UNIVERSITA’ DI PISA
Facoltà di Scienze Matematiche, Fisiche e Naturali
Corso di Laurea Specialistica in SCIENZE BIOLOGICHE MOLECOLARI
Indirizzo: SCIENZE FISIOPATOLOGICHE GENERALI
IL RUOLO DEL PEPTIDE NATRIURETICO DI TIPO C
(CNP) NELL’INSUFFICIENZA CARDIACA CRONICA:
STUDIO IN UN MODELLO SPERIMENTALE ANIMALE
Candidata
Manuela Cabiati
.....................................
Relatori
Dott.ssa Silvia Del Ry
Dott.ssa Daniela Giannessi
.....................................
.....................................
Sessione di Laurea 23 Novembre 2006
Anno Accademico 2005/2006
INDICE
SOMMARIO
……………………………….………………………..
pag.
1
ABSTRACT
………………………………………………………….
»
.5
pag.
8
1.1 Cenni storici ………………………………………….
»
8
1.2 Struttura peptidica e biosintesi intracellulare ………..
»
10
1.3 Secrezione dei peptidi natriuretici …………………..
»
19
1.4 Struttura genica ed espressione tessutale ……………
»
21
1.5 Recettori e metabolismo ………………………….....
»
24
1.6 Degradazione dei peptidi natriuretici ………………
»
31
1.7 Azioni biologiche dei peptidi natriuretici ……………
»
32
CAPITOLO PRIMO
I PEPTIDI NATRIURETICI
CAPITOLO SECONDO
LO SCOMPENSO CARDIACO
pag. 43
2.1 Eziologia dello scompenso cardiaco ………………....
»
44
2.2 Fisiopatologia dello scompenso cardiaco
……………
»
45
…………………………..
»
46
2.4 Il sistema chemorecettoriale e barorecettoriale ……...
»
52
2.5 Il sistema renina-angiotensina
……………………….
»
55
2.6 Sistema dell’Endotelina ………………………………
»
56
2.7 Sistema della vasopressina ……………………………
»
57
2.3 Il modello neuroendocrino
CAPITOLO TERZO
CNP E SCOMPENSO CARDIACO
pag. 59
CAPITOLO QUARTO
DISFUNZIONE ENDOTELIALE E
MALATTIA CARDIOVASCOLARE
pag. 66
CAPITOLO QUINTO
SCOPO DEL LAVORO DI TESI
pag. 71
CAPITOLO SESTO
pag. 73
MATERIALI E METODI
»
73
……...
»
77
………..
»
78
…….....................………………
»
78
6.5 Protocollo Sperimentale ………………………………
»
79
6.5.1 Estrazione dell’RNA totale ……………………….
»
79
6.5.2 Determinazione dell’RNA totale
…………………
»
80
6.5.3 Estrazione tessutale delle proteine …..……………
»
82
»
83
»
86
»
86
……………………... »
86
6.1 Modello sperimentale animale
...…………………….
6.2 Campioni biologici: tessuto cardiaco di maiale
6.3 Campioni biologici: tessuto cardiaco di topo
6.4 Campioni plasmatici
6.6 Analisi di RT-PCR (Reverse Transcription-Polymerase
Chain Reaction) semiquantitativa
…………………...
6.7 Saggio radioimmunologico per la misura del CNP ….
6.7.1 Estrazione dei campioni ………………………….
6.7.2 Saggio Radioimmunologico
6.8 Analisi statistica dei dati ……………………………..
»
87
CAPITOLO SETTIMO
RISULTATI
pag. 88
»
88
…………………….…..
»
88
7.1.2 Reazioni di RT-PCR ………………………………
»
89
………………………………..
»
91
7.2.1 RT-PCR su tessuto cardiaco di topo ……………..
»
91
7.2.2 RT-PCR su tessuto cardiaco di maiale …………...
»
91
7.2.3 Sequenziamento e analisi dei dati ………………..
»
94
7.2.4 Sottomissione alla GenBank ....…………………...
»
96
7.3 Risultati relativi al modello sperimentale …………….
»
98
7.1 Risultati metodologici ………………………………...
7.1.1 Quantizzazione dell’ RNA
7.2 Sequenziamento dell’mRNA codificante
per l’NPR-B di maiale
CAPITOLO OTTAVO
CONCLUSIONI
pag. 104
ELENCO DEGLI ACRONIMI ……………………………..
»
108
BIBLIOGRAFIA
»
.111
…………………………………...……..
RINGRAZIAMENTI
Sommario
SOMMARIO
Titolo
della tesi: Il ruolo del peptide natriuretico di tipo C (CNP)
nell’insufficienza cardiaca cronica: studio in un modello sperimentale
animale.
Introduzione: Il peptide natriuretico di tipo C (CNP) appartiene alla
famiglia dei peptidi natriuretici e mostra proprietà strutturali e fisiologiche
simili a quelle del peptide natriuretico atriale (ANP) e del peptide
natriuretico di tipo B (BNP). Il CNP è prodotto principalmente
dall’endotelio e solo recentemente è stata evidenziata una sua produzione
a livello cardiaco in pazienti con scompenso cardiaco (SC). In questa
situazione anche i livelli plasmatici di CNP risultano elevati in relazione
alla severità della malattia, analogamente a quanto osservato per ANP e
BNP, ma, mentre il ruolo patofisiologico dell’ANP e del BNP è
ampiamente dimostrato, quello del CNP non è ancora completamente
definito.
Lo scompenso cardiaco cronico, una sindrome clinica determinata
dall’incapacità del cuore di esercitare la sua funzione di pompa in maniera
adeguata alle richieste metaboliche dell’organismo, dà origine a una serie
di alterazioni a livello emodinamico e neuroormonale che includono
l’attivazione di molti sistemi neuroendocrini, principalmente il sistema
nervoso adrenergico e il sistema renina-angiotensina-aldosterone e altri
sistemi vasoattivi, quali quello delle endoteline e dei peptidi natriuretici.
1
Sommario
Sebbene sia stata dimostrata una produzione miocardica di CNP in
pazienti con SC, non è ancora stato determinato il sito di produzione del
peptide e le eventuali differenze tra produzione atriale e ventricolare.
Scopo del lavoro di tesi: scopo di questa tesi è stato valutare il ruolo del
CNP nello SC in un modello sperimentale animale di maiale con
insufficienza cardiaca cronica indotta da stimolo con pace-maker ad alta
frequenza; per questo sono stati determinati i livelli plasmatici di CNP e
l’espressione cardiaca, a livello di mRNA e di proteina, del peptide e del
suo recettore biologico specifico, NPR-B.
Materiali e Metodi: sono stati studiati complessivamente 10 animali da
esperimento (minipig, maschi adulti, 35-40 kg). Ad ogni animale
sottoposto a esperimento (n=5) è stato applicato un pace-maker unipolare
nella parete ventricolare sinistra e lo scompenso cardiaco è stato indotto
stimolando l’animale per 4 settimane (180 battiti/min). La condizione
clinica è stata valutata tramite tecniche ecocardiografiche, Tomografia ad
Emissione di Positroni e Risonanza Magnetica. Come controllo (n=5)
sono stati utilizzati minipig adulti, non sottoposti ad alcun trattamento e
mantenuti nelle stesse condizioni degli animali sottoposti a stimolazione
cardiaca.
I campioni di sangue sono stati raccolti in EDTA e aprotinina prima
(basale) e a 10 minuti, 1, 2, 3 e 4 settimane dall’applicazione dello
stimolatore cardiaco mentre i campioni di tessuto cardiaco (atrio e
ventricolo,
destro
e
sinistro),
sono
stati
raccolti
al
termine
dell’esperimento. Le proteine e l’mRNA sono stati estratti dallo stesso
2
Sommario
campione di tessuto cardiaco con il metodo del fenolo/guanidinatiocianato/cloroformio. L’espressione cardiaca dell’mRNA codificante per
il CNP, per l’ NPR-B e, come controllo, per il BNP (di cui è ben nota
l’espressione a livello cardiaco) è stata determinata con RT-PCR dopo
opportuna messa a punto del metodo. I livelli plasmatici di CNP, nel
plasma e negli estratti cardiaci, sono stati determinati con un metodo
radioimmunologico specifico dopo preliminare estrazione in fase solida su
Sep-Pak C18.
Risultati: l’affidabilità dell’analisi tramite RT-PCR semiquantitativa nel
tessuto cardiaco di maiale è stata valutata per ciascun effettore con prove
sperimentali dedicate. L’espressione dello mRNA codificante per il CNP è
risultata indotta dopo scompenso cardiaco in parallelo con quella del BNP,
sebbene i livelli di CNP siano circa 10 volte inferiori.
Nel corso di questo lavoro di tesi è stato sequenziato l’mRNA codificante
per il recettore NPR-B di maiale (GenBank DQ 487044; 1-396 pb)
mancante nella GenBank, usando primer di Mus musculus per l’NPR-B,
grazie alla alta omologia fra specie. Il recettore NPR-B è risultato espresso
nelle diverse camere cardiache sia negli animali di controllo che in quelli
scompensati, confermando la presenza di questo recettore nel tessuto
cardiaco di maiale.
I livelli plasmatici di CNP sono risultati significativamente aumentati
rispetto ai controlli a partire dalla prima settimana 36,9 ± 10,4 pg/ml vs.
16,7 ± 1,1 pg/ml, media ± sem, p=0.013. Per quanto riguarda gli estratti
tissutali, in condizioni basali livelli misurabili di CNP sono stati trovati in
tutte le camere cardiache; la concentrazione osservata negli atrii (dx: 13,7
3
Sommario
± 1,9 pg/mg; sin: 8,7±3,8 pg/mg) è risultata circa 10 volte più elevata
rispetto ai ventricoli (dx: 1,07 ± 0,33 pg/mg; sin: 0,93 ± 0,17 pg/mg).
Dopo 4 settimane di stimolazione con pace-maker ad alta frequenza, i
livelli miocardici di CNP sono risultati aumentati particolarmente nel
ventricolo sinistro (15,8 ± 9,9 pg/mg vs. 0,9 ± 0.17 pg/mg, p=0,01).
Conclusioni: questi dati indicano che il CNP e il suo recettore biologico
hanno un ruolo rilevante nella patofisiologia dello scompenso cardiaco,
sebbene ulteriori studi siano necessari per la completa definizione della
funzione di questo peptide nello SC. In particolare, la conoscenza della
sequenza del NPR-B nel maiale potrebbe costituire un importante
strumento per studiare il coinvolgimento di questo recettore nella
regolazione di varie patologie che possono essere studiate con questo tipo
di modello animale.
4
Abstract
ABSTRACT
Title: Myocardial C-type natriuretic peptide in an experimental model of
pacing-induced heart failure.
Background: C-type natriuretic peptide (CNP), a member of the family of
natriuretic peptides, was recently found to be produced in the
myocardium, but its cellular source and the possible difference between
atrium and ventricle production are so far lacking.
Aim: The main goal of this study was to evaluate, in an experimental
model of pacing induced heart failure (HF), the role of CNP in this
condition by measuring the circulating levels of this peptide and the
expression, at mRNA and protein level, of CNP as well as of its specific
receptor, NPR-B. For this latter issue it will be necessary to sequence and
determine the cardiac expression of NPR-B in Sus Scrofa, lacking in
GenBank.
Methods: Adult male minipigs (n=5) were chronically instrumented with a
unipolar pacemaker connected to the anterior left ventricular (LV) wall.
HF was induced by rapid pacing (180 beats/min) for 4 weeks. End-stage
HF occurred at 24±2 days of pacing when the LV end-diastolic pressure
was ≥25 mmHg. As control, we studied 5 adult male minipigs.
5
Abstract
Blood samples were collected (aprotinin and EDTA) at rest (control) and
at 10 min, 1, 2, 3 and 4 weeks of pacing stress. Myocardial samples were
collected at the end of the experiment (4 weeks). Both mRNA and proteins
were extracted from a same sample, obtained from the different cardiac
chambers,
by
using
the
method
of
phenol/guanidine-
thiocyanate/chloroform.
CNP levels in plasma and in cardiac extracts were determined by a
radioimmunoassay after a preliminary extraction on Sep-Pak C18, while
the expression of mRNA coding for CNP in myocardial tissue by RTPCR. As overall control, a parallel RT-PCR assay for BNP mRNA
expression was carried out in the same samples.
Results: Compared to controls, plasmatic CNP levels were increased after
1 week of pacing stress (36.9±10.4 pg/ml vs. 16.7±1.1, p=0.013,
mean±sem). As to myocardial extract, at rest CNP was found in all cardiac
chambers and its content was ten fold higher in atria than in ventricles
(RA: 13.7±1.9 pg/mg; LA: 8.7±3.8 pg/mg; RV: 1.07±0.33 pg/mg;
LV:0.93±0.17). At 4 weeks of pacing stress, myocardial levels of CNP
were higher in left ventricle than in controls (15.8±9.9 pg/mg vs. 0.9±0.17
pg/mg, p=0.01). The expression of mRNA coding for CNP was observed
at 4 weeks of pacing althought CNP gene expression appears to be
noticeable lower than that of BNP.
During this degree thesis we also sequenced Sus scrofa natriuretic peptide
receptor 2 mRNA, 1-396 pb, that was submitted to GenBank (accession
number DQ487044) and made available to all researchers that study this
widely used animal model. Bands obtained with pig cardiac tissue shared a
6
Abstract
93% sequence homology with a region of mouse NPR-B and 95% with
Homo Sapiens. NPR-B receptors were found in all the cardiac regions,
both in controls and in HF animals.
Conclusion: In this study we demostrated that CNP and its biological
receptor have an important role in chronic heart failure phatophysiology,
although
further
studies
are
necessary
to
better
clarify
the
pathophysiological function of this peptide in chronic heart failure. In
addition, the knowledge of the sequence of NPR-B receptor in the pig
could constitute an important tool to study the involvment of this receptor
in the regulation of several diseases that could be studied with this animal
model.
7
I peptidi natriuretici
1
I PEPTIDI NATRIURETICI
Capitolo
1.1 Cenni storici
Fino a pochi anni fa, si riteneva che la pressione sanguigna ed il volume
dei fluidi extracellulari fossero regolati esclusivamente dal rene e dal
sistema nervoso autonomo in associazione con fattori umorali come il
sistema renina-angiotensina-aldosterone ed arginina-vasopressina.
Sebbene numerose sperimentazioni fisiologiche avessero già da tempo
individuato un legame umorale tra l’apparato cardiocircolatorio e quello
renale soltanto nel 1979 un gruppo di studiosi americani, sotto la guida di
Adolfo De Bold, rilevarono la funzione fisiologica di questi granuli e la
natura del secreto prodotto (De Bold A.J. et. al., 1981). Essi identificarono
in estratti atriali di ratto un fattore ad azione natriuretica ed ipotensiva: il
peptide natriuretico atriale od ANP e descrissero come i granuli atriali
venissero modificati dai cambiamenti del bilancio osmotico ed elettrolitico
operato a livello renale. De Bold e colleghi scoprirono che
somministrando intravena a ratti un omogenato atriale, veniva indotto un
rapido aumento della pressione sanguigna, accompagnato da una rapida e
massiva natriuresi.
In seguito a questa preliminare osservazione, vennero purificati, a livello
del tessuto atriale, numerosi peptidi, di varie dimensioni, dotati di attività
natriuretica e capaci di indurre rilassamento delle fibre muscolari lisce. A
questi peptidi sono stati assegnati diversi nomi, tra cui cardionatrine,
cardiodilatine, atriopeptine, ma attualmente il più utilizzato è quello di
peptide natriuretico atriale (ANP).
8
I peptidi natriuretici
Tuttavia la dimostrazione dell’attività endocrina del cuore poggia anche su
osservazioni precedenti.
Infatti già nel 1847 Harthshorne registrò l’effetto diuretico ed ipotensivo
conseguente all’immersione totale del corpo in acqua, ed ipotizzò
l’esistenza di recettori di volume sensibili all’incremento del riempimento
cardiaco (Harsthone H., 1847).
Nel 1935 Peters e collaboratori riproposero la stessa teoria che fu
convalidata, nel 1956, dai successivi esperimenti di Henry, Gauer e Reevs
(Henry J.P. et al., 1965). Nello stesso anno Kish, utilizzando la
microscopia elettronica nell’analisi istolologica, riportò la prima evidenza
diretta dell’esistenza, in miociti atriali di ratto, di “specifici corpi”
elettrondensi rivestiti di membrana ed inseriti fra gli elementi di un
voluminoso complesso del Golgi, in prossimità dei mitocondri (Kish B.,
1956), con caratteristiche morfologiche analoghe ai granuli secretori
presenti nelle cellule endocrine
Nel 1982 De Bold confermò la relazione esistente tra i granuli atriali e il
fattore natriuretico e nel 1983 Flynn e collaboratori disegnarono la
struttura attuale dell’ANP.
Nel 1988, Sudoh e colleghi isolarono dal tessuto cerebrale porcino un
peptide con attività biologica simile all’ANP e denominato peptide
natriuretico cerebrale o di tipo B (BNP) sebbene venisse prodotto
ampiamente a livello dei ventricoli cardiaci (Sudoh T. et al., 1988; Sudho
T. et al., 1990). A distanza di due anni dalla scoperta del BNP, Sudoh
identificò, a livello del sistema nervoso centrale di suino, un terzo peptide
strutturalmente simile all’ANP e BNP, chiamato CNP (peptide natriuretico
di tipo C). Queste osservazioni furono estese a tutti i mammiferi da altri
9
I peptidi natriuretici
ricercatori che evidenziarono, e continuano tuttora a rilevare, la natura
proteica del contenuto vescicolare e la variabilità numerica dei granuli in
relazione alla specie di appartenenza ed al contenuto di sodio nella dieta
(De Bold AJ. et al., 1983; Huet M. et al., 1974; Jamieson JD. et al.,1964).
1.2 Struttura peptidica e biosintesi intracellulare
Nei
mammiferi
attualmente
si
riconoscono
numerosi
membri
strutturalmente e funzionalmente correlati, appartenenti a questa famiglia:
il peptide natriuretico atriale (ANP), il peptide natriuretico di tipo
cerebrale (BNP), il peptide natriuretico di tipo C (CNP), DNP
(dendroaspis natriuretic peptide) e l’urodilatina (De Bold AJ. et. al., 1981;
Baxter GF., 2004) (Fig. 1). Analisi sull’evoluzione di BNP e ANP
dimostrano che tali peptidi derivano da eventi di duplicazione all’interno
del gene codificante per il CNP.
Tutti i membri della famiglia dei peptidi natriuretici sono strutturalmente
correlati tra loro; ogni ormone viene sintetizzato a partire da un
precursore, il preproormone, che viene processato attraverso vari passaggi
fino al raggiungimento della forma biologicamente attiva del peptide che
costituisce la parte COOH-terminale del precursore.
L’attività biologica dei peptidi natriuretici risiede in una struttura ad anello
di 17 aminoacidi, formata da un ponte disolfuro intramolecolare dal quale
si dipartono una terminazione amminica ed una coda carbossilica (assente
solo nel CNP); la distruzione dell’anello mediante un taglio idrolitico
comporta la perdita dell’attività biologica.
10
I peptidi natriuretici
Figura 1: Struttura dei peptidi natriuretici
11
I peptidi natriuretici
L’ANP è considerato il prototipo fra gli ormoni della famiglia dei peptidi
natriuretici nell’uomo, la forma circolante che possiede attività biologica è
costituita da 28 aminoacidi.
La sua sequenza amminoacidica è altamente conservata fra le varie specie:
nei mammiferi la variabilità della sequenza da una specie ad un’altra si
limita al solo residuo in posizione 110, che nell’uomo, nel maiale, nel
cane, nel bovino e nell’ovino è una metionina, mentre nel coniglio, nel
ratto e nel topo è una isoleucina.
L’ANP deriva da una grande molecola precursore, di 15 kD e 151
amminoacidi (uomo, cane, mucca) o di 152 amminoacidi (ratto e topo),
chiamata prepro-ANP (Kangawa A. et. al.,1984; Nakayama K. et.
al.,1984; Oikawa S. et. al.,1985; Zivin RA. et. al.,1984).
Quest’ultima include, all’estremità amminica, una sequenza segnale
idrofobica N-terminale, necessaria per la traslocazione del peptide
nascente dal ribosoma al reticolo endoplasmatico ruvido, dove una
endopeptidasi di membrana idrolizza il peptide segnale e forma il proANP di 126 aminoacidi (γANP o ANP1-126), che si trova principalmente
contenuto nei granuli atriali.
Durante l’accumulo nei granuli il pro-ANP, viene scisso (Michener ML.,
1986, Levin ER. et. al.,1998; Ruskoaho H.,1992) nel frammento attivo
COOH-terminale, di 3 kD e 28 amminoacidi (αh ANP1-28 o ANP99-126)
ed in un frammento ammino terminale (pro ANP1-98).
Da quest’ultimo frammento si formano, dopo la secrezione in circolo ad
opera di serinproteasi transmembrana (atrioactivasi), chiamate corine, altri
12
I peptidi natriuretici
tre fattori ad azione natriuretica, kaliuretica od ipotensiva (Vesely DL. et.
al.,1994; Gower WR. et. al.,1994 ):
1) pro ANP1-30 o stimolatore di sodio ad azione duratura;
2) pro ANP31-67 o dilatore vasale;
3) pro ANP79-98 od ormone kaliuretico (Fig. 2).
Se l’enzima processante è attivato nei granuli, deve esistere un
meccanismo che ne garantisca l’attivazione solo al momento opportuno.
È probabile che ad elevate concentrazioni di calcio ed elevata forza ionica
la serin-proteasi sia inattiva, mentre in risposta all’adeguato stimolo
fisiologico le condizioni intravescicolari (Ca++, forza ionica, pH)
verrebbero alterate variando l’attività enzimatica, così da produrre
l’ormone immediatamente prima o durante la fusione esocitotica dei
granuli con la membrana plasmatici.
Il processamento può subire delle modifiche in tessuti extracardiaci: nel
cervello si ottengono i peptidi ANP102-126 ed ANP103-126, mentre il rene
genera un’unica forma, detta urodilatina (proANP95-126), che promuove
con modalità paracrina, l’escrezione tubulare del sodio (Drummer C. et
al.,1996; Goetz KL., 1991).
Le proprietà biologiche intrinseche dell’ANP si riscontrano anche nel
BNP che pur mantnendo il distintivo modello ad anello di 17
amminoacidi, è il peptide meno conservato tra le specie quanto a
lunghezza e sequenza amminoacidica (Davidson NC. et al., 1994).
Esiste infatti una maggiore omologia tra l’ANP e il BNP intraspecifici, che
non fra le molecole di BNP appartenenti a specie diverse.
Il BNP fu inizialmente purificato da estratti cerebrali di suino e per tale
motivo venne identificato come peptide natriuretico cerebrale (brain
13
I peptidi natriuretici
natriuretic pepide) (Masashi M. et. al.,1991; Ruskoaho H.,1992; Levin ER.
et. al.,1998; Stein BC. et al., 1998; Sagnella GA., 1998).
Successivamente venne rilevato un aumento della sua concentrazione
principalmente nel ventricolo cardiaco sinistro di pazienti e di animali da
esperimento affetti da patologie cardiache come lo scompenso cardiaco
cronico o nell’infarto miocardio; per questo venne ritenuto opportuno
riferirsi al BNP come peptide natriuretico di tipo B piuttosto che peptide
natriuretico cerebrale.
Il BNP umano è sintetizzato a partire da un prepro-ormone di 134
aminoacidi (Sagnella GA., 1998), contenente una sequenza segnale di 26
amminoacidi, il proBNP, dalla cui eliminazione deriva un proormone di
108 amminoacidi (proBNP1-108) che viene racchiuso in granuli solo negli
atri insieme alla proteina biologicamente attiva. Una seconda proteasi
media la scissione del pro-BNP in due residui, un residuo amminoterminale costituito da 76 amminoacidi NT-proBNP, che sembra non
possedere attività ormonale (Mair J. et al., 2001) e un residuo carbossiterminale di 32 amminoacidi dotato di attività biologica (Fig. 2).
Il BNP è conservato in granuli a livello atriale assieme all’ANP, ma non
nei ventricoli dove la sua produzione è regolata dallo stiramento della
parete cardiaca.
Sebbene originariamente isolato, come il CNP, da estratti cerebrali di
maiale, il BNP è stato identificato come il normale prodotto del tessuto
cardiaco e nonostante i primi studi avessero ipotizzato una sua produzione
soprattutto nei miociti ventricolari, ora è ben noto che il BNP è prodotto e
secreto sia a livello atriale che ventricolare.
14
I peptidi natriuretici
Come l’ANP e il BNP, il DNP esercita un’azione vasodilatante, ma il suo
ruolo e le sue funzioni sono stati fino ad oggi scarsamente determinati,
sebbene si sia dimostrata l’immunoreattività del DNP a livello plasmatico
(Schirger JA. et al., 1999; Best PJ. et al., 2002).
Figura 2: Produzione e secrezione di ANP e BNP
15
I peptidi natriuretici
L’urodilatina è un peptide carbossi-terminale di 32 amminoacidi derivante
dal taglio alternativo, mediato da proteasi, del pro-ANP a livello dei tubuli
renali (Schulz-Knappe P. et al., 1988), e può essere considerato come un
peptide natriuretico di derivazione renale. Questo peptide fu inizialmente
isolato in campioni umani di urina e vennero in seguito dimostrate sia la
sua importanza nella regolazione dell’escrezione renale di sodio e acqua
che i suoi effetti benefici nello scompenso cardiaco (Schulz-Knappe P. et
al., 1988; Elsner D. et al., 1995).
Limitate sono ancora le conoscenze riguardanti il peptide natriuretico di
tipo C. Il CNP, analogamente all’ANP, presenta una sequenza
amminoacidica altamente conservata nelle diverse specie.
Il CNP, venne inizialmente isolato da cervello porcino nel 1990 (Sudoh T.
et al., 1990) e sebbene venga considerato come neurotrasmettitore a livello
del sistema nervoso centrale, ha un’ampia distribuzione che include
l’endotelio vascolare, il miocardio e i tratti gastrointestinale e urogenitale,
dove gioca un ruolo importante nel controllo del tono vascolare in
associazione con i sistemi locali (Abassi Z. et al., 2004; Barr CS. et al.,
1996; Scotland RS. et al., 2005).
E’ stato infatti osservato che il CNP viene prodotto nelle cellule
endoteliali dove è in grado di indurre vasorilassamento attraverso
iperpolarizzazione delle cellule muscolari lisce (Barton M. et al., 1998),
suggerendo che il CNP può rappresentare un fattore iperpolarizzante di
origine endoteliale, EDHF (endothelium derived hyperpolarizing factor),
ed avere azione paracrina, come altri mediatori vasorilassanti endoteliali
quali il monossido di azoto (NO) e la prostaciclina. Studi recenti hanno
inoltre dimostrato che nel letto vascolare coronarico il CNP può agire
16
I peptidi natriuretici
come un EDHF, confermando il suo ruolo prioritario nella regolazione del
tono vascolare e del flusso sanguigno (Chauhan SD. et al., 2003; Griffith
TM., 2004).
Alte concentrazioni di CNP sono state trovate nel sistema nervoso centrale
di ratto, maiale e uomo e in minore quantità in altri distretti tessutali come
l’intestino, il rene e il cuore (Komatsu JB. et al., 1991; Vollmar AM. et al.,
1993). Inoltre nella circolazione periferica umana sono presenti quantità
misurabili di CNP la cui origine è probabilmente endoteliale o cardiaca.
Il CNP è un peptide costituito da 22 amminoacidi (peso molecolare di
circa 2200) (Sudoh T. et al., 1990) e presenta omologie fisiologiche e
strutturali con ANP e BNP, ma si differenzia per una più breve emivita
circolatoria. Rispetto ai peptidi natriuretici ANP e BNP è altamente
conservato tra le diverse specie (incluse uomo, roditori, rettili, pesci e
specie primitive) (Barr CS. et al., 1996; Takei Y., 2001; Fowkes RC.,
2000). L’attività biologica di tutti i peptidi natriuretici risiede nella
struttura centrale ad anello di 17 amminoacidi, generata da un ponte
disolfuro intramolecolare, che conferisce alla molecola la sua attività
biologica (Schiller PW. et al., 1986; Furuya M.).
Nell’uomo il gene del CNP è localizzato sul cromosoma 2 a differenza del
BNP edell’ANP entrambi localizzati sul cromosoma 1.
Il CNP umano è sintetizzato a partire da un prepro-ormone di 126
aminoacidi, chiamato prepro-CNP (Fig. 3), contenente una sequenza
segnale dalla cui eliminazione, mediata da una peptidasi segnale, deriva un
proormone (NTpro-CNP). Una successiva scissione del pro-CNP
catalizzata dalla furina, una protein-convertasi ubiquitaria dell’Apparato
del Golgi, porta alla formazione del CNP53 (Wu C. et al., 2003; Tawaragi
17
I peptidi natriuretici
Y. et al., 1991). Questo peptide può essere secreto e/o processato,
attraverso un meccanismo non ancora identificato, per ottenere la forma
attiva del CNP, il CNP22.
Il CNP53 ha un peso molecolare superiore al CNP22 e si ritrova
prevalentemente nei tessuti, a differenza del CNP22 che si rileva
maggiormente nel plasma e nel liquido cerebrospinale (Stingo AJ. et al.,
1992; Togashi K. et al., 1992).
Figura 3: Secrezione e produzione di CNP
18
I peptidi natriuretici
1.3 Secrezione dei peptidi natriuretici
Contrariamente al CNP, l’ANP e il BNP rappresentano un duplice sistema
di ormoni circolanti i cui livelli sono influenzati dalla secrezione cardiaca
(De Bold A.J. et al. , 1996; Ruskoaho H. et al., 1997).
Si possono distinguere due modalità di secrezione: la prima, detta
regolatoria, viene modulata attraverso l’immagazzinamento del peptide in
granuli ed il suo rilascio avviene in risposta ad un secondo messaggero,
solitamente il calcio; la seconda, detta costitutiva, consente la rapida
secrezione dell’ormone subito dopo la sua sintesi (Lingappa V.R., 1989;
De Bold A.J. et al. , 1996; Ruskoaho H. et al., 1997).
L’ANP è secreto prevalentemente dai cardiomiociti atriali, dove è
conservato in granuli, in risposta allo stiramento locale della parete
cardiaca, dovuto ad un aumento del volume intravascolare basali
(Ruskoaho H.,1992).
Una volta secreto l’ANP si comporta come un ormone e perfonde nel
seno coronario e passa in circolo a raggiungere gli organi bersaglio dove
esplica la sua azione.
Numerosi stimoli, fisiologici e/o patologici, possono incrementare la
secrezione dell’ormone, fra cui: la postura supina, gli stati di aumentata
volemia, l’ipossia, l’ischemia del miocardio e l’aumento della frequenza
cardiaca.
È stata osservata una correlazione lineare positiva fra la secrezione di
ANP e l’apporto dietetico di sodio, ma anche la temperatura ambientale
interviene nel processo raddoppiando la secrezione per un aumento di
10°C. Svariati fattori neuroendocrini, come l’angiotensina II, le
catecolamine, la vasopressina e l’endotelina-1, gli antagonisti α19
I peptidi natriuretici
adrenergici, il monossido di azoto e le prostaglandine possono influenzare
la produzione di ANP (Ruskoaho H., 1992; Espiner E.A., 1994; Ruskoaho
H. et al., 1997).
Il meccanismo di traduzione del segnale secretorio è mediato
probabilmente dall’attivazione di proteine chinasi che aumentano i livelli
intracellulari di calcio. Sia la distensione della parete atriale che i fattori
chimici sopra citati stimolano la cascata del fosfatidil inositolo,
promuovendo l’attivazione della protein-chinasi C e innalzando la
concentrazione citoplasmatica di calcio.
Anche il contenuto cellulare di cAMP svolge un ruolo importante nel
rilascio ormonale in quanto attiva le protein-chinasi cAMP-dipendenti che,
fosforilando determinate subunità del canale del calcio voltaggio
dipendente, le converte dalla forma latente a quella responsabile
dell’entrata di calcio. Perciò tutti gli antagonisti β-adrenergici, che
attivano la via dell’adenilato ciclasi, agiscono sinergicamente con la
distensione delle pareti atriali.
Studi recenti hanno dimostrato che il CNP è prodotto prevalentemente
dalle cellule dell’endotelio vascolare (Stingo AJ. et al., 1992; Totsune K.
et al., 1994; Mattingly MT. et al., 1994) in associazione ad altre sostanze
come l’endotelina (ET-1), l’ adrenomedullina (AM), il monossido di azoto
(NO) e le prostacicline (PGI2) e che tal peptide gioca un ruolo importante
nel modulare la funzionalità piastrinica, la contrazione della contrazione
della muscolatura liscia (Zhihong Y. et al., 2002), la proliferazione e la
migrazione cellulare.
Molti fattori regolano la trascrizione del CNP, tra questi numerose
citochine come il transforming growth factor (TGF), il tumor necrosis
20
I peptidi natriuretici
factor (TNF), l’interleuchina-1 (IL-1) e il fibroblast growth factor (FGF)
(Suga S. et al., 1992; 1993).
Il rilascio di CNP in risposta allo stimolo di queste citochine suggerisce un
possibile interscambio tra produzione di citochine macrofagiche e tessuto
vascolare e ciò può indicare un ruolo importante del CNP in numerose
patologie (Mantovani A. et al., 1992; Ross R., 1991). Inoltre, mentre
l’endotelina-1 e l’angiotensina II hanno scarsi effetti sulla secrezione
basale del CNP, l’ANP e il BNP stimolano fortemente la produzione e la
secrezione di CNP attraverso la guanilato-ciclasi (GC) espressa dalle
cellule endoteliali e in particolare la stimolazione promossa dal BNP
risulta 20 volte maggiore di quella attuata dall’ANP (Nazario B. et al.,
1995).
I livelli plasmatici di CNP alterati sono stati identificati in numerose
condizioni patologiche, come l’ipossia e lo scompenso renale cronico.
1.4 Struttura genica ed espressione tessutale
Nell’uomo il gene per l’ANP è localizzato sul braccio corto del
cromosoma 1 (banda 36) ed è organizzato, come in tutte le specie di
mammiferi, in tre esoni separati da due introni. Il primo esone codifica la
regione 5’non tradotta, la sequenza leader ed i primi 16-20 amminoacidi.
del pre-pro-ormone, mentre il secondo esone codifica il resto della
molecola eccetto la tirosina terminale (uomo, cane) o il tripeptide
terminale tiroxina-arginina-arginina (ratto, topo, coniglio e mucca), che
sono invece codificati dall’ultimo esone insieme ad una regione 3’ non
tradotta.
21
I peptidi natriuretici
Il gene per l’ANP porta elementi caratteristici dei geni eucariotici
(Rosenzweig A.et al.,1991) che comprendono:
1) una TATAA box 30pb a monte del sito di inizio della trascrizione;
2) sequenze GT-AC di splicing;
3) segnali di poliadenilazione.
I geni per il BNP ed il CNP hanno la stessa organizzazione strutturale già
messa in evidenza per l’ANP, ma gli mRNA di questi due peptidi
possiedono, nella regione non tradotta, una serie di ripetizioni di una
sequenza conservata (AUUUA) destabilizzante che sembra accelerare il
turn-over del messaggero riducendone il tempo di emivita (Masayasu K.,
1990; Yandle T.G.,1994).
L’espressone dell’ANP è massima negli atri, in particolare in quello
destro, mentre nei ventricoli l’espressione è generalmente bassa, ma tende
a salire notevolmente in condizioni patologiche come lo scompenso
cardiaco (Ruskoaho H., 1992).
Trascritti di ANP sono presenti anche in tessuti extracardiaci fra cui: il
sistema nervoso (bordo antero-laterale del terzo ventricolo, corteccia
cerebrale, ipotalamo, e gangli del sistema nervoso autonomo), il lobo
anteriore dell’ipofisi, i polmoni, gli occhi, il timo,il tratto gastroenterico, il
rene, la midollare surrenale ed il tessuto vascolare (Gardner DG. et al.,
1986; Gutkowska J. et al., 1989).
Analogamente all’ANP, il gene per il BNP è espresso massivamente nel
cuore, sebbene ad un ritmo inferiore rispetto all’ANP (Davidson NC. et
al., 1994). La produzione di BNP sembra influenzata soprattutto dalla
distensione delle pareti ventricolari ed aumenta bruscamente in casi di
insufficienza cardiaca acuta e cronica, in funzione della gravità della
22
I peptidi natriuretici
malattia (Davidson NC. et al., 1994; Nicholls MG., 1995; De Bold AJ. et
al., 1996; Ruskoaho H. et al., 1997). La sintesi extra-cardiaca è stata
riscontrata nei tessuti midollare e surrenale bovino, cerebrale di maiale,
cerebrale ed amniotico umano, surrenale e renale di ratto (Espiner EA. et
al., 1995).
Come l’ANP anche il BNP umano è localizzato sul cromosoma 1.
I trascritti di CNP sono presenti prevalentemente nel tessuto nervoso,
soprattutto nel cervelletto e nel midollo spinale, e in misura minore nel
tessuto cardiaco (Davidson NC et al., 1994). mRNA codificanti per il CNP
sono stati ritrovati anche nell’endotelio vascolare a sostegno di un
probabile ruolo paracrino nella regolazione del tono e della crescita
cellulare vasale (Davidson NC., 1996; Komatsu Y. et al., 1992; Porter JD.
et al., 1992), dovute alle sue azioni inibitorie sulla vasocostrizione e la
mitogenesi (Suga S. et al., 1992; Komatsu Y. et al., 1996).
Numerosi studi riportano inoltre che la sintesi del CNP a livello cardiaco e
polmonare non cambi ne si riduca durante l’ipossia, mentre aumentano i
livelli circolanti del peptide.
Recenti studi hanno basato le loro indagini sul ruolo patofisiologico del
CNP nello scompenso cardiaco al fine di valutare se in questa condizione
patologica la sua produzione è alterata come avviene per l’ANP e il
BNP.
Inoltre è stato indicato che il CNP può avere effetti antitrombotici e
antinfiammatori, i quali contribuiscono al suo potente effetto inibitorio
sulla formazione neointimale (Qian JY. et al., 2002).
23
I peptidi natriuretici
1.5 Recettori e metabolismo
Gli effetti biologici dei peptidi natriuretici sono mediati dal legame ad alta
affinità con i loro recettori di membrana specifici situati nella maggior
parte dei tessuti di mammifero (Maack T., 1992), inclusi il cuore, l’aorta,
la corteccia surrenale, i glomeruli renali, i dotti collettori della surrenale
midollare (Kahana L. et al.,1995), i polmoni, il cervello, le piastrine, il
timo, la milza e la placenta.
Il numero di recettori varia con il tipo cellulare: le fibrocellule lisce della
muscolatura vasale sembrano possederne il maggior numero (fino a
500000 per cellula), seguite dai fibroblasti polmonari (circa 80000 per
cellula), dalle cellule della corticale surrenale (circa 50000 per cellula),
dalle cellule dell’endotelio dell’aorta (circa 14000 per cellula) ed infine
dalle cellule mesangiali renali (Ballerman BJ. et al., 1985; Leitman DC.,
1988; Schenk DB., 1985).
Come avviene per la maggior parte degli ormoni, il numero di recettori per
i peptidi natriuretici è inversamente proporzionale alla concentrazione
plasmatica ormonale (Ballerman BJ. et al., 1985; Gauquelin GR. et al.,
1988).
Studi effettuati con tecniche di clonaggio molecolare hanno identificato in
organi bersaglio almeno tre diversi recettori specifici per i peptidi
natriuretici (NPRs): NPR-A, NPR-B, NPR-C (Maack T., 1992, Jamieson
RL. et al., 1992 ; Koller KJ. et al., 1992; Ruskoaho H. 1992; Espiner EA.,
1995; Lin X. et al., 1995), anche conosciuti come GC-A, GC-B, e recettori
di clearance o come NPR-1, NPR-2 e NPR-3 rispettivamente (Fig. 4).
24
I peptidi natriuretici
Figura 4: Recettori dei peptidi natriuretici
25
I peptidi natriuretici
Il recettore NPR-A (natriuretic peptide receptor A/guanylate cyclase A) è
specifico per l’ANP e BNP, mentre l’NPR-B (natriuretic peptide receptor
B/guanylate cyclase B) mostra affinità di legame tre volte maggiore con il
CNP rispetto agli altri due peptidi (Suga S. et al.,1993; Koller KJ. et al.,
1991), l’NPR-A e l’NPR-B possiedono attività guanilato-ciclasica e
determinano l’accumulo intracellulare di cGMP. Il terzo recettore, NPR-C,
presenta elevata affinità di legame per tutti e tre i peptidi natriuretici, è il
più abbondante ed ampiamente distribuito nei tessuti, ma non risulta in
grado di formare cGMP. L’NPR-C agisce come recettore di clearance
poiché rimuove i peptidi natriuretici dal circolo (Maack T. et al., 1993;
Ruskoaho H., 1992; Espiner EA., 1995); i infatti il ruolo primario
dell’NPR-C è quello di modulare la disponibilità di questi effettori in
circolo attraverso il legame e la rimozione degli stessi dalla circolazione
periferica.
I livelli plasmatici e tessutali degli ormoni natriuretici sono il risultato del
bilancio fra produzione e secrezione, da un lato, ed estrazione d’organo
dall’altro.
Nell’uomo le velocità di scomparsa dal plasma (t1/2) dei peptidi natriuretici
sono diverse. Infatti, poiché l’NPR-C ha una bassa affinità per il BNP, il
tempo di emivita di quest’ultimo (circa 21 minuti) risulta 6-7 volte
superiore rispetto a quello dell’ANP e del CNP, che mostrano maggiore
affinità per il recettore di clearance.
La rimozione del peptide da parte dell’NPR-C prevede diversi passaggi,
come l’internalizzazione del complesso recettore-ormone, l’idrolisi
lisosomiale dell’ormone e il recupero del recettore, che viene condotto dal
26
I peptidi natriuretici
citoplasma alla membrana dove si rende disponibile per un nuovo ciclo di
attività
Oltre alla clearance mediata dall’NPR-C, la disponibilità dei peptidi
natriuretici è regolata da un’endopeptidasi neutra, ectoenzima di
membrana appartenente alle zinco-metallopeptidasi, detta E.C.24.11
(encefalinasi o atriopeptidasi) ampiamente distribuita nella muscolatura
vascolare e in molti tessuti, inclusi l'endotelio e le fibrocellule liscie
vascolari, i tubuli renali e le cellule del sangue (PMN) (Llorens-Cortes C.,
1992; Nortier J. et al., 1995; Yandle TG., 1994), e capace di degradare
numerosi peptidi differenti. Sebbene la sua esatta localizzazione rispetto ai
recettori dei peptidi natriuretici risulti poco conosciuta, è possibile che
l’endopeptidasi si trovi giustapposta all’NPR-A sulla membrana cellulare
(Okamoto A. et al.,1994).
Il sito iniziale di azione dell'E.C.24.11. sull'ANP é rappresentato dal
legame Cys105-Phe106 che viene scisso, portando all'apertura dell'anello
con successiva inattivazione ormonale.
L’endopeptidasi neutra mostra un’elevata capacità di idrolisi del CNP
rispetto ad ANP e BNP, questo suggerisce un ruolo importante
dell’enzima nella regolazione della disponibilità in circolo del CNP
rispetto al suo legame con il recettore di clearance (Kenny A. et al., 1993;
Brandt RR. et al., 1997).
Per quanto concerne l’attività dell’NPR-C sono emerse, accanto a quella
principale di clearance dei peptidi natriuretici dalla circolazione, evidenze
di svariate altre azioni biologiche, che potrebbero essere mediate dal
recettore C attraverso meccanismi trasduzionali diversi, fra cui l’inibizione
27
I peptidi natriuretici
dell’adenilato ciclasi e l’attivazione della fosfolipasi C (Levin E.R., 1993;
Madhu B. et al., 1993; Savoie P. et al., 1995).
Il gene umano dell’NPR-A (NPR-1) ha una sequenza approssimativa di16
kb contenente 22 esoni e 22 sequenze introniche ed è localizzato sul
cromosoma 1q21-22 (Lowe DG. et al., 1989). Il relativo mRNA ha una
sequenza di 3.805 pb e codifica per una proteina di 1,061 pb.
Il gene dell’ NPR-B (NPR-2) è localizzato sul cromosoma 9p21-12
(Rehemudula D. et al., 1999), ha una sequenza di 17.303 pb (16,5 kb),
contenente 22 esoni.
Il gene codificante per l’NPR-C, denominato NPR-3, è localizzato sul
cromosoma 5p14-13 e ha una sequenza di 74.698 pb (contenente 8 esoni),
il relativo mRNA ha una sequenza di 1.753 pb e codifica per una proteina
di 540 amminoacidi.
NPR-A e NPR-B sono due proteine monomeriche di 1030 amminoacidi. e
115 kD con un dominio transmembrana di 21 amminoacidi.
La porzione amminoterminale, extracellulare, di 441 amminoacidi,
costituisce il sito che lega gli ormoni; per questa attività sembrano
importanti gli amminoacidi: Leu364 dell’NPR-A e Glu332 dell’NPR-B.
La porzione citoplasmatica possiede, in prossimità della membrana, un
dominio con attività chinasica ed, all’estremità carbossilica, un dominio
intracellulare con attività guanilato-ciclasica. Un’altra regione importante
del recettore, è il dominio chinasico che inibisce normalmente la guanilato
ciclasi.
Il legame del peptide natriuretico col suo recettore specifico, NPR-A o
NPR-B, (Fig. 6) promuove il rilascio di una guanilato ciclasi dal dominio
chinasico e la conseguente conversione in guanosin monofosfato ciclico
28
I peptidi natriuretici
della guanosina trifosfato (GTP → cGMP) (Suga S. et al., 1992; Chinkers
M. et al., 1989), la quale agisce come secondo messaggero mediando gli
effetti biologici dei peptidi natriuretici.
Studi sui recettori A e B (Chinkers M., 1989; Duda T. e al., 1995; Koller
KJ. et al., 1992) fanno supporre che il dominio chinasico svolga un
importante ruolo nella trasduzione del segnale e che l'ATP sia essenziale
per potenziare l'attività guanilato-ciclasica. Infatti, recettori mutanti con
una delezione della regione chinasica, producono cGMP costitutivamente
in assenza di ATP ed indipendentemente dal legame dell'ormone al sito
extracellulare.
Stando a questi dati si potrebbe concludere che il dominio chinasico agisca
come possibile regolatore negativo dell'attività ciclasica e che il legame
dell'ormone rimuova l'inibizione.
Nell’uomo il recettore per il CNP, l’NPR-B, è stato rilevato nella
muscolatura vascolare liscia in stretto contatto con il sito di produzione
endoteliale del CNP (Komatsu Y. et al., 1992). La localizzazione del CNP
e dei suoi recettori in questi siti suggerisce il coinvolgimento del peptide
nella regolazione della fisiologia cardiaca e renale; inoltre la presenza
dell’NPR-B a livello cardiaco indica che il CNP può avere importanti
azioni cardiache locali (Nunez DJR. et al., 1992; Wei CM. et al., 1993).
Studi sui recettori A e B fanno supporre che il dominio chinasico svolga
un’importante ruolo nella traduzione del segnale e che l’ATP sia
essenziale per potenziare l’attività guanilato-ciclasica. Infatti recettori
mutanti con una delezione della regione chinasica, producono cGMP
costitutivamente in assenza di ATP ed indipendentemente dal legame
dell’ormone al sito extracellulare. Stando a questi dati si potrebbe
29
I peptidi natriuretici
concludere che il dominio chinasico agisca come possibile regolatore
negativo dell’attività ciclasica e che il legame dell’ormone rimuova
l’inibizione.
Il recettore di clearance NPR-C, è un dimero costituito da due subunità
legate fra loro da due ponti di solfuro. La porzione extracellulare di 496
amminoacidi. lega l’ormone e presenta una certa omologia con i recettori
NPR-A e NPR-B (30%), mentre la porzione citoplasmatica, priva
dell’attività chinasica e guanilato-ciclasica, è composta da soli 37
amminoacidi. Si ritiene che la caratteristica strutturale responsabile della
discriminazione del sito recettoriale sia una sequenza di 7 amminoacidi.
(Gly108-Gly114) o di 5 amminoacidi. (Arg109-Ile113), contenuta
nell’anello centrale degli ormoni.
Sebbene molti degli effetti biologici del CNP siano attribuiti
all’attivazione dell’NPR-B, vi sono numerose evidenze che il recettore di
clearance, quando stimolato dal CNP, possa esercitare i suoi effetti in
maniera Gi dipendente (Maack T. et al., 1987).
È stato dimostrato che l’NPR-C agisce non soltanto come recettore di
clearance, ma che la sua azione sia anche legata all’iperpolarizzazione
della muscolatura vascolare liscia indotta dal CNP (Chauhan SD. et al.,
2003).
Il coinvolgimento dell’NPR-C nella regolazione del tono vascolare sembra
essere prevalente a livello delle arteriole (Chauhan SD. et al., 2003;
Steinmetz M. et al., 2004). Questo è in accordo con l’identificazione del
CNP come EDHF, il mediatore endoteliale che esplica il vasorilassamento
attraverso una caratteristica iperpolarizzazione delle cellule muscolari
lisce (Ahluwalia A. et al., 2005). Inoltre è stata evidenziata la possibilità
30
I peptidi natriuretici
che molte altre azioni biologiche siano mediate dal recettore C attraverso
meccanismi trasduzionali diversi, fra cui l’inibizione dell’adenilato ciclasi
e l’attivazione della fosfolipasi C (Levin ER. et al., 1993).
La localizzazione dei recettori a livello tissutale è stata ampiamente
studiata sia nei primati che nell’uomo usando la tecnica dell’ibridazione in
situ: l’NPR-A è altamente espresso a livello di reni, porzione terminale
dell’ileo, polmoni e aorta; l’NPR-B è stato identificato prevalentemente
nel cervello ma anche a livello della ghiandola surrenale, dei polmoni e di
utero e ovaie.
1.6 Degradazione dei peptidi natriuretici
I livelli plasmatici e tessutali degli ormoni natriuretici sono il risultato del
bilancio fra produzione e secrezione, da un lato, ed estrazione d’organo
dall’altro.
Nell’uomo il tasso di clearance metabolica (MCR) per i tre peptidi
natriuretici è simile (circa 2-4 l/min), mentre differiscono le velocità di
scomparsa dal plasma (t1/2). Infatti, poiché l’NPR-C ha una bassa affinità
per il BNP, il tempo di emivita di quest’ultimo (circa 21 minuti) (Holmes
SJ. et al., 1993) risulta 6-7 volte superiore rispetto a quello dell’ANP
(Yandle T.G. et al., 1986) e del CNP, che mostrano maggiore affinità per
il recettore di clearance.
La rimozione da parte dell’NPR-C prevede diversi passaggi, come
l’internalizzazione del complesso recettore-ormone, l’idrolisi lisosomiale
dell’ormone e il recupero del recettore, che viene condotto dal citoplasma
alla membrana dove si rende disponibile per un nuovo ciclo di attività
(Morel GS. et al., 1988).
31
I peptidi natriuretici
1.7 Azioni biologiche dei peptidi natriuretici
È importante notare che i recettori specifici per i peptidi natriuretici sono
stati trovati nella maggior parte dei tessuti di mammifero, sebbene in
differenti concentrazioni, e ciò suggerisce che tali peptidi giocano un ruolo
biologico importante in questi distretti.
Molti degli effetti biologici dei peptidi natriuretici sembrano essere dovuti
alla loro azione autocrina/paracrina piuttosto che a quella ormonale.
Gli ormoni natriuretici cardiaci esercitano potenti effetti fisiologici sul
sistema cardiovascolare, sulla regolazione dei liquidi corporei, sul
mantenimento dell’omeostasi elettrolitica (McGrath MF. et al., 2005;
Clerico A. et al., 2004; 2002; De Lemons JA. et al., 2003; Ruskoaho H.,
2003).
Inoltre essi esercitano un’azione natriuretica, diuretica, vasodilatante e di
inibizione della contrazione dei miociti ventricolari (Zang Q. et al., 2005)
oltre che un’azione antiipertrofica, di rimodellamento (Nagaya N. et al.,
1998; Hayashi M. et al., 2001; Hardt SE., 2004) e antinfiammatoria sui
cardiomiociti e sulle cellule muscolari lisce.
Esercitano anche un effetto protettivo sulla funzione endoteliale
promuovendo la diminuzione dello “shear stress”, la modulazione della
cascata coagulativa e della fibrinolisi e l’inibizione dell’attivazione
piastrinica. I peptidi possono inibire i processi di rimodellamento
vascolare, come avviene, ad esempio, nella ristenosi coronarica postangioplastica (Ma KK. et al., 2004; Chen HH. et al., 1998; QianJY. et al.,
2002; Nakanishi M. et al., 2005). Mostrano, altresì, un azione inibitoria
del sistema neuroormonale e immunitario, ma anche di numerosi fattori di
32
I peptidi natriuretici
crescita (McGrath MF. et al., 2005; Clerico A. et al., 2004; 2002;
DeLemons JA. et al., 2003; Ruskoaho H. 2003; Ventura RR. et al., 2002;
Vatta MS. et al., 1996; Kapoun AM. et al., 2004; Vollmar AM., 2005): è
stato recentemente dimostrato che il ruolo centrale dei peptidi natriuretici
(soprattutto
dell’ANP)
risiede
nella
modulazione
della
risposta
immunitaria (Kapoun AM. et al., 2004). I primi studi sul loro
coinvolgimento a livello del sistema immunitario si basavano sulla
presenza dei recettori biologici specifici dei peptidi che sono espressi in
numerosi organi immunitari; successivamente nuove ricerche indicarono
che il sistema dei peptidi natriuretici, presente nelle cellule immunitarie,
dipendeva da specifici meccanismi regolatori. In particolare l’ANP
promuove l’aumento dell’attività fagocitaria e induce la risposta
immunitaria innata attraverso la regolazione dell’attività dei macrofagi a
vari stadi dell’infiammazione. L’ANP inoltre riduce la produzione di
mediatori pro-infiammatori attraverso l’inibizione della ossido-nitrico
sintasi (iNOS) e della cicloossigenasi-2 (COX2) e inibisce la sintesi del
tumor necrosis factor (TNF)-α. Il peptide atriale limita la permeabilità
endoteliale e l’adesione delle molecole infiammatorie mediate dal TNF-α
indotto.
L’ANP influisce sulla timopoiesi e sulla maturazione dei linfociti T,
agendo sulla maturazione delle cellule dendritiche e sulle risposte
immunitarie mediate dai linfociti T helper (TH) 1 e 2 (Vollmar AM.,
2005).
I sopraindicati effetti sul sistema cardiovascolare, sul bilancio corporeo di
liquidi, sul mantenimento dell’omeostasi elettrolitica possono essere
spiegati, almeno in parte, attraverso il meccanismo di inibizione dei
33
I peptidi natriuretici
sistemi di controllo, quali: diminuzione dell’attività del simpatico e delle
catecolamine, incremento dell’attività parasimpatica, inibizione del
rilascio e/o dell’azione di aldosterone, renina, angiotensina II e
vasopressina; inibizione del sistema dell’endotelina, delle citochine e dei
fattori di crescita (Ventura RR. et al., 2002; Vatta MS. et al., 1996;1997;
Tsukagoshi H. et al., 2001; Kapoun AM. et al. 2004; Vollmar AM., 2005).
L’azione endocrina, mostrata dall’ANP e BNP plasmatici, può essere
migliorata
dall’azione
paracrina
dei
peptidi
natriuretici
prodotti
localmente. Invece le cellule endoteliali sintetizzano il CNP, il quale
mostra un effetto paracrino nello spessore della parete vascolare in
associazione con i sistemi locali (Woodard GE. et al., 2002; Chen HH. et
al., 1998; Qian JY. et al., 2002; Morishige K. et al., 2000); inoltre le
cellule dei tubuli renali producono l’urodilatina, ormone appartenente alla
famiglia dei peptidi natriuretici, dotata di una forte azione diuretica e
natriuretica (Vesely DL. et al., 2003) (Fig. 5).
Figura 5: Principali effetti dei peptidi natriuretici sul sistema cardiovascolare
34
I peptidi natriuretici
Geni codificanti per i peptidi natriuretici sono anche espressi a livello del
sistema
nervoso,
dove
agiscono
come
neurotrasmettitori
e/o
neuromodulatori (Vatta MS. et al., 1996;1997; Vesely DL. et al., 2003;
Imura H. et al., 1992). In particolare, è stato dimostrato che nell’uomo
l’ANP somministrato per via nasale agisce come inibitore nervoso centrale
del sistema adrenergico-ipotalamo-ghiandola pituitaria (Perras B. et al.,
2004); in fine la co-espressione dei peptidi natriuretici e dei recettori
specifici è stata osservata nelle cellule timiche e nei macrofagi di ratti
(Vollmar AM. et al., 1995;1995), suggerendo che i peptidi possono avere,
nei mammiferi, funzioni immunomodulatorie ed anti-infiammatorie
(Vollmar AM. et al., 2001).
Un ultimo studio dettagliato (Waschek JA., 2004) ha messo in luce un
possibile ruolo per i peptidi natriuretici nello sviluppo di sistemi
importanti, come lo scheletro, il cervello e i vasi, ma anche nella
regolazione della proliferazione, sopravvivenza e crescita dei neuriti,
osservata in culture di cellule neuronali e/o gliali (Kiefer F. et al., 2004).
Numerosi studi hanno dimostrato che i peptidi natriuretici stimolano la
sintesi e il rilascio di testosterone in maniera dose-dipendente, in cellule di
Leydig normali isolate e purificate (Bex F. et al., 1985; Pandey KN.,
2005).
È stato suggerito che questo effetto sulle cellule normali di Leydig non
coinvolge i classici meccanismi di regolazione dell’attività steroidogenica
mediati dal cAMP (Pandey KN., 2005). L’aumentata produzione di
testosterone stimolata da ANP, BNP è comparabile a quella mediata dalle
gonadotropine, a differenza del CNP, che risulta essere un debole
35
I peptidi natriuretici
stimolatore della produzione di testosterone nelle cellule di Leydig (da
parte delle cellule di Leydig).
Inoltre nelle cellule testicolari, è notevolmente espresso il recettore NPR-A,
specifico per l’ANP (Pandey KN., 2005): queste osservazioni suggeriscono
un’azione autocrina/paracrina a livello dei testicoli.
La presenza di ANP e del suo recettore specifico è stata anche rilevata
nelle cellule ovariche, questa prova sostiene il fatto che i peptidi
natriuretici sono presenti, e probabilmente sintetizzati, nelle cellule
ovariche di differenti specie di mammiferi, ma anche che giocano un ruolo
fisiologico importante nella sintesi e secrezione di estradiolo da parte delle
gonadi femminili (Pandey KN., 2005; Vollmer AM. et al., 1988; Ivanova
MD. et al., 2003). Comunque sono necessari ulteriori studi per chiarire
completamente l’azione dei peptidi natriuretici nella regolazione della
funzione delle gonadi e anche valutare l’inter-relazione tra la funzione
endocrina del cuore e delle gonadi nell’uomo.
È stato ormai fermamente dimostrato il coinvolgimento dei peptidi
natriuretici come molecole attive nel controllo dei sistemi nervoso,
endocrino ed immunitario; secondo questi studi il cuore non può essere
considerato soltanto come un automa passivo regolato da stimoli nervosi
endocrini o emodinamici.
Quindi
i
peptidi
natriuretici,
in
associazione
con
altri
fattori
neuroormonali, regolano l’emodinamica cardiovascolare, l’omeostasi di
fluidi ed elettroliti corporei e, probabilmente modulano la risposta
infiammatoria in diversi distretti, incluso il sistema cardiovascolare.
Queste ipotesi indicano che sono presenti due sistemi antagonisti per la
regolazione della fisiologia corporea: un sistema svolge azioni sodio
36
I peptidi natriuretici
ritentive, vasocostrittive, trombofiliche, pro-infiammatorie e ipetrofiche,
mentre il secondo promuove la natriuresi e la vasodilatazione, ma inibisce
la trombosi, l’infiammazione e l’ipertrofia. I peptidi natriuretici sono i
principali effettori di quest’ultimo sistema ed interagiscono con diverse
citochine vasoattive, come l’monossido di azoto, alcune prostaglandine e
altri peptidi vasodilatatori (ad esempio le bradichinine) (Chatterjee PK. et
al., 1999; Ruskoaho H. et al., 1997; Booz GW., 2005).
In condizioni normali gli effetti di questi due sistemi sono bilanciati da
meccanismi di feed-back in grado di regolare la funzione cardiaca e la
pressione sanguigna in risposta a stimoli sia endogeni che esogeni.
Tutte queste considerazioni riguardanti i peptidi natriuretici, suggeriscono
che vi è un continuo ed intenso scambio di informazioni tra il sistema
cardiovascolare, il sistema nervoso, il sistema immunitario e gli altri
organi corporei, inclusi reni, ghiandole endocrine, fegato, tessuto adiposo
e cellule immuno-competenti (Fig. 6).
Dal punto di vista fisiopatologico, lo stretto legame tra il sistema dei
peptidi natriuretici e il sistema contro-regolatorio può essere spiegato
dall’aumento dei livelli circolanti di tali peptidi in patologie che non
coinvolgono l’apparato cardiaco, infatti è stato osservato l’incremento o la
diminuzione dei livelli di BNP nelle malattie respiratorie acute o croniche
(Kruger S. et al., 2004; Kucher N. et al., 2003; Ando T. et al., 1996;
Leuchte HH. et al., 2004), in diverse patologie metaboliche e del sistema
endocrino (Khono M. et al., 1993; Parlapiano C. et al., 1998; Schultz M. et
al., 2004; Fujio N. et al., 1989; Straub RH. et al., 1996; Bhalla MA. et al.,
2004), nella cirrosi epatica (Henriksen JH. et al., 2003), nello scompenso
37
I peptidi natriuretici
Figura 6: Il sistema neuro-endocrino: l'intima correlazione tra sistema
dei peptidi natriuretici altri organi e sistemi di regolazione
38
I peptidi natriuretici
renale (Ogawa Y. et al., 1994, Henriksen JH. et al., 2003), nello shock
anafilattico, nelle malattie infiammatorie croniche (McCullough PA. et al.,
2004; Castillo JR. et al., 2004; Palladini G. et al., 2003 ), nelle emorragie
subaracnoidee (McGirt MJ. et al., 2004; Fukui S. et al., 2004) e in diverse
sindromi paraneoplastiche (Mimura y. et al., 1996; Johson BE. et al.,
1997). È stato riportato, in aggiunta, un aumento delle concentrazioni
plasmatiche dei peptidi natriuretici in caso di danni al miocardio, legati al
rilascio di costituenti sarcoplasmatici nei liquidi extracellulari e dovuti, ad
esempio, ad agenti cardiotossici (Hayakawa H. et al., 2001; Suzuki T. et
al., 1998; Okumura H. et al., 2000) o a necrosi ischemiche (Mukoyama M.
et al., 1991; Morita E. et al., 1993).
Inoltre, la correlazione tra i peptidi natriuretici e le citochine proinfiammatorie suggerisce che questi ormoni cardiaci giocano un ruolo
importante nei meccanismi responsabili dell’adattamento cardiovascolare
e del rimodellamento in risposta a vari stimoli fisiopatologici
(Tenhunen O. et al., 2005; Walther T. et al., 2003; Glembotski CC. et al.,
1993; Mukoyama M. et al., 1991).
Elevati livelli di BNP in patologie extra-cardiache, rivelano una risposta
cardiaca endocrina allo “stress cardiovascolare”.
In effetti, recenti studi hanno riportato che le concentrazioni plasmatiche
di BNP rappresentano un fattore di rischio indipendente di mortalità
nell’embolia polmonare (Kruger S. et al., 2004; Kucher N. et al., 2003),
nell’ipertensione (Nagaya N. et al., 1998), nello scompenso renale
(Clerico A. et al., 2004; Vesely DL., 2003; Henriksen JH. et al., 2003),
nello shock anafilattico (McCullough PA. et al., 2004), nell’amiloidosi
(Palladini G. et al., 2003) e nel diabete mellito (Bhalla MA. et al., 2004).
39
I peptidi natriuretici
In base a queste ipotesi, il dosaggio del BNP può essere considerato come
un marker di stress cardiaco.
La funzione endocrina dell’ANP e del BNP è potenziata, a livello
periferico, dall’azione paracrina del CNP (nel tessuto vascolare) e
dell’urodilatina (nel tessuto renale).
Infine, numerosi studi sperimentali condotti su topi knock-out
suggeriscono un distinto ruolo patofisiologico per il BNP rispetto all’ANP
(LaPointe MC., 2005). I topi knock-out per il BNP non presentano
differenze rispetto ai controlli nella pressione sanguigna, nel volume
urinario e nell’escrezione urinaria di elettroliti, mentre mostrano una più
estesa fibrosi ventricolare accompagnata da un aumento dell’espressione
dell’mRNA codificante per TGF (transforming growth factor)-β3e per il
collagene.
Questi dati suggeriscono che il BNP può funzionare sia come inbitore
autocrino/paracrino della crescita cellulare nel cuore, mentre l’ANP può
essere considerato come un tradizionale ormone circolante con marcati
effetti diuretici, natriuretici e antiipertensivi.
Infatti l’ANP, agendo sul sistema cardiovascolare, determina ipotensione,
vasodilatazione, aumento della permeabilità capillare, rimodellamento
vasale ed inibizione della mitogenesi (Morishita R. et al., 1994).
Per quanto riguarda il CNP, sin dalla sua scoperta nel cervello porcino, è
stato identificato, come anche il suo recettore specifico NPR-B, in diverse
porzioni dei tessuti umani come il sistema nervoso centrale, i tubuli renali
e l’endotelio. L’osservazione che il CNP si ritrova in colture di cellule
endoteliali umane è di notevole importanza poiché il recettore NPR-B è
presente in elevate concentrazioni sulla membrana delle cellule muscolari
40
I peptidi natriuretici
vascolari lisce adiacenti (Komatsu Y. et al., 1992), ed inoltre suggerisce
l’ipotesi che il CNP possa avere un effetto paracrino nello spessore della
parete vascolare (Nunez DJR. et al., 1992; Wei CM. et al., 1993)
Sebbene molti degli effetti biologici del CNP siano attribuiti
all’attivazione dell’NPR-B, esistono numerose evidenze che la clearance
del recettore NPR-C, quando stimolato dal CNP, possa esercitare effetti in
maniera Gi-dipendenti (Maack T. et al., 1987). Il coinvolgimento dell’
NPR-C nella regolazione del tono vascolare appare prevalente nelle arterie
a bassa resistenza (arteriole) (Chauhan SD. et al., 2003; Steinmetz M. et
al., 2004).
Esistono numerosi studi che dimostrano sia l’importanza del sistema di
traduzione CNP/NPR-C nella regolazione della circolazione periferica che
dell’azione del CNP analoga all’EDHF. Da questi dati si evince che il
CNP è un importante mediatore della circolazione coronarica e che i suoi
effetti possono essere mediati principalmente dal recettore NPR-C
(Scotland RS. et al., 2005). La presenza del recettore NPR-B nel cuore
umano, suggerisce che il CNP possa avere un importante azione locale.
Inoltre numerosi studi riportano che la sintesi del CNP nel cuore e nel
polmone diminuisce durante l’ipossia mentre aumenta in circolo, e ciò
suggerisce un ruolo fondamentale del peptide nella modulazione del tono
vascolare polmonare in associazione con l’ANP (Klinger JR. et al., 1998;
Mitani Y. et al., 2000). È stato indicato che il CNP può avere effetti
antitrombotici e antinfiammatori, che possono contribuire al suo potente
effetto inibitorio sulla formazione della neointima (Qian JY. et al., 2003).
L’espressione locale del CNP nelle arterie danneggiate aumenta
41
I peptidi natriuretici
l’espressione genica dell’iNOS e la produzione di NO
riducendo
l’infiammazione e gli effetti indotti dallo shear stress.
42
Lo scompenso cardiaco
2
LO SCOMPENSO CARDIACO
Capitolo
Lo scompenso cardiaco (SC) è la malattia cardiovascolare con più elevato
impatto epidemiologico (Schocken DD., 1992) e costo socio sanitario,
associata ad una prognosi infausta che solo recenti progressi terapeutici
stanno migliorando sensibilmente. Le acquisizioni fisiopatologiche
dell’ultimo decennio hanno fatto emergere come il quadro clinico, la
risposta alla terapia e la prognosi dipendano non soltanto dal tipo ed entità
del danno cardiaco primitivo, ma anche dal coinvolgimento di altri organi
e apparati (in particolare circolo sistemico e polmonare, rene, fegato e
apparato locomotore) tanto da indurre a pensare allo scompenso cardiaco
come a una malattia “multiorgano”; ed ancora, dal livello di attivazione di
una serie di sistemi neuroormonali e paracrini, tanto da poter definire il SC
come una “malattia dei sistemi di regolazione”.
Oggigiorno esistono molte definizioni di scompenso cardiaco, la
definizione che viene considerata come più appropriata è quella data da
Braunwald nel suo trattato: "Lo scompenso cardiaco si verifica quando
un'anomalia della funzione cardiaca fa si che il cuore non sia in grado di
pompare sangue in quantità sufficiente per soddisfare i bisogni metabolici
dell'organismo o possa farlo solo a spese di un aumento della pressione di
riempimento. L’incapacità del cuore a soddisfare i fabbisogni tessutali
può essere dovuta a riempimento inefficace e insufficiente e\o ad una
anomala contrazione e successivo svuotamento" (Braunwald E., 1997).
Frequentemente l'insufficienza cardiaca è determinata da un difetto di
contrazione del miocardio, che può essere il risultato di patologie
43
Lo scompenso cardiaco
extramiocardiche come l'aterosclerosi coronarica e le malattie valvolari o
secondario ad anomalie primitive del muscolo cardiaco.
A causa dell'efficacia del trattamento della cardiopatia ischemica e
dell'infarto miocardico in fase acuta, lo scompenso cardiaco rappresenta
oggi una causa maggiore e crescente di mortalità e morbilità.
2.1 Eziologia dello scompenso cardiaco
Lo scompenso cardiaco cronico è il risultato di molteplici concause e
fattori scatenanti, ma si può affermare che nel mondo occidentale la
cardiopatia ischemica costituisce attualmente il fattore più frequente che
facilita l'insorgenza dell'insufficienza cardiaca, mentre in diminuzione
appare il ruolo dell'ipertensione arteriosa, verosimilmente per la maggiore
attenzione diagnostica ed i miglioramenti terapeutici.
Circa il 20% di tutti i casi di scompenso presenta un'eziologia
misconosciuta, dando luogo così ad una cardiomiopatia dilatativa definita
"idiopatica", mentre in netta diminuzione nei paesi occidentali appare la
patologia secondaria a valvulopatie reumatiche.
Nella restante parte del mondo la malattia reumatica e la malattia di
Chagas rappresentano ancora la principale causa di insufficienza cardiaca
(Kannel WB. et al., 1991).
I fattori di rischio per lo sviluppo di scompenso cardiaco sono dovuti a
ipertrofia ventricolare sinistra, fumo di sigaretta, aumento del rapporto
colesterolo totale/colesterolo HDL, ipertensione, diabete e obesità.
Molti dei fattori di rischio per lo scompenso cardiaco sono anche fattori di
rischio per la cardiopatia ischemica, che sottolinea il suo ruolo come
elemento causale dello scompenso stesso (Sutton JH., 1990).
44
Lo scompenso cardiaco
Vista la genesi multifattoriale di questa patologia e visto che molti di
questi fattori sono eliminabili o reversibili, è importante identificarli per
pianificare una strategia di trattamento ottimale.
2.2 Fisiopatologia dello scompenso cardiaco
Lo scompenso cardiaco può essere considerato come una malattia
progressiva generata da un evento che provoca un danno miocardio con
perdita di cardiomiociti funzionanti e che altera quindi la capacità del
muscolo cardiaco di generare forza, risultando in una riduzione della
capacità di pompa del cuore.
L'inizio dell'insufficienza cardiaca si può far risalire al momento in cui i
miociti sono sottoposti ad un aumento di carico emodinamico eccessivo
che dura nel tempo; tale aumento può essere relativo (incapacità dei
miociti di eseguire un lavoro in rapporto all'aumento delle richieste), o
assoluto (come nell'infarto miocardico dove i miociti residui sono costretti
a sostenere il carico emodinamico totale) (Rostagno, 1996)
A livello patogenetico si possono distinguere disturbi primitivi della
contrattilità, disturbi del riempimento diastolico, alterazioni persistenti del
carico emodinamico e fattori extracardiaci che modificano la funzione
diastolica.
Il danno cardiaco che deriva può avere insorgenza brusca o lenta (dovuta
ad un sovraccarico volumetrico o pressorio) o può essere ereditario (come
nel caso di cardiomiopatie congenite) (Emdin M. et al., 2001); ciò porta ad
un aumento insufficiente delle pressioni nel circolo polmonare a monte del
ventricolo sinistro e alla riduzione della gittata sistolica.
45
Lo scompenso cardiaco
Le modifiche strutturali (rimodellamento) a carico del ventricolo
contribuiscono alla morbilità e alla mortalità nello scompenso cardiaco
(Pfeffer MA., et al., 1990). Questo processo si associa con un aumento
della massa del miocardio, un aumento del volume ventricolare sinistro, e
una variazione nella forma del ventricolo. Il rimodellamento influenza il
consumo miocardio di ossigeno, contribuendo alla progressione dello
scompenso o alla comparsa di aritmie (Francis GS., 1985).
L’ esito finale dello scompenso è l’incapacità del cuore di assicurare una
portata cardiaca necessaria ai bisogni metabolici e funzionali dei diversi
organi, anche se i pazienti possono rimanere asintomatici o minimamente
sintomatici dopo l’ inizio della disfunzione, e sviluppare disturbi
tardivamente. Al fine di preservare la funzionalità cardiaca, intervengono
infatti meccanismi compensatori che sono capaci di modulare la funzione
ventricolare in un appropriato range omeostatico e fisiologico, in modo
tale da preservare la capacità funzionale cardiaca del paziente o di
deprimerla solo in minima parte. In questo modo i pazienti potranno
rimanere asintomatici o minimamente sintomatici per un determinato
periodo di tempo.
2.3 Il modello neuroendocrino
Storicamente la sindrome dello scompenso cardiaco è stata descritta
usando diversi modelli, sulla base dell’esperienza clinica:
1. il modello renale, quando i clinici “vedevano” lo scompenso cardiaco
come un problema di eccessiva ritenzione idro-salina che provocava
anormalità nel flusso sanguigno renale (Packer M., 1993);
46
Lo scompenso cardiaco
2. il modello cardiocircolatorio, quando, grazie alla possibilità di
effettuare accurate valutazioni emodinamiche, questa patologia risultò
associata a una riduzione della portata cardiaca e a un aumento della
vasocostrizione periferica (Massie BM. et al, 1998);
3. il modello neuroendocrino, quando l’attenzione è stata rivolta ai
potenziali meccanismi biochimici responsabili della progressione della
malattia. La scelta dei vari modelli ha progressivamente indirizzato anche
le varie strategie terapeutiche per questa patologia, dai diuretici agli agenti
inotropi e vasodilatatori a farmaci in grado di modulare il sistema
neuroormonale
(ACE
inibitori,
β-bloccanti,
anti-aldosteronici,
anti-endotelinici) (Mann DL. et al., 1999).
I meccanismi compensatori che intervengono in seguito a danno cardiaco
e a riduzione della portata cardiaca includono l’attivazione di sistemi
neuro-endocrini quali il sistema adrenergico e il sistema reninaaldosterone.
La transizione paziente asintomatico/paziente sintomatico è accompagnata
da un'ulteriore attivazione del sistema neuroormonale e citochinico e da
una
serie
di
modificazioni
a
livello
del
miocardio,
indicate
complessivamente come fenomeno di rimodellamento ventricolare.
(Pfeffer MA., 1990)
È stato così ipotizzato che la fisiopatologia dello scompenso cardiaco
potesse essere parzialmente spiegata usando un modello neuroendocrino in
cui la progressione della patologia è dovuta alla sovraespressione di
molecole biologicamente attive in grado di esercitare effetti negativi a
livello della circolazione, del cuore e del rimodellamento ventricolare.
(Mann DL., 1999).
47
Lo scompenso cardiaco
Il danno miocardico si associa ad attivazione adrenergica cronica,
ipertrofia ventricolare sinistra e ad un successivo aumento nel volume
ventricolare
sinistro.
Il
miocardio
non
danneggiato
produce
RNAmessaggero (mRNA) codificante per l’angiotensinogeno; viene
attivato il sistema endotelinico con proprietà proliferative sia a livello
miocardico che vascolare.
Ricopre un ruolo fondamentale l’attivazione del sistema nervoso simpatico
(Eisenhofer G.et al., 1996) in fuzione cronotropa (aumento della frequenza
cardiaca che contribuisce al mantenimento della portata in presenza di una
riduzione del volume sistolico) ed inotropa.
Le elevate concentrazioni di noradrenalina provocano un aumento delle
resistenze vascolari periferiche con il conseguente aumento del postcarico
e si associano ad un rischio crescente di aritmie ventricolari.
Il
grado
di
incremento
delle
concentrazioni
plasmatiche
della
noradrenalina è inversamente proporzionale alla frazione di eiezione ed
alla sopravvivenza e si accompagna ad una marcata riduzione del numero
dei recettori adrenergici periferici, che può essere visto come meccanismo
di protezione contro l'eccessiva stimolazione adrenergica (Hall S. et al.,
1995).
L'elevata attivazione simpatica è in parte conseguenza della ridotta
capacità dei barorecettori arteriosi di inibire i centri vasomotori, evenienza
che si verifica precocemente nei pazienti con scompenso cardiaco (Gafney
TE. et al., 1963).
Altri sistemi vasoattivi e idrosalino-ritentivi endocrini e paracrini (reninaangiotensina,
aldosterone,
endoteline,
vasopressina)
si
associano
all’attivazione adrenergica con lo scopo di preservare la portata cardiaca e
48
Lo scompenso cardiaco
la pressione arteriosa media e hanno come risultato un’alterazione nel
bilancio fra agenti vasocostrittori e agenti vasodilatatori (Mann DL.,
1999). L’attivazione di questi sistemi ha come risultato ritenzione
idrosalina e vasocostrizione periferica, due indici clinici di scompenso
cardiaco.
La ritenzione idrosalina che deriva da tale attivazione avviene:1. per
vasocostrizione dell’arteriola efferente; 2. per aumento della filtrazione
glomerulare; 3. per riassorbimento di sodio a livello del tubulo prossimale,
sotto l’azione dell’angiotensina; 4. per riassorbimento di sodio mediato
dall’aldosterone; 5. per stimolazione dei centri della sete e diminuzione
dell’eliminazione dell’acqua libera mediata dalla vasopressina.
L'aumento delle concentrazione di angiotensina II plasmatica stimola la
contrazione delle cellule muscolari lisce vasali, la secrezione di
aldosterone, il riassorbimento di sodio a livello tubulare, le risposte
tachicardica e pressoria ed il senso della sete.
L'aumento della concentrazione locale di angiotensina II favorisce inoltre
le risposta ipertrofica del miocardio, favorendo l'espressione di protooncogeni.
Anche le concentrazioni di vasopressina sono aumentate nell'insufficienza
cardiaca, specialmente nelle fasi più avanzate. Questo ormone viene
fisiologicamente liberato in seguito a stimolazione da parte di
osmorecettori per aumento dell'osmolarità plasmatica o in seguito alla
stimolazione di meccanorecettori di volume atriali.
La vasopressina (ADH) agisce sul rene promuovendo il riassorbimento di
acqua libera e sulle cellule muscolari delle arteriole periferiche
stimolandone la contrazione.
49
Lo scompenso cardiaco
L’attivazione di questi sistemi è controbilanciata da sistemi vasodilatatori
(peptidi natriuretici, prostaglandine e monossido di azoto) con azione
natriuretica, diuretica ed antimitogena che, stimolati dall’aumento di stress
a livello parietale e dalla distensione atriale, modulano la disfunzione
cardiaca nelle prime fasi di comparsa, sia opponendosi agli effetti deleteri
dell’attivazione adrenergica che viene inibita, sia esercitando un’azione
natriuretica e vasodilatatrice diretta, sia riducendo il carico emodinamico
del cuore.
Il sovraccarico cronico impartisce segnali meccanici che vengono
interpretati come uno stimolo all’ipertrofia ventricolare eccentrica,
all’attivazione di collagenasi interstiziali, con degradazione della matrice
di collageno extracellulare che porta alla dilatazione del cuore e allo
sviluppo della sindrome clinica. Il processo di “remodeling” ventricolare
corrisponde alla fase preclinica, può essere bloccato dalla terapia ACEinibitrice.
Nella sindrome clinica dello scompenso cardiaco cronico il danno non è
solo a livello miocardico ma si instaura una sorta di alterazione
metabolica/strutturale anche a livello del muscolo scheletrico. Alcuni
autori hanno addirittura proposto l’esistenza di una miopatia da
scompenso cardiaco indipendente dal flusso ematico (Lipkin DP., 1988)
Tuttavia le cause che possono indurre la miopatia da scompenso sono
ancora ignote. Recentemente, per spiegare questo fenomeno, sono state
chiamate in causa alterazioni del sistema endocrino (incremento delle
catecolamine e del cortisolo circolante, alterazioni del rapporto
insulina/glucagone, fenomeni di insulino resistenza) e del sistema
infiammatorio (elevati valori circolanti di Tumor Necrosis Factor (TNF) e
50
Lo scompenso cardiaco
l’Interleuchina (IL)-6). Tutte queste molecole hanno marcati effetti
catabolici sul cuore e sul circolo. In particolare le citochine e il TNF hanno
effetto
proinfiammatorio
ed
isotropo
negativo
e
contribuiscono
all’aggravamento della disfunzione ventricolare sinistra.
Diverse osservazioni suggeriscono che esiste un legame tra sistema
neuroendocrino e sistema infiammatorio; è ormai noto che fin dalle prime
fasi della disfunzione ventricolare sinistra viene attivato il sistema
neuroendocrino e che esso è responsabile del peggioramento della
condizione clinica dello scompenso cardiaco (Ruffolo R., 1998). D’altro
canto è ormai ben documentato che il sistema infiammatorio è stimolato
nelle fasi avanzate dello scompenso; infatti alcuni studi hanno dimostrato
un incremento nel siero di pazienti con scompenso severo di neopterina
(marker di attivazione dei monociti/macrofagi), Interleuchina (IL)-1,
Interleuchina (IL)-2, IL-6 e suoi recettori solubili (sIL-6R), fattore di
necrosi tumorale (TNF) e suoi recettori solubili (sTNF-R1, sTNF-R2)
(Murger MA., 1996).
Il prolungato utilizzo di questi meccanismi di compenso comporta
l’esaurimento o la perdita dei loro effetti fisiologici favorevoli: durante
questa fase si passa dalla disfunzione ventricolare sinistra asintomatica
allo scompenso cardiaco conclamato e tali meccanismi compensatori
divengono motori di aggravamento della patologia.
Il sovraccarico cronico ventricolare conduce a fibrosi ed assottigliamento
della parete ventricolare (Dzau VJ. et al., 1984) ; il sovraccarico cronico
atriale porta al resetting dei meccanocettori, che divengono incapaci di
contrastare l’attivazione adrenergica, ed alla riduzione della produzione di
ANP (Iervasi G. et al., 1995).
51
Lo scompenso cardiaco
L’iperattività adrenergica e dei sistemi vasocostrittori e sodio-ritentivi
conduce ad una dilatazione ventricolare progressiva legata ad una attività
del sistema nervoso simpatico persistente e alla riduzione della riserva
inotropa, dovuta sia a meccanismi cellulari (perdita dell’efficienza
contrattile dei sarcomeri stirati) che recettoriali (“down-regolation” dei
recettori β-adrenergici) che favoriscono le aritmie ventricolari (Bristow
MR. et al., 1990).
2.4 Il sistema chemorecettoriale e barorecettoriale
I soggetti affetti da scompenso cardiaco presentano una sintomatologia
caratterizzata soprattutto da dispnea ed intolleranza allo sforzo fisico, che
risultano scarsamente relazionabili al deficit di pompa del cuore e
maggiormente correlabili alle modificazioni del sistema neurovegetativo e
respiratorio, oltre che all’indebolimento fisico. Studi recenti hanno
evidenziato come, associato ad un problema puramente cardiaco, vi sia,
rispetto ai soggetti sani, anche una modificazione dell’attività simpatica e
vagale ed un’alterazione della funzionalità di alcuni importanti sistemi
recettoriali, tra cui i barocettori, i chemocettori e gli ergocettori.
I
pazienti
scompensati
presentano
un’alterazione
del
sistema
neurovegetativo caratterizzata da un aumento dell’attività ortosimpatica e
da una diminuzione di quella parasimpatica (Francis GS. et al., 1984;
Floras JS., 1993).
Parallelamente a questa alterazione si osserva una modificazione
funzionale di alcuni importanti sistemi recettoriali presenti nell’organismo
umano: i chemocettori centrali e periferici (rispettivamente ubicati a
livello bulbare, i primi, e del glomo carotideo e dell’arco aortico, i
52
Lo scompenso cardiaco
secondi) presentano un’aumentata sensibilità; le aree barocettive
cardiopolmonari (a bassa pressione) e quelle aortiche e carotidee (ad alta
pressione) mostrano una diminuita sensibilità; gli ergocettori muscolari,
infine, esibiscono un’ipersensibilità ed iperattività (Floras JS., 1993;
Thames MD. et al., 1993).
Non è tuttavia possibile determinare con certezza se le variazioni
funzionali di questi sistemi recettoriali rappresentino la causa od originino
successivamente alle alterazioni neurormonali, in quanto esistono rapporti
di stretta interrelazione fra questi sistemi (Ponikowski P. et al., 1997)
Per quanto riguarda i barocettori (responsabili del mantenimento
dell’omeostasi cardiocircolatoria, in condizioni normali) la loro ridotta
sensibilità potrebbe essere dovuta ad una diminuita distensibilità della
parete vascolare, ad un malfunzionamento dell’integrazione riflessa a
livello centrale, ad una combinazione di entrambi o, ancora, ad altri
meccanismi meno conosciuti (Kingwell BA. et al., 1995).
La disfunzione del baroriflesso impedisce la corretta risposta riflessa di
fronte ad una riduzione della portata cardiaca e quindi della pressione
arteriosa,
al
fine
di
ripristinare
l’omeostasi
cardiocircolatoria.
Normalmente i barocettori esercitano un’azione inibitoria sul sistema
nervoso simpatico, ma a causa della loro alterazione funzionale si
stabilisce una diminuzione sia della loro azione inibitoria che della loro
influenza eccitatoria sul sistema vagale a livello cardiocircolatorio. Da ciò
deriva la comparsa di uno stato di iperattività simpatica e di diminuita
attività vagale, con conseguente tachicardia e vasocostrizione periferica.
L’influenza della variazione della sensibilità chemorecettoriale su alcuni
aspetti fisiopatologici dello scompenso cardiaco diviene comprensibile per
53
Lo scompenso cardiaco
gli effetti che opera sul sistema respiratorio, cardiocircolatorio e
neurormonale.
Nei soggetti con scompenso cardiaco l’aumentata attività chemoriflessa
può essere vista come un meccanismo di compenso, atto, attraverso un
aumento della ventilazione, a garantire una saturazione arteriosa di
ossigeno adeguata alle necessità metaboliche.
Essa ha tuttavia effetti deleteri, essendo, in parte, responsabile
dell’iperventilazione, della dispnea e dell’iperattività simpatica che si
osservano in questa condizione (Chugh SS. et al., 1996).
Diversi sono i fattori che, nello scompenso, concorrono a determinare
l’ipersensibilità dei chemocettori. L’aumento del tono simpatico,
innanzitutto, è in grado di indurre un aumento della sensibilità dei
chemocettori
periferici
probabilmente
provocando,
mediante
vasocostrizione, una riduzione del flusso sanguigno arterioso a livello
centrale e attraverso i glomi carotidei (Chugh SS. et al., 1996). Ad una
riduzione di flusso attraverso i glomi carotidei e aortici contribuisce anche
l’allungamento del tempo di circolo (Coats AJS., 1997), dovuto alla
ridotta gittata cardiaca che si osserva in questi soggetti. Anche i
cambiamenti neurormonali, che hanno luogo nello scompenso cardiaco
cronico, come l’aumento del livello di catecolamine, potrebbero far
aumentare la chemosensibilità (Coats AJS., 1997). Infine, viene meno
l’azione inibitoria dei barocettori sui chemocettori a causa del
malfunzionamento del baroriflesso (Ponikowski P. et al., 1997).
Nei soggetti con scompenso cardiaco, infatti, vi è una ridotta sensibilità ed
attività delle aree barocettive, carotidee ed aortica, valutata come
regolazione riflessa della pressione arteriosa e della frequenza cardiaca
54
Lo scompenso cardiaco
(Creager MA., 1994). È stato tuttavia dimostrato che, in condizioni
fisiologiche, la stimolazione baroriflessa è in grado di ridurre
significativamente la risposta simpatica alla stimolazione ipossica dei
chemocettori periferici (Somers VK. et al., 1991).
Risulta pertanto chiaro come nei soggetti con scompenso cardiaco la
diminuzione della sensibilità e dell’attività baroriflessa possa facilitare,
attraverso una minore inibizione, l’iperattivazione chemorecettoriale.
2.5 Il sistema renina-angiotensina
La renina e l’angiotensina costituiscono un sistema endocrino di notevole
importanza nella regolazione dell’equilibrio idrosalino e delle resistenze
vascolari, fondamentale per il ripristino del volume dei fluidi (Michel JB.
et al.).
In presenza di disfunzione ventricolare, queste funzioni contribuiscono
alla ritenzione idosalina e alla vasocostrizione al fine di ripristinare il
volume plasmatico e la pressione arteriosa media.
L’attivazione del sistema renina-angiotensina e dell’aldosterone nello
scompenso cardiaco è ciclica: è marcata ad ogni ricaduta congestizia della
malattia e nello scompenso terminale; è facilitata dal trattamento diuretico,
che è uno stimolo potente del sistema, mentre è frenata da un meccanismo
di “feedback” negativo quando la ritenzione idrosalina diminuisce la
secrezione di renina; inoltre induce direttamente ritenzione idrosodica ed
ipopotassiemia.
Tale sistema ha un’azione vasocostrittiva sistemica e sul tono coronario,
stimola inoltre l’attivazione del sistema nervoso simpatico con vari
meccanismi, che includono il blocco del “re-uptake” della noradrenalina e
55
Lo scompenso cardiaco
la facilitazione del suo rilascio, favorisce l’ipertrofia miocardia e delle
cellule lisce vascolari.
La riduzione della portata cardiaca provoca, in collaborazione con il
sistema adrenergico, un aumento della renina plasmatica e della
formazione di angiotensina II.
L’angiotensina II stimola inoltre la liberazione di vasopressina e
aldosterone, al fine di incrementare il tono vasomotore e la ritenzione
idrosalina.
2.6 Sistema dell’Endotelina
Le endoteline sono una famiglia di peptidi prodotta da un'ampia varietà di
tessuti; il principale sito di generazione è costituito dalle cellule
endoteliali, da cui tali proteine traggono il nome (Inoue A. et al., 1989) .
L'ET-1 è l'isoforma predominante a livello vascolare e esercita diverse
azioni biologiche: è il più potente agente vasocostrittore endogeno ed ha
effetti inotropi e cronotropi positivi.
L'ET-1 presenta anche proprietà mitogene; è in grado di influenzare
l'omeostasi idro-salina e di stimolare vari sistemi, tra cui il sistema reninaangiotensina.
L'ET-1 contribuisce alla fisiopatologia dello scompenso cardiaco agendo
come fattore paracrino e come ormone circolante nella regolazione del
tono vascolare. La generazione endogena dell'ET-1 sembra contribuire al
mantenimento del tono vascolare basale nei soggetti sani e alla resistenza
vascolare periferica nei pazienti scompensati.
L'ET-1 può, altresì, aumentare l'azione di alcuni fattori neuroendocrini
promuovendo la conversione dell’angiotensina I in angiotensina II, come
56
Lo scompenso cardiaco
osservato nelle colture cellulari o aumentando la sintesi di adrenalina e
aldosterone. Inoltre, concentrazioni fisiologiche di ET-1 aumentano la
risposta biologica alle catecolamine.
È stato osservato che alti livelli di ET-1 plasmatica, in pazienti
scompensati, sono fondamentalmente correlati ad elevati livelli di Big-ET1, il suo precursore biologicamente inattivo.
L’attivazione del sistema dell’endotelina, che potrebbe rappresentare un
meccanismo di compenso che si oppone alla riduzione dell’inotropismo e
contribuisce
al
mantenimento
della
funzionalità
cardiaca,
è
controbilanciato da effetti sfavorevoli, quali la riduzione del flusso
coronario o l’induzione di ipertrofia cardiaca e rimodellamento. Questo è
confermato dagli effetti positivi ottenuti in pazienti con scompenso
cardiaco dopo trattamento con antagonisti recettoriali dell’endotelina e
della parallela attivazione del sistema dei peptidi natriuretici cardiaci (Del
Ry S. et al., 2001; Wei CM. et al., 1993).
2.7 Sistema della vasopressina
La vasopressina (arginino vasopressina, ormone antidiuretico, AVP,
ADH) è un ormone nonapeptidico vasomotore ed antidiuretico, rilasciato
dalla
neuroipofisi
sotto
il
controllo
dei
nuclei
sopraottico
e
paraventricolare ipotalamici. Questo ormone gioca un ruolo importante
nella regolazione del volume dei fluidi extracellulari e dell’osmolalità
plasmatica, modulando la “clearance” renale dell’acqua libera.
Lo scompenso cardiaco è caratterizzao da una attivazione attraverso il
baroriflesso dei differenti sistemi ormonali vasoattivi e idrosalino ritentivi
57
Lo scompenso cardiaco
(sistema simpatico, sistema renina-angiotensina, aldosterone, endotelina,
vasopressina) (Arnolda L. et al., 1986).
Nei modelli sperimentali di scompenso l’aumento della sensibilità
recettoriale vascolare precede insieme con l’attivazione del sistema reninaangiotensina e l’attivazione adrenergica, l’incremento dei valori di
vasopressina (Johnston CI. et al., 1986); infatti in questa patologia si
osserva un elevato aumento della vasopressina, che dovrebbe essere inibita
dall’aumento della pressione atriale e del volume ematico. L’attivazione
simpatica e angiotensinica supera la modulazione delle afferente dei
volocettori e barocettori cardiaci a bassa pressione e l’osmoregolazione;
ciò può contribuire all’inadeguata capacità di eliminare l’acqua libera e
alla ipoosmolarità.
La vasopressina elevata esercita inoltre un effetto additivo vasocostrittore
ed idroritentore nello scompenso.
58
CNP e scompenso cardiaco
3
CNP E SCOMPENSO CARDIACO
Capitolo
Il ruolo del CNP nella patofisiologia cardiaca, e specialmente nello
scompenso cardiaco (Clerico A. et al., 2004; Richards AM., 2003; Jortani
SA. et al., 2004) non è ancora completamente definito, al contrario di
quello di ANP e BNP che è ampiamente dimostrato.
Come già detto in precedenza l’ANP e il BNP regolano l’omeostasi
circolatoria e svolgono la loro azione in risposta allo stiramento locale
della parete cardiaca, dovuto ad un aumento del volume intravascolare
determinando massiva diuresi e natriuresi. Inoltre influenzano la
vasodilatazione locale e la regolazione della crescita vascolare,
interagendo con numerose citochine vasoattive e con il CNP stesso,
determinando sia la riduzione della pressione sanguigna che del volume
circolatorio (Kalra PR. et al., 2001).
Per quanto riguarda i livelli circolanti di CNP, sono stati condotti molti
studi per valutare variazioni nei livelli di questo peptide nello scompenso
cardiaco, ma i risultati ottenuti sono discordanti. Questa discrepanza
potrebbe essere dovuta sia al numero limitato di pazienti analizzati in
singoli studi, sia a differenze nei metodi di determinazione di questo
peptide (Wei CM. et al., 1993; Totsune K. et al., 1994; Cargill RI. et al.,
1994; Del Ry S. et al., 2005; Del Ry S. et al., 2006) (Tab 1).
59
CNP e scompenso cardiaco
Tabella 1: Studi su CNP e scompenso cardiaco
60
CNP e scompenso cardiaco
I primi studi sul CNP non individuarono la sua presenza nè a livello
cardiaco nè a livello plasmatico in soggetti scompensati (Wei CM. et al.,
1993; Totsune K. et al., 1994; Cargill RI. et al., 1994), ritardando così il
riconoscimento dell’importanza del CNP a livello cardiovascolare.
Tali osservazioni, dissuasero inizialmente i ricercatori dall’investigare
quali fossero le potenziali correlazioni tra il CNP e i peptidi
cardioregolatori conosciuti, sebbene fosse stata evidenziata sia la presenza
di mRNA codificante per il recettore di tipo B, specifico per il CNP a
livello miocardico umano e murino (Nunez DJR. et al., 1992), sia di
trascritti per il CNP negli atrii e nei ventricoli di ratti (Vollmar AM. et al.,
1993) (recentemente sono state ritrovate basse concentrazioni di mRNA
codificante per il CNP nel cuore di topi sia neonati che adulti) (Stepan H.
et al., 2000).
La concentrazione di CNP a livello miocardico è stata determinata
attraverso
tecniche
immunoistochimiche
e
radioimmunometriche,
risultando più elevata nei pazienti scompensati rispetto ai soggetti normali
(Wei CM. et al., 1993).
Nel 1994 è stato rilevato un aumento significativo del CNP a livello
urinario in pazienti scompensati (Mattingly MT. et al., 1994) che ha
suggerito un suo potenziale coinvolgimento a livello del sistema dei
peptidi natriuretici renali, i quali sono coinvolti nella regolazione del
bilancio di acqua e sodio e nella circolazione renale.
Il gruppo di Kalra (Kalra PR. et al., 2003) ha mostrato recentemente, che
nei pazienti scompensati i livelli di CNP sono significativamente
aumentati nel seno coronario rispetto a quelli dell’aorta, dimostrandone la
produzione cardiaca. Questi risultati sono stati confermati da uno studio
61
CNP e scompenso cardiaco
effettuato nel nostro laboratorio (Del Ry S. et al., 2006). Come si può
notare
un significativo step-up è stato osservato nelle concentrazioni
plasmatiche del CNP prelevato dall’aorta rispetto a uello prelevato dal
seno coronarico, confermando che il CNP è prodotto direttamente nel
miocardio di pazienti con scompenso cardiaco.
Questo suggerisce un ruolo autocrino/paracrino cardiaco in accordo con
altri studi che riportano che il CNP inibisce l’ipertrofia dei cardiomiociti e
che è sintetizzato e secreto dai fibroblasti cardiaci (Horio T. et al., 2003;
Tokudome T. et al., 2004), dove l’NPR-B sembra essere il recettore
maggiormente espresso. Gli effetti inibitori del CNP sulla proliferazione e
produzione del collagene nei fibroblasti cardiaci, probabilmente mediati
dal recettore di tipo B, suggeriscono che il CNP possa funzionare come un
regolatore autocrino negativo contro l’eccessiva fibrosi cardiaca degli stati
patologici.
Sebbene ulteriori studi siano necessari per chiarire il ruolo fisiologico e
patofisiologico del CNP endogeno nel cuore, questi dati, presi nel loro
insieme, fanno ipotizzare che il CNP sia prodotto nel cuore durante lo
scompenso,
dove
può
svolgere
un
ruolo
compensatorio
sul
rimodellamemento ventricolare.
Questo è in accordo con alcuni studi in cui è stato osservato un aumento
dei livelli plasmatici del precursore NT-proCNP e del CNP nei soggetti
scompensati rispetto ai controlli (Prickett TCR. et al., 2001; Wright SP. et
al., 2004; Del Ry S. et al., 2005).
L’aumento della secrezione osservata, sembra essere in relazione al grado
di severità della patologia (classe NYHA), come è indicato dal progressivo
incremento dei livelli plasmatici di CNP durante il peggioramento dei
62
CNP e scompenso cardiaco
sintomi e dalla correlazione negativa osservata tra i livelli di CNP
plasmatico e la frazione di eiezione del ventricolo sinistro (Del Ry S. et
al., 2005).
È infatti noto che il CNP esercita importanti azioni cardiovascolari
indirette come la riduzione della pressione cardiaca, dovuta alla
vasodilatazione e alla diminuzione del ritorno venoso (Chen HH. et al.,
1998). Inoltre, la produzione cellulare di CNP è aumentata sia in vitro in
presenza di effettori come il BNP e citochine, che in vivo, in modelli
animali in seguito ad infusione con ANP e BNP. Queste osservazioni,
insieme con i risultati sperimentali, vanno a sostenere il ruolo del CNP
nella fisiopatologia dello scompenso (Potter LR., 2004).
Questo è anche confermato dall’incremento dei livelli plasmatici di CNP
e del frammento N-terminale del suo precursore NT-proCNP nello
scompenso cardiaco, che sono in accordo con altri studi in cui è stata
osservato, nell’animale, un’interazione tra i livelli circolanti di ANP, BNP
e CNP e dimostrano come l’aumento dei livelli plasmatici di uno di questi
peptidi porti all’aumento dei livelli circolanti degli altri due (Charles CJ. et
al., 1996).
Assumendo la co-secrezione dell’NT-proCNP e del CNP, la “upregulation” e/o la ridotta clearance di questo vasodilatatore locale nello
scompenso cardiaco può essere interpretata come una risposta
compensatoria con effetti positivi che si oppone all’aumento delle
resistenze periferiche vascolari caratteristiche dello scompenso cardiaco
(Wright SP. et al., 2004).
La conoscenza del ruolo dei peptidi natriuretici nello scompenso cardiaco
è importante in quanto può suggerire nuove strategie terapeutiche rivolte a
63
CNP e scompenso cardiaco
modularne i livelli, tra queste particolare interesse è stato rivolto
all’inibizione dei meccanismi di clearance attraverso le endopeptidasi:
sono stati osservati sia nell’uomo che su animali sperimentali gli effetti
terapeutici dovuti alla somministrazione di endopeptidasi inibitorie, in
associazione
ad
altri
trattamenti
farmacologici,
come
gli
ACE
(angiotensin-converting enzyme) e ECE (endothelin-converting enzyme)
inibitori (Margulies KB. et al., 1991; Cruden NL. et al., 2004; Abassi ZA.
et al., 2005; Emoto N. et al., 2005; Mellin V. et al., 2005; Solomon SD. et
al., 2005)
Per quanto riguarda il meccanismo d’azione, se ANP e BNP sono in grado
di regolare la produzione e secrezione di CNP in vivo, come osservato
nelle cellule endoteliali in coltura, alcune importanti azioni dei peptidi
natriuretici nei vasi, potrebbero essere in parte dovute alla stimolazione e
successiva azione del CNP. Questo può costituire una base razionale per
l’uso farmacologico a scopo terapeutico del BNP, l’agente più potente
nello stimolare la produzione di CNP nella sindrome dello scompenso
cardiaco al fine di migliorare i sintomi clinici dei pazienti colpiti
(Fitzgerald RL. et al., 2005; Burger AJ. et al., 2005). È stato comunque
dimostrato che il nesiritide (farmaco con caratteristiche identiche al BNP
endogeno),
utilizzato
nello
scompenso
cardiaco
acuto,
aumenta
significativamente il rischio di scompenso renale e di morte.
Sebbene ulteriori studi siano richiesti per definire meglio il ruolo del CNP
nella patofisiologia dello scompenso cardiaco, diverse osservazioni
sperimental indicano che il CNP può rappresentare un nuovo ed
importante mediatore locale autocrino/endocrino a livello cardiaco.
64
CNP e scompenso cardiaco
In aggiunta il CNP potrebbe rappresentare un ormone contro-regolatore
del sistema neuroormonale vasocostrittore, che è noto essere attivato
durante lo scompenso cardiaco, dove contribuisce al bilancio e alla
modulazione dell’omeostasi circolatoria.
65
Disfunzione endoteliale e malattia cardiovascolare
4
DISFUNZIONE ENDOTELIALE E
MALATTIA CARDIOVASCOLARE
Capitolo
Le cellule endoteliali producono molti mediatori vasoattivi, generalmente
denominati
“endothelium-derived
vasorelaxant
mediators”,
che
contribuiscono al controllo del tono e della crescita delle cellule muscolari
lisce e hanno effetti sulla funzione delle cellule circolanti (globuli bianchi,
eritrociti e piastrine) (Ahluwalia A. et al., 2005).
L’alterazione nella capacità da parte dell’endotelio di produrre alcuni
mediatori in risposta a stimoli patofisiologici (la così definita “disfunzione
endoteliale”) sembra sia il principale fattore comune di molte patologie
cardiovascolari.
I più importanti tra questi mediatori sono la prostaciclina (PGI2) e il
monossido di azoto (NO), ma recentemente è stato descritto anche un
nuovo mediatore vasorilassante di origine endoteliale, denominato EDHF
(endothelium-derived hyperpolarizing factor) che esercita un caratteristico
effetto di iper-polarizzazione e rilassamento sulle cellule muscolari
(Ahluwalia A. et al., 2005).
Anche i peptidi natriuretici cardiaci (e specialmente il CNP) sembra
possano agire come EDHF in alcuni letti vascolari (Ahluwalia A. et al.,
2005; Houben AJ. et al., 2005; Scotland RS. et al., 2005; Han B. et al.,
2003). Infatti, numerosi studi hanno dimostrato che ANP, BNP e CNP
legandosi ai recettori biologici (NPR-A e NPR-B) sulle cellule muscolari
lisce vascolari, stimolano l’accumulo di cGMP causando una dilatazione
dose-dipendente (Zeidel ML., 2001; Houben AJ. et al., 2005; Scotland RS.
et al., 2005; Han B. et al., 2003). Questo aumento di cGMP causa
66
Disfunzione endoteliale e malattia cardiovascolare
vasodilatazione attraverso la riduzione dei livelli di calcio intracellulare,
come avviene quando l’accumulo di cGMP è stimolato dal monossido di
azoto (NO) e dai suoi analoghi (Zeidel ML., 2001).
E’ ipotizzabile che l’ANP e il BNP agiscano come ormoni a livello del
tessuto vascolare raggiungendo le cellule muscolari lisce dalla
circolazione dopo essere stati secreti dal cuore, a differenza del CNP che
esercita un azione paracrina essendo secreto direttamente dalle cellule
endoteliali (Woodard GE. et al., 2002; Chen HH. et al., 1998; Qian JY. et
al., 2002).
Studi con metodi di ibridazione in situ e di immunocitochimica hanno
dimostrato l’esistenza di un completo sistema dei peptidi natriuretici
cardiaci (inclusa produzione e secrezione di ANP, BNP e CNP) nei vasi
coronarici aterosclerotici umani (Rahmutula D. et al., 2001). In particolare
l’espressione di mRNA codificante per ANP, BNP e CNP , misurata con
RT-PCR, tende ad essere aumentata in arterie microscopicamente malate
rispetto ai vasi normali, sebbene solo l’espressione del BNP risulti
significativamente aumentata. (Casco VH. et al., 2002).
I risultati di questo studio suggeriscono che il sistema dei peptidi
natriuretici è coinvolto sia nella patobiologia della formazione della placca
che nei processi di rimodellamento vascolare dell’uomo.
È stato dimostrato che tra i
peptidi natriuretici ci sono complesse
interazioni: l’ANP e il BNP possono stimolare la produzione di CNP
attraverso un recettore guanilato ciclasico sulle cellule endoteliali (Nazario
B. et al., 1995); la vasodilatazione e gli effetti anti-mitogenici dell’ANP e
del BNP a livello del sistema vascolare sono in parte mediati dalla
67
Disfunzione endoteliale e malattia cardiovascolare
produzione di CNP. Queste osservazioni indicano che ANP/BNP e CNP
possono avere un’azione sinergica sul tessuto vascolare.
Sono state anche dimostrate complesse interazioni tra peptidi natriuretici e
altri mediatori vasorilassanti di derivazione endoteliale (Ahluwalia A. et
al., 2005). In particolare il CNP mima molte delle azioni anti-aterogeniche
della prostaciclina e dell’monossido di azoto (Ahluwalia A. et al., 2005).
Questo fa ipotizzare che il CNP, in aggiunta alle sue azioni anti-adesive e
anti-aggreganti, possa compensare la riduzione di questi mediatori nelle
patologie cardiovascolari al fine di ristabilire le capacità vasodilatatorie
dell’endotelio.
I peptidi natriuretici a livello del tessuto vascolare intergiscono anche con
gli effettori del sistema contro-regolatore (McGrath MF. et al., 2005;
Clerico A. et al., 2002; Clerico A., 2004; Ruskoaho H., 2003; De Lemos
JA. et al., 2003; Ventura RR. et al., 2002; Vatta MS. et al., 1996; Vatta
MS. et al., 1997; Fermepin M. et al., 2000; Tsukagoshi H. et al., 2001;
Kapoun AM. et al., 2004; Vollmar AM. et al., 2005), in particolare con
l’endotelina (ET)-1, i cui effetti vascolari sono direttamente opposti a
quelli dei peptidi natriuretici (Ahluwalia A. et al., 2005; Zeidel ML., 2001;
Han B. et al., 2003). Le azioni diuretiche e natriuretiche del CNP sono
meno marcate rispetto a quelle di ANP e BNP e il CNP stesso è in grado
di modulare gli effetti vascolari del sistema renina-angiotensinaaldosterone.
Inoltre il CNP non solo antagonizza l’ET-1 e l’angiotensina II, ma modula
direttamente la sintesi di entrambe; sua volta l’ET-1 induce l’aumento del
numero di cellule endoteliali che secernono CNP (Khono M. et al., 1992;
Davidson NC. et al., 1996). La parallela produzione e attivazione di
68
Disfunzione endoteliale e malattia cardiovascolare
effettori con azione vasodilatante, come il CNP, e di effettori con azione
vasocostrittiva, come l’ET-1 e angiotensina II, permette la fine
regolazione locale dell’espressione di questi peptidi e quindi del flusso
sanguigno (Ahluwalia A. et al., 2005; Zeidel ML., 2001; Han B. et al.,
2003; Nazario B. et al., 1995; Casco VH. et al., 2005; Scotland RS. et al.,
2005; Khono M. et al., 1992; Davidson NC. et al., 1996 ).
Infine, la inter-relazione tra il sistema dei peptidi natriuretici e le citochine
pro-infiammatorie, suggerisce che tali ormoni cardiaci esercitano un ruolo
importante nei meccanismi responsabili di rimodellamento e adattamento
sia cardiaco che vascolare, in risposta a vari stimoli fisiologici e patologici
(Tenhunen O. et al., 2005; Walther T. et al., 2003; Glembotoski CC. et al.,
1993; Mukoyama M. et al., 1991). L’identificazione del CNP come EDHF
associato alla sua espressione nelle cellule endoteliali, indica che il CNP è
adatto a modulare l’attività delle cellule circolanti, soprattutto di leucociti
e piastrine. In particolare, stimoli infiammatori, come l’interleuchina (IL)11 e il tumor necrosis factor (TNF)-α, inducono la produzione di CNP da
cellule endoteliali isolate e, come risultato, la modulazione dell’attività
biologica del CNP ha una profonda influenza sullo sviluppo della risposta
infiammatoria (Ahluwalia A. et al., 2005; Houben AJ. et al., 2005;
Scotland RS. et al., 2005; Han B. et al., 2003; Suga S. et al., 1993).
Molte evidenze indicano che i peptidi natriuretici, e in particolare il CNP,
sono importanti mediatori endogeni con effetti anti-aterogenici: il CNP è
un potente inibitore della migrazione e proliferazione delle cellule
muscolari lisce (Khono M. et al., 1992) ed è in grado di bloccare
l’aggregazione
piastrinica,
esercitando
così
effetti
anti-trombotici
(Ahluwalia A. et al., 2005).
69
Disfunzione endoteliale e malattia cardiovascolare
Tutti gli studi sopra citati indicano che i peptidi natriuretici, e
specialmente il CNP, esercitano un potente effetto sulla funzione
endoteliale in quanto riducono lo shear stress, modulano le vie di
coagulazione e fibrinolisi e inibiscono l’attivazione piastrinica e sono
coinvolti nei processi di rimodellamento e nella ristesosi postangioplastica; per queste loro importanti azioni, i peptidi natriuretici hanno
un ruolo rilevante in tutte quelle situazioni caratterizzate da disfunzione
endoteliale e rimodellamento (Ma KK. et al., 2004; Chen HH. et al., 1998;
Qian JY. et al., 2002; Nakanishi M. et al., 2005; Ahluwalia A. et al., 2005;
Ohno N. et al., 2002; Kiemer AK. et al., 2005).
70
Scopo del lavoro di tesi
5
SCOPO DEL LAVORO DI TESI
Capitolo
Il CNP, per la sua origine prevalentemente endoteliale e per le sue azioni
biologiche, sembra essere coinvolto in situazioni fisiopatologiche
associate a disfunzione endoteliale e a processi di rimodellamento, quali lo
scompenso cardiaco.
Nello scompenso cardiaco, infatti, i livelli plasmatici di CNP sono
risultati elevati in relazione alla severità della malattia, analogamente a
quanto osservato per ANP e BNP, ma mentre il ruolo patofisiologico
dell’ANP e del BNP è ampiamente dimostrato, quello del CNP non è
ancora completamente definito. Recentemente è stata evidenziata una
produzione di questo peptide a livello cardiaco in pazienti con scompenso
cardiaco, tuttavia non è ancora stato determinato il sito di produzione del
peptide e le eventuali differenze tra produzione atriale e ventricolare.
Scopo di questa tesi è quello di valutare il ruolo del CNP nello scompenso
cardiaco, in un modello sperimentale animale di maiale, che utilizza
minipigs con insufficienza cardiaca cronica, indotta da stimolo con pacemaker ad alta frequenza. In questo modello verranno determinati i livelli
plasmatici di CNP e l’espressione cardiaca, a livello di mRNA e di
proteina, del peptide e del suo recettore biologico specifico NPR-B.
Infatti, per quanto riguarda il meccanismo di azione del CNP, non è
completamente nota la regolazione dei recettori specifici a livello
cardiaco, indispensabile per definire il ruolo paracrino di questo effettore e
le sue interazioni con i peptidi natriuretici cardiaci, ANP e BNP.
71
Scopo del lavoro di tesi
Al fine di valutare le possibili variazioni nell’espressione dell’mRNA
codificante per l’NPR-B nel tessuto cardiaco di maiali normali e
scompensati sarà necessario sequenziare (almeno in parte) il gene per il
recettore NPR-B nel tessuto cardiaco di Sus Scrofa, attualmente mancante
nella GenBank (data base internazionale delle sequenze genetiche del
National Institutes of Health, NIH). Il sequenziamento del gene per
l’NPR-B di maiale fornirà, a noi e a tutti i ricercatori che utilizzano questo
modello animale, uno strumento aggiuntivo ed importante per valutare il
ruolo del recettore biologico specifico in condizioni fisiologiche e
patologiche oltre alla risposta ad eventuali interventi terapeutici.
Nonostante ulteriori studi siano necessari per chiarire le proprietà
patofisiologiche di questo peptide e l’inter-scambio con l’attivazione
neuro-ormonale, questo lavoro di tesi potrà essere importante in quanto,
analizzando il sistema da più punti di vista contemporaneamente (livelli
plasmatici, tessutali, mRNA per CNP, mRNA per NPR-B) ci permetterà di
capire meglio il ruolo patofisiologico del CNP nello scompenso cardiaco.
72
Materiali e metodi
6
MATERIALI E METODI
Capitolo
I protocolli a cui sono stati sottoposti tutti gli animali utilizzati in questo
studio sono conformi alle norme proposte dalle linee guida dell’NIH per la
cura e l’utilizzo degli animali da laboratorio (Nih Guide for Care and Use
of Laboratory Animal), ed è stato approvato dall’Istituto per le cure
animali (Institutional Animal Care).
6.1 Modello sperimentale animale
Lo studio è stato condotto in collaborazione con il Centro di Biomedicina
Sperimentale dell’Istituto di Fisiologia Clinica, che si occupa del modello
sperimentale. Sono stati utilizzati 10 minipig adulti (di cui 5 normali,
utilizzati come controllo, e 5 in cui è stato indotto lo scompenso cardiaco
cronico). A seguito delle difficoltà correlate alla stabulazione ed
all’utilizzo sperimentale di suini di fattoria adulti, nel nostro studio
abbiamo utilizzato “minipig” adulti. Sotto tale denominazione vengono
comprese alcune razze di suini di taglia ridotta (altezza di 20-30
centimetri; peso che non supera i 30-40 chilogrammi); questi animali
seppur di più facile gestione, hanno caratteristiche anatomiche (grugno
molto breve, orecchie piccole con trama vascolare poco accessibile) che
rendono ancora più complesse le manualità anestesiologiche.
Gli animali sono stati sedati (Zoletil®6.3 mg/kg i.m.), anestetizzati
(isoflurane, Forane, MAC 1%, aria/ossigeno: 60/40%), assistiti mediante
ventilazione meccanica e sottoposti ad intervento chirurgico in condizioni
73
Materiali e metodi
sterili. Durante la procedura parametri vitali venivano monitorati in
continuo (pressione arteriosa, elettrocardiogramma, temperatura corporea,
PaO2, PaCO2, pH).
Dopo toracotomia, eseguita a livello del quinto spazio intercostale di
sinistra, è stato inciso il sacco pericardico e il cuore è stato strumentato
mediante l’impianto dei seguenti cateteri e sonde (Fig. 7):
• sonda di flusso doppler, posizionata a livello della coronaria discendente
anteriore, per misurare il flusso coronarico medio;
• sonda di pressione tipo Konisberg, posizionata nella camera ventricolare
sinistra, per la misurazione della pressione ventricolare;
• elettrocatetere unipolare a vite, posizionato a livello della parete libera
del ventricolo sinistro e collegato a sua volta ad un pacemaker
posizionato nel sottocute dorsale, per la stimolazione elettromeccanica
del cuore;
• catetere di poliuretano eparinato per la misura della pressione arteriosa;
• catetere di Tygon, posizionato nella vena giugulare esterna di sinistra per
la somministrazione di liquidi e farmaci dall’esterno.
Figura 7: Schema di strumentazione chirurgica del cuore
74
Materiali e metodi
Al termine della procedura, dopo aver provveduto alla rimozione dello
pneumotorace chirurgico, si è provveduto ad una terapia sia antibiotica
(Cefamezine®1gr al giorno, per 5 giorni) che del dolore post-operatorio
(Toradol® 30mg/ml al giorno, per 5 giorni) .
A dieci giorni dal ricovero post-operatorio, durante il quale sono stati
monitorati i parametri vitali, i minipig sono stati sottoposti al seguente
protocollo sperimentale:
• Tomografia ad Emissione di Positroni (PET), per quantificare la
perfusione miocardia, mediante previa somministrazione intravenosa di
13
NH3 (ammoniaca radiomarcata), e l’uptake miocardico di glucosio,
mediante
previa
somministrazione
intravenosa
di
18
FDG
(18-
fluorodesossiglucosio) (Fig. 8);
• Risonanza Magnetica (RM), per quantificare i volumi ventricolari, la
contrattilità globale (frazione di eiezione, EF%), e la contrattilità
regionale (Tagging-RMN);
• Ecocardiografia, per quantificare la funzione cardiaca globale e lo
spessore delle pareti ventricolari (Fig. 9);
• Emodinamica: frequenza cardiaca e respiratoria (FC e FR), PaO2,
PaCO2, pH, pressione arteriosa sistolica (SAP), diastolica (DAP) e
media (MAP), prelievi arteriosi.
75
Materiali e metodi
Figura 8: Ecotomografia ad Emissione di Positroni (PET)
1A
1A
1B
2A
2B
Figura 9: Esame ecocardiografico e RMN cuore normale (1A e 1B)
e cuore con scompenso cardiaco (2A e 2B)
76
Materiali e metodi
L’evoluzione progressiva della malattia è stata valutata come di seguito
riportato:
⇒ I° settimana di scompenso cardiaco: RMN - PET - esami
emodinamici, ECG, emogas, prelievi arteriosi (per la
valutazione bio-umorale)
⇒ II° settimana di scompenso cardiaco: esami emodinamici,
ECG, emogas, prelievi arteriosi
⇒ III° settimana di scompenso cardiaco: esami emodinamici,
ECG, emogas, prelievi arteriosi
⇒ IV° settimana di scompenso cardiaco (termine del protollo
sperimentale): RMN – PET - esami emodinamici, ECG,
emogas, prelievi arteriosi.
6.2 Campioni biologici: tessuto cardiaco di maiale
Alla IV° settimana di scompenso cardiaco i minipig venivano sacrificati e
le quattro camere cardiache (atrio, destro e sinistro; ventricolo, destro e
sinistro) venivano prelevate e conservate in RNAlater (RNA stabilization
reagent, Qiagen GmbH, Hilden D).
Questa procedura di raccolta e conservazione del campione è importante
in quanto la stabilizzazione dell’RNA è un requisito fondamentale per
l’analisi dell’espressione genica. L’immediata stabilizzazione dell’RNA
nei materiali biologici è necessaria in quanto, immediatamente dopo la
raccolta del campione, si possono avere cambiamenti dei pattern di
espressione genica dovuti ad una degradazione specifica o aspecifica
dell’RNA sia ad un’induzione della trascrizione.
77
Materiali e metodi
I campioni di tessuto cardiaco sono stati conservati a -80°C per la
determinazione dell’espressione dell’mRNA dei peptidi natriuretici in
esame e del recettore specifico per il CNP (NPR-B).
6.3 Campioni biologici: tessuto cardiaco di topo
Per il sequenziamento dell’mRNA codificante per il recettore dei peptidi
natriuretici di tipo B è stato utilizzato tessuto cardiaco di topi BALB/c
maschi, non affetti da patologie, di sei settimane e 30 g circa di peso.
I topi sono stati anestetizzati con un’iniezione intraperitoneale di 250
mg/kg Avertin (0,1 ml 2,5% Avertin/10 g peso corporeo) e sacrificati; il
cuore prelevato è stato posto in RNAlater e conservato a -80°C
6.4 Campioni plasmatici
I campioni di sangue dei minipig (8-10 ml), prelevati durante le quattro
settimane dello studio, venivano raccolti in tubi di propilene contenenti
EDTA (1mg/ml) e aprotinina (500 kIU), un inibitore delle proteasi. Il
prelievo veniva effettuato in ghiaccio, dopo di che i campioni plasmatici
venivano rapidamente centrifugati a +4°C a 1000 g per 15 minuti e
conservati in aliquote a -20°C.
I prelievi venivano effettuati dopo l’intervento (basale), a 10 minuti
dall’accensione del pacemaker e dopo 1, 2, 3, 4 settimane dall’intervento.
78
Materiali e metodi
6.5 Protocollo Sperimentale
6.5.1 Estrazione dell’RNA totale
L’RNA totale è stato estratto da campioni tessutali cardiaci di topo e di
maiale con il metodo del fenolo-guanidina tiocianto (GTC) (TRIREAGENT: Molecular Research Center, Cincinnati USA), modificato al
fine di ottenere buone rese di estrazione da piccole quantità di tessuto.
Tale procedura permette l’isolamento simultaneo di mRNA e proteine
(RNeasy Fibrous tissue Midi kit, Qiagen Milano) dallo stesso campione.
I tessuti sono stati omogenati in TRI-REAGENT (1 ml per frammenti
tessutali di peso inferiore a 100 mg) utilizzando Mixer Mill MM300
(Qiagen S.p.A. Milano). Questo strumento permette una rapida ed efficace
frammentazione del campione biologico in tempi molto brevi. L’alta
concentrazione di guanidina tiocianto presente nel TRI-REAGENT
provoca una rapida inattivazione dell’attività delle RNAsi endogene e
completa la dissociazione delle componenti cellulari dal RNA; inoltre
l’aggiunta di cloroformio, dopo l’omogenizzazione, e la centrifugazione
del campione a 12000g per 15 minuti a 4°C, permettono la separazione
della fase acquosa incolore, contenente prevalentemente RNA, dalla fase
organica, contenente per la maggior parte proteine e lipidi.
La fase acquosa viene isolata, dalla fase organica, che viene conservata in
aliquote a -20°C, previa aggiunta di etanolo 96%,
fino al momento
dell’uso.
La fase acquosa, dopo aggiunta di quantità appropriate di etanolo, viene
caricata su una micro-colonna di gel di silice (Midi Spin Column RNeasy,
Qiagen, Milano) in grado di legare l’RNA presente nel campione.
79
Materiali e metodi
Dopo un primo lavaggio con una soluzione di guanidina tiocianto ed
etanolo (tampone RW1) e successiva centrifugazione a 3000 g per 5
minuti a 25°C, viene aggiunta una soluzione di DNasi (DNaseStock
solution RNasi free) che permette di eliminare le eventuali interferenze
dovute al DNA e quindi per purificare il campione biologico.
Seguono ulteriori tre lavaggi e successive centrifugazioni a 3000 g e
+25°C, l’RNA viene poi eluito con piccole quantità (100-150 μl) di acqua
RNase free e dopo la prima eluizione, raccolto e caricato nuovamente sulla
micro-colonna, al fine di concentrare al massimo il campione, il quale
viene conservato in aliquote a -80° fino al momento dell’utilizzo.
6.5.2 Determinazione dell’RNA totale
La concentrazione di RNA negli estratti tessutali cardiaci è stata
determinata utilizzando il metodo spettrofotometrico. Il campione viene
diluito 1:100 con acqua RNase free, caricato in cuvette di quarzo, e
l'assorbanza viene letta alle lunghezze d'onda di 260 nm e di 280 nm,
mediante l’utilizzo di uno spettrofotometro (Beckman DU, 640).
Ogni lettura viene normalizzata rispetto al valore di assorbanza relativo
alla soluzione di acqua priva di RNAsi che è stata usata come bianco.
Una volta misurata l’assorbanza a 260 nm, si valuta la concentrazione di
RNA totale attraverso la formula, ricavata dalla legge di Lambert-Beer
(Aλ= ελ · l · c), dove ελ rappresenta il coefficiente di estinzione molare, l
indica il cammino ottico e c la concentrazione del campione:
[RNA]campione = (A260nm) campione · D · (40 μg/ml)
Nella formula sopra riportata D è il fattore di diluizione e 40 μg/μL è il
valore di concentrazione che è associato ad una soluzione standard di
80
Materiali e metodi
RNA che presenti un'assorbanza pari a 1 quando λ= 260nm (ovvero
1/(ΣRNA(260nm)×l)= 40 μg/mL).
Il rapporto tra l'assorbanza del campione a 260 nm (A260nm) e quella del
campione a 280 nm (A280nm) fornisce un'indicazione della purezza dello
RNA contenuto nel campione estratto. Si considera pura una soluzione di
RNA il cui rapporto (A260nm / A280nm) sia compreso nell'intervallo 1,8-2,1.
L’integrità dell’RNA totale estratto è stata valutata attraverso una corsa
elettroforetica su gel d’agarosio, in condizioni denaturanti e in presenza di
un agente intercalante, quale il bromuro d’etidio.
I campioni vengono prelevati da -80°C e mantenuti a +55°C, per eliminare
l’eventuale presenza di RNasi endogene; in seguito viene aggiunta, in
rapporto 1:3, una soluzione contenente formaldeide e blu di bromofenolo
(The NorthernMax Formaldehyde Load Dye, Ambion Inc. Austin, USA),
(1 μl di campione + 3 μl di Load Dye).
I campioni così preparati vengono incubati per 15 minuti a +65°C, per
permettere la denaturazione delle strutture secondarie dell’RNA, poi
centrifugati e caricati sul gel.
L’elettroforesi è effettuata a 50 Volt per 50 minuti e il gel analizzato al
transilluminatore (260 nm, UV Transilluminator
2000,
Bio-Rad
Laboratories, CA) per verificare la comparsa di due bande nette in
corrispondenza delle subunità 18 e 28 Svedberg (S) dell’RNA
ribosomiale.
Per confermare la concentrazione del campione, valutata dalla lettura allo
spettrofotometro, le bande, ottenute dalla corsa elettroforetica, sono state
analizzate con un programma specifico per analisi densitometriche di gel
(Quantity One Software, Bio-Rad) in grado di determinare l’intensità della
81
Materiali e metodi
banda stessa. Il rapporto tra l’intensità ottenuta e quella di un campione
standard di RNA totale di topo (Mouse Total RNA, Ambion Inc. Austin,
USA; 1 μg/μl) permette di determinare la concentrazione di RNA nel
campione incognito.
6.5.3 Estrazione tessutale delle proteine
Le proteine vengono isolate dalla fase organica, di cui al paragrafo 6.5.1.
L’aggiunta di etanolo e la successiva centrifugazione permettono di
eliminare la componente lipidica; la successiva aggiunta di acetone al
surnatante e la centrifugazione a 12000 g per 5 minuti a +4°C portano alla
formazione di un pellet proteico. Il pellet così ottenuto viene lavato e
centrifugato, per tre volte successive, con una soluzione di lavaggio
(guanidina HCl 0,3 M, glicerolo al 25%, etanolo). Viene effettuato un
quarto e ultimo lavaggio con una soluzione contenente glicerolo al 25% in
etanolo.
Il pellet proteico derivante è risospeso con un tampone Tris (idrossimetilamminometano) HCl 4 mM (pH 7,4), contenente NaCl (154 mM),
fenilmetilsulfonil-fluoruro (PMSF 0,1 mM), sodiododecilsolfato (SDS
2%) e conservato a -20°C fino al momento dell’utilizzo.
La concentrazione totale dell’estratto tissutale viene misurata con una
modificazione del metodo di Lowry (Lowry OH. et al., 1951), usando
albumina bovina come proteina standard. Viene così costruita una curva di
taratura in base alla quale determinare, per interpolazione, il valore della
concentrazione proteica totale del campione estratto.
82
Materiali e metodi
6.6 Analisi di RT-PCR (Reverse Transcription-Polymerase Chain
Reaction) semiquantitativa
Il metodo della RT-PCR è basato sulla capacità dell’enzima trascrittasi
inversa di generare un frammento di cDNA a partire da un templato di
mRNA. Tale enzima è una DNA polimerasi DNA/RNA dipendente, che,
in presenza di una coda di mRNA-poliA a singolo filamento e di un
innesco di oligonucleotidi poli-dT, è in grado di retrotrascrivere tutti gli
mRNA nei relativi filamenti di DNA complementari a doppia elica ed
eteroduplici (cDNA).
Dopo denaturazione con calore, il frammento di cDNA rimanente è usato
come templato dalla polimerasi, nei successivi passaggi di amplificazione.
Sia il processo di trascrizione inversa, che la reazione di PCR sono state
effettuate utilizzando un kit dedicato (Ready to Go RT-PCR Beads,
Amersham Biosciences; Freiburg Germany) e, come strumento di
amplificazione, un termociclatore (Thermal-cycler, Perkin Elmer,
Emerville, CA, USA).
L’RNA totale è inversamente trascritto dall’enzima trascriptasi inversa
Moloney Murine Leukaemia Virus (M-MuLV) in presenza di 0,5 μg di
oligo (dT)12-18 per reazione.
Come controllo positivo viene utilizzato l’mRNA della globina di
coniglio, parallelamente come controllo negativo viene preparata una
provetta contenente tutti i reagenti escluso il campione, al fine di valutare
le possibili contaminazioni di DNA.
I campioni vengono incubati a +42°C per 20 minuti, per la
retrotrascrizione, e successivamente a +95°C per 5 minuti, per inattivare
l’enzima M-MuLV e denaturare completamente il templato di mRNA.
83
Materiali e metodi
La reazione di PCR è catalizzata dalla Taq DNA polimerasi, un enzima
capace di resistere a temperature molto elevate (+95°C) ed in grado di
funzionare a partire da temperature di +65°C.
Il cDNA derivante, viene amplificato in presenza di 1μM di ogni primer
specifico, in un volume finale di 50 μl.
Le RT-PCR per mRNA codificante per CNP e BNP di maiale, e NPR-B di
topo, sono state messe a punto utilizzando i seguenti primers (SygmaGenosys):
• CNP senso: 5’-GCTGCTCACGCTCCTCTC-3’
• CNP anti-senso: 5’-GTCTTGTCGCCCTTCTTCTG-3
• BNP senso: 5’-GTGCTCCTGCTCCTGTTCTT-3’
• BNP anti-senso: 5’-TCCCAGGCTTCTGTGAGG-3’
• NPR-B senso: 5’-TGTACCATGACCCCGACCTT-3’
• NPR- B anti-senso: 5’-CCCGTTGGCTCTGATGAAGT-3’
I primer per tutti gli mRNA sono stati costruiti su Primer 3. La sequenza
dei primer utilizzati nello studio di RT-PCR, è stata valutata con il
programma BLAST e verificata sul database della GenBank.
Come gene di riferimento è stata utilizzata la gliceraldeide-3fosfatodeidrogenasi (GAPDH), in quanto dalla letteratura è noto che è
espresso in tutte le cellule e non subisce modificazioni nella specifica
situazione studiata (GAPDH senso: 5’-ACCACAGTCCATGCCATCAC3’;
GAPDH
anti-senso:
5’-TCCACCACCCTGTTGCTGTA-3’)
(Amersham Pharmacia Biotech) (Barber RD. et al., 2005; Hsiao LL. et al.,
2001).
84
Materiali e metodi
Per questo i dati relativi ai livelli di espressione dei diversi mRNA di
interesse vengono normalizzati rispetto a quelli dei trascritti del gene
codificante per il GAPDH.
Le condizioni per la reazione di PCR sono state ottimizzate per ciascun
peptide analizzato.
I prodotti della PCR sono stati stati trattati
in rapporto 1:5 con la
soluzione “Loading Buffer” (contenente glicerolo e blu di bromofenolo) e
caricati su un gel di agarosio in parallelo ad un DNA Ladder 100pb
(Amersham Biosciences). I campioni così preparati e il DNA Ladder sono
stati separati per elettroforesi (Sub-Cell GT Electrophoresis Cell, Bio-Rad
Laboratories, CA) su gel d’agarosio all’1.5% in tampone TBE (Tris/Acido
Borico/EDTA), colorati con bromuro di etidio e visualizzati per mezzo
della luce ultravioletta a 260 nm (UV Transilluminator 2000, Bio-Rad
Laboratories, CA). Le immagini ottenute sono state fotografate con una
sistema fotografico digitale (Panasonic LC 40) e elaborate con un
programma dedicato (Quantity One, Bio-Rad).
La banda relativa all’mRNA del CNP è risultata a 574 pb, quella
dell’mRNA codificante per il BNP a 197 pb, quella per l’NPR-B a 398 pb
e quella dell’mRNA per il GAPDH a 451 pb.
85
Materiali e metodi
6.7 Saggio radioimmunologico per la misura del CNP
6.7.1 Estrazione dei campioni
L'estrazione del CNP dal plasma e dagli estratti tessutali è necessaria per
eliminare le interferenze aspecifiche e per concentrare il campione da
determinare.
Il procedimento di estrazione è il seguente: 2mL di plasma o di estratto
tissutale vengono acidificati con 2mL di acido trifluoracetico (TFA) 1% e
in seguito centrifugati a 1500 g x 10 min a +4°C.
Il surnatante viene caricato su una piccola colonna Sep-Pak C18 attivata
con 1mL di soluzione contenente acetonitrile al 60% in TFA 1% e, per 3
volte, con 3mL di una soluzione di TFA 1%. La colonna viene poi lavata
con 3mL di TFA 1% (per 3 volte) e il CNP eluito con 3mL di soluzione
contenente acetonitrile al 60% in TFA 1%.
Il residuo secco (ottenuto con un concentratore centrifugo) viene risospeso
con 500µL del tampone usato nel saggio immunometrico e 100µL di
questa soluzione sono utilizzati per il saggio immunometrico.
6.7.2 Saggio Radioimmunologico
Il CNP è stato misurato utilizzando un sistema radioimmunologico
commerciale
(Kit
RIA:
C-type
peptide-22
human
Phoenix
Pharmaceuticals, Belmount, CA, USA).
Rispetto alla procedura consigliata dalla ditta produttrice, sono state
modificate le concentrazioni della curva standard (1,25-2,5-5-10-20-4080-160 pg/tubo), dalla quale, per interpolazione, può essere determinata la
concentrazione del CNP nel campione sconosciuto.
86
Materiali e metodi
Ogni saggio è stato effettuato in duplicato e l'intero esperimento è stato
condotto in ghiaccio.
Utilizzando la preparazione standard di CNP, fornita dal kit, sono stati
preparati due campioni di riferimento da misurare in ogni prova per il
controllo di qualità interno.
6.8 Analisi statistica dei dati
L’analisi statistica dei risultati è stata effettuata tramite il programma StatView 5.0.1 per Machintosh computers (1992-98, SAS Istitute Inc., SAS
campus Drive, Cary, NC, USA).
I valori relativi alle concentrazioni di CNP, ottenute dal saggio
radioimmunologico, sono stati calcolati con un programma dedicato su
computer che usa, per l’interpolazione della curva dose-risposta, una
funzione logistica a 4 parametri (Pilo A. et al., 1982). Poiché i livelli
plasmatici del CNP non sono distribuiti normalmente, per l’analisi
statistica, quando necessario, viene utilizzata la trasformazione logaritmica
naturale dei dati.
I risultati sono espressi come media ± SEM.
Per i confronti multipli è stato utilizzato il test di Fisher, dopo analisi della
varianza (ANOVA).
87
Risultati
RISULTATI
7
Capitolo
7.1 Risultati metodologici
7.1.1 Quantizzazione RNA
Il rapporto tra l’assorbanza alla lunghezza d’onda di 260 nm e 280 nm
(A260nm/A280nm) dei campioni di RNA ottenuti dal tessuto cardiaco è
risultato compreso nell'intervallo 1.8-2.1 (Fig. 10a). Tutti i campioni sono
stati sottoposti a elettroforesi su gel d’agarosio, per provarne l’integrità
(Fig. 10b).
Infine, per confermare il valore della concentrazione di campione, ottenuto
dalla lettura allo spettrofotometro, le bande, ottenute dalla corsa
elettroforetica, sono state analizzate con il programma specifico Quantity
One Software, utilizzando come controllo l’RNA totale di topo a
concentrazione nota (1 μg/μl). I due differenti metodi hanno dato risultati
sovrapponibili in tutti i casi analizzati.
a
b
Figura 10: Integrità dell'RNA: (a) quantizzazione; (b) corsa elettroforetica su gel
d'agarosio
88
Risultati
7.1.2 Reazioni di RT-PCR
Per valutare la sensibilità e la linearità dell’amplificazione, sono state
effettuate reazioni di PCR, variando il numero di cicli, la temperatura di
annealing e la concentrazione di RNA e primer.
Negli esperimenti di messa a punto del metodo sono state utilizzate le
seguenti condizioni:
• concentrazioni di RNA: 0,05-0,1-0,44-0,88 μg
• temperature di annealing per CNP: 56°C-61°C-63°C-65°C
• temperature di annealing per BNP: 56°C; 59°C; 61°C; 63°C; 65°C
• concentrazioni di primer per CNP e BNP: 1-0,1-0,01 μM
• numero di cicli per CNP e BNP: 25-30-35-40-45
Nelle figure (Fig. 11 e 12) sono riportati come esempio i gel e le relative
curve dose-risposta in funzione, rispettivamente, della temperatura di
annealing e della concentrazione di RNA, sia per il CNP che per il BNP.
Tali curve sono state ottenute analizzando le bande, risultanti dalla corsa
elettroforetica, con il programma specifico Quantity One Software.
Le condizioni ottimali di reazione scelte per la valutazione dei campioni
biologici corrispondono ad una temperatura di annealing per il CNP di
+63°C e un numero di cicli di 40; mentre per quanto riguarda il BNP le
condizioni scelte sono state: temperatura di annealing +59°C e numero di
cicli pari a 35. Sia per il BNP che per il CNP la concentrazione di primer
scelta è stata 1 μM.
Le condizioni utilizzate per il GAPDH sono state: 56°C per 30 cicli, come
già stabilito da uno studio precedente (Del Ry S. et al., 2004).
La fase di estensione, impiegata per tutti gli effettori, è stata di 72°C per
10 minuti.
89
Risultati
Figura 11: Corse elettroforetiche su gel d'agarosio e relative curve
dose-risposta in funzione di temperatura di annealing e
concentrazione di RNA per il CNP (574 pb)
Figura 12: Corse elettroforetiche su gel d'agarosio e relative curve
dose-risposta in funzione di temperatura di annealing e
concentrazione di RNA per il BNP (197 pb)
90
Risultati
7.2 Sequenziamento dell’mRNA codificante per l’NPR-B di maiale
7.2.1 RT-PCR su tessuto cardiaco di topo
Le condizioni per la reazione della RT-PCR sono state ottimizzate
utilizzando i primers per l’NPR-B di topo. Per valutare la sensibilità e la
linearità dell’amplificazione, la PCR è stata condotta in un range diverso
di numero di cicli (25-30-35-40), temperature di annealing (59°C-61°C63°C-65°C), concentrazione di RNA e primers (1-0,1-0,01 μM).
In figura (Fig. 13 a) è riportata la curva di calibrazione per l’NPR-B nel
tessuto cardiaco di topo, ottenuta usando concentrazioni crescenti di RNA
(0,07-0,146-0,293-0,56-1,05μg). La densità ottica delle bande è riportata
in figura 13 b).
Le condizioni di reazione ottimali scelte per l’NPR-B murino sono state:
0,1/0,2 μg di RNA, 60 secondi a 63°C (tempo e temperatura di annealing),
35 cicli.
7.2.2 RT-PCR su tessuto cardiaco di maiale
Per sequenziare l’mRNA codificante per il recettore biologico di tipo B
nel maiale, finora mancante nella GenBank, sono state utilizzate le
condizioni e i primer scelti per la messa a punto delle RT-PCR su tessuto
cardiaco di topo.
Lavorando in queste condizioni si sono ottenute bande in corrispondenza
di circa 400 pb (Fig. 14a) e, come si può osservare dalla curva doserisposta, costruita in funzione della concentrazione di RNA (Fig. 14b),
hanno mostrato un andamento dose-crescente. Questa osservazione ci ha
permesso di ipotizzare che questa banda potesse essere attribuibile
all’espressione dell’NPR-B nel maiale.
91
Risultati
a
b
Figura 13: Corsa elettroforetica su gel d'agarosio (a) e relativa curva dose-risposta (b) in funzione
della concentrazione di RNA per l'NPR-B, utilizzando tessuto cardiaco di topo
a
b
Figura 14: Corsa elettroforetica su gel d'agarosio (a) e relativa curva dose-risposta (b) in funzione
della concentrazione di RNA per l'NPR-B, utilizzando tessuto cardiaco di maiale
92
Risultati
Per confermare questa ipotesi è stato necessario sequenziare le bande
ottenute dalla corsa elettroforetica. Sono state effettuate ulteriori reazioni
di RT-PCR, utilizzando campioni diversi di tessuto cardiaco porcino
(n=20) e, al fine di aumentare il grado di purezza del DNA abbiamo
proceduto all’estrazione delle bande ottenute su gel con un kit dedicato
(Gel Extraction Kit Protocol, QIAquick Spin Handbook, Qiagen, Milano).
Le bande sono state tagliate accuratamente dal gel e caricate in tubi a cui è
stato aggiunto una soluzione di guanidina tiocianato (QG Buffer, Qiagen,
Milano) e un indicatore di pH. I campioni sono stati incubati per 10 minuti
a +50° per permettere il completo dissolvimento delle bande e caricati su
micro-colonne di gel di silice. Dopo varie centrifugazioni e lavaggi il
campione è stato eluito con acqua RNasi free e al termine dell’estrazione il
DNA purificato è stato fatto correre ulteriormente su gel d’agarosio (Fig.
15), in presenza di un marker di DNA a concentrazione nota (100 pb step
Ladder, Promega, Madison, USA) in modo da poterne determinare l’esatta
concentrazione.
L’analisi con il programma Quantity-One, ci ha permesso di esprimere
l’intensità delle bande ottenute rispetto a quella del marker a
concentrazione nota; infatti la banda corrispondente a 400 pb del marker,
aveva una concentrazione nota di 38ng/5μl. Da questa analisi è emerso che
ciascuna banda di DNA purificato, risultava di circa 3-8ng/5μl. Questa
valutazione è stata essenziale per inviare concentrazioni note di campione
al centro di sequenziamento.
93
Risultati
Figura 15: Corsa elettroforetica su gel d'agarosio di DNA purificato, in presenza di
marker di DNA a concentrazione nota
7.2.3 Sequenziamento e analisi dei dati
Il sequenziamento dell’mRNA codificante per l’NPR-B, è stato effettuato
dal centro CRIBI dell’Università di Padova (.BMR Bio Molecular
Research, CRIBI, Padova), a cui sono stati inviati i campioni essicati di
DNA ottenuto da tessuto cardiaco di maiale.
Per il sequenziamento sono state richieste quantità precise di campione (16
ng) e primer (6,4 pmol).
Il sequenziamento è stato effettuato con uno strumento dedicato (Applyed
Biosystem, ABI PRISM 3730 XL DNA Sequencer) che utilizza il metodo
di Sanger (Sanger F. et al., 1977).
Come si può osservare in figura 16, per ogni campione ci sono pervenuti i
risultati come: 1. cromatogramma; 2. sequenza del campione incognito in
forma FASTA; 3. numero totale di ciascuna base presente nel campione
inviato.
94
Risultati
Figura 16:
Risultati del sequenziamento: 1.
cromatogramma, 2. sequenza in
forma FASTA, 3. numero
complessivo di ciascuna base
presente nella sequenza.
95
Risultati
Dall’elaborazione delle sequenze ricevute per i 20 campioni inviati,
mediante programmi specifici quali BLAST, FASTA e per verifica
GenBank, abbiamo ottenuto la sequenza parziale di mRNA per il recettore
di tipo B di maiale che è risultata di 396 pb (Fig. 17).
Dall’analisi di questa sequenza con BLAST abbiamo osservato
un’omologia con 37 sequenze codificanti per il recettore di tipo B dei
peptidi natriuretici, ed in particolare un’omologia del 93% con l’NPR-B
del Mus Musculus e del 95% con l’ NPR-B di Homo Sapiens.
Figura 17: Sequenza parziale dell'mRNA codificante per l'NPR-B di maiale
7.2.4 Sottomissione alla GenBank
La sequenza parziale di mRNA codificante per l’NPR-B è stata inserita
nella GenBank (numero identificativo del gene: Sus Scrofa Natriuretic
peptide receptor-2 mRNA, 1-395 pb; accession number DQ487044- Fig.
18); la GenBank ha inoltre automaticamente determinato la sequenza
proteica, a cui è stato assegnato il numero identificativo ABF 19801.1.
Poiché i domini delle proteine sono unità distinte tridimensionali,
associate a particolari e specifiche funzioni molecolari, come binding e
catalisi, l’identificazione di tali caratteristiche, può dare indicazioni sulla
funzione molecolare e cellulare della proteina a cui appartengono.
96
Risultati
La sequenza proteica ottenuta nel nostro lavoro (ABF 19801.1) appartiene
alla famiglia pFAM (Bateman A. et al., 2004) degli ANF-receptor (Pfam
01094.12- ANF_receptor), gruppo di recettori specifici per il binding
proteico.
Figura 18: GenBank: DQ487044 Sus Scrofa natriuretic peptide receptor 2 mRNA, partial cds
97
Risultati
7.3 Risultati relativi al modello sperimentale
I livelli plasmatici di CNP sono risultati significativamente aumentati nei
minipig con insufficienza cardiaca cronica indotta da stimolo con
pacemaker ad alta frequenza, rispetto agli animali di controllo. Questo
incremento è osservabile a partire dalla prima settimana di trattamento
(36,9 ± 10,4 vs. 16,7 ± 1,1 pg/ml, media ± sem; p=0,013), come riportato
in figura 19.
Per quanto riguarda la presenza di CNP nel tessuto cardiaco, negli animali
di controllo sono stati trovati livelli misurabili di CNP in tutte le camere
cardiache. In figura 20 sono riportati i livelli tessutali medi relativi a atrio
e ventricolo, destro e sinistro rispettivamente (atrio destro: 13,7 ± 1,9
pg/mg; atrio sinistro: 8,7 ± 3,8 pg/mg; ventricolo destro: 1,07 ± 0,33
pg/mg; ventricolo sinistro: 0,93 ± 0,17 pg/mg). La concentrazione
osservata negli atrii è risultata circa 10 volte più elevata rispetto a quella
osservata nei ventricoli.
Dopo 4 settimane di stimolazione con pace-maker ad alta frequenza, i
livelli tessutali cardiaci di CNP nel ventricolo sinistro sono risultati
significativamente aumentati rispetto ai quelli del ventricolo sinistro degli
animali di controllo (15,8 ± 9,9 pg/mg vs. 0,9 ± 0,17 pg/mg; p= 0,01) (Fig.
21).
Questi dati sono stati confermati dagli studi di espressione dell’mRNA
codificante per il CNP. In tutte le camere cardiache dei minipig
scompensati è stata osservato un aumento dell’espressione dell’mRNA
codificante per il peptide natriuretico di tipo C rispetto agli animali di
controllo. L’aumento di tale espressione è risultato particolarmente
evidente nel ventricolo sinistro (Fig. 22).
98
Risultati
In tutti gli animali studiati è stato osservato anche un aumento di
espressione a livello di mRNA per il BNP, come atteso in presenza di
scompenso cardiaco.
Per quanto riguarda il recettore NPR-B è risultato espresso in tutte le
camere cardiache sia nei minipig di controllo che in quelli con SC come si
può osservare in figura 23, dove sono riportati in maniera riassuntiva i
livelli di espressione, ottenuti nelle quattro camere cardiache, dell’mRNA
codificante per CNP, NPR-B, BNP e GAPDH, rispettivamente, per un
singolo caso sperimentale.
Questi dati preliminari sembrano indicare una diminuita espressione dei
recettori NPR-B in parallelo con l’aumento dell’espressione di CNP nei
minipgs con SC.
La possibilità di una down-regulation di questo recettore, che deve essere
valutata su un numero più ampio di esperimenti, è in accordo con quanto
riportato precedentemente in letteratura, relativamente alle variazioni dei
recettori biologici dei peptidi natriuretici nello scompenso cardiaco
(Tsutamoto T. et al., 1993).
99
Risultati
Figura 19: Livelli plasmatici di CNP in minipig di controllo e
in minipig con scompenso cardiaco (SC) indotto da una settimana
100
Risultati
Figura 20: Livelli tessutali di CNP in atrio (in alto) e in ventricolo (in basso)
destro e sinistro rispettivamente
101
Risultati
Figura 21: Livelli tessutali di CNP in ventricolo sinistro di minipig di controllo e
di minipig alla IV° settimana di scompenso cadiaco
Figura 22: Espressione dell'mRNA codificante per CNP, BNP e GAPDH
nel ventricolo sinistro di minipig normali e minipig con SC
102
Risultati
Figura 23: Livelli di espressionedell’mRNA codificante per CNP, NPR-B, BNP e GAPDH rispettivamente,
ottenuti nelle quattro camere cardiache
103
Conclusioni
8
CONCLUSIONI
Capitolo
Lo studio svolto in questa tesi deve essere considerato innanzitutto come
uno studio propedeutico atto a sviluppare metodologie applicabili a
protocolli specifici, rivolti a caratterizzare la biosintesi e il meccanismo di
azione del CNP e le sue azioni autocrine/paracrine sia in condizioni
normali che in condizioni patologiche caratterizzate da disfunzione
endoteliale e rimodellamento.
Il modello animale utilizzato ci ha permesso di valutare il ruolo del CNP
nella fisiopatologia dello scompenso cardiaco in maniera più completa di
quanto sia possibile effettuare sull’uomo proprio grazie alla quantità di
materiale biologico disponibile e alla versatilità del modello.
I dati sui livelli plasmatici hanno confermato quanto gia valutato
nell’uomo e sono risultati essere in accordo con i dati della letteratura
sull’aumento dei livelli del CNP e del suo precursore NT-proCNP in
funzione della severità della malattia (Prickett TCR. et al., 2001; Wright
SP. et al., 2004; Del Ry S. et al., 2005).
In questo lavoro di tesi, per la prima volta, è stata misurata la
concentrazione del peptide negli estratti miocardici che è risultato presente
in tutte le camere cardiache dei minipig normali. Il contenuto tessutale di
CNP degli atrii è risultato circa 10 volte più elevato di quello dei
ventricoli. Inoltre, dopo 4 settimane di stress da pacing, i livelli tessutali
del ventricolo sinistro sono risultati significativamente aumentati nei
minipig scompensati rispetto ai minipig normali in parallelo con un
104
Conclusioni
aumento dei livelli tessutali del BNP che risulta particolarmente elevato
nel ventricolo dei soggetti scompensati (Luchner A. et al., 1998).
La dimostrazione che il CNP è presente nel tessuto cardiaco è un dato
importante per i possibili effetti sinergici con gli altri peptidi natriuretici,
anche a livello cardiaco.
Infatti è noto che ANP e BNP possono stimolare la produzione di CNP e
avere un’azione sinergica sul tessuto vascolare (Nazario B. et al., 1995).
Sono state inoltre dimostrate interazioni complesse tra peptidi natriuretici
e altri mediatori vasorilassanti di derivazione endoteliale (Ahluwalia A. et
al., 2005); questo fa ipotizzare che il CNP, in aggiunta alle sue azioni antiadesive e anti-aggreganti, possa compensare la riduzione di tali mediatori
in
patologie
cardiovascolari,
al
fine
di
ristabilire
le
capacità
vasodilatatorie.
Avremo quindi un bilanciamento tra il sistema dei peptidi natriuretici e
altri effettori del sistema neuroormonale con azione contrapposta quali il
sistema angiotensina/aldosterone e l’endotelina (ET)-1. Quest’ultima
stimola la produzione del BNP nei cardiomiociti ventricolari e promuove
l’aumento delle cellule endoteliali che secernono CNP. Questi risultati
sostengono l’ipotesi che l’ET-1 endogena delle cellule vascolari
endoteliali umane agisca in maniera autocrina/paracrina al fine di
modulare il rilascio basale di CNP e adrenomedullina (AM) (Evans JJ. et
al., 2002).
Un risultato di particolare importanza ottenuto nel corso di questo lavoro
di tesi, è stato il sequenziamento del recettore specifico per l’NPR-B.
Il minipig è infatti molto usato come modello animale per le sue analogie
con la fisiopatologia dell’uomo, quindi l’identificazione della sequenza
105
Conclusioni
dell’NPR-B, fornisce un nuovo strumento per meglio investigare il
meccanismo di azione del CNP in condizioni fisiologiche e patologiche.
ABF 19.801.1, la sequenza proteica associata all’NPR-B di maiale,
appartiene alla famiglia delle Pfam, gruppo di recettori specifici per il
binding proteico, come precedentemente descritto (Bateman A. et al.,
2004).
Non dobbiamo inoltre dimenticare che il recettore di tipo B si lega,
sebbene con affinità minore rispetto al CNP, anche al BNP; quindi, data
l’importanza, ormai da tutti riconosciuta, per il BNP nello SC, la
possibilità di valutare le variazioni di questo recettore può contribuire a
meglio definire il ruolo dei peptidi natriuretici in questa patologia.
Per quanto riguarda la funzione dell’NPR-B a livello del sistema
cardiovascolare, uno studio recente effettuato sia in vivo che in vitro, che
prevedeva l’utilizzo di ratti transgenici con sovraespressione del recettore
B mutante, ha dimostrato, per la prima volta, che tale recettore è coinvolto
nella regolazione dell’ipertrofia cardiaca (Langenikel TH. et al., 2006).
Questi risultati sono in accordo con i recenti studi in cui è stata dimostrata
la capacità del CNP di inibire l’ipertrofia dei cardiomiociti (Tokudome T.
et al., 2004). E’ stato inoltre osservato che il CNP è sintetizzato e secreto
dai fibroblasti cardiaci dove il recettore B sembra essere l’isoforma
predominante (Horio T. et al., 2003).
La co-localizzazione del CNP e del suo recettore suggerisce un ruolo
importante del CNP nella fisiologia cardiovascolare, dove può avere un
importante azione locale.
Dalla letteratura è inoltre emerso che gli effetti inibitori del CNP sulla
proliferazione e produzione di collagene da parte dei fibroblasti cardiaci
106
Conclusioni
sono probabilmente mediati dal recettore NPR-B (Horio T. et al., 2003;
Tokudome T. et al., 2004).
Queste osservazioni indicano che il CNP può funzionare come regolatore
autocrino negativo contro l’eccessiva fibrosi cardiaca caratteristica di
alcuni stati patologici.
La sequenza parziale dell’mRNA codificante per l’NPR-B di maiale e la
sequenza proteica, ottenute nel lavoro di tesi, sono risultate simili, sebbene
non identiche a quelle di numerose altre specie, suggerendo che l’NPR-B
possa svolgere un ruolo simile nelle diverse specie.
Sebbene ulteriori studi siano necessari per chiarire il ruolo patofisiologico
del CNP endogeno nel cuore, i nostri dati, presi nel loro insieme, fanno
ipotizzare che il peptide possa svolgere un ruolo compensatorio sul
rimodellamento ventricolare. È infatti noto che il CNP esercita importanti
azioni cardiovascolari indirette come la riduzione della pressione cardiaca
dovuta a vasodilatazione e diminuzione del ritorno venoso.
La valutazione delle azioni del CNP nel modello sperimentale proposto
potrebbe
quindi
contribuire
a
meglio
comprendere
situazioni
fisiopatologiche diverse, caratterizzate da disfunzione endoteliale e da
processi di rimodellamento.
Infine, il sequenziamento parziale del recettore B, effettuato in questo
lavoro di tesi, ha contribuito, seppur in piccolissima parte, al
sequenziamento del genoma di maiale che è ormai in via di definizione.
107
Elenco degli acronimi
ELENCO DEGLI ACRONIMI
AC: adenilato ciclasi (Adenylate Cyclase)
ACE: Angiotensin-Converting Enzyme
ADH/AVP: ormone anti-diuretico o vasopressina (Antidiuretic Hormone,
Vasopressin)
AM: adrenomedullina (Adrenomedullin)
ANF_receptor: Receptor Family Ligand Binding Region
ANOVA: analisi della varianza (Analysis of Variance)
ANP: peptide natriuretico atriale (Atrial Natriuretic Peptide)
BNP: peptide natriuretico cerebrale, o di tipo B (Brain Natriuretic Peptide)
cAMP: adenosinmonofosfato ciclico (Cyclic adenosine monophosphate)
cDNA: DNA complementare
cGMP: guanosinmonofosfato ciclico (Cyclic guanosine monophosphate)
CNP: peptide natriuretico di tipo C (C-type Natriuretic Peptide)
COX2: cicloossigenasi-2 (Cyclooxygenase-2)
DAP: pressione arteriosa diastolica (Diastolic Arterial Pressure)
DNA: acido deossiribonucleico (DeoxyriboNucleic Acid)
DNP: dendroaspis natriuretic peptide
E.C.24.11: encefalinasi o atriopeptidasi (Endoprotease/Endopeptidase)
EC: ecocardiografia (Echocardiography)
ECE: Endothelin-Converting Enzyme
ECG: elettrocardiogramma (Echocardiograms)
EDHF: fattore iperpolarizzante di origine endoteliale (endothelium derived
hyperpolarizing factor)
108
Elenco degli acronimi
EF%: frazione di eiezione (Ejection Fraction)
ET-1: endotelina-1 (Endothelin-1),
FC: frequenza cardiaca (Cardiac Frequency)
18
FDG: 18fluor-desossiglucosio ([18]F-flourodeoxyglucose)
FGF: fattore di crescita fibroblastico (fibroblast growth factor)
FR: frequenza respiratoria (RespiratoryFrequency)
GAPDH: gliceraldeide-3-fosfatodeidrogenasi (Glyceraldehyde-3phosphate dehydrogenase)
GC: guanilato ciclasi (Guanylate Cyclase)
HDL: lipoproteine ad alta densità (High-Density Lipoprotein)
125
I: 125-Iodio (125-Iodine)
IL-1: interleuchina-1 (Interleukin-1)
iNOS: ossido-nitrico sintasi inducibile (Inducible Nitric Oxide Synthase)
MAP: pressione arteriosa media (Medium Arterial Pressure)
MCR: tasso di clearance metabolica (Metabolic Clearance Rate)
mRNA: RNA messaggero (Messenger RNA)
13
NH3: ammoniaca radiomarcata (13N-labeled Ammonia)
NIH: National Institute of Healt
NO: monossido di azoto (Nitric Oxide)
NPR-A: recettore dei peptide natriuretici di tipo A (natriuretic peptide
receptor/guanylate cyclase A)
NPR-B: recettore dei peptide natriuretici di tipo B (natriuretic peptide
receptor/ guanylate cyclase B)
NPR-B: recettore dei peptide natriuretici di tipo C (clearance receptor)
NYHA: New York Heart Association
PaCO2: pressione parziale di anidride carbonica (Pressure of Carbon
109
Elenco degli acronimi
Dioxide)
PaO2: pressione parziale di ossigeno (Pressure Oxygen)
PET: tomografia ad emissione di positroni (Positron Emission
Tomography)
Pfam: Protein Families Database
PGI2: prostaciclina (Prostacyclin)
PMSF: fenil-metilsolfonil-fluoruro (Phenyl-methylsulphonyl-fluoride)
RIA: dosaggio radioimmunologico (RadioImmuno Assay)
RMN: Risonanza Magnetica Nucleare (Nuclear Magnetic Resonance)
RNA: acido ribonucleico (Ribonucleic Acid)
rRNA: RNA ribosomiale (Ribosomal RNA)
RT-PCR: Reverse Translation-Polymerase Chain Reaction
SAP: pressione arteriosa sistolica (Systolic Arterial Pressure)
SC: scompenso cardiaco (Heart Failure)
sIL-6R: recettori solubili per l’Interleuchina-6 (soluble Interleukin 6
Receptor)
sTNF-R: recettori solubili per il fattore di necrosi tumorale (soluble Tumor
Necrosis Factor- Receptor)
TBE: Tris/Acido borico/EDTA
TFA: acido trifluoracetico (Trifluoroacetic acid)
TGF: transforming growth factor
Th: linfocita T helper (T helper lynphocyte)
TNF: fattore di necrosi tumorale (tumor necrosis factor)
Tris: idrossimetil amminometano
110
Bibliografia
BIBLIOGRAFIA
•
ABASSI Z., KARRAM T., ELLAHAM S., WINAVER J.,
HOFFMAN A. (2004). “Implications of the natriuretic peptide
system in the pathogenesis of heart failure: diagnostic and
therapeutic importance.” Pharm Ther 102: 223-41.
•
ABASSI ZA., YAHIA A., ZEID S., KARRAM T., GOLOMB
E., WINAVER J., HOFFMAN A. (2005). “Cardiac and renal
effects of omapatrilat, a vasopeptidase inhibitor, in rats with
experimental congestive heart failure.” Am J Physiol Heart Circ
Physiol 288: H722-8.
•
AHLUWALIA A., HOBBS AJ. (2005). “Endothelium-derived
C-type natriuretic peptide: more than just a hyperpolarizing
factor.” TRENDS in Pharmacological Sciences 26: 162-7.
•
ANDO T., OGAWA K., YAMAKI K., et al. (1996). “Plasma
concentrations of atrial, brain and C-type natriuretic peptides and
endothelin-1 in patients with chronic respiratory disease.” Chest
110: 462-468.
•
ARNOLDA L., McGRATH BP., COCKS M., JOHNSTON CI.
(1986). “Vasoconstrictor role for vasopressin in experimental
heart failure in the rabbit.” J Clin Invest 78 (3): 674-679.
•
BALLERMAN BJ., HOOVER RL., KARNOVSKY MJ.,
BRENNEN BM. (1985). “Physiologic regulation of atrial
peptide receptors in rat renal glomeruli”. J Clin Invest 76: 20492056.
111
Bibliografia
•
BARBER RD., HARMER DW., COLEMAN RA., CLARK BJ.
(2005). “Regulation of neuronal type genes in congestive heart
rats.” Physiol Genomics 21: 389–395.
•
BARR C S., RHODES P., STRTUHERS AD. (1996). “C-Type
natriuretic peptide.” Peptides 17: 1243-51.
•
BARTON M., BENY JL., D’USCIO LV., WYSS T., NOLL G.,
LUSCHER TF. (1998) “Endothelium-independent relaxation and
hyperpolarization to C-type natriuretic peptide in porcine
coronary arteries.” J Cardiovasc Pharmacol 31 (3): 377-83.
•
BATEMAN A., COIN I., DURBIN R., FINN RD., HOLLICH
V., GRIFFITHS-JONES S., KHANNA A., MARSHALL M.,
MOXON S., SONNHAMMER ELL., STUDHOLME DJ.,
YEATS C., EDDY SR. (2004). “The PFam protein families
database.” Nucleic Acids Research 32: D138-D141.
•
BAXTER GF. (2004) “The natriuretic peptides.” Basic Res
Cardiol 99: 71-75.
•
BEST PJ., BURNETT JC., WILSON SH., HOLMES JR. DR.,
LERMAN A. (2002). “Dendroaspis natriuretic peptide relaxes
isolated human arteries and veins.” Cardiovasc Res 55: 375-384.
•
BEX F., CORBIN A. (1985). “Atrial natriuretic factor stimulates
testosterone production by mouse interstitial cells.” Eur J
Pharmacol 115: 125-126.
•
BHALLA MA., CHIANG A., EPSHTEYN VA., et al. (2004).
“Prognostic role of B-type natriuretic peptide levels in patients
with type 2 diabetes mellitus.” J Am Coll Cardiol 44: 1047-1052.
112
Bibliografia
•
BOOZ GW. (2005). “Putting the brakes on cardiac hypertrophy:
exploiting
the
NO-cGMP
counter-regulatory
system.”
Hypertension 45: 341-346.
•
BRANDT
RR.,
MATTINGLY
MT.,
CLAVELL
AL.,
BARCLAY PL., BURNETT JC. (1997). “Neutral endopeptidase
regulates C-type natriuretic peptide metabolism but does not
potentiate its bioactivity in vivo.” Hypertension 30: 184-90.
•
BRANDT RR., SCOTT WRIGHT R., REDFIELD M.M.,
BURNETT J.C. (1993). “Atrial natriuretic peptide in heart
failure.” J Am Coll Cardiol 22 (4 Suppl A): 86A-92A.
•
BRAUNWALD E. (1997). “Heart Failure: An overview.” New
York: Mc Graw Hill.
•
BRISTOW MR., HERSHBERGER RE., PORT JD., ET AL.
(1990). “Beta-adrenergic pathways in nonfailing and failing
human ventricular myocardium.” Circulation 83 (suppl 1): 12-25.
cardiovascular medicine. Eur Heart J 2001 22: 997-1007.
•
BURGER AJ., BURGER MR. (2005). “Nesiritide: past, present,
and future.” Minerva Cardioangiol 53: 509-22.
•
CARGILL RI., BARR CS., COUTIE WJ., STRUTHERS AD.,
LIPWORTH BJ. (1994). “C-type natriuretic peptide levels in cor
pulmonare and in congestive cardiac failure.” Thorax 49: 1247-9.
•
CASCO VH., VEINOT JP., KUROSKI DE BOLD ML., et al.
(2002). Natriuretic peptides system gene expression in human
coronary arteries.” J Histochem Cytochem 50: 799-809.
113
Bibliografia
•
CASTILLO JR., ZAGLER A., CARRILLO-JIMENEZ R.,
HENNEKENS CH. (2004). “Brain natriuretic peptide: a potential
marker for mortalityin septic shock.” Int J infect Dis 8: 271-274.
•
CHARLES CJ., ESPINER EA., RICHARDS AM., NICHOLLS
MG., YANDLE TG. (1996). “Comparative bioactivity of atrial,
brain and C-type natriuretic peptides in conscious sheep.” Am J
Physiol 270: R1324-31.
•
CHATTERJEE PK., HAWKSWORTHY GM., McLAY GS.
(1999). “Cytochine-stimulated nitric oxide production in the
human renal proximal tubule and its modulation by natriuretic
peptides: a novel immunomodulatory mechanism?” Exp Nephrol
7: 438-448.
•
CHAUHAN SD., NILSSON H., AHLUWALIA A., HOBBS AJ.
(2003). “Release of C-type natriuretic peptide accounts for the
biological activity of endothelium-derived hyperpolarizing
factor.” PNas 100: 1426-31.
•
CHEN HH., BURNETT JC. (1998). “C-type natriuretic peptide:
the endothelial component of the natriuretic peptide system.” J
Cardiovasc Pharmacol 32: S22-8.
•
CHINKERS M., GARBERS D.L. (1989). “The protein kinase
domain of the ANP receptor is required for signaling.” Science
245: 1392-1394.
•
CHUGH SS., CHUA TP., COATS AJ. (1996). “Peripheral
chemoreflex in chronic heart failure: friend and foe.” Am Heart J
132: 900-4.
114
Bibliografia
•
CLERICO A. (2002). “Pathophysiological and clinical relevance
of circulating levels of cardiac natriuretic hormones: is their
assay merely a marker of cardiac disease?” Clin Chem Lab Med
40: 752-760.
•
CLERICO A., EMDIN M. (2004). “Diagnostic accuracy and
prognostic relevance of the measurement of cardiac natriuretic
peptides: a review.” Clin Chem 50: 33-50.
•
COATS AJS. (1997). “Origins of symptoms in heart failure.”
Cardiovasc Drugs Ther 11: 265-72.
•
COGAN MG. (1991). “Atrial Natriuretic Peptide.” Kidney Intern
37: 1148-1160.
•
CREAGER MA., CREAGER SJ. (1994). “Arterial baroreflex
regulation of blood pressure in patients with congestive heart
failure.” J Am Coll Cardiol 23: 401-5.
•
CRUDEN NL., FOX KA., LUDLAM CA., JOHNSTON NR.,
NEWBY
DE.
(2004).
“Neutral
endopeptidase
inhibition
augments vascular actions of bradykinin in patients treated with
angiotensin-converting enzyme inhibition.” Hypertension 44:
913-8.
•
DAVIDSON NC., BARR CS., STRUTHERS AD. (1996). “CType Natriuretic Peptide An Endogenous Inhibitor of Vascular
Angiotensin-Converting Enzyme Activity.” Circulation
93:
1155-1159.
•
DAVIDSON NC., NAAS AA., HANSON JK., KENNEDY
NSJ., CUTIE WJ., STRUTHERS AD. (1996). “Comparison of
atrial natriuretic peptide, B-type natriuretic peptide, and n115
Bibliografia
terminal proatrial natriuretic peptide as indicators of left
ventricular systolic dysfunction.” Am J Cardiol 77: 828-831.
•
DAVIDSON NC., STRTHERS AD. (1994). “Brain Natriuretic
peptide.” J Hypert 12: 329-336.
•
DE BOLD A.J. (1985). “Atrial natriuretic factor: a hormone
produced by the heart.” Science 230: 767-70.
•
DE
BOLD
AJ.,
BORENSTEIN
HB.,
VERESS
AT.,
SONNENBERG H. (1981). “A rapid and potent natriuretic
response to intravenous injection of atrial miocardial extracts in
rats.” Life Sci 28: 89-94.
•
DE BOLD AJ., BRUNEAU BG., KUROSKI DE BOLD ML.
(1996). “Mechanical
and neuroendocrine regulation of the
endocrine heart.” Cardiovasc Res 31: 7-18.
•
DE BOLD AJ., SALERNO TA. (1983). “Natriuretic activity of
extract obtained from heart of different species and from various
rat tissues.” Can J Physiol Pharmacol 61: 127-130.
•
DEL RY S., ANDREASSI MG., GIANNESSI D., CLERICO A.,
ET AL. (2001). “Endothelin-1, Endothelin-1 receptors and
cardiac natriuretic peptides in failing human heart.” Life
Sciences 68(24): 2715-30.
•
DEL RY S., VITALE RL., COLOTTI C., PIERSIGILLI A.,
GIANNESSI D. (2004). “Methodological assesment of mRNA
expression of endothelin receptors in pig heart and coronary
arteries.” SiBioc 2004, Padova (Abstract 22).
•
DEL RY S., MALTINTI M., EMDIN M., PASSINO C.,
CATAPANO G., GIANNESSI D. (2005). “Radioimmunoassay
116
Bibliografia
for
plasma
C-type
natriuretic
peptide
determination:
a
methodological evaluation.” Clin Chem Lab Med 43: 641-5.
•
DEL RY S., MALTINTI M., PIACENTI M., PASSINO C.,
EMDIN M., GIANNESSI D. (2006). “Cardiac production of Ctype natriuretic peptide in heart failure.” J Cardiovasc Med 7(6):
397-9.
•
DEL RY S., PASSINO C., MALTINTI M., EMDIN M.,
GIANNESSI D. (2005). “C-type natriuretic peptide plasma
levels increase in patients with congestive heart failure as a
function of clinical severity.” Eur J Heart Failure 7: 1145-8.
•
DE LEMONS JA. McGUIRE DK. DRAZNER MH. ( 2003). “Btype natriuretic peptide in cardiovascular disease.” Lancet 362:
316-322.
•
DRUMMER C., FRANCK W., HEER M., FORSSMANN W.G.,
GERZER R., GOETZ K. (1996). “Postprandial natriuresis
evidence that urodilatin, not ANP modulates sodium excretion.”
Am J Physiol 270: F301-F310.
•
DUDA T., SHARMA R. (1995). “ATP bimodal switch that
regulates the ligand binding and signal transduction activities of
the atrial natriuretic factor receptor guanylate cyclase.” Biochem
Biophys Res Commun 209: 286-292.
•
DZAU VJ., PACKER M., LILLY LS., ET AL. (1984).
“Prostaglandins in severe heart failure: relation to activation of
the renin-angiotensin system and hyponatriemia.” N Engl J Med
310: 347-352. edizione, De Groot L.J. editore, W.B. Saunders
Company, Philadelphia. Pag.2897-2916.
117
Bibliografia
•
EISENHOFER G., FRIBERG P., RUNDQVIST B., ET AL.
(1996). “Cardiac sympathetic nerve function in congestive heart
failure.” Circulation 93: 1667-1676.
•
ELSNER D., MUDERS F., MUNTE A., KROMER EP.,
FORSSMANN WG., RIEGGER GA. (1995). “Efficacy of
prolonged infusion of urodilatin [ANP-(95-126)] in patients with
congestive heart failure.” Am Heart J 129: 766-73.
• EMDIN M., PASSINO C. (2001). “Il modello neuroendocrino
dello scompenso cardiaco.” Ed. La Primula 2001; cap.1: 15-16.
•
EMOTO N., RAHARJO SB., ISAKA D., MASUDA S.,
ADIARTO S., JENG AY., YOKOVAMA M. (2005). “Dual
ECE/NEP inhibition on cardiac and neurohumoral function
during the transition from hypertrophy to heart failure in rats.”
Hypertension 45: 1145-52.
•
ESPINER E.A. (1994). “Physiology of natriuretic peptides.” J
Intern Med 235: 527-541.
•
ESPINER EA. (1995). “Hormones of the cardiovascular
system.” In Endocrinology, Vol. 3 Terza edizione, De Groot L.J.
editore, W.B. Saunders Company, Philadelphia. Pag.2897-2916.
•
ESPINER EA., MARK RICHARDS A., YANDLE TG.,
NICHOLLS MG. (1995). “Natriuretic hormones.” Met Clin
North Am 24: 481-509.
•
EVANS JJ., YOUSSEF AH., YANDLE TG., LEWIS LK.,
NICHOLLS MG. (2002). “Effects of endothelin-1 on release of
adrenomedullin and C-type natriuretic peptide from individual
human vascular endothelial cells.” J Endocrinol 175: 225-232.
118
Bibliografia
•
FERMEPIN M., VATTA MS., PRESAS M., et al. (2000). “Btype and C-type natriuretic peptides modify norepinephrine
uptake in discrete encephalic nuclei of the rat.” Cell Mol
Neurobiol 20: 763-771.
•
FITZGERALD RL., CREMO R., GARDETTO N., CHIU A.,
CLOPTON P., BHALLA V., MAISEL AS. (2005). “Effect of
nesiritide in combination with standard therapy on serum
concentrations of natriuretic peptides in patients admitted for
decompensated congestive heart failure.” Am heart J 150: 471-7.
•
FLORAS JS. (1993). “Clinical aspects of sympathetic activation
and parasympathetic withdrawal in heart failure.” J Am Coll
Cardiol 4: 72A-84A.
•
FOWKES RC., MCARDLE CA. (2000). “C-type natriuretic
peptide:an important neuroendocrine regulator?” TEM 11: 333338.
•
FRANCIS GS. (1985). “Neurohumoral mechanism involved in
congestive heart failure.” Am J Cardiol 55: 15-21.
•
FRANCIS GS., GOLDSMITH SR., OLIVARI MT., LEVINE
TB., COHN JN. (1984). “The neurohumoral axis in congestive
heart failure.” Am Intern Med 101: 370-377.
•
FUJIO N., OHASHI M., NAWATA H., et al. (1989).
“Cardiovascular, renal and endocrine effects of ahuman atrial
natriuretic peptide in patiens with Cushing’s sindrome and
primary aldosteronism.” J Hypertens 7: 653-659.
119
Bibliografia
•
FUKUI S., KATOH H., TSUZUKI N., et al. (2004). “Focal brain
edema and natriuretic peptides in patients with subarachnoid
hemorrage.” J Clin Neurosci 11: 507-511.
•
FURUYA M. (1993). “Structural requirements of C-type
natriuretic peptide for elevation of cyclic GMP in cultured
vascular smooth muscle cells.” Biophys Res Commun 138: 88086.
•
GAFFNEY TE., BRAUNWALD E. (1963).“Importance of
adrenergic nervous system in support of circultory function.” Am
J Med 34: 320-324.
•
GARDNER DG., DESHEPPER CF., GANONG WF., HANE S.,
FIDDES J., BAXTER JD., LEWICKI J. (1986). “Extra atrial
expression of the gene for atrial natriuretic factor.” Proc Natl
Acad Sci 83: 6697-6701.
•
GAUQUELIN GR., GARCIA R., CARRIER F., CANTIN M.,
GUTKOWSKA J., THIBAULT G., SCHIFFRIN EL. (1988).
“Glomerular ANF receptor regulation during changes in sodium
and water metabolism.” Am J Physiol 254: F51-F55.
•
GLEMBOTSKI CC., IRONS CE., KROWN KA., et al. (1993).
“Myocardial a-thrombin receptor activation induces hypertrophy
and increases atrial natriuretic factor gene expression.” J Biol
Chem 268: 20646-20652.
•
GOETZ KL. (1991). “ Renal natriuretic peptide (urodilatin?) and
atriopeptin evolving concepts.” Am J Physiol 261: F921-F932.
120
Bibliografia
•
GOWER WR. JR, CHIOU S., SKOLNICK KA., VESELY DL.
(1994). “Molecular forms of circulating atrial natriuretic peptides
in human plasma and their metabolites.” Peptides 15: 861-867.
•
GRIFFITH TM. (2004). “Endothelium-dependent smooth muscle
hyperpolarization: do gap junctions provide a unifying
hypothesis?” British J Pharm 141: 881-903.
•
GUTKOWSKA J., NEMER M. (1989). “Stucture, expression,
and function of atrial natriuretic peptide in extra atrial tissues.”
Endocrine Rev 10: 516-535.
•
HALL S., CIGARROA GC., MARXOUX L., et al. (1995).
“Time course of improvment in left venticular function, mass
and geometry in patient with congestive heart failure e treated
with b-adrenergic blockade.” J Am Coll Card 25: 1544-60.
•
HAN B., HASIN Y. (2003). “Cardiovascular effects of
natriuretic peptides and their interrelation with Endothelin-1.”
Cardiovasc Drugs Ther 17: 41-52.
•
HARDT SE., SADOSHIMA J. (2004). “Negative regulators of
cardiac hypertrophy.” Cardiovasc Res 63: 500-509.
•
HARTHSHORNE H. (1847). “Water versus hydrotherapy.”
Lloid P. Smith Press, Philadelphia pag. 28.
•
HAYAKAWA H., KOMADA Y., HIRAYAMA M., et al.
(2001). “Plasma levels of natriuretic peptides in relation to
doxorubicin-induced cardiotoxicity and cardiac function in
children with cancer.” Med Pediatr Oncol 37: 4-9.
•
HAYASHI M., TSUTAMOTO T., WADA A., et al. (2001).
“Intravenous atrial natriuretic peptide prevent left ventricular
121
Bibliografia
remodelling in patients with first anterior acute myocardial
infarction.” J Am Coll Cardiol 37: 1820-1826.
•
HENRIKSEN JH., GOTZE JP., FUGLSANG S., et al.(2003).
“Increased circulating pro-brain natriuretic peptide (proBNP) and
brain natriuretic peptide (BNP) in patients with cirrhosis: relation
to cardiovascular dysfunction and severity of disease.” Gut 52:
1511-1517.
•
HENRY JP.,GAUER OH., REEVES JL. (1956). “Evidence of
the atrial location of receptor influencing urine flow.” Circ Res
4: 85-90.
•
HOLMES SJ., ESPINER EA. (1993). “Renal endocrine and
hemodinamic effects of human brain natriuretic peptide in
normal man.” J Clin Endocrinol Metab 76: 91-96.
•
HORIO T., TOKUDOME T., MAKI T., YOSHIHARA F.,
SUGA S., NISHIKIMI T., KOJIMA M., KAWANO Y.,
KANGAWA K. (2003) “Gene expression, secretion, and
autocrine action of C-type natriuretic peptide in coltured adult rat
cardiac fibroblast.” Endocrinology 144: 2279-84.
•
HOUBEN AJ., VAN DER ZANDER K., DE LEEUW PW.
(2005). “Vascular and renal actions of brain natriuretic peptide in
man: physiology and pharmacology.” Fundam Clin Pharmacol
19: 411-419.
•
HSIAO LL., DANGOND F., YOSHIDA T., HONG R., JENSEN
RV., MISRA J., DILLON W., LEE KF., CLARK KE.,
HAVERTY P., GULLANS SR., et al. (2001). “A compendium
122
Bibliografia
of gene expression in normal human tissues.” Physiol Genomics
7: 97–104.
•
HUET M., CANTIN M. (1974). “Ultrastructural cytochemistry
of atrial muscle cells II. Characterization of the protein content of
specific granules.” Lab Invest 30: 525-532.
•
IERVASI G., CLERICO A., BERTI S., PILO A., BIAGINI A.,
BIANCHI R., DONATO L. (1995). “Altered tissue degradation
and distribution of atrial natriuretic peptide in patients with
idiopathic dilated cardiomyopathy and its relationship with
clinical severity of the disease and sodium handling.” Circulation
91: 2018-27.
•
IMURA H., NAKAO K., ITOH H. (1992). “The natriuretic
peptide system in the brain: implications in the central control of
cardiovascular
and
neuroendocrine
functions.”
Front
Neuroendocrinol 13: 217-249.
•
INOUE A., YANAGISAWA M., KIMURA S., KASUYA Y.,
MIYAUCHI T., GOTO K., MASAKI T. (1989). “The human
endothelin family: three structurally and pharmalogically distinct
isopeptides predicted by three separate genes.” Proc Natl Acad
Sci USA 86: 2863-7.
•
IVANOVA MD., GREGORASZCZUK EL., AUGUSTOWSKA
K., et al. (2003). “Localization of atrial natriuretic peptide in pig
granulosa cells isolated from ovarian follicles of ovarious size.”
Reprod Biol 3: 173-181.
•
JAMIESON JD., PALADE GE. (1964). “Specific granules in
atrial muscle cells.” J Cell Biol 23: 151-162.
123
Bibliografia
•
JAMIESON RL., CANAAN-KUL S., PRATT R. (1992). “The
natriuretic peptides and their receptors.” Am J Kidney Dis 5:
519-530.
•
JOHNSTON CI., ARNOLDA L., ABRAHAMS J., McGRATH
BP. (1986). “Role of vasopressin in experimental congestive
cardiac failure.” J Cardiovasc Pharmacol 8 suppl 7: S96-100.
•
JOHSON BE., DAMOARAN A., RUSHIN J., et al.(1997).
“Ectopic production and processing of atrial natriuretic peptide
in a small cell.” Cancer 79: 35-44.
•
JORTANI SA., PRABHU S., VALDES RV. (2004) “Strategies
for developing biomarkers of heart failure.” Clin Chem 50: 26578.
•
KAHANA L., YECHIELY H., MECZ Y., LURIE A. (1995).
“Identification and quantification of human kidney atrial
natriuretic peptide receptors.” Eur J Endocrinol 132: 465-471
•
KALRA PR., ANKER SD., STRUTHERS AD., COATS AJS.
(2001) “The role of C-type natriuretic peptide in C-type
natriuretic peptide in cardiovascular medicine.” Eur Heart J 22:
997-1007.
•
KALRA PR., CLAGUE JR., BOLGER AP., ANKER SD.,
POOLE-WILSON PA., STRUTHERS AD., COATS AJ. (2003)
“Myocardial production of C-type natriuretic peptide in chronic
heart failure.” Circulation 107: 571-3.
•
KANGAWA KY., TAWARAGI Y., OIKAWA S., MIZUNO A.,
SAKURAGAWA
Y.,
NAKAZATO
H.,
FUKUDA
A.,
MINAMINO N., MATSUE H. (1984). “Identification of rat
124
Bibliografia
gamma atrial natriuretic polipeptide and characterization of
cDNA encoding its precursor.” Nature 312: 152-155.
•
KANNEL WB., BELANGER AJ. (1991). “Epidemiology of
heart failure.” Am Heart J 121: 951-7
•
KAPOUN AM., LIANG F., O’YOUNG G., et al. (2004). “B
type natriuretic peptide exerts broad functional opposition to
transforming growth factor-beta in primary human cardiac
fibroblast: fibrosis, myofibroblast conversion, proliferation and
inflammation.” Circ Res 94: 453-461.
•
KENNY AJ., BOURNE A., INGRAM J. (1993). “Hydrolysis of
human and pig brain natriuretic peptides, urodilatin, C-type
natriuretic
peptide
and
some
C-receptor
ligands
by
endopìeptidase-24.11.” Biochem J 291 (1): 83-88.
•
KHONO M., HORIO T., TOKOKAWA K. (1992). “C-type
natriuretic peptide inhibits thrombin- and angiotensin IIstimulated endothelin release via cyclic guanosine 3’,5’monophosphate.” Hypertension 19: 320-325.
•
KIEFER F., WIEDEMANN K. (2004). “Neuroendocrine
pathways of addictive behaviour.” Addict Biol 9(205): 12
•
KIEMER AK., LEHNER MD., HARTUNG T., VOLLMAR
AM. (2002). “Inhibition of cyclooxygenase-2 by natriuretic
peptides.” Endocrinology 143: 846-852.
•
KINGWELL BA., CAMERON JD., GILLIES KJ., JENNINGS
GL., DART AM. (1995). “Arterial compliance may influence
baroreflex function in athletes and hypertensives.” Am J Physiol
26: H411-H418.
125
Bibliografia
•
KISCH B. (1956). “Electron microscopy of the atrium of the
heart.I Guinea Pigs.” Exp Med Surg 14: 99-112.
•
KLINGER JR., SIDDIQ FM., SWIFT RA., JACKSON C.,
PIETRAS L., WARBURTON RR., ALIA C., HILL NS. (1998)
“C-type
natriuretic
peptide
expression
and
pulmonary
vasodilation in hypoxia-adapted rats.” Am J Physiol 19: L64552.
•
KOHNO M., HORIO T., YASUNARI K., et al. (1993).
“Stimulation of brain natriuretic peptide release from heart by
thyroid hormone.” Metabolism 42: 1059-1064.
•
KOLLER KJ., GOEDDEL DV. (1992). “Molecular biologyof
the natriuretic peptide and their receptor.” Circulation 86 (4):
1081-1088.
•
KOLLER KJ., LOWE DG., BENNET GL., MINAMOTO N.,
KANGAWA K., MATSUO H., GOEDDEL DV. (1991).
“Selective activation of B natriuretic peptide receptor by C-type
natriuretic peptide.” Science 252 (5002): 120-123.
•
KOMATSU JB., NAKAO K., SUGA S., OGAWA Y.,
MUKOYAMA M., ARAI H., SHIRAKAMI G., HOSODA K.,
NAKAGAWA O., HAMA N. (1991). “C-type natriuretic peptide
(CNP) in rats and humans.” Endocrinology 129: 1104-6.
•
KOMATSU Y., ITOH H., SUGA S., OGAWA Y., HAMA N.,
KISHIMOTO I., NAKAGAWA O., IGAKI T., DOI K.,
YOSHIMASA
T.,
NAKAO
K.
(1996).
“Regulation
of
endothelial production of C-type natriuretic peptide in coculture
with vascular smooth muscle cells:role of the vascular natriuretic
126
Bibliografia
peptide system in vascular growth inhibition.” Circ Res 78: 606614.
•
KOMATSU Y., NAKAO K., ITOH H., SUGA S., OGAWA Y.,
IMURA H. (1992). “Vascular natriuretic peptide syatem in
human.” Lancet 340: 622.
•
KRUGER S., GRAF J., MERX MW., et al. (2004). “Brain
natriuretic peptide predicts right heart failure in patiens with
acute pulmonary embolism.” Am Heart J 147: 60-65.
•
KUCHER N., PRINTZEN G., GOLDHABER SZ. (2003).
“Prognostic role of brain natriuretic peptide in acute pulmonary
embolism.” Circulation 107: 2545-2547.
•
LANGENIKEL TH., BUTTGEREIT J., PAGEL-LANGENIKEL
I., LINDNER M., MONTI J., BEUERLEIN K., AL-SAADI N.,
POPOLA E., TANK J., DIETZ R., WILLENBROCK R.,
BADER M. (2006). “Cardiac Hypertrophy in transgenic rats
expressing a dominant-negative mutant of the natriuretic peptide
receptor B.” Pnas 103: 4735-4740.
•
LAPOINTE MC. (2005). “Molecular regulation of brain
natriuretic peptide gene.” Peptides 26: 944-956.
•
LEITMAN
DC.,
ANDRESEN
JW.,
CATALANO
RM.,
WALDMAN SA., TUAN JJ., MURAD F. (1988). “Atrial
natriuretic petide binding, cross linking, and stimulation of cyclic
GMP accumulation and particulate guanylate cyclase activity in
cultured cells.” J Biol Chem 263 (8): 3720-3728.
•
LEUCHTE HH., HOLZAPFEL M., BAUMGARTNER RA., et
al. (2004). “Clinical significance of brain natriuretic peptide in
127
Bibliografia
primary pulmonary hypertension.” J Am Coll Cardiol 43: 764770.
•
LEVIN E.R. (1993). “Natriuretic peptide C-receptor: more than a
clearance receptor.” Am J Physiol 264 (4): F483-F489.
•
LEVIN ER., GARDNER DG., SAMSON WK. (1998).
“Natriuretic peptides.” N Eng J Med 321-328.
•
LIN X., HANZE J., HEESE F., SADMAN R., LANG RE.
(1995). “Gene expression of Natriuretic peptide receptors in
Myocardial Cells.” Circ Res 77 (4): 750-758.
•
LINGAPPA VR. (1989). “Intracellular traffic of newly
sinthesized proteins: current understanding and future prospect.”
J Clin Invest 83 (3): 739-751.
•
LIPKIN DP. (1988). “Abnormalities of skeletal muscle in
patients with chronic heart failure.” Int J Cardiol 18: 187-95.
•
LLORENS-CORTES C., HUANG H., VICART P., GASC JM.,
PAULIN
D.,
CORVOL
P.
(1992).
“Identification
and
characterization of neutral endopeptidase in endothelial cells
from venous or arterial origins.” J Biol Chem 267 (20): 1401214018.
•
LOWRY OH., ROSEBROUGH NJ., FARR AL., RANDALL
RJ. (1951). “Protein measurement with the Folin phenol
reagent.” J Biol Chem 193: 265-275.
•
LUCHNER A., STEVENS TL., BORGESON DD., REDFIELD
M., WEI CM., PORTER JG., BURNETT JC
•
MA KK., OGAWA T., DE BOLD AJ. (2004). “Selective
upregulation of
cardiac brain natriuretic peptide at the
128
Bibliografia
transcriptional and translational levels by pro-inflammatory
cytokines and by conditioned medium derived from mixed
lymphocyte reactions via p38 MAPkinase.” J Mol Cell Card 36:
505-513.
•
MAACK T. (1992). “Receptor of atrial natriuretic peptide.” Ann
Rev Physiol 54: 11-27.
•
MAACK T., OKOLICANY J., KOH G.Y., PRICE D.A. (1993).
“Functional properties of atrial natriuretic factor receptors.” Sem
Nephrol 13 (1): 50-60.
•
MAACK T., SUZUKI M., ALMEIDA FA., NUSSENZVEIG D.,
SCARBOROUGH RM., MCENROE GA., LEWICKI JA.
(1987). “Physiological role of silent receptors of atrial natriuretic
factor.” Science 238: 675-8.
•
MADHU B., SRIVASTA A., CANTIN M. (1986). “Atrial
natriuretic factor receptors are negatively coupled to adenylate
cyclase in cultured atrial and ventricular cardiocyte.” Bioch
Biophys Res Commun 138 (1): 427-436.
•
MADHU B., SRIVASTAVA A., TACHTE G.T. (1993). “Atrial
Natriuretic
Factor
Receptor
and
signal
transduction
mechanisms.” Pharmacol Rev 45 (4): 455.
•
MANN DL. (1999). “Mechanism and models in Heart Failure.”
Circulation 1999; 31;100(9): 999-1008.
•
MANTOVANI A., BUSSOLINO F., DEJANA E. (1992)
“Cytokine regulation of endothelial function.” FASEB J 6: 25912599.
129
Bibliografia
•
MARGULIES
KB.,
PERELLA
MA.,
MCKINLAY
LJ.,
BURNETT JC. (1991). “Angiotensin inhibition potentiates the
renal responses to neutral endopeptidase inhibition in dogs with
congestive heart failure.” J Clin Invest 88: 1636-42.
•
MASASHI M., NAKAO K., HOSODA K., SUGA S.I., SAYTO
Y., OGAWA Y., SHIRAKAMI G., JOUGASAKI M., OBATA
K., YASUE H., KAMBAYASHI Y., INOUYE K., IMURA H.
(1991). “ Brain natriuretic peptide as a novel cardiac hormone in
humans”. J. Clin. Invest. 87 (4): 1402-1412.
•
MASAYASU K., MINAMINO N., KANGAWA K., MATSUO
H. (1990):”Cloning and sequence analysis of a cDNA encoding a
precursor for rat C- type natriuretic peptide (CNA)”. FEBS 276
(1, 2): 209-213.
•
MASSIE BM. (1998). “15 years of heart-failure trias: what have
we learned?” Lancet 352 (suppl 1): 29-33.
•
MATTINGLY MT., BRANDT RR., HEUBLEIN DM., WEI C.,
NIR A., BURNETT JC. (1994). “Presence of C-type natriuretic
peptide in human kidney and urine.” Kidney Int; 46: 744-747.
•
McCULLOUGH PA., KUNCHERIA J., MATHUR VS. (2004).
“Diagnostic and therapeutic utilità of B-type natriuretic peptide
in patients with renal insufficiency and decompensated heart
failure.” Rev Cardiovasc Med 5: 16-25.
•
McGIRT MJ., BLESSING R., NIMJEE SM., et al. (2004).
“Correlation
of
serum
brain
natriuretic
peptidewith
hyponatriemia and delayed ischemic neurological deficits after
subarachnoid hemorrage.” Neurosurgery 54: 1369-1373.
130
Bibliografia
•
MCGRATH MF., DE BOLD AJ. (2005). “Determinants of
natriuretic peptide gene expression.” Peptides 26: 933-943.
•
MELLIN V., JENG AY., MONTEIL C., RENNET S., HENRY
JP., THUILLEZ C., MULDER P. (2005). “Triple ACE-ECENEP inhibition in heart failure: a comparison with ACE and dual
ECE-NEP inhibition.” J Cardiovasc Pharmacol 46: 390-397.
•
MICHEL JB., BONVALET JP. “Système rénine-angiotensine,
aldostérone, coeur et vaisseaux.” In Hormones, coeur et
vaisseaux. Hittinger L., Berthezéne, Castaigne, Dubois-Randé,
Plouin PF. 171-226.
•
MICHENER ML., GIERSE JK., SLETHARAM R., FOK KF.,
OLINS P.O. (1986). “Proteolytic processing of atriopeptin prohormone.” Mol. Pharmacol. 30 (6): 552-557.
•
MIMURA Y., KANAUCHI H., OGAWA T., OOHARA T.
(1996). “Resistance to natruresis in patients with peritonitis
carcinomatosa.” Horm Metab Res 28: 183-186.
•
MITANI Y., MARUYAMA J., YOKOCHI A., MARUYAMA
K., YOSHIMOTO T., NARUSE M., SAKURAI M. (2000)
“Modulated vasodilator responses to natriuretic peptides in rats
exposed to chronic hypoxia.” Eur Respir J 15: 400-6.
•
MOREL G., HEISLER S. (1988). “Internalization of endogenous
and exogenous atrial natriuretic peptide by target tissues.”
Electron Microsc Rev 1 (1): 221-231.
•
MORISHIGE K., SHIMOKAWA H., YAMAWAKI T., et al.
(2000). “Local adenovirus-mediated transfer of C type natriuretic
131
Bibliografia
peptide suppresses vascular remodelling in porcine coronary
arteries in vivo.” J Am Coll Cardiol 35: 1040-1047.
•
MORISHITA R., GIBBONS G.H., PRATT E.R., TONITA N.,
KANEDA Y., OGIHOUA T., DZON WG. (1994). “Autocrine
and paracrine effects of atrial natriuretic peptide gene transfer on
vascular smooth muscle and endothelial cells.” J Clin Invest 94
(2): 824-829.
•
MORITA E., YASUE H., YOSHIMURA M., et al. (1993).
“Increased plasma levels of Brain natriuretic peptide in patients
with acute myocardial infarction.” Circulation 88: 82-91.
•
MUKOYAMA M., NAKAO K., OBATA K., et al. (1991).
“Augmented secretion of brain natriuretic peptide in acute
myocardial infarction.” Biochem Biophys Res Commun 15: 431436.
•
MURGER
MA.
(1996).
“Circulating
concentrations
of
proinflammatory cytokines in mild or moderate heart failure
secondary to ischemic or idiopathic dilated cardiomyopathy.”
Am J Cardiol 77: 723-727.
•
NAGAYA N., NISHIKIMI T., GOTO Y., et al. (1998). “Plasma
brain natriuretic peptide is a biochemical marker for the
prediction of progressive ventricular remodelling after acute
myocardial infarction.” Am Heart J 135: 21-28.
•
NAKAIAMA K., OHKUDO H., HIROSE T., INAYAMA S.,
NAKANISHI S. “mRNA sequence for human cardiodilatin-atrial
natriuretic factor precursor and regulation of precursor mRNA in
rat atria.” Nature 310: 699-701.
132
Bibliografia
•
NAKANISHI M., SAITO Y., KISHIMOTO I., et al. (2005).
“Role of natriuretic peptide receptor guanylyl cyclase-A in
myocardial infarction evaluated using genetically engineered
mice.” Hypertension 46: 1-27.
•
NARUKO T., ITOH A., HAZE K., EHARA S., FUKUSHIMA
H., SUGAMA Y., SHIRAI N.,
IKURA Y., OHSAWA M.,
UEDA M. (2005). “C-type natriuretic peptide and natriuretic
peptide receptors are expressed by smooth muscle cells in the
neointima
after
percutaneous
coronary
intervention.”
Atherosclerosis 181: 241-250.
•
NAZARIO B., HU R., PEDRAM A., BRUCE P., LEVIN ER.
(1995). “Atrial and brain natriuretic peptides stimulate the
production and secretion of C-type natriuretic peptide from
bovine aortic endothelial cells.” J Clin Invest 95: 1151-1157.
•
NICHOLLS MG. (1994). “The natriuretic peptide in heart
failure.” J Intern Med 235: 516-526.
•
NORTIER J., PAUWELL S., DE PREZ E., LANCKMAN MD.
(1995). “Human neutrophil and plasma endopeptidase 24.11.:
quantification and respective roles in atrial natriuretic peptide
hydrolysis.” Eur J Clin Invest 25: 206-212.
•
NUNEZ DJR., ICKSON CM., BROWN KJ. (1992) “Natriuretic
peptide receptor mRNAs in the rat and human heart.” J Clin
invest 90: 1966-1971.
•
OGAWA Y., ITOH H., YOSHIAKE Y., et al. (1994).
“Molecular cloning and chromosomal assignment of the mouse
133
Bibliografia
C-type natriuretic peptide (CNP) gene (Nppc): comparison with
the human CNP gene (NPPC).” Genomic 24: 383-387.
•
OHNO N., ITOH H., IKEDA T., et al. (2002). “Accelerated
reendothelialization with suppressed thrombogenic property and
neointimal hyperplasia of rabbit jugular vein grafts by
adenovirus-mediated gene transfer of C-type natriuretic peptide.”
Circulation 105: 1623-1626.
•
OIKAWA S., IMAI M., INUSUKA C., TAWARAGI Y.,
NAKAZATO H., MATSUO H. (1985). “Structure of a dog and
rabbit precursor of atrial natriuretic polipeptide deduced from
nucleotide sequence of cloned cDNA.” Biochem Biophys Res
Commun 132: 892-899.
•
OKAMOTO A., LOVETT M., PAYAN DG., BUNNETT NW.
(1994).
“Interaction
between
neutral
endopeptidase
(EC
3.4.24.11.) and the substance P (NK1) receptor expressed in
mammalian cells.” Biochem J 299: 683-693.
•
OKUMURA H., IUCHI K., YOSHIDA T., et al. (2000). “Brain
natriuretic peptide is a predictor of anthracycline-induced
cardiotoxicity.” Acta Haematol 104: 158-163.
•
PACKER M. (1992). “The neurohormonal hypothesis: a theory
to explain the mechanisms of disease progression in heart
failure.” J Am Coll Cardiol 20: 248-254.
•
PALLADINI G., CAMPANA C., KLERSY C., et al. (2003).
“Serum N-Terminal-pro-brain natriuretic peptide is a sensitive
marker of myocardial dysfunction in AL amyloidosis.”
Circulation 107: 2440-2445.
134
Bibliografia
•
PANDEY KN. (2005). “Biology of natriuretic peptide and their
receptors.” Peptides 26: 901-932.
•
PARLAPIANO C., CAMPANA E., ALESSANDRI N., et al.
(1998). “Plasma atrial natriuretic hormone in hyperthyroidism.”
Endocr Res 24: 105-112.
•
PERRAS B., SCHULTES B., BEHN B., et al. (2004).
“Intranasal atrial natriuretic peptide acts as central nervous
inhibitor of the hypothalamo-pituitary-adrenal stress system in
humans.” J Clin Endocrinol Metab 89: 4642-4648.
•
PFEFFER MA., BRAUNWALD E. (1990). “Ventricular
remodeling
after
myocardial
infarction.
Experimental
observation and clinical implications.” Circulation 81: 17-23.
•
PILO A., ZUCCHELLI GC., MALVANO R., MASINI S.
(1982). “Main features of computer algorithms for RIA data
reduction. Comparison of same different approaches for the
interpolation of dose-response curve.” J Nucl Med Allied Sci 26:
235-248.
•
PONIKOWSKI P., CHUA TP., PIEPOLI M., ET AL. (1997).
“Augmented peripheral chemosensitivity as a potential input to
baroreflex impairment and autonomic imbalance in chronic heart
failure.” Circulation 96: 2586-94.
•
PORTER JD., CATALANO R., MC ENROE G., LEWICKI JA.,
PROTTER AA. (1992). “C-type natriuretic peptide inhibits
growth factor-dependent DNA synthesis in smooth muscle
cells.” Am J Physiol 263 (5): C1001-C1006.
135
Bibliografia
•
POTTER LR. (2004). “CNP, Cardiac natriuretic peptide?”
Endocrinology 145(5): 2129-30.
•
PRICKETT TCR., YANDLE TG. NICHOLLS MG., ESPINER
EA., RICHARDS AM. (2001). “Identification of amino-terminal
pro-C-type natriuretic peptide in human plasma.” Biochem
Biophys Res Commun 286: 513-7.
•
QIAN JY., HARUNO A., ASADA Y., et al., (2002). “Local
expression
of
C-type
natriuretic
peptide
suppresses
inflammation, eliminates shear stress-induced thrombosis, and
prevents neointima formation through enhanced nitric oxide
production in rabbit injured carotid arteries.” Circ Res 91:10631069.
•
QIAN JY., HARUNO A., ASADA Y., NISHIDA T., SAITO Y.,
MATSUDA T., UENO H. (2002). “Local expression of C-type
natriuretic peptide suppresses inflammation, eliminates shear
stress- induced thrombosis, and prevents neointima formation
through enhanced nitric oxide production in rabbit injured
carotid arteries.” Circ Res 91: 1063-9.
•
RAHMUTULA D., NAKAYAMA T., SOMA M., et al. (2001).
“Association study between the variants of the human ANP gene
and essential hypertension.” Hypertens Res 24: 291-294.
•
RICHARDS AM. (2003). “Outpatient management of heart
failure.” Heart Failure Reviews 8:345-8.
•
ROSENZWEIG A., SEIDMAN C.E. (1991). ”Atrial natriuretic
factor and related peptides hormones.” Annu Rev Biochem 60:
229-255.
136
Bibliografia
•
ROSS
R.
(1991).
“Polypeptide
growth
factors
and
atherosclerosis.” Trends Cardiovasc Med 1:277-282.
•
ROSTAGNO (1996). “Lo Scompenso Cardiaco.” Ed. Sei Firenze
•
RUFFOLO R. (1998). “Neurohormonal activation, oxygen free
radicals and apoptosis in pathogenesis of congestive heart
failure.” J Cardiovasc Pharmacol 32(Suppl. 1): S22-S30.
•
RUSKOAHO H. (1992). “Atrial Natriuretic Peptide:Synthesis,
release, and metabolism.” Pharmacological Rew 44 (4): 479602.
•
RUSKOAHO H. (2003). “Cardiac hormones as diagnostic tools
in heart failure.” Endocr Rev 24: 341-356.
•
RUSKOAHO H., LESKINEN H., MAGGA J., TASKINEN P.,
MANTYMAA P., VUOLTEENAHO O., LEPPALUOTO J.
(1997).
“Mechanisms
of
mechanical
load-induced
atrial
natriuretic peptide secretion: role of endothelin, nitric oxide, and
angiotensin II.” J Mol Med 75: 876-885.
•
SAGNELLA GA. (1998). ”Measurement and significance of
circulating natriuretic peptides in cardiovascular diseases.” Clin
Sci 95: 519-529.
•
SANGER F., NICKLEN S., COULSON AR. (1977). “DNA
sequencing with chain-terminating inhibitors” Proc Natl Acad
Sci USA 74:5463-5467.
•
SAVOIE P., DE CHAMPLAIN J., MADHU B., SRIVASTAVA
A. (1995). ”C-type natriuretic peptide and brain natriuretic
peptide inhibit adenylyl cyclase activity: interaction with ANFR2/ANP-C receptors.” FEBS Lett 370 (1,2): 6-10.
137
Bibliografia
•
SCHENK DB., PHELPS MN., PORTER JG, SCARBOROUGH
LM., MC ENROE GA., LEVINKI JA. (1985). ”Identification of
the receptor for atrial natriuretic factor on cultured vascular
cells.” J Biol Chem 260 (28): 14887-14890.
•
SCHILLER PW., BELLINI F., DIONNE G., MAZIAK LA.,
GARCIA R., DE LEAN A., CANTIN M. (1986). “Synthesis and
activity profiles of atrial natriuretic peptide (ANP) analogs with
reduced ring size.” Biochem Biophys Rs Commun 138: 880-86.
•
SCHIRGER JA., HEUBLEIN DM., CHEN HH., LISY O.,
JOUGASAKI M., WENNBERG, BURNETT JR JC. (1999).
“Presence
of
Dendroaspis
natriuretic
peptide-like
immunoreactivity in human plasma and its increase durino
human heart failure.” Mayo Clin Proc 74: 126-130.
•
SCHOCKEN DD., ARRIETA MI., LEAVERTON PE., ET AL.
(1992). “Prevalence and mortalità rate of congestive heart failure
in the United States.” J Am Coll Cardiol 20: 301-6.
•
SCHULTZ M., FABERJ., KISTORP C., et al. (2004). “NTerminal-pro-B-type
natriuretic
peptide
(NT-proBNP)
in
different thyroid function states.” Clin Endocrinol 60: 54-59.
•
SCHULZ-KNAPPE P., FORSSMANN K., HERBEST F.,
HOCK D., PIPKORN R., FORSSMANN WG. (1988). “Isolation
and structural analysis of “urodilatin”, a new peptide of the
cardiodilatin-(ANP)-family, extracted from human urine.” Klin
Wochenschr 66: 752-59.
138
Bibliografia
•
SCOTLAND RS., AHLUWALIA A., HOBBS AJ. (2005). “Ctype natriuretic peptide in vascular physiology and disease.”
Pharm Ther 105: 85-93.
•
SOLOMON SD., SKALI H., BOURGOUN M., FANG J.,
GHALI
JK.,
MARTELET
M.,
WOJCIECHOWSKI
D.,
ANSMITE B., SKARDS J., LAKS T., HENRY D., PACKER
M., PFEFFER MA. (2005) OVERTURE Investigators. “Effect of
angiotensin-converting enzyme or vasopeptidase inhibition on
ventricular size and function in patients with heart failure: the
omapatrilat versus enalapril randomized trial of utility in
reducing events (OVERTURE) echocardiographic study.” Am
Heart J 150: 257-62.
•
SOMERS VK., MARK AL., ABBOUD FM. (1991). “Interaction
of baroreceptor and chemoreceptor reflex control of sympathetic
nerve activity in normal humans.” J Clin Invest 87: 1953-7.
•
STEIN BC., LEVIN RI. (1998). ” Natriuretic peptides:
physiology, therapeutic potential, and risk stratification in
ischemic heart disease.” Am Heart J 135, 914-923.
•
STEINMETZ M., POTTHAST R., SABRANE K., KUHN M.
(2004). “Diverging vasorelaxing effects of C-type natriuretic
peptide in renal resistance arteries and aortas of GC-A-deficient
mice.” Regul Pept 119: 31-37.
•
STEPAN H., LEITNER E., BADER M., WALTHER T. (2000).
“Organ specific mRNA distribution of C-type natriuretic peptide
in neonatal and adult mice.” Regulatory Peptides 95: 81-85.
139
Bibliografia
•
STINGO AJ., CLAVELL ALL., HEUBLEIN DM., WEI CM.,
PITTELKOW MR., BURNETT JC. (1992). “Presence of C-type
natriuretic peptidein cultured human endothelia cells and human
plasma.” Am J Physiol; 263: H1318-1321.
•
STRAUB RH., HALL C., KRAMER BK., et al. (1996). “Atrial
natriuretic factor and digoxin-like immunoreactive factor in
diabetic patients: their interrelation and the influence of the
autonomic nervous system.” J Clin Endocrinol Metab 81: 33853389.
•
SUDOH T., KANGAWA K., MINAMINO N., MATSUO H.
(1988). ”A new natriuretic peptide in porcine brain.” Nature 332
(6159): 78-81.
•
SUDOH T., MINAMINO N., KANGAWA K., MATSUO H.
(1990). “C-type natriuretic peptide (CNP): a new member of the
natriuretic peptide family identified in porcine brain.” Biochem
Biopys Res Comm 168: 863-870.
•
SUGA S., ITHOH H., KOMATSU Y., OGAWA Y., HAMA N.,
YOSHIMASA T., NAKAO K. (1993). “Cytokine-induced Ctype natriuretic peptide (CNP) secretion from vascular
endothelial cells-evidence for CNP as a novel autocrine/paracrine
regulator from endothelial cells.” Endocrinology 133: 3038-41.
•
SUGA S., ITHOH H., KOMATSU Y., OGAWA Y., HAMA N.,
YOSHIMASA T., NAKAO K. (1993). “Cytokine-induced Ctype natriuretic peptide (CNP) secretion from vascular
endothelial cells-evidence for CNP as a novel autocrine/paracrine
regulator from endothelial cells.” Endocrinology 133: 3038-41.
140
Bibliografia
•
SUGA S., NAKAO K., HOSODA K., MUKOYAMA M.,
OGAWA Y., SHIRAKAMI G., ARAI H., SAITO Y.,
KAMBAYASHI Y., INOUYE K., IMURA H. (1992). “
Receptor selectivity of natriuretic peptide family, atrial
natriuretic peptide, brain natriuretic peptide, and C-type
natriuretic.” Endocrinology 130: 229-39.
•
SUGA S., NAKAO K., HOSODA K., MUKOYAMA M.,
OGAWA Y., SHIRAKAMI G., ARAI H., SAITO Y.,
KAMBAYASHI Y., INOUYE K. (1992). ”Receptor selectivity
of natriuretic peptide family, atrial natriuretic peptide, brain
natriuretic
peptide
and
C-type
natriuretic
peptide.”
Endocrinology 130 (1): 229-339.
•
SUGA S., NAKAO K., ITHOH H., KOMATSU Y., OGAWA
Y., HAMA N., IMURA H. (1992) “Endothelial production of Ctype natriuretic pepide and its marked augmentation by
transforming growth factor–β. Possible existence of “vascular
natriuretic peptide system.” J Clin Invest 90:1145-1149.
•
SUTTON (1990). “Epidemiological aspects of heart failure.” Am
Heart J 120: 1538-1540
•
SUZUKI T., HAYASHI D., YAMAZAKI T., et al. (1998).
“Elevated B-type natriuretic peptide levels after anthracycline
administration.” Am Heart J 136: 362-363.
•
TAKEI Y. (2001). “Does the natriuretic peptide system exist
throughout the animal and plant kingdom? Comp Biochem
Physiol B Biochem Mol biol; 129:559-73.
141
Bibliografia
•
TAWARAGI Y., FUCHIMURA K., TANAKA S., MINAMINO
N., KANGAWA K., MATSUO H. (). “Gene and precursor
structures of human C-type natriuretic peptide. Biochem Biophys
Res commun 1991; 175: 645-51.
•
TENHUNEN O., SZOKODI I., RUSKOAO H., et al. (2005).
“Posttranscriptional activation of BNP gene expression in
response to increased left ventricular wall stress: role of
calcineurina and PKC.” Regul Pept 128:187-196.
•
THAMES MD., KINUGAWA T., SMITH ML., DIBNERDUNLAP ME. (1993). “Abnormalities of baroreflex control in
heart failure.” J Am Coll Cardiol 22: 56A-60A.
•
TOGASHI K., KAMEYA T., KUROSAWA T., HASEGAWA
N., KAWAKAMI M. (1992). “Concentrations and molecular
forms of C-type natriuretic peptide in brain and cerebrospinal
fluid.” Clin Chem 38: 2136-2139.
•
TOKUDOME T.,
HORIO T., SOEKI T., MORI K.,
KISHIMOTO I., SUGA S., YOSHIHARA F., KAWANO Y.,
KOHNO M., KANGAWA K. (2004). “Inhibitory effect of Ctype natriuretic peptide (CNP) on cultured cardiac myocite
hypertrophy:interference
between
CNP
and
endothelin-1
signaling pathways.” Endocrinology 145: 2131-40.
•
TOTSUNE K., TAKAHASHI .K, OHNEDA M., ITOI K.,
MURAKAMI O., MOURI T. (1994). “C-type natriuretic peptide
in the human central nervous system: distribution and molecular
form.” Peptides 15: 37-40.
142
Bibliografia
•
TOTSUNE K., TAKAHASHI K., MURAKAMI O., SATOH F.,
SONE M., MOURI T. (1994). “Elevated plasma C-type
natriuretic peptide concentrations in patients with chronic renal
failure.” Clin Sci 87: 319-22.
•
TSUKAGOSHI H., SHIMIZU Y., KAWATA T., et al. (2001).
“Atrial natriuretic peptide inhibits tumor necrosis factor-α by
interferonγ-activated macrophages via suppression of p38
mitogen-activated protein kinase and nuclear factor–K B
activation.” Reg Pept 99: 21-29.
•
TSUTAMOTO
T.,
KANAMORI
T.,
MORIGAMI
N.,
SUGIMOTO Y., YAMAOKA O., KINOSHITA M. (1993).
“Possibility of down-regulation of atrial natriuretic peptide
receptor couplet to guanylate cyclase in peripheral vascular beds
of patients with chronic severe heart failure.” Circulation 87: 7075.
•
VATTA MS., PRESAS M., BIANCIOTTI LG., et al. (1996). “B
and C types natriuretic peptides modify norepinephrine uptake
and release in the rat hypothalamus.” Regul Pept 16: 175-184.
•
VATTA MS., PRESAS M., BIANCIOTTI LG., et al. (1997). “B
and C types natriuretic peptides modify norepinephrine uptake
and release in the rat adrenal medulla.” Peptide 18: 1483-1489.
•
VENTURA RR., GOMES DA., REIS WL., et al. (2002).
“Nitrergic modulation of vasopressin, oxytocin and atrial
natriuretic peptide secretion in response to sodium intake and
hypertonic blood volume expansion.” Braz J Med Biol Res 35:
1101-1109.
143
Bibliografia
•
VESELY DL. (2003). “Natriuretic peptides and acute renal
failure.” Am J Physiol Renal Physiol 285: F167-177.
•
VESELY DL., DOUGLASS MA., DIETZ JR., GOWER WR.,
MC CORMICK MT., RODRIGUEZ G., SHOKEN DD. (1994).
”Three peptide from the atrial natriuretic factor prohormon
amino teminus lower blood pressure and produce diuresis,
natriuresis and/or Kaliuresis in humans.” Circulation 90 (5):
1129-1140.
•
VOLLMAR AM. (2005). “The role of atrial natriuretic peptide in
the immune system.” Peptides 26: 1086-1094.
•
VOLLMAR AM., GERBES AL., NEMER M., SCHULZ R.
(1993). “Detection of C-type natriuretic peptide (CNP) trascript
in the rat heart and immune organs.” Endocrinology 134 (4):
1872-74.
•
VOLLMAR AM., KIEMER AK. (2001). “Immunomodulatory
and cytoprotective function of atrial natriuretic peptide.” Crit
Rev Immunol 21: 473-485.
•
VOLLMAR AM., SCHULZ R. (1995). “Expression and
differential regulation of the natriuretic peptides in mouse
macrophages.” J Clin Invest 95: -2450.
•
VOLLMAR AM., WOLF R., SCHULZ R. (1995). “Coexpression of the natriuretic peptides (ANP, BNP, CNP) and
their receptors in normal and acutely involuted rat thymus.” J
Neuroimmunol 57: 117-127.
144
Bibliografia
•
VOLLMAR AM., MYTZKA C., ARENDT RM., SCHULZ R.
(1988). “Atrial natriuretic peptide in bovine corpus luteum.”
Endocrinology 123: 762-767.
•
WALTHER T., KLOSTERMANN K., HERING- WALTHER
S., et al. (2003). “Fibrosis rather than blood pressure determines
cardiac BNP expression in mice.” Regul Pept 116: 95-100.
•
WASCHEK JA. (2004). “Developmental actions of natriuretic
peptides in the brain and skeleton.” Cell Mol Life Science 61:
2332-2342.
•
WEI CM., HUBLEIN D., PERRELLA MA., LERMAN A.,
RODEHEFFER R., McGREGOR CGA., EDWARDS WD.,
SCHAFF HV., BURNETT JC. (1993). “Natriuretic peptide
system in human heart failure.” Circulation 88: 1004-1009.
•
WOODARD
GE.,
ROSADO
JA.,
BRAWN
J.
(2002).
“Expression and control of C type natriuretic peptide in rat
vascular smooth muscle cells.” Am J Phisiol Reg Int Comp
Phisiol 282: R156-165.
•
WRIGHT SP., PRICKETT TC., DOUGHTY RN., FRAMPTON
C., GAMBLE GD., YANDLE TG., SHARPE N., RICHARDS
M. (2004). “Amino-terminal pro-C-type natriuretic peptide in
heart failure.” Hypertension 43: 94-100.
•
WU C., WU F., PAN J., MORSER J., WU Q. (2003). “Furinmediated processing of pro- C-type natriuretic peptide.” J Biol
Chem 278: 25847-25852.
•
YANDLE TG. (1994). ”Biochemistry of natriuretic peptides”. J.
Intern. Med. 235 (6): 561-576.
145
Bibliografia
•
ZANG Q., MOALEM J., TSE J., et al. (2005). “Effects of
natriuretic peptides on ventricular myocite contraction and role
cyclic GMP signalling.” Eur J Pharm 510: 209-215.
•
ZEIDEL ML. (2000). “Physiological responses to natriuretic
hormones.” In: FRAY JCS., GOODMAN HM (eds) Handbook
of physiology, Section 7, The endocrine system, Volume III:
Endocrine regulation of water and electrolyte balance. New
York, Oxford University Press, pp 410-435.
•
ZHIHONG Y., LUSCHER T. (2002). “Vascular endothelium.”
in: Lanzer P., Topol JE.; eds. Pan vascular medicine. Integrated
Clinical Management.
•
ZIVIN RA., CONDRA JS., DIXON RA., SEIDOH NG.,
CHRETIEN M., NEMER M., CHAMBERLAND M., DRONIN
J. (1984). ”Molecolar cloning and characterization of DNA
sequence encoding rat and human atrial natriuretic factor.” Proc
Natl Acad Sci USA 81 (20): 6325-6329.
146
RINGRAZIAMENTI
Vorrei ringraziare la Dott. Silvia Del Ry e la Dott. Daniela Giannessi
per l’arricchimento culturale che mi hanno fornito nel corso di questi anni
e che mi ha permesso di arrivare alla Laurea di Primo Livello e in seguito
alla Laurea Specialistica, con un bagaglio di conoscenze maggiormente
consolidato ed approfondito;
per la pazienza e la costanza dimostrate nell’insegnarmi le tecniche di
laboratorio che hanno accompagnato i miei studi;
per le opportunità che mi hanno concesso in un ambiente di ricerca così
qualificato e che mi hanno pemesso di introdurmi in modo “sereno ed
indolore” dal mondo teorico-universitario a quello reale e pratico del
lavoro;
ma soprattutto per l’insostituibile sostegno morale sempre vivo e presente
all’occorrenza, per l’umanità e la saggezza dimostrata, che mi hanno fatto
ritrovare, in un’equipe di lavoro, il calore di una famiglia allargata.
Ringrazio inoltre Maristella e Chiara per la loro preziosa collaborazione e
costante disponibilità, per avermi insegnato a capire cosa vuol dire essere
“un gruppo” e a collaborare in armonia, ma anche per avermi fatto
sorridere nei momenti più difficili ed essermi sempre state vicine.
Un grazie speciale al gruppo del Centro di Biomedicina Sperimentale della
Scuola Superiore Sant’Anna coordinato dal Prof. Fabio Recchia, che, mi
ha permesso di accostarmi in modo critico e responsabile ad un modello
animale di scompenso cardiaco cronico, rivelatosi poi fondamentale nella
validazione del nostro studio.
Ancora un ringraziamento a Valeria, la mia migliore compagna di
università e laboratorio, per i bellissimi momenti che abbiamo trascorso
insieme sostenendoci a vicenda, condividendo ogni successo ed
insuccesso e raggiungendo così un solido e duraturo rapporto d’amicizia.
Infine un ringraziamento particolare va alla mia mamma ai miei nonni, a
Matteo e a Patrizia e Fabio per essermi stati sempre vicini con affetto e
discrezione, dimostrandomi tutta la loro fiducia e permettendomi di
arrivare a questo traguardo per me così importante.



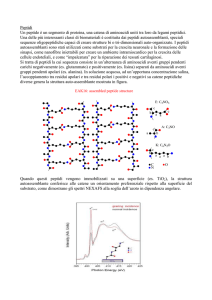


![Scompenso cardiaco- attività dell`Asl di Nuoro [file]](http://s1.studylibit.com/store/data/005106553_1-2acc9f03391e8aa6792037a95036da21-300x300.png)