 selected haplotype mismatched
transplants. Curr Opin Hematol 2008; 15:561-567
18.
Aversa F, Martelli MF. Transplantation of haploidentically mismatched stem cells for
the treatment of malignant diseases. Springer Semin Immunopathol 2004; 26:155-168
19.
Hambach L, Spierings E, Goulmy E. Risk assessment in haematopoietic stem cell
trasplantation: Minor histocompatibility antigens. Best Pract Res Clin Haematol 2007;
20:171-187
20.
Goulmy E, Termijtelen A, Bradley BA, van Rood JJ. Y-antigen killing by T cells of
women is restricted by HLA. Nature 1977; 266:544-545
21.
Dickinson AM. Risk assessment in haematopoietic stem cell trasplantation: Pre-
transplant patient and donor factors: non-HLA genetics. Best Pract Res Clin Haematol 2007;
20:189-207
22.
Reddy P, Arora M, Guimond M, Mackall CL. GVHD: a continuing barrier to the safety
of allogeneic transplantation. Biol Blood Marrow Transplant 2008; 15:162-168
23.
Kolb HJ. Graft-versus-leukemia effects of transplantation and donor lymphocytes.
Blood 2008; 112:4371-4383
24.
Lee SJ, Klein JP, Barrett AJ, Ringden O, Antin JH, Cahn JY, Carabasi MH, Gale RP,
Giralt S, Hale GA, Ilhan O, McCarthy PL, Socie G, Verdonck LF, Weisdorf DJ, Horowitz
MM. Severity of chronic graft-versus-host disease: association with treatment-related
mortality and relapse. Blood 2002; 100:406-414
25.
Zhang C, Todorov I, Zhang Z, Liu Y, Kandeel F, Forman S, Strober S, Zeng D. Donor
CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune
manifestation. Blood 2006; 107:2993-3001
223
26.
Mineo D, Ricordi C, Xu X, Pileggi A, Garcia-Morales R, Khan A, Baidal DA, Han D,
Monroy K, Miller J, Pugliese A, Froud T, Inverardi L, Kenyon NS, Alejandro R. Combined
islet and hematopoietic stem cell allotransplantation: a clinical pilot trial to induce chimerism
and graft tolerance. Am J Transplant 2008; 8:1262-1274
27.
Bonnet D. Hematopoietic stem cells. Birth Def Res (Part C) 2003; 69:219-229
28.
Andrews RE, Singer JW, Bernstein ID. Precursors of colony-forming cells in humans
can be distinguished from colony-forming cells by expression of CD33 and CD34 antigen and
light scatter. J Exp Med 1989; 169:1721-1731
29.
Civin CI, Strauss LC, Browall C. Antigenic analysis of hematopoiesis III. A
hematopoietic progenitor cell surface antigen defined by a monoclonal antibody against KG1a cells. J Immunol 1985; 133:157-164
30.
Bhatia M, Bonnet D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human
hematopoietic cells with SCID-repopulating activity. Nat Med 1998; 4:1038-1044
31.
Vogel W, Scheding S, Kanz L, Brugger W. Clinical Applications of CD34+ Peripheral
Blood Progenitor Cells (PBPC). Stem cells 2000; 18:87-92
32.
Urbano-Ispizua A. Risk assessment in haematopoietic stem cell trasplantation: Stem cell
source. Best Pract Res Clin Haematol 2007; 20:265-280
33.
Perez-Simon JA, Diez-Campelo M, Martino R, Sureda A, Caballero D, Canizo C,
Brunet S, Altes A, Vazquez L, Sierra J, Miguel JF. Impact of CD34+ cell dose on the
outcome of patients undergoing reduced-intensity-conditioning allogeneic peripheral blood
stem cell transplantation. Blood 2003; 102:1108-1113
34.
Todd G, Hang JS, Brown R, DiPersio JF. The effects of G-CSF mobilitation on
lymphocyte subsets, monocytes, NK cells, RBCs, platelets and CD34+/LIN-progenitors in
normal allogeneic PBSC donors. Blood 2001; 88:679a
35.
Hassan HT, Stockschlader M, Schleimer B, Kruger W, Zander AR. Comparison of the
content and subpopulation of CD3 and CD34 positive cells in bone marrow harvests and GCSF-mobilized peripheral blood leukoapheresis products from healthy adult donors. Transpl
Immunol 1996; 4:319-323
36.
Chao NJ, Emerson SG, Weinberg KI. Stem cell transplantation (Cord blood
Transplants). Hematology Am Soc Hematol Educ Program 2004:354-371.
37.
Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of
chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp
Hematol 2002; 30:973-981
224
38.
Larochelle A, Krouse A, Metzger M. Durable engraftment of AMD3100-mobilized
autologous and allogeneic peripheral-blood mononuclear cells in a canine transplantation
model. Blood 2006; 107:3772-3778
39.
Martínez C, Urbano-Ispizua A, Marín P. Efficacy and toxicity of high dose G-CSF
schedule for peripheral blood progenitor cell mobilization in healthy donors. Bone Marrow
Transplant 1999; 24:1273-1278
40.
Brunstein GC, Setubal DC, Wagner JE. Expanding the role of umbilical cord blood
transplantation. Br J Haematol 2007; 137:20-35
41.
Barker JN, Krepski TP, DeFor TE, Davies SM, Wagner JE, Weisdorf DJ. Searching for
unrelated donor hematopoietic stem cells: availability and speed of umbilical cord blood
versus bone marrow. Biol Blood Marrow Transplant 2002; 8:257-260
42.
Ballen KB. New trends in umbilical cord blood transplantation. Blood 2005; 105:3786-
3792
43.
Goldstein G, Toren A, Nagler A. Human Umbilical Cord Blood Biology,
Transplantation and Plasticity. Curr Med Chem 2006; 13:1249-1259
44.
Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, Storb R.
Antileukemic effect of graft-versus-host-disease in human recipients of allogeneic-marrow
grafts. N Engl J Med 1979; 300:1068-1073
45.
Urbano-Ispizua A, Rozman C, Pimentel P, Solano C, de la Rubia J, Brunet S, Pérez-
Oteiza J, Ferrá C, Zuazu J, Caballero D, Carvalhais A, Díez JL, Espigado I, Martínez C,
Campilho F, Sanz MA, Sierra J, García-Conde J, Montserrat E. The number of donor CD3+
cells is the most important factor for graft failure after allogeneic transplantation of CD34+
selected cells from peripheral blood from HLA-identical siblings. Blood 2001; 97:383-387
46.
Brunstein CG, Wagner JE. Umbilical cord blood transplantation and banking. Ann Rev
Med 2006; 57:403-417
47.
de Lima M, Shpall E. Strategies for widening the use of cord blood in hematopoietic
stem cell transplantation. Haematologica 2006; 91:584-587
48.
Brunstein CG, Laughlin MJ. Extending cord blood transplant to adults: dealing with
problems and results overall. Semin Hematol 2010; 47:86-96
49.
Schoemans H, Theunissen K, Maertens J, Boogaerts M, Verfaillie C, Wagner J. Adult
umbilical cord blood transplantation: a comprehensive review. Bone Marrow Transplant
2006; 38:83-93
50.
Tse W, Laughlin MJ. Umbilical Cord Blood Transplantation: A New Alternative
Option. Hematology Am Soc Hematol Educ Program 2005:377-383
225
51.
Mills KC, Gross TG, Varney ML, Heimann DG, Kessinger A. Immunologic phenotype
and function in human bone marrow, blood stem cells and umbilical cord blood. Bone
Marrow Transplant 2001; 18:53-61
52.
Rubinstein P. Why Cord Blood? Human Immunol 2006; 67:398-404
53.
Hofmeister CC, Zhang J, Knight KL, Le P, Stiff PJ. Ex vivo expansion of umbilical
cord blood stem cells for transplantation: growing from the hematopoietic niche. Bone
Marrow Transplant 2007; 39:11-23
54.
Aschan J. Risk assessment in haematopoietic stem cell trasplantation: Conditioning.
Best Pract Res Clin Haematol 2007; 20:295-310
55.
Barnes DW, Corp MJ, Loutit JF. Treatment of murine leukaemia with X rays and
homologous bone marrow; preliminary communication. Br Med J 1956; 2:626-627
56.
Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on
engraftment, graft- versus-tumor, and graft-versus-host responses. Biol Blood Marrow
Transplant 2009; 15:765-776
57.
Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA,
Ringdén O, Rozman C, Speck B. Graft-versus-leukemia reactions after bone marrow
transplantation. Blood 1990; 75:555-562
58.
Kolb HJ, Simoes B, Schmid C. Cellular immunotherapy after allogeneic stem cell
transplantation in hematologic malignancies. Current Opin Oncol 2004; 16:167-173
59.
Geddes M. Storek J. Immune reconstitution following hematopoietic stem-cell
transplantation. Best Pract Res Clin Haematol 2007; 20:329-348
60.
Fry TJ, Mackall CL. Immune reconstitution following hematopoietic progenitor cell
transplantation: challenges for the future. Bone Marrow Transplant 2005; 35 Suppl 1:S53-57
61.
Loren AW, Porter DL. Donor leukocyte infusions after unrelated donor hematopoietic
stem cell transplantation. Curr Opin Oncol 2006; 18:107-114
62.
Kolb HJ, Schmid C, Chen X, Woiciechowski A, Roskrow M, Weber M, Guenther W,
Ledderose G, Schleuning M. Adoptive immunotherapy in chimeras with donor lymphocytes.
Acta Haematol 2003; 110:110-120
63.
Drobyski WR, Majewski D. Donor γδ T lymphocytes promote allogeneic engraftment
across the major histocompatibility barrier in mice. Blood 1997; 89:1100-1109
64.
Graubert TA, Russell JH, Ley TJ. The role of granzyme B in murine models of acute
graft-versus-host disease and graft rejection. Blood 1996; 87:1232-1237
65.
Ferrara JL, Cooke KR, Pan L, Krenger W. The immunopathophysiology of acute graft-
versus-host-disease. Stem Cells 1996; 14:473-489
226
66.
Chakraverty R, Côté D, Buchli J, Cotter P, Hsu R, Zhao G, Sachs T, Pitsillides CM,
Bronson R, Means T, Lin C, Sykes M. An inflammatory checkpoint regulates recruitment of
graft-versus-host reactive T cells to peripheral tissues. J Exp Med 2006; 203:2021-2031
67.
Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ,
Emerson SG. Prevention of graft versus host disease by inactivation of host antigenpresenting cells. Science 1999; 285:412-415
68.
Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik
WD. Donor APCs are required for maximal GVHD but not for GVL. Nat Med 2004; 10:987992
69.
Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-
versus-host disease, each with distinct end-organ targets. J Clin Invest 2000; 105:1289-1298
70.
Duffner U, Lu B, Hildebrandt GC, Teshima T, Williams DL, Reddy P, Ordemann R,
Clouthier SG, Lowler K, Liu C, Gerard C, Cooke KR, Ferrara JL. Role of CXCR3-induced
donor T-cell migration in acute GVHD. Exp Hematol 2003; 31:897-902
71.
Foster AE, Marangolo M, Sartor MM, Alexander SI, Hu M, Bradstock KF, Gottlieb DJ.
Human CD62L-memory T cells are less responsive to alloantigen stimulation than CD62L
naive T cells: potential for adoptive immunotherapy and allodepletion. Blood 2004;
104:2403-2409
72.
Iori AP, Torelli GF, De Propris MS, Milano F, Pupella S, Gozzer M, Mancini F, Milani
ML, Intoppa S, Cerretti R, Lucarelli B, Valle V, Malandruccolo L, Iannella E, Arleo E,
Guarini A, Foà R. B-cell concentration in the apheretic product predicts acute graft-versushost disease and treatment-related mortality of allogeneic peripheral blood stem cell
transplantation. Transplantation 2008; 85:386-390
73.
Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends
Mol Med 2007; 13:108-116
74.
Kolb HJ, Schmidt C, Barret AJ, Schendel DJ. Graft-versus-leukaemia reactions in
allogeneic chimeras. Blood 2004; 103:767-776
75.
Chinen J, Buckley RH. Transplantation immunology: solid organ and bone marrow. J
Allergy Clin Immunol 2010; 125 Suppl 2:S324-335
76.
Mastaglio S, Stanghellini MT, Bordignon C, Bondanza A, Ciceri F, Bonini C. Progress
and prospects: graft-versus-host disease. Gene Ther 2010; May 27 [Epub ahead of print]
77.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S,
Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer
cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295:2097-2100
227
78.
Ruggeri L, Mancusi A, Burchielli E, Capanni M, Carotti A, Aloisi T, Aversa F, Martelli
MF, Velardi A. NK cell alloreactivity and allogeneic hematopoietic stem cell transplantation.
Blood Cells Mol Dis 2008; 40:84-90
79.
Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by
killing donor antigen-presenting cells. J Exp Med 2006; 203:1851-1858
80.
Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell
repertoire reactive to a self peptide: a large fraction of T cell clones escapes clonal deletion.
Immunity 2000; 13:829-840
81.
Aluvihare VR, Betz AG. The role of regulatory T cells in alloantigen tolerance.
Immunol Rev 2006; 212:330-343
82.
Read S, Powrie F. CD4(+) regulatory T cells. Curr Opin Immunol 2001; 13:644-649.
83.
Sakaguchi S, Powrie F. Emerging challenges in regulatory T cell function and biology.
Science 2007; 317:627-630
84.
Rouse BT. Regulatory T cells in health and disease. J Int Med 2007; 262:78-95.
85.
Nielsen J, Lindebo Holm T, Claesson MH. CD4+CD25+ Regulatory T cells: II. Origin,
disease models and clinical aspects. APMIS 2004; 112:642-650
86.
Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu
Rev Immunol 2003; 21:139-176
87.
Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A,
Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist selfpeptide. Nat Immunol 2001; 2:301-306
88.
Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, Sakaguchi S.
Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T
cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol
1999; 162:5317-5326
89.
Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, Migliorati,
Riccardi C. A new member of the tumor necrosis factor/nerve growth factor receptor family
inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci 1997; 94:6216-6221
90.
Sakaguchi S. Policing the regulators. Nat Immunol 2001; 2:283-284
91.
Taams LS, Vukamanovic-Stejic M, Smith J, Dunne PJ, Fletcher JM, Plunkett FJ,
Ebeling SB, Lombardi G, Rustin MH, Bijlsma JWJ, Lafeber FPJG, Salmon M, Akbar AN.
Antigen-specific cell suppression by human CD4+CD25+ regulatory T cells. Eur J Immunol
2002; 32:1621-1630
228
92.
Salomon B, Lenschow J, Rhee L, Ashourian N, Singh B, Sharp A, Bluestone JA.
B7/CD28
costimulation
is
essential
for
the
homeostasis
of
the
CD4+CD25+
immunoregulatory T cells that control autoimmune diabetes. Immunity 2000; 12:431-440
93.
Shevach EM. CD4+CD25+ suppressor T cells: More questions than answers. Nat Rev
Immunol 2002; 2:389-400
94.
Banham AH, Powrie FM, Suri-Payer E. FOXP3+ regulatory T cells: current
controversies and future perspectives. Eur J Immunol 2006; 36:2832-2836
95.
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of
CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat
Immunol 2002; 3:135-142
96.
Munn DH, Sharma MD, Mellor AL. Ligation of B7-1/B7-2 by human CD4+ T cells
triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol 2004; 172:41004110
97.
Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan
catabolism. Nat Rev Immunol 2004; 4:762-774
98.
Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J.
Regulatory T cells selectively express toll-like receptors and are activated by
lipopolysaccharide. J Exp Med 2003; 197:403-411
99.
Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T
cellmediated suppression by dendritic cells. Science 2003; 299:1033-1036
100. Daglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K,
Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. Adenosine generation catalyzed by
CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med
2007; 204:1257-1265
101. Blair PJ, Bultman SJ, Haas JC, Rouse BT, Wilkinson JE, Godfrey VL. CD4+CD8- T
cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J Immunol 1994;
153:3764-3774
102. Kanangat S, Blair P, Reddy R, Daheshia M, Godfrey V, Rouse BT, Wilkinson E.
Disease in the scurfy (sf) mouse is associated with overexpression of cytokine genes. Eur J
Immunol 1996; 26:161-165
103. Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol 2006; 24:209-226
104. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic
autoimmunity throughout the lifespan of mice. Nat Immunol 2007; 8:91-97
229
105. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T,
Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008;
322:271-275
106. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function
of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330-336
107. Ramsdell F. Foxp3 and Natural Regulatory T cells: Key to a Cell Lineage? Immunity
2003; 19:165-168
108. Jiang, H. and Chess, L. Regulation of immune responses by T cells. N Engl J Med
2006; 354:1166-1176
109. Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of
human Tregs. J Clin Invest 2006; 116:1473-1475
110. Tarbell KV, Yamazaki S, Steinman RM. The interaction of dendritic cells with antigenspecific regulatoty T cells that suppress autoimmunità. Sem Immunol 2006; 18:93-102
111. Wilson NS, El-Sukkari D, Villadangos JA. Dendritic cells constitutively present self
antigens in their immature state in vivo and regulate antigen presentation by controlling the
rates of MHC class II synthesis and endocytosis. Blood 2004; 103:2187-2195
112. Fehervari Z, Sakaguchi S. Control of Foxp3+ CD25+ CD4+ regulatory cell activation
and function by dendritic cells. Int Immunol 2004; 16:1769-1778
113. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress
polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 1998;
188:287-296
114. Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for
CD4(+)CD25(+) T regulatory cell development during the neonatal period. J Exp Med 2005;
201:769-777
115. Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon
BL. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state.
J Exp Med 2003; 198:737-746
116. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological cell death triggers
priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med 2003;
198:1527-1537
117. Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, Steinman RM. Direct
expansion of functional CD25+ CD4+ regulatory T cells by antigen processing dendritic cells.
J Exp Med 2003; 198:235-247
230
118. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig, MCSteinman RM.
Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state
leads to antigen presentation on major histocompatibility complex class I products and
peripheral CD8+ T cell tolerance. J Exp Med 2002; 196:1627-1638
119. D‟Ambrosio D, Sinigaglia F, Adorini L. Special attraction for suppressor T cells.
Trends Immunol 2003; 24:122-126
120. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance
maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a
single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995;
155:1151-1164
121. Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev
Immunol 2003; 3:199-210
122. Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) Treg
cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity
2002; 16:183-191
123. Sakaguchi, S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance
and negative control of immune responses. Annu Rev Immunol 2004; 22:531-562
124. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the
transcription factor Foxp3. Science 2003; 299:1057-1061
125. Paust S, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T
cells prevents autoimmune disease. Proc Natl Acad Sci 2004; 101:10398-10403
126. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S,
Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA. Role of LAG-3 in
regulatory T cells. Immunity 2004; 21:503-513
127. Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent
graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol
2002; 168:1080-1086
128. Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, Protschka M, Galle PR,
Neurath MF, Blessing M. Cutting edge: TGF-β signaling is required for the in vivo expansion
and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol 2004;
173:6526-6531
129. Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Ray P, Ray A. Treg-mediated
immunosuppression involves activation of the Notch–HES1 axis by membrane-bound TGF-β.
J Clin Invest 2006; 116:996-1004
231
130. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in
Foxp3-expressing regulatory T cells. Nat Immunol 2005; 6:1142-1151
131. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall
E, Canning C, Soiffer RJ, Frank DA, Ritz J. IL-2 regulates FOXP3 expression in human
CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the
expansion of these cells in vivo. Blood 2006; 108:1571-1579
132. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contactmediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent,
perforin-independent mechanism. J Immunol 2005; 174:1783-1786
133. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol 2005;
6:338-344
134. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF,
Lauber J, Frey O, Przybylski GK, Niesner U, de la Rosa M, Schmidt CA, Bräuer R, Buer J,
Scheffold A, Hamann A. Developmental stage, phenotype, and migration distinguish naiveand effector/memory-like CD4+ regulatory T cells. J Exp Med 2004; 199:303-313
135. Thornton AM. Signal transduction CD4+CD25+ regulatory T cells. CD25 and IL2.
Front Biosci 2006; 11:921-927
136. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J,
Nomura T, Chiba T, Sakaguchi S. Treatment of advanced tumors with agonistic anti-GITR
mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med
2005; 202:885-891
137. Shevach EM, Stephens GL. The GITR–GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol 2006; 6:613-618
138. Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad
el-B, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld
JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF,
Galle PR, Finotto S. The IL-6R α chain controls lung CD4+CD25+ Treg development and
function during allergic airway inflammation in vivo. J Clin Invest 2005; 115:313-325
139. Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol
2005; 6:353-360
140. Kang SM, Tang Q, Bluestone JA. CD4CD25 regulatory T cells in transplantation:
progress, challenger and prospects. Am J Trasplant 2007; 7:1457-1463
141. Joffre O, van Meerwijk JPM. CD4+CD25+ regulatory T lymphocyte in bone marrow
transplantation. Sem Immunol 2006; 18:128-135
232
142. Modigliani Y, Thomas-Vaslin V, Bandeira A, Coltey M, Le Douarin NM, Coutinho A,
Salaün J. Lymphocytes selected in allogeneic thymic epithelium mediate dominant tolerance
toward tissue grafts of the thymic epithelium haplotype. Proc Natl Acad Sci 1995; 92:75557559
143. Nguyen VH, Zeiser R, Negrin RS. Role of Naturally Arising Regulatory T cells in
Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant 2006; 12:995-1009
144. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL.
Recipient-type specific CD4+CD25+ regulatory T cells favour immune reconstitution and
control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest 2003;
112:1688-1696
145. Taylor PA, Noelle RJ, Blazar BR. CD4+CD25+ Immune Regulatory Cells Are
Required for Induction of Tolerance to Alloantigen via Costimulatory Blockade. J Exp Med
2001; 193:1311-1318
146. Johnson BD, Konkol MC, Truitt RL. CD25+ immunoregulatory T-cells of donor origin
suppress alloreactivity after BMT. Biol Blood Marrow Transplant 2002; 8:525-535
147. Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded
CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood
2002; 99:3493-3499
148. Rezvani K, Mielke S, Ahmadzadeh M, Kilical Y, Savani BN, Zeilah J, Keyvanfar K,
Montero A, Hensel N, Kurlander R, Barrett AJ. High donor FOXP3-positive regulatory T-cell
(Treg) content is associated with a low risk of GvHD following HLA-matched allogeneic
SCT. Immunobiology 2006; 108:1291-1297
149. Clark FJ, Gregg R, Piper K, Dunnion D, Freeman L, Griffiths M, Begum G, Mahendra
P, Craddock C, Moss P, Chakraverty R. Chronic graft-versus-host disease is associated with
increased numbers of peripheral blood CD4+CD25high regulatory T cells. Blood 2004;
103:2410-2416
150. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, Bellucci R, Alyea EP,
Antin JH, Soiffer RJ, Ritz J. Reduced frequency of FOXP3 CD4+ CD25+ regolatory T cells
in patients with chronic graft-versus-host disease. Transplantation 2005; 106:2903-2911
151. Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, Negrin RS,
Fathman CG, Strober S. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells
protects from lethal acute GVHD. Blood 2005; 105:2220-2226
233
152. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, Blazar
BR, Serody JS. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T
cells during acute graft-versus-host disease. Blood 2005; 106:3300-3307
153. Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, van Meerwijk
JP. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T
lymphocytes. Nat Med 2008; 14:88-92
154. Wing K, Larsson P, Sandstrom K, Lundin SB, Suri-Payer E, Rudin A.
CD4+CD25+FOXP3+ regulatory T cells from human thymus and cord blood suppress
antigen-specific T cell responses. Immunology 2005; 115:516-525
155. Fisson S, Djelti F, Trenado A, Billiard F, Liblau R, Klatzmann D, Cohen JL, Salomon
BL. Therapeutic potential of antigen specific CD4+CD25+ regulatory T cells selected in vitro
from a polyclonal repertoire. Eur J Immunol 2006; 36:817-827
156. Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, Levine BL, June
CH, Serody JS, Blazar BR. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory
cells are potent inhibitors of GVHD and BM graft rejection. Blood 2004; 104:3804-3812
157. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+)
Immunoregulatory T Cells: New Therapeutics for Graft-Versus-Host Disease. J Exp Med
2002; 196:401-406
158. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type
CD4(+)CD25(+) Regulatory T Cells Suppress Lethal Acute Graft-Versus-Host Disease after
Allogeneic Bone Marrow Transplantation. J Exp Med 2002; 196:389-399
159. Blazar BR, Lees CJ, Martin PJ, Noelle RJ, Kwon B, Murphy W, Taylor PA. Host T cell
resist graft-versus-host disease mediated by donor lymphocytes infusions. J Immunol 2000;
165:4901-4909
160. Wing K, Ekmark A, Karlsson H, Rudin A, Suri-Payer E. Characterization of human
CD25+CD4+ T cells in thymus, cord and adult blood. Immunology 2002; 106:190-199
161. Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, Boussiotis VA. CD4+CD25+
regulatory T-cell lines from human cord blood have functional and molecular properties of Tcell anergy. Blood 2005; 106:3068-3073
162. Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, Levine BL, June CH, Blazar BR,
Porter SB. Cord blood CD4+CD25+-derived T regulatory cell lines express FOXP3 protein
and manifest potent suppressor function. Blood 2005; 105:750-758
234
163. Porter SB, Liu B, Rogosheske J, Levine BL, June CH, Kohl VK, Wagner JE, Miller JS,
Blazar BR. Suppressor function of umbilical cord blood-derived CD4+CD25+ T-regulatory
cells exposed to graft-versus-host disease drugs. Transplantation 2006; 82:23-29
164. Randolph DA, Fathman CG. CD4+CD25+ regulatory T cells and their therapeutic
potential. Annu Rev Med 2006; 57:381-402
165. Maggio R, Peragine N, Calabrese E, De Propris MS, Intoppa S, Della Starza I, Ariola C,
Vitale A, Foà R, Guarini A. Generation of functional dendritic cells (DC) in adult acute
lymphoblastic leukemia: Rationale for a DC-based vaccination program for patients in
complete hematological remission. Leuk Lymphoma 2007; 48:302-310
166. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS.
CD4+CD25+ regulatory T cell preserve graft-versus-tumor activity while inhibiting graftversus-host disease after bone marrow transplantation. Nat Med 2003; 9:1144-1150
167. Vogtenhuber C, Bucher C, Highfill SL, Koch LK, Goren E, Panoskaltsis-Mortari A,
Taylor PA, Farrar MA, Blazar BR. Constitutively active Stat5b in CD4+ T-cells inhibits graftversus-host disease (GVHD) lethality associated with increased regulatory T-cell (Treg)
potency and decreased T effector cell (Teff) responses. Blood 2010; May 4 [Epub ahead of
print]
168. Paczesny S, Choi SW, Ferrara JL. Acute graft-versus-host disease: new treatment
strategies. Curr Opin Hematol 2009; 16:427-436
169. Dutt S, Tseng D, Ermann J, George TI, Liu YP, Davis CR, Fathman CG, Strober S.
Naive and memory T cells induce different types of graft-versus-host disease. J Immunol
2007; 179:6547-6554
170. Hoffmann P, Boeld TJ, Eder R, Albrecht J, Doser K, Piseshka B, Dada A, Niemand C,
Assenmacher M, Orsó E, Andreesen R, Holler E, Edinger M. Isolation of CD4+CD25+
regulatory T cells for clinical trials. Biol Blood Marrow Transplant 2006; 12:267-274
171. Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The
lifestyle of naturally occurring CD4+CD25+Foxp3+ regulatory T cells. Immunol Rev 2006;
212:60-73
172. Giorgini A, Noble A. Blockade of chronic graft-versus-host disease by alloantigeninduced CD4+CD25+Foxp3+ regulatory T cells in nonlymphopenic hosts. J Leukoc Biol
2007; 82:1053-1061
173. Knoechel B, Lohr J, Zhu S, Wong L, Hu D, Ausubel L, Abbas AK. Functional and
Molecular Comparison of Anergic and Regulatory T Lymphocytes. J Immunol 2006;
176:6473-6483
235
174. Escalón MP, Komanduri KV. Cord blood transplantation: evolving strategies to
improve engraftment and immune reconstitution. Curr Opin Oncol 2010; 22:122-129
175. Feng G, Wood k. Regulatory T cells – an emerging role in transplantation. Yonsei Med
J 2004; 45:968-977
176. Singh RK, Varney ML, Leutzinger C, Vose JM, Bierman PJ, Buyukberber S, Ino K,
Loh K, Nichols C, Inwards D, Rifkin R, Talmadge JE. Immune reconstitution after
autologous hematopoietic transplantation with Lin-, CD34+, Thy-1lo selected or intact stem
cell products. Int Immunopharmacol 2007; 7:1033-1043
177. Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T
cells in cancer. Adv Cancer Res 2010; 107:57-117
178. Sakaguchi S, Wing K, Miyara M. Regulatory T cells - a brief history and perspective.
Eur J Immunol 2007; 37 Suppl 1:S116-123
179. Nakamura S, Suzuki M, Sugimoto A, Tsuji-Takayama K, Yamamoto M, Otani T, Inoue
T, Harashima A, Okochi A, Motoda R, Yamasaki F, Orita K, Kibata M. IL-2-independent
generation of FOXP3(+)CD4(+)CD8(+)CD25(+) cytotoxic regulatory T cell lines from
human umbilical cord blood. Exp Hematol 2007; 35:287-296
180. Lombardi G, Sidhu S, Batchelor R, Lechler R. Anergic T cells as suppressor cells in
vitro. Science 1994; 264:1587-1589
181. Yamazaki S, Steinman RM. Dendritic cells as controllers of antigen-specific Foxp3+
regulatory T cells. J Dermatol Sci 2009; 54:69-75
182. Loeffler J, Ok M, Morton OC, Mezger M, Einsele H. Genetic Polymorphisms in the
Cytokine and Chemokine System: Their Possible Importance in Allogeneic Stem Cell
Transplantation. Curr Top Microbiol Immunol 2010; Apr 14 [Epub ahead of print]
183. Resende RG, Correia-Silva JD, Arão TC, Silva TA, Abreu MH, Bittencourt H, Gomez
RS. Investigation of Functional IL-10 Gene Polymorphism and IL-10 Levels in Acute GraftVersus-Host Disease. J Clin Immunol 2010; 30:465-473
236