SIMULAZIONE MOLECOLARE DELLE INTERAZIONI
RECETTORE/SUBSTRATO: IL CASO DELL’ALFA-CHIMOTRIPSINA
E I BETA-CARBOETOSSI-GAMMA-LATTAMI
Sabrina Pricl1, Fulvia Felluga2
1
Dipartimento di Ingegneria Chimica, dell’Ambiente e delle Materie Prime, Università degli Studi di Trieste,
Piazzale Europa 1, 34127 Trieste
2
Dipartimento di Scienze Chimiche, Università degli Studi di Trieste, Via Giorgeri 1, 34127 Trieste
INTRODUZIONE
Il nucleo gamma-lattamico (o 2-pirrolidinonico) si trova in molti composti che
presentano importanti attività biologiche e farmacologiche. Gamma-lattami diversamente
sostituiti sono utilizzati, infatti, come agenti psicotropici, antagonisti dell’acido muscarinico, e
anti-ipertensivi (1,2). Di particolare interesse è la funzionalizzazione in posizione beta con un
gruppo carbossilico, poiché i corrispondenti derivati lattamici, a potenziale attività biologica,
sono poco caratterizzati in letteratura, particolarmente nelle loro forma otticamente attiva.
Esteri alchilici di questi derivati, sostituiti all’azoto con diversi raggruppamenti, sono
stati sottoposti a risoluzione cinetica con enzimi idrolitici quali esterasi, lipasi e proteasi.
L’alfa-chimostripsina (alfa-CT), in particolare, è in grado di idrolizzare con elevata
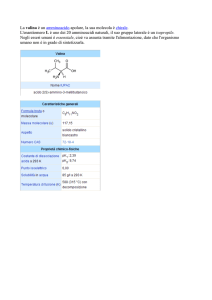
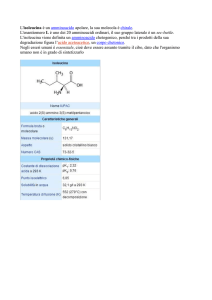
enantioselettività, e in maniera enantiocomplementare, i derivati 1 (metil 1-benzil-5-oxo-3pirrolidincarbossilato), 2 (metil 1-(2-idrossietil)-5-oxo-3-pirrolidincarbossilato) e 3 (metil 1isopropil-5-oxo-3-pirrolidincarbossilato), consentendo l’ottenimento dell’enantiomero (3S)-()-1 con 95% ee, (3R)-(+)-2 con 99% ee, e (3R)-(+)-3 con 96% ee, rispettivamente. Lo stesso
enzima, al contrario, non ha rivelato alcuna enantioselettività nei confronti del composto 4
(metil 5-oxo-3-pirrolidincarbossilato), che è stato recuperato dall’idrolisi in forma racema. La
Figura 1 riporta i modelli molecolari dei quattro gamma-lattami derivati appena descritti.
Figura 1: Modelli molecolari dei quattro gamma lattami sostituiti considerati in
questo lavoro. Partendo da sinistra: 1, 2, 3 e 4.
Precedenti studi delle relazioni tra struttura e reattività in reazioni di idrolisi di beta-arilalfa-aminoacidi e loro derivati da parte dell’alfa-CT hanno permesso di ottenere delle
informazioni sulle dimensioni del sito attivo, sulle interazioni enzima-substrato che sono in
grado di influenzare la reattività e la stereoselettività, nonché sulla orientazione reattiva e la
conformazione del substrato all’interno del sito attivo (3,4). Studi di diffrazione ai raggi X,
accoppiati alla determinazione della sequenza aminoacidica, hanno portato, sempre nel caso
citato in precedenza, alla dettagliata descrizione delle interazioni enzima-inibitore e enzimasubstrato (5,6). Così, il sito di binding coinvolge i frammenti 189-194 e 214-220, mentre il
gruppo da idrolizzare viene diretto verso l’essenziale legame idrogeno –O-H-N tra la Ser 195
e l’Hys 57.
In questo lavoro presentiamo i risultati dell’applicazione delle tecniche di simulazione
molecolare come mezzo di indagine microscopica delle origini della diversa enantioselettività
dei gamma-lattami 1, 2, 3 e 4 da parte dell’alfa-chimotripsina.
DETTAGLI COMPUTAZIONALI
Le simulazioni molecolari sono state condotte su una workstation Silicon Graphics
Origin 200. Per la predizione dell’interazione estere-proteina è stato usato il Software
AutoDock v. 3.0 (7), mentre le simulazioni di dinamica molecolare sono state effettuate
usando il Software Discover (Molecular Simulations Inc., USA) e varie routines FORTRAN
sviluppate in-house. La struttura 3-D dell’alfa-chimotripsina è stata ottimizzata partendo dalle
relative coordinate cristallografiche con un approccio meccanico-molecolare (MM) usando il
force field COMPASS (8) e ricorrendo ad un metodo misto di ottimizzazione gradiente
coniugato/Newton Raphson. La soglia di convergenza è stata posta uguale a 0.01 Kcal/mole
per Å. In ogni caso, una costante dielettrica effettiva ε = 4 è stata usata per tener conto della
schermatura dielettrica.
I dettagli strutturali delle molecole isolate, così come quelli dei complessi proteinaestere, sono stati ottenuti da simulazioni di dinamica molecolare (MD) nell’insieme canonico
(NVT) a 25C. Ogni simulazione MD è stata iniziata assegnando un set di velocità atomiche
iniziali in accordo con una distribuzione di tipo Maxwell-Boltzmann a 2xT, essendo T la
temperatura della simulazione. La temperatura è stata controllata con un accoppiamento
debole ad un bagno (9), con costante di accoppiamento T=0.01 ps. Le equazioni Newtoniane
del moto sono state integrate usando l’algoritmo di leapfrog Verlet (10), usando un intervallo
di integrazione pari a 1 fs. Il tempo totale di simulazione era pari a 20 ps. Le energie di
complessazione sono state calcolate a partire dalle componenti dell’energia ottenute dalla
simulazione dinamica per il complesso, la proteina ed i vari enantiomeri dei gamma-lattami
nonleg
nonleg
usando la seguente relazione: E compl = E cnonleg
omplesso − E alfa -CT − E estere .
RISULTATI E DISCUSSIONE
Come primo caso sono stati presi in considerazione i complessi alfa-CT con i due
enantiomeri del gamma-lattame 4, i cui risultati sperimentali dell’idrolisi enzimatica hanno
rivelato la non-enantioselettività della alfa-CT nei confronti di tale composto. La Figura 2
mostra i risultati del processo di docking dei due enantiomeri. L’analisi del sito di binding ha
rivelato che effettivamente, anche nel caso dei gamma-lattami ciclici, tale sito risiede proprio
nella tasca compresa tra i frammenti 189-194 e 214-220. I due enantiomeri assumono due
orientazioni diverse all’interno del sito di binding, in modo da accomodare il gruppo estereo
all’interno della tasca proteica; tuttavia, in entrambi i casi, il gruppo COOMe è rivolto verso i
residui Ser 195 e Hys 57, come nel caso degli analoghi aciclici o dei beta-lattami.
Figura 2: Confronto tra il modello molecolare risultante dal processo di docking
dell’enantiomero R del composto 4 (a sinistra) e dell’enantiomero S del composto 4 (a destra).
La struttura di equilibrio del complesso alfa-CT/enantiomero R del metil 5-oxo-3pirrolidincarbossilato (composto 4) è caratterizzata da un pattern di legami idrogeno
intermolecolari che interessa, rispettivamente, l’ossigeno del gruppo OCH3 dell’estere e l’NH
del legame peptidico tra la Ser 195 e l’ASP194, l’ossigeno del gruppo CO dell’estere e l’NH
del legame peptidico tra la Gly 193 e la Met 192, e infine l’ossigeno del gruppo CO
dell’anello lattamico e l’NH del legame peptidico tra la Gly 126 e il Trp 125. La
corrispondente struttura di equilibrio del complesso alfa-CT/enantiomero S del metil 5-oxo-3pirrolidincarbossilato è anch’essa caratterizzata da un pattern di legami H simile, che
coinvolgono tuttavia l’ossigeno del gruppo OCH3 e l’H del gruppo OH della Ser 195,
l’idrogeno dell’NH dell’anello lattamico e l’ossigeno del legame peptidico tra la Ser 190 e la
Cys 191 e, infine, l’ossigeno del gruppo CO dell’anello lattamico e l’NH del legame peptidico
tra Cys 191 e Ser 190. Da un punto di vista energetico, l’analisi dell’energia di binding
dell’alfa-CT con i due enantiomeri del composto 4 ha rivelato che i due complessi sono
caratterizzati da valori molto simili, essendo pari a –6.5 kcal/mole per l’enantiomero R e –6.2
kcal/mole per l’enantiomero S. Tali evidenze sono decisamente consistenti con l’evidenza
sperimentale della non enantioselettività dell’alfa-CT nei confronti del composto 4.
Diverso è il caso, ad esempio, dei due enantiomeri del derivato 1 (metil 1-benzil-5-oxo3-pirrolidincarbossilato). Ancora una volta l’analisi di docking ha rivelato che il binding site
per entrambi gli enantiomeri è lo stesso descritto in precedenza, e i relativi modelli molecolari
sono riportati in Figura 3.
Figura 3: Confronto tra il modello molecolare risultante dal processo di docking dell’enantiomero R
del derivato 1 (a sinistra) e dell’enantiomero S del derivato 1 (a destra).
Tuttavia, nel caso dei due enantiomeri del derivato 1, l’analisi della simulazione di
dinamica molecolare ha rivelato che vi è una sostanziale differenza tra il numero di legami
idrogeno che si instaurano tra il sito attivo e le molecole ospiti. Infatti, nel caso
dell’enantiomero 1-R, la traiettoria rivela la formazione di legami H solamente tra il gruppo
OCH3 dell’estere e l’NH del legame peptidico tra la Ser 195 e l’Asp194 e ancora tra lo stesso
gruppo dell’estere e il gruppo NH del legame peptidico tra la Gly 193 e la Met 192.
L’enantiomero 1-S, viceversa, presenta, oltre allo stesso pattern di legami H coinvolgente il
gruppo OCH3, un ponte H tra il gruppo CO dell’anello lattamico e il gruppo NH del legame
peptidico tra la Gly 126 ed il Trp 125. Infine, in quest’ultimo caso, le interazioni idrofobiche
tra la tasca formata dai residui 189-194 e 214-220 e l’anello aromatico del gamma-lattone
risultano più favorevoli, in virtù della migliore orientazione di quest’ultimo all’interno del sito
di binding.
Questo trova riscontro nelle relative energie di binding calcolate, che risultano essere
pari a –13.6 kcal/mole per l’isomero 1-S ed a +6.8 kcal/mole nel caso dell’enantiomero1-R.
L’elevata affinità dell’alfa-CT per l’enantiomero 1-S è il risultato di un effetto sinergico tra le
componenti dell’energia di binding di natura elettrostatica e/o di dispersione e le interazioni
idrofobiche. L’energia di interazione notevolmente minore del complesso alfa-CT/(3S)-(-)(metil 1-benzil-5-oxo-3-pirrolidincarbossilato) può essere considerata come un’indicazione
della formazione di un complesso di inclusione termodinamicamente più favorito, e tale
risultato è consistente con le evidenze sperimentali della idrolisi enantioselettiva dell’alfa-CT
nei confronti di tale enantiomero.
RIFERIMENTI BIBLIOGRAFICI
(1) Nilsson, B.M., Ringdahl, B., Hacksell, U., J. Med. Chem., 33, 580 (1990).
(2) Bergmann, R., Gericke, R., J. Med. Chem., 33, 492 (1990).
(3) Cohen, S.G., Trans. N.Y. Acad. Sci., 31, 705 (1969).
(4) Silver, M.S., Stoddard, M., Stone, T., Matta, M.S., J. Amer. Chem. Soc., 42, 3151 (1970).
(5) Steitz, T.A., Henderson, R., Blow, D.M., J. Mol. Biol., 46, 337 (1969).
(6) Blow, D.M., Israel J. Chem. 12, 483 (1974).
(7) Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., Olson,
A.J., J. Comp. Chem., 19, 1639 (1998).
(8) Sun, H., J. Phys. Chem., 102, 7338 (1998).
(9) Berendsen, H.J.C., Postma, J.P.M:, Di Nola, A., van Gunsteren, W.F. and Haak, J.R., J.
Chem. Phys., 81, 3684 (1984).
(10) Verlet, L., Phys. Rev., 159, 98 (1967).