Le malattie neuromuscolari
Dossier informativo
Edizione primavera 2001
Dr Thierry Kuntzer, Divisione di Neurologia,
CHUV, 1011 Losanna
Editore ASRIM
Associazione della Svizzera Romanda e Italiana contro le Miopatie
Le malattie neuromuscolari
Dossier informativo
Edizione primavera 2001
Sommario
Impressum
Editore
Associazione della Swizzera Romanda
e Italiana contro le Miopatie ASRIM
Chemin de la Traverse 12
Case postale 179
CH-1170 Aubonne
Téléphone 021 808 74 11
Téléfax
021 808 81 11
E-mail asrim @planet.ch
Internet : http : //www.asrim.ch
CCP 10-15136-6
Redazione
Dr Thierry Kuntzer
Concepimento
Peter Blaser, Claude Sansonnens
Fondation Battenberg
2504 Bienne
Veste grafica della pagina di copertina
Calandra Pierre-Marie
graphiste atelier de publicité
2034 Peseux
Stampa
Fondation Battenberg
2504 Bienne
Tiraggio
100 esempi
Avvertenze
2
Definizioni
4
Le distrofie muscolari
17
Distrofia muscolare di Duchenne
20
Distrofia muscolare di Becker
23
Distrofia muscolare facioscapolomerale
25
Distrofie muscolari dei cingoli
28
Distrofia muscolare di Emery-Dreifuss
32
Le miopatie distali
33
Miopatie congenite
39
Distrofie muscolari congenite
42
Miopatie miotoniche
45
Miopatie metaboliche
57
Miopatie infiammatorie
64
Malattie della giunzione neuromuscolare
66
Neuropatie periferiche e polineuropatie
72
Malattie della corna anteriore
82
2
Dr Thierry Kuntzer
Divisione di Neurologia, CHUV, 1011 Losanna
e-mail:[email protected]
Avvertenze:
Questo testo presenta la definizione e una descrizione succinta delle varie
malattie neuromuscolari.
Trattandosi di affezioni rare la cui manifestazione spesso varia da un
paziente all’altro, é indispensabile considerare con cautela le modalità
evolutive che potrebbero non concernere la malattia di vostro interesse.
Le informazioni sulle malattie descrivono un insieme di elementi clinici
e biologici che potrebbero impressionare alcuni pazienti e le loro famiglie e riguardare determinati casi: è doveroso ricordare che ogni caso
dev’essere considerato singolarmente. Solo il medico curante è in grado
di fornire un’informazione personalizzata e mirata sulla problematica
del singolo paziente. Inoltre, la comprensione dei meccanismi patologici
e le scelte terapeutiche variano in funzione del flusso costante di nuove
conoscenze.
Questo testo può quindi fungere da punto di partenza per una discussione tra il paziente, la sua famiglia e il medico incaricato del consulto neuromuscolare, ma non deve essere considerato una referenza a sé stante.
3
Le informazioni di questo scritto provengono dai corsi seguiti presso
l’Istituto di Miologia di Parigi, da sintesi di congressi della World Muscle
Society; da estratti di corsi tenuti presso la Facoltà di Medicina di
Losanna. Un riconoscimento particolare va senza dubbio all’European
Neuromuscolar Center che ha pubblicato alcuni dei criteri diagnostici
riportati in questo testo. Allo stesso modo, alcuni passaggi del sito
dell’Associazione Francese contro le Miopatie (http://www.afmfrance.org) hanno permesso di riassumere in maniera ottimale alcune
patologie complesse.
L’attività della consultazione neuromuscolare del servizio di neurologia
del CHUV di Losanna è stata descritta in un articolo pubblicato in francese nella Revue Médicale de la Suisse Romande (Kuntzer T., LettryTrouillat R., Bogousslavsky J, «Epidémiologie des maladies neuromusculaires chez l’adulte». Rev Med Sui Rom 2000, ottobre; 120(10):725-31).
Per coloro che desiderano approfondire l’argomento e comprendere
meglio la complessità delle malattie neuromuscolari, citiamo le referenze bibliografiche seguenti:
– Delaporte F., Pinell P. – Histoire des myopathies. Edition Payot et Rivages,
Parigi, 1998.
– Serratrice G., Pouget J.,Azulay J.-Ph. – Exercice intolerance and muscle contracture. Springer, Parigi, 1999.
– Emery AEH – Neuromuscolar disorders.Wiley, Chichester, 1998.
- Schapira AHV, Griggs RC. Muscle diseases. Butterworth-Heinemann,
Boston, 1999.
– Lane RJM – Handbook of muscle disease. Marcel Dekker, Basilea, 1996.
Definizioni
4
Le miopatie causano un deficit motorio
Il termine utilizzato di miopatia (pathos designa una malattia e myos il
muscolo) descrive, nell’uso corrente, tutte le malattie dell’unità motoria
(vedere sopra) che si esprimono attraverso una debolezza muscolare.Tale
indebolimento muscolare (o motorio o paresi1) è variabile, e va dalla
semplice asimmetria dei piccoli muscoli facciali senza conseguenze funzionali fino allo sviluppo di un deficit completo delle funzioni motorie.
Nell’ambito delle miopatie, la debolezza muscolare dura nel tempo
dando origine ad un’incapacità o ad un handicap, che può arrivare a
compromettere lo svolgimento delle attività della vita quotidiana o della
vita professionale.
La compromissione può essere progressiva, mantenersi stabile per
numerosi anni o regredire anche a seconda delle possibilità di trattamento farmacologico. Il deficit motorio può causare una serie di problemi
secondari, diversi a seconda del tipo di miopatia, come l’indebolimento
del muscolo cardiaco o la presenza secondaria di una deformazione
osteoarticolare. La gravità della debolezza muscolare e la sua evoluzione
variano da un tipo di miopatia all’altra e questa è la ragione per cui è
importante conoscere bene la natura della malattia, cioè la sua causa,
diversa per ogni tipo di miopatia.
Dal punto di vista della terminologia, la disfunzione (o impairment in
inglese) si riferisce al deficit di funzionamento di un organo o di un
sistema, la disabilità (disability in inglese) ne descrive le ripercussioni
funzionali, mentre l’handicap (stesso termine in inglese) si riferisce alle
1
Paralisi o paresi: deficit totale o parziale dei movimenti volontari in una
regione del corpo, dovuta ad un’affezione muscolare o, più sovente, ad una
lesione nervosa centrale o periferica.
5
sue ripercussioni sociali. La disfunzione muscolare può causare una disabilità motoria che, a sua volta, può condurre ad un handicap.
La debolezza muscolare è conseguenza di una disfunzione dell’unità motoria
La struttura anatomica e funzionale terminale del sistema motorio è
denominata unità motrice (Figura 1): essa comprende il neurone motore (si tratta della cellula nervosa localizzata nel midollo spinale), il suo
prolungamento cellulare (o assone), che si fa strada nella rete dei nervi
periferici, e l’insieme delle fibre muscolari che dipendono dal neurone.
Qualsiasi disfunzione delle singole strutture dell’unità motoria comporta una diminuzione della forza muscolare. Le malattie che ne sono alla
base vengono raggruppate con il termine di malattie neuromuscolari.
Motoneurone A1
Radice nervosa ventrale
Nucleo
motore A
Nucleo
motore B
Figura 1: schema dell’unità
motoria, con sezioni del midollo spinale, dei nervi periferici e
del muscolo.
Muscolo B
Muscolo A
6
La classificazione delle malattie neuromuscolari
È possibile classificare le malattie neuromuscolari in diversi modi, a
seconda per esempio:
• dell’acuità del manifestarsi (ad es. con malattie acute, che sopraggiungono nel giro di pochi giorni, o malattie croniche, che si esprimono
nell’arco di svariate settimane o mesi);
• della severità dei deficit neurologici (per es. malattie benigne, diagnosticabili unicamente da un esaminatore esperto, ma senza conseguenze
funzionali nella vita di tutti i giorni, o malattie gravi, che si ripercuotono sulla vita di tutti i giorni);
Polineuropatie:
assonopatie e
mielinopatie
Miopatie
Malattie delle
corna anteriori
Malattie della
giunzione NM
Figura 2: le malattie dell’unità motoria secondo la ripartizione anatomica;
midollo spinale (all’estrema sinistra), i nervi periferici, la giunzione neuromuscolare e l’insieme delle fibre muscolari che costituiscono il muscolo
(all’estrema destra).
7
• della causa (ad es. miopatia acquisita secondaria ad una malattia infiammatoria o geneticamente determinata, con un deficit della struttura o
della funzione delle cellule del nervo o del muscolo).
Nel 1968, la Federazione Mondiale di Neurologia ha emanato una classificazione basata sull’elemento dell’unità motoria (Figura 2) che viene
colpito: miopatie per le malattie dei muscoli scheletrici, malattie della
giunzione neuromuscolare per quelle che interessano la placca motrice2,
neuropatie periferiche per quelle che interessano il nervo periferico3,
malattie delle corna anteriori per quelle a carico dei neuroni motori.
Inoltre, queste forme sono ulteriormente classificate in congenite4, ereditarie5 o acquisite. Questo tipo di classificazione viene costantemente
aggiornata in funzione delle nuove conoscenze dei meccanismi che stanno all’origine delle varie malattie.
In questa sede verranno prese in considerazione solo le malattie che portano ad un deficit durevole, escludendo quindi alcune malattie neuromuscolari acute, come la sindrome di Guillan-Barré, il botulismo, la poliomielite o altre malattie infettive.
Placca motrice : struttura anatomica e funzionale, corrisponde alla parte terminale del nervo periferico e alla zona eccitabile della fibra muscolare; è proprio in
seno a questa placca che un neurotrasmettitore (una molecola) viene emesso per
escrezione dalla terminazione nervosa: essa da poi luogo all’eccitazione della
fibra muscolare. Il neurotrasmettitore del sistema locomotore è l’acetilcolina.
3 Distale: parte che è più lontana dal centro del corpo, dall’origine nell’ambito
di una struttura anatomica.
4 Congenito: che esiste già dalla nascita.
5 Ereditario: che si trasmette per ereditarietà, di generazione in generazione.
6 Malattia rara: malattia che colpisce meno di una persona su duemila, vedere
EURORDIS all’indirizzo http://www.eurordis.org/index-fr.asp.
2
8
Come si arriva a porre la diagnosi di una malattia neuromuscolare?
Si tratta di un cammino per tappe, comprendente l’anamnesi, l’esame
clinico e eventuali esami di laboratorio. Questi ultimi sono individualizzati e dipendono dal tipo di malattia neuromuscolare sospettata. Le
malattie neuromuscolari sono rare6, ed è quindi utile rivolgersi ad un
centro che svolge abitualmente questo tipo di procedura. I vari esami
clinici e di laboratorio saranno descritti in seguito.
• Mediante l’anamnesi (o intervista) si ricercano nella storia del paziente
indizi atti a definire una pista da seguire nelle ulteriori indagini
• assunzione di medicamenti che potrebbero avere interferito con le
funzioni chimiche del muscolo (medicamenti ipolipidemici ad es.
all’origine di dolori muscolari o mialgie);
7
Mitocondrio: organulo citoplasmatico costante in qualsiasi cellula, di forma,
dimensioni e numero variabili, costituito da una doppia membrana limitante
una matrice amorfa, che svolge un ruolo essenziale in tutti i fenomeni di ossidazione. Immagazzina l’energia cellulare sotto forma di ATP ed è suscettibile
di stoccare determinate sostanze. Questo organulo è presente nelle cellule vegetali e animali, indipendentemente dal ciclo nucleare. I mitocondri costituiscono
un sistema multienzimico organizzato, cioè ogni enzima è localizzato secondo una disposizione funzionale: in questo modo le reazioni a catena possono
svolgersi secondo un ordine appropriato. I mitocondri sono in grado di suddividersi, aumentando il loro numero all’interno della cellula. Essi contengono
degli RNA-r(mitoribosoma), RNA-t e RNA-m ed il loro DNA specifico,
capace di duplicarsi, e la cui struttura è molto simile al DNA dei Batteri. I
mitocondri sono sede di numerose attività biochimiche: respirazione, ossidazione degli acidi grassi, concentrazione di sostanze (ferro, lipidi, proteine). I
mitocondri possono svolgere determinate sintesi proteiche.
9
• associazione con una malattia nota (un diabete può condurre a una
polineuropatia o accompagnare una malattia a carico del muscolo, ad
es. una malattia dei mitocondri7);
• presenza di una predisposizione famigliare, che farebbe pensare ad
una malattia geneticamente determinata;
• modalità di esordio della debolezza muscolare. Per esempio un attacco acuto, associato a febbre fa pensare ad una poliomielite, un’intolleranza al sole ad una dermatomiosite, una deformazione osteoarticolare ad una malattia cronica, eventualmente congenita;
• evoluzione della debolezza muscolare, che può essere variabile,
remittente o progressiva;
• eventuale presenza e tipo di limitazioni nelle attività professionali o
sociali. Difficoltà ad inghiottire, ad articolare (disartria), a vocalizzare
(disfonia), ad alzare le braccia, a correre, salire o scendere le scale,
alzarsi dalla posizione accovacciata, etc. I deficit motori asimmetrici e
acuti allertano rapidamente il paziente mentre i deficit progressivi e
simmetrici possono passare inosservati per un lungo periodo.
• L’esame neurologico, nella sequenza logica dell’esame generale, si
effettua chiedendo al paziente di restare in abbigliamento intimo, per
non tralasciare l’esame delle parti prossimali delle estremità e del tronco.
• L’esame dei nervi cranici, la condizione dei riflessi, l’esame del sistema locomotore, della coordinazione e della sensibilità appartengono
all’esame di routine.
• Successivamente, si procede all’ispezione dei muscoli (diminuzione
del volume muscolare o amiotrofia o, al contrario, aumento del volu-
10
me o ipertrofia) quindi alla palpazione, per individuare un’alterazione della consistenza. Si può osservare un’eventuale deformazione
osteoarticolare o muscolare, una modifica dell’espressione del viso
(abbassamento delle palpebre superiori o ptosi, bocca a V rovesciata,
mascella cadente), una piega anomala dei muscoli pettorali, un’orizzontalizzazione delle clavicole, una deformazione della colonna vertebrale, dei piedi, una ritrazione tendinea (difficoltà a flettere la testa,
a toccarsi lo sterno, a stendere le braccia, a congiungere le mani in
posizione di preghiera, o a mantenere i piedi appoggiati a terra a
causa della retrazione dei tendini di Achille); questi segni indicheranno una malattia neuromuscolare cronica.
• I test funzionali permettono di conoscere le ripercussioni dei deficit
sulle attività della vita quotidiana: studio dell’andatura, uso delle scale
ed altre manovre. Lo studio dell’andatura permette di riconoscere
uno steppaggio, indicativo di un deficit dei muscoli elevatori del
piede, o un tallonamento dovuto al deficit dei flessori dei piedi, o
ancora una retroflessione delle ginocchia, in relazione con un deficit
dei muscoli quadricipiti. Una andatura dondolante risulta da un deficit
dei muscoli glutei: il bacino dondola dall’alto in basso quando il peso
del corpo si sposta da un arto all’altro. Il salire le scale diventa difficile
quando si ha un deficit dei muscoli ileopsoas (o muscoli psoasiliaci),
lo scendere con un deficit dei muscoli quadricipiti (rischio di cedimento improvviso delle ginocchia e caduta). Oltre all’andatura,
occorre osservare la possibilità di gonfiare le guance (come quando si
gonfia un pallone), di sollevare la testa stando distesi sul dorso (una
spinta del capo in avanti contro resistenza dell’esaminatore è sempre
possibile se non vi è debolezza dei muscoli sternocleidomastoidei), di
spingere il braccio sollevato in avanti (uno scollamento delle scapole
- scapola alata - è osservato in presenza di una debolezza dei muscoli
serrato anteriore e grande dorsale), osservare anche come la persona
si alza dopo aver assunto la posizione accovacciata; l’appoggiare le
mani sulle cosce (segno di Gowers) indica la debolezza dei muscoli
11
prossimali degli arti inferiori. Osservare come si alza da una sedia
(non deve aiutarsi con le braccia), o si porta in posizione seduta partendo dal decubito dorsale (sempre possibile in assenza di debolezza
dei muscoli addominali)..
• Lo studio dell’alternanza contrazione-rilassamento (fare il pugno,
battere il piede, aprire e chiudere le palpebre) permette di riconoscere le anomalie della decontrazione muscolare in particolare il ritardo
nel rilassamento muscolare delle miopatie miotoniche (la miotonia
riflette questo fenomeno) o la rigidità muscolare della malattia di
Brody.
• La misurazione della forza muscolare si effettua con l’aiuto di una
scala di forza. Si tratta di una valutazione in parte soggettiva, ma l’impiego della resistenza fornita dalla gravità costituisce un buon metodo
di misura. Un gruppo di ricerca inglese (Medical Research Council)
ha definito la scala denominata MRC utilizzata a livello internazionale: assenza di contrazione (forza MRC 3/4), traccia di contrazione
senza attivazione del movimento (forza MRC 1/5), contrazione che
realizza un movimento ma non contro gravità (forza MRC 2/5), contrazione tale che il segmento esaminato può essere mobilizzato contro gravità (forza MRC 3/5), contrazione buona, il segmento può essere mobilizzato contro gravità e una certa resistenza dell’esaminatore
(forza MRC 4/5), contrazione normale (forza MRC 5/5). La gravità
fornisce quindi per la maggioranza dei muscoli una resistenza adatta
per la valutazione muscolare. Il deficit motorio può avere una localizzazione predominante in determinate malattie: ad es. facio-scapoloomerale significa che il deficit predomina nei muscoli del viso, della
cintura scapolare e prossimali delle braccia; dei cingoli equivale ad un
deficit motorio predominante nei muscoli del cingolo scapolare e
pelvico. Un bilancio preciso della forza muscolare fa emergere elementi che aiutano a porre una diagnosi e permettere di seguire il
decorso della malattia nel tempo.
12
• L’esame dei riflessi si effettua con una breve percussione del tendine
con un martelletto; il muscolo subisce quindi un brusco allungamento che determina una contrazione riflessa. In linea di massima,
sono oggetto di esame i riflessi dei tricipiti, dei bicipiti, degli stiloradiali e dei flessori delle dita per gli arti superiori, dei rotulei e degli
achillei per gli arti inferiori. I riflessi possono essere globalmente
ridotti (ad es. nel caso di neuropatie diffuse), ridotti prossimalmente
(determinate miopatie), o unilateralmente e isolatamente (neuropatia focale o malattia delle corna anteriori). La percussione muscolare
attiva una contrazione (risposta idiomuscolare), che può essere
assente in determinate miopatie ma può anche in altre evidenziare
un’anomalia di distensione del muscolo (descritta sotto il termine di
miotonia).
• Esami del sangue. Sovente è appropriato richiedere un esame ematologico per es. alla ricerca di un’anemia o di una sindrome infiammatoria, l’esame dei parametri renali, epatici e pancreatici per individuare la presenza di un’anomalia metabolica latente. In determinate
situazioni, può risultare opportuno dosare la funzione tiroidea e delle
surrenali, le vitamine, richiedere l’elettroforesi e l’immunoelettroforesi o altri test. Il test biochimico richiesto con maggiore frequenza
nelle malattie neuromuscolari è la misurazione della concentrazione
di creatinfosfochinasi (CPK), l’enzima che permette di trasformare
l’ADP in ATP (sistema fornitore di energia muscolare immediata). La
creatinchinasi è presente in quantità considerevoli nel muscolo e
viene liberato nel sangue quando le fibre muscolari sono lese. Il valore della creatinchinasi dipende dal sesso e dall’età del soggetto, ma
anche dall’esercizio muscolare effettuato nelle ore che precedono l’analisi.Altre cause non neuromuscolari possono elevare la sua concentrazione nel sangue (ad es. assunzione di certi medicamenti, disfunzione tiroidea). La creatinchinasi é elevata in diverse miopatie. Un
tasso normale non esclude alcune miopatie in cui non vi é la distruzione di fibre muscolari.
13
• Quando si sospetta una malattia geneticamente determinata, si procede semplicemente ad un prelievo di sangue per un’analisi genetica
specifica qualora questa sia disponibile. Deve essere eseguita con il
consenso informato8 del paziente.
• Esami neurofisiologici: EMG o ENG9. È l’esplorazione funzionale dei
nervi periferici e dei muscoli che aiuta a localizzare la patologia (nelle
corna anteriori, nei nervi periferici, nella giunzione neuromuscolare o
nei muscoli) e a stimarne la gravità e le caratteristiche. Come altri
esami neurofisiologici (ECG o EEG10), l’EMG si basa sulla registrazione di piccoli campi elettrici generati dalla depolarizzazione/contrazione delle fibre muscolari e delle fibre nervose. Questi potenziali
vengono registrati da sensori (elettrodi) di superficie, applicati sui
muscoli o sui nervi periferici (ENG) o intramuscolari (elettrodi ad
ago sterili) (EMG) in risposta ad una contrazione volontaria o ad uno
stimolo elettrico indotto da stimolatori di superficie. I potenziali registrati vengono amplificati e riprodotti sullo schermo di un computer,
che effettua le misurazioni.
• Esami cardiologici. L’ECG e l’ecografia cardiaca sono esami da prendere in considerazione per escludere una compartecipazione cardiaca.
• Esami radiologici. L’immagine ottenuta dalla risonanza magnetica o
dallo scanner permette di visualizzare le strutture delle estremità e di
valutare il volume, il contorno e la consistenza del muscolo.
• Biopsia del muscolo o del nervo periferico: si tratta di prelevare in ane-
Consenso informato: il consenso viene dato dopo avere preso conoscenza del
significato e degli obiettivi delle analisi.
9 EMG: elettromiogramma, ENMG: elettroneuromiografia.
10 ECG: elettrocardiogramma, EEG: elettroencefalografia.
8
14
stesia locale un piccolo frammento di tessuto (frammento bioptico) per
effettuare un esame istologico e biochimico e, più raramente, un’analisi
genetica. Il sito del prelievo (braccio, coscia o gamba) varia in funzione
del territorio dei deficit, esso viene comunque sempre discusso con il
paziente. La fasciatura e i punti da rimuovere dopo pochi giorni sono i
soli fastidi della biopsia muscolare. Per la biopsia del nervo periferico possono insorgere dei dolori nel sito del prelievo in 1 caso su 10.
Spetta al medico specialista in malattie neuromuscolari realizzare l’integrazione dei dati clinici e degli esami complementari; talvolta, il lavoro è
facilitato dalla conferma data da un test specifico, ma in altre situazioni si
tratta di completare il bilancio effettuando esami più mirati, quali test
fisiologici, biochimici o genetici.
Quando la diagnosi non può essere stabilita con chiarezza, è importante
discuterne nell’ambito di un gruppo di specialisti in neurologia o miologia. La trasmissione delle informazioni e gli scambi di opinioni sono
una realtà frequente nel campo delle malattie neuromuscolari, in cui la
comprensione dei meccanismi patologici e le scelte terapeutiche vengono continuamente aggiornate.
Nelle malattie croniche, sovente ereditarie, il trattamento curativo mirerebbe a ridare alle cellule il potere di sintesi perso a causa di una mutazione genetica. Questo trattamento che modificherebbe un deficit
genetico non è ancora disponibile, ma è l’oggetto di un’intensa ricerca
fondamentale. Nell’attesa di questi sviluppi, é importante che il paziente
sia seguito regolarmente, in modo da offrirgli una somma di trattamenti
palliativi efficaci. A questo proposito si può citare la prevenzione delle
contratture muscolotendinee attraverso massaggi regolari ed un esercizio muscolare periodico, il trattamento delle complicazioni cardiache,
l’efficacia delle operazioni ortopediche nel miglioramento delle deformazioni toraciche e delle estremità, il miglioramento della rigidità e
delle difficoltà nel rilassamento muscolare mediante l’assunzione di
15
medicamenti adeguati, senza dimenticare l’insieme dei molteplici aiuti
nelle attività fisiche di tutti i giorni. L’interesse che il paziente può manifestare alle visite periodiche deriva anche dalla possibilità di condividere
le sue difficoltà familiari o socio-professionali: le associazioni di pazienti
possono costituire un notevole aiuto. La presa a carico globale di una
malattia neuromuscolare cronica si articola così idealmente tra il medico
curante, il gruppo di esperti in malattie neuromuscolari e l’associazione
dei pazienti. Il gruppo di esperti diventa quindi il centro specialistico in
Nervo periferico: neuropatie
Giunzione presinaptica:
sindrome di Lambert-Eaton
Canale
Canali sodici: miotonia,
paralisi periodiche
Giunzione postsinaptica:
miastenia
Pompa del calcio: sindrome di
Brody
Recettore DPH: paralisi
periodiche
Recettore per la rianodina:
ipertermia maligna
Figura 3a: la giunzione neuro-muscolare e i canali ionici della membrana del
nervo, della giunzione neuro-muscolare e della fibra muscolare. Questi
canali permettono l’eccitabilità di queste strutture. La loro disfunzione è
all’origine di un deficit del sistema locomotore, riportato in grassetto nella
leggenda.
16
grado di (i) orientare correttamente la diagnosi, (ii) informare il paziente e la sua famiglia e (iii) fornire un piano terapeutico con l’instaurazione di un rapporto regolare.
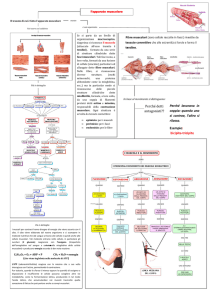
Nelle figure 3a e 3b sono descritte le costituenti terminali del sistema
locomotore.
Cisterne
Tubuli trasversali
Reticolo
sarcoplasmico
Sarcolemma: membrana della
fibra muscolare
Filamenti
Mitocondrio
Disco Z
Filamento sottile
Sarcomero
Disco Z
Tropomiosina
Troponina
Actina
Figura 3b: schema delle strutture delle fibre muscolari e delle proteine contrattili del muscolo.
Le distrofie muscolari
Con questo termine vengono raggruppate le miopatie di origine genetica e la cui evoluzione è progressiva. Le distrofie muscolari costituiscono il gruppo più significativo a livello numerico delle affezioni muscolari primitive, e quello con le conseguenze funzionali o vitali più gravi. Si
capisce quindi che queste sono state le prime ad essere storicamente
riconosciute. La classificazione delle distrofie si basa su criteri clinici e
genetici.
Prima di affrontare le caratteristiche proprie alle principali distrofie, è
possibile elencare un certo numero di caratteri comuni:
Sul piano clinico, la compromissione muscolare si traduce in una diminuzione di forza, eventualmente con una modifica del volume, della
consistenza, dell’estensibilità e della contrattilità del muscolo (amiotrofia
o al contrario ipertrofia). La diminuzione della forza è l’elemento essenziale che interessa numerose regioni muscolari e si ripercuote precocemente nell’esecuzione di attività sportive o abituali; la predominanza dei
deficit sui muscoli prossimali degli arti fa sì che risultino compromessi
inizialmente movimenti come il sollevarsi da terra o da una sedia (segno
di Gowers, Figura 4), salire le scale, arrampicarsi sulla corda, fare gli esercizi sulle sbarre parallele. Diventa inoltre difficile sollevare le braccia e la
statica della colonna vertebrale può trasformarsi in iperlordosi.Allo stesso modo, si modifica l’attitudine delle spalle o dei piedi. Le conseguenze
funzionali variano a seconda della gravità, della topografia e dell’evoluzione della malattia. Il tipo di deficit motorio è specifico per ogni distrofia. Alcune toccano in maniera diffusa l’insieme della muscolatura, altre
ne colpiscono selettivamente determinate aree. La disfunzione dei
muscoli oculo-motori, facciali, faringei è specifica di alcune distrofie.
Manovra di Gowers
• Abitualmente, la diminuzione della forza progredisce di pari passo con
la riduzione del volume del muscolo. L’atrofia può essere difficile da
17
18
individuare, poiché può essere mascherata dall’adiposità sottocutanea.
Alcune aree possono presentare un’ipertrofia vera o, più sovente, falsa,
dovuta allo sviluppo del tessuto connettivo-adiposo intramuscolare che
prende il posto del muscolo (Figura 5). La consistenza può essere modificata, per esempio, palpando determinati muscoli si può osservare una
consistenza e una resistenza insolita. Possono costituirsi delle retrazioni
che limitano il gioco delle articolazioni.
• Poiché la patologia è puramente muscolare, l’atrofia e il deficit muscolare non si associano a disturbi della sensibilità, o ad alterazione dei riflessi.Tuttavia, è possibile che insorgano dei dolori, che sono sovente secondari ad uno squilibrio statico legato al deficit muscolare.
• L’attività elettrofisiologica muscolare (elettromiogramma) è alterata
dalla riduzione del numero di fibre per unità motoria. I parametri della
conduzione nervosa motoria e sensitiva restano invece normali.
Manovra di
Gowers
Figura 4: il sollevamento del tronco viene realizzato mediante l’estensione
delle braccia che appoggiano sugli arti inferiori, segno di un deficit motorio
prossimale degli arti inferiori.
19
• La malattia del muscolo può portare in un aumento del tasso degli enzimi serici di origine muscolare, il più sensibile dei quali è la creatinchinasi.
• L’evoluzione è variabile a seconda del tipo di distrofia. Alcune hanno
un’evoluzione più rapida, mettendo addirittura a rischio la vita dopo 15
o 20 anni. Per contro, altre presentano lunghi periodi di stabilità.
Le principali distrofie muscolari sono: la Distrofia muscolare (DM)
di Duchenne o DMD, la Distrofia muscolare di Becker o DMB, la
Distrofia muscolare facio-scapolo-omerale (DMFSH) e la
Distrofia muscolare dei cingoli.
In questo dossier sono descritte anche distrofie muscolari più rare.
Figura 5: a sinistra, sezione istologica del muscolo normale. A destra,
muscolo distrofico. Si nota una diminuzione del numero di fibre muscolari,
alcune delle quali sono sul punto di degenerare, mentre altre compensano
la carenza con un’ipertrofia. Tra le fibre, si osserva la presenza di un tessuto
fibroso e anomalo di grasso (il diametro medio di una fibra muscolare è di
80 micrometri).
Distrofia muscolare
di Duchenne o DMD
20
La distrofia inizialmente descritta da Duchenne di Boulogne nel 1868 è
frequente e grave.
Dal 1986, le nuove conoscenze nel campo genetico hanno trasformato i
nostri concetti, evidenziando il rapporto tra la distrofia di Duchenne, la
distrofia di Becker (DMB) e le distrofie muscolari che colpiscono la
donna.Trasmessa secondo un modo recessivo legato al cromosoma X, la
DMD si esprime clinicamente in modo severo nei ragazzi e più raramente e in forma più lieve nelle ragazze. Nei ragazzi, l’incidenza è di 1 su
5’000. La prevalenza dell’affezione è stimata globalmente a 3,5 per
100’000.
Il gene responsabile, il più grande gene umano, chiamato DYS e localizzato sul cromosoma X in Xp21, codifica per una proteina chiamata distrofina; quest’ultima permette l’ancoraggio dei filamenti contrattili della fibra
muscolare alla loro membrana. La DMD nel 70% dei casi è legata ad una
delezione all’interno del gene, la cui ricerca è facilitata dalla localizzazione in alcuni punti del gene. Nel 30% dei casi circa, la ricerca è più difficile, trattandosi di mutazioni puntiformi del genoma DYS.
La frequenza della malattia è stabile nonostante che gli uomini malati non
procreino: si ammette quindi un tasso di mutazione spontanea elevata. La
comparsa di una nuova mutazione è stimata ad un terzo dei casi sporadici
di DMD, mentre le donne sono portatrici nei 2/3 restanti. Nella DMD,
esiste un’assenza totale o quasi totale della distrofina rispetto ad un
muscolo normale. L’anomalia genetica porta o ad un’assenza totale della
proteina o alla sintesi di una proteina molto instabile.
La malattia, con un’evoluzione progressiva ed inesorabile, comporta in
maniera schematica due periodi dal punto di vista funzionale: il primo
decennio, durante il quale la marcia è conservata, il secondo decennio,
21
durante il quale la posizione eretta diventa impossibile e che culmina
con il decesso.
Fase preclinica: la malattia si sviluppa nel periodo prenatale, come sembra
indicare il tasso degli enzimi sierici talvolta elevati e la presenza di lesioni istologiche già nelle prime settimane di vita. Nella fase neonatale al
contrario di quanto avviene nelle miopatie congenite, non si osserva
alcuna anomalia clinica. La data esatta di inizio della malattia non può
essere stabilita con precisione. Si osserva sovente con un certo ritardo
nel camminare che diventa possibile solo verso i 15–18 mesi; tutti i
bambini comunque camminano all’età di due anni.A questa età, la marcia viene sovente effettuata sulla punta dei piedi, segno che viene riconosciuto e percepito come anomalo solo nelle famiglie in cui un altro
bambino ha già avuto la malattia.
Fase iniziale (dai 3 ai 6 anni): i disturbi alla marcia diventano evidenti
verso l’età di tre anni e comportano una certa difficoltà a correre e a saltare. Le cadute sono frequenti, salire le scale diventa difficoltoso mentre la
discesa non comporta problemi. L’uso degli arti superiori è normale.
Progressivamente, sopraggiunge una deformazione vertebrale (lordosi
lombare) con accentuazione della tendenza alla marcia in punta di piedi e
retrazione dei tendini d’Achille. Il deficit muscolare si manifesta soprattutto nella difficoltà nel passare da una posizione accovacciata o bassa alla
stazione eretta (segno di Gowers), questo segno implica l’utilizzo delle
mani che si appoggiano sulle ginocchia e sulle cosce per mettersi in piedi.
Un’ipertrofia muscolare si instaura verso i 5‒6 anni, essenzialmente a
livello dei polpacci. I muscoli sono sodi alla palpazione. Fino all’età di sei
anni, il deficit è soprattutto prossimale e i muscoli degli arti inferiori e del
tronco sono nettamente più colpiti rispetto agli arti superiori.
Seconda fase (6‒10 anni); la perdita della deambulazione autonoma si colloca in media verso i 10,5 anni, ma in questo vi sono delle variazioni individuali molto importanti. Una volta persa la facoltà di camminare, il bam-
22
bino trascorre la maggior parte della giornata in poltrona; il deficit
muscolare progredisce, le retrazioni si estendono con il costituirsi di una
cifoscoliosi ad un’evoluzione rapida. Le difficoltà motorie diventano
maggiori agli arti inferiori, il controllo della posizione eretta del capo
diventa precaria. Verso i 15-16 anni subentrano difficoltà respiratorie.
Durante tutta questa evoluzione i muscoli oculomotori non vengono
toccati, mentre i muscoli facciali vengono colpiti in maniera diffusa e tardiva. La deglutizione non è alterata.
Oltre alla compromissione dei muscoli scheletrici, la DMD è caratterizzata da anomalie all’elettrocardiogramma (ECG) costanti dopo i 10-12
anni, ma non esiste un’insufficienza cardiaca manifesta prima dell’età di
10 anni. Le infezioni respiratorie e l’insufficienza respiratoria costituiscono complicazioni frequenti.
Che cosa si può fare? Anche se non esiste ancora un trattamento curativo
per questa malattia, è possibile rallentarne l’evoluzione, grazie alla presa a
carico pluridisciplinare di tipo ortopedico, respiratorio, cardiaco e nutrizionale. Questa presa a carico indispensabile, precoce, regolare e permanente permette al bambino di conservare la sua qualità di vita limitando
le conseguenze della malattia.
L’esistenza di modelli animali (topi, cani, ...) colpiti dalla DMD ha permesso di realizzare nel 1997 i primi tentativi di terapia genica nell’animale. Una prova di tolleranza e di fattibilità della terapia genetica è allo
studio nell’uomo in Francia.Altri approcci terapeutici (terapia cellulare
e terapia farmacologica) rappresentano promettenti vie di ricerca.
Distrofia muscolare
di Becker o DMB
La DMB, descritta nel 1955, è una variante della DMD; é una forma che
progredisce più lentamente, ha prognosi migliore con un’età media di
decesso tra i 40 e i 50 anni e una modalità di trasmissione simile.
L’anomalia genetica, nell’85% dei casi il deficit di una grossa parte del
gene, induce in questo caso una perdita parziale della distrofina della fibra
muscolare. L’incidenza annuale è di 1 su 20 o 30’000 ragazzi. Gli studi
genetici sistematici hanno ampliato lo spettro clinico dei deficit della
distrofina, individuando altri fenotipi come l’innalzamento isolato della
creatinchinasi, forme pseudometaboliche con crampi e mioglobinuria
sotto sforzo o cardiomiopatie dilatative.
L’età di esordio della DMB è assai variabile, dai 2 ai 45 anni. L’età media
d’esordio è di 12 anni. La diagnosi può essere sospettata in una persona che
cammina sulla punta dei piedi, che ha una pseudoipertrofia dei polpacci e
che ha dei precedenti in famiglia di miopatia di Becker.Tuttavia, l’incidenza di casi sporadici non è trascurabile e resta da stabilire attraverso lo studio
sistematico della distrofina. L’incidenza attuale è stimata a 6 su 100’000.
Il deficit iniziale si concentra principalmente sui muscoli del cingolo pelvico. Il cingolo scapolare, viene coinvolto solo più tardi, dopo un lasso di
tempo che varia da 1 a 30 anni. Esiste una selettività per determinati
muscoli: cingolo pelvico, quadricipiti, tibiali anteriori, pettorali, serrati,
bicipiti e muscoli periscapolari. I muscoli distali degli arti vengono risparmiati. Una pseudoipertrofia dei polpacci è costante; più raramente, essa è
presente a livello dei muscoli deltoidi. I riflessi osteotendinei sovente sono
rispettati, ma sono destinati a scomparire con l’evolversi della malattia. I
crampi da sforzo sono frequenti. L’incapacità alla marcia subentra in
media verso i 30 anni. L’età del decesso è in media di 42 anni. Come nel
corso della forma di Duchenne, le cause di morte sono attribuibili ad
infetti polmonari e all’insufficienza cardiaca.
Talvolta si osservano delle miopatie con presentazione clinica analoga alla
DMD e alla DMB nelle ragazze e nelle donne: lo studio della storia fami-
23
24
liare, la diagnosi genetica e lo studio istologico permettono poi di ricondurre queste distrofie muscolari ad un’anomalia del gene DMD. Le
donne portatrici dell’anomalia genetica e che la trasmettono alla generazione successiva hanno un tasso elevato di CK nel 70% dei casi. L’8% di
esse presenta segni clinici di miopatia, anche se in generale, con una gravità media e poco evolutiva. L’esordio è sovente precoce, il deficit, predominante ai cingoli, è talvolta asimmetrico; la pseudoipertrofia muscolare è
classica, in particolare a livello dei polpacci.
Il consiglio genetico si basa, da una parte, sulla storia familiare e sullo studio del tasso di CK e, dall’altra parte, sugli studi genetici familiari, dove
sovente è possibile seguire la presenza del gene patogeno. Lo studio del
liquido amniotico permette di determinare il sesso dell’embrione. In caso
di anomalia genetica identificata, lo studio di una biopsia trofoblastica
permette di stabilire la presenza o l’assenza di questa anomalia in vista di
una eventuale interruzione di gravidanza.
Che cosa si può fare? La presa a carico ortopedica comprende la fisioterapia, che deve essere precoce, permanente e personalizzata, e la necessaria
apparecchiatura ausiliaria. Permette di rallentare l’evoluzione della malattia, conservando per esempio la mobilità delle articolazioni (la perdita
della forza muscolare può comportare delle deformazioni articolari). Un
monitoraggio medico precoce e regolare è indispensabile per depistare la
partecipazione cardiaca e proporre una trattamento medicamentoso. Si
dovrà pure monitorare la capacità respiratoria. Numerose ricerche vengono condotte per compensare la mancanza di distrofina nei muscoli: una
cura medicamentosa è difficile da concepire e i ricercatori pensano piuttosto a trapianti di cellule muscolari capaci di ricostituire il muscolo sano
(terapia cellulare) o a trasferire al muscolo una versione corretta del gene
DYS (terapia genica). La terapia genica è oggetto, nella distrofia muscolare di Duchenne, di uno studio clinico in Francia. I risultati permetteranno
di determinare se potrà essere presa in considerazione anche nella distrofia muscolare di Becker.
Distrofia muscolare
facio-scapolo-omerale
(DM facio-scapolo-omerale di Lamdouzy e Déjérine)
Nel proseguire gli studi di Duchenne sull’atrofia muscolare progressiva,
Landouzy e Dejerine descrissero nel 1884 e 1885 dei casi di miopatia che
colpiscono il viso e i muscoli del tronco e degli arti. Questa fu la prima
miopatia nella quale venne dimostrata l’integrità anatomica del sistema
nervoso centrale e periferico.
Si tratta di un’entità autonoma, la più selettiva delle distrofie muscolari,
che è bilaterale ma mai simmetrica. L’affezione è trasmessa con una
modalità autosomica dominante, colpendo quindi in ugual misura ragazze e ragazzi. La penetranza del gene (= il manifestarsi clinicamente del
difetto genetico) è molto marcata, ma l’espressione fenotipica è variabile
da un soggetto all’altro anche nella stessa famiglia. L’incidenza della miopatia, stimata tra 3 e 10 per milione, é sicuramente sottodiagnosticata visto
il grande numero di forme leggere e poco appariscenti.
In una forma d’intensità media, l’affezione può essere scoperta relativamente presto, fin dai primi anni, quando la malattia è nota nella famiglia.
Segni sono una chiusura incompleta delle palpebre durante il sonno, il
sorriso asimmetrico, la difficoltà a fischiare, e durante la lezioni di ginnastica difficoltà ad arrampicare sulla corda o praticare il salto in alto. La
diagnosi nella maggior parte dei casi viene posta tra i 12 e i 20 anni, di
fronte ad un distacco anomalo delle scapole (scapola alata), la difficoltà a
sollevare le braccia, ad alzarsi dalla posizione seduta o alla comparsa di
uno steppaggio.
All’esame clinico, il deficit dei muscoli del viso risulta più o meno marcato: esso può essere assai discreto con indebolimento degli orbicolari,
asimmetria del sorriso o dell’espressione, labbra superiori rovesciate in
modo anomalo.A livello del collo sovente vengono colpiti i muscoli sternocleidomastoidei. Agli arti superiori, i muscoli più presi di mira sono i
25
26
fissatori della scapola (grande dentato e grande dorsale), i pettorali, i bicipiti e i tricipiti brachiali, i supinatori e gli estensori della mano e delle
dita. La malattia è quindi selettiva, risparmia i deltoidi, i rotatori interni
della spalla e i flessori delle dita. Il deficit è bilaterale, ma sovente resta
asimmetrico, prevalendo per esempio su un bicipite da un lato e i fissatori
della scapola dall’altro. Il sollevare le braccia sulla linea orizzontale mette
solitamente in evidenza questa asimmetria del deficit muscolare.
A livello del tronco, sovente vengono colpiti in maniera asimmetrica
anche i muscoli addominali.Agli arti inferiori, la patologia è pure selettiva, bilaterale e asimmetrica, interessando soprattutto i glutei, gli ileopsoas,
i quadricipiti e gli elevatori del piede. Solitamente, i muscoli del polpaccio vengono risparmiati.
La malattia porta unicamente a un’atrofia, senza retrazioni tendinee, i
muscoli oculomotori e il velo palatino non vengono colpiti. I muscoli
respiratori sono normali così come il cuore e lo sviluppo intellettuale.
L’evoluzione è molto lenta, può apparire stabile per numerosi anni. Il
paziente si adatta alle difficoltà e sovente conduce una vita pressoché normale, a condizione che svolga un’attività professionale compatibile con il
suo handicap. L’aspettativa di vita non è accorciata.
Sono frequenti forme della malattia molto discrete, sovente scoperte nella
famiglia solo nel corso delle indagini genetiche; alcune volte possono
subire un’esacerbazione tardiva.
Sono inoltre possibili forme più gravi, con un’evoluzione più rapida. Il
loro esordio clinico è sovente precoce, prima dei 10 anni di età. La distribuzione dei deficit è simile a quella delle altre forme, ma la diffusione è
molto più marcata. Questo può causare la formazione di una cifoscoliosi
e di una iperlordosi. Questi adolescenti sono costretti in poltrona a partire
dai 20-25 anni.
27
Gli esami complementari sono d’interesse modesto per la diagnosi di
questa miopatia. Il valore della CK è leggermente aumentato.
L’elettromiogramma è anomalo e l’esame dei muscoli alla risonanza
magnetica dimostra chiaramente la selettività della malattia solo per alcuni gruppi muscolari. La biopsia muscolare evidenzia due tipi di lesioni,
entrambe non specifiche: vi possono essere delle necrosi, eventualmente
circondate da elementi infiammatori, e un’atrofia predominante inizialmente sulle fibre di tipo I con un rimaneggiamento dell’architettura
interna delle fibre.Vi è una scomparsa progressiva delle fibre muscolari
rimpiazzate dal tessuto adiposo. La proliferazione connettiva è moderata.
Le informazioni genetiche attualmente disponibili permettono di esprimere una consulenza genetica. La penetranza del gene é importante, pari
al 95% all’età di 25 anni. Pochissimi portatori sono clinicamente normali
in età adulta. Un terzo dei portatori è colpito in forma media, e circa un
quinto dei portatori deve ricorrere all’uso di una sedia a rotelle al di sopra
dei 40 anni. È possibile effettuare una diagnosi prenatale e presintomatica
attraverso l’impiego di una marcatore DNA sul cromosoma 4 (4Q).
Che cosa si può fare? Anche qui è essenziale la fisioterapia regolare e adattata alle condizioni del soggetto. Una stecca per il piede pendulo, un
bastone o una sedia a rotelle possono essere proposte per compensare le
difficoltà alla marcia.
In alcuni casi, viene proposto un intervento chirurgico per fissare le scapole alla gabbia toracica, intervento che permette di sollevare le braccia.
Distrofie muscolari dei cingoli
28
(distrofia muscolare giovanile d’Erb)
Sotto questo nome sono raggruppate delle miopatie aventi il carattere di
distrofia muscolare sul piano clinico, elettrofisiologico e istologico, una
topografia che ha predilezione per i muscoli dei cingoli, degli arti e del
tronco, un inizio abituale nella seconda infanzia o nell’adolescenza e una
trasmissione autosomale recessiva o dominante.
Questo gruppo ha subito numerose vicissitudini. Infatti, il numero di casi
identificati come distrofie a carico dei cingoli e successivamente riconosciuti come amiotrofie spinali (malattie delle corna anteriori), miopatie
congenite o metaboliche o ancora come polimiositi è elevato.
Tuttavia, a questo gruppo appartengono determinate distrofie muscolari
piuttosto rare.
Le distrofie muscolari dei cingoli («Limb Girdle Muscolar Distrophy» o
LGMD) costituiscono un gruppo eterogeneo di malattie muscolari. Si
tratta di malattie genetiche che si distinguono a secondo della loro modalità di trasmissione, autosomica dominante o autosomica recessiva:
– Le forme dominanti comprendono attualmente LGMD1A, la
LGMD1B, la LGMD1C e la LGMD1D.
– Le forme recessive attualmente comprendono la LGMD2A, la
LGMD2B, la LGMD2C e la LGMD2D, la LGMD2E, la LGMD2F, la
LGMD2G e la LGMD2H. Le stesse vengono classificate anche secondo
l’anomalia proteica in causa, quando quest’ultima è nota. Si distinguono
così le calpainopatie (LGMD2A), le sarcoglicanopatie (LGMD2C,
LGMD2D, LGMD2E, LGMD2F) e la disferlinopatia (LGMD2B).
Queste malattie interessano globalmente da 5 a 6 persone su 1 mio. Le
calpainopatie possono essere 10 volte più frequenti in alcune zone del
mondo (isola di Réunion, Paesi Baschi, comunità Amish negli Stati
Uniti).
29
Le distrofie muscolari dei cingoli si manifestano con una perdita progressiva della forza dei muscoli del bacino (cingolo pelvico) e dei muscoli
delle spalle (cingolo scapolare).
Nelle forme recessive, i primi segni compaiono sovente prima dei 20
anni. Nelle forme dominanti, l’esordio è più tardivo.
I primi segni consistono principalmente in difficoltà a correre, salire le
scale e sollevarsi da terra. Le cadute sono più frequenti. I polpacci possono
apparire molto muscolosi (ipertrofia dei polpacci). Una partecipazione
della muscolatura respiratoria non evolutiva è possibile. La partecipazione
cardiaca è rara.
L’evoluzione di queste malattie è assai variabile. Può essere lenta o rapida.
Si accompagna sovente a deformazioni delle colonna vertebrale (iperlordosi) e ad un accorciamento dei tendini di Achille. Camminare diventa
difficoltoso. La prognosi dipende dalla malattia in causa. Certe sarcoglicanopatie presentano un’evoluzione paragonabile a quella della distrofia
muscolare di Duchenne.Altre distrofie dei cingoli sono decisamente più
benigne, con carattere scarsamente o per nulla evolutivo.
La conoscenza della modalità di trasmissione è un elemento essenziale
della diagnosi. Per le forme autosomiche recessive, la biopsia muscolare
permette di studiare con grande precisione la composizione e di ricercare
la proteina in causa. Il prelievo di sangue permette poi di procedere all’analisi molecolare del gene in questione, partendo dal DNA dei globuli
bianchi.
Nelle forme recessive, attualmente si distinguono le seguenti forme.
- le calpainopatie (LGMD2A), dovute all’alterazione o all’assenza di un
enzima specifico del muscolo scheletrico: la calpaina muscolare (gene
CAPN3 sul cromosoma 15);
30
- le sarcogliconopatie (LGMD2C, LGMD2D, LGMD2E, LGMD2F),
dovute ad anomalie in una delle proteine che, con la distrofina, partecipano alla struttura delle fibre muscolari: i sarcoglicani (SG) (geni della
gSG, della aSG, della bSG e della dSG, rispettivamente sui cromosomi
13, 17, 4 e 5). In uno dei modelli animali messi a punto di recente (criceto siriano «Bio 14.6», carente di dSG) si tenta un trasferimento del
gene della dSG per via intra/arteriosa;
- la disferlinopatia (LGMD2B) è dovuta ad una deficienza della disferlina, una proteina localizzata nella membrana della fibra muscolare che si
esprime assai presto nello sviluppo dell’embrione. Questo tesso gene
è nuovamente in causa in una miopatia distale, la miopatia di Myoshi
(gene della disferlina sul cromosoma 2);
- la LGMD2G è dovuta ad una deficienza di teletonina, una proteina del
muscolo (gene della teletonina sul cromosoma 17);
- le cause della LGMD2H non sono ancora note, ma sembra che la
malattia sia legata al cromosoma 9;
Tra le forme dominanti attualmente sono note le anomalie in causa nella
LGMD1B e 1C: si tratta di una deficienza di lamina A/C, proteina della
membrana del nucleo delle cellule (il cui gene si colloca sul cromosoma
1) nel caso della LGMD1B, e di caveolina 3, una componente specifica
del muscolo (il cui gene si trova nel cromosoma 3) per la LGMD1C.
Non si conosce invece ancora la causa della LGMD1A e della LGMD1D,
ma si sa che le stesse sono legate ad anomalie genetiche che interessano
rispettivamente i cromosomi 5 e 7.
Che cosa si può fare? Si raccomanda un monitoraggio annuale per fare un
bilancio del quadro muscolare, ortopedico, cardiaco e respiratorio. Il trattamento dal punto di vista ortopedico comprende l’apparecchiatura ausiliaria, nonché la chinesiterapia, che deve essere precoce, permanente e
31
personalizzata. Questo permette di rallentare l’evoluzione della malattia,
mantenendo ad esempio la mobilità delle articolazioni (la perdita della
forza muscolare può comportare l’insorgere di deformazioni articolari).
Gli ausili tecnici possono anche compensare la perdita di alcune possibilità motorie. La sedia a rotelle permette di riacquistare l’autonomia negli
spostamenti.
Distrofia
Distrofia
muscolare
muscolare
(EMD)
(EMD)
di Emery-Dreifuss
di Emery-Dreifuss
32
Si tratta di una malattia genetica molto rara, che progredisce lentamente, nella maggior parte dei casi a trasmissione recessiva legata al cromosoma X. E’ una distrofia muscolare che inizia durante l’infanzia o l’adolescenza. I criteri d’inclusione si fondano sulla presenza degli elementi
seguenti (non obbligatoriamente presenti nel singolo paziente):
1) Retrazioni precoci del tendine di Achille,del gomito e del rachide che si
sviluppano abitualmente prima della comparsa del deficit muscolare; 2)
Amiotrofia lenta e progressiva, con un deficit predominante a distribuzione scapoloperoneale,a grandi linee simmetrico.In aggiunta,può verificarsi
anche un deficit del cingolo scapolare, del cingolo pelvico e delle cosce. 3)
Disturbi della conduzione cardiaca (bradicardia, extrasistoli, blocco atrioventricolare, blocco di branca destra) e/o segni di cardiomiopatia (cardiomegalia, diminuzione della funzione ventricolare sinistra). 4) Distrofia
muscolare evidente o alterazioni miopatiche alla biopsia muscolare. 5)
Genealogia compatibile con una trasmissione legata al cromosoma X.
Solitamente la malattia esordisce nell’infanzia, raramente dopo i 20
anni e l’evoluzione è lentamente progressiva.
La forma della malattia legata a mutazioni sul cromosoma X è dovuta
all’assenza di emerina, una proteina normalmente situata nella membrana di tutti i nuclei cellulari. Il suo ruolo è ancora sconosciuto. La
forma autosomica dominante legata a mutazioni di uno dei due cromosomi 1 è dovuta alla mancanza della lamina A/C, proteina localizzata sotto la faccia interna della membrana nucleare.
Che cosa si può fare? E’basilare effettuare un monitoraggio della funzione
cardiaca, ogni 6 mesi, alfine di valutare la necessità di impiantare un elettrostimolatore. La chinesiterapia è mirata a limitare le retrazioni. Un intervento chirurgico è talvolta necessario per allungare i tendini di Achille
diventati troppo corti e permettere così di camminare normalmente.
Le miopatie distali
Le miopatie distali costituiscono un gruppo eterogeneo di malattie del
muscolo. Queste affezioni presentano la particolarità di colpire le estremità degli arti (gambe, piedi, avambracci, mani), ed è questa la ragione
per cui esse vengono dette «distali» (temine dal significato opposto a
«prossimali»).
Si distinguono quattro forme principali:
- una forma tardiva dell’adulto che inizia alle mani (miopatia distale di
Welander, descritta in Svezia);
- una forma tardiva dell’adulto che inizia alle gambe (miopatia distale di
Markesbery-Griggs, descritta negli Stati Uniti e successivamente in
Finlandia da Udd);
- una forma precoce dell’adulto che inizia alle gambe e colpisce in particolare i muscoli della parte anteriore della gamba (miopatia distale di
Nonaka, descritta in Giappone).
Si tratta di malattie genetiche che si trasmettono secondo la modalità
autosomica dominante (forme tardive) o autosomica recessiva (forme
precoci).
In Europa, la forma di miopatia distale più frequente è la miopatia di
Myoshi.
Miopatia distale di Welander (Myopathia distalis tarda hereditaria)
Questa affezione, descritta nel 1951 da Welander in 249 casi appartenenti a famiglie svedesi, è chiamata myopathia distalis tarda hereditaria.
Esordisce in media all’età di 50 anni (34-82 anni). In 90 casi su 100, il
deficit inizia alle mani, e secondariamente, vi è un’estensione agli arti
inferiori in 58 casi su 100, ma solamente 7 pazienti su 100 hanno difficoltà a camminare.
Tuttavia, questo gruppo di pazienti non è perfettamente omogeneo,
poiché in alcuni di loro l’esordio della malattia è avvenuto ai 4 arti o agli
arti inferiori. Il deficit predomina generalmente sui muscoli estensori
33
34
delle mani. La biopsia muscolare non evidenzia anomalie specifiche.
L’evoluzione è lenta e l’attesa di vita non è diminuita. La localizzazione
del gene di questa affezione è ancora sconosciuta. Quattro famiglie di
«myopathia distalis tarda hereditaria» sono state testate per il locus 2q31
con esito negativo.
Miopatia distale di Markesbery-Griggs, Udd, di Seze
La miopatia tibiale è una miopatia distale con esordio tardivo e trasmissione autosomica dominante, inizialmente descritta unicamente in
Finlandia. Un’affezione vicina per caratteristiche è stata descritta negli
Stati Uniti da Markesbery nel 1974.
I recenti progressi della ricerca genetica nelle miopatie distali hanno
permesso di localizzare la miopatia tibiale sul cromosoma 2 in posizione
2q31. Si tratta di un progresso maggiore. La miopatia tibiale può ormai
essere considerata un’entità a sé di miopatia distale con esordio tardivo
da differenziare dalle altre miopatia distali. Nonostante il debole handicap legato alla miopatia tibiale nella sua forma eterozigota, come nelle
altre patologie neurologiche ereditarie, la presa a carico delle famiglie e
la consulenza genetica, giocano un ruolo importante. Le prossime tappe
nella comprensione della patogenesi della miopatia tibiale riguardano
l’individuazione del gene e la conferma dell’implicazione della proteina
titina.
Miopatia di Miyoshi
Questa miopatia a trasmissione autosomica recessiva è stata descritta inizialmente in Giappone e successivamente in numerose altre regioni e
sembra essere frequente. I primi segni clinici compaiono tra i 20 e i 35
anni con un deficit distale degli arti inferiori, che comporta difficoltà a
35
correre e cadute. Esiste una predominanza del deficit sui muscoli flessori (soprattutto i polpacci), con notevoli difficoltà alla marcia sulla punta
dei piedi rispetto alla marcia sui talloni, più sovente colpita nelle altre
forme. Frequentemente, vi è l’estensione secondaria ad altri gruppi
muscolari, dapprima agli arti inferiori e poi agli arti superiori, al cingolo
scapolare e, infine, al viso. Gli enzimi muscolari sono generalmente assai
elevati (da 20 a 100 volte la norma), anche nei soggetti portatori asintomatici, contrariamente alle altre forme di miopatie distali. Non esiste
alcun segno patognomonico alla biopsia muscolare.Tuttavia, essa mostra
frequentemente una necrosi delle fibre muscolari associata a macrofagi e
a fibre muscolari degenerate. Il locus del gene della miopatia di Myoshi
è stato individuato sul braccio corto del cromosoma 2 in 2p12-14.
Miopatia di Nonaka
Questa miopatia a trasmissione autosomica recessiva si distingue dalla
miopatia di Myoshi per un esordio generalmente più precoce (15-25
anni), localizzato nei muscoli anteriori delle gambe. I compartimenti
muscolari posteriori vengono colpiti solo in un secondo tempo. Gli
enzimi muscolari risultano normali o leggermente aumentati e la biopsia muscolare mette in evidenza alcuni vacuoli bordati, senza infiltrato
infiammatorio. Il gene di questa miopatia è stato individuato sul braccio
corto del cromosoma 9. Questa entità deve essere raffrontata alla miopatia ereditaria a inclusione, che colpisce i muscoli prossimali e descritta in
Iran. L’affezione è localizzata nello stesso locus e si tratta solo di varianti
alleliche.
Altri attacchi distali
Miopatia con deposito di desmina
Questa affezione, identificata per la prima volta da Fardeau e collabora-
36
tori nel 1978, è anch’essa una miopatia distale autosomica dominante
con esordio tardivo (III-IV decade).Essa differisce dalle precedenti per
la frequenza della partecipazione cardiaca che impone l’impianto rapido
di uno stimolatore cardiaco. Infatti, blocchi della conduzione cardiaca
insorgono precocemente. Il deficit muscolare esordisce agli arti inferiori, ma si estende frequentemente e in tempi rapidi anche agli arti superiori e poi al viso. La partecipazione dei muscoli velo-faringei è classica.
Gli enzimi muscolari possono essere normali o leggermente aumentati.
La biopsia muscolare mostra alcuni vacuoli bordati, associati ad inclusioni granulo-filamentose contenente depositi di desmina, distrofina e
vimentina nel citoplasma. L’evoluzione è generalmente più rapida (da 15
a 20 anni) rispetto ad altri casi di miopatie con esordio tardivo. Il monitoraggio cardiaco è fondamentale, non solo nei soggetti colpiti, ma
anche nei membri asintomatici delle famiglie, poiché in alcuni soggetti
la malattia esordisce con gravi disturbi del ritmo o della conduzione, che
necessitano l’inserimento di uno stimolatore cardiaco.
Caso particolare: miosite e miopatia a inclusioni
Queste entità, difficili da diagnosticare, necessitano di una distinzione.
Da una parte, vi sono le miositi a inclusioni, forme sporadiche il cui quadro clinico iniziale può essere simile a quelli precedenti. L’età di esordio
è frequentemente tardiva; verso i 40-50 anni. L’estensione ai muscoli
quadricipiti è rapida e si può anche osservare un esordio prossimale. In
49 casi su 100 esiste una disfagia, sintomo che si ritrova solo in via eccezionale nelle altre miopatie distali. La diagnosi è confermata dalla biopsia
muscolare con presenza di infiltrati infiammatori localizzati preferibilmente nell’endomisio. Dall’altra parte, vi sono le miopatie a inclusioni,
insieme eterogeneo di affezioni a trasmissione autosomica dominante o
recessiva, che differiscono dal gruppo precedente per l’assenza di infiltrati infiammatori endomisiali. Nelle forme recessive possono essere
distinti due sottogruppi, con o senza interessamento dei muscoli quadricipiti. Il secondo è il più vicino alla miopatia tibiale. Esordisce nell’adulto giovane, colpendo i muscoli estensori per poi estendersi agli altri
37
muscoli della gamba e, successivamente, ai muscoli prossimali, risparmiando i quadricipiti. Non vi è differenza istologica tra questa forma di
miopatia e le altre forme di miopatia a inclusioni.
La distrofia muscolare scapoloperoneale
La distrofia muscolare scapoloperoneale è stata descritta nella tesi di
Brossard nel 1886. La trasmissione è nella maggior parte dei casi autosomica dominante. I primi segni clinici compaiono nell’adolescenza o nel
giovane adulto, più raramente verso i 40 anni, con un deficit dei muscoli peronei, che in un primo tempo può far pensare ad una neuropatia.
L’estensione ai muscoli del cingolo scapolare con un’atrofia dei muscoli deltoidi, trapezi, serrati anteriori e sternocleidomastoidei è caratteristica. La partecipazione dei bicipiti è pure frequente, quella facciale si
ritrova in 70 casi su 100, e quest’incidenza suggerisce una relazione tra
questa patologia e la distrofia facio-scapolo-omerale (DFSH). Nella
maggior parte dei casi, gli enzimi muscolari sono normali o leggermente
aumentati e la biopsia muscolare, senza specificità, permette semplicemente di confermare la patologia muscolare.Tuttavia,un quadro clinico di
miopatia scapoloperonea è stato descritto, in cui la biopsia muscolare evidenzia corpi ialini sottosarcolemmici in circa il 20% delle fibre di tipo I.
L’evoluzione è lenta, con periodi di stabilizzazione. Il ricorso alla sedia a
rotelle è tardivo e incostante. L’attesa di vita non viene ridotta.
Amiotrofia spinale scapoloperoneale (Sindrome di Stark-Kaeser)
Per la prima volta nel 1959 Stark utilizza il termine di amiotrofia spinale
scapoloperoneale per caratterizzare un’affezione a trasmissione autosomica dominante con esordio tra i 30 e 50 anni che presenta due particolarità: i piccoli muscoli del piede sono risparmiati e a livello del cingolo
scapolare, il deficit predomina sull’atrofia. I riflessi osteotendinei sono
aboliti e non vi sono disturbi sensitivi. Le velocità di conduzione nervosa sono normali, l’EMG mostra una attività spontanea con fibrillazioni e
fascicolazioni, soprattutto nei muscoli degli arti inferiori.Tuttavia, può
38
risultare difficoltosa l’interpretazione, in particolare nei muscoli del cingolo scapolare, in presenza di potenziali brevi, di bassa ampiezza e sovente polifasici. La biopsia muscolare mette in evidenza degli aspetti neurogeni con un’atrofia fascicolare e presenza di parti chiare centrali.
L’evoluzione è lenta, scarsamente invalidante con estensione secondaria
ai muscoli del cingolo pelvico e, talvolta, ai muscoli bulbari, il che rende
indiscutibile il termine di amiotrofia spinale. Il locus di questa affezione
è stato individuato sul cromosoma 12 in posizione 12q24.1-q24-31.
Neuropatia scapoloperoneale (Sindrome di Davidenkow)
Nel 1929 Davidenkow ha descritto una forma particolare di amiotrofia
che egli considera una variante della malattia di Charcot-Marie. Questa
entità è estremamente rara e mal documentata. L’evoluzione è lunga,
senza diminuzione dell’attesa di vita. Per alcuni, la sindrome di StarkKaeser e la sindrome di Davidenkow costituiscono un’unica entità, cioè
una forma di amiotrofia spinale che si manifesta con sintomi differenti.
Miopatie congenite
Queste miopatie hanno in comune una rivelazione precoce, alla nascita
o nella prima infanzia, un’evolutività clinica variabile e lesioni istopatologiche con anomalie morfologiche particolari. Per queste miopatie si
tratta di un blocco dello sviluppo della fibra muscolare.
Miopatia congenita a «central core»
La miopatia congenita a «central core» si manifesta in modo differente a
seconda dell’età in cui la stessa insorge: nel bambino si traduce con un’ipotonia generalizzata (bambino flaccido), un ritardo dello sviluppo
motorio e deformazioni ortopediche (lussazione dell’anca, torace o
piedi deformati). Nell’adulto si manifesta attraverso un indebolimento
muscolare diffuso e talvolta, anche con delle deformazioni ortopediche.
Essa può essere scoperta in occasione di una crisi di ipertermia maligna
durante un’anestesia.
La miopatia congenita a «central core», in linea di massimo è benigna e
stabile: questa affezione è compatibile con una scolarità e una vita sociale normali. Il rischio maggiore concerne l’anestesia: le persone affette da
miopatia congenita a «central core» presentano spesso una predisposizione all’ipertermia maligna, contrattura muscolare generalizzata associata ad un innalzamento della temperatura corporea che si manifesta in
occasione di un’anestesia generale con determinati prodotti.
La miopatia congenita a «central core» è dovuta ad un’anomalia genetica situata sul cromosoma 19. Il gene in questione è l’RYR1. Esso codifica il recettore della rianodina, un canale del calcio che permette di
fare transitare il calcio indispensabile al muscolo scheletrico attraverso
la membrana della cellula muscolare. Questo stesso RYR1 è uno dei
geni in causa nella suscettibilità dell’ipertermia maligna. Tuttavia, esistono altri casi di miopatia congenita a «centrale core» che non sono
dovuti ad un’anomalia di questo gene e questo rende difficile la diagnosi genetica.
39
40
Miopatia a bastoncelli (nemaline myopathy)
Anche la miopatia nemalinica si manifesta in modo differente a seconda
dell’età di esordio della malattia. Nelle forme più gravi si manifesta con
un’ipotonia generalizzata (bambino flaccido), retrazioni (accorciamento
del tendine-che attacca il muscolo allo scheletro-comportando una
limitazione dei movimenti), importanti deformazioni articolari, disturbi
della deglutizione e della funzionalità respiratoria.
Nella forma tardiva la miopatia a bastoncelli può presentarsi con difficoltà nell’attività sportiva e deformazioni dei piedi e della colonna vertebrale (cifoscoliosi).
A livello generale, più precoce è l’esordio, più la malattia si aggraverà nel
corso della sua evoluzione. Nei soggetti più piccoli esistono delle forme
assai gravi con insufficienza dei muscoli respiratori. Nei bambini più
grandi e negli adulti si manifesta in genere con effetti meno invalidanti.
A tutt’oggi esistono almeno 3 geni responsabili della miopatia nemalinica:
- alcune forme sono dovute ad un’anomalia genetica situata sul cromosoma 2, il gene in causa è quello della nebulina;
- altre forme sono dovute ad un’anomalia genetica situata sul cromosoma 1, il gene in causa codifica la tropomiosina 3 (TPM-3);
- altre forme ancora sono dovute ad un’anomalia genetica situata sul
cromosoma 1, sul gene dell’alfa-actina (ACTA1).
Questa molteplicità dei geni in questione rende difficile la consulenza
genetica per questa malattia.
Miopatia congenita miotubulare
La miopatia miotubulare è una malattia presente fin dalla nascita (congenita), distinta dalla miopatia centro-nucleare con la quale essa è stata a
lungo confusa. È una malattia genetica ereditaria classicamente dovuta
ad un’anomalia del cromosoma X. Si trasmette generalmente secondo
modalità recessiva legata al cromosoma X.Tuttavia, esistono delle forme
che non sono correlate al cromosoma X.
41
Prima della nascita si osserva sovente una riduzione dei movimenti fetali,
e la presenza di una quantità molto importante di liquido amniotico
(hydramnios). Al momento della nascita il bambino è di norma flaccido
(ipotonia neonatale) e presenta difficoltà respiratorie. Spesso questi
disturbi sono associati a disturbi della deglutizione, paralisi dei muscoli del
viso (diplegia facciale), palpebre cadenti (ptosi), paralisi dei muscoli degli
occhi (oftalmoplegia), deformazioni del torace e dei piedi.
L’evoluzione è nella maggior parte dei casi fatale in tempi brevi. Se il
bambino con una rianimazione respiratoria supera lo scoglio neonatale, egli riuscirà con molto ritardo ad assumere la posizione seduta, la
posizione eretta e vi saranno ritardi anche nell’imparare a camminare. Il
soggetto presenta una debolezza muscolare notevolmente marcata e
una paresi dei muscoli degli occhi (oftalmoplegia). Lo sviluppo intellettivo è normale.
La miopatia congenita miotubulare è classicamente legata ad un’anomalia del gene MTM1, che codifica un enzima, la miotubularina.
Questo enzima, carente nelle persone colpite, potrebbe condurre all’arresto precoce della maturazione della fibra muscolare, spiegando così la
persistenza dei miotubi. L’indentificazione del gene MTM1 permette
in questo caso di precisare la diagnosi e di proporre una consulenza
genetica ai genitori. Esistono poi altre forme che non sono correlate al
cromosoma X.
Che cosa si può fare? Le forme neonatali gravi impongono una presa a
carico con rianimazione immediata e ventilazione assistita nei casi più
gravi.
Nelle forme ad apparizione più tardiva, la fisioterapia permette di prevenire la comparsa di retrazioni, mantenendo i muscoli nelle migliori
condizioni possibili. I pazienti devono rimanere il più possibile attivi e
sorvegliare il peso, per limitare il carico imposto ai muscoli.
Distrofie muscolari
congenite (DMC)
42
Il termine di distrofia muscolare congenita è stato largamente utilizzato
per un gruppo di lattanti che presentavano, alla nascita o nei primi mesi
di vita un deficit muscolare associato ad un processo distrofico alla biopsia muscolare. Sovente, nel corso dell’esame clinico è stata osservata
un’ipotonia, ma altri casi si presentano con una artrogriposi o con retrazioni a carico di svariate articolazioni. Questa affezione ha la tendenza a
restare relativamente stabile, ma alcuni casi possono essere lentamente
evolutivi.Alcuni pazienti, tuttavia, possono migliorare sul piano funzionale, fare dei progressi sul piano motorio e acquisire la capacità di camminare. Possono esserci dei disturbi respiratori e delle difficoltà di deglutizione all’esordio, e la paresi del diaframma può comportare un’insufficienza respiratoria nell’infanzia avanzata o nell’adolescenza.
Negli ultimi anni, sono state osservate diverse sindromi di distrofia
muscolare congenita associate ad una partecipazione del sistema nervoso centrale.Attualmente si distinguono i fenotipi clinici seguenti:
DMC «pura». I segni principali sono deficit muscolare con ipotonia o
artrogriposi, alterazioni istologiche di tipo distrofico, con proliferazione
frequente del tessuto adiposo o del tessuto connettivo, ma senza fenomeni importanti di necrosi-rigenerazione. CPK normali o moderatamente in aumento. In linea di massima, nessun interessamento sul piano
intellettivo. L’immagine cerebrale (mediante scanner o RM cerebrale)
può mettere in evidenza o un’immagine normale o delle anomalie della
sostanza bianca.
DMC di tipo Fukuyama. Oltre al deficit muscolare e all’aspetto distrofico alla biopsia muscolare, questa forma di distrofia muscolare congenita
è caratterizzata da un ritardo mentale sovente importante, valori di CPK
costantemente elevati, alterazione delle strutture cerebrali all’autopsia o
agli esami neuroradiologici; vi è incostantemente una partecipazione
43
oculare, l’associazione frequente con un’epilessia (nel 40% dei casi
circa). La sopravvivenza supera abitualmente l’infanzia e talvolta, l’adolescenza.
La sindrome Muscolo-Occhio-Cervello (Muscle-Eye-Brain Disease,
MEB). Oltre al deficit muscolare e all’aspetto distrofico del muscolo vi
è una partecipazione degli occhi e del sistema nervoso centrale. Si osserva un ritardo mentale grave nella maggior parte dei casi. La miopia è l’anomalia oculare più costante, ma si osserva talvolta l’insorgenza di strabismo, un glaucoma, un’opacità della cornea, o un’atrofia nel nervo ottico e della retina. Frequentemente si registra anche l’associazione con
un’epilessia e l’EEG risulta sempre anomalo dopo l’età di un anno. La
maggioranza dei casi presenta un’idrocefalia. I valori della CPK possono
essere normali durante il primo anno di età, ma successivamente sono
sempre elevati.
La sindrome Walker-Warburg. Questa sindrome è caratterizzata da alterazioni della struttura del cervello, da un ritardo mentale associato al
deficit motorio e dall’aspetto distrofico del muscolo. Le anomalie
costantemente ritrovate a livello del sistema nervoso centrale sono una
lissencefalia di tipo II con anomalie varie delle circonvoluzioni cerebrali, una corteccia di spessore anomalo. Possono esserci altre anomalie
strutturali del sistema nervoso centrale.Anomalie oculari sono anch’esse
frequenti, ma sono meno gravi rispetto a quelle della sindrome muscolo-occhio-cervello.
Nel 1992, il gene di Fukuyama è stato localizzato sul cromosoma 9q e
nel 40% circa dei casi di DM classica è stato scoperto un deficit della
catena alfa-2 della laminina, la merosina. Studi complementari hanno
mostrato che si trattava proprio di un deficit primario e che questi casi
erano legati al locus del gene corrispondente (LAMA2) sul cromosoma
6q. È risultato chiaramente che si potevano distinguere le forme merosina-negative e merosina-positive, che le forme con carenza di merosina
44
avevano un fenotipo molto più grave con deambulazione impossibile, al
contrario delle forme merosina-positive che nella maggior parte dei casi
permettono la deambulazione. Tutti i casi con anomalie a carico della
sostanza bianca nelle sequenze T2 della risonanza magnetica cerebrale
presentano una carenza di merosina.
In un numero ristretto di casi di DMC esiste un deficit di alfa-actina.
Non è noto se si tratti di un deficit primario o secondario e quale proporzione di DMC merosina-positiva è interessata.
Per quanto concerne la sindrome di Walker-Warburg e la sindrome
MEB, non è ancora stato possibile individuarne una localizzazione genica, né di sapere se si tratti di entità separate oppure no. Questo principalmente a causa della rarità di queste sindromi e per la mancanza di famiglie informative. I dati attualmente disponibili hanno permesso di escludere una correlazione con il cromosoma 6q o 9q, in modo tale che probabilmente si tratta di forme distinte rispetto a quella di Fukuyama e alle
DMD classiche.
Miopatie miotoniche
Quando la contrazione volontaria si effettua normalmente, ma non si
allenta immediatamente e si constata un ritardo alla distensione si parla
di miotonia. Per metterla in evidenza si chiede al soggetto di effettuare
una contrazione volontaria abbastanza forte, per esempio chiudere la
mano a pugno; la riapertura della mano avviene solo lentamente, sovente con una contrazione attiva e visibile dei muscoli antagonisti. La percussione del muscolo è un altro modo di metterla in evidenza; al posto
di una breve contrazione idiomuscolare, si osserva una contrazione
sostenuta per diversi secondi e un lento rilassamento di tutto il corpo
muscolare (Figura 6). Le registrazioni elettromiografiche forniscono allo
stesso tempo un’interpretazione ed un’oggettivazione della miotonia
sottoforma di uno scroscio di potenziali d’azione che accompagna tutta
la fase di decontrazione lenta delle fibre muscolari, potenziali la cui
ampiezza e frequenza diminuiscono progressivamente, producendo il
rumore caratteristico dell’aereo in picchiata.
Figura 6: la percussione del muscolo provoca, invece di una breve contrazione muscolare, una contrazione sostenuta per diversi secondi con un
lento rilassamento dell’intero corpo muscolare.
45
46
La miotonia può essere osservata in diverse condizioni sperimentali o
tossiche in diverse stirpi di animali mutanti (capra). È stato quindi possibile stabilire che si tratta di un’anomalia della membrana e della fibra
muscolare, sottoforma di conduzione anomala dello ione cloro.
La miotonia è un elemento maggiore di svariate malattie muscolari.Tre
grandi tipi di affezione ne sono interessati: le distrofie miotoniche, le
miotonie congenite e la paramiotonia.
Distrofia miotonica (miotonia atrofica, malattia di Steinert)
La distrofia miotonica o malattia di Steinert è una malattia che coinvolge diversi organi (malattia multisistemica): i muscoli, gli occhi, il sistema
nervoso, l’apparato cardio-respiratorio, l’apparato digerente e le ghiandole endocrine.
Si tratta di una malattia genetica che si trasmette secondo modalità autosomica dominante: una persona colpita ha 1 probabilità su 2 di trasmettere l’anomalia responsabile ad ognuno dei suoi figli. La malattia può
avere un decorso differente da una persona all’altra, anche nell’ambito
della stessa famiglia.
Questa distrofia possiede numerose designazioni, poiché diversi autori
hanno contribuito in maniera importante alla sua descrizione a partire
dall’inizio del XX secolo. La malattia è frequente, la sua incidenza è
valutata a 13,5 x 10-5 e la sua prevalenza a 5 x 10-5. La sua trasmissione è
autosomica dominante con una penetranza assai forte ed un’espressione
variabile. I dati di genetica molecolare hanno confermato la localizzazione della mutazione sul cromosoma 19.
Nella forma abituale, di espressione media, la distrofia miotonica è riconosciuta nel secondo o nel terzo decennio. La miotonia è importante e
47
predominante nelle mani; la stretta di mano è caratteristica. La percussione del muscolo permette di mettere in evidenza con facilità la miotonia sui muscoli distali degli avambracci, sui polpacci e sulla lingua. La
miotonia scompare progressivamente con la ripetizione del movimento,
mentre si aggrava con il freddo.
L’atrofia e il deficit muscolare procedono di pari passo e sono predominanti al viso, al collo e sui segmenti distali degli arti. La mimica facciale è
ridotta con ptosi moderata bilat, labbra inferiori rovesciate. Il paziente
assume un’aria triste. Il rilievo dei muscoli masticatori scompare, al collo
i muscoli sternocleidomastoidei diventano atrofici. I deficit a carico dei
muscoli degli avambracci e delle mani è diffuso, non selettivo e simmetrico.Agli arti inferiori, i deficit risultano altrettanto diffusi e simmetrici
e interessano i polpacci e gli elevatori del piede, determinando sovente
uno steppage. Possono estendersi ai muscoli delle radici degli arti e possono essere coinvolti anche i muscoli addominali. Possono toccare
anche i muscoli del velo palatino e della faringe dando un carattere
disfonico. La mobilità oculare non è toccata. Non vi sono retrazioni, né
crampi, né fascicolazioni. I riflessi sono normali, diminuiti o aboliti.
La partecipazione multisistemica si manifesta clinicamente a livello delle
fanere (calvizie precoce e testicoli atrofici), in una cataratta particolare
all’esame oftalmologico, cataratta posteriore con numerosi corpi colorati. L’ECG mostra delle anomalie della conduzione atrio-ventricolare o
intraventricolare. I dosaggi endocrini confermano l’ipogonadismo.
L’iperglicemia provocata da una curva elevata e prolungata in modo
anomalo. Infine, il tasso di immunoglobulina gamma nel sangue è sovente diminuito.
L’affezione ha un’evoluzione lenta. I disturbi si aggravano e la malattia
può comportare altre manifestazioni come obesità, disturbi della digestione, insufficienza respiratoria, disturbi caratteriali e del sonno.Tuttavia
la malattia sovente si manifesta in forma minore e praticamente stabile,
associando per esempio una cataratta ad una miotonia delle mani.
48
Il trattamento della distrofia miotonica mira innanzitutto a migliorare di
disturbi viscerali associati, la cataratta (accessibile al trattamento chirurgico), il blocco atrio-ventricolare, i disturbi endocrini (per esempio
somministrazione di androgeni).La miotonia risponde al chinino, in
minor misura alla difenilidantoina e alla procainamide, ma questi trattamento sono scarsamente utilizzati per i loro inconvenienti specifici ed il
carattere solitamente moderato della miotonia.
La prevenzione della malattia passa per una consulenza genetica appropriata. È possibile effettuare una diagnosi prenatale. Infatti, la distrofia
miotonica è dovuta ad un’anomalia genetica situata sul cromosoma 19.
Si tratta della ripetizione da 50 a più di 300 volte di una tripletta di
nucleotidi (CTG), mentre nella persona sana il numero delle ripetizioni
è compreso tra 5 e 37. Sembra che più tripletta viene ripetuta, più la
malattia assume una forma grave. La tripletta CTG è situata sul gene
DMPK, che codifica la miotonina, una proteina localizzata soprattutto
nelle cellule muscolari scheletriche e cardiache. La ripetizione della tripletta altera l’espressione dei due geni vicini; il gene N9, la cui proteina
si esprime soprattutto nei testicoli e nel cervello, e il gene DMAHP, che
sembra essere un fattore regolatore di altri geni.
Recentemente un’altra forma di distrofia miotonica, non legata al cromosoma 19, è stata scoperta. Si tratta della miopatia miotonica prossimale (DM2 o PROMM per proximal myotonic myopathy). L’anomalia
genetica sarebbe situata sul cromosoma 3.
Che cosa si può fare? Occorre effettuare regolarmente un bilancio
muscolare, ortopedico, cardiaco, respiratorio, uditivo, oftalmologico e
dietetico (rischio di diabete). Una fisioterapia regolare e sostenuta è
apprezzata dai pazienti. La miotonia, i dolori, i disturbi dell’umore possono beneficiare di trattamenti medicamentosi efficaci.L’inserimento di un
pace-maker può essere indicato per prevenire complicanze cardiache.
Nella forma congenita, i disturbi neonatali necessitano di un servizio di
49
cure intensive pediatriche. Dopo il ritorno a casa, il bambino dovrà essere seguito a livello pluridisciplinare medico e psico-pedagogico.
Miotonie non distrofiche, paralisi periodiche e ipertermia maligna
È grazie alla biologia molecolare che è stato possibile raggruppare le
miotonie congenite, le paralisi periodiche primitive famigliari e l’ipertermia maligna.
Le paralisi periodiche e le miotonie congenite sono state descritte con
precisione alla fine del XIX secolo partendo dapprima da criteri clinici
e poi biologici. Si tratta di malattie ereditarie rare, con una prevalenza
dell’ordine di 1/100’000.
Le loro anomalie genetiche si collocano su geni che codificano per i
canali ionici muscolari che consentono il passaggio del cloro, sodio e
calcio. È importante precisare che la miotonia, non è che un sintomo di
svariate malattie.
La miotonia può essere evidenziata in modo ottimale con l’elettromiogramma sotto forma di scariche miotoniche. La diagnosi differenziale
tra le varie forme di miotonie non distrofiche non è possibile sulla semplice base di questi profili EMG. La biopsia muscolare contribuisce alla
diagnosi.
Il gruppo di malattie legate al canale del cloro comprende la miotonia
congenita dominante (Thomsen), e la miotonia generalizzata recessiva
(Becker). Il gruppo di malattie legate al canale del sodio comprende la
paralisi periodica ipocaliemica (hyperPP), la paralisi periodica normocaliemica (NormoPP), la paramiotonia congenita (PC) o malattia di
Eulenburg, e la miotonia legata al canale del sodio.
50
Oltre ai segni classici, episodi di debolezza e di rigidità muscolare, esistono delle forme di transizione. Da un punto di vista clinico sembrerebbe
logico continuare a distinguere l’HyperPP (malattia di Gamstorp o adinamia episodica ereditaria) dalla PC, poiché le misure terapeutiche sono
differenti nelle due sindromi. L’HyperPP implica talvolta un deficit permanente e progressivo, cosa che invece non avviene nella PC. Nella
miotonia legata al canale del sodio, il segno principale è la rigidità
muscolare simile alla miotonia incontrata nella miotonia congenita.
Questa miotonia può essere debole (myotonia fluctuans), moderata o
grave (myotonia permanens).
Miotonie congenite (MC di Thomsen e Becker)
La MC è una miopatia che si manifesta con una difficoltà al rilassamento muscolare in relazione con uno stato di ipereccitabilità della membrana della fibra muscolare. La descrizione originale della malattia è
opera di Thomsen che, nel 1876, ha osservato la malattia sulla propria
persona e nella sua famiglia. A quel tempo, l’affezione è stata descritta
secondo un modo di trasmissione autosomica dominante.Alla fine degli
anni ’50, Becker, analizzando un gruppo di famiglie di MC di origine
tedesca, ha isolato una forma a trasmissione autosomica recessiva. Le
MC hanno un esordio precoce e sovente i portatori sono riconosciuti
dalla famiglie fin dai primi mesi di vita. La miotonia ha la particolarità di
migliorare sotto sforzo (fenomeno di riscaldamento o «warm up»). Il
termine di MC dovrebbe essere riservato unicamente alle forme di
Thomsen e di Becker.
Gli studi elettrofisiologici hanno mostrato che la rigidità muscolare nelle
MC é dovuta ad una diminuzione della conduzione del cloro a livello della
membrana muscolare.Il gene CHLCN1 che codifica per il canale del cloro
muscolare è stato localizzato sul cromosoma 7 in 7q32-ter,e si è potuto stabilire un legame per le famiglie colpite da MC di Becker o di Thomsen.
51
MC dominante (malattia di Thomsen)
Benché rara è la forma abituale di MC. L’eredità è autosomica dominante e la penetranza completa, è del 100%.
I segni clinici comprendono una rigidità muscolare, soprattuto dopo il
riposo. La funzionalità muscolare migliora con l’esercizio. In generale la
miotonia viene facilmente messa in evidenza a livello clinico, poiché si
manifesta in maniera spontanea e in seguito a percussione. Non vi è evolutività e l’ipertrofia muscolare è frequente (aspetto pseudoatletico). La
rigidità si accentua con il freddo. La diagnosi clinica è facilmente confermata dall’esame EMG, che mostra delle scariche miotoniche in relazione con lo stato di ipereccitabilità della membrana della fibra muscolare. Diagnosi differenziale: esistono casi di malattia legata al canale del
sodio con miotonia senza alcuna debolezza muscolare. La miotonia può
essere presente senza fenomeno di accentuazione al freddo.
Il trattamento non è sistematico e varia in funzione dell’intensità della
miotonia. Possono essere utilizzati medicamenti appartenenti al gruppo
degli antiaritmici di classe I (mexiletina o difenilidantoina).
Miotonia generalizzata recessiva di Becker
L’eredita è autosomica recessiva. Alcuni eterozigoti presentano scrosci
miotonici all’EMG. Casi di questo genere non devono essere confusi
con una miotonia dominante e la biologia molecolare talvolta è necessaria per differenziarli da una distrofia miotonica.
La malattia esordisce a volte nella prima infanzia, ma piu spesso nel
corso del primo decennio di vita. Molti pazienti sviluppano, dopo un
periodo di riposo, una debolezza muscolare transitoria, che migliora
dopo diversi minuti di esercizio continuo. Il deficit muscolare è marcato
52
a livello degli arti superiori, mentre la rigidità è più significativa a livello
delle gambe. In numerosi casi, i muscoli della coscia e della gamba risultano ipertrofici. I sintomi sono solitamente progressivi nei primi anni
dall’esordio e successivamente si mantengono stabili per il resto della
vita. La malattia è permette comunque ai pazienti una vita normale. Il
trattamento è identico a quello della forma dominante.
Paralisi periodiche, paramiotonia congenita e miotonia aggravata
dal potassio
Paralisi periodiche ipokaliemiche. Due mutazioni del gene
(CACLN1A3) che codificano la sottounità alfa-1 del canale del calcio di
tipo L (recettore della diidropiridina) sono responsabili del quadro clinico
in più del 50% delle famiglie. La trasmissione è di tipo autosomico dominante con penetranza del gene piu debole nelle donne (il rapporto M:F è
di 3-4 : 1). È la forma di paralisi periodica più frequente nella popolazione
europea.
Alcune forme gravi esordiscono nella prima infanzia, circa il 60% prima
dei 16 anni, mentre le più benigne verso il terzo decennio. I pazienti sviluppano episodi di debolezza muscolare che si presentano generalmente
nella seconda parte della notte o di prima mattina. . La gravità varia da
un deficit limitato ad un solo gruppo muscolare fino ad una paralisi
generalizzata. Abitualmente, la forza muscolare aumenta progressivamente nell’arco della giornata, ma a volte l’attacco può durare due o tre
giorni. Inizialmente, gli attacchi sono rari, ma la loro frequenza aumenta con il passare dei mesi o degli anni, fino a sopraggiungere quotidianamente.Tra i fattori scatenanti dell’attacco vanno menzionati un’attività
fisica importante o pasti ricchi in carboidrati la sera prima. Nell’arco
della giornata, gli attacchi possono essere provocati o aggravati dall’assunzione di sodio o dall’eccitazione. Una debole attività fisica può talvolta ritardare o prevenire gli attacchi. Durante gli attacchi severi, il
53
potassio sanguigno diminuisce e questo può comportare una bradicardia
sinusale e segni di ipokaliemia all’ECG. Non vi è miotonia, né clinica né
elettrica.
Indipendentemente dalla gravità o dalla frequenza degli attacchi di paralisi, il 30% dei malati sviluppa una miopatia prossimale progressiva con
deficit motorio persistente.
Gli attacchi di paralisi rispondono bene alla somministrazione di potassio per via orale. Per evitare la comparsa di crisi, si raccomanda di seguire un regime povero di sale (2-3 g al giorno) e di glucidi (50-60 g al
giorno). Si devono inoltre evitare esercizi fisici intensi e l’esposizione al
freddo. Se le crisi diventano troppo frequenti, si può proporre un trattamento preventivo con acetazolamide (Diamox).
Paralisi periodiche iperkaliemiche. L’eredità è di tipo autosomico
dominante con una penetranza completa, ma di gravità variabile. L’età di
esordio varia tra la prima infanzia e il secondo decennio. I segni clinici
consistono in episodi di debolezza muscolare, solitamente alla mattina,
che durano dai 10 minuti ad un’ora circa; solo eccezionalmente sono più
lunghi (da 1 a 2 giorni). Alcuni pazienti hanno rare crisi nel corso della
loro vita, mentre altri presentano episodi di debolezza generalizzata quotidianamente o quasi. Durante gli attacchi, la kaliemia è al limite superiore o addirittura largamente al di sopra della norma. Alcuni segni clinici
possono trarre in inganno.Alla fine di una crisi paralitica, la kaliemia può
scendere al di sotto dei valori normali. Un prelievo di sangue effettuato
in questo momento potrebbe quindi suggerire una paralisi periodica
ipokaliemica. Il riposo dopo l’esercizio, il digiuno o l’assunzione di
potassio sono fattori scatenanti delle crisi e possono servire da test diagnostici. Alcuni pazienti presentano fenomeni miotonici tra le crisi e
all’inizio delle stesse, alcuni sviluppano segni di paramiotonia; in altri
casi, i segni miotonici sono assenti. Non si osserva rigidità miotonica
Il trattamento è soprattutto preventivo con pasti ricchi di glucidi, un
esercizio muscolare moderato e assenza di esposizione al freddo.
54
L’acetazolamide (Diamox) può essere utilizzato come trattamento preventivo. Il trattamento delle crisi non è sempre necessario, vista la brevità
delle stesse. Bere una bevanda zuccherata all’insorgere della crisi può
prevenire il suo sviluppo. In caso contrario, è opportuno fare ricorso ad
un’iniezione d’insulina ad azione immediata.
Paralisi periodiche normokaliemiche. Concerne un numero assai
ristretto di famiglie. Si tratta di una variante della paralisi periodica
iperkaliemica che non si accompagna da un aumento della kaliemia
durante gli attacchi. Gli attacchi possono durare diversi giorni.
Paramiotonia congenita. La forma classica è stata descritta da
Eulenburg e in maniera indipendente da Rich. Svariate mutazioni del
gene del canale del sodio sono responsabili del quadro clinico classico.
L’eredità è autosomica dominante con penetranza completa. La malattia
esordisce già alla nascita.
I segni clinici comprendono una rigidità muscolare che aumenta con
l’esercizio (miotonia paradossale). In diverse famiglie la paramiotonia è
notevolmente aggravata dall’esercizio fisico svolto in ambienti freddi.
Alcune famiglie presentano una rigidità indotta dal freddo o dall’esercizio fisico, ma senza deficit muscolare permanente. Sovente questi segni
vengono confusi con quelli di una miotonia congenita. Esistono famiglie in cui i soggetti colpiti presentano segni classici di paramiotonia
congenita e spesso sviluppano anche episodi di paralisi iperkaliemica.
Un deficit muscolare permanente non è stato osservato nella paramiotonia congenita. Il trattamento si basa sui medicamenti che migliorano
la miotonia: mexiletina o difenilidantoina. Si può anche somministrare
l’acetazolamide.
Miotonie peggiorate dal potassio. L’eredità è di tipo autosomica
dominante.
La miotonia con crampi dolorosi e le miotonie fluttuanti e permanenti
sono malattie assai rare. Si tratta di malattie legate ad alterazioni del gene
55
che codifica la sottounità alfa del canale di sodio localizzato sul cromosoma 17. I sintomi sono scatenati o aggravati dalla somministrazione di
potassio.
La particolarità della miotonia con crampi è il suo carattere doloroso. La
miotonia fluttuante («Myotonia fluctuans») è particolare poiché i segni
clinici, contrariamente a quanto avviene per le altre miotonie, sono
variabili da un momento all’altro. La rigidità muscolare può accompagnarsi da dolori muscolari indotti dall’esercizio, ma la comparsa di fenomeni miotonici è ritardata rispetto allo sforzo. L’ingestione di potassio
aggrava la miotonia, ma non comporta alcun deficit come nella paralisi
periodica iperkaliemica. Alcuni agenti depolarizzanti, come il suxamethonium, possono scatenare o aggravare la miotonia al punto da provocare gravi problemi di ventilazione qualora il soggetto venisse sottoposto ad un’anestesia generale.Anche in assenza di miotonia clinica, con
l’EMG può essere messa in evidenza una miotonia latente in maniera
costante.
La miotonia persistente generalizzata («Myotonia permanens») è grave
ed è abbinata ad un’ipertrofia muscolare a carico del collo e delle spalle.
Gli attacchi di importante rigidità muscolare a livello dei muscoli del
torace possono causare disturbi di respirazione a volte gravi, in particolare nei bambini. Il trattamento si basa sull’acetazolamide o sui medicamenti che agiscono efficacemente sulla miotonia.
Ipertermia maligna (HM), anni fa considerata come una malattia familiare osservata unicamente dagli anestesisti, ha visto ampliare il proprio
quadro a sintomi di intolleranza muscolare.
L’HM è un modello di malattia causata da una desincronizzazione dei
processi di depolarizzazione del sarcolemma e contrazione muscolare,
caratterizzato da una liberazione eccessiva di calcio dal reticolo sarcoplasmico. Una mutazione nel gene che codifica le subunità dei canali del
calcio muscolari (recettori della rianodina o della diidropiridina) provo-
56
ca l’apertura prolungata del canale, innescando una cascata di eventi che
sfocia nella produzione eccessiva di calore. Questo fenomeno può verificarsi in forma lieve in seguito all’esercizio o dolorosa con rabdomiolisi, con o senza ipertermia.
L’HM è una sindrome clinica che si osserva in seguito ad interventi eseguiti in anestesia generale che comporta la somministrazione di agenti
depolarizzanti come alotano o di curaro. L’attacco di HM associa una
rigidità muscolare, una ipertermia e segni di acidosi metabolica. La rigidità è predominante dapprima nei muscoli masticatori, originando un
trisma. Una rigidità generalizzata sopraggiunge successivamente. Prima
della scoperta del trattamento a base di dantrolene per via endovenosa, la
mortalità era considerevole, mentre attualmente è del 10%. L’ipertermia
nel corso di anestesie puo sopraggiungere anche in altre miopatie, come
la miopatia congenita «central core», le paralisi periodiche e le miotonie, nonché in alcune distrofie muscolari. Una diagnosi di suscettibilità
all’HM è stata proposta da un test di contrazione in vitro.
Sindrome di Brody. Questa sindrome è vicina all’HM sul piano patofisiologico. Essa è caratterizzata da un deficit della ricaptazione del calcio
intracitoplasmico dovuto ad una mutazione del gene che codifica per
l’ATP-asi sarcoplasmatica e quindi non appartiene alle canalopatie. La
malattia è caratterizzata da una rigidità muscolare legata all’esercizio,
non progressiva, senza indebolimento muscolare, che permette di condurre una vita normale.Talvolta i pazienti sviluppano episodi di febbre
inspiegabili. Il verapamil e il dantrolene migliorano la rigidità.
Miopatie metaboliche
Poiché tutte le miopatie genetiche sono «metaboliche», questo termine
viene riservato per convenzione alle affezioni nelle quali l’anomalia
metabolica è localizzata su uno dei grandi assi metabolici, o legata ad un
disturbo di permeabilità della membrana della fibra muscolare.
Il grado di precisazione dell’anomalia è variabile; a volte il deficit enzimatico viene identificato, come nella maggior parte delle glicogenosi, in
certe lipidosi e in certe mitocondriopatie.
Glicogenosi
Le glicogenosi muscolari interessano unicamente il muscolo oppure
sono associate ad’un coinvolgimenti di altri organi come il cervello,
il fegato, il cuore. Sono definite nove affezione diverse:
– la malattia di Pompe (tipo II)
– la malattia di Forbes (tipo III)
– la malattia di Andersen (tipo IV)
– la malattia di Mc Ardle (tipo V)
– la malattia di Tarui (tipo VII)
– il deficit di fosforilasi chinasi
– il deficit di fosfoglicerato chinasi (PKG)
– il deficit di fosfoglicerato mutasi (PGAM)
– il deficit di lattato-deidrogenasi (LDH)
Si tratta di malattie genetiche. Nella maggior parte dei casi si trasmettono secondo la modalità autosomica recessiva. Il deficit di fosforilasi chinasi e il deficit di PKG sono legati al cromosoma X.
Le glicogenosi muscolari si manifestano frequentemente con un’intolleranza allo sforzo, dolori muscolari (mialgie) o crampi. I sintomi vengono
scatenati dall’esercizio fisico. Contemporaneamente possono insorgere
sintomi di un coinvolgimento di altri organi come fegato o cuore. Gli
sforzi inabituali possono causare una necrosi delle fibre muscolari, una cui
57
58
proteina, detta mioglobina (proteina del muscolo che contiene ferro e il
cui ruolo è quello di fissare l’ossigeno, come l’emoglobina del sangue)
viene liberata nel sangue ed eliminata con le urine causando la loro colorazione scura.
Le glicogenosi muscolari sono malattie del metabolismo caratterizzate da
un difetto nella catena di reazioni chimiche che trasformano gli zuccheri
da noi introdotti attraverso il cibo in energia utilizzabile dal nostro corpo.
Questi «zuccheri» sono depositati nelle cellule sotto forma di glicogeno.
Durante l’esercizio fisico, di norma il glicogeno viene trasformato in
energia (ATP) utilizzabile dai muscoli, grazie ad una serie di reazioni biochimiche che fanno intervenire svariati enzimi. Se uno di queste viene a
mancare, il glicogeno si accumula nelle cellule senza poter essere utilizzato, e questo è ciò che avviene nel caso delle glicogenosi muscolari. I deficit interessano la maltasi acida per la malattia di Pompe (tipo II), l’enzima
della deramificazione per la malattia di Forbes (tipo III), l’enzima ramificatore per la malattia di Andersen (tipo IV), la fosforilasi muscolare per la
malattia di Mc Ardle, la fosfofructochinasi per la malattia di Tarui (tipo
VII), mentre le altre glicogenosi prendono il nome direttamente dai deficit enzimatici in questione.
Le anomalie genetiche sono conosciute nella maggior parte dei casi e
permettono di intravedere delle prospettive nella terapia genica, in particolare nella glicogenosi di tipo II. I risultati di alcuni studi sugli animali,
che impiegano enzimi ricombinanti (sintetizzati mediante ingegneria
genetica) per tamponare l’enzima carente/mancante, sono incoraggianti.
Malattia di Mc Ardle (deficit di fosforilasi muscolare)
Questa affezione, descritta nel 1951, comporta una tensione muscolare
dolorosa con rigidità, che sopraggiunge nel corso o all’inizio di un esercizio muscolare, bloccandolo momentaneamente. In un secondo tempo,
59
dopo un breve riposo, è possibile una ripresa dell’esercizio. La vita quotidiana è sovente del tutto normale. In alcune condizioni di sforzo violento, al freddo, può invece comparire una rabdomiolisi estesa con mioglobinuria e insufficienza renale acuta.
L’esame fisico è il più delle volte normale, ma, in condizioni ischemiche
(per. es con un laccio stretto al braccio che impedisce afflusso di sangue) il
soggetto è incapace di ripetere il movimento di apertura e chiusura del
pugno per piu di 40 secondi. La misurazione dell’acido lattico nel sangue
non mostra, come avviene nelle persone sane in queste condizioni, alcun
aumento della sua concentrazione, che normalmente aumenta da 4 a 5
volte. Infatti, nella malattia di Mc Ardle vi è un blocco della degradazione
del glicogeno dovuto all’assenza di fosforilasi muscolare. Questa anomalia
può essere rilevata biochimicamente o istochimicamente su un frammento bioptico del muscolo, così come l’accumulo di glicogeni che ne risulta.
La glicogenosi di tipo V (malattia di Mc Ardle) è dovuta al deficit di fosforilasi muscolare. La trasmissione è autosomica recessiva. Il gene è stato
localizzato sul cromosoma 11 (11q13) e successivamente sono state identificate le sue mutazioni. La mutazione R49X é più frequente (piu del 40%)
nella popolazione caucasica. Il trattamento consiste in un programma di
esercizio fisico controllato per sviluppare le capacità ossidanti mitocondriali del muscolo e in un apporto glucidico programmato in funzione
dell’esercizio. I regimi iperproteici hanno dato risultati variabili.
Deficit di maltasi acida (malattia di Pompe e forma ad esordio
tardivo)
Il deficit di maltasi acida si traduce in quadri clinici assai differenti dai
precedenti, poiché esso comporta un deficit muscolare permanente e
grave. La forma più grave (malattia di Pompe, 1932) si manifesta alla
nascita con ipotonia massiccia , macroglossia, epatomegalia e cardiomiopatia e il decesso avviene nel giro di 6 mesi per insufficienza cardiaca e
60
respiratoria. La malattia può manifestarsi più tardi, nel bambino piccolo,
dando vita ad un quadro che assomiglia ad una distrofia muscolare; la
macroglossia e l’epatomegalia sono rare e non vi è un coinvolgimento
cardiaco. La malattia puo anche esordire tardivamente nel terzo o nel
quarto decennio con un coinvolgimento simmetrico dei muscoli dei
cingoli e del tronco. Spesso vi è una marcata compromissione dei
muscoli respiratori, intercostali e diaframma, ma cuore e fegato vengono abitualmente risparmiati. Il tasso di CK è elevato, il tracciato elettromiografico mostra delle scariche dette «pseudomiotoniche» associate a
modifiche della morfologia dei potenziali di unità motorie compatibili
con una miopatia.
Il quadro istologico del muscolo è identico nelle 3 forme ed è caratterizzato dall’accumulo di vacuole piene di glucogeno nelle fibre muscolari.
La trasmissione è autosomica recessiva. Gli eterozigoti hanno sovente un
tasso ridotto di maltasi acida nei leucociti o nell’urina. Poiché il deficit è
presente anche nei fibroblasti e nelle cellule del liquido amniotico, nelle
famiglie a rischio è possibile effettuare una diagnosi prenatale.
Deficit di enzimi deramificante del glicogeno (glicogenosi tipo III):
la degradazione completa del glicogeno richiede la presenza di un’enzima deramificante l’assenza di questo enzima causa una minore disponibilità di glicogeno e quindi di energia. La presentazione clinica consiste
in un coivolgimento del fegato e del cuore, mentre il coinvolgimento
muscolare è costante, ma relativamente benigno e puo scomparire, come
altri segni della malattia, dopo la pubertà. In età adulta puo svilupparsi
una debolezza muscolare progressiva.
Deficit di enzima ramificante: il coinvolgimento muscolare può essere
importante, ma generalmente è in secondo piano rispetto alla compromissione cardiaca ed epatica che possono portare ad una grave disfunzione di questi organi e alla morte del bambino.
61
Lipidosi muscolari
Le lipidosi muscolari sono malattie dovute ad un accumulo di lipidi
(grassi) nei muscoli e in altri tessuti. Si distinguono, tra gli altri, il deficit di carnitina, il deficit di carnitina palmatiltrasferasi di tipo II
(CTPII) ed altre lipidosi secondarie ad anomalie della catena respiratoria delle cellule.
Queste malattie sono caratterizzate da anomalie del metabolismo dei
lipidi, ovvero sono causate da un difetto della catena di reazioni chimiche che trasformano i grassi da noi introdotti con il cibo in energia utilizzabile dal nostro corpo. Questi «grassi» (lipidi) normalmente sono trasformati in energia (ATP) utilizzabile dai muscoli durante lo sforzo
muscolare prolungato. Nelle lipidosi, i grassi si accumulano nelle cellule
muscolari e non possono essere utilizzati.
Le cause di queste malattie non sono ancora ben conosciute. Si sa che
alcune di esse sono associate a deficit di carnitina, molecola implicata nel
metabolismo dei grassi o di carnitinapalmatil-trasferasi di tipo II
(CTPII), che costituisce un vettore della carnitina. Il deficit di CTPII è
dovuto ad un’anomalia genetica localizzata sul cromosoma 1 che ha ereditarietà di tipo autosomico dominante.
Nella maggior parte dei casi, i sintomi si manifestano durante un esercizio fisico o quando si è a digiuno. Durante l’esercizio fisico sopraggiungono di frequente dolori muscolari. Talvolta, questi dolori insorgono
dopo svariate ore dall’interruzione dell’esercizio fisico.
I sintomi possono anche consistere in uno stato di confusione mentale,
vomito, disturbi cardiaci o una colorazione rossa delle urine (in seguito a
mioglobinuria, perdita nell’urina di mioglobina, una proteina del
muscolo che contiene ferro, il cui ruolo è quello di fissare l’ossigeno,
come l’emoglobina del sangue).
62
Miopatie mitocondriali
Le malattie mitocondriali sono malattie dovute a delle anomalie della
catena respiratoria mitocondriale. I mitocondri sono le «centrali energetiche» della cellula. Ogni cellula dell’organismo ne contiene svariate
centinaia o addirittura migliaia. La disfunzione dei mitocondri comporta il malfunzionamento della cellula stessa e del rispettivo organo.
La catena respiratoria è una catena di reazioni chimiche che produce
energia per la cellula, grazie alle molecole che sono codificate o dal
DNA mitocondriale, o dal DNA del nucleo della cellula (DNA nucleare). Infatti, i mitocondri contengono i loro geni specifici (genoma mitocondriale), ma, per funzionare, essi hanno bisogno di altri geni della cellula (genoma nucleare). Sono state identificate numerose mutazioni
responsabili di miopatie mitocondriali, tanto nel genoma mitocondriale, quanto nel genoma nucleare. La loro trasmissione è complessa: trasmissione puramente materna (i mitocondri in principio vengono trasmessi unicamente dalla mamma), trasmissione autosomica recessiva,
trasmissione autosomica dominante. Le ricerche si orientano verso
nuovi approcci: si immagina si poter agire sulla proporzione di mitocondri normali e mutati, stimolando la replica del genoma mitocondriale
normale, o inibendo quella del genoma mitocondriale mutato. Si tratta
quindi di malattie ereditarie con modalità di trasmissione che non
rispettano le leggi classiche di ereditarietà mendeliana.
Le miopatie mitocondriali che interessano più particolarmente i
muscoli comprendono in particolare le sindromi MELAS, MERFF e di
Kearns-Sayre.
Storicamente, le prime scoperte di anomalie dei mitocondri identificate
sui prelievi muscolari dei malati con disturbi oculomotori hanno successivamente permesso di individuare numerose forme cliniche, in funzione dell’area colpita: muscolatura faringea, facciale, cervicale. Vi si
associano possibili segni di compromissione extramuscolare, per es. della
retina, disturbi di conduzione cardiaca (sindrome di Kearns-Sayre), della
63
maturità sessuale, dello sviluppo intellettuale. Nell’insieme, più precoce
è l’esordio, più ricco è il quadro clinico e più grave l’evoluzione.
La gravità delle malattie mitocondriali è assai variabile a seconda degli
individui. Nelle malattie mitocondriali dovute ad un’anomalia del
DNA mitocondriale, la compromissione di una cellula (e quindi di un
tessuto, e di un organo) dipende dalla proporzione di mitocondri
«malati» in essa contenuti, questo perché in ogni cellula coesistono dei
mitocondri sani e dei mitocondri malati (questa coesistenza si chiama
eteroplasmia). Il coinvolgimento di un organo dipende anche dalle sue
esigenze energetiche abituali: il muscolo, accanto al cervello, è l’organo
che consuma più energia nell’organismo.
Nel bambino la malattia muscolare (ipotonia, acidosi lattica) coesiste
assai di frequente con la compromissione di altri organi, come cervello,
fegato, reni o cuore.
Nell’adulto, il coinvolgimento muscolare si manifesta con uno stato di
affaticamento, un affanno o dei dolori durante l’esercizio fisico (intolleranza allo sforzo). Spesso a questi sintomi si associano anche una debolezza permanente dei muscoli degli occhi (oftalmoplegia) e una caduta
delle palpebre.
Miopatie infiammatorie
64
Polimiosite e dermatomiosite
Le miopatie infiammatorie (miositi) comportano principalmente:
– la dermatoniosite (DM) che associa inoltre un attacco a carico
della pelle,
– la polimiosite (PM).
La polimiosite forma un gruppo di malattie acquisite del muscolo, caratterizzate da una debolezza muscolare prossimale e da segni infiammatori nella biopsia muscolare. Si tratta di malattie di origine sconosciuta, ma
sicuramente di tipo autoimmune. L’incidenza è valutata intorno a 0,5 –
1 su 100’000 l’anno.
L’esordio è subacuto (da settimane a mesi), la malattia interessa dapprima la muscolatura prossimale degli arti inferiori e poi quella degli arti
superiori. Sono frequenti i dolori muscolari. A volte può svilupparsi
anche una difficoltà a deglutire (disfagia) o un disturbo dell’articolazione (disartria), legati alla compromissione della muscolatura faringea. Nei
casi piu gravi può esserci un coinvolgimento della muscolatura respiratoria.
Il termine di dermatomiosite è impiegato quando segni cutanei accompagnano la compromissione muscolare. Spesso esiste una ipersensibilità
alla luce (colpo di sole dopo una breve esposizione) che segue o, più
sovente, precede lo sviluppo della debolezza muscolare. La manifestazione cutanea più caratteristica è un eritema che colpisce la faccia, la regione orbitale e le guance ad «ala di farfalla».
In 2/3 dei casi gli esami di laboratorio evidenziano una sindrome infiammatoria, con innalzamento della velocità di sedimentazione.Tra gli enzimi muscolari, quello più sensibile è la creatinchinasi, il cui valore può
essere fino a 50 volte superiore rispetto al tasso normale.
L’elettromiogramma è abbastanza caratteristico e mette in evidenza un
quadro di tipo miopatico associato ad una attività spontanea a riposo
65
sotto forma di potenziali di fibrillazione e di scariche ripetitive. La biopsia muscolare deve essere effettuata in zone clinicamente coinvolte, ma
non atrofiche.
Il quadro istologico é caratterizzato da atrofia e necrosi delle fibre
muscolari, ed infiltrati infiammatori perivascolari (fig.7)
Il trattamento delle miopatie infiammatorie è largamente empirico e si
basa essenzialmente sulla prescrizione di derivati del cortisone a dosaggi
elevati, da assumere per settimane.Attualmente, un trattamento ben eseguito permette di ottenere un netto miglioramento del deficit muscolare nell’80% dei casi, e una guarigione una volta su due.
Dermatomiosite
Muscolo normale
Polimiosite
Figura 7: le frecce indicano la presenza di un’infiltrazione infiammatoria.
Malattie della giunzione
neuromuscolare
66
Miastenia grave (MG)
La MG è una malattia cronica legata ad un difetto di trasmissione dell’impulso nervoso tra il nervo e il muscolo. Questa anomalia è limitata ai
muscoli stirati, ad innervazione volontaria e non automatica. Essa colpisce entrambi i sessi e può esordire a qualsiasi età, ma una maggiore frequenza è osservata nella donna tra i 20 e i 40 anni, e nell’uomo sopra i 40
anni.
Si tratta di una malattia rara autoimmune, con un’incidenza di 2-5
nuovi casi all’anno, ed una prevalenza di 43-64 soggetti per milione di
abitanti, o 5 individui per 100’000. La maggior parte dei soggetti è portatore di autoanticorpi che si fissano sui recettori dell’acetilcolina
(Ach).
L’origine della MG è oggetto di discussioni: è secondaria ad un’alterazione primitiva del recettore dell’Ach o ad un controllo immunologico
difettoso nei confronti di un antigene normale, o ancora è dovuta alla
combinazione di entrambi questi meccanismi? Una suscettibilità genetica è molto verosimile, come è attestato dalla esistenza di forme familiari di MG, dal riscontro nei parenti prossimi sani di anomalie elettrofisiologiche o di un innalzamento degli anticorpi contro il recettore
dell’Ach, infine dai gruppi HLA particolarmente frequenti. Infine, ora è
stabilito che il timo svolge un ruolo scatenante nella comparsa della
MG.
La MG è polimorfa ed è caratterizzata da: i) affaticamento, ii) debolezza
muscolare simmetrica o asimmetrica di una parte o della totalità dei
muscoli volontari, iii) assenza di deficit sensitivo e normalità dei riflessi
tendinei e iv) miglioramento dei sintomi con i medicamenti anticolinesterasici.
67
Le caratteristiche cliniche rilevanti ai fini diagnostici sono:
• Le fluttuazioni dell’intensità dei disturbi nel corso della giornata e
delle settimane.
• L’aggravamento della debolezza in seguito all’esercizio ripetuto o
prolungato, innalzamento della temperatura corporea, stress psicologico, infezioni virali intercorrenti e nei periodi mestruali.
L’esposizione alla luce intensa può aggravare i segni oculari.
L’evoluzione è variabile nel corso della gravidanza.
Il coinvolgimento della muscolatura oculare, che si manifesta con una
diplopia e/o la caduta delle palpebre (ptosi), è presente nella maggior
parte dei casi. Possono essere colpiti anche altri muscoli innervati dai
nervi cranici: facciali, masticatori, linguali, faringei, laringei. La malattia si
manifesta inizialmente con debolezza dei muscoli degli arti nel 20% dei
casi circa, solitamente coinvolgendo i muscoli prossimali. Anche la
dispnea può costituire una manifestazione inaugurale.
Esistono inoltre forme progressive, con coinvolgimento progressivo dei
muscoli oculari, facciali, bulbari e, infine del tronco e delle estremità. Se
la compromissione respiratoria comporta la ventilazione artificiale, si
parla di «crisi miastenica».
Gli esami complementari servono a confermare la diagnosi clinica e a
ricercare eventuali patologie associate. Comprendono la determinazione
degli autoanticorpi, e in particolare degli anticorpi antirecettore
dell’Ach, gli studi elettrofisiologici, la TAC o la RMN del torace (stato
del timo?) e la tipizzazione HLA. Un test farmacologico al Tensilon può
confermare la diagnosi.
L’evoluzione è assai variabile. I primi segni possono restare isolati, in particolare i segni oculari. Una forma oculare o oculo-palpebrale isolata,
anche unilaterale, viene osservata all’incirca nel 15% dei casi. Sovente, la
68
MG si aggrava a ondate e si arricchisce di nuovi sintomi che portano ad
uno stato di debolezza generalizzata nel giro di alcuni mesi. In altri casi,
l’evoluzione è intervallata da remissioni più o meno complete e di durata
più o meno variabile, che possono andare da alcuni mesi a qualche anno.
Solitamente è nei primi cinque anni che la MG raggiunge la sua massima
gravità. La persona affetta da MG ha una malattia potenzialmente grave: è
minacciata dall’insorgere di episodi di insufficienza respiratoria, non
sempre prevedibile.
La presa a carico dei malati comporta un trattamento medicamentoso e
un monitoraggio in ambito specialistico.Anche se esistono tuttora delle
forme gravi e invalidanti della malattia, la timectomia, i trattamenti
immunosoppressori e i cicli di plasmaferesi o di immunoglobuline i.v.
hanno permesso di migliorare la loro prognosi. Il trattamento viene
scelto a seconda del paziente, in funzione di uno score muscolare che
somma le sue funzioni motorie.
Nella MG, alcuni medicamenti sono controindicati poiché possono
aggravare la debolezza muscolare: D-penicillamnina, miorilassanti curarizzanti, anestetici, antiaritmici (chinidina, procainamide, clorochina),
gli antibiotici aminoglicosidi, i betabloccanti, la fenitoina, il trimetadione, il dandrolene. In linea generale, si dovrà prescrivere con cautela qualsiasi medicamento. I pazienti e i medici devono essere consapevoli della
possibilità di un aggravamento dopo l’assunzione di qualsiasi nuovo
medicamento.
Il trattamento sintomatico si basa innanzitutto sugli inibitori della acetilcolinesterasi (piridostigmina o Mestinon) che prolungano l’effetto della
acetilcolina, trasmettitore tra il nervo e il muscolo. Il prednisone ed altri
medicamenti di tipo immunosoppressivo costituiscono il trattamento di
fondo della malattia.Esso può essere completato con la somministrazione
di immunoglobuline per via endovenosa o con plasmaferesi. La timectomia è generalmente indicata prima dei 40 anni.
69
Il rischio di esacerbazioni periodiche della malattia puo essere ridotto
grazie al trattamento adeguato delle infezioni,al considerare la malattia in
occasione di un intervento chirurgico, all’attenzione per i fattori emozionali e all’educazione del paziente nell’uso dei medicamenti. Una crisi
colinergica (sovradosaggio degli anticolinesterasici) può essere evitata
informando i pazienti sugli effetti che accompagnano il sovradosaggio
dei medicamenti, come l’aumento della debolezza muscolare che
sopraggiunge nel giro di una o due ore dall’assunzione.
Sindrome miastenica di Lambert-Eaton (SMLE)
La SMLE consiste in un blocco acquisito della trasmissione neuromuscolare, caratterizzato da una riduzione della liberazione dell’Ach a livello
della membrana presinaptica, all’origine di una paresi solitamente areflessica della muscolatura prossimale, associata a segni di disautonomia colinergica. Si tratta di un’affezione rara e si conta circa 1 caso di SMLE ogni
100 casi di miastenia. Essa è considerata come una patologia autoimmune , associata dei carcinomi o a delle malattie autoimmuni.
La SMLE si contraddistingue per: i) affaticamento, ii) debolezza muscolare, iii) disturbi disautonomici e iv) una risposta variabile agli anticolinesterasici. Gli elementi della diagnosi sono: i) comparsa insidiosa, progressiva, talvolta fluttuante di manifestazioni cliniche che peggiorano al caldo
ii) la predominanza della debolezza agli arti inferiori. Il disturbo principale consiste così in una difficoltà a camminare in salita od a salire le scale.
Sono possibili episodi di diplopia, ma a carattere transitorio. In primo
piano può esserci una dispnea, iii) la disautonomia colinergica caratterizzata da una secchezza degli occhi e della bocca, talvolta da una disgeusia,
da impotenza nell’uomo, da una costipazione e da un’anidrosi, iv) riflessi
osteotendinei deboli o assenti.
La debolezza muscolare e i riflessi osteotendinei possono migliorare
dopo uno sforzo sostenuto v) L’associazione, in 2/3 dei casi ad un carcino-
70
ma, il più delle volte polmonare a piccole cellule, più raramente ad un’altra malattia autoimmune (compresa la miastenia) o un’altra malattia neoplastica. La ripetizione delle indagini mirate alla ricerca di un tumore
occulto è giustificata visto che la SMLE può precedere fino a 4 anni lo
sviluppo di una malattia neoplastica.
L’EMG ha un aspetto caratteristico.
Nella metà dei casi vengono riscontrati anticorpi diretti contro il canale
del calcio della membrana presinaptica della giunzione neuromuscolare.
Il trattamento si basa sulla cura del tumore sottogiacente o della malattia
associata, sui medicamenti che migliorano la trasmissione neuromuscolare (Mestinon e 3-4-diaminopiridina), sull’immunosoppressione (prednisone e azatioprina) e l’immunomodulazione (plasmaferesi e perfusioni di
immunoglobuline i.v.).
Sindromi miateniche congenite (SMC)
Il termine di SMC si riferisce ad un insieme di affezioni genetiche congenite che interessano la giunzione neuromuscolare. Come nella miastenia gravis vi è un decremento d’ampiezza delle risposte alla stimolazione
elettrica ripetitiva con la frequenza di 3 Hz. Gli studi clinici sono stati
limitati ad un piccolo numero di malati. Gli anticolinesterasici migliorano i sintomi.
Miastenia familiare infantile (tipo Ia).Autosomica recessiva.Esordio:dalla
nascita fino alla prima infanzia, con ptosi fluttuante e coinvolgimento dei
muscoli bulbari. Nell’infanzia, affaticamento muscolare di intensità
variabile con ptosi o oftalmoparesi. Occasionalmente episodi di aggravamento con eventuali difficoltà respiratorie in concomitanza con stati
febbrili o forti emozioni. Nel corso degli anni la sintomatologia diventa
meno pronunciata.
71
Miastenia dei cingoli (tipo Ib). Autosomica recessiva. Età di esordio:
secondo decennio. Il segno cardinale è un affaticabilità muscolare simmetrica dei quattro arti.
Sindrome di deficit di acetilcolinesterasi (tipo Ic).Autosomica recessiva.
Esordio dalla nascita fino all’età di 2 anni. Segni clinici cardinali costituiti
da affaticabilità muscolare dei muscoli oculari, facciali o bulbari, ritardo
di sviluppo motorio, coinvolgimento selettivo dei muscoli assiali con
sviluppo di scoliosi. All’EMG si puo riscontrare una doppia risposta
motoria ad uno stimolo elettrico unico.
SMC con deficit di recettori dell’Ach (tipo Id). Autosomica recessiva.
Esordio dalla nascita fino all’età di due anni, con ptosi, coinvolgimento
dei muscoli bulbari e affaticabilità di intensità variabile. Generalmente,
l’evoluzione è benigna, ma i sintomi possono persistere anche in età
adulta.
Sindrome del canale lento (tipo II). Autosomica dominante. Età d’esordio, grado di affaticamento muscolare e distribuzione variabili.
L’evoluzione è progressiva, o a scalini fino allo sviluppo di un quadro
caratteristico con un coinvolgimento dei muscoli della testa,delle spalle e
degli estensori delle dita e del polso. Compromissione variabile dei
muscoli oculari, facciali e bulbari. All’EMG presenza di una doppia
risposta motoria in risposta ad una stimolazione elettrica unica.
SMC di tipo III. Include i malati senza storia familiare affetti da un’affaticabilità muscolare focale o generalizzata con un’età di esordio prima dei
12 anni, associato ad anomalie elettrofisiologiche della trasmissione neuromuscolare.
Neuropatie periferiche e
polineuropatie
72
Con neuropatia periferica si intende una malattia dei nervi periferici. I
nervi periferici sono le strutture che collegano il midollo spinale con i
muscoli (movimento) e la pelle (sensibilità). Si tratta di una polineuropatia se la malattia è diffusa, di una mononeuropatia multipla se si manifesta in modo simultaneo e in omogeneo su più singoli nervi.
Le polineuropatie possono avere meccanismi fisiopatologici differenti.
Schematicamente, la sofferenza della fibra nervosa può risiedere nel
corpo neuronale (neuronopatia motoria o sensitiva), nell’assone (assonopatia) o nelle guaine di mielina degli assoni, prodotte dalle cellule di
Schwann (mielinopatia).
Descriveremo unicamente le forme croniche: le neuropatie geneticamente determinate e le poliradiculoneuropatie infiammatorie croniche.
Neuropatie geneticamente determinate
Il termine di malattia di Charcot-Marie-Tooth (CMT) designa un
gruppo di almeno una quindicina di malattie croniche dei nervi periferici.Appartengono al gruppo delle neuropatie sensitivo-motorie ereditarie (HSMN, di cui fa parte anche la malattia di Déjérine-Sottas o
HSMN3).
Queste malattie colpiscono 1 persona su 2’500. Si tratta di malattie
genetiche che si trasmettono in svariati modi: autosomico dominante
(CMT1), autosomatico recessivo (CMT2) o dominante legato al cromosoma X (CMTX). La classificazione attuale è basata sul tipo di ereditarietà, sulla velocità di conduzione nervosa del nervo mediano e sulle
anomalie genetiche.
73
La malattia si manifesta nell’infanzia o nell’adulto giovane, talvolta
anche più tardi, anche dopo i 60 anni. L’amiotrofia esordisce lentamente,
colpendo dapprima i muscoli dei piedi ed è sovente preceduta da crampi. Si estende poi insidiosamente sui muscoli anteroesterni delle gambe,
dando luogo a difficoltà a correre e successivamente ad uno steppaggio.
Solitamente, diventa impossibile flettere dorsalmente il dorso del piede,
fatto che viene risconosciuta da alcuni pazienti. Spesso il deficit è concomitante a deformazioni dei piedi.Agli arti superiori l’amiotrofia inizia più tardivamente, qualche anno dopo gli arti inferiori. L’amiotrofia si
concentra sui muscoli delle mani, con possibilità motorie preservate a
lungo. I riflessi achillei sono solitamente aboliti. Nella metà dei casi, vi è
una areflessia rotulea. I riflessi degli arti superiori vengono solitamente
preservati. Una ipoestesia distale è frequente. L’evoluzione è per lo più
lentamente progressiva. La prognosi vitale non è modificata.
Le neuropatie del tipo Charcot-Marie-Tooth sono dovute a delle anomalie che possono localizzarsi sulla guaina che circonda il nervo, la mielina (forme demielinizzanti) o nel nervo stesso (forme assonali).Tre sono
attualmente i geni indentificati: PMP22, PO e Cx 32, che codificano
rispettivamente le proteine PMP22, PO e connexina 32. Queste proteine intervengono nella composizione della mielina.
Forme demielinizzanti. La forma autosomica dominante con demielinizzazione (con una velocità di conduzione nervosa <30 m/sec normale >50 m/sec.) è probabilmente la più frequente. Nell’80% dei casi vi è
una duplicazione della regione p11,2 del cromosoma 17, che contiene il
gene della proteina PMP22 (CMT1a). Per la metà si tratta di nuove
duplicazioni. Negli altri casi esistono delle mutazioni nel gene addetto
alla codifica della PMP22 (chiamata anche CMT1a) o della proteina PO
(CMT1 o forma autosomica dominante con VCNM <35 m/sec, individuata sul cromosoma 1q23) o della connexina 32 (CMTX o forma
intermedia legata al cromosoma X). Esistono delle forme non ancora
assegnate ad un cromosoma (CMT1C). La forma CMTX è sovente dif-
74
ficile da riconoscere perché può manifestarsi sia nell’uomo che nella
donna, seppur in forma più grave negli uomini. Non vi è trasmissione di
padre in figlio.
Le rare forme autosomiche recessive sono raggruppate attualmente
sotto il termine CMT4. Il tipo CMT4a è definito da una ipomielinizzazione del nervo periferico, il tipo 4b da ammassi globulari di mielina; la
forma dei gitani o CMT Lom è degna di nota per la sua associazione ad
una sordità progressiva.
Esistono diverse varietà cliniche di CMT demielinizzanti: sindrome di
Davidenkow (amiotrofia con distribuzione scapoloperoneale), sindrome
di Roussy Lévy (manifestazione artrocinetica e tremore d’attitudine) e
neuropatia di Déjérine e Sottas (vecchia forma HSMN II della classificazione di Dyck, dall’esordio precoce e dall’handicap grave), che tuttavia presenta le stesse anomalie genetiche delle forme CMT1a o 1b.
Forme assonali. Le forme dominanti assonali, con una VCNM > 40
m/sec, mettono in gioco diversi cromosomi, ma i geni non sono ancora
noti: 1p (CMT2), 3q (CMT2b) e 7p (CMNT2d). La PN CMTX entra
nella diagnosi differenziale. La forma CMT2c associa PN e paresi vocale
progressiva.
Che cosa si può fare? La cura ortopedica comprende la fisioterapia, che
deve essere precoce, regolare e personalizzata, con l’aggiunta dell’apparecchiatura ortopedica opportuna. Essa permette di rallentare l’evoluzione della malattia, in particolare mantenendo la mobilità delle articolazioni (la perdita della forza muscolare può provocare la comparsa di
deformazioni articolari).Talvolta, per migliorare le difficoltà di deambulazione si può ricorrere a calzature ortopediche, a delle stecche e in certi
casi ad un intervento chirurgico.
75
Neuropatia troncolare recidivante familiare (o HNPP per «hereditary neuropathy with liability to pressure palsy»)
Presenta una trasmissione ereditaria autosomica dominante ed è stata
riconosciuta dal 1947 da de Jong in Olanda. Legata al cromosoma 17, l’anomalia genica è stata localizzata nella stessa regione della neuropatia
CMT1a, ma si tratta in più del 90% dei casi di una delezione e non di
una duplicazione. La sua prevalenza è stimata a 16/100’000.
Si manifesta generalmente nel giovane adulto, ma l’età di esordio può
andare dalla nascita (plessopatia branchiale) alla sesta decade. Il deficit è
di tipo motorio e parestetico e sovente indolore. Questo spiega perché
sovente non venga riconosciuta. Evolve per episodi regressivi di gravità
variabile, talvolta scatenati da un trauma banale. I tronchi nervosi maggiormente colpiti sono quelli che passano attraverso delle strettoie anatomiche, per ordine di frequenza: il nervo peroneo, l’ulnare, il radiale, il
plesso brachiale, il nervo mediano, in nervi delle dita ed eccezionalmente i nervi cranici e in particolare il nervo facciale e il nervo ricorrente.
L’esame EMG mette in evidenza segni di una neuropatia generalizzata
infraclinica, con segni non omogenei di demielinizzazione. La biopsia
del nervo evidenzia dei rigonfiamenti focalizzati di determinati segmenti della guaina di mielina detti tomaculi. Il meccanismo di costituzione dei tomaculi viene ritenuto secondario ad una ipermielinizzazione, in relazione con una anomalia di funzionamento della cellula di
Schwann. A causa del carattere indolente e talvolta cumulato delle
mononeuropatie, può risultare difficoltoso distinguere questa neuropatia da una polineuropatia (PN) cronica.
In assenza di un trattamento causale le persone colpite devono evitare
professioni e occupazioni che comportano il mantenimento prolungato
di determinate posizioni, dei movimenti ripetitivi o l’utilizzo di apparecchi vibranti. Devono evitare stiramenti e compressioni dei tronchi
76
nervosi (gomiti, ginocchia). L’aspettativa di vita non é modificata da
questa neuropatia considerata tutto sommato generalmente benigna.
Malattia di Refsum
Si tratta di una malattia autosomica recessiva rara dovuta ad un disturbo
del metabolismo dei perossisomi. Ne deriva un deficit dell’ossidazione
alfa dell’acido fitanico, acido grasso saturo assunto con il cibo.
Vi sono delle manifestazioni sistemiche presenti prima dell’età di 20 anni
con una cecità notturna (emeralopia della retinite pigmentosa, criterio
obbligatorio), una anosmia e una sordità progressiva, un difetto della crescita delle ossa della mano o del piede (un terzo dei casi, con piccola falange terminale del pollice e del quarto metatarsale), una pigmentazione
incostante della cute e disturbi del ritmo cardiaco. La polineuropatia è
progressiva e cronica,e si manifesta con atassia,areflessia e amiotrofia distale. Si esacerba in relazione con il digiuno o l’aumento del catabolismo
(infezione intercorrente, chirurgia). L’ENMG dimostra una PN assonomielinica, con caratteristica di demielinizzazione durante gli episodi di
esacerbazione, che evocano le anomalie della sindrome di Guillain-Barré.
Esiste sovente una dissociazione albuminocitologica all’esame del LCR.
La biopsia del nervo è non specifica, con presenza di bulbi di cipolla.
La diagnosi della malattia di Refsum si pone con la dimostrazione di un
aumento della concentrazione dell’acido fitanico (norma < a 30
umol/l).Altre malattie dei perossisomi hanno anch’esse un tasso elevato
di acido fitanico.
Il tasso di acido fitanico può essere tenuto sotto controllo grazie ad una
dieta appropriata. Una sua riduzione rapida con delle plasmaferesi è
indicata in caso di episodi di aggravamento. I disturbi visivi e sensoriali
non vengono modificati dal trattamento.
77
Neuropatie amiloidi ereditarie (NAH)
Si tratta di polineuropatie autosomiche dominanti. Le manifestazioni
non si limitano al nervo periferico perché vi è deposito di sostanza amiloide in altri tessuti. La natura biochimica delle proteine che costituiscono i depositi è stata precisata per le differenti varietà cliniche: il tipo I,
che è anche il più frequente, associa polineuropatia (PN), disautonomia
e insufficienza renale. Il tipo II si contraddistingue per una sindrome del
canale carpale che precede una PN o una cardiomiopatia. Il tipo III,
raro, associa PN, disautonomia e insufficienza renale. Il tipo IV associa
deficit a carico dei nervi cranici e una distrofia della cornea. L’età di
esordio è variabile, tra il terzo e il settimo decennio.
La PN si manifesta con una sintomatologia sensitivo-motoria, e in particolare con disturbi della sensibilità termica e dolorosa. La disautonomia
può essere un segno indicatore della malattia, con disturbi digestivi in
primo piano: diarree alternate a episodi di stitichezza. Una ipotensione
ortostatica può essere responsabile di lipotimie. Si verifica un’impotenza
precoce. Si osservano dei disturbi della sudorazione con ipoidrosi degli
arti.Talvolta sono presenti anche disturbi pupillari. La NAH è un’affezione evolutiva, responsabile di uno stato di infermità dopo una evoluzione media di 10 anni.
I geni responsabili delle proteine associate alle NAH sono stati clonati e
sono state dimostrate diverse mutazioni: il gene che codifica la transtiretina (TTR) sul cromosoma 18 (tipo I e II), l’apoliporproteina A1 sul cromosoma 11(forme di tipo III), la gelsolina sul cromosoma 9 (forme di
tipo IV). Nel siero è possibile individuare la TTR anomala per la variante Met 30. La diagnosi si basa sulla dimostrazione di depositi di amiloidi
sul nervo o altri tessuti, sulla pelle o sul canale digerente. La diagnosi differenziale si pone con le neuropatie amiloidi nell’ambito delle disglobulinemie.
Il trattamento è sintomatico. Il trapianto epatico costituisce l’unico trat-
78
tamento specifico che può permettere di interrompere la progressione
della PN.
Porfirie
Si tratta di un disturbo metabolico della sintesi dell’eme. I portatori sani
scompensano il deficit metabolico nel corso di una induzione epatica, in
seguito all’assunzione di alcuni medicamenti (antiepilettici e diuretici in
primo luogo), di alcool, in caso di digiuno e in occasione del ciclo.
Le manifestazioni si esprimono mediante attacchi che possono durare
da alcune ore ad alcuni giorni con sintomi di disautonomia (vomito,
costipazione dolorosa, tachicardia e labilità ella pressione arteriosa, raramente disuria), psichiatrici (agitazione, stati confusionali) e neurologici
(deficit muscolare, crisi epilettiche).
La porfiria acuta intermittente è la forma più frequente. La NP è soprattutto motoria e asimmetrica, con paresi prevalentemente prossimale
degli arti superiori. Gli ultimi nervi cranici sono sovente colpiti. I
disturbi sensitivi comprendono disestesie asimmetriche, «a corazza».
Le porfirie acute si trasmettono mediante modalità autosomica dominante (solo la carenza di acido delta aminolevulinico è autosomica recessiva), con una penetranza debole, perché fino al 90% dei portatori é asintomatico. La porfiria acuta intermittente è associata ad un calo dell’attività della deaminasi PBG (porfobilinogeno), con un deficit localizzato
sul cromosoma 11q e sono state identificate 100 mutazioni del gene. La
diagnosi resta difficile e si basa sull’aumento dei metaboliti dell’eme:
porfobilinogeno, coproporfirina, acido delta aminolevulinico nelle urine
e coproporfirina nelle feci. Il tasso di escrezione dei metaboliti può essere
normale e in questo caso si impone la ripetizione degli esami.
79
Il trattamento degli attacchi comprende la perfusione di glucosio e di
ematina, che prevengono l’induzione dell’enzima ALA sintetizzato nel
fegato.
Poliradiculonevrite (PRN) cronica o CIDP
Questa neuropatia periferica molto eterogenea non possiede alcun eponimo. Il concetto di PRN cronica (Chronic Inflammatory
Demyelinating Polyradiculoneuropathy) è stato sviluppato da Dyck a
partire dal 1975. Si tratta di una NP demielinizzante acquisita di origine
disimmune potenzialmente curabile, che si manifesta attraverso diverse
forme evolutive sovente intricate: forme croniche progressive e subacute recidivanti. La sua incidenza è stimata a 0,5 su 100’000.
Nel 1991 un comitato ad hoc ha proposto dei criteri diagnostici che
successivamente hanno avuto numerosi sviluppi. Delle neuropatie puramente sensitive e delle neuropatie motorie multifocali sono state associate alle PRN croniche, seppur con le loro specificità. È stata migliorata
la conoscenza di fenomeni elettrofisiologici come i blocchi di conduzione e la dispersione temporale.
Questa polineuropatia può insorgere a qualsiasi età. Si tratta di una
malattia cronica e la sua diagnosi non può essere posta prima delle otto
settimane di evoluzione. L’evoluzione è progressiva, con stabilizzazione
e ricadute. I segni clinici comprendono un deficit sensitivo-motorio
abbastanza simmetrico. I sintomi sono piuttosto distali, talvolta localizzati prevalentemente agli arti superiori. Sono stati descritti dei deficit
dei muscoli del collo (sindrome della testa cascante). Possono essere colpiti tutti i nervi cranici. I disturbi sensitivi riguardano essenzialmente le
fibre sensitive di grosso calibro. I riflessi sono ridotti o assenti nella maggior parte dei casi.
80
Aspetti pratici. Una PRN cronica deve essere evocata nel caso di polineuropatie croniche o ricorrenti, anche dopo svariati anni. L’esame
ENMG svolge un ruolo determinante alla ricerca di anomalie subcliniche. Consiste nella misurazione di parametri della conduzione nervosa
su diversi tronchi nervosi agli arti superiori e inferiori. Le risposte tardive (onde F) sono sovente assai ritardate. All’occorrenza, l’ENMG dove
essere ripetuto al fine di dimostrare delle anomalie compatibili con una
demielinizzazione. Il bilancio clinico e sanguigno deve permettere di
escludere altre forme, soprattutto la neuropatia troncolare famigliare
(HNPP) e le forme secondarie di PRN croniche (associate all’infezione
HIV, ad una gammopatia nell’ambito di un MGUS o di un’altra discrasia
sanguigna, diabete, malattie sistemiche). L’analisi del liquido cefalorachidiano mette in evidenza una dissociazione albuminocitologica nel 90%
dei casi.
La malattia migliora sotto Prednisone nel 65-95% dei casi. Tuttavia,
sovente è necessario assumere dosi elevate per dei mesi, con ricadute
frequenti o comparsa di effetti indesiderati. È possibile associare
dell’Azatioprina. Gli scambi plasmatici (plasmaforesi) e le perfusioni di
immunoglobuline endovena sono efficaci.
Neuropatia motoria a blocco di conduzione
Si tratta di una neuropatia periferica puramente motoria che interessa
con predilezione gli arti superiori, in modo asimmetrico.
La malattia è rara e colpisce prevalentemente gli uomini giovani.
Esordisce in modo asimmetrico con una debolezza muscolare.
L’affezione può essere sospettata su base clinica: paresi limitata inizialmente a uno o più territori troncolari, amiotrofia poco accentuata, in
netto contrasto con l’importanza della debolezza, e attività muscolare
spontanea. I crampi sono frequenti e solitamente costituiscono il solo
81
elemento doloroso. L’esame del LCR è normale. L’affezione è lentamente progressiva: tuttavia, l’evoluzione può variare dalla remissione
spontanea senza trattamento allo sviluppo di una tetraplegia entro qualche anno.
L’esame ENMG mette in evidenza delle anomalie localizzate della conduzione nervosa (blocchi di conduzione o BC).
La patogenesi dell’affezione è incerta.Tuttavia, diversi elementi suggeriscono un’origine disimmunitaria. È soprattutto la frequente presenza di
anticorpi antigangliosidi GM1, la selettività dell’attacco delle fibre
motrici, la relativa efficacia dei trattamenti che interferiscono con la
risposta immunitaria o delle immunoglobuline.
Occorre segnalare l’assenza di effetto del Prednisone. L’attacco preferenziale delle fibre motrici risulta verosimilmente da una antigenicità differente delle fibre motorie e sensitive. La persistenza del BC si spiega forse
con l’assenza di remielinizzazione o la presenza di un fattore indeterminato che blocca i canali ionici. Le rare biopsie effettuate su un nervo
motorio hanno rilevato segni di demielinizzazione e remielinizzazione
cronica.
Malattie della
corna anteriore
82
Amiotrofia spinale o SMA (spinal muscular atrophy)
Le amiotrofie spinali sono malattie neuromuscolari ereditarie a trasmissione autosomica recessiva.
Esse sono dovute alla degenerazione dei motoneuroni del midollo spinale: i nervi motori non sono più in grado di trasmettere l’ordine di movimento ai muscoli. Questi ultimi si indeboliscono a causa dell’inattività e si
ritraggono.
Si distinguono diversi tipi di amiotrofie spinali a seconda dell’età di esordio e dell’evoluzione della malattia:
• Amiotrofia spinale di tipo I o forma acuta della malattia di WerdnigHoffmann, che compare prima dell’età di sei mesi; è la forma più grave
• Amiotrofia spinale di tipo II o forma intermedia della malattia di
Werdnig-Hoffmann, che compare all’età di 6-18 mesi
• Amiotrofia spinale di tipo III o malattia di Kugelberg-Welander, che
compare dopo che il bambino ha imparato a camminare (18 mesi-2 anni)
• Amiotrofia spinale di tipo IV, che si manifesta in età adulta.
La distinzione di queste forme secondo l’età d’esordio deve tener conto
del fatto che si tratta di termini orientativi e delle sovrapposizioni sono
possibili.
Il gene dell’amiotrofia spinale infantile (SMA) è stato localizzato sul braccio lungo del cromosoma 5 (5q).
Il deficit muscolare riguarda il tronco e le estremità (i muscoli prossimali
più che quelli distali, gli arti inferiori più degli arti superiori), é simmetrico, e risparmia i muscoli oculari, il diaframma, il miocardio. Non vi è partecipazione importante della muscolatura del viso.
83
Nella SMA di tipo I e II si osserva un arresto dello sviluppo motorio: i
bambini affetti da SMA di tipo II non sono in grado di camminare o di
stare in piedi in maniera autonoma. Pazienti affetti da SMA di tipo III
sono riusciti a camminare o a mantenersi in piedi.
L’evoluzione di queste malattie è variabile ed è difficile fare una prognosi
sicura. Tuttavia più l’inizio è precoce (fin dalla nascita o nel corso dei
primi mesi di vita), più la prognosi è grave.
Indipendente dall’importanza del deficit neurologico, l’evoluzione spontanea in assenza di cure ortopediche sfocia in deformazioni destinate a
diventare dolorose e a compromettere la qualità di vita. Per questa ragione è fondamentale seguire una terapia mirata. Essa permette ad alcuni dei
bambini colpiti di arrivare all’età adulta.
Questi tipi diversi si amiotrofia spinale sono dovuti a delle anomalie dello
stesso gene situato sul cromosoma 5. Questo gene è stato scoperto nel
1995 ed ora si può proporre alle coppie che hanno già un bambino colpito da questa malattia di effettuare una diagnosi prenatale in previsione di
una nuova gravidanza.
La messa a punto di modelli animali (topi affetti dalla malattia) permette
di comprendere in maniera più approfondita i meccanismi della malattia e
di ricercare delle strade terapeutiche innovative.
Sclerosi laterale amiotrofica
La sclerosi laterale amiotrofica è una malattia legata alla progressiva alterazione dei neuroni motori, le cellule che comandano ai muscoli volontari.
La malattia si manifesta tra i 45 e i 75 anni. Si tratta di una affezione invalidante dall’aggravamento progressivo, la cui evoluzione media è di circa 36
mesi. L’incidenza (50 nuovi casi all’anno in Svizzera) sembra in aumento
nei paesi industrializzati.
84
La cura consiste in un’azione coordinata di medici, di esperti in rieducazione e personale infermieristico. Essa associa un trattamento dei principali sintomi ad una rieducazione di mantenimento e alla considerazione
degli handicap.
Svariate scoperte fatte di recente hanno messo in evidenza il ruolo di
diversi fattori nella malattia: stress ossidante, aminoacidi eccitanti, fattori
di crescita cellulare. Queste scoperte permettono di ipotizzare nuove possibilità terapeutiche. Diversi test clinici sono in corso per valutare l’efficacia di molecole suscettibili di rallentare la malattia.
Associazione della Svizzera Romanda e Italiana contro le Miopatie
Chemin de la Traverse 12
Casella postale 179
CH-1170 AUBONNE
Tel. 021 808 74 11
Fax 021 808 81 11
E-mail [email protected]
Internet www.asrim.ch
CCP 10-15136-6