Dispensa di Cardiologia e Cardiochirurgia
A.Fusco
1
Presentazione
Questa dispensa è stata creata con lo scopo di semplificare lo
studio della cardiologia ai fini dell’esame di Cardiologia per gli
studenti di Medicina.
Lungi dall’avere la pretesa di poter competere con i testi aggiornati
sull’argomento, la dispensa può però essere uno strumento di
studio valido per lo studente, oltre che un rapido mezzo di
consultazione di singoli argomenti.
Si è cercato di fornire uno schema completo di ogni singolo
argomento, dando cenni di epidemiologia, patogenesi, riassumendo
le principali caratteristiche cliniche, gli algoritmi diagnostici e le
terapie, comprese manovre chirurgiche. Vi sono inoltre cenni e
richiami di fisiologia, semeiotica, anatomia patologica.
Le nozioni sono state tratte dai migliori testi di cardiologia
consigliati e integrate con appunti delle lezioni.
Ne è risultata un’impostazione consona ad un apprendimento
approfondito e indirizzato ai fini dell’esame.
Con la speranza, ma anche la convinzione, che se usata
saggiamente possa essere un valido supporto per molti.
A.Fusco
2
Scompenso cardiaco
Definizione: detto anche insufficienza cardiaca, è la situazione fisiopatologica in cui il cuore non
è in grado di pompare una quantità di sangue adeguata alle richieste metaboliche
dell’organismo, oppure ci riesce solo tramite un forte aumento della pressione venosa
(aumentato ritorno venoso). Può essere causato da una ridotta portata e quindi meno sangue
pompato oppure da un aumento delle esigenze. Si hanno due conseguenze principali:
ipoperfusione periferica e congestione venosa. Se sono presenti sintomi è reale scompenso, se
il paziente è asintomatico si parla di disfunzione ventricolare. Si può avere una riduzione della
portata ossia disfunzione sistolica (ridotta capacità contrattile) oppure un difetto di
riempimento ossia disfunzione diastolica (ventricolo poco distendibile e meno compliance =
maggiore rigidità).
Epidemiologia: molto frequente, con 20 milioni di persone al mondo coinvolte. La prevalenza
totale è dell’1-2%, più comune negli uomini, per quanto vi sia in pratica un’equivalente numero
di donne affette data la maggiore aspettativa di vita. La sua incidenza aumenta infatti
progressivamente con l’età e colpisce quasi il 10% delle persone sopra i 65 anni, risultando sopra
questa età la prima causa di ricovero. Questo avviene poiché l’invecchiamento favorisce
condizioni come la l’ipertensione e la fibrosi. L’incidenza è in aumento (3-4 volte negli ultimi 25
anni) a causa della maggiore sopravvivenza dei pazienti affetti da IM (necrosi -> fibrosi ->
scompenso). Si può sviluppare in condizione di FE ridotta o preservata. La maggior parte delle
cardiopatie (specie coronaropatie) terminano nello scompenso. Lo scompenso diastolico è più
femminile e la sua incidenza aumenta con l’età (in età giovanile pochi scompensi diastolici, negli
anziani molti, forse a causa della comune ipertensione). L’ipertrofia ventricolare sinistra è più
comune nello scompenso diastolico così come IM e DM sono più spesso associati a scompenso
sistolico.
Eziologia: è in genere dovuto a condizioni che alterano la struttura o la funzione ventricolare,
quasi sempre si ha un’insufficienza miocardica che può essere dovuta a: 1) perdita anatomica o
funzionale di parte del tessuto contrattile (tipo IM), 2) compromissione diffusa delle fibre
miocardiche (cardiomiopatie) 3) sovraccarico cronico di pressione (ipertensione, stenosi) 4)
sovraccarico cronico di volume: per esempio per una quota di sangue rigurgitata a causa di
un’insufficienza valvolare. Queste quattro condizioni sono associate a portata ridotta,
tipicamente associata a cause cardiache. 5) scompenso ad alta gittata: in pratica una forma con
sovraccarico di volume perché determinata da condizioni che impongono un maggiore flusso
ematico. Principalmente sono dovute ad un aumento delle “esigenze” e pertanto a cause
extracardiache. Queste sono ad esempio: a) Ipertiroidismo: aumento del MB e della gittata
cardiaca, inoltre sintesi di diverse proteine contrattili, si ha tachicardia e fibrillazione atriale b)
Deficit di B12: come nel beri-beri o alcolismo cronico. Si notano sintomi neurologici e una
marcata vasodilatazione periferica che aumenta il ritorno venoso e pertanto la gittata (Starling).
Si può avere anche compromissione del metabolismo cardiaco e con l’alcol una cardiomiopatia
dilatativa. C) Fistole artero-venose: poichè vengono saltati i capillari, per migliorare la perfusione
il cuore aumenta la gittata (o portata). D) Anemia: per mantenere il trasporto di ossigeno c’è
aumento del flusso ematico. Si ha anche ipossia miocardica. E) altre condizioni: esercizio fisico
3
intenso, stress, dieta, infezioni, gravidanza. Si può avere scompenso anche per un lavoro acuto
improvviso del cuore come nell’ipertensione cardiaca o nel distacco di un lembo valvolare in
un’endocardite, così come a causa di un ostacolo improvviso al riempimento (tamponamento
cardiaco). Altra cosa è l’insufficienza circolatoria, ipoperfusione causata da altre componenti del
sistema circolatorio.
Patogenesi: Si distinguono un po’ artificiosamente (poiché spesso sono associate) due forme di
scompenso: sistolico o anterogrado (portata inadeguata) e diastolico o retrogrado (elevate
pressioni di riempimento) che sono rispettivamente un deficit di pompa o di distensione. Nel
25-40% la causa è diastolica. In una persona normale durante l’esercizio c’è molto riempimento
rapido e poca diastasi, poiché la pressione protodiastolica diminuisce e per il riempimento non
serve un aumento della pressione atriale. In una persona con scompenso cardiaco la pressione
proto diastolica in esercizio è quasi la stessa che a riposo e pertanto il riempimento avviene, ma
al prezzo di un notevole aumento della pressione atriale. I due scompensi possono coesistere e
l’uno può esitare anche nell’altro. Nella maggior parte dei casi entrambi i meccanismi sono
attivati in maniera variabile. Ad esempio lo scompenso diastolico provoca una riduzione
dell’afflusso di sangue all’atrio che causerà una ridotta gittata sistolica e dunque uno scompenso
anterogrado. Lo scompenso causa l’ipoperfusione periferica con minore irrorazione dei tessuti e
congestione venosa che comporta un aumento della pressione venosa la quale nei capillari
comporta una fuoriuscita di liquido dall’interstizio (cambiamento di equilibrio per la legge di
Starling in seguito all’aumento della pressione idrostatica) con conseguente formazione di
edemi.
Un’altra distinzione, anche questa prevalentemente didattica è tra scompenso destro e sinistro.
Nella maggior parte dei casi le cause di scompenso agiscono per lo più sulle cavità sinistre del
cuore perché: la pressione è più elevata nel circolo sistemico (è comune l’ipertensione e un
sovraccarico cronico di pressione sul ventricolo sinistro), le patologie valvolari acquisite sono più
frequenti nelle sezioni sinistre così come la cardiopatia ischemica (ventricolo sinistro). Il cuore
destro è quindi meno colpito a meno che non ci sia patologia polmonare (cuore polmonare) o
sin da subito uno scompenso globale. È una divisione abbastanza didattica, infatti spesso uno
scompenso sinistro comporta anche uno scompenso destro e comunque il miocardio dei due
ventricoli è separato da un setto comune (interventricolare) e le alterazioni biochimiche e
strutturali di uno finiscono per coinvolgere anche l’altro. Ha senso nei casi acuti o iniziali. I
sintomi sono però diversi: Sinistro difficoltà di scarico delle vene polmonari edema
polmonare. Destro difficoltà vene cave edemi periferici. Il cuore risponde ad una
riduzione della gittata con meccanismi di compenso. Si parla di scompenso quando questi sono
insufficienti, prima è solo disfunzione ventricolare.
Meccanismi di adattamento o rimodellamento: Alle eventuali alterazioni il cuore risponde con
un rimodellamento (principalmente ventricolare) che permette di raggiungere un nuovo
equilibrio (disfunzione ventricolare) che con il tempo può evolvere in scompenso e sintomi.
Legge di Frank-Starling: se la lunghezza della fibra miocardica aumenta, aumenta la forza di
contrazione (massima forza a 2,2 micron). Così nel cuore se aumenta il ritorno venoso e quindi il
volume telediastolico aumenta la gittata cardiaca (aumento volume tele diastolico -> il cuore si
dilata -> aumento forza di contrazione).
Legge di D & G.Hill: più basso è il post-carico maggiore è la velocità di accorciamento della fibra
e viceversa.
Legge di Pierre Laplace: lo stress di parete s=P*r/2h (P è la pressione endocavitaria, r il raggio,
ha lo spessore di parete). Pertanto un aumento di pressione o di volume (dimensioni) possono
aumentare lo stress di parete che come conseguenza comporta un maggiore consumo di
4
ossigeno e produzione di radicali liberi che inducono ipertrofia e fibrosi cardiaca. Per rispondere
all’aumentato stress di parete si può aumentare lo spessore di parete (l’ipertrofia-anche di
fibroblasti e quindi fibrosi- agisce in questo senso).
Ipertrofia miocardica: le fibre muscolari non potendo iperproliferare divengono ipertrofiche in
risposta a condizioni di maggiore carico. Lo stress ossidativo sarebbe il primum movens. Il TGF
beta pare essere il principale mediatore di ipertrofia e fibrosi. Vengono attivati una serie di geni
come quello per esprime l’isoforma beta della miosina, normalmente fetale (contrazione più
lenta ma con meno energia), o la pompa del calcio AT-dipendente (porta il calcio nel reticolo
sarcoplasmatico più lentamente). Vi è contemporaneamente una maggiore produzione di
collagene da parte dei fibroblasti, si ha fibrosi e minore distensibilità. Con l’aumento dello
spessore si cerca di mantenere costante lo stress di parete che aumenterebbe con l’aumentare
del raggio o della pressione. Nel caso in cui vi sia un sovraccarico cronico di volume si ha
ipertrofia eccentrica ossia con cellule che divengono più lunghe e con proliferazione in serie dei
sarcomeri che produce un aumento di raggio e di spessore (riduzione della gittata, scompenso
sistolico) Nel caso in cui vi sia un sovraccarico cronico di pressione si ha ipertrofia concentrica
ossia con cellule che divengono più grosse e con proliferazione in parallelo dei sarcomeri che
produce un aumento di spessore (aumento della pressione, scompenso diastolico). Dopo un po’
ipertrofia e fibrosi, che all’inizio sostengono il maggior carico, compromettono la funzione
cardiaca.
Meccanismi di adattamento extracardiaci: Aumento dell’estrazione dell’ossigeno: si ha in tutte
le forme di insufficienza cardiocircolatoria con aumento del 2,3 bifosfoglicerato negli eritrociti
che riduce l’affinità dell’Hb per l’O2 e pertanto ne facilita il rilascio nei tessuti (ma fa diminuire la
saturazione nel sangue arterioso). Attivazione dei barocettori: presenti nell’arco aortico e seno
carotideo, si attivano con il cambiamento di pressione e ne inducono la modifica ad esempio
attivando e il vago e inibendo il centro vasocostrittore del bulbo ( abbassando così la pressione,
ma nello scompenso comportano vasocostrizione). Non svolgono un ruolo a lungo termine nella
regolazione della pressione arteriosa. Attivazione del simpatico: comporta un aumento della
frequenza e contrattilità cardiaca (stimolazione adrenergica) e maggiore vasocostrizione (però
non omogenea, infatti organi nobili come cervello, reni e cuore hanno un controllo autonomo
delle resistenze vascolari). In sostanza ha un effetto inotropo e cronotropo positivo. L’aumento
della frequenza normalmente è legato anche ad un aumento della forza di contrazione (effetto
Treppe o Bowdich) però nei pazienti con scompenso questa relazione forza/frequenza si
deprime. Pur essendo una risposta del nostro organismo per riequilibrare il sistema, l’attivazione
adrenergica risulta essere dannosa, in modo evidente in termini di prognosi. Attivazione del
sistema renina-angiotensina: meno flusso ematico renale comporta rilascio di renina da parte
dell’apparato iuxtaglomerulare. Con la formazione di angiotensina II si ha vasocostrizione
arteriolare (in tutti i distretti) e produzione di aldosterone (recupero di sodio e acqua). AT1 è più
nei vasi dando vasocostrizione, AT2 è più nel cuore dando ipertrofia e fibrosi. Questo sistema
favorisce una maggiore perfusione degli organi vitali, ma porta anche un aumento del postcarico e quindi un maggiore lavoro cardiaco (aumento del flusso ematico) e per la legge di Hill
anche una minore velocità di contrazione. L’aumento del pre-carico, per la legge di Starling
favorisce una maggiore gittata almeno fino al raggiungimento di un plateau oltre il quale
l’aumento della volemia è dannoso). L’ipoperfusione induce il rena anche a produrre EPO che a
lungo termine aumenta il numero di eritrociti.
Sistemi di contro regolazione: Liberazione di ormoni e fattori neuroendocrini: servono per
contrastare il simpatico e il sistema renina-angiotensina, sono detti fattori natriuretici atriali
(ANP e BNP), sostanze vasoattive prodotte in risposta allo stiramento delle cellule atriali
(aumento del precarico) che inducono vasodilatazione ed eliminazione di sodio e acqua. ANP è
5
prodotto soprattutto nell’atrio, BNP più nel ventricolo. Nello scompenso c’è un notevole
aumento di BNP, ma pare che non basti in quanto le esigenze sono maggiori e perché aumenta
soprattutto il pro-BNP, fisiologicamente meno attivo. Le prostaglandine inducono
vasodilatazione soprattutto a livello delle arteriole renali. Sottoregolazione recettoriale: o down
regulation, per bilanciare l’effetto dei fattori ormonali vi è un sistema di riduzione del numero e
della sensibilità di recettori stimolati in modo prolungato. Altro: Endotelina: scarso effetto
fisiologico, comporta vasocostrizione e fibrosi. TNF alpha: diminuisce inotropismo, porta
apoptosi e cachessia.
Clinica: è possibile riscontrare una sintomatologia varia, intensa o sfumata.
Sintomi respiratori: Dispnea: è il sintomo più comune oltre all’affaticabilità e consiste in una
sensazione di sforzo a respirare e mancanza di respiro (fame d’aria). È dovuta alla congestione
venosa polmonare che comporta accumulo di liquidi interstiziali e intra-alveolari con attivazione
dei recettori J iuxtacapillari (che provocano una respirazione rapida e superficiale), aumento
delle resistenze delle vie aeree, riduzione della compliance polmonare e affaticamento dei
muscoli respiratori (danneggiati anche dall’ipoperfusione periferica). È meno caratteristica nello
scompenso destro e insufficienza tricuspidale. All’inizio si manifesta solo sotto sforzo poi anche
a riposo. Ortopnea: è una dispnea che si manifesta se il paziente è in posizione supina, ma non
se è in posizione eretta perché in questo caso il sangue è nella circolazione e negli arti inferiori.
Quando è sdraiato c’è un maggiore ritorno venoso e si ha ipertensione polmonare. Spesso si
manifesta con tosse notturna e con la necessità di dormire con molti cuscini. Dispnea
parossistica notturna: episodi acuti di grave mancanza del respiro e di tosse che si manifestano
la notte, in genere dopo 1-3 ore di sonno e costringono il malato ad alzarsi con fame d’aria e ad
esempio andare alla finestra. Si possono avere tosse e ansimi e a differenza dell’ortopnea spesso
non basta assumere la posizione eretta. È dovuta al prevalere nel sonno del vago sul simpatico e
alla depressione del centro del resprio oltre che all’aumentato ritorno venoso per la posizione
supina. Si associa all’asma cardiaca (affanno secondario a broncospasmo). Respiro di CheyneStokes: spesso nella fase finale dello scompenso, si ha una minore sensibilità alla PCO2. Si ha
ciclicamente apnea seguita da iperventilazione. Nei casi ancora più gravi si possono avere crisi e
arresto del respiro. Edema polmonare: si manifesta quando c’è un’importante congestione
polmonare. Si ha dispnea e tosse con escreato schiumoso. Classificazione funzionale dei
pazienti con scompenso cardiaco di NYHA (associazione cardiologica di New York): Classe I:
pazienti asintomatici per normale attività (sani) II: pazienti che stanno bene a riposo, ma con
sintomi (dispnea per lo più) per sforzi di ordinaria intensità. III: bene a riposo, sintomi per sforzi
lievi. IV: sintomatici anche a riposo.
Sintomi urinari: Nicturia: più del 66% del volume urinario è notturno. Di giorno l’attività fisica
accentua l’ipoperfusione renale, la notte si smaltiscono i liquidi e aumenta la gittata. Nelle fasi
avanzate si ha costante ipoperfusione e quindi oliguria.
Altri sintomi: sintomi digestivi come nausea vomito e gonfiore sono dovuti alla stasi della porta
e delle mesenteriche. Per riduzione del flusso cerebrale (specie anziani) si può avere
confusione, cefalea e insonnia. Per ipoperfusione muscolare si ha debolezza e facile
affaticabilità. Ci può essere per ipossia cardiaca anche un maggiore rischio di aritmie.
Esame obiettivo: Il paziente si presenta affaticato, con difficoltà a respirare e necessità di
mantenere una posizione verticale. Cuore: ingrandimento dell’aia cardiaca (si ha un itto
prolungato e palpabile su due interspazie per ipertrofia del ventricolo sinistro). A causa
dell’ipertono simpatico si ha tachicardia e ritmo di galoppo. Si ha infatti un terzo tono
protodiastolico. Il polso è piccolo, frequente e a volte alternante (pulsazione forte poi debole).
Nelle fasi finali si ha bradicardia. La PA sistolica è ridotta (meno gittata) e la diastolica è
6
aumentata (aumento resistenze periferiche). La PVC è anche sopra i 20. Cute: la cute appare
fredda e pallida per la vasocostrizione. Possono comparire edemi, all’inizio con fovea. Poi
possono indurirsi e pigmentarsi (specie alle caviglie). Nei casi gravi si ha anasarca. Gli edemi
sono nelle parti declivi e se il paziente è a letto nella regione presacrale. Giugulari: con il
paziente semidisteso, a 45° con la testa sollevata, si nota un anormale riempimento delle
giugulari, che collabiscono più in alto del normale (stima della PVC= altezza sangue giugulari
dallo sterno + 5cm). Si ha forte distensione delle giugulari con pressione prolungata
all’ipocondrio destro (riflesso epatogiugulare). Torace: si possono avvertire rantoli crepitanti alle
basi polmonari, e sempre a causa dell’edema interstiziale e la congestione bronchiale si possono
avere ronchi e asma cardiaca. A volte si ha un versamento pleurico (spesso bilaterale, se
unilaterale per lo più destro). Addome: per aumento della PVC si può avere epatomegalia
(fegato anche dolente) e anche ascite (tardiva). Con il tempo anche atrofia e insufficienza
epatica.
Diagnosi: Si basa all’inizio sul quadro clinico, valutando i sintomi in ordine di importanza.
Sintomi e segni maggiori e minori: Criteri di Framingham: Maggiori (cioè specifici di
scompenso): DPN e ortopnea (che è in pratica lo stesso), rantoli, distensione giugulari e REG +,
cardiomegalia, edema polmonare, ritmo di galoppo S3, aumento PVC. Minori (cioè aspecifici):
edemi (declivi), epatomegalia, dispnea da sforzo, tosse notturna, versamento pleurico, forte
perdita di peso dopo diuretici (da edemi). Inoltre c’è pallore, sudorazione, tachicardia,
affticabilità, nicturia, segni di ridotto flusso cerebrale. C’è bisogno che siano soddisfatti almeno
un criterio maggiore e due minori.
Indagini: Analisi di laboratorio: si guardano elettroliti che si alterano a causa di
iperaldosteronismo secondario (iponatriemia da trattenimento di liquidi-non bisogna
somministrare sodio, ma togliere i liquidi- e ipokaliemia da eccesso di diuretici), azoto ureico,
creatinina, enzimi epatici (aumento AST e ALT se c’è epatomegalia e aumento del PT per
riduzione di trombina che sono segni epatici aspecifici), urine, ma anche glicemia, lipidi, ormoni
tiroidei.
ECG: può verificare la presenza di anomalie, ipertrofia, blocchi. Radiografia del torace: si può
notare cardiomegalia e edema interstiziale dei polmoni, strie di Kerley (stasi linfatica) e segni di
versamento pleurico.
Ecocardiografia: può valutare alcune cause di cardiopatia e soprattutto la funzione e lo stato
ventricolare (si vedrà dilatazione, ridotta frazione di eiezione - % di gittata sul volume tele
diastolico -, ipertrofia). Si possono cercare i fattori natriuretici atriali.
Esistono anche alcuni test da sforzo: Una misura “soggettiva” è il 6 minutes walk test: si fa
camminare il paziente il più veloce che può per 6 minuti e si vede quanta strada fa (dipende
molto dalla compliance). Più indaginoso ma più oggettivo è il test del massimo consumo di O2:
la % di O2 che una persona inala dipende dalla % presente nell’aria ed è pertanto costante, la %
di CO2 dipende da quanto ossigeno consumiamo perché è il prodotto del metabolismo aerobico
dell’ossigeno (con fosforilazione ossidativa). Questo vale a riposo così come durante un’attività
fisica moderata, ma quando i nostri muscoli sono sollecitati a tal punto che il nostro cuore non è
più in grado di dar loro un apporto di sangue adeguato a fornire tutto l’ossigeno di cui hanno
bisogno usano la glicolisi anaerobica (e poi la fermentazione con formazione di acido lattico). In
questo caso CO2>O2. Si applica una maschera ad un paziente posto sotto sforzo e si misura
l’entrata di O2 e l’uscita di CO2. Si nota un aumento di O2 sino ad un livello di plateau oltre il quale
il cuore non è più in grado di pompare abbastanza sangue (non aumenta più la portata), infatti il
consumo di ossigeno è funzione diretta della portata cardiaca. Misuriamo dunque il massimo
consumo di O2 al plateau e valutiamo la portata. Questo test può servire anche a regolamentare
7
le liste per il trapianto di cuore. Una persona sana ha un consumo di ossigeno massimo pari a
30-40 ml/kg/min (anche di più) e un consumo a riposo di circa 7-8 ml. In base all’ossigeno
consumato si possono tracciare delle curve di sopravvivenza ad un anno. Per i pazienti con
consumo basso, ma ancora >18 la mortalità ad un anno è normale; 14-18= moderatamente
aumentata; 10-14= 25%; <10= 75% (è quasi come il consumo a riposo, insomma questi pazienti
non sono in grado di aumentare affatto la portata). In lista di trapianto entrano pazienti con
consumo massimo <14.
Algoritmo diagnostico: Anamnesi (criteri maggiori e minori) sintomi compatibili con
scompenso. Si cercano prima cause extracardiache, se ci sono si curano. Se non ci sono
valutazione cardiaca: si cercano cardiopatie (pericarditi, valvulopatie, etc.) valutazione della
frazione di eiezione (funzione sistolica): se >40% scompenso diastolico ; se<40%
scompenso sistolico. Diagnosi differenziale: ECG può aiutare a distinguere ipertrofie da IM,
bradi aritmie, pericarditi. Le analisi di laboratorio (sangue, urine, creatinina) possono aiutare ad
escludere altre cause (anemia: scompenso da ridotto trasporto di ossigeno).
Prognosi: Dipende da molti fattori e patologie associate. Dipende anche dalla classificazione
NYHA al momento della diagnosi . In genere comunque non è buona con probabilità di morte in
4-5 anni maggiore del 50% più alta negli uomini che nelle donne, nonostante il miglioramento
della terapia.
Terapia: Dipende dallo stato del paziente. In generale bisogna rallentare il rimodellamento
ventricolare e la progressione della malattia. È comunque fondamentale la prevenzione dato che
in genere la patologia è preceduta da un lungo stato di disfunzione ventricolare. Le misure
generali (terapia igienico-dietetica) prevedono: test di screening e controlli periodici; sono da
evitare stress fisici e psicologici, ma un’attività fisica modesta negli stati non avanzati è utile per
aumentare la tolleranza alo sforzo e sfavorire trombi; la dieta deve essere ipocalorica per il
sovrappeso e comunque con pochi sali (3-4g di sodio e non 8-10).
Terapia farmacologica: Per la terapia dello scompenso si può immaginare il cuore in difficoltà
come dei cavalli che trainano una carrozza in salita e che non ce la fanno.
Cavalli = cuore; Salita = Post-carico (resistenze periferiche per lo più, pressione arteriosa);
Carrozza con persone a bordo = Pre-carico (volemia, ossia quantità di liquidi in circolo). Noi
siamo il nocchiere. Possiamo:
1) Frustare i cavalli = farmaci inotropo-positivi. Inotropo positivi: Un tempo erano molto
utilizzati questi farmaci come la digitale che aumentano la contrattilità. Essi hanno però effetti
collaterali come nausea, vomito e aritmie e importanti interazioni. Non servono per trattare lo
scompenso cronico, magari solo in casi di emergenza. La digitale (dalla pianta digitalis prupurea,
si usa da secoli per curare “l’idropisia” cioè edemi da scompenso) trova leggero impiego per il
suo effetto vago mimetico (pur essendo intoropa). Si è visto che gli inotropo-positivi aumentano
la mortalità a lungo termine.
2) Scegliere la strada più lunga, ma meno ripida = vasodilatatori: ACE-inibitori: (tipo captopril)
in pazienti con ridotta FE sono fondamentali. Riducono il post-carico (cioè riducono la salita).
Bloccano la conversione in angiotensina II e upregolano la bradichinina. Possono essere
inefficaci se non si è risolta la ritenzione idrica. Come effetti collaterali ci può essere ipotensione
e aumento azotemia. Il rafforzamento delle chinine può dare angioedema e tosse non
produttiva. I pazienti intolleranti possono fare uso di ARB (o sartani, tipo valsartan) cioè inibitori
del recettore dell’angiotensina II. L’impiego di ACE inibitori e sartani ha diminuito la mortalità di
¼.
3) Facciamo rallentare i cavalli = Beta-bloccanti: (cavedilolo, bisoprololo) Riducono il post-carico
(diminuiscono il lavoro rallentando la frequenza e la diminuendo la gittata) e migliorano la
8
sintomatologia del paziente scompensato, che in buona parte è dovuta all’ipertono adrenergico.
Sono inotropo negativi. Questo può sembrare un controsenso in pazienti con ridotta gittata (con
scompenso, cioè che non ce la fanno) eppure la stimolazione adrenergica è un fattore
prognostico negativo. I vantaggi pertanto superano gli svantaggi. Devono però essere dosati con
cura perché possono portare peggioramento della ritenzione idrica per mancanza del supporto
adrenergico a cuore e circolazione e anche bradicardia, quindi si usano solo nei pazienti stabili e
con un aumento lento della posologia.
4) Facciamo scendere le persone = Diuretici: Diminuiscono il pre carico (scendono le persone =
meno liquidi). Diuretici d’ansa (furosemide, torsemide) e tiazidici (idroclorotiaziede). Quelli
d’ansa agiscono sull’ansa di Henle, i tiazidici agicono sul tubulo contorto distale. I diuretici d’ansa
hanno un maggiore effetto (che consiste principalmente nella riduzione del pre-carico e
pertanto riduzione dei sintomi congestizi e accumulo di liquido negli interstizi), i tiazidici
possono a volte essere associati se persiste ritenzione idrica (ma sono meno potenti e inefficaci
in caso di insufficienza renale). Principale effetto collaterale è l’ipokaliemia (aumento del rischio
di aritmie) e il peggioramento dell’azotemia. Bisognerebbe sempre iniziare con bassi dosaggi e
poi somministrare dosi crescenti fino a stabilizzazione della diuresi. Si usano solo nel paziente
sintomatico con ritenzione idrica. Antagonisti dell’aldosterone: tipo spironolattone, sono
diuretici risparmiatori di potassio. Nei pazienti a lungo a letto si fa uso anche di anticoagulanti e
antiaggreganti.
Complicanze: le più comuni sono aritmie e edema polmonare acuto.
Aritmie: la fibrillazione atriale è molto comune in pazienti scompensati. Il farmaco di prima
scelta per ripristinare il ritmo sinusale è l’amiodarone (antiaritmico di classe III). La morte
improvvisa per fibrillazione ventricolare rappresenta quasi il 50% delle cause di morte (il resto
per lo più muore per insufficienza della pompa cardiaca) pertanto bisogna si possono usare i
defibrillatori cardiaci impiantabili (ICD) in pazienti con NYHA II-III sotto terapia. In pazienti con
ritmo sinusale può essere utile (NYHA III-IV) un pacing biventricolare (terapia di
risincronizzazione cardiaca, CRT) che permette di evitare una condizione come la contrazione
ventricolare asincrona che comporta un QRS>120 ms.
Edema polmonare acuto: il paziente è agitato con fame d’aria e ansia, dispnoico (dispnea spesso
improvvisa), tachipneico, tachicardico con diastolica aumentata e differenziale diminuita, cute
fredda, sudata e pallida ed estremità cianotiche. Si possono avere su tutti campi polmonari
rantoli inspiratori e tosse con escreato schiumoso e/o roseo. Si ha ipossia e acidosi,
compromissione anche cardiaca, attivazione adrenergica, aumento resistenze e ulteriore
compromissione cardiaca.1
1
Edema polmonare acuto: EPA, è un aumento di liquidi nello spazio extravascolare del polmone
per trasudazione o essudazione di liquido sieroematico nell’interstizio, alveoli o bronchioli
polmonari. Esiste un edema polmonare cardiogeno (descritto su) e un edema polmonare non
cardiogeno. I meccanismi che possono comportare EPA sono tutti quelli che variano le forze
della legge di Starling o le condizioni della parete capillare. Un aumento del flusso può infatti
essere dovuto ad aumento della pressione nei capillari (pressione capillare di incuneamento
polmonare elevata è pressione in atrio sinistro o telediastolica ventricolare, in genere modificata
nell’EPA cardiogeno), riduzione nella pressione oncotica del plasma, ma anche aumento della
permeabilità della parte capillare e insufficiente drenaggio linfatico. EPA cardiogeno: è associato
a scompenso sinistro ed è dovuto ad aumento della pressione idrostatica capillare. È più
frequente ed è spesso correlato ad eventi acuti, come IMA (almeno 85%), ma anche valvulopatie
e cardiomiopatie. EPA non cardiogeno: può essere dovuto a diminuzione della pressione
9
L’IC con FE conservata non ha uno schema terapeutico ben definito e si tende per lo più a curare
la patologia sottostante. L’IC acuta è una condizione di emergenza in cui si hanno alterazioni
emodinamiche, principalmente: elevata pressione di riempimento del VS (detto dry o wet) e
ridotta gittata cardiaca (con aumento delle resistenze periferiche, detto warm o cold). I pazienti
possono presentarsi con entrambe le alterazioni o solo una delle due (warm and wet, cold and
wet, etc…). La terapia consiste nell’uso di vasodilatatori (arteriosi o venosi per ridurre pre-carico
o post-carico) come la nitroglicerina, e agenti inotropi come la dobutamina.
osmotica (malattie renali, enteropatie, malnutrizione) o ad alterazione della membrana alveolocapillare (ARDS). vi è anche un EPA multifattoriale.
Diagnosi dell’edema polmonare acuto: esame clinico (la clinica è quella già descritta),
emogasanalisi (ipossiemia e all’inizio ipocapnia da iperventilazione, poi ipercapnia). Radiografia
al torace: segni di congestione polmonare e opacità nei campi superiori o anche confluenti
periilari ad ali di farfalla. Altri esami come ECG e laboratorio possono far luce sulle cause.
Terapia: EPA non cardiogeno: trattare patologia di base. Nel cardiogeno: far sedere il paziente,
somministrare nitroglicerina ed inotropi come dobutamina, un diuretico d’ansa (furosemide),
ossigeno (PO2 almeno >60) ed eventualmente amminofillina (vasodilatatore) se c’è
broncospasmo. Morfina per dominare l’ansia e la vasocostrizione adrenergica. Eventuale salasso
ed emodialisi.
10
Ipertensione polmonare e cuore polmonare
Ipertensione polmonare
Definizione: IP, è caratterizzata da un cronico incremento della pressione in arteria polmonare
(PAP) e incremento delle resistenze polmonari (PVR) che inducono ipertrofia e dilatazione del
ventricolo destro. È definita da una pressione arteriosa media >25mmHG a riposo (valori medi
12-16mmHg), >30 mmHg durante lo sforzo, o una PAP sistolica >35mmHg (picco sistolico
normale 18-25 mmHg). L’IP si definisce lieve quando la PAP media è tra 19-25, moderata se 2640mmHg, severa se >40mmHg. L’IP nella sua forma arteriosa è un’affezione rara (5/100000), ma
l’IP in senso generale può essere associata ad affezioni cardiache e respiratorie croniche molto
comuni, pertanto la sua incidenza è decisamente superiore.
Classificazione: si distingue un’ipertensione polmonare arteriosa, un’ipertensione associata a
malattie del cuore sinistro (che interessano atrio o ventricolo, valvulopatie), un’ipertensione
associata a malattie polmonari e/o ipossiemia (BPCO, malattia interstiziale polmonare, sleep
apnea, ipoventilazione alveolare), un’ipertensione dovuta a malattia trombotica cronica o
embolia, e miscellanea (da sarcoidosi, tumori e adenopatie comprimenti i vasi polmonari, etc).
Ipertensione polmonare arteriosa: PAH, 1-2 casi per milione all’anno. Si distingue in:
1) Idiopatica (IPAH): cause ignote, forse sono concausa condizioni congenite.
2) Familiare (FPAH): ereditarietà autosomica dominante con penetranza incompleta. Nel 50%
dei casi di FPAH e nel 25% di IPAH ci sono mutazioni su di un gene sul cromosoma 2 che codifica
per il recettore BMPR2 per il fattore di crescita TGF-, che normalmente inibisce la
proliferazione dell’endotelio e della muscolatura liscia vascolare, per cui la sua alterazione
incrementa le resistenze vascolari periferiche e la pressione arteriosa polmonare.
3) Associata a fattori di rischio o malattie (APAH): le condizioni che facilitano lo sviluppo della
malattia sono: Sindrome di Eisenmerger: sindrome causata da difetti congeniti cardiaci che
inizialmente comportano uno shunt sinistro-destro con seguente aumento delle pressione
nell’arteria polmonare e delle resistenze periferiche con sovraccarico ventricolare destro, fin
quando le resistente polmonari non superano le sistemiche e lo shunt si inverte (causando
cianosi naturalmente). Cirrosi ed ipertensione portale: questi pazienti hanno resistenze
polmonar più basse che nelle altre forme, vi è scarsa saturazione di ossigeno. Collagenopatie:
LES, AR, Sjögren, sclerosi sistemica con CREST (16% di CREST presenta IP). Altre cause sono HIV,
alcuni farmaci e tossine, fattori di rischio come obesità, gravidanza ed ipertensione sistemica,
tireopatie, glicogenosi, emoglobinopatie, malattie croniche mieloproliferative, splenectomia.
4) Associata a malattie che coinvolgono vene o capillari polmonari: malattia veno-occlusiva
polmonare, emangiomatosi capillare polmonare, ipertensione polmonare persistente del
neonato. Inserite in classificazione perchè presentazione clinica e fattori di rischio sono comuni.
Patogenesi: in parte ancora ignota. Sicuramente sono coinvolti: Vasocostrizione polmonare:
soprattutto nelle fasi iniziali dell’ipertensione, forse causata da alterazione della funzione e
ridotta espressione dei canali del K+ voltaggio dipendenti. nelle cellule muscolari lisce vascolari.
Si ha un ridotto efflusso di K+ con relativa depolarizzazione, apertura dei canali del Ca 2+ voltaggio
dipendenti e quindi entrata di Ca 2+, contrazione e relativa vasocostrizione. Si ha inoltre minore
vasodilatazione a causa della ridotta produzione di sostanze vasodilatatrici come NO; PGI 2,
endotelina, che causa anche un rimodellamento vascolare (che coivolge tutti gli strati della
11
parete vacolare e le cellule endoteliali, muscolari lisce e fibroblasti). Infiammazione: si è notato
nei pazienti con PAH un aumento delle citochine infiammatorie, e alterazione del rilascio della
serotonina da parte delle piastrine (che induce vasocostrizione e iperplasia del cellule muscolari
lisce). Importante è anche la trombosi a carico del microcircolo. Questi fattori, associati ad un
danno vascolare polmonare (che comporta disfunzione endoteliale, attivazione di cellule
infiammatorie e piastrine, disfunzione delle cellule muscolari lisce) scatenato da predisposizione
genetica e fattori di rischio, sembrano quelli che fanno progredire verso l’ipertensione.
Anatomia patologica: nelle varie forme il quadro istologico è simile. Si ha ispessimento
dell’intima, e ipertrofia della media (muscolarizzazione) con aumento delle cellule muscolari,
aumento di fibroblasti e matrice, ispessimento avventizia e formazione di lesioni plessiformi con
dilatazione vascolare e numerosi canali vascolari con aspetto a bulbo di cipolla.
Fisiopatologia: L’aumento delle resistenze vascolari causa alterazioni: Respiratorie: si ha
aumento delo spazio morto funzionale (vi sono unità ad alto rapporto v/p) e limitazione nella
diffusione alveolo capillare con conseguente ipossiemia e ipocapnia con aumento della
ventilazione necessaria ad eliminare CO2. Cardiache: ipertrofia delle sezioni destre con aumento
della pressione di riempimento del ventricolo destro, ipertensione atriale destra e stasi del
circolo venoso sistemico. Può esserci un progressivo deterioramento della portata sistemica con
ipotensione ed ipoperfusione dei microcircoli renale, coronario e cerebrale (conseguenze
sistemiche).
Clinica: all’inizio asintomatica, comparsa dei sintomi in media dopo due anni. Si ha: dispnea da
sforzo, sintomo freuquente all’esordio, poi anche a riposo. Si può avere astenia, angina, sincope
(40%) in rapporto a diminuzioni della portata cardiaca. Ortopnea e DPN suggeriscono
ipertensione polmonare (da scompenso sinistro). Eventuali segni tipo fenomeno di Raynaud e
artralgie indicano connettivopatie. L’anamnesi può orientare verso una sindrome ostruttiva da
apnee notturne (apnee, russamento, sonnolenza, cefalee al mattino). Si può utilizzare una
classificazione dello stato funzionale (riadattamento di quella del NYHA per scompenso sinistro).
Classe I: no limitazioni nell’attività fisica ordinaria. II: sintomi di dispnea e fatica con attività fisica
ordinaria. III: sintomi con attività fisica minore dell’ordinaria. IV: sintomi anche a riposo.
All’esame obiettivo nel 90% dei pazienti c’è un’accntuazione del II tono sulla polmonare dovuta
all’incremento della pressione arteriosa polmonare, soffio diastolico da insufficienza in area
polmonare, soffio sistolico da rigurgito tricuspidalico per l’eventuale dilatazione destra. Anche
epatomegalia, edemi, distensione delle giugulari a causa dello scompenso destro.
Diagnosi: dopo l’anamnesi e l’esame obiettivo le indagini strumentali prevedono: ECG: poco
sensibile e specifico, ci può essere ipertrofia ventricolare destra. Radiografia dl torace: nel 90%
dei casi dilatazione dei vasi polmonari in regione ilare con aspetto ad albero potato dei vasi
periferici. Può evidenziare patologie responsabili di un’IP secondarie. Ecocardiografia transtoracica: TTE, forte correlazione con I risultati ottenuti con cateterismo cardiaco destro.
Misurando la velocità di reflusso tricuspidalico si stima la pressione sistolica in arteria polmonare
PAPs che è uguale alla pressione sistolica nel ventricolo destro (RVSP) in assenza di ostruzioni
dell’arteria polmonare. RVSP= 4v2+RAP. La RAP è la pressione nell’atrio destro considerata come
valore standard di 5mmHg oppure stimata con doppler in base alla distenzione delle giugulari
nel respiro spontaneo. Un collasso <50% o >50% pare indichino RAP<10mmHg o >10mmHg (per
non sbagliare stima usando solo un valore fisso). Valori di PAPs tra 36 e 50 (ancora ipertensione
lieve) corrispondono a RVSP di 2,8-3,4m/s. L’età e il BMI possono comportare aumenti della
12
PAPs (6% dei soggetti sani con età >50 e BMI>30PAPs>40). Esami funzionali respiratori:
spirometria e emogasanalisi escludono patologie polmonari o bronchiali. Si ha una riduzione
della diffusione alveolo capillare DLCO (per le alterazioni di capillari e piccole arterie) mentre
capacità vitale forzata e indice di Tiffenau sono normali. Riduzione di DLCO e incremento del
gradiente transtricuspidalico sono segni di IP iniziale. La PaO 2 è un po’ ridotta così come pal PCO2
per iperventilazione. Nelle connettivopatie risuzione isolata di DLCO. Se all’IP è associata fibrosi
polmonare ci può essere indice di Tiffenau alto. Polisonnografia per valutare le apnee notturne.
Scintigrafia ventilo-perfusiva polmonare: molto sensibile e specifica (oltre il 90%) per
distinguere tra IPAH e IP post-TEP (nella prima è normale, ella seconda difetti perfusivi). TC: un
aspetto a vetro smerigliato con ispessimento dei setti interlobulari con adenopaite o velature
pleuriche indica malattia polmonare veno-occlusiva. TC con mezzo di contrasto: distingue IP
arteriosa e post-TEP (si vedono I trombi). Test del cammino in 6 minuti: 6MWT, correla con la
prognosi. Cateterismo cardiaco destro: glod standard per individuare IP e valutarne la severità.
Bisogna valutare PAP (s,d e media, >25mmHg), RAP, PWP (pressione di incuneamento capillare
≤15mmHg per istinguere IP arteriosa e venosa), portata cardiaca e test di vasoreattività a
vasodilatatori (caduta dei valori di PAPm di almeno 10mmHg. BNP: prodotto dai ventricoli
quando sottoposti a sovraccarichi di volume e pressione. Aumenta nell’IP perchè il ventricolo
destro è sottoposto a sovraccarichi di pressione. I valori plasmatici di BNP correlano con lo stato
funzionale (NYHA e 6MWT), valori pressori e prognosi. Altri esami sono l’angiografia (TEP),
ecografia addome (IP portale), dosaggio anticoripi nelle collagenopatie.
Terapia: prima si riteneva non ci fosse terapia. Si possono usare diuretici (furosemide) per
riduerre il precarico, anticoagulanti per evitare fenomeni di trombosi in situ, calcio antagonisti
se il test acuto di vasodilatazione è positivo (si rischia vadoilatazione sistemica e morte). Farmaci
che hano effetti su mortalità e qualità di vita sono I prostanoidi (epoprostenolo) e altri
vasodilatatori (antagonisti recettore endotelina, adenosina, sildenafil).
Cuore polmonare
Definizione generale: dilatazione o ipertrofia del ventricolo destro in risposta ad aumento del
post-carico (ipertensione polmonare) causata da affezioni del parenchima polmonare, della
gabbia toracica e del controllo respiratorio. Le patologie che possono causare CPC sono
vasculopatie polmonari (ipertensione polmonare primitiva, tromboembolia, arteriti), patologie
neuromuscolari e deformità ossee, malattie polmonari che alterano gli scambi gassosi (malattie
parenchimali ostruttive quali brochite cronica eed enfisema polmonare e restrittive quali deficit
neurologici e muscolari, obesità, ostruzione delle vie aeree, fibrosi). Inoltre ostruzioni,
infiammazioni, compressioni dei vasi polmonari e sindromi da ipoventilazione alveolare cronica.
Si definisce ipertensione polmonare (da lieve in su) un aumento della PAP (pressione arteria
polmonare) oltre i 20mmHg. Quest’aumento determinerà modificazioni del ventricolo destro, le
quali possono essere acute o lente a seconda che il sovraccarico di pressione sia rapido (come
nell’embolia polmonare, cuore polmonare acuto) o lento (cuore polmonare cronico). Spesso si
usa cuore polmonare come sinonimo di cuore polmonare cronico. Ipertensione polmonare e
cuore polmonare non collimano sempre in quanto possono esserci condizioni cardiache alla
base dell’ipertensione polmonare (scompenso sinistro, cardiopatie congenite) e anche
un’ipertensione non così grave da causare alterazioni del VD. Non ci può essere cuore
polmonare senza ipertensione.
13
Cuore polmonare cronico
Definizione: CPC, consiste nel 10-20% delle ospedalizzazioni cardiache in età adulta, e il 40% dei
pazienti con BPCO (con VEMS<1L) presenta cuore polmonare.
Patogenesi: il ventricolo destro, a differenza del sinistro (ellissoidale e concentrico) ha la forma
di una piramide triangolare con gli osti arterioso e venoso alla base. Ha un tratto di afflusso
detto seno e uno di efflusso detto infundibulo o cono. La massa muscolare del VD è 1/6 del VS, il
VD è più dilatabile e pertanto mantiene bassa la pressione venosa, ma ha scarso adattamento al
sovraccarico pressorio (accorciamento limitato delle fibre). Da qui la frase: “il ventricolo sinistro
è una camera a pressione, il ventricolo destro è una camera a volume”. Il post-carico del VD
dipende dalle resistenze dei vasi polmonari e dalla compliance delle arterie polmonari (70% del
carico per rispondere al carico meccanico delle geometria ventricolare e alle resistenze vascolari
polmonari, il 30% per distendere le arterie elastiche polmonari). Le patologie che causano
ipertensione polmonare possono anche causare cuore polmonare. Normalmente, già in
inspirazione si ha aumento della pressione tele diastolica del ventricolo destro a seguito del
maggiore ritorno venoso e della minore capacità Del VD rispetto al VS di sfruttare la legge di
Starling. I fattori patogenetici più importanti sono vasocostrizione arteriolare (da ipossia
alveolare), riduzione della superficie del letto-arterioso capillare (patologia respiratoria
cronica), rimodellamento della parete vascolare con ipertrofia e fibrosi. La circolazione
polmonare però è a bassa resistenza ed infatti il gradiente pressorio tra arteria polmonare
(15cmH2O) ed atrio sinistro (10) è di soli 5cmH 2O, eppure consente il transito dell’intera gittata
cardiaca (5L/min, anche il triplo sotto sforzo). Anche in condizioni di sforzo la PAP aumenta di
poco perché sono possibili grandi variazioni del letto capillare (vi è una grande riserva di arterie
chiuse a riposo), e persino con un solo polmone la pressione non presenta grosse variazioni.
Pertanto nella maggior parte dei casi, più che modificazioni anatomiche dei vasi, è la
vasocostrizione (in genere secondaria ad un’ipossia alveolare che in genere avviene a seguito di
un’ipoventilazione, per compenso, cioè alveoli meno ventilati vengono meno perfusi) ad essere
determinante. Altri fattori determinanti oltre vasocostrizione da compenso, aggravata da
ipercapnia e acidosi (esaltano la vasocostrizione) sono: aumento della viscosità del sangue
(poliglobulia, a seguito di produzione di eritropoietina da parte del rene), aumento della portata
cardiaca (da compenso), liberazione di ormoni e citochine (aumentano le sostanze
vasodilatatrici e diminuiscono i vasocostrittori). Alcuni fattori come il fumo (sposta l’equilibrio
verso agenti vasocostrittori e pro infiammatori), patologie interstiziali (che squilibrano fattori
angiogenetici e angiostatici) possono aggravare l’ipertensione. In una prima fase si osserva
dilatazione ed ipertrofia del cuore destro con incapacità del ventricolo di superare il progressivo
aumento di post-carico che comporta aumento della pressione tele diastolica del VD con
ipertensione atriale destra, aumento PVC e stasi venosa sistemica. Inoltre la patologia può
aggravarsi con uno scompenso destro con edemi declivi, epatomegalia e ascite. Con il tempo
l’ipertrofia destra disturba anche la funzione del cuore sinistro (bassa portata ed ipotensione).
Clinica: è una patologia insidiosa con manifestazioni aspecifiche e sintomi spesso coperti o
attribuiti a quelli della patologia polmonare di base. In genere rispetto a questa c’è
un’accentuazione della tosse e della dispnea da sforzo. A volte disfonia, dolore similanginoso.
Con il tempo e l’avanzamento verso lo scompenso destro si ha epatomegalia, ascite, edemi
declivi e a volte lieve ittero, inoltre si ha una ritenzione idro-salina (alterazioni renali) e con il
peggioramento dell’ipossiemia e a causa della bassa portata si possono avere disturbi cerebrali.
14
Si può ascoltare sdoppiamento del II tono (comparsa III solo se c’è scompenso). Quadri clinici
particolari sono: sindrome di Pickwich: ipoventilazione alveolare in pazienti obesi e sonnolenti
con cianosi e poliglobulia (l’obesità di per sé provoca un aumento del lavoro respiratorio) che
produce un cuore polmonare; sleep apnea: il paziente si sveglia spesso di notte per periodi di
apnea anche di 10 secondi.
Diagnosi: La diagnosi della patologia di base è comunque in genere precedente. Esame
obiettivo: è possibile ascoltare un’accentuazione della componente polmonare del II tono ed in
genere uno sdoppiamento, inoltre sono comuni tachicardia ed un soffio di rigurgito tricuspidale
olosistolico per insufficienza della valvola (oltre ai segni dello scompenso destro se presente).
ECG: un’onda P di maggiore ampiezza e durata è un segno di ipertensione polmonare
(dilatazione atrio destro), inoltre vi sono segni di ipertrofia ventricolare destra. È possibile che si
sviluppino aritmie da blocco della conduzione (fino al BBD completo) ed extrasistole atriali o
anche tachicardia sopraventricolare e fibrillazione atriale. Radiografia del torace: segni di
ipertensione sono dilatazione dei vasi polmonari con aspetto ad “albero potato”.
Ecocardiografia trans toracica: rileva dilatazione dell’atrio e del ventricolo destro oltre a poter
stimare la PAP in base alla velocità di rigurgito tricuspidalico. Cateterismo cardiaco: è raramente
richiesto perché invasivo, ma serve a documentare la pressione in arteria polmonare, atrio
sinistro e vene polmonari. Esami di laboratorio: si può avere poliglobulia, all’emogasanalisi
aumento PCO2 e diminuzione PO2.
Prognosi e terapia: la prognosi dipende dalla patologia polmonare di base, ma spesso è grave
(50% di mortalità a cinque anni). La terapia si basa principalmente sulla risoluzione della
patologia respiratoria di base. I pazienti possono trovare giovamento dall’ossigenoterapia (con
PO2<60mmHg), diuretici se c’è ritenzione idrosalina, anticoagulanti per evitare trombo embolie.
Se sfocia nello scompenso è bene usare diuretici, e vasodilatatori usati nella terapia dello
scompenso per ridurre pre e postcarico.
Cuore polmonare acuto
Definizione: CPA, in pratica si tratta di Tromboembolia polmonare. Infatti è una condizione
nella quale vi è una brusca dilatazione del ventricolo destro dovuta ad una grave ed improvvisa
ipertensione polmonare. La causa più frequente è l’embolia polmonare, ma anche altre
condizioni (come atelettasia massiva e pneumotorace), se gravi possono causarlo. L’embolia
polmonare, EP, è un’ostruzione dei vasi polmonari da parte di emboli (materiali estranei,
generalmente provenienti dal sistema venoso profondo). È una condizione di emergenza
respiratoria con alto rischio di mortalità senza terapia giusta (10% a 30 giorni, 25% senza terapia
anticoagulante). A volte è complicato porre diagnosi per mancanza di segni clinici patognomici.
Epidemiologia ed eziologia: ha una frequenza di 70-100/100000 all’anno, nel 75-80% dei casi
l’embolo viene a formarsi a seguito di TVP (trombosi venosa profonda) agli arti inferiori. 1 TVP
su 10 si complica con EP. È facilitato da alcuni fattori di rischio quali: trombofilie (fattori primari,
rischio tromboembolico fino a 20 volte superiore) e fattori cumulativi (gravidanza, cateteri
venosi centrali, fumo, obesità, chirurgia, traumi, scompenso, contraccettivi orali, lunghi viaggi).
Patogenesi: non sempre l’embolia causa cuore polmonare acuto in quanto il polmone ha una
notevole riserva funzionale di vasi e pertanto solo un grosso embolo o tanti piccoli emboli
possono causare il CPA. In effetti le ripercussioni respiratorie e cardiocircolatorie dipendono
15
dalle dimensioni dell’embolo e dell’area ostruita (oltre che dalle condizioni del paziente) e si
distinguono pertanto embolie massive (più del 50% del letto vascolare polmonare è interessato)
e non massive. Nell’EP massiva si riscontra instabilità emodinamica con una pressione arteriosa
sistemica <90mmHg, instabilità respiratoria con dispnea ingravescente a riposo con ipossiemia
grave. La EP sub massiva è caratterizzata da stabilità emodinamica, ma da segni ecocardiografici
di disfunzione del ventricolo destro (dilatazione del settore destro con sbandamento del setto
interventricolare destro). La ripercussione cardiocircolatoria è l’aspetto più grave dell’EP (anche
se dipende oltre che dal grado di ostruzione anche dalle condizioni antecedenti). EP con più del
55% di ostruzione comporta: caduta della frazione di eiezione del VD, tachicardia, aumento del
precarico del VD, diminuzione del flusso coronarico con possibile ischemia. Le alterazioni del VD
comportano automaticamente una riduzione del flusso anche al VS con conseguente:
diminuzione della pressione arteriosa, riduzione del volume ventricolare sinistro e della gittata
cardiaca. La riduzione della gittata comporta ipoperfusione sistemica e ipoperfusione dei vari
microcircoli (anche cerebrale) che possono causare dolore anginoso (coronarico), vertigine,
sincope e anche shock. L’embolia però si associa solo raramente ad infarto polmonare in quanto
la perfusione dell’organo è garantita dal circolo arterioso bronchiale (a meno che non ci sia una
pregressa cardiopatia sinistra). Questo è ciò che accade a valla dell’ostruzione, mentre a monte,
nel cuore destro si ha progressiva disfunzione ventricolare. Questa è dovuta all’ipertensione
polmonare, quindi aumento del post-carico a cui all’inizio il ventricolo destro risponde
dilatandosi, poi (a causa del minore meccanismo di Starling rispetto al VS) si giunge alla
disfunzione ventricolare e talvolta all’insufficienza tricuspidale che aggravano la stasi venosa. Dal
punto di vista respiratorio si ha ipossiemia a causa della comparsa di un notevole spazio morto
funzionale (>>V/P) e di un conseguente squilibrio (<V/P) dei territori ancora normoperfusi.
L’iperventilazione reattiva comporta anche ipocapnia in quanto la CO2 è eliminata più
facilmente poiché ha una diffusione più rapida. La tachipnea non è in grado però di mantenere
un’adeguata ossigenazione. Si riscontra un alto gradiente alveolo-arterioso e meno CO 2 nell’aria
ispirata (perché l’aria che entra nello spazio morto ne esce con la stessa concentrazione di CO 2).
Clinica: Anche se l’EP è generalmente sintomatica, i sintomi variano con il grado di ostruzione, il
tempo intercorso dall’esordio, malattie cardiorespiratorie concomitanti. Nel 95% dei pazienti
sono presenti dispnea improvvisa (in genere da cause non evidenti), dolore toracico trafittivo in
genere dorsale che si esacerba con gli atti respiratori. I sintomi sono prevalentemente
respiratori nella non massiva e cardiaci nella massiva (più di due arterie lobari ostruite). Si può
avere esordio con ipotensione, polso accelerato, pallore e dispnea, sudorazione e , quando il VD
è insufficiente, epatomegalia e segni di stasi venosa. Si può avere un arresto cardiaco associato a
emoftoe (addensamenti polmonari) o sincope (sindrome neurologica)
Diagnosi: non è quasi mai facile in quanto l’EP non presenta segni e sintomi specifici. Una buona
anamnesi ed un corretto esame del paziente possono indirizzare verso la soluzione (e
contribuiscono nella definizione della probabilità clinica di EP). Il paziente si presenta in genere
con dolore e dispnea e a volte con palpitazioni, tosse, emoftoe, shock, sudorazione, polso
accelerato, etc. Importante verificare la presenza di fattori di rischio quali: TVP, obesità,
allettamento, intervento chirurgico recente, trombofilie. All’ascoltazione si avverte la
componente polmonare del II tono in ritardo (sdoppiamento) e rinforzata, possibile soffio
sistolico da insufficienza tricuspidale.
Tra gli strumenti diagnostici si distinguono test di primo livello e indagini diagnostiche di
certezza. Queste ultime non possono essere però adoperate per tutti i pazienti presentanti i
sintomi aspecifici e possono essere adoperate solo in condizioni di forte sospetto di EP o al
termine di un determinato percorso diagnostico.
16
I test di primo livello sono: Dosaggio dei dimeri D della fibrina: test semplice e rapido, che
permette di escludere con buona sicurezza l’EP (alto valore predittivo negativo) anche se ha
scarsa specificità. Dosa i prodotti della recente attivazione di un processo emocoagulativo. Altri
biomarker talvolta utili sono la troponina (indice di danno miocardico) e il BNP (indice di stress
di parete del ventricolo destro). Radiografia del torace: alcune alterazioni come la
sopraelevazione dell’emidiaframma, la dilatazione dell’arteria polmonare discendente dx,
trasparenza dei campi polmonari possono essere sospette. ECG: tachicardia sinusale in genere
associata a sindrome S1Q3T3 ossia con S allargata in D1, Q allargata e T invertita in D3 e inoltre ST
sottoslivellato in D2, DAD e rotazione destra e T invertite da V 1 a V4. Emogasanalisi: ipossiemia
ed ipocapnia. Se normale però non esclude EP. Già in base ai dati raccolti in questo modo si
classifica la probabilità clinica di EP in alta, intermedia e bassa, in modo da poter poi valutare
l’eventuale esecuzione di ulteriori procedure diagnostiche. Probabilità clinica di EP: Alta: (80100%) presenza di fattore di rischio e dispnea e/o dolore toracico non spiegabile da altre cause +
alterazioni radiografiche ed emogasanalitiche non spiegabili da altre cause. Bassa: fattori di
rischio assenti e sintomi toracici e alterazioni spiegabili da altre cause. Intermedia: né alta né
bassa. Ecocardiografia: esame aggiuntivo utile soprattutto nelle fasi iniziali perché alcuni segni
possono sparire dopo alcune ore. Difficile però che venga usato come vero e proprio esame di
primo livello. Valuta la morfologia del VD e può stimare la PAP. Raramente possono essere
direttamente visibili trombi intraluminali nelle arterie polmonari o nelle cavità cardiache destre.
Ecocolordoppler: viene usata per le vene degli arti inferiori per valutare la presenza di TVP.
Indagini diagnostiche di certezza: Scintigrafia polmonare di perfusione: alta sensibilità e buona
specificità, ma può essere limitata da condizioni che limitano la visibilità (fibrosi, neoplasie,
BPCO). Visualizza zone con deficit di perfusione zone (non ostruite) con iperperfusione. TC
spirale: permette di vedere trombi nelle polmonari, ma è meno sensibile dell’angiografia.
Angiografia polmonare: è il gold standard dell’EP, ma dovrebbe essere riservata ai pazienti per i
quali non è stato possibile formulare una diagnosi senza. Permette riconoscere presenza e
distribuzione degli emboli. Si introduce un catetere in vena periferica e lo si spinge prima nel
cuore destro, poi nell’arteria polmonare dove si inietta il mezzo di contrasto (per le radiografie
che si fanno in successione). È invasiva e non priva di rischi. Talvolta è difficile la diagnosi
differenziale con altre partologie cardiache e respiratorie come l’IM (ECG tipico, dolore
precordiale, biomarker), dissezione aortica (shock), pneumotorace (basta la radiografia per
vederlo), polmoniti, esordi neoplasie, scompenso, affezioni dolorose della parete toracica. Molto
utili nelle diagnosi differenziali i dimeri D e la scintigrafia (no lesioni).
Prognosi: severa. Il 90% dei pazienti con embolia massiva muoiono entro le prime due ore.
Terapia: la terapia più indicata è l’eparina (non frazionata per ev o a basso peso molecolare e
per via sottocutanea), anticoagulante in genere somministrato per non più di una settimana,
incrociandola magari con dicumarolici (ipoprotrombinizzanti) per non meno di 6 mesi. La
profilassi nei pazienti con importanti fattori di rischio come trombofilie o TVP. L’eparina si
somministra in base al peso corporeo e bisogna giungere ad un tempo di tromboplasmina
parziale, PTT di 1,5-2,5 il normale e misurare l’attività (AP) e il tempo di protrombina (PT)
espressi con l’indice INR (da tenere tra 2 e 3). Nell’EP massiva, o nella sub-massiva è indicata la
trombo lisi farmacologica (con uro o streptochinasi e attivatore tissutale del plasminogeno t-PA
ricombinante). Si deve inoltre correggere l’instabilità emodinamica con vasopressori e
integrazione di liquidi, l’insufficienza respiratoria con ossigenoterapia (se necessario). Per
eliminare i trombi vi è la possibilità dell’approccio interventistico con angio-jet (dispositivi che
per via angiografica usano getti di soluzione salina), o l’intervento chirurgico di embolectomia (o
anche tromboendoarterectomia in un centro specializzato che asporta anche trombi
17
cronicamente adesi alle pareti dei grossi vasi).
Cardiopatie congenite
Definizione: sono anomalie cardiache o dei grossi vasi presenti sin dalla nascita. Gli errori
durante il processo di sviluppo cardiaco si concentrano principalmente tra la 3 a e l’8a settimana
di gestazione. In genere difetti compatibili con la vita intrauterina (ma che comunque nel 50%
dei casi si manifestano con sintomi entro il primo anno di vita) sono quelli che coinvolgono
singole camere o regioni cardiache.
Eziologia: le principali cause note sono anomalie genetiche sporadiche, che per lo più
coinvolgono fattori di trascrizione (per i quali pare basti una mutazione in eterozigosi). Sindromi
che comportano significative lesioni cromosomiche o aneuploidie sono spesso associate (Di
George, Turner, trisomie 13, 18, 21). Un fattore di rischio è certamente la familiarità. Fattori
eziologici ambientali noti sono patologie quali la rosolia (aumento dell’incidenza di PDA), diabete
gestazionale, esposizione ad alcuni farmaci quali la difenildantoina, barbiturici, antitumorali.
Si distinguono principalmente cardiopatie congenite cianogene e non cianogene.
Le cardiopatie possono infatti comportare ostruzioni o shunt. Lo shunt è una comunicazione
anomale tra camere o vasi sanguigni. Queste comunicazioni consentono il flusso di sangue
secondo il gradiente pressorio vigente (dal vaso o camera con pressione maggiore a quello con
pressione minore ovviamente) da destra a sinistra o viceversa.
Quando c’è uno shunt destro-sinistro si ha ipossiemia e cianosi (vengono saltati i polmoni) e si
ha una cardiopatia congenita cianogena.
Queste si dividono ulteriormente in cardiopatie congenite cianogene a flusso polmonare ridotto
(come la tetralogia di Fallot) e a flusso polmonare aumentato (come la trasposizione dei grandi
vasi e il tronco arterioso persistente). Con lo shunt destro-sinistro può anche avvenire che
emboli dalle vene periferiche provochino embolia paradossa nel circolo sistemico (infarti e
ictus) oltre a ippocratismo digitale e policitemia.
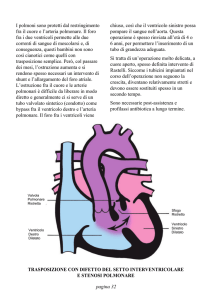
Quando c’è uno shunt sinistro-destro si ha un aumento del flusso ematico polmonare e all’inizio
non c’è cianosi e pertanto si ha una cardiopatia congenita non cianogena (come nel DIA, DIV e
PDA). In questo caso però c’è un aumento del flusso polmonare e quindi della pressione nel
piccolo circolo che può condurre a ipertrofia ventricolare destra e aterosclerosi dei vasi
polmonari. I vasi polmonari vanno infatti incontro a vasocostrizione (anche per evitare l’edema
polmonare) che alla lunga comporta come nell’ipertensione sistemica lesioni dell’intima e
ostruzioni.
Quando le resistenze polmonari raggiungono i livelli sistemici si produce uno shunt destrosinistro che comporta una cardiopatia congenita cianogena tardiva (sindrome di Eisenmenger).
Quando si sviluppa un’ipertensione polmonare irreversibile i difetti della cardiopatia sono
considerati irreparabili.
Vi è anche un altro tipo di cardiopatie non cianogene, le cardiopatie congenite ostruttive
(ostruzione completa è detta atresia) che comportano restringimenti di vasi, camere o valvole e
che sono a flusso polmonare normale (stenosi aortica, coartazione dell’aorta, stenosi
polmonare).
Cardiopatie non cianogene (flusso polmonare aumentato, shunt sinistro-destro):
Difetto del setto interatriale: DIA, è un’apertura anomala e stabile del setto interatriale per
formazione incompleta di tessuto che consente un passaggio di sangue tra gli atri.
Non bisogna confonderlo con forame ovale pervio: questo è un piccolo foro dovuto ad un
18
lembo aperto nella fossa ovale. Il forame ovale è normalmente del tutto pervio in vita
intrauterina per permettere il bypass del circolo polmonare (l’ossigenazione del sangue avviene
tramite la placenta). Dovrebbe chiudersi in alla nascita ma in una grossa percentuale (20%) il
lembo non è sigillato e può aprirsi quando aumenta la pressione del lato destro (ipertensione
polmonare transitoria come durante tosse o starnuti possono portare brevi shunt destro-sinistro
ed embolie paradosse).
I difetti del setto invece sono distinti in: ostium primum (5%, in vicinanza delle valvole AV);
ostium secundum (90% a livello della fossa ovale); seno-venoso: (quasi 5%, in prossimità dello
sbocco della cava superiore) ; seno-coronarico (in prossimità dello sbocco del seno coronarico).
L’ostium secundum è nettamente il più comune e può associarsi anche ad alterazione di lembi e
corde tendinee della mitrale. Lo shunt fa sì che durante la diastole vi sia passaggio di sangue
dall’atrio sinistro al destro, questo aumenta la gittata del ventricolo destro e dunque il flusso
polmonare (che da 1,5 volte il normale è emodinamicamente significativo, ma può essere anche
2-4 volte il normale). Con le decadi si ha sempre maggiore ipertensione polmonare e aumento
delle resistenze polmonari fino a che queste non superano quelle periferiche comportando
un’inversione dello shunt (comunque l’ipertensione polmonare irreversibile è poco frequente).
Clinica: La sintomatologia dipende dalle dimensioni del difetto, che se medio comporta
comparsa dei sintomi solo intorno al ventesimo anno di età (i pazienti spesso non sono
sintomatici prima dei 30 anni). I primi sintomi sono dispnea da sforzo, affaticabilità. La malattia
vascolare pomonare irreversibile, scompenso cardiaco e embolia paradossa sono in genere
tradivi. Se il difetto è grande può esserci distensione atriale e aumentato rischio di insorgenza di
tachiaritmie sopraventricolari. All’esame obiettivo gli adulti appaiono normali con polsi periferici
a volte piccoli (ridotta gittata sinistra) e all’ascoltazione può apprezzarsi uno sdoppiamento del
secondo tono (aumentato ritardo della tricuspide) e un soffio sistolico da eiezione in area
polmonare (ipertensione polmonare).
Diagnosi: All’ECG si nota ipertrofia ventricolare destra e blocco di branca destra incompleto.
All’esame radiologico si può evidenziare una dilatazione del tronco arterioso e delle arterie
polmonari con aumentata vascolarizzazione polmonare. L’indagine fondamentale è
l’ecocardiografia con color-doppler che permette di localizzare il difetto, valutarne le dimensioni
e, con il doppler, di valutare l’entità dello shunt.
Terapia: la chiusura spontanea può avvenire solo entro il primo anno. La terapia è chirurgica per
qualunque shunt emodinamicamente significativo con PQ/PS (portata polmonare/sistemica)
>1,5. Si fa sutura diretta del difetto oppure, quando i margini non sono facilmente accollabili si
può applicare un patch di pericardio prelevato dallo stesso paziente o utilizzando materiale
protesico (dacron).
Difetto del setto interventricolare: DIV, è la cardiopatia congenita più comune, ma solo nel 2030% dei casi si presenta da sola e non associata ad altre anomalie. Consiste in una
comunicazione tra i due ventricoli per pervietà del setto interventricolare.
Si distingue in: difetto membranoso (parte membranosa del setto, 80-90% dei casi); difetto
infundibolare (nel tratto di efflusso del ventricolo sinistro, sotto polmonare); difetto muscolare
(parte muscolare dl setto). In genere sono difetti singoli, i muscolari possono essere a groviera.
La shunt all’inizio è sinistro-destro, ma con il tempo sono comuni episodi di ipertrofia e fibrosi
delle arteriole polmonari. La gravità dipende dalle dimensioni del difetto (in media quanto
l’ostio aortico). Se il difetto è di piccole dimensioni (malattia di Roger) lo shunt è trascurabile.
Nel caso il difetto sia grande si ha rapidamente forte ipertensione polmonare e aumento del
carico di lavoro sia per il ventricolo destro che per il sinistro con frequente ipertrofia bi
ventricolare.
19
Clinica: Si ha praticamente in tutti i pazienti con grande DIV sindrome di Eisenmenger e quindi
vascolopatia arteriolare polmonare irreversibile con obliterazione del letto polmonare, cianosi e
morte. I sintomi iniziali sono dispnea da sforzo e affaticabilità. All’esame obiettivo si nota bozza
precordiale (nell’infanzia la parete toracica è più cedevole e una notevole ipertrofia cardiaca
può causarla) segno di cardiomegalia (comunque più comune nei grandi DIV), itto ipercinetico e
anche fremito sistolico (anche il ventricolo sinistro lavora di più). Si può apprezzare un soffio
olosistolico di 3-4/6 sulla scala di Levine (quindi con possibile fremito) con irradiazione verso
destra e verso la base cardiaca. Si ha uno sdoppiamento del secondo tono e anche possibile S3
(ritmo di galoppo proto diastolico).
Diagnosi: l’ECG è normale nei piccoli difetti, indica ipertrofia, anche bi ventricolare, nei grandi.
All’esame radiologico si ha ingrandimento dell’ombra cardiaca e dilatazione dell’arteria
polmonare. L’indagine d’elezione è sempre l’ecocardiogramma color-doppler (sede e dimensioni
difetto, entità dello shunt).
Terapia: la malattia di Roger in oltre il 50% dei casi comporta chiusura del difetto durante
l’infanzia. Nei DIV grandi è necessario l’intervento chirurgico (urgente se PQ/PS>2, ma
controindicato se ormai si ha già sindrome di Eisenmerger in qual caso è irreparabile).
L’intervento si esegue in ipotermia moderata e circolazione extracorporea. Si pratica atriotomia
o ventricolotomia destra per raggiungere il difetto che viene corretto con patch pericardica o
materiale sintetico, prestando attenzione ad evitare danneggiamenti del sistema al sistema di
conduzione ventricolare.
Pervietà del dotto arterioso di Botallo: PDA, è un’anomalia che si presenta per lo più isolata,
costituisce il 10-15% delle cardiopatie congenite, ed è più femminile. Il dotto di Botallo è una
comunicazione tra l’aorta discendente e il tronco arterioso polmonare struttura che resta pervia
durante tutta la vita fetale e funge da bypass del 90% della gittata ventricolare destra dato che
non è necessario il passaggio per i polmoni. Durante la vita fetale chiaramente lo shunt
attraverso il dotto è destro-sinistro in quanto le resistenze polmonari sono elevate ( i polmoni
sono pieni di liquido). Con il primo respiro le resistenze polmonari diminuiscono
drammaticamente e lo shunt attraverso il dotto diviene sinistro-destro, anche se per poco
tempo perché, a seguito della mancanza di prostaglandine (E per lo più) prodotte dalla placenta,
dopo la nascita il dotto va incontro a vasocostrizione in 10-15 ore dalla nascita, poi in qualche
giorno si ha trombosi e fibrosi con trasformazione del dotto nel legamento arterioso di Botallo,
teso sopra l’arco aortico. Cause che possono determinare mancata chiusura del dotto sono
l’ipossia cronica e la rosolia contratta in gravidanza (primo trimestre).
Clinica: Le conseguenze dipendono dalle dimensioni del dotto e dalle resistenze polmonari
(direzione dello shunt):
Calibro ridotto: lo shunt è lieve, il flusso dall’aorta all’arteria polmonare è continuo, sia in sistole
che in diastole. I pazienti non hanno sintomi (comunque in genere nel PDA non ci sono sintomi
alla nascita, no cianosi), ma all’ascoltazione si avverte un soffio continuo irradiato alla regione
sottoclavicolare sinistra (soffio aspro e continuo, definito a locomotiva o di Gibson), con
massima intensità al focolaio aortico.
Medio-grosso calibro: aumentato flusso polmonare con sovraccarico del cuore sinistro. Con il
tempo le resistenze polmonari aumentano fino a causare la sindrome di Eisenmenger con
desaturazione in ossigeno del sangue arterioso discendente (che naturalmente per l’inversione
dello shunt fa mischiare il sangue) e vascolopatia polmonare irreversibile. Il paziente evolve
spesso verso lo scompenso cardiaco già nel primo anno di vita, presenta in genere dispnea con
lo sforzo fisico. Si ha aumento della gittata sistolica, ma ridotto flusso diastolico e perciò si ha
aumento della pressione differenziale per incremento della pressione sistolica e riduzione della
20
diastolica. Si ha un itto ipercinetico. Se le resistenze polmonari aumentano il classico soffio si
presenta per lo più in sistole e il secondo tono risulta sdoppiato (con componente polmonare
rinforzata).
Diagnosi: radiologicamente si nota un’aorta ascendente dilatata (riceve anche lo shunt, se le
resistenze sono aumentate) così come sono dilatati atrio e ventricolo sinistro per sovraccarico di
volume. L’ecocardiografia bidimensionale color-doppler evidenzia direttamente il dotto e valuta
la continuità dello shunt in sistole e diastole. Con il cateterismo cardiaco si riscontra: saturazione
di ossigeno del sangue prelevato in arteria polmonare superiore di 5-10% rispetto al ventricolo
destro (differenza tra prima e dopo lo shunt); nella sindrome di Eisenmerger desaturazione di
ossigeno in aorta discendente rispetto all’ascendente.
Terapia: il dotto dovrebbe essere chiusa il prima possibile, molto urgente se c’è già scompenso
cardiaco (anche primi mesi di vita), del tutto controindicato in caso di sindrome di Eisenmenger
con inversione dello shunt (suppongo perché a quel punto il ventricolo destro non sarebbe più in
grado di rispondere da solo a quella pressione molto alta una volta chiuso lo shunt). Si pratica
toracotomia postero-laterale sinistra all’altezza del IV spazio intercostale, si apre la pleura, si fa
attenzione a vago e frenico e si raggiunge il dotto che può essere legato direttamente oppure
prima resecato e poi successivamente vengono suturati i due monconi. Il dotto di Botallo può
risultare una risorsa terapeutica in quei pazienti ad esempio con atresia valvolare aortica in
quanto è l’unico modo per sostenere il flusso sistemico (si somministra infatti prostaglandina E
per mantenerlo pervio). Esistono anche difetti del setto atrioventricolare.
Cardiopatie congenite ostruttive (no shunt, flusso polmonare normale):
Coartazione dell’aorta: fra le più comuni malformazioni strutturali, può essere isolata ma anche
associata ad altre (nel 50% ad una valvola aortica bicuspide, ma anche una stenosi aortica),
frequente nella sindrome di Turner, ma più comune nei maschi. Consiste in un restringimento
dell’aorta. Vi è una forma infantile, già sintomatica dalla prima infanzia (spesso prossimale ad un
PDA) e una forma adulta (distale ad un dotto chiuso) asintomatica nell’infanzia e che può
passare inosservata sino all’età adulta. Normalmente si localizza a livello dell’istmo aortico
(giunzione tra arco e aorta discendente) e può essere associata ad anomalie della succlavia (se
prossimale all’origine la succlavia sarà coinvolta nella coartazione, se distale sarà dilatata).
Patogenesi: la principale conseguenza è l’ipertensione arteriosa nel distretto vascolare
superiore alla coartazione. Questa è dovuta all’incremento della resistenza al flusso e
all’attivazione del sistema renina-angiotensina-aldosterone per ridotta perfusione renale. La
risposta del ventricolo sinistro all’aumento della pressione è l’ipertrofia, si può giungere allo
scompenso sinistro. Inoltre, per il gradiente pressorio tra distretto vascolare prossimale e distale
alla coartazione si ha lo sviluppo di un circolo collaterale arterioso delle arterie che originano
dalla succlavia come le mammarie interne e le intercostali per mantenere un flusso adeguato
all’aorta discendente. Durante gli sforzi aumenta ancora la pressione perché aumenta ancora il
gradiente. La gravità della condizione dipende anche dal tempo in cui il restringimento peggiora:
se rapido non permette che si venga a creare un adeguato circolo collaterale e che il ventricolo
sinistro possa adattarsi.
Clinica: si ha ipertensione arteriosa, anisosfigmia tra arti superiori e inferiori (differenza di 20
mmHg a riposo) con reale iposfigmia del polso femorale che può anche scomparire. Si possono
avere pulsazioni a livello delle arterie palpabili nel distretto superiore. Sono da esaminare i polsi
ad entrambe le braccia e contemporaneamente il radiale e il femorale per apprezzare la
differenza della forza e del tempo di comparsa dell’onda sfigmica. Ci possono essere soffi
continui o sistolici. I pazienti possono avere sintomi aspecifici come cefalea, epistassi, estremità
21
fredde e claudicatio sotto sforzo. Il rischio è quello di sviluppare aneurismi ed emorragie (anche
cerebrali, circolo di Willis), dissecazione e rottura aortica, aterosclerosi coronarie, scompenso
sinistro.
Diagnosi: l’ecocardiografia permette di fare diagnosi di certezza (segno del 3: dilatazione pre e
post stenotica con incisura al centro), anche se è visibile anche con RM e TC. Radiologicamente
possono essere visibili erosioni (incisure), dovute alle arterie del circolo collaterale dilatate, alle
superfici inferiori delle coste (3a e 9a).
Terapia: l’intervento chirurgico si esegue con toracotomia postero-laterale sinistra all’altezza del
IV-V spazio intercostale. Si può resecare il tratto aortico coartato con anastomosi terminoterminale tramite un tratto protesico intermedio. Se la stenosi è lieve e breve oppure se l’aorta
non è isolabile per la resezione a causa di vasi collaterali emergenti in stretta vicinanza, si può
fare una plastica di allargamento mediante l’innesto di un patch di materiale sintetico.
Stenosi polmonare: costituisce il 5-10% delle anomalie cardiache riscontrabili dopo l’infanzia.
Rappresenta un’ostruzione della valvola polmonare oppure dell’infundibulo sottovalvolare o
anche in sede sopravalvolare. Sì può riscontrare nella sindrome da rosolia materno-fetale.
L’ostruzione valvolare è dovuta alla sostituzione della semilunare con un diaframma con
un’apertura centrale, l’ostruzione dell’infundibulo è spesso dovuta alla presenza di tessuto
fibroso sottovalvolare o ad un’ipertrofia massiva infundibolare. Può essere isolata o associata a
malformazioni complesse.
Patogenesi: si ha un sovraccarico pressorio con ipertrofia del ventricolo destro proporzionale al
grado di stenosi (una stenosi completa è detta atresia). C’è una dilatazione post-stenotica del
tronco polmonare e del suo ramo sinistro (dovuta anche ai vortici che escono a getto
dall’ostruzione) e un’ipertrofia dell’infundibulo sottovalvolare. Viene a crearsi un grande
gradiente pressorio sistolico con pressione nel ventricolo destro che può persino superare
quella del sinistro (pressione ipersistemica). Se è associato a tetralogia di Fallot la polmonare
non è dilatata perché coesiste una stenosi sottopolmonare per cui il gradiente non è trasmesso
alla valvola ed il tronco polmonare può essere persino ipoplastico. Soprattutto in caso di atresia
vi è un sovraccarico pressorio anche nell’atrio destro per cui (poiché mancherebbe la
comunicazione tra ventricolo destro e polmone) si associa ad apertura della fossa ovale o
comunque a un DIA con sangue che può raggiungere i polmoni solo tramite un dotto arterioso
pervio.
Clinica: la sintomatologia dipende dalla gravità dell’ostruzione. Se lieve il paziente è
asintomatico, se di media entità i sintomi si manifestano in età adulta. Se severa il bambino è già
sintomatico. Si ha principalmente affaticabilità e dispnea, se vi è inversione del flusso attraverso
la fossa ovale si ha facilmente cianosi. È rilevabile un soffio sistolico da eiezione in area
polmonare (II-III spazio parasternale sinistro nelle forme valvolari , IV-V parasternale nelle
infundibolari) del tipo crescendo-decrescendo (a diamante). Il soffio si genera a causa del
passaggio attraverso lo valvola stretta e a causa del flusso che da laminare diviene turbolento e
ha intensità proporzionale alla gravità dell’ostruzione. La massima intensità (apice del diamante)
corrisponde al massimo gradiente pressorio tra ventricolo destro e arteria polmonare. Se il
gradiente massimo è precoce l’ostruzione sarà lieve (basta poco), se è tardivo sarà severa. Si
avverte anche uno sdoppiamento del secondo tono per ritardo nella chiusura della polmonare (il
ventricolo destro si svuota più lentamente). Il polso giugulare ha un’onda “a” prominente per
aumento della pressione di riempimento del ventricolo destro. In area precordiale è avvertibile
un fremito che si irradia verso l’area polmonare. L’ipertrofia del ventricolo destro si instaura
quando la pressione sistolica destra supera i 60mmHg e si manifesta all’ECG con onde R>7mm in
V1 e V2 e deviazione assiale destra.
22
Diagnosi: radiologicamente è evidente la dilatazione dell’arteria polmonare e quella del
ventricolo destro. All’ecocardiografia è possibile valutare lo stato di valvola e ventricolo e con il
color-doppler si misura il gradiente pressorio trans valvolare (più preciso con cateterismo
cardiaco).
Terapia: le ostruzioni lievi consentono una normale aspettativa di vita, le gravi devono essere
trattate chirurgicamente perché tendono ad evolvere in scompenso cardiaco (principale causa di
morte, ma c’è pure maggiore rischio di endocarditi infettive). L’intervento è di correzione
chirurgica della stenosi o valvuloplastica con palloncino.
Esiste anche la stenosi aortica in cui è possibile la vita solo se il dotto arterioso resta pervio.
Cardiopatie congenite cianogene (a flusso polmonare ridotto o aumentato)
Tetralogia di Fallot: Cianogena a flusso polmonare ridotto.
TF, ha quattro caratteristiche fondamentali:
1) Stenosi polmonare: in genere sottopolmonare, ossia nell’infundibulo del ventricolo destro.
2) DIV
3) Deviazione a destra dell’aorta: ossia aorta a cavaliere del DIV.
4) Ipertrofia ventricolare destra.
Il DIV in genere è ampio, il cuore ha spesso aspetto “a scarpa” per l’ipertrofia destra. Alla stenosi
sottopolmonare può associarsi stenosi valvolare polmonare (o atresia) con eventuale stenosi
anche delle arterie polmonari, cosicchè per la sopravvivenza è necessario un PDA e/o arterie
bronchiali dilatate. Il setto interventricolare infundibolare in pratica è deviato anteriormente e a
destra determinando la stenosi polmonare e la deviazione a destra dell’ostio aortico. Si può
infatti avere anche deviazione estrema del setto con ostruzione completa della via polmonare
(Fallot estremo). Nel 30% dei casi ci sono anomalie coronariche. Il Fallot estremo è
naturalmente un Fallot dotto-dipendente.
Patogenesi: nel Fallot viene a crearsi uno shunt attraverso il DIV la cui direzione ed entità
dipendono dalle resistenze sistemiche e polmonari e dal grado di ostruzione polmonare. Se
abbiamo infatti un’ostruzione polmonare lieve con resistenze polmonari (inizialmente almeno)
inferiori a quelle sistemiche, si comporta quasi come un DIV isolato e pertanto lo shunt sarà
prevalentemente sinistro-destro e la forma sarà non cianogena (tetralogia rosa). Se le resistenze
polmonari superano le periferiche (ostruzione grave), ci sarà uno shunt destro-sinistro e quindi
la forma sarà cianogena (tetralogia di Fallot classica). Con l’aumentare dell’ostruzione le arterie
polmonari divengono ipoplasiche e l’aorta aumenta di diametro. L’ostruzione peggiora con la
crescita del bambino perché l’orifizio polmonare non si espande proporzionalmente al cuore. La
prognosi è determinata dalla severità dell’ostruzione. L’unico vantaggio è che la stenosi
protegge il circolo polmonare dal sovraccarico e lo scompenso ventricolare destro è raro.
Clinica: Nella maggior parte dei casi il bambino nasce già cianotico o lo è subito dopo. Esistono
casi di sopravvivenza fino a 20 anni (10%) o 40 anni (3%). La cianosi è in genere sempre presente
e si accompagna a ippocratismo digitale e a policitemia (la riduzione della PO 2 stimola
l’emopoiesi con ematocrito anche del 65-70%). Durante lo sforzo fisico l’incremento della
frequenza e l’aumento del ritorno venoso inducono un aumento dello shunt destro-sinistro con
aggravamento della cianosi. Spesso si ha il fenomeno dello squatting o accovaccia mento del
paziente dopo uno sforzo in quanto il paziente dispnoico tende a sistemarsi in posizione
genupettorale per ridurre il ritorno venoso sistemico e aumentare le resistenze periferiche (con
conseguente riduzione dello shunt destro-sinistro).
Diagnosi: all’ascoltazione è presente un soffio sistoli coda eiezione in area polmonare, in
genere accompagnato da un fremito palpabile, direttamente proporzionale al grado di stenosi.
23
All’ECG è rilevabile l’ipertrofia ventricolare destra, radiologicamente è visibile il coeur en sabot
(a scarpa) e all’ecocardiografia si nota bene la stenosi così come il DIV.
Terapia: neonati cianotici con ematocrito superiore a 65 necessitano di trattamento chirurgico.
Se la situazione non consente un intervento definitivo si realizza la correzione chirurgica in due
tempi, ossia si pratica prima un intervento palliativo che consiste nell’anastomosi succlavioarteria polmonare sinistra (intervento di Blalock-Taussing). La correzione definitiva consiste
nella chiusura del DIV tramite patch in dacron e nella risoluzione della stenosi polmonare
tramite patch sul tratto di efflusso ventricolare destro. La correzione chirurgica è più complessa
per i pazienti che presentano atresia polmonare. I risultati sono confortanti, ma permane il
rischio di insufficienze valvolari in età adulta così come di un’endocardite infettiva.
Un’altra anomalia complessa cianogena con ridotto flusso polmonare è l’Anomalia di Ebstein,
che consiste in uno spostamento verso il basso della valvola tricuspide con ventricolo destro che
risulta ipoplastico.
Trasposizione dei grandi vasi: Cianogena con aumentato flusso polmonare. Detta anche
trasposizione delle grandi arterie, TGA, è caratterizzata dall’inversione dell’origine di aorta e
arteria polmonare. La polmonare origina dal ventricolo sinistro e l’aorta dal destro. Questo
ovviamente comporta una separazione del circolo sistemico da quello polmonare e pertanto una
mancata ossigenazione del sangue in circolo, una condizione incompatibile con la vita. Per poter
sopravvivere il neonato dovrà avere delle alterazioni strutturali che permettono il
rimescolamento del sangue. Lo shunt è stabile nei pazienti con DIV (35%, cianosi più lieve) e
instabile nei pazienti con PDA o DIA (65%) (o forame ovale pervio) che pertanto necessitano di
un altro shunt (ottenuto con settostomia atriale con catetere a palloncino, si può anche pensare
ad una anastomosi tra arteria polmonare ed aorta). Si può associare ad anomalie come stenosi
sottopolmonare o della valvola polmonare (trasposizione complessa, in genere con cianosi
severa). Il ventricolo destro è notevolmente ipertrofico e il sinistro ipotrofico. All’ascoltazione si
nota un secondo tono unico e con intensità aumentata e all’ECG c’è ipertrofia ventricolare
destra (con marcata deviazione assiale destra).
Terapia: senza intervento la maggior parte dei pazienti muore nei primi mesi di vita. Vi sono più
possibilità di intervento chirurgico: Intervento di detrasposizione: (di Jatene) l’aorta e la
polmonare vengono resecate e ricollocate al posto giusto. Nel primo mese di vita questo si può
fare in un unico tempo. Successivamente c’è bisogno di abituare prima il ventricolo sinistro a
sopportare resistenze maggiori (cioè normali, periferiche) a quelle a cui si era abituato
(polmonari) e lo si realizza con la tecnica di bendaggio dell’arteria polmonare. Nei pazienti con
DIV e grave ostruzione si ricorre ad un bypass ventricolare e ad un condotto protesico
extracardiaco che sostituisce la polmonare (intervento di Rastelli, ???). Intervento di inversione
venosa: tecnica di Mustard: si pratica l’escissione del setto interatriale e un patch pericardico
viene suturato a livello atriale in modo da dirigere il sangue venoso sistemico verso la mitrale e il
sangue venoso polmonare (ossigenato) verso la tricuspide. Tecnica di Senning: stessa finalità,
ma anziché la patch utilizza la parete dell’atrio destro per dirigere il sangue venoso sistemico
verso la mitrale.
24
Elettrofisiologia cardiaca e aritmologia
Elettrofisiologia
Generalità: la contrazione dei miociti è permessa dalla depolarizzazione della cellula cardiaca.
L’impulso elettrico che comporta la depolarizzazione dei miociti viene generato da cellule
pacemaker che hanno la capacità di generare stimoli in modo ritmico senza bisogno a loro volta
di una stimolazione. Il pacemaker cardiaco dominante è il nodo senoatriale (NSA) che ha la
capacità di emettere stimoli con attività ritmica detta sinusale (automatismo). Vi sono anche
altri focolai automatici. Le cellule del miocardio, come i neuroni, sono normalmente polarizzate,
ossia sono maggiormente elettronegative all’interno a causa della differente permeabilità della
membrana cellulare ai vari ioni. A partire dalle cellule pacemaker l’onda di depolarizzazione si
propaga attraverso il sistema di conduzione cardiaco, fino al miocardio ventricolare. Dapprima,
attraverso i tratti internodali, che collegano il NSA al NAV (nodo atrioventricolare), avviene la
depolarizzazione atriale (ossia l’entrata di cariche positive, per lo più NA+ e K+, attraverso la
membrana divenuta più permeabile a questi ioni). Dopo l’impulso si propaga più lentamente,
rallentando, attraverso il NAV (che è l’unico punto di connessione elettrica tra atri e ventricoli,
per il resto isolati elettricamente tramite le valvole atrio-ventricolari). La depolarizzazione atriale
rappresenta l’onda P positiva dell’ECG, il passaggio attraverso il NAV è rappresentato dalla linea
isoelettrica che segue l’onda P. Lo stimolo procede poi nel setto interventricolare attraverso il
fascio di His e poi si distribuisce alle branche di destra e di sinistra. Queste così come il fascio
sono costituite da cellule del Purkinje (che pare costituiscano praticamente tutta questa parte
del sistema di conduzione e non solo, come spesso viene detto, i filamenti terminali). Il sistema
termina poi attraverso i filamenti terminali che distribuiscono l’impulso a tutti i miociti
ventricolari permettendone la contrazione. La depolarizzazione del miocardio ventricolare è
rappresentata dal complesso QRS. Successivamente nell’ECG si può notare un’onda T di
ripolarizzazione ventricolare (e anche un’onda U papillare) mentre la ripolarizzazione atriale è
mascherata dal complesso QRS. Il sistema di conduzione è pertanto costituito da due differenti
tipi di cellule. Delle cellule lente nel NSA e NAV per lo più CA2+ dipendenti, e delle cellule veloci
che sono le cellule del Purkinje per lo più con correnti di NA +.
Il potenziale di azione cardiaco è notevolmente più lungo di quello neuronale. Infatti può durare
anche 200 msec contro i 2-5 del potenziale d’azione delle fibre nervose. Il potenziale di
membrana a riposo di una cellula del nodo seno-atriale è di -55-60mV, di un miocita ventricolare
è di -90. Il p.d.a. delle due cellule ha una forma e un meccanismo diversi (diversa funzione dei
canali e diverse correnti ioniche). In realtà ad esempio nella cellula del NSA non c’è un reale
potenziale di riposo dal momento che appena queste raggiungono un valore di circa -60mV
intervie la corrente If (cationica mista, soprattuto di NA +) che comporta l’inizio di una nuova
depolarizzazione. Successivamente a -50 si aprono i canali del Ca 2+ transienti e subito dopo (-40)
quelli long lasting che permettono una depolarizzazione fino a +10 (e accumulo di Ca 2+ al’interno
della cellula. Queste cellule depolarizzano grazie al Ca 2+. Dopo +10 attraverso i canali del
potassio (che esce) si ha ripolarizzazione. Questa sequenza è spesso detta tri-potenziale (prepotenziale -65 -> -40; depolarizzazione -40->+10; ripolarizzazione fino a -65). Le cellule
miocardiche ventricolari hanno invece un ben potenziale di membrana a riposo di -90 che si
mantiene tale fino a quando non sono raggiunte da un p.d.a. che determina principalmente
l’apertura di canali del Na+. Si ha all’inizio una fase 0, detta overshoot perché c’è inversione del
25
potenziale, con un aumento della G Na all’interno della cellula e passaggio da -90 a +30. A questa
fase segue la ripolarizzazione che comincia con una fase 1: da +30 a +10 e quasi fino a 0
caratterizzata da una Gk in uscita (corrente transient outward Kto costituita da due componenti
Ito1 e Ito2). Fase 2: detta di plateau con otenziale che oscilla intorno allo 0 tra -10 e +10 dovuta ad
una corrente delayed rectifier (IKv, ritardata) di K+ verso l’esterno e ad una corrente slow inward
(lenta, verso l’interno), del Ca2+ (attraverso canali long lasting). Fase 3: da -10 a +90 con
l’inattiazione della corrente di Ca2+ e successivamente l’entrata in gioco di una massiccia
corrente KIR inward rectifier di K+ verso l’esterno. Nella fase 4 si ha invece la restitutio ad
integrum con ripristino delle precedenti concentrazioni di ioni tramite varie pompe ioniche
come la Na+/K+ ATPasi, lo scambiatore Na+/Ca2+ e la Ca2+ ATPasi (anche se le concentrazioni non
cambiano molto perché il passaggio di ioni è sempre un fenomeno di limitato alla parte di
citoplasma più vicina alla membrana). Prima di poter essere di nuovo eccitabili, le cellule del
miocardio hanno bisogno di un periodo di riposo (refrattarietà). Il periodo di refrattarietà
assoluta (ARP) è il periodo durante il quale nessuno stimolo, di qualunque intensità, uò essere in
grado di indurre una risposta (fasi 1,2, e inizio 3). Il periodo di refrattarietà relativa (RRP) è un
periodo in cui la soglia di stimolazione è pi elevata del normale (c’è bisogno di uno stimolo più
intenso del solito per poter indurre depolarizzazione fasi 3 e 4). Segue spesso un periodo di
ipereccitabilità.
Aritmie
Aritmie: le aritmie sono alterazioni del normale ritmo sinusale, stabilito dal NSA, il pacemaker
dominante. Il ritmo sinusale causato dall’automatismo (cellule in grado di auto-depolarizzarsi
ritmicamente) del NSA è detto regolare, in quanto vi è equidistanza tra onde identiche
(frequenza costante). Può essere presente una “aritmia sinusale” (accelerazione e
decelerazione del normale ritmo sinusale controllato dal NSA) in genere del tutto fisiologica.
Questa viene distinta nella forma respiratoria con variazione lieve della frequenza in rapporto
alla fase della respirazione, con una lieve accelerazione durante l’ispirazione e lieve
decelerazione durante l’espirazione, e nella forma non respiratoria indipendente dagli atti.
Viene sempre conservata la sequenza P-QRS. È comune in giovani sani, ma anche in anziani con
problemi respiratori o con malattia degenerativa del nodo del seno.
[Questa artimia è da imputare all’azione del SNA, simpatico e parasimpatico, che agisce
regolando la frequenza cardiaca e quindi il numero di depolarizzazioni che partono dal NSA
(avendo invece un’azione decisamente ridotta sugli altri focolai ectopici e sulle altre cellule
miocardiche). Il sistema nervoso simpatico (anche inspirazione) porta tachicardia, tramite nervi
simpatici che usano come neurotrasmettitore la noradrenalina (che agisce sui recettori betaadrenergici legati a proteina Gs. Il parasimpatico porta bradicardia, tramite il vago che usa
acetilcolina che agisce sul recettore muscarinico (cellule pacemaker) legato a proteina G i.
Noradrenalina: la proteina Gs attiva l’adenilatociclasi (aumentoc cAMP) che attiva la PKA che
fosforila e attiva i canali del Ca2+ che dunque entra nella cellula aumentando la velocità di salita
del potenziale e dunque: aumento della velocità di contrazione e di rilasciamento (perché il Ca 2+
in più attiva anche le SERCA) e aumentando la velocità della corrente I to2 di Cl- velocizzando la
ripolarizzazione.
L’acetilcolina tramite il dimero betagamma della G i prolunga l’apertura dei canali del K+
causando iperpolarizzazione, per cui la corrente If impiegherà più tempo per depolarizzare.
Inoltre la subunità alpha attiva una fosfatasi che inibendo l’adenilato ciclasi riduce l’entrata di
Ca2+ con effetti opposti alla noradrenalina. Pertanto rallenta la frequenza cardiaca.
26
Bisogna ricordare che le cellule miocardiche sono eccitabili (batmotropismo), capaci di
trasmettere la depolarizzazione (dromotopismo), di depolarizzarsi un certo numero di volte
(cronotopismo) e di contrarsi con una certa forza (inotropismo). Il vago non interferisce con la
forza di contrazione perché riducendo la frequenza aimenta il tempo di riempimento e dunque il
volume telediastolico che per la legge di Starling si accompagna ad un aumento della forza
contrattile. Inoltre sul ventricolo non ci sono recettori muscarinici a sufficienza.
Pertanto il simpatico comporta 4 risposte positive, il parasimpatico 3 negative e 1 positiva
(inotropa).]
Le aritmie sono il risultato di alterazioni della generazione dell’impulso, della conduzione o di
entrambi. Le aritmie si distinguono in ipercinetiche, con numero di impulsi superiore al normale
a livello atriale (sopraventricolari) o ventricolare (ventricolari), e ipercinetiche, con numero di
impulsi minore.
I meccanismi con cui si possono generare le aritmie ipercinetiche (o tachiaritmie) sono:
1) Aumento della frequenza intrinseca del NSA, 2) Attivazione di un pacemaker ectopico con
frequenza superiore al NSA, 3) Meccanismo di rientro, 4) Triggered activity.
Automatismo (1-2): è la capacità di depolarizzarsi in maniera spontanea che possiede il tessuto
miocardico specifico. È propria del NSA e di altri focolai ectopici. Il NSA emette impulsi ad una
frequenza di 60-100 bpm, i focolai atriali in genere di 60-80, giunzionali 40-60, ventricolari 2040. Poiché il NSA ha una frequenza intrinseca maggiore, normalmente è dominante (pacemaker
primario) e sopprime (per overdrive) tutti gli altri focolai. Un aumento di eccitabilità di un
focolaio ectopico può far sì che questo si sostituisca al NSA nel generare il ritmo (acquisisce
frequenza maggiore). Oltre all’SNA, altri fattori possono influenzare la frequenza dei pacemaker
cardiaci, come l’ipokaliemia o l’ischemia (scarsità di ossigeno), ma anche sostanze tossiche o
stimolanti i recettori beta-adrenergici, ipertiroidismo etc.
Meccanismo di rientro (3): alterazione nella propagazione dell’impulso. Si conoscono
principalmente due tipi di rientro: il rientro da gap eccitabile (anatomicamente determinato) e il
rientro con circuito principale (funzionale).
Il primo (e più comune, [l’unico citato dalla CLU]) è possibile quando vi è uno sdoppiamento
della propagazione dell’impulso in due vie (A e B) intorno ad un ostacolo ineccitabile. Queste vie
aggirato l’ostacolo si ricongiungono (in una via C). Quando una delle due vie (A e B) è bloccata da
una refrattarietà, l’impulso giunto alla via C solo tramte la via A può avere conduzione
retrograda ttraverso la via B non più refrattaria e generare un circuito di rientro che passa
attraverso la via B e poi la A intorno all’ostacolo ineccitabile e generando inoltre propagazione di
impulsi lungo la via comune C dando luogo ad una aritmia (reciprocante).
Si può avere un micro e un macrorientro, (quest’ultimo in grado di generare tachiaritmie
reciprocanti come nella sindrome di Wolf-Parkinson-White). Il meccanismo è definito da gap
eccitabile perché all’interno del circuito di rientro, fra il fronte d’onda che avanza e la coda di
recupero vi è un gap (intervallo) che permette che il rientro sia sostenuto (continui).
Un meccanismo che permette di estinguere la tachicardia (terminazione del rientro) si ha
quando per modificazione delle caratteristiche di conduzione del circuito il gap si perde e il
fronte d’onda coincide con la coda.
Il rientro con circuito principale non ha una un circuito anatomico fisso, ma è funzionale e
pertanto anche meno stabile. La patologia del cuore a volte modifica la conduzione e la
27
refrattarietà aumentando il rischio di aritmie da rientro. Un miocardio ischemico evidenzia ad
esempio una down-regulation della proteina connessina 43 delle gap junction.
Triggered activity (4): è un fenomeno che non si verifica spontaneamente, ma è scatenato da
variazioni della frequenza e determinato dalla presenza di potenziali d’azione aggiuntivi, definiti
postdepolarizzazioni. Queste possono essere postdepolarizzazioni precoci e sovrapporsi alla
fase finale di un p.d.a. (PDP, fasi 2-3) oppure possono essere posdepolarizzazioni tardive e
insorgere dopo la fase finale di un p.d.a. (PDT, fase 4). In pratica le PDP interrompono la
ripolarizzazione, le PDT si presentano al valore di riposo. Possono essere causa di aritmie nel
momento in cui la triggered activity si verifica (ossia quando queste postdepolarizzazioni
raggiungono la soglia per poter generare un p.d.a. che a sua volta può essere seguito da altre
postdepolarizzazioni). Le basi molecolari delle due sembravano un tempo diverse in quanto è
noto da tempo che le PDT siano in relazione ad un sovraccarico di Ca 2+ intracellulare, mentre è
un’acquisizione recente il fatto che anche le PDP ne sono influenzate.
In particolare, le PDP sembrano essere causate da un aumento delle correnti verso l’interno del
calcio e da una diminuzione delle correnti verso l’esterno di potassio. Questo comporta un
allungamento del p.d.a. e un conseguente prolungamento dell’intervallo QT (come nella
sindrome del QT lungo, in cui in effetti il p.d.a. è prolungato, e questo aumenta il rischio di PDP).
Le condizioni che aumentano la durata del potenziale d’azione aumentano il rischio di PDP.
Pertanto condizioni (ipokaliemia, ipossia e acidosi) e farmaci (chinidina ad esempio) che
inibiscono le correnti di potassio verso l’esterno e condizioni che promuovono le correnti di
calcio verso l’interno (bradicardia, aumento del tono simpatico). Pare che le PDP siano alla base
della torsades des pointes, osservata ad esempio spesso in pazienti affetti da SQTL.
Le PDT sembrano facilitate da transitorie correnti di calcio verso l’interno. Le catecolamine e
l’ischemia, l’ipokaliemia e l’ipercalcemia aumentano il rischio di PDT, e in minor misura anche la
digitale. Pare che la triggered activity da PDT sia facilitata da un aumento della frequenza e sia
alla base di aritmie come RIVA e TV da sforzo. I calcio-antagonisti che riducono la
concentrazione intracellulare di calcio, come il verapamil, sono particolarmente utili nel
sopprimere TV da sforzo.
Bradiaritmie
I meccanismi con cui si possono generare aritmie ipocinetiche (o bradiaritmie) sono: 1)
Diminuzione della frequenza del NSA che resta comunque il pacemaker dominante: varie cause
di abbassamento della frequenza oppure mancata conduzione. 2) Sostituzione del NSA con un
altro pacemaker con frequenza più bassa: ossia ritmo di scappamento. Normalmente l’NSA è
sostituito da un focolaio ectopico giunzionale (ritmo di scappamento giunzionale, ma esiste
anche atriale o ventricolare). A volte il NSA recupera subito e si ha solo un battito di
scappamento. 3) Alterazioni della conduzione: principalmente atrio-ventricolare.
Bradiaritmie: Bradicardia sinusale, Blocco seno-atriale (1-2-3), Blocco atrio-ventricolare (1-2-3).
Bradicardia sinusale: riduzione della frequenza di scarica del nodo del seno inferiore a 60 bpm.
È una aritmia che può manifestarsi in seguito a ipotiroidismo, iperstimolazione vagale che può
aversi in malattie sistemiche, ma anche durante il vomito. La cardiopatia ischemia e la sindrome
del nodo del seno possono rallentare la frequenza così come farmaci come digitale, betabloccanti ed amiodarone. La frequenza in genere non scende sotto i 40 bpm e la sequenza PQRS è conservata. Facilmente è fisiologica in giovani sani, spesso se atleticamente allenati (per
28
iperstimolazione vagale, ad esempio durante il sonno), ed è difficile distinguerla da una
patologica. In genere è considerata anormale quando è inferiore a 40 bpm in stato di veglia e
senza allenamento fisico. Comune in pazienti allenati giovani e in anziani, anche una pausa del
ritmo sinusale (arresto sinusale) fino a 3s (nel sonno anche più lunga).
Blocco seno-atriale: ritardo della conduzione dello stimolo elettrico dal NSA all’atrio destro,
abbastanza raro. Viene anche detto: “blocco d’uscita del seno” e ne vengono distinti tre tipi:
Blocco seno-atriale di I grado: semplice allungamento del tempo di conduzione seno-atriale che
può anche non essere evidente all’ECG (in genere non si vede).
Blocco seno-atriale di II grado: pause sinusali intermittenti con mancata conduzione di alcuni
impulsi e ritmo atriale “regolarmente irregolare”. Nel tipo I l’ECG mostra un progressivo
allungamento dell’intervallo P-R e accorciamento dell’R-R seguito da una pausa, nel tipo II
l’intervallo P-R non cambia e ci sono solo pause che comportano una bradicardia sinusale.
Blocco seno-atriale di III grado: assenza di onde P sinusali per periodi di durata varaibile. È
completo, nell’ECG non si evidenziano onde P (mancata depolarizzazione atriale). Dopo la pausa
la frequenza riprende in genere come prima del blocco. La pausa può dare origine ad un battito
di scappamento (se c’è mancato funzionamento a lungo, anche a ritmo di scappamento) dovuto
a focolai ectopici che escono dalla soppressione normale del NSA.
[ECG: Ritmo di scappamento atriale: 60-80 bpm. Si hanno onde P’ diverse dalle onde P del NSA.
Ritmo di scappamento giunzionale,idiogiunzionale: 40-60 bpm. In genere non ci sono onde P
né P’ anche se può esserci un’onda P’ retrograda e invertita prima, dopo o durante il complesso
QRS.
Ritmo di scappamento ventricolare, idioventricolare: in genere se c’è un forte blocco AV o un
totale downward displacement of the pacemaker, 20-40 bpm. Enorme QRS, le onde P possono
esserci ma possono essere ovunque.
Battito di scappamento atriale: onda P’ diversa.
Battito di scappamento giunzionale: No onde P o invertita.
Battito di scappamento ventricolare: enorme QRS.]
Tutti i tipi di blocco seno-atriale possono essere associati a malattia del nodo del seno malato. In
questo caso nelle pause di attività elettrica può inserirsi un’attivazione di focus ectopici dando
una sindrome bradicardia-tachicardia. In genere i ritmi che insorgono come segnapassi
secondari in questo caso, anzicchè ritmi o battiti di scappamento, sono tachicardia
sopraventricolare, flutter atriale e fibrillazione atriale. [ECG: vedi tachiaritmie].
Malattia del nodo del seno (malato): SSS (sick-sinus syndrome), si distingue in estrinseca ed
intrinseca.
Estrinseca: dovuta a cause extracardiache come farmaci, SNA, ipotiroidismo, ipertensione
endocranica, manovre vagali, apnea da sonno, etc. (deve essere curata prima la patologia di
base per evitare un inutile impianto di pacemaker). Intrinseca: ha carattere degenerativo,
spesso per sostituzione con tessuto fibroso. Può essere anche associata a malattia coronarica,
infiammazione (tipo pericarditi e miocarditi, ma anche da LES), amiloidosi senile. Può raramente
essere anche ereditaria (AD).
Clinica: dal punto di vista ECGgrafico si manifesta con bradicardia sinusale, arresto sinusale,
blocco seno-atriale d’uscita (già visti) oppure una tachicardia sopraventricolare alternata a
bradicardia (sindrome tachicardia-bradicardia, da 1/3 a ½ dei pazienti). I sintomi possono essere
associati in questo caso alla tachicardia (palpitazioni, angina pectoris, insufficienza cardiaca) o a
bradicardia (sincope per riduzione della gittata, astenia, affaticamento). La tachicardia si
29
manifesta il più delle volte come fibrillazione ( o anche flutter) atriale con associato rischio di
tromboembolia (anticoagulanti soprattuto per pazienti a rischio). Se associata ad altre patologie
cardiovascolari, la sintomatolagia e la mortalità sono aumentate. Diagnosi: in genere con
anamnesi ed ECG. Serve però spesso un ECG a lungo termine (monitor ECGgrafici impiantabili) e
assocazione con la sintomatologia per poter fare la diagnosi di disfunzione del nodo seno-atriale.
Può servire alla diagnosi una valutazione invasiva dell’attività del nodo del seno, oppure una
valutazione della inadeguatezza cronotropa (incapacità di aumentare la frequenza sotto sforzo).
Terapia: non c’è terapia farmacologica particolarmente efficace, anzi bisogna sospendere
eventuali calcio-antagonisti e beta-bloccanti che possono causare la sindrome. La bradicardia
sinusale snon richiede terapia specifica e in generale la SSS non aumenta la mortalità. Nel caso
sia sintomatica, con sintomi ECG correlati, la terapia di prima scelta è l’impianto di un
pacemaker permanente.
Blocco atrio-ventricolare (BAV): disturbo di propagazione degli impulsi atriali ai ventricoli. Ne
vengono distinti tre tipi (gradi), e inoltre possono essere distinte sulla base della localizzazione
(NAV, fascio di His, branche di conduzione). L’eziologia è varia, può essere funzionale o organica.
Le eziologie funzionali (praticamente estrinseche) sono principalmente l’ipersensibilità del seno
carotideo, iperkaliemia, ipotiroidismo (endocrino-metaboliche), beta-bloccanti, calcio
antagonisti e digitale (farmacologiche). Le cause organiche sono infettive (come endocardite,
malattia di Lyme e di Chagas, Sifilide etc.), ereditarie/congenite (distrofie miotoniche),
infiammatorie (come LES e AR), infiltrative (come amiloidosi, sarcoidosi, emocromatosi),
degenerativa (con fibrosi progressiva idiopatica del sistema di conduzione che può
accompagnarsi all’invecchiamento). Anche la malattia coronarica, l’infarto miocardico, può
causare un blocco AV, per lo più transitorio (in un 15-20% di pazienti). Il blocco può verificarsi a
vari livelli nel sistema di conduzione AV, a livello del NAV (tipica), ma anche del fascio di His o
nel sistema His-Purkinje.
[ECG: In generale per distinguere a che livello è avvenuto il blocco bisogna guardare i complessi
QRS, se sono larghi è per lo più distale, se restano stretti è facile che sia a monte, magari proprio
a livello del NAV. Un QRS molto allargato è infatti tipico dei blocchi di branca, in quanto spesso
derivante da un QRS “riunito” (dei due ventricoli) che hanno le onde R (R e R’) fuori fase, perché
uno si depolarizza prima ed uno dopo (a causa del blocco sono desincronizzati). In un cuore
normale il QRS si verifica in un tempo inferiore a 0,12 s.]
Blocco AV di I grado: aumento del tempo di conduzione nel NAV con rallentamento e quindi
prolungamento dell’intervallo PR che supera i 0,2 s (200ms corrispondono ad un quadrato
grande) e resta costante in ogni ciclo. Spesso è asintomatico, si riscontra in soggetti sani con
aumentato tono vagale, o può essere dovuto a farmaci (ad esempio digitalici, beta-bloccanti,
calcio-antagonisti) o anche ad alcune cardiopatie (ischemia ad esempio con ostruzione della
coronaria destra che di norma vascolarizza il NAV, spesso causando un blocco transitorio ad
esempio per edema).
Blocco AV di II grado: assenza intermittente della conduzione degli impulsi dall’atrio al
ventricolo. Alcuni impulsi atriali vengono trasmessi, altri no, generando un ritmo ventricolare
irregolare con alcune onde P che restano da sole, non associate ad un QRS. Se ne distinguono
due tipi:
Mobitz tipo 1: o Wenckebach, in genere tipico del nodo AV. Si hanno una serie di cicli in cui
l’impulso viene progressivamente ostacolato fino al punto in cui l’onda P non è seguita da alcun
complesso QRS. Si può avere un rapporto P:QRS uniforme del tipo 3:2; 4:3; 5:4. Si ha un
allungamento progressivo dell’intervallo P-R (periodismo di Luciani-Wenckebach, fenomeno
fisiologico ad alte frequenze, ma patologico in questo caso perché si presenta anche a basse
30
frequenze) e un accorciamento di quello R-R. Dei due è quello associato ad una prognosi
migliore, spesso dovuto ad un eccesso di parasimpatico che inibisce il NAV o a medicinali che
hanno effetto simile. Può anche essere causato da cardiopatia ischemica, principalmente infarto
miocardico della parete inferiore.
Mobitz tipo 2: o Mobitz, in genere tipico di un blocco della conduzione distale o sotto-Hissiano,
spesso associato a ritardi della conduzione ventricolare come blocchi di branca. Ha maggiori
probabilità di progredire verso un blocco AV di grado completo. Un certo numero di
depolarizzazioni atriali (onde P) vengono bloccate del tutto prima di essere condotte al
ventricolo. Pertanto si producono normali cicli P-QRS-T senza alterazione degli intervalli P-R e RR, ma preceduti da una serie di onde P che non producono complessi QRS. Nella maggior parte
dei casi è di tipo Mobitz 2:1 con due onde P per ogni QRS, ma può essere anche 3:1 o anche con
rapporti maggiori (che indicano un aggravamento e blocchi come 4:1 e 5:1 sono indicati come
Mobitz avanzati. Le onde P sono regolari e puntuali, mai premature (questo per non confonderlo
con extrasistoli atriali). Ha una prognosi peggiore, ed è più frequentemente associato ad infarto
miocardico della parete anteriore.
[ECG: può essere molto difficile distinguere un Mobitz da un Wenckebach, soprattutto il Mobitz
2:1 che in effetti potrebbe anche trattarsi di un Wenckebach breve, con una pausa ogni due cicli
(2:1). Le differenze principali sono due: 1) nel Wenckebach noteremo un intervallo P-R
allungato, mentre nel Mobitz sarà del tutto normale. 2) Poiché il Mobitz è più distale, anzi
spesso associato a blocchi di branca, il complesso QRS sarà probabilmente più allargato. Per
distinguere i due tipi di blocco di II grado si può ricorrere anche a manovre manuali o
stimolazioni farmacologiche. Il nodo AV è fortemente innervato dal parasimpatico, mentre il
tessuto infranodale lo è molto meno. Manovre vagali, come il massaggio del seno carotideo
possono rallentare ulteriormente la conduzione nel NAV e non influenzare o addirittura
migliorare la conduzione nel sistema di conduzione ventricolare. Pertanto il nostro blocco AV
2:1, a seguito di un massaggio del seno carotideo: 1) Se Wenckebach: aumentano il numero di
cicli/serie e pertanto si avranno 2 complessi QRS ogni 3 onde P. 2) Se Mobitz: rimarrà di tipo 2:1
o addirittura la conduzione migliorerà e tornerà ad essere 1:1. Atropina, isoproterenolo ed
esercizio fisico hanno l’effetto opposto delle manovre vagali, bloccando il tessuto infranodale e
favorendo la conduzione del NAV, quindi ugualmente possono aiutarci a differenziare e i due tipi
di blocco].
A volte il blocco di II grado può dare anche un blocco AV parossistico, ossia una serie di onde P
non condotte.
Blocco AV di III grado: o blocco AV completo, nessun impulso atriale viene condotto al sistema
ventricolare. Pertanto focolai ectopici ventricolari dovranno intervenire con il proprio
automatismo, un’attività di pacemaker che impone un ritmo idioventricolare, con frequenza tra
20 e 40 bpm. In realtà se il blocco colpisce solo la parte superiore del NAV può rimanere un
focolaio giunzionale. Se coinvolge tutto il NAV o è sotto il fascio di His allora funzionerà un
focolaio ventricolare. Pertanto più alto è il blocco (sopra anziché sotto-hissiano) maggiore e la
frequenza e migliore è la prognosi (inoltre il QRS sarà meno slargato). È indice di una patologia
avanzata del sistema di conduzione. Deve essersi verificato un blocco completo del sistema di
conduzione. Poiché il blocco è bidirezionale, la frequenza degli atri resta decisa dal NSA. Nel
tracciato risulterà quindi evidente una frequenza dei complessi QRS determinata dal pacemaker
ventricolare non più soppresso, ma contemporaneamente c’è un ritmo sinusale delle onde P. Si
ha pertanto una dissociazione AV. Non tutte le dissociazioni sono blocchi. È un blocco AV solo se
la frequenza atriale è maggiore di quella ventricolare (si ha infatti dissociazione anche nel ritmo
si scappamento ventricolare o nell’idioventricolare). Nel caso le frequenze di atrio e ventricolo
siano uguali (dissociazione AV isoritmica) può essere difficile porre diagnosi soprattutto se le
31
onde P precedono (per caso) i QRS.
[ECG: Per distinguere a che livello è avvenuto il blocco completo (che in genere è determinato
da un aggravarsi delle stesse condizioni che determinano gli altri blocchi) si notano due
differenze: 1) se il blocco è sopra la giunzione AV si ha l’instaurarsi di un ritmo idiogiunzionale
(40-60), se invece è al disotto il ritmo è idioventricolare (20-40) 2) se il blocco è sopra la
giunzione inoltre è più facile che i QRS siano stretti anzicchè allargati e simili a extrasistoli
ventricolari (PVC)].
A causa della notevole bradicardia che viene a crearsi, sì può avere una riduzione della gittata
cardiaca che può portare ischemia cerebrale e sincope, una condizione definita sindrome di
Morgagni-Stokes-Adams.
Diagnosi: oltre all’ECG, ed alle eventuali manovre vagali o somministrazioni di farmaci che
influenzano il SNA vi sono valutazioni diagnostiche aggiuntive, che possono ad esempio essere
indicati in pazienti con sincope e sospetto blocco AV di alto grado. Registrazione
dell’elettrocardiogramma del fascio di His: tramite catetere si misurano attività atriale,
elettrogramma del fascio di His e attivazione ventricolare.
Si valutano: Intervallo PA: tempo tra onda P e deflessione atriale sul fascio di His= o < 50ms.
Intervallo AH: tempo tra deflessione atriale sul fascio di His ed elettrogramma di His (tempo di
conduzione attraverso il NAV)< o =130ms. Intervallo HV: tempo tra elettrogramma di His ed
esordio del QRS (tempo di conduzione attraverso il sistema HIs-Purkinje)< o =55ms. Anomalie in
questi tempi possono facilitare la diagnosi specie nelle D.D. tra Mobitz e Wenckeback. Un blocco
nell’His-Purkinje è associato ad un rischio maggiore di progressione verso gradi più elevati di
blocco.
Terapia: il trattamento migliore nel BAV è il pacing artificiale temporaneo o permanente.
Bisogna escludere prima le cause reversibili di blocco. Un blocco nel NAV può trarre beneficio da
atropina o isoproterenolo che riducono il tono vagale (e dunque l’inibizione sul NAV). Le terapie
farmacologiche possono richiedere tempo e nel frattempo si può usare un pacemaker
temporaneo transcutaneo o transvenoso. In genere un blocco di conduzione distale richiede un
impianto di pacemaker permanente.
Pacemaker: dispositivo con la capacità di generale stimoli elettrici ritmici, mediati attraverso
cateteri inseriti in genere per via venosa che raggiungono le camere cardiache. Due tipi di
cateteri: unipolari con un unico elettrodo a contatto con l’endocardio della camera stimolata, e
bipolari con due elettrodi stimolatori. Tutti i cateteri funzionano come conduttori a due vie una
per trasmettere impulsi ed una per trasmettere al pacemaker l’attività elettrica intrinseca del
cuore (sensing). La batteria è al litio è si scarica in 5-10 anni, dopo i quali è sostituito. È
identificato da un codice a 5 lettere (le prime tre sono praticamente importanti). La prima e la
seconda sono A(atrio), V(ventricolo), D(atrio e ventricolo), O(nessuna), S(singola) e indicano la
prima la camera stimolata, la seconda la camera in cui avviene il sensing. La terza è la risposta
all’evento rilevato O(nessuna), I(inibizione), T(stimolazione ossia triggered), D(inibizione e
stimolazione). La quarta si riferisce alla programmabilità, la seconda alla risposta alla
tachiaritmia. L’elettrostimolazione è usata sia per bradi che tachiaritmie. I pacemaker sono
affidabili, ma possono dare problemi come fastidi, infezioni e la sindroma da pacemaker (vari
sintomi cardiaci, risolta con modifiche della modalità di pacing).
Stimolazione temporanea: Transvenosa: in genere con elettrocatetere inserito dalla vena
femorale. Transcutanea: elettrodi sulla parete toracica anteriore e posteriore. Epicardica:
temporanea durante intervento cardochirurgico. La stimolazione temporanea è indicata in
pazienti con blocchi AV o disfunzioni del NSA, asistolia in corso di IM, magari aspettando
l’effetto di un trattamento farmacologico o l’impianto di un pacemaker permanente.
32
Stimolazione permanente: pacemaker applicato per via venosa dalla cefalica, con tasca di
alloggiamento preparata nel grande pettorale.
È indicato prevalentemente in: 1) Disfunzione del nodo SA: disfunzione con bradicardia
sintomatica, inadeguatezza cronotropa sintomatica e forse altri casi con bpm<40. 2)
Ipersensibilità del seno carotideo e sincope vasovagale: al massimo pacing temporaneo 3) Difetti
di conduzione AV: Blocco AV di terzo grado o alto grado (intermedio tra secondo e terzo),
sintomatico o con asistolia o associato a patologie ereditarie, blocco di secondo grado con
bradicardia sintomatica (specie se con arresti superiori a 3 s.) o con QRS ampio con o senza
sintomi (forse terzo grado asintomatico e alcuni di secondo). 4) Infarto miocardio: se associato a
blocco AV (anche se spesso è transitorio, specie nell’IM inferiore) di II o III grado più se
sintomatico, o se associato anche a blocco di branca. Nella stimolazione permanente dei
bicamerali sono introdotti due cateteri uno che stimola l’atrio destro e l’altro che stimola il
ventricolo destro. Nel blocco AV: se associato a buona attività del NSA ha sempre due elettrodi
uno nell’atrio destro l’altro nel ventricolo destro, ma quello nell’atrio ha solo funzione di
sensing, trasmettendo al ventricolo l’onda di depolarizzazione spontanea del NSA. Se l’NSA non
funziona c’è bisogno di due stimolatori.
I pacemaker più usati sono VVI ed i DDD:
VVI: pacemaker ventricolare a domanda. Evita alle frequenze di scendere sotto il valore
impostato, ma non stimola se l’attività spontanea è sufficiente. È semplice e piccolo, ma non
mantiene una grande sincronizzazione AV né risponde molto con aumenti di frequenza allo
sforzo. Si applica soprattutto nella fibrillazione atriale cronica a bassa risposta ventricolare.
Singolo catetere in ventricolo (V); sensing in ventricolo (V); risponde al sensing con un’inibizione
della stimolazione (I).
DDD: stimolazione bicamerale (D), sensing bicamerale (D) e sequenzialmente all’attività atriale
spontanea risponde con inibizione o stimolazione (D).
Blocchi di branca: alterazioni nella trasmissione di impulsi elettrici attraverso la branca destra e
sinistra del fascio di His. Gli stimoli giungono nai ventricoli tramite vie più lente, ossia fibre del
muscolo ventricolare. Il ventricolo non bloccato si depolarizza prima. Si hanno così dei complessi
QRS slargati perché non sincronizzati. Il QRS allargato è >120ms nel blocco completo, tra 100120 ms nel blocco incompleto (che però presenta in genere morfologia simile).
[ECG: Se c’è forte tachicardia la rapida successione di QRS allargati può far pensare ad una
tachicardia ventricolare. Normalmente il vettore del complesso QRS è orientato nella direzione
in cui si verifica il ritardo nella depolarizzazione, avanti a destra nel BBD e indietro a sinistra nel
BBS. A volte l’ago utilizzato nella rilevazione per problemi meccanici se c’è grande ampiezza può
ritardare e presentare QRS più slargati del reale, pertanto è bene valutare soprattutto le
derivazioni degli arti, che sono a minore ampiezza, nel valutare i blocchi di branca].
Blocco di branca destra, BBD: 1) Aumento della durata del QRS 2) Onda S nella derivazione D1
(ossia orizzontale con elettrodo positivo al braccio sinistro) e profonde S nella derivazione V6. 3)
In V1 c’è una forma RSR’e qRS in V6 (sempre con netta onda S, forma a M) 4) Inversione
secondaria dell’onda T da V1 a V6 (negativa nelle derivazioni in cui c’è RSR’). In realtà in generale
nei blocchi l’onda T ha andamento opposto all’ultima deflessione del QRS per alterazione della
ripolarizzazione dovuta ad alterazioni miocardiche 5) Aumento della deflessione intrinsecoide
ossia dell’intervallo tra l’inizio del QRS e il primo picco del QRS. Dal punto di vista clinico il BBD
non implica sempre una patologia cardiaca., c’è anche in soggetti sani. È comune in caso di
embolia polmonare a seguito del sovraccarico del ventricolo destro. Non è indicato un
intervento terapeutico.
33
Blocco di branca sinistra, BBS: 1) Aumento durata del QRS 2) Assenza di onde q in D1 e in V4, V5
e V6 (si parte subito in alto, senza onda negativa. 3) In V1 il complesso è QS (senza onda positiva
R, quindi tutto negativo) con stessa forma del complesso in D1 e V6 (che invece ha onde tutte
positive, senza q). 4) Inversione secondaria dell’onda T (negativa nelle deflessioni con ampia R e
senza Q cioè D1 e V6). Il BBS è riscontrato per lo più in cardiopatici per lo più con coronaropatie,
cardiopatia ipertensiva, patologie aortiche valvolari e miocardiopatie (qundi IM, SC, miocarditi
etc.). Non richiede interventi terapeutici speciali oltre alla risoluzione della cardiopatia di base.
Mobitz intermittente: blocco di branca permanente di un lato e intermittente di un altro
provoca un blocco AV intermittente con un QRS che salta ad intervalli intermittenti.
Emiblocchi: La branca sinistra del fascio di His è costituita da due fascicoli: anteriore e
posteriore. Il blocco di uno di questi due fascicoli si definisce emiblocco. Normalmente non
comporta uno slargamento del QRS bensì una sua deviazione assiale (modificano la via di
depolarizzazione del ventricolo) e in parte un cambio di morfologia.
Emiblocco anteriore sinistro: il ventricolo sinistro viene attivato solo dal fascicolo posteriore con
impulsi che diffondono nella parte anteriore a partire dalla posteriore. 1) Deviazione assiale
sinistra maggiore di -40° 2) Complesso qR in D1 3) rS in D2, D3, aVF 4) QRS normale o
leggermente allargato. (in generale anche Q 1S3 con onda Q in I e S larga e sottoslivellata in III).
Tipici di cardiomiopatie e cardiopatia ischemica.
Emiblocco posteriore sinistro: l’attivazione ventricolare sinistra si realizza solo nella regione del
fascicolo anteriore. 1) Marcata deviazione assiale destra 2) Piccole onde q con alte onde R in D2,
D3 e aVF. QRS è normale o leggermente allargato. Aspetto S 1Q3. È più raro del destro. Gli
emiblocchi avvengono spesso in concomitanza con infarti in quanto anche i fasci di conduzione
ventricolari sono normalmente irrorati dalle coronarie. La coronaria destra irrora NSA, NAV,
fascio di His e il ramo posteriore della branca sinistra. La branca sinistra è irrorata anche dalla
coronaria sinistra. Il ramo discendente anteriore della coronaria sinistra irrora anche lui parte
del ramo posteriore della branca sinistra, il ramo anteriore della branca sinistra e la branca
destra. Ostruzioni possono quindi causare un blocco di un solo fascio o di più fasci (bi
fascicolari). Ad esempio un’ostruzione del ramo discendente anteriore può causare BBD +
emiblocco anteriore. Da quanto detto si capisce pure perché l’emiblocco posteriore è più raro
(è irrorato sia dalla coronaria destra che dal ramo discendente della sinistra), ma anche più
grave, soprattutto se associato a BBD (forte tendenza a progredire in blocco AV). Inoltre si
capisce che il BBS può essere dovuto a emiblocco anteriore + posteriore e che il blocco AV può
essere dovuto a 2 blocchi di branca (o addirittura BBD + emiblocco anteriore + posteriore). Le
combinazioni di blocchi bi fascicolari sono spesso intermittenti, connun blocco fisso ed uno non
sempre visibile. Esistono anche blocchi trifascicolari i quali se continui costituiscono un blocco
AV, se intermittenti danno luogo ad un fenomeno di Mobitz intermittente.
Anche dei blocchi bi fascicolari non sono ad alta mortalità a meno che non siano (come spesso
però accade) associati ad un infarto del miocardio.
Tachiaritmie
Definizione: o aritmie ipercinetiche. Si tratta quindi di ritmi tendenzialmente superiori a 100
bpm, considerando però alcune eccezioni in cui il ritmo atriale non corrisponde a quello
ventricolare. Sono comprese tachicardie da focolai ectopici, circuiti di rientro, forme con battiti
prematuri isolati. Il paziente può essere asintomatico, avvertire palpitazioni e polso irregolare, a
volte anche vertigini e sincope per riduzione della gittata cardiaca, o affanno per aumento della
pressione di riempimento cardiaco. Anche malessere toracico, ma la gravità dei sintomi è
34
comunque determinata dalla patologia cardiaca di base.
Tachicardia sinusale: aumento della frequenza degli impulsi, a partire dal NSA, con aumento
della frequenza oltre i 100 bpm. Spesso è una risposta ad uno stimolo fisiologico, come
l’esercizio fisico, ma è soprattutto comune in soggetti ansiosi. È possibile anche in caso di febbre,
anemia o ipotensione, tireotossicosi, nelle intossicazioni da caffè o tabacco, digestione uso di
farmaci come l’atropina (blocco parasimpatico), amine simpatico-mimetiche. Raro che superi i
150 bpm. Se è associata a cardiopatia organica e si manifesta in condizioni di riposo indica
insufficienza cardiaca. L’onda P è normale, perché c’è in genere normale attività atriale e
normale conduzione. Può essere comune nelle donne giovani, spesso ansiose, con prolasso della
valvola mitrale e cosiddetta sindrome da iperbetastimolazione adrenergica (sindrome di
Barlow?), con aumento della frequenza in ortostatismo. Si ha un’ottima risposta ai betabloccanti. La sequenza P-QRS è conservata e raramente supera i 130 bpm. La terapia è in genere
evitare sostanze eccitanti, correggere un’eventuale patologia di base (tipo ipertiroidismo), fare
uso di beta-bloccanti solo nel caso in cui ci sia pericolo come con un paziente che abbia una
cardiopatia ischemica.
Tachicardia sinusale inappropriata: la frequenza cardiaca aumenta spontaneamente e in modo
inappropriato. È rara spesso si presenta dopo un’infezione virale, si risolve spontaneamente
entro 3-12 mesi. I sintomi possono essere di vario grado, anche gravi come sincope e
palpitazione.
Fibrillazione atriale: o FA, è l’aritmia sopraventricolare più frequente. Si ha un’attivazione
elettrica atriale scoordinata con contrazioni deboli e incomplete ed un ritmo ventricolare del
tutto irregolare. Probabilmente la causa consiste aree multiple di (micro) rientro (secondo Dubin
si hanno scariche provenienti da numerosi focolari ectopici parasistolici, e quindi che agiscono in
modo indipendente l’uno dall’altro) le quali inviano continui impulsi al nodo AV che, sempre in
considerazione del proprio periodo di refrattarietà, conduce queste scariche ai ventricoli
provocando un’attivazione irregolare e in genere più frequente di essi (120-160, bpm fino a
200). In questo caso bisognerà valutare la frequenza “media”, e quindi magari i QRS presenti in 6
s (30 quadrati grandi) moltiplicati per 10. La frequenza di stimolazione atriale è invece di 350450 (e oltre) bpm.
Eziopatogenesi: La FA è più comune con l’avanzare dell’età (5% sopra i 70 anni). Talvolta può
essere dovuta a ipertiroidismo e tireotossicosi (FA parossistica). È frequente nel periodo
successivo ad interventi chirurgici maggiori, a seguito di tachicardie sopraventricolari, ma anche
durante un IM, cardiomiopatia dilatativa, difetto del setto interatriale e sindrome braditachicardica. La stenosi mitralica è una delle cause più frequenti di FA.
Clinica: si può avere riduzione della gittata cardiaca per ridotta capacità degli atri fibrillanti di
contrarsi, ma soprattutto per la riduzione del tempo di riempimento ventricolare, con volume
tele diastolico ridotto (aumento frequenza anche ventricolare). Se ne distingue una forma
parossistica e una stabile.
Parossistica: da alcune ore a qualche giorno, con conversione spontanea in ritmo sinusale. In
genere si presenta con cardiopalmo. Il polso è aritmico e frequente. Con l’ascoltazione
contemporanea dei toni cardiaci e del polso si nota una dissociazione centro-periferia poiché
non tutte le contrazioni cardiache producono un’onda sfigmica rilevabile. Alcuni sono
asintomatici, e comunque le palpitazioni possono essere più o meno intense. Si può avere anche
sincope o vertigini.
[ECG: c’è assenza di onde P, con onde F, più visibili in V1 perché più vicina all’atrio, che sono
35
praticamente oscillazioni della linea isoelettrica. I QRS sono irregolari con ampiezza varia]
Uno dei problemi principali della FA è il controllo della coagulazione del sangue. Si verifica
infatti una stasi ematica per mancata contrazione dell’atrio che può portare alla formazione di
trombi murali (nell’atrio) i quali possono staccarsi e dar vita a fenomeni embolici, ictus, ischemie
(soprattutto nei pazienti >65 anni e con rischio aumentato). Se c’è concomitante dilatazione
atriale poiché c’è più sangue il rischio aumenta. Bisogna porre attenzione perché il restaurarsi
del ritmo sinusale può far staccare trombi.
Terapia: deve badare a 1) controllare la frequenza per stabilizzare il paziente sotto il profilo
emodinamico e 2) controllare la coagulazione. 1) si usano calcio-antagonisti: verapamil o
diltiazem 2) con eparina o warfarin (terapia cronica).
Se le condizioni del paziente indicano di interrompere immediatamente la FA, si può operare la
conversione a ritmo sinusale in due modi, ma sempre dopo aver scoagulato il paziente (con
eparina per 5-7 giorni):
Farmacologico: amiodarone, verapamil, procainamide, digossina. Elettrico: cardioversione trans
toracica con shock bifasico a 200 J somministrato in maniera sincrona al QRS.
[Farmaci antiarimici: Classe I: agenti che bloccano la corrente verso l’interno del Na. IA:
prolungano inoltre la durata del potenziale d’azione: Procainamide, Chinidina, Disopiramide. Ic:
?. Classe II: agenti antisimpatici (suppongo beta-bloccanti): Acebutololo, Atenololo,
Metoprololo, Nadololo. Altri ancora sono carvedilolo, bisoprololo, metoprololo succinato.
Classe III: agenti che prolungano principalmente la durata del potenziale d’azione: Amiodarone,
Dofetilide, Sotalolo. Classe IV: calcio-antagonisti: Diltiazem, Verapamil].
Flutter atriale: (detta anche tachicardia atriale macrorientrante) aritmia comune, caratterizzata
da una regolare attivazione atriale dovuta ad un circuito di macrorientro spesso sviluppatosi a
seguito di un intervento chirurgico, attorno all’annulus tricuspidale. Secondo alcuni è dovuto
all’attività di un unico focolaio ectopico atriale. Anche il flutter può essere parossistico o
permanente e le cause sono simili a quelle della fibrillazione atriale (compresa la tireotossicosi,
la stenosi mitralica, interventi chirurgici, ma anche patologie polmonari). Nell’ECG,
principalmente nelle derivazioni D2, D2 e aVF si notano le onde flutter, ossia onde sempre
uguali, con frequenza in genere tra 250-300 (e oltre) bpm. La risposta ventricolare in genere
avviene 1:3 o 1:2 onde di flutter (130-150bpm) e può essere regolare o irregolare. Il polso
appare ritmico e frequente. Quando le onde di flutter sono <200 bpm, la situazione può
aggravarsi con risposta ventricolare 1:1 e gravi conseguenze emodinamiche. Il flutter rispetto
alla fibrillazione ha una maggior probabilità di far scaturire un’insufficienza cardiaca.
Terapia: come per la fibrillazione bisogna tenere sotto controllo la frequenza con calcioantagonisti e tentando la conversione farmacologica a ritmo sinsusale con amiodarone,
verapamil, procainamide, digossina, o meglio ancora con cardioversione elettrica a 50-100J.
Anche nel flutter c’è bisogno di una terapia anticoagulante.
Ritmi ectopici sopraventricolari:
Extrasistoli: l’extrasistole è una contrazione cardiaca che insorge in risposta ad un impulso
proveniente da una sede ectopica che può essere l’atrio, il NAV o il ventricolo.
Sopraventricolari sono definite le extrasistoli atriali e giunzionali.
Extrasistole atriale: può provenire da un qualsiasi punto degli atri e depolarizzare il tessuto
circostante. All’ECG è rilevata come un’onda P’ che indica una depolarizzazione atriale “fuori
tempo”. La depolarizzazione prodotta da un focolaio ectopico, diverso dal NSA, può essere
condotta o non condotta attraverso il NAV e quindi provocare o meno la comparsa di un
complesso QRS. Inoltre potrebbe anche non essere in grado di condurre bene per tutto
36
l’apparato di conduzione ventricolare e dare in questo modo un complesso QRS leggermente
allargato, si ha cioè un’extrasistole con conduzione ventricolare aberrante. Ad ogni modo, la
depolarizzazione è in grado di depolarizzare anche il NSA il quale si risincronizza in base al tempo
in cui è avvenuta l’extrasistole. La sua attività di pacemaker riparte in sincronia con il battito
prematuro. L’intervallo tra la sistole normale e l’extrasistole è definito copula, quello tra
l’extrasistole e la successiva sistole è la pausa. Questa pausa può essere compensatoria o non
compensatoria. Nell’extrasistole sopraventricolare è dovuta alla risincronizzazione del NSA,
nell’extrasistole ventricolare è in genere dovuta al fatto che il ventricolo che è stato attivato
dall’extrasistole non è in grado di rispondere subito alla stimolazione normale che segue. Le
onde P’ nelle extrasistoli atriali hanno forma diversa da quelle del NSA perché provengono da
parti diverse degli atri. A volte, se provengono dal basso (così come vale per le extrasistoli
giunzionali) possono dare una conduzione retrograda all’interno dell’atrio e pertanto dare
un’onda P’ invertita (depolarizzazione atriale dal basso all’alto).
Extrasistole giunzionale: è molto più facile che ci sia conduzione retrograda ed anche che ci sia
conduzione ventricolare aberrante, a causa della posizione più bassa del focolaio ectopico. Le
extrasistoli sopraventricolari possono presentarsi anche con uno schema che si ripete,
alternandosi con il ritmo sinusale. Il ciclo che contiene l’extrasistole assieme al ciclo a cui si
associa viene detto distico. In pratica quando un’extrasistole si associa alla fine di ciascun ciclo
abbiamo bigeminismo atriale, rapporto 1:2. Ogni tre cicli = trigeminismo atriale, etc. Clinica:
quando tendono a presentarsi in maniera sporadica le extrasistoli sopraventricolari hanno un
significato benigno e sono comuni nei soggetti sano. Se molteplici possono rischiare di causare
aritmie sopraventricolari come la fibrillazione atriale o la tachicardia sopraventricolare
parossistica.
Terapia: extrasistoli sporadiche in giovani sani non richiedono terapia. Se insorgono in sindromi
da stimolazione adrenergica o in ipertiroidismo traggono notevole beneficio da beta-bloccanti.
Tachicardia parossistica sopraventricolare: è un ritmo accelerato, tra i 150 e i 200 (e oltre)bpm,
che genera da un focolaio ectopico (o da un circuito di rientro) che ha origine negli atri (atriale) o
nel nodo AV (giunzionale). È definita parossistica perché si genera improvvisamente e non come
una risposta graduale (tipo tachicardia sinusale) all’esercizio fisico o ad altri stimoli. Il NSA,
poiché la frequenza di scarica è più alta, viene soppresso. Le onde P’ generate sono diverse dalle
onde P normali. Possono essere anche invertite, ancor più facilmente se provengono dal basso
atrio o dal NAV, questo perché l’attivazione atriale avviene per via retrograda. Ogni impulso di
depolarizzazione atriale viene condotto ai ventricoli. Le onde P’ anomale hanno un rapporto
fisso con i QRS. In genere sono tachicardie reciprocanti, ossia avvengono in seguito alla presenza
di un circuito di rientro che può essere rappresentato dal NAV o da una via di conduzione
anomala(WPW). All’interno del NAV si può avere un rientro a causa della presenza di due vie di
conduzione, una più lenta e una più rapida (inoltre nel momento in cui si verifica un’extrasistole
atriale (battito ectopico) una via può essere ancora refrattaria e l’altra no, generando un
rientro). La tachicardia atriale e quella giunzionale hanno aspetti ECGgrafici molto simili in
quanto spesso le onde P’ della atriale sono mescolate con le onde T, così come le onde P’
invertite della giunzionale possono essere prima, dopo o durante (e quindi coperte) un QRS.
Pertanto si considerano insieme nel termine generico di sopraventricolare il che non è un
problema perché la clinica e la terapia sono ampiamente sovrapponibili e urge per lo più
distinguerle da quella ventricolare. I complessi QRS, soprattutto nella giunzionale, possono
essere più ampi sempre a causa di una conduzione ventricolare aberrante.
Clinica: l’esordio così come la fine dalla tachicardia è appunto improvviso e brusco. In genere
l’eziologia è benigna, ed è comune nei bambini (più femmine) e in giovani con cuore sano. Le
37
crisi posono anche essere scatenate da un grande pasto o da una forte emozione e la sensazione
è come di un forte cardiopalmo come un “frullio di ali sul petto”, ma sono in genere ben
tollerate.
Terapia: Spesso, soprattutto per le tachiaritmie da rientro del NAV, può bastare la manovra
vagale o l’adenosina. Come terapia farmacologica, il farmaco che ha avuto più effetto è il
verapamil, nei soggetti con insufficienza cardiaca la digitale (digossina).
Tachicardia atriale con blocco: (blocco AV naturalmente). È una forma di tachicardia atriale,
quindi con frequenza di scarica di un focolaio ectopico atriale tra 150 e 200 (e oltre) bpm. Non è
molto frequente, ma si verifica solo in soggetti malati. In genere è dovuta ad un sovradosaggio e
quindi tossicità da digitalici, specialmente se avviene in un paziente con ipopotassiemia. La
digitale irrita un focolaio atriale a tal punto da emettere impulsi ad una frequenza maggiore del
NSA. Nel frattempo la digitale inibisce il NAV e lo blocca impedendogli di condurre impulsi a
quella velocità, perché il NAV non è in grado di recuperare l’eccitabilità in tempo. La frequenza
ventricolare è più bassa e il grado di questo blocco può essere fisso (regolare rapporto tra onde
P e QRS) o variabile. All’ECG si hanno onde P’ in rapida successione alte e appuntite (quasi come
dei QRS) di cui alcune sono seguite da un QRS (in genere normale). Comunemente il rapporto
P’:QRS è di 2:1.
Clinica: in genere avvertono cardiopalmo improvviso. Se c’è alta risposta ventricolare la
tachicardia può condurre a scompenso e angina.
Terapia: sono utili infusioni ev di potassio.
Ritmo giunzionale accelerato: detta anche tachicardia nodale non parossistica. È associata ad
intossicazione digitalica o IM inferiore. Normalmente i focolai ectopici del NAV scaricano ad una
frequenza di 40-60 bpm, ma in questo caso scaricano ad una frequenza maggiore (tra 70 e 140)
e quindi sono anche in grado di sopprimere l’NSA. Normalmente infatti il ritmo idiogiunzionale è
una bradicardia che può instaurarsi solo come ritmo di scappamento quando non funziona il
pacemaker dominante o la conduzione da esso. L’ECG mostra QRS normali preceduti in genere
da onde P’ anomale che si presentano in genere invertite e dunque negative in II, III e aVF a
causa della depolarizzazione atriale retrograda. In genere non dà sintomi particolari e non serve
terapia a meno che non si debba curare la condizione che l’ha causata. Si deve sospendere la
digitale o utilizzare (come per altri casi da intossicazione digitalica) frammenti di anticorpi antidigitalici.
Sindrome di Wolff-Parkinson-White: WPW. Detta anche sindrome da preeccitazione
ventricolare. È caratterizzata dalla presenza di una o più vie accessorie che forniscono diretta
connessione AV saltando il NAV. Una via accessoria è detta fascio di Kent. Il ventricolo viene
pertanto eccitato attraverso due vie, questo fa sì che l’impulso atriale per la conduzione
ventricolare scavalca il NAV e attiva rapidamente i ventricoli dando luogo ad una sorta di
preeccitazione ventricolare. Questo perché è molto importante ricordare che la via accessoria è
generalmente più rapida del NAV, mentre può avere un periodo refrattario più lungo o più
breve. In questo modo l’intervallo P-R è accorciato e inoltre il complesso QRS risulta allargato e
con un tratto iniziale ascendente (onda delta) dovuto al fatto che la conduzione attraverso la via
accessoria comincia prima (P-R più piccolo) ma è più lenta (onda delta) e graduale ( poiché
avviene a partire dalla via accessoria attraverso il muscolo) di quella avvenuta tramite il fascio di
His-Purkinje. La WPW è, dopo le alterazioni della conduzione nel NAV, la seconda causa più
comune di tachicardia parossistica sopraventricolare, sempre a causa di un circuito di rientro.
Nella maggior parte dei casi infatti la via accessoria ha un periodo refrattario più lungo del NAV.
38
Pertanto può accadere che un battito ectopico atriale (CPA) possa essere condotto nel NAV e
restare bloccato nella via accessoria ancora refrattaria, e poi percorrere quest’ultima in modo
retrogrado attivando gli atri (attivazione retrograda degli atri = echo beat, battito eco). Se ciò si
ripete si può sviluppare tachicardia.
Questa tachicardia circolare è detta ortodromica, ed è associata ad un complesso QRS normale
(perché la via accessoria conduce ora solo in senso retrogrado e quindi scompare l’onda delta).
Più raramente a via accessoria ha un periodo refrattario più breve e dunque attraverso i fasci di
Kent si avrà conduzione anterograda e si avrà andamento retrogrado lungo il NAV, generando
una tachicardia parossistica sopraventricolare antidromica. Questa è associata ad un QRS
allargato (perché ora è la via accessoria che conduce in senso anterogrado e l’onda delta dovuta
alla lenta e graduale conduzione attraverso il muscolo resta).
Clinica: si possono manifestare episodi aritmici già nell’infanzia oppure in età adulta. Si può
avere tachicardia parossistica sopraventricolare, flutter atriale o fibrillazione atriale. Più
comunemente tachicardia parossistica, al secondo posto FA ( 50% dei pazienti con vie accessorie
è a rischio per FA). Queste aritmie da rientro sono maggiormente sintomatiche nei pazienti con
WPW che in quelli con classiche aritmie da rientro nel NAV, si ha spesso lipotimia e sincopi.
Questo avviene perché, a causa della presenza anche della via accessoria, molto spesso si ha
conduzione rapida ai ventricoli, e quindi una risposta ventricolare alla fibrillazione atriale molto
maggiore di quella che si ha nei pazienti con aritmie da rientro del NAV. Si ha spesso una
tachicardia sopraventricolare parossistica con frequenze tra 150 e 250 bpm e QRS più spesso
stretti (via accessoria con periodo refrattario più lungo) che allargati. Lo stesso avviene per la
fibrillazione atriale, in cui la risposta ventricolare è persino maggiore (anche 1:1, con frequenze
pericolosamente elevate). In generale la diagnosi necessita dell’ECG e comunque un ritmo
irregolare ed una frequenza ventricolare media>200bpm (QRS larghi o stretti) può far
propendere per una WPW.
Sono distinti due tipi di sindrome:
WPW di tipo A: via accessoria localizzata a sinistra, vicino la mitrale (si avrà onda delta e QRS
verso l’alto in V1 e V6)
WPW di tipo B: via accessoria localizzata a destra (QRS negativo in V1 e positivo in V6).
Terapia: come nelle aritmie da rientro la stimolazione vagale che rallenta il NAV può essere
sufficiente a porre fine alla tachicardia da rientro. In questo può servire anche l’adenosina così
come pura calcio-antagonisti (diltiazem o verapamil) o propranololo (beta-bloccante) o
procainamide. Nei casi di WPW con FA con conduzione anterograda per la via anomala i farmaci
d’elezione sono quelli che aumentano la refrattarietà e riducono la conduzione lungo la via
anomala, ossia quelli di classe I come procainamide (IA) chinidina, flecainide (Ic). Pazienti con
episodi frequenti di aritmie sintomatiche dovrebbero essere trattati con ablazione trans
catetere della via accessoria, sicura nel 95% dei casi, ma con rischi di arresto cardiaco che vanno
soppesata con i rischi emodinamici, con il rischio di FV e con la gravità dei sintomi.
Aritmie ventricolari
Extrasistoli ventricolari: o complessi prematuri ventricolari (PVC). Sono battiti ectopici a
provenienza ventricolare, contrazioni ventricolari premature. Sull’ECG appaiono come complessi
QRS di ampio voltaggio non preceduti da un’onda P, se provengono da destra sono simili al BBS
(negativi in V1), se provengono da sinistra sono simili al BBD (positivi in V1). Sono più ampi dei
QRS normali perché la conduzione è disordinata. Due CPV successivi sono definiti paia o coppie.
Si parla di tachicardia ventricolare quando ci sono almeno tre CPV successivi ad una frequenza
>100 bpm (se si fermano spontaneamente è TV non sostenuta).
39
L’intervallo tra un battito ectopico e il battito normale è la pausa extrasistolica che
comunemente è compensatoria (anche se non compensa nulla) perché la somma della copula
più la pausa è uguale a 2 R-R normali. La pausa compensatoria è dovuta al fatto che
normalmente il CPV non raggiunge il NSA e non è in grado di farlo risincronizzare alla sua
frequenza come fa un’extrasistole sopraventricolare [in cui allora la pausa è soprattutto non
compensatoria] pertanto il NSA genera un impulso che viene condotto attraverso il NAV e
raggiunge i ventricoli quando non sono ancora in grado di rispondere, e pertanto il ritmo
sinusale riparte dopo il secondo impulso del NSA (per cui pausa + copula= 2 battiti).
Se il CPV è in grado di risincronizzare l’NSA allora si ha una pausa non compensatoria.
Talvolta, più raramente, i CPV non sono affatto seguiti da una pausa e pertanto si definiscono
interpolati. Le extrasistoli ventricolari, comuni anche nelle persone sane, sono provocate da
molteplici cause quali acidosi e ipokaliemia, stress fisici e psichici, farmaci e droghe, cardiopatie.
Al polso appaiono come battiti prematuri meno forti (sistole ventricolare che avviene quando il
cuore non è del tutto riempito) seguiti una pausa a cui fa seguito un battito più forte. Sono
frequenti in pazienti (soprattutto donne, forse sindrome di Barlow) con prolasso della mitrale
oppure in caso di IM o cardiopatia ischemica e in questo caso hanno una prognosi sfavorevole.
Se sono dovute a stimolazione beta-adrenergica possono essere usati beta-bloccanti, ma
normalemente non richiedono terapia se presenti in soggetti sani e hanno prognosi benigna. In
caso di infarto acuto del miocardio si utilizza lidocaina (anche se ci sono studi controversi sul
reale beneficio alla sopravvivenza, infatti terapie che rallentano la conduzione miocardica o
aumentano la refrattarietà possono aumentare il rischio di aritmie fatali). Quando una PVC si
associa con un ciclo normale e lo schema si ripete per ogni ciclo successivo si parla di
bigeminismo, se il rapporto e 1:3 è trigeminismo.
Ritmo idioventricolare accelerato: RIVA, è un ritmo con frequenza tra 40 e 120bpm generato da
un focolaio ectopico ventricolare. Normalmente i centri di automatismo ventricolari sono
dominati dal NSA in quanto questo genera stimoli ad una frequenza più elevata. Un focolaio
ectopico ventricolare può al massimo produrre un ritmo idioventricolare alla sua frequenza (2040bpm) solo quando c’è una totale inefficienza dei focolai sopraventricolari o un totale blocco
(ritmo di scappamento ventricolare).In questo caso il focolaio ha un ritmo accelerato che gli
permette di uscire dalla soppressione del NSA (si parla di automatismo anomalo). È
sovrapponibile (per frequenze tra 90 e 120) ad una tachicardia ventricolare lenta anche se
questa ha implicazioni terapeutiche differenti. Normalmente RIVA è considerata un’aritmia a
decorso benigno in genere breve e autolimitante. Si verifica nelle prime ore dopo un infarto
miocardico, dopo un intervento chirurgico o per intossicazioni da digitale.
Terapia: ci può essere buona risposta all’atropina o pacing atriale che accelerano la frequenza
atriale. Anche disopiramide.
Tachicardia ventricolare: TV, tachicardia che ha origine al di sotto del fascio di His in genere con
frequenza > 100 bpm, fino a 200 e oltre (250). È possibile avere anche una TV lenta, a frequenza
tra 90 e 120, confondibile facilmente con un ritmo idioventricolare accelerato, anche se la TV ha
una probabilità maggiore di verificarsi in un quadro d’infarto cronico o miocardiopatia, piuttosto
che nell’ifarto acuto o miocardite come RIPA. In genere rappresenta infatti un ampio circuito
macrorientrante generatosi in un miocardio con patologia cronica. La TV normale, ha come
causa più frequente l’infarto miocardico acuto, ma può essere causata anche da ipopotassiemia
o da intossicazione digitalica. In effetti può essere di due tipi:
TV monomorfica: con complesso QRS uniforme. Riproducibile con pacing e stimolazione
ventricolare programmata, in genere avviene in assenza di cardiopatia strutturale, generata da
40
un focolaio ectopico (irritato da ipopotassiemia o digitale, etc).
TV polimorfica: con complesso QRS vario. Non riproducibile e causata da cardiopatia. Una TV si
definisce sostenuta quando dura più di 30 secondi. Il pazienti si mostra con polso piccolo,
ritmico e di ampiezza irregolare. Possono esserci segni di scompenso dovuti alla cardiopatia di
base.
[ECG: può essere difficile distinguere una TV ventricolare da una TV sopraventricolare in
particolar modo se questa si presenta con QRS allargati dovuti ad esempio ad un BBD o BBS. Vi
sono alcune caratteristiche distintive. La TV presente spesso dissociazione AV, è possibile
raramente vedere anche le onde P generate dal NSA che sono sparse all’interno dei QRS. Questi
ultimi sono in genere allargati e >140ms (cosa rara nella TSV a meno che non ci sia un reale
blocco di branca). La dissociazione AV può portare a dei battiti di cattura, ossia QRS normali
all’interno dei QRS allargati (dovuti a rari casi in cui la conduzione dal NSA riesce a depolarizzare
i ventricoli) e battiti di fusione, ossia QRS diversi perché dovuti alla fusione di un QRS normale
con uno allargato. Questi sono segni che mancano nella TSV. Nella TV c’è anche in genere una
deviazione estrema destra dell’asse cardiaco sul piano frontale, fino a +180°. Inoltre c’è scarsa
definizione della parte iniziale del QRS e un pattern QRS che non corrisponde né a BBS né a BBD]
Torsione di punta: o torsades des pointes, è una forma di TV polimorfica caratterizzata da una
frequenza molto elevata (250-350 bpm) e da complessi QRS che variano progressivamente la
loro forma, da positivi a negativi e viceversa, dando l’impressione di una torsione, un
capovolgimento, un nastro attorcigliato. In genere si associa alla sindrome del QT lungo e tende
a risolversi spontaneamente.
Flutter ventricolare: frequenza tra 250 e 350 bpm. I QRS hanno aspetto sinusoidale con
ampiezza simile tra loro. Evolve quasi invariabilmente in una fibrillazione ventricolare. Il
riempimento ventricolare è già insufficiente così come la gittata cardiaca.
Fibrillazione ventricolare: FV, grave aritmia che comporta collasso emodinamico del paziente. Si
ha un’attivazione dei cardiomiociti totalmente alterata e con contrazioni irregolari e inefficaci,
ECG del tutto caotico con “frequenza” (se così si può ancora dire ) ventricolare anche >400 bpm.
In genere c’è progressione verso l’arresto cardiaco con ampiezza delle deflessioni che diminuisce
sempre più. Il paziente rischia la vita in pochi minuti se non si interviene con un defibrillatore
(cardioversione elettrica). Spesso è preceduta da extrasistoli ventricolari precoci, ossia CPV che
cadono nella fase di ripolarizzazione ventricolare, un periodo considerato di maggiore
vulnerabilità (tratto discendente dell’onda T). Questo è il fenomeno R/T.
Arresto cardiaco: esistono due forme di arresto cardiaco: Asistolia: non funziona NSA e nessun
focolaio (nessun ritmo di scappamento); Dissociazione elettromeccanica: come nella FV, il cuore
produce segnali (sempre più deboli) di attività elettrica, ma non vi è risposta meccanica. Terapia:
è noto che la TV polimorfica, il flutter ventricolare e la FV portano sempre a collasso
emodinamico. In questi casi è indicato l’uso di un defibrillatore (scarica asincrona a 200J, se non
funziona scariche ritmiche). Se c’è TV monomorfica con compromissione emodinamica
defibrillatore sincrono all’onda R, se c’è TV monomorfica tollerata trattamento farmacologico
con lidocaina o amiodarone e se non funziona scarica sincrona sull’onda R. Esistono anche
defibrillatori computerizzati che individuano la FV e producono una scarica che provoca
cardioversione. C’è l’AED che si applica sulla parete del torace e l’ICD che si impianta sotto il
torace di pazienti a rischio ed è in gradi di defibrillare e anche di funzionare da pacemaker. Può
essere associato a terapia antiaritmica.
41
Anomalie genetiche che predispongono alle aritmie ventricolari polimorfiche
Sindrome del QT lungo: SQTL, forma congenita è dovuta a mutazioni nei canali ionici che
comportano un’aumentata durata della ripolarizzazione. Le otto mutazioni identificate sono in
genere ai canali del K+ o del Na+. Le aritmie ventricolari potrebbero essere innescate da
postdepolarizzazioni precoci favorite da un accumulo intracellulare di calcio dovuto ad un
allungamento del plateau. C’è predisposizione pertanto ad aritmie ventricolari polimorfiche
come la torsades des pointes. L’intervallo QTc nei pazienti con SQTL pare essere da 400 a 460
uomini e 400-480 donne. Vi sono però condizioni, associate a rischio di aritmie molto alto, con
QT>500ms. Spesso il QT non è in grado di accorciarsi durante lo sforzo ed è quindi lungo a
intermittenza. Alcuni manifestano la sindrome solo se gli si dà il sotalolo (classe III, aumenta la
durata del p.d.a.). Il genotipo influenza la prognosi. Le mutazioni LQT 1-2-3 sono il 99% delle
SQTL clinicamente rilevanti.
LQT1: non riescono ad abbreviare il QT sotto sforzo. Onda T ampia. No attività sportiva. Terapia
beta-bloccante.
LQT2: onda T con un’incisura e quindi bifida. Terapia con beta-bloccanti
LQT3: onde T tardive bifasiche, è mutato il canale del sodio. Prognosi peggiore, spesso aritmie
durante il sonno. No beta bloccanto.
Terapia: nei pazienti con QT rilevata e sintomatici si impianta ICD, così come in quelli con LQT3 o
con QT>500 ms, considerati molto a rischio.
SQTL acquisita: per polimorfismi e/o mutazioni sporadiche ci sono soggetti predisposti
all’allungamento del QT in risposta ad alcuni farmaci e peggiorati da condizioni di ipokaliemia o
da bradicardia.
SQT breve: rara, associata a mutazioni che velocizzano la depolarizzazione, portano QT<320ms,
onde T alte e appuntite (che possono essere confuse con QRS e contate due volte dall’ICD).
Rischio di FA e FV, si impianta l’ICD.
Sindrome di Brugada: Importante causa di morte improvvisa nei giovani. Per un difetto del
canale del sodio (mutazione SCN5A) si ha minore entrata di sodio all’interno e p.d.a che dura di
meno nell’epicardio del tratto di efflusso del ventricolo destro. Si ha pertanto un’ampia
differenza di potenziale tra l’epicardio della zona di efflusso del VD e l’endocardio. Nell’ECG si ha
sopraslivellamento evidente del tratto ST in V1-V2-V3 e aspetto del QRS tipico del BBD. Si
impianta ICD nei sintomatici o con episodi aritmici. È più comune nei maschi asiatici.
Procainamide e fleicanide bloccano il canale del sodio e possono esacerbare la sindrome (e
anche renderla evidente, trovano utilizzo nei test diagnostici).
42
Infezione reumatica
Definizione: detta anche febbre reumatica (FR) o reumatismo articolare acuto, è una malattia
infiammatoria acuta diffusa, spesso associata a cardiopatia reumatica (CR). Con l’avvento della
terapia antibiotica e antinfiammatoria e con la prevenzione delle recidive, attualmente è molto
meno comune di un tempo nei paesi industrializzati, ma resta ancora molto comune nel terzo
mondo.
Eziologia e patogenesi: sono coinvolti due fattori intimamente collegati:
1) Infezione iniziale da streptococco beta-emolitico di gruppo A.
2) Suscettibilità specifica del soggetto colpito.
La febbre reumatica acuta colpisce prevalentemente i soggetti tra i 5 e i 15 anni ed è molto rara
sotto i 2 e sopra i 25. La patologia insorge a seguito di un’infezione da streptococco che ha
causato faringite o faringotonsillite (evidenti nei 2/3 dei casi, sono però riscontrabili anticorpi
negli altri). L’incidenza di febbre reumatica sembra più comune nelle epidemie di infezioni
faringotonsillari (in comunità). Pare comunque che il batterio debba rimanere a lungo
nell’organismo per poterla causare, infatti la cura entro tre giorni con penicillina ne impedisce
l’insorgenza.
Alcune delle manifestazioni (anche la cardite) possono essere mediate direttamente dalle
tossine (come la streptolisina A, cardiotossica) prodotte dal batterio, ma per la maggior parte la
patogenesi è di tipo immunitario. Pare infatti che ad esempio uno dei maggiori costituenti della
parete del batterio (polisaccaride streptococcico A) sia simile ad una glicoproteina valvolare,
potendosi quindi generare un danno dovuto ad una risposta inappropriata.
La patogenesi immunitaria troverebbe conferma nel periodo di latenza di 2-3 settimane tra
infezione e manifestazioni. La suscettibilità individuale è fondamentale, infatti l’infezione
faringea comporta FR solo nel 0,3-3% dei casi, e pare sia associata anche ad una predisposizione
genetica (la suscettibilità individuale è confermata dalla tendenza a recidivare).
Anatomia patologica:
Fase acuta: dominano le lesioni infiammatorie prevalentemente cardiache, vascolari, cutanee e
articolari. Le lesioni infiammatorie assumono in genere l’aspetto della degenerazione fibrinoide
di Neumann con comparsa nel collageno di una sostanza simile alla fibrina, ma nel cuore hanno
un aspetto specifico ossia sono corpi (o noduli) di Aschoff in genere in endocardio valvolare,
miocardio e più raramente aorta e tessuto di conduzione. Questi noduli sono focolai (tipo
granulomi) di linfociti (per lo più T), plasmacellule e grossi macrofagi attivati detti cellule di
Anitschkow (patognomiche) che hanno citoplasma abbondante e nucleo ovalare con cromatina
centrale in un nastro sottile e ondulato (cellule bruco). Alcuni macrofagi (giganti) possono essere
multinucleati. Le lesioni anatomiche interessano:
A) Cuore: interessato sia il miocardio che l’endocardio, meno frequentemente anche il
pericardio (pancardite o cardite). Una vera cardite si riscontra in circa il 50% dei malati (ma
spesso ci sono sempre segni ECG).
Miocardio: noduli di Aschoff e infiltrazione infiammatoria. Il miocardio specifico (di conduzione)
è molto raramente interessato.
Endocardio: soprattutto lesioni valvolari.
Si ha una infiammazione che esita in una necrosi fibrinoide all’interno delle cuspidi o delle corde
tendinee. Sopra questi focolai, lungo le linee di chiusura dei lembi valvolrai si trovano piccole
vegetazioni (verruche). Nell’atrio sinistro, spesso a causa del reflusso possono esserci
43
ispessimenti irregolari sub endocardici detti placche di McCallum. La mitrale è interessata da
sola nel 50% dei casi, insieme all’aortica nel 20%, con anche la tricuspide nel 7%, tutte e quattro
solo nel 3%. La FR causa il 10% circa delle stenosi aortiche e una gran parte delle mitraliche. Se la
stenosi mitralica è grave, l’atrio sinistro può dilatarsi e al suo interno possono formarsi trombi. Il
tessuto fibroso che si forma tra le commessure valvolari e le calcificazioni dà un aspetto a “bocca
di pesce” delle stenosi. Le alterazioni della valvola mitrale comprendono ispessimento dei lembi,
fusione delle commessure, accorciamento, e ispessimento e fusione delle corde tendinee. La
pericardite compare circa nel 10% dei casi.
B) Vasi: l’aortite è rara, spesso sopravalvolare. Sono più frequenti arterite, capillarite, flebite.
C) Articolazioni: ispessite ed edematose le strutture articolari e periarticolari, con congestione e
flogosi della sinovia, ma le lesioni guariscono spontaneamente (Lasegue: “il reumatismo
articolare acuto è come un cane che lecca le articolazioni e morde il cuore).
D) Noduli sottocutanei: più frequenti sopra tendini, legamenti e fasce, sono con zona centrale di
necrosi fibrinoide con attorno macrofagi e fibroblasti.
E) Corea (detta minor o di Sydenham): 15% dei casi, più femmine, emorragie, trombosi
arteriolare, edema e infiltrazioni di linfociti nei nuclei della base.
F) Polmonite: rara.
G) Glomerulonefrite: 1/5 dei casi.
Clinica: le infezioni streptococciche cutanee e renali non producono FR.
La faringite o la faringotonsillite recedono dopo 5-7 giorni e sono seguite da un periodo di
latenza asintomatico di 7-10 giorni.
Si ha poi la fase iniziale della malattia reumatica: con febbre elevata continua, sudorazione,
affaticabilità, anemia, artrite. Poi per due settimane anche solo febbricola.
L’artrite si manifesta come una poliartrite migrante che interessa le grandi articolazioni (una o
due per volta per una o due settimane), con lesioni che tendono a guarire spontaneamente.
Eritema marginato: manifestazione eritematosa fugace a tronco e parte prossimale degli arti.
Noduli sottocutanei: non dolenti né arrossati, di 2-20mm.
Corea di Sydenham: nel 20% dei casi, tardiva (entro 2 mesi), più femminile, si manifesta con
movimenti bruschi, involontari, incontrollati e finalistici.
Fase florida: segue la precedente (ma va!) e si manifesta con pancardite (asintomatica nel 40%
dei casi). La CR è la manifestazione più grave di FR ed il suo marchio è l’interessamento valvolare
con comparsa di soffi. I segni sono: Soffio olostistolico puntale (2-3/6, mitralico) legato a
interessamento della mitrale. Soffio mesodiastolico apicale (con il III tono) dovuto a stenosi
mitralica. Soffio diastolico da insufficienza aortica (raramente reperto isolato). Vi sono poi
sfregamenti pericardici che possono variare con la pressione dello stetoscopio e le fasi del
respiro (pleuropericardici), ridotta intensità del I tono, comparsa di un III tono (ritmo di galoppo
proto diastolico).
Più rare sono la cardite reumatica fulminante (coinvolgimento delle coronarie con
restringimento acuto del lume e necrosi miocardica), polmonite reumatica, glomerulonefrite.
Diagnosi:
Esami di laboratorio: vi sono alterazioni aspecifiche, dovute alla flogosi, come l’aumento della
VES, anemia e moderata leucocitosi neutrofila, PCR, aumento enzimi miocardici. C’è però anche
aumento della TAS (titolo antistreptolisinico, o meglio ancora TASLO, anti-O-streptolisine) e
anti-streptochinasi, anti-ialuronidasi, etc. oltre a tampone faringeo positivo per streptococco
beta-emolitico di gruppo A.
ECG: nel 60% dei casi appare PR>0,2 s (blocco AV di 1°grado), e a volto anche un blocco AV di 2°
44
grado di tipo Mobitz 1. Sono possibili anche aritmie sopraventricolari.
Radiografia ed ecocardio non sono specifiche anche se possono rilevare alterazioni.
La diagnosi è per lo più clinica tramite i criteri di Jones:
Maggiori: poliartrite migrante, pancardite, corea minor, eritema marginato, noduli sottocutanei.
Minori: precedente FR, segni di infezioni streptococcica precedente, febbre, laboratorio,
artralgie, allungamento PR. Per la diagnosi sono necessari almeno due criteri maggiori,
probabile se c’è una manifestazione maggiore e due o tre minori.
Prognosi: nella fase acuta c’è guarigione spontanea (tranne lesioni valvolari) nel 75%, il restante
25% muore soprattutto di scompenso congestizio. Se si fa terapia si guarisce al 95%, e senza
lesioni cardiache nel 70%.
Terapia: Riposo a letto: dovrebbe iniziare nel periodo febbrile e della poliartrite ed essere
prolungato di 3-4 settimane nel caso di pancardite.
Terapia antibiotica: si basa su penicillina G intramuscolare (o pennicilinna V orale o eritromicina
se allergico) per almeno 2 settimane.
Terapia antinfiammatoria: acido acetilsalicilico e corticosteroide se c’è cardite (prednisone). Si
possono aggiungere farmaci per trattare problemi cardiaci.
Profilassi: sulfadiazina o penicillina ritardo (a lento rilascio) va continuata circa una volta al
mese anche per 10 anni nei pazienti sotto i 20 anni (per 5 o 3 sopra i 20 anni). La profilassi serve
a prevenire la recidiva reumatica che tende a seguire i futuri episodi di reinfezioni
streptococciche faringee (FR avviene nel 3% al primo episodio,nel 20% al secondo e 50% al
terzo) con inoltre maggiore rischio di danni valvolari e in generale cardiaci (che tra l’altro paiono
essere cumulativi).
45
Malattie valvolari
Valvulopatie acquisite: possono essere di tipo restrittivo (stenosi) o di tipo incontinenza
(insufficienze). Entrambe le disfunzioni possono colpire tutti gli apparati valvolari e possono
essere isolati (stenosi o insufficienza di una sola valvola), combinati (steno-insufficienza della
stessa valvola) o composti (più valvole interessate). Le stenosi sono solitamente a insorgenza
progressiva, le insufficienze possono essere acute o croniche. Le valvulopatie sono spesso
evidenti all’esame obiettivo (auscultazione di soffi) e all’ecocardiogramma (soprattutto con
color-doppler) e causano conseguenze emodinamiche nelle camere cardiache per il tentativo di
compenso. Le stenosi causano conseguenze soprattutto a monte, con ipertrofia e a volte
dilatazione delle camere, le insufficienze causano conseguenze sia a monte che a valle con
dilatazione e a volte ipertrofia delle camere che si trovanoa manipolare maggiori volumi di
sangue. Quando questi meccanismi si esauriscono può manifestarsi scompenso.
Le affezioni della valvola polmonare sono per lo più congenite o connesse a ipertensione
polmonare o (di rado, a endocardite). Per il resto sono rare.
Stenosi mitralica
Definizione: malattia cronica progressiva della valvola mitrale, caratterizzata da restringimento
dell’orifizio mitralico che crea ostacolo al flusso dell’atrio sinistro durate il riempimento
diastolico ventricolare. L’orifizio nell’adulto ha una superficie media normale di 4-6cm 2.
Se la stenosi è lieve la superficie è ristretta, ma comunque superiore a 2cm 2, se è media è tra 1 e
2cm2, se è severa è <1cm2.
Eziologia: la causa è prevalentemente una cardite reumatica postreptococcica pregressa (ed è
più comune nella donne). Il processo infiammatorio danneggia la valvola con un meccanismo
autoimmune nella fase acuta e può comportare, anche dopo la guarigione, un progressivo
aggravarsi della stenosi (forse perché il processo reumatico è subdolo e progressivo, o perché la
valvola già danneggiata risulta più sensibile ai traumi del flusso turbolento del sangue).
Il processo di cicatrizzazione della valvulite reumatica determina ispessimento e fibrosi delle
cuspidi valvolari con aree di aderenza e fusione della commessure valvolari.
La valvola stenotica assume una conformazione a imbuto e l’ostio ha la tipica forma a “bocca di
pesce”. Nelle forme a lunga durata possono essere presenti calcificazioni con depositi calcarei.
Le cuspidi possono restare flessibili o divenire rigide e calcifiche. Talvolta i lembi sono talmente
aderenti e rigidi da non riuscire né ad aprirsi né a chiudersi (steno-insufficienza mitralica).
Altre cause di stenosi mitralica sono formazioni neoplastiche (tipo mixoma atriale sinistro) o
trombotiche o proliferazione eccessiva di vegetazioni endocarditiche. La stenosi mitralica
congenita è piuttosto rara. Si definisce sindrome di Lutembacher una stenosi mitralica associata
ad un difetto interatriale di tipo ostium secundum.
Fisiopatologia: La riduzione dell’orifizio valvolare mitralico inferiore a 2,5cm 2 determina un
aumento della pressione atriale sinistra e viene a determinarsi un gradiente pressorio tra atrio
e ventricolo sinistro necessario alla propulsione del sangue. Il gradiente non dipende però solo
dalle dimensioni della valvola, ma anche dal flusso e dal tempo di riempimento diastolico
(equazione di Gorlin). Pertanto, se il flusso è ancora basso e il tempo di riempimento diastolico
ancora alto, come accade a riposo, basta che l’area valvolare sia maggiore di 1,5cm 2 per non
produrre sintomi. Però un aumento del flusso o una diminuzione del tempo di riempimento
46
(stress fisico o emotivo, infezioni, gravidanza) comportano un aumento della pressione atriale
sinistra. Pertanto non vi è una proporzionalità diretta tra grado di stenosi mitralica e entità della
pressione atriale sinistra, mentre vi è proporzionalità inversa tra pressione e volume atriale
(spesso grandi volume e bassa pressione). Nell’atrio sinistro ingrandito possono formarsi trombi
e dunque possono aversi eventi embolici.
Pertanto l’ipertensione atriale sinistra aumenta la pressione all’interno di vene e capillari
polmonari con conseguente incremento del contenuto idrico polmonare dapprima
intravascolare e poi extravascolare (trasudato nell’interstizio nel momento in cui la pressione
idrostatica supera la pressione oncotica, successivamente) e quindi edema polmonare (con
possibile drenaggio linfatico). Il liquido trasudato diminuisce la compliance polmonare
accrescendo il lavoro respiratorio e causando dispnea (aumenta con lo sforzo, ma anche a
riposo) e anche possibile ortopnea o dispnea parossistica notturna.
Un marcato aumento può anche causare emottisi (rottura delle venule nel parenchima).
Si ha pertanto ipertensione polmonare che è il risultato di vari fattori:
1) trasmissione retrograda dell’incremento pressorio atriale sinistro sull’arteria polmonare
(ipertensione polmonare passiva).
2) Vasocostrizione reattiva delle arteriole polmonari innescata dall’ipertensione atriale sinistra e
venosa (ipertensione polmonare reattiva).
3) Modificazioni organico-obliterative del letto vascolare polmonare.
A causa della portata cardiaca ridotta e dell’aumento della resistenza arteriolare polmonare, un
paziente con stenosi mitralica serrata può restare asintomatico a lungo.
All’inizio, se l’ipertensione polmonare è moderata, il ventricolo destro funziona normalmente ,
ma se questa sale (sopra i 70mmHg) si ha insufficienza cardiaca destra con aumento della
pressione tele diastolica ventricolare e atriale destra.
L’insufficienza cardiaca destra causa congestione sistemica (turgore giugulare, ascite, edema,
marcato affaticamento).
La funzione ventricolare sinistra è normale, ma a causa del diminuito riempimento diastolico
(per la stenosi) e quindi del diminuito pre-carico, la performance del ventricolo e quindi la
gittata sono ridotte. La contrattilità del ventricolo sinistro può essere ridotta dalla malattia
reumatica. Si hanno frequentemente aritmie atriali, prevalentemente fibrillazione atriale (60%).
Questa aumenta ancora di più il rischio di emboli.
Clinica: I sintomi più comuni sono: dispnea, ortopnea, dispnea parossistica notturna, edema
polmonare che rappresentano le conseguenze dell’ipertensione polmonare. Edema polmonare
e DPN sono più comuni in pazienti con aritmie atriali ipercinetiche. La dispnea si aggrava con lo
sforzo, ma può essere presente anche a riposo.
Bronchiti invernali: causate dalla congestione polmonare e dall’ipersecrezione di muco.
Emottisi: può verificarsi in seguito a: rottura delle venule bronchiali, o edema polmonare
(escreato roseo), o infezioni (escreato striato di sangue), o embolia polmonare.
Astenia e affaticamento: spesso in pazienti con stenosi serrata (<1cm 2) per ridotto flusso trans
mitralico.
Palpitazioni: correlate alla fibrillazione atriale (flutter e tachicardia sono meno comuni)
Embolie sistemiche: a causa della formazioni di trombi nell’atrio sinistro dilatato e a volte con
FA, dovuti alla stasi ematica. Una volta si verificavano nel 20% dei pazienti, ora 1,5-5%. Il 75%
colpisce il cervello. Si può avere un ictus localizzato o infarto di grosse parti dell’emisfero
cerebrale. Può colpire arti superiori o inferiori, biforcazione iliaca (embolia a sella), arterie della
retina, renali, mesenterica, coronarie. Raramente anche trombo “a palla”che ostruisce il flusso
attraverso la valvola mitralica.
47
Tromboembolia polmonare: emboli provenienti dall’atrio destro, ma più comunemente dagli
arti inferiori.
Dolore toracico: da insufficienza ventricolare destra o angina.
Raucedine e voce bitonale (sindrome di Ortner): dovuta alla compressione del nervo laringeo
ricorrente sinistro da parte dell’atrio sinistro e dell’arteria polmonare dilatata.
Diagnosi: come in tutte le valulopatie, l’esame obiettivo resta fondamentale.
Esame obiettivo: il paziente si presenta con facies mitralica ossia aspetto di paradossale
benessere con zigomi rosso-cianotici e cianosi dei prolabi che in realtà indica stenosi severa
(possono esserci anche mani fredde e cianotiche). Se c’è scompenso destro si possono notare
ascite, turgore delle giugulari e edemi declivi. Al polso giugulare c’è un evidente onda
presistolica (onda a), che però è assente nei pazienti con fibrillazione atriale. Se c’è insufficienza
tricuspidale è evidente l’onda v.
L’itto può essere ridotto e in decubito laterale sinistro si può apprezzare un fremito diastolico
all’apice (corrispondente di un forte soffio mitralico) e si potrebbe apprezzare un impulso del
parasternale in caso di ipertrofia ventricolare destra.
Il I tono è aumentato d’intensità, si riduce solo se la valvola mitrale è rigida perché molto
calcifica.
Il II tono è normale o aumentato, e può apparire singolo e non sdoppiato.
Segno di ipertensione polmonare è anche un tono polmonare d’eiezione (click). Si può avvertire
uno schiocco d’apertura della valvola mitrale che si avverte come un suono forte subito dopo il
II tono (tanto più vicino quanto più è alta la pressione atriale sinistra perché la mitrale si apre
prima). Questo è dovuto alla brusca tensione del lembo anteriore mitralico nella massima
escursione della sua apertura.
Si avverte in genera all’apice un soffio diastolico a carattere rullante che ha intensità massima
durante all’inizio della diastole e durante la presistole. La sua durata è indice di gravità. Il soffio è
dovuto al movimento delle cuspidi valvolari e quindi manca se c’è stenosi mitralica severa con
valvola calcifica.
Può esserci anche un soffio diastolico tricuspidale nel caso di insufficienza tricuspidale (da
insufficienza ventricolare destra).
ECG: non particolarmente specifico, si leggono segni di ingrandimento atriale sinistro e ipertrofia
ventricolare destra (quindi con relative alterazioni evidenti all’ECG), può verificarsi FA.
Radiografia del torace: è visibile la configurazione mitralica che ha un aspetto triangolare
nell’immagine posteroanteriore per la dilatazione del ventricolo destro e la protrusione
dell’atrio sinistro e arteria polmonare. Sono visibili le strie di Kerley indicative di ipertensione
polmonare. Ci può anche essere edema alveolare con aspetto ad ali di farfalla a partenza
dall’ilo.
Ecocardiogramma: ci permette di rilevare l’aspetto a bastone di hockey dei lembi della mitrale e
di verificare la compromissione anatomica della valvola. È in base all’ecocardio che è possibile
distinguere, misurando l’area valvolare mitralica, una stenosi mitralica lieve, moderata o
serrata (severa). È possibile stimare il gradiente valvolare mitralico con l’ecocardiografia
doppler con buona correlazione rispetto ai dati valutati con cateterismo cardiaco. È inoltre
possibile notare l’aumento di volume dell’atrio sinistro, l’eventuale riduzione del ventricolo
sinistro e l’eventuale aumento del ventricolo destro. Con l’ecocardiografia transesofagea si
possono in modo ancora più preciso rilevare trombi nell’atrio sinistro e valutare l’anatomia della
48
valvola. L’ecocardio resta quindi sicuramente l’esame calcolare con di elezione, anche per il
follow-up.
Cateterismo cardiaco: serve, soprattutto nella valutazione preoperatoria, per calcolare con
precisione il gradiente tra atrio e ventricolo sinistro. Questo si ottiene misurando
simultaneamente le curve pressorie nel ventricolo sinistro (raggiunto con cateterismo arterioso
retrogrado) e nei capillari polmonari (catetere in un’arteriola polmonare). Si può poi rilevare la
consistenza dell’ipertensione polmonare con catetere in arteria polmonare. Tramite l’Equazione
idraulica di Gorlin è possibile valutare l’area valvolare mitralica con precisione (anche se solo se
non c’è insufficienza aortica e la portata non è né troppo alta né troppo bassa) infatti
MVA=CO[portata cardiaca]/38 * FC [frequenza cardiaca] * DFT [tempo di riempimento
diastolico] * radice quadrata del gradiente pressorio atrio sx-ventricolo sx). Può essere
necessario eseguire una coronarografia.
Terapia medica: la malattia diviene progressivamente più severa, indipendentemente dalle
recidive reumatiche (per le quali comunque serve fare profilassi antibiotica). È in generale
raccomandato di evitare eccessive attività fisiche. Si può somministrare un beta-bloccante per
garantire il controllo della frequenza, un diuretico (furosemide) se c’è ipertensione venosa
polmonare (scompenso). La digitale può essere utile nel diminuire la frequenza di risposta
ventricolare nei pazienti con fibrillazione atriale. Nel caso di fibrillazione atriale bisogna fare un
trattamento anticoagulante con warfarin (e tutta la terapia per FA) per prevenire le trombo
embolie. In genere è possibile programmare una cardioversione elettrica (in genere dopo 3
settimane di trattamento anticoagulante).
Terapia chirurgica: è indicata quando il paziente ha area valvolare inferiore a1,5cm 2 ed è almeno
nella II classe NYHA, o ha FA, ha avuto episodi embolici, oppure ha insufficienza ventricolare
destra. In questo caso si fa prima una valutazione tramite ecocardiografia ed ecocardiografia
trans esofagea per determinare lo “score ecocardiografico” ossia per verificare il grado di
ispessimento, di calcificazione e di mobilità dei lembi, mentre tramite la trans esofagea si
determina la presenza o meno di trombi nell’atrio sinistro. Uno score basso è indicazione per
eseguire la PMBV, ossia valvuloplastica con palloncino (di Inoue). Questa è la tecnica di prima
scelta quando può essere eseguita da un operatore esperto in un grande centro (è sempre utile
avere a disposizione una sala operatoria cardiochirurgia nei rari casi di rottura dei lembi
valvolare o dell’anello mitralico) perché ha una minore mortalità della chirurgia e gli effetti a
lungo termine sono paragonabili. Per poter eseguire questa tecnica i lembi valvolari devono
ancora essere abbastanza duttili con poche o assenti calcificazioni commissurali.
Controindicazioni relative sono insufficienza mitralica significativa e trombi interatriali. Le
complicanze (1%) cono principalmente embolia cerebrale e perforazione cardiaca.
Procedura: si utilizza un catetere che viene portato in atrio sinistro tramite puntura transettale
(si entra dal sistema venoso) e, una volta raggiunto l’ostio mitralico si gonfia un singolo
palloncino che separa le commessure e rompe le calcificazioni nodulari. Dopo l’intervento c’è
notevole riduzione delle resistenze polmonari e dei sintomi, e la sopravvivenza libera da eventi è
dell’80-90% a cinque anni. La procedura chirurgica è indicata nei pazienti che presentano
ristenosi o in cui la PBMV non ha avuto successo.
Il primo approccio chirurgico è la valvulotomia (o commissurotomia) che può essere effettuata
sia a cuore aperto che a cuore chiuso. Quest’ultima, procedura, eseguita trans catetere, è stata
quasi completamente sostituita dalla PMBV. Nella procedura a cuore aperto si effettua
sternotomia mediana e bypass cardiopolmonare, si incidono e si aprono le commessure, si
49
liberano i muscoli papillari e le corde tendinee, si rimuovono le calcificazioni e gli eventuali
trombi atriali. È un intervento che nella metà dei casi deve essere ripetuto dopo dieci anni, e ha
una mortalità perioperatoria del 2%, infatti, a meno che non si verifichi grave ipertensione
polmonare o un’embolia, non deve essere effettuato in pazienti completamente asintomatici
e/o affetti da una stenosi con valvola >1,5cm 2.
La sostituzione valvolare mitralica con protesi biologica o protesi meccanica è un intervento
che si effettua solo se il paziente è gravemente sintomatico (classe III di NYHA) ed ha una stenosi
serrata, o le altre procedure non hanno portato benefici o non sono state possibili (lembi solo
moderatamente fibrotici, corde di normale lunghezza e apparato sottovalvolare conservato
rendono possibile una valvuloplastica o una commissurotomia). Infatti l’intervento ha una
mortalità del 5% (maggiore negli anziani e in caso di comorbilità), e una sopravvivenza del 70% a
dieci anni. Vengono rimossi i trombi nell’atrio sinistro cercando di non frantumarli, si pratica
l’escissione della valvola incidendo i muscoli papillari all’apice (anche se attualmente si cerca di
conservare l’attacco delle corde tendinee per favorire il recupero funzionale del ventricolo. Si
applica poi la protesi con punti ad U di Tevlek 2-0 e tra un punto ed il successivo non bisogna
lasciare spazi per non creare leak perivalvolari.
Insufficienza mitralica
Definizione: IM, è caratterizzata da un reflusso di sangue dal ventricolo sinistro (camera ad
alta pressione) all’atrio sinistro (bassa pressione) durante la sistole ventricolare, a causa di
un’incompleta chiusura dei lembi mitralici. Comporta un grado variabile di dilatazione di
entrambe le cavità. Si distingue in acuta e cronica, ma anche in primaria (alterazione
primitiva dell’apparato valvolare) o secondaria (disfunzione dell’apparato a causa della
dilatazione del ventricolo sinistro. Le alterazioni possono essere a carico dei lembi,
dell’anello mitralico, delle corde tendinee o dei muscoli papillari.
Eziologia: diverse alterazioni possono causare insufficienza:
1) Lembi valvolari: sono alterati prevalentemente a causa della malattia reumatica che
provoca fenomeni di fibrosi, accorciamento, deformità e retrazione di uno o più lembi che
possono riguardare anche le corde e i papillari che comportano una mancata chiusura e
anche una possibile steno-insufficienza mitralica (insieme alla stenosi). Le cuspidi possono
essere alterate anche dall’endocardite infettiva, per perforazione a causa di vegetazioni.
Attualmente è comune la degenerazione a causa di mixomi.
2) Anello mitralico: può andare incontro a dilatazione ad esempio a causa di una
miocardiopatia dilatativa, o a calcificazione, frequente nelle persone anziane, spesso
idiopatica.
3) Corde tendinee: possono rompersi in malattie degenerative del connettivo o nel mixoma,
ma anche nella FR e endocardite.
4) Muscoli papillari: possono essere danneggiati in caso di cardiopatia ischemica (il
posteriore è irrorato dall’IVP, l’anterolaterale da IVA o circonflessa, rami diagonali). In
questo caso l’insufficienza può essere transitoria o duratura a seconda dell’ischemia.
Disfunzione ventricolare: è la causa di insufficienza mitralica secondaria perché altera i
rapporti tra muscoli papillari, corde tendinee e lembi. Il rigurgito è in genere meno
importante rispetto alle forme primarie.
Patogenesi: la malattia dipende dall’entità del rigurgito e dal tempo in cui si instaura (acuta
50
o cronica). In generale, in caso di insufficienza, la contrazione isovolumetrica del ventricolo
sinistro quasi si annulla, in quanto sin dall’inizio il ventricolo è in grado (già in fase
preespulsiva aortica, quando ancora la pressione ventricolare deve raggiungere l’ampiezza
adatta ad aprire la semilunare) di far passare del sangue attraverso l’ostio mitralico nell’atrio
sinistro. Quando si apre la valvola aortica c’è una competizione tra zona di afflusso (ostio
mitralico, passaggio in atrio sinistro) e zona di efflusso (aorta). Nelle insufficienze mitraliche
gravi la frazione di rigurgito (in percentuale sulla gittata) può essere anche del 65-70%.
Insufficienza cronica: si ha adattamento del ventricolo sinistro in quanto i rigurgiti sono di
intensità graduale. Il sovraccarico di volume cronico comporta progressiva dilatazione delle
cavità ed ipertrofia eccentrica (tipica di insufficienza mitralica e aortica, perché c’è
sovraccarico di volume) che permette alla gittata cardiaca di mantenersi buona anche in
caso di una discreta frazione di rigurgito.
Nelle forme lievi e moderate anche sotto sforzo la gittata si mantiene buona, ma quando c’è
insufficienza importante si ha una progressiva depressione della contrattilità (che non
sempre si traduce subito in una riduzione della frazione di eiezione) dovuto all’aumento
sempre maggiore del volume e della pressione tele diastolica (alla normale quantità di
sangue si aggiunge nel ventricolo anche la quantità di sangue rigurgitato in sistole nell’atrio).
La componente principale della malattia è pertanto la ridotta funzione ventricolare sinistra
(che provoca affaticabilità) e lo scompenso che è quindi conseguenza della ridotta
contrattilità e inoltre del fatto che l’ipertrofia è speso inadeguata (ossia c’è più dilatazione
che ipertrofia). Si possono avere pertanto insufficienza ventricolare, ridotta portata,
ipertensione atriale che comporta ipertensione polmonare. In dipendenza dell’entità della
compliance atriale si può avere dilatazione e ipertensione atriale sinistra.
Insufficienza mitralica acuta: il ventricolo si trova all’improvviso a far fronte ad un
sovraccarico di volume. Questo può verificarsi nelle endocarditi acute e più spesso nella
rottura del muscolo papillare a causa di un infarto. Il ventricolo non si dilata né si ipertrofizza
e si ha un marcato aumento pressorio atriale (grande onda v nel polso giugulare). Si verifica
rapidamente un’ipertensione polmonare con aumento della P-wedge (pressione di
incuneamento nei capillari polmonari) e relativo edema polmonare. Situazione critica che
può causare shock cardiogeno e morte senza un repentino intervento chirurgico. Tramite
ecocardio con doppler si può valutare l’entità del rigurgito e anche la funzione ventricolare
sinistra. A causa del diminuito postcarico all’inizio la FE può sembrare normale e anche con
contrattilità già diminuita anche se dopo un po’ può scendere (sotto 50-40%). Il volume tele
sistolico che se molto aumentato (normalmente è <30) indica forte disfunzione ventricolare.
Clinica:
Insufficienza mitralica cronica: i sintomi sono attenuati e si sviluppano lentamente. Nelle
forme lievi i pazienti possono rimanere asintomatici anche per più di dieci anni. I sintomi
possono essere correlata alla ridotta gittata (affaticabilità) o in generale allo scompenso
ventricolare sinistro e alla progressiva ipertensione polmonare (dispnea all’inizio lieve, poi
ingravescente da sforzo, poi anche dispnea parossistica notturna). Si può avere cardiopalmo,
segno di extrasistoli sopraventricolari o fibrillazione atriale (poco frequente e in genere ben
tollerata). Fenomeni trombo embolici sono rari anche nell’atrio sinistro dilatato(perché
comunque c’è ricambio di sangue) tranne in caso di fibrillazione atriale.
Insufficienza mitralica acuta: sintomi di dispnea che esordisce improvvisamente ed è
rapidamente ingravescente, anche edema polmonare acuto e progressione rapida verso lo
51
shock cardiogeno.
Esame obiettivo: l’aia cardiaca può essere ingrandita (dipende dalla dilatazione
ventricolare).
Il I tono risulta diminuito (chiusura incompleta e quindi blocco incompleto del flusso), il II
tono può risultare maggiormente sdoppiato (componente aortica anticipata a causa della
ridotta portata anterograda). Si può ascoltare un III tono per la brusca messa in tensione
delle corde tendinee all’apertura della valvola.
La caratteristica principale è uno soffio sistolico alla punta dovuto al rigurgito (tele sistolico,
ma anche olosistolico se grave) intorno a 4/6. Può essere debole se la valvola è già troppo
deteriorata per resistere al flusso. È ridotto dalla manovra di Valsalva e dai nitrati che
riducono il precarico. È possibile persino la presenza di un soffio diastolico da stenosi
mitralica, non solo nei casi di steno-insufficienza, ma anche quando vi è un notevole
passaggio di sangue in diastole (quantità normale più rigurgito) che comporta una stenosi
mitralica relativa. In caso di IM acuta può essere presente un IV tono, e il soffio sistolico è
meno riconoscibile.
Diagnosi: l’esame obiettivo resta essenziale in quanto gli esami di routine sono poco
specifici.
All’ECG è visibile un’ipertrofia atriale sinistra.
Alla radiografia del torace si può vedere (se evidente) una dilatazione ventricolare sinistra o
segni di stasi polmonare (le calcificazioni si vedono meno spesso rispetto alla stenosi
mitralica).
L’ecocardiogramma è un esame fondamentale. Oltre a rilevare eventuali dilatazioni
permette di evidenziare con metodica doppler il rigurgito e di valutarlo quantitativamente
da 1 a 4: 1-2=lieve (o lieve-media); 3=moderata; 4=severa o rilevante. Si possono notare
anche vegetazioni e la rottura di una corda tendinea (ampie oscillazioni del lembo valvolare),
oltre a eventuali calcificazioni.
Se questi esami non si sono rivelati diagnostici si può eseguire un cateterismo cardiaco, che
permette di rilevare pressione atriale sinistra aumentata così come quella diastolica
ventricolare sinistra. Il modo migliore per visualizzare l’IM è però la ventricolo grafia con
mezzo di contrasto iodato che permette di valutare precisamente la dilatazione del
ventricolo sinistro e in base al grado di opacizzazione dell’atrio sinistro e al tempo di lavaggio
del m.d.c. dallo stesso atrio sinistro, è possibile classificare il rigurgito in gradi. Vi sono le
ovvie differenze di evidenze tra forma acuta e cronica.
Prognosi: i pazienti restano stazionari molto a lungo. Peggioramenti della prognosi si hanno
nelle forme con degenerazione connettivale (progressive) e dal momento in cui si verifica
scompenso cardiaco sinistro.
Terapia medica: a parte la terapia con warfarin per eventi di FA, si può solo effettuare
profilassi per endocardite. Bisogna evitare l’esercizio fisico isometrico. Terapia per lo
scompenso nel caso si presenti.
Terapia chirurgica: l’intervento chirurgico è indicato dal momento in cui cominciano a
comparire sintomi. Mentre un tempo venivano indirizzati alla chirurgia solo pazienti con
classe NYHA IV, ora anche pazienti in classe II o III se la terapia medica non basta e
presentano segni come cardiomegalia o volume tele sistolico elevato sono candidati
52
all’intervento. Questo perché l’istituzione di una terapica chirurgica conservativa, con
riparazione della valvola ha ridotto di molto i rischi legati alla chirurgia con sostituzione
protesica che aveva una maggiore mortalità e più complicanze.
Pazienti che sicuramente devono essere indirizzati alla chirurgia sono quelli con FE<60% o
con volume tele sistolico >60ml/m2 (30 è normale) o con rilevazione all’ ecocardiogramma di
diametro tele sistolico ventricolare sinistro >40mm. Nelle forme lievi di rigurgito non c’è
indicazione alla chirurgia che è invece utile per evitare la progressione dello scompenso.
Vi è un forte aumento del rischio (sia di mortalità intraoperatoria, sia di mancato recupero
funzionale ventricolare) nei pazienti con FE<30-35 e VTS>90ml/m 2 o diametro tele sistolico
>50mm. Però la terapia conservativa ha scarso valore in questi casi.
In caso di insufficienza acuta massiva (magari da infarto che coinvolge il papillare)
l’intervento è obbligatorio (così come per le endocarditi, ma dopo pretrattamento
antibiotico. In ogni caso, prima dell’intervento può essere necessaria una valutazione della
presenza di IM severa (se segni clinici ed ecocardio non combaciano) tramite cateterismo
con ventricolo grafia.
La terapia chirurgica può essere di tipo sostitutivo (protesi) o conservativo (riparativo, circa il
90% delle IM sono trattate così):
Sostituzione valvolare: provoca una discontinuità tra muscolo papillare e anulus,
interferendo sulla funzione ventricolare sinistra (attualmente però viene conservato spesso
il lembo posteriore con mantenimento dei relativi papillari). Ha una mortalità
intraospedaliera che varia dal 2 al 7% , più bassa in caso di danno da cardiopatia ischemica.
L’uso di vasodilatatori (e altri farmaci contro lo scompenso) in fase post-operatoria ha
ridotto la mortalità che spesso avveniva per scompenso cardiaco subito dopo.
Riparazione valvolare: la chirurgia conservativa è il motivo di un’indicazione chirurgica per
IM così aggressiva. L’intervento infatti (particolarmente utile nell’insufficienza mixomatosa)
ha una mortalità intraoperatoria inferiore all’1% e durata ottima con indicazione al re
intervento solo dell’1% in dieci anni. È indicato nel caso in cui non vi sia sclerosi calcifica dei
lembi o vegetazioni infettive. Si può attuare una valvuloplastica o una plastica dell’anulus
valvolare consistente nell’applicazione di anelli protesici che servono a restringere l’anulus
restituendo continenza. Ce ne sono di due tipi:
Anello di Carpentier: montato come fosse una valvola, infatti i punti vengono prima passati
nell’anulus valvolare originale con ampiezza si circa 1cm, poi sono passati nell’anello
protesico con un’ampiezza minore. In questo modo, abbassando l’anello protesico, anulus
valvolare si stringe.
Anello di Puig-Massana: viene fissato direttamente sull’anulus valvolare, all’estremità ha
due capi che, una volta tirati e legati riducono il diametro dell’anulus valvolare.
Un altro intervento molto diffuso è la resezione quadrangolare che consiste
nell’asportazione del segmento centrale del lembo posteriore mitralico (scallop M2, il lembo
posteriore è costituito da 3 scallops: M1-M2-M3) e poi accostamento degli scallops laterali.
L’intervento si effettua in circolazione extracorporea, ma si può praticare ministernotomia
mediana. Questa tecnica permette anche di effettuare sostituzioni valvolari. Si incide a
partire dall’incisura giugulare dello sterno fino al 3°-4° spazio intercostale poi si devia
trasversalmente verso destra. Si avvia la circolazione extracorporea dopo cannulazione
dell’aorta ascendente e (con altre due cannule) delle vene cave. L’auricola destra viene
aperta e si entra nell’atrio destro. Si incide il setto interatriale e la mitrale, ora esposta, può
essere ora sostituita o riparata. Viene poi chiuso il setto interatriale mediante sutura con
53
prolene 4-0 e viene suturata anche l’auricola destra per poi procedere alla decannulazione.
Possono essere necessarie altre tecniche riparative:
Edge to edge: (di Alfieri) apposizione di un punto di sutura tra la parte centrale di un lembo
prolassante con il margine corrispondente dell’altro lembo prolassante (si vien a creare un
ostio a otto).
Creazione di nuove corde tendinee: può essere necessaria, si impiega politetrafluoroetilene
espanso (goterex).
Accorciamento delle corde tendinee: se le corde troppo lunghe sono responsabili di un
prolasso (del lembo anteriore mitralico) con conseguente insufficienza si seziona il muscolo
papillare ed all’interno di esso viene invaginato il tratto di corda da eliminare, con successiva
sutura dell’incisione e conseguente accorciamento della corda.
Trasposizione delle corde tendinee: se le corde non sono adatte all’accorciamento perché
molto sottili (potrebbero rompersi) si trasferiscono le corde dal lembo posteriore al lembo
anteriore.
Prolasso mitralico
Definizione: alterazione della valvola mitrale che consiste in lembi mitralici ridondanti che i
estroflettono (prolassato) anche cospicuamente (a volte sbandierando) oltre l’anello si
inserzione durante la sistole ventricolare (spinti dalla pressione ventricolare). Può
interessare fino al 6% della popolazione, specialmente femminile. Solo nel 10% causa
insufficienza mitralica,che solo in un terzo di questi è rilevante.
Vi sono forme primarie (familiari, sindrome di Marfan, altre patologie connettivali), e
secondarie (FR, cardiopatia ischemica, cavità ventricolare ridotta come nella miocardiopatia
ipertrofica, difetti interatriali, anoressia, ipertensione polmonare).
Di solito avviene che lo strato spongioso intermedio dei lembi valvolari è aumentato a causa
di infiltrazione sempre maggiore (anche a soverchiare il tessuto fibroelastico atriale e lo
strato di collageno rivolto verso il ventricolo) dei mucopolisaccaridi (tessuto mixomatoso) e
causa perdita di consistenza delle cuspidi. Possono essere coinvolte anche le corde tendine
spesso allungate.
Nella sindrome di Marfan quest’alterazione può coinvolgere anche la valvola tricuspide e
aortica. Le malattie da disordini connettivali come la sindrome di Marfan comportano spesso
queste alterazioni (pertanto la sindrome di Marfan costituisce anche un maggiore rischio di
insufficienza mitralica).
Patogenesi: nella maggior parte dei casi la patologia è diagnosticata tramite un
ecocardiogramma, ma non comporta nessun sintomo o comunque nessun sintomo grave o
specifico. L’insufficienza mitralica può insorgere per esagerata fluttuazione dei lembi o
eccessiva lunghezza delle corde o per sovrapposta endocardite. Ancora meno comune che
evolva verso insufficienza massiva.
Clinica: il paziente è spesso asintomatico. I sintomi possono essere una certa astenia e
ipotensione ortostatica. Può esserci notevole ansia per sintomi come vertigini, palpitazioni e
fugaci dolori toracici non associati allo sforzo. Possono esserci aritmie sopraventricolari e
lieve dispnea. L’ischemia cerebrale è leggermente più comune in questi pazienti. Se compare
IM si associano i sintomi relativi.
Esame obiettivo: si può rilevare un click mesosistolico (alla punta) dovuto alla massima
54
tensione dei lembi. Può essere seguito da un soffio mesotelesistolico da insufficienza
mitralica. Il click si sente prima in ortostatismo (riduzione della pressione e del postcarico e
quindi più rapido svuotamento sistolico del ventricolo) e si sente dopo nel clinostatismo (per
i motivi opposti) in cui tra l’altro aumenta anche l’intensità dell’eventuale soffio.
Diagnosi: oltre all’esame obiettivo, si possono rilevare all’ECG alterazioni nella
depolarizzazione (tratto ST sottoslivellato e onde T invertite) e all’Holter possono comparire
aritmie sopraventricolari. All’ecocardio, soprattutto bidimensionale, è possibile vedere segni
del prolasso e anche verificare la presenza di IM. Può confermare l’obiettività.
Prognosi: in genere benigna con decorso asintomatico anche per tutta la vita. Da discutere
l’idoneità sportiva, specie in caso di aritmie.
Terapia: i pazienti vanno rassicurati sulla benignità della condizione. Se c’è storia di
endocardite si può fare profilassi. I beta-bloccanti possono in alcuni casi alleviare i sintomi
(possono anche essere indicati ansiolitici). L’indicazione chirurgica c’è solo nei casi di IM che
la richiedono.
Stenosi aortica
Definizione: malattia congenita o acquisita delle semilunari aortiche caratterizzata da
restringimento valvolare che provoca ostruzione all’efflusso di sangue dal ventricolo sinistro
con sviluppo di gradiente pressorio trans valvolare e ipertrofia concentrica del ventricolo. In
un soggetto normale la valvola è tricuspide (lembo destro, sinistro e posteriore) con ostio tra
i 3 e i 4cm2. È considerata stenosi una riduzione sotto i 1,5cm 2. Talvolta si associa ad una
dilatazione post-stenotica dell’aorta. Anche altre condizioni possono portare blocco
dell’efflusso ventricolare sinistro, come stenosi sotto o sopravalvolari.
Eziologia:
1) Degenerazione fibro-calcifica di una valvola aortica congenitamente malformata: è stato
calcolato che circa l’1-2% della popolazione possiede una valvola aortica bicuspide (in
qualche caso addirittura monocuspide con un piccolo orifizio centrale) e solo 1/3 di queste
non svilupperà né stenosi né insufficienza aortica. Nella maggior parte dei casi questi
individui sono più suscettibili al trauma meccanico e forse anche alla deposizione di trombi.
Spesso si manifesta tra 4° e 5° decade di vita. In alcuni può essere in associazione con la
coartazione dell’aorta.
2)Malattia fibrocalcifica postinfiammatoria: la malattia reumatica può coinvolgere anche la
semilunare aortica con ispessimento delle cuspidi (dovuto alla flogosi) e fusione delle
commessure con restringimento dell’orifizio. Si può avere anche una retrazione dei bordi
con conseguente steno-insufficienza aortica.
3) Degenerazione fibro-calcifica senile: rappresenta forse lo stesso processo che si verifica
più rapidamente nei soggetti con valvola bicuspide. Si ha flogosi e poi progresiva
calcificazione dei lembi (e può interessare anche aorta, e anche mitrale). Si associa agli stessi
fattori di rischio dell’aterosclerosi (processo comune) e all’ipertensione e aumento della
lipoproteina A. è la causa più frequente di stenosi nella 7°-8° decade di vita.
Patogenesi: una stenosi che riduce l’orifizio del 50% genera resistenza all’eiezione
ventricolare con aumento della pressione intraventricolare sinistra e sviluppo di un
55
gradiente pressorio tra ventricolo sinistro e aorta (di entità relativa al grado di stenosi).
Poiché la stenosi aortica si sviluppa gradualmente il ventricolo sinistro può innescare
meccanismi di adattamento.
Il sovraccarico pressorio comporta ipertrofia concentrica ossia aumento del numero di
sarcomeri in parallelo (anziché in serie, come nell’eccentrica) con conseguente aumento
dello spessore miocardico senza dilatazione della cavità. Grazie a questa ipertrofia adeguata,
ciascuna miofibrilla è posta alla stessa tensione di un cuore normale cosicché il paziente
resta a lungo asintomatico.
Questo meccanismo però comporta un notevole dispendio energetico con relativo aumento
del fabbisogno di ossigeno da parte del miocardio (a causa della maggiore massa, e anche
del maggiore tempo di eiezione).
Contemporaneamente però nel cuore ipertrofico si ha una riduzione del flusso coronarico
per grammo di muscolo per aumento della pressione diastolica e quindi compressione delle
arterie coronariche e quindi minore riserva e capacità di dilatazione, ma anche per il ridotto
tempo di diastole, la ridotta pressione aortica per la perfusione coronarica (le coronarie
sboccano a livello dei seni di Valsalva, dopo la valvola stenotica) e per riduzione della densità
vascolare capillare (più massa, ma stesso numero di vasi).
Questo squilibrio nella perfusione miocardica comporta l’insorgenza di ischemia miocardica
con possibile angina, ma anche aritmie (focolai eccitati da ipossia), sincopi e anche morte
improvvisa.
Uno squilibrio cronico può produrre fibrosi e compromissione della funzione ventricolare.
L’ipertrofia concentrica comunque è per molti ani un sistema di adattamento che garantisce
un compenso perfetto. Però se poi la contrattilità ventricolare diviene ridotta (per esempio
per fibrosi), se si verifica un incremento progressivo della stenosi e semplicemente la riserva
contrattile (capacità di ipertrofia compensatoria) si esaurisce, il volume tele sistolico del
ventricolo sinistro aumenta e si produce una dilatazione con stiramento delle fibre.
Si ha alla lunga disfunzione sistolica perché le fibre si dilatano e, non potendo più
ipertrofizzare, non sono più in grado di contrarsi efficacemente. Inoltre l’ipertrofia
concentrico comporta anche alla lunga una riduzione della compliance ventricolare (rigidità)
limitando il riempimento, comportando cioè disfunzione diastolica.
Questo vuol dire che il cuore non è in grado di aumentare il ritorno venoso (precarico) in
modo adeguato alla necessità della massima (incapacità di reclutamento del precarico), a
causa della fibrosi e dell’ipertrofia (la disfunzione diastolica è spesso più precoce, senza
disfunzione sistolica nel 50% dei casi).
Pertanto per ottenere certi volumi c’è bisogno di un’elevata pressione di riempimento
(causa dei sintomi come dispnea ed edema polmonare). L’atrio sinistro svolge in questo un
ruolo fondamentale, aumentando la pressione al momento della presistole (calcio atriale)
per favorire il riempimento. Pertanto aritmie sopraventricolari, come la fibrillazione atriale
che fanno venir meno questo meccanismo causano un rapido peggioramento.
Se la contrattilità è conservata, e l’ipertrofia e la fibrosi non sono ancora troppo massicce,
l’intervento chirurgico permette di normalizzare rapidamente la situazione. La depressione
di contrattilità, potrebbe essere dovuta ad anomalie nel ciclo del calcio o espressione di
miosina beta V3 (a lenta contrazione).
Clinica: può restare asintomatica per anni (fase di latenza) anche se c’è una stenosi serrata.
La diagnosi avviene in fase sintomatica per presenza di soffio o di ECG anormale, oppure non
appena si presentano i sintomi, cosa che spesso accade tra i 50 e i 60 anni. Dopo la comparsa
dei sintomi la prognosi è spesso inferiore ai 5 anni.
56
La morte improvvisa senza sintomatologia è minore dell’1% all’anno.
La triade classica è:
1) Angina pectoris: comune a causa della minore perfusione, con possibile comparsa di
infarti soprattutto sub endocardici, ma anche trans murali. È il sintomo più frequente (5070%) anche senza lesioni evidenti alla coronarografia, cosa che comunque è più comune in
questi pazienti. Può essere accentuata da stress fisici e tachicardia.
2) Sincope: a causa della ridotta gittata, la perfusione cerebrale, soprattutto sotto sforzo è
ridotta (come la perfusione coronarica). Una vasodilatazione periferica a livello muscolare
sotto sforzo causa ipotensione che non viene compensata da una gittata cardiaca che risulta
fissa. Le aritmie che possono manifestarsi (ipercinetiche) potrebbero essere una concausa.
3) Dispnea: a volte anche ortopnea, DPN ed edema polmonare, dovuti alla congestione
polmonare secondaria a scompenso sinistro (prognosi di 2 anni).
Diagnosi:
Esame obiettivo: l’itto può essere intenso per ipertrofia ventricolare sinistra, il polso
arterioso piccolo e tardo (per ridotta ampiezza e aumento del tempo di eiezione).
In genere c’è diminuzione della differenziale (per diminuita sistolica, ma non negli anziani).
Si ha un’onda a prominente al polso giugulare per ridotta compliance del ventricolo destro
per ipertrofia del setto (effetto Bernheim).
Si può avvertire un fremito sistolico in genere nel II spazio intercostale destro (area aortica).
All’auscultazione si può avere un click sistolico (tono di eiezione ad alta intensità nell’area
aortica).
Si può avere scomparsa dello sdoppiamento del II tono oppure in espirazione addirittura
sdoppiamento paradosso del II tono entrambi a causa del ritardo della chiusura della
componente aortica. Nelle forme calcifiche la componente aortica può anche scomparire.
Si ha un soffio sistolico a diamante in area aortica ossia con morfologia “crescendodecrescendo”, spesso aspro (specie se calcifica, ma nell’anziano può essere musicale) con
intensità variabile tra 3/6 e 6/6 (infatti è spesso associato al fremito). Può anche esserci un
soffio proto diastolico alla base per lieve insufficienza aortica.
Può comparire un IV tono per ridotta complicanze e forte contrazione atriale.
ECG: possono esserci segni di ipertrofia ventricolare sinistra e segni come onde T invertite
simmetriche o modificazioni del tratto ST che sono segni prognostici infausti.
Radiografia del torace: si ha configurazione aortica con aumentato diametro di profondità
(rx laterale) e maggiore sporgenza a sinistra. Si può notare dilatazione post-stenotica
dell’aorta e possibili calcificazioni.
Ecocardiogramma: è estremamente utili come in tutte le valvulopatie perché permette: di
distinguere una reale stenosi aortica da una stenosi sotto o sopravalvolare; identifica
ispessimento, ipomobilità, dilatazione post-stenotica dell’aorta e ipertrofia concentrica del
ventricolo sinistro. Con l’eco-doppler si può valutare il gradiente trans valvolare tra
ventricolo sinistro ed aorta (con il teorema di Bernoulli P=4*V 2) e si può anche valutare con
buona precisione l’area valvolare aortica oltre a tutte le condizioni morfofunzionali. Analisi
pratica, non invasiva e ripetibile.
Cateterismo cardiaco: procedure diagnosti preoperatoria, obbligatoria (con coronarografia)
se si vuole escludere una coronaropatia. Permette di valutare bene la patologia e con grande
accuratezza l’entità della stenosi. Una compromissione significativa del flusso si ha
normalmente con area valvolare di 0,8cm2 (riduzione del 75%), che crea infatti un gradiente
anche superiore a 50mmHg. Nel valutare il gradiente trans valvolare bisogna però
compararlo alla portata cardiaca, se bassa un gradiente minore (30mmHg) può indicare
57
stenosi severa, se alta un gradiente maggiore può indicare stenosi lieve.
Nel valutare la gravità della stenosi si usa perciò una classificazione in base all’area valvolare
media (misurata tramite l’equazione di Gorlin): lieve (>1,5cm2); moderata (1,5-1); medioserrata (1-0,75); serrata (<0,75).
Terapia medica: nei pazienti senza stenosi severa e asintomatici non è necessaria alcuna
terapia, data la tendenza al rimanere stabili. Se vi è stenosi severa (<1cm 2) va evitata
l’attività fisica strenua. La nitroglicerina e i nitrati, usato con prudenza possono aiutare i
pazienti con angina pectoris. Segni di scompenso o ipertensione arteriosa possono far
somministrare beta-bloccanti o calcio-antagonisti. Si è visto che la stenosi calcifica evolve più
lentamente se sono utilizzati inibitori dell’HMG-CoA reduttasi (statine).
Terapia chirurgica: nei pazienti con stenosi severa, ma senza disfunzione ventricolare
sinistra (FE>50%) e asintomatici è bene procrastinare l’intervento ed effettuare controlli
periodici. In tutti i pazienti con area valvolare <1cm 2, gradiente medio maggiore di 40mmHg,
velocità massima di flusso trans valvolare >4m/s che: devono essere sottoposti a bypass
coronarico o altri interventi cardiochirurgici, presentano sintomatologia di scompenso,
hanno frazione di eiezione <50%, e (secondo alcuni) hanno calcificazione valvolare severa e
rapida progressione sono candidati alla sostituzione valvolare aortica (previa coronarografia
di controllo). La mortalità nei pazienti senza scompenso è inferiore al 3%, e la sopravivenza a
dieci anni è maggiore del 70% (anche se anziani), mentre la mortalità sale al 20% se vi sono
importanti segni di scompenso (del resto l’intervento rimane l’unica scelta, data in questi
casi l’inefficacia della terapia medica).
La valvuloplastica aortica percutanea con palloncino non ha dato grandi risultati e può
essere indicata solo per gruppi ristretti di pazienti, come quelli con alto rischio per
intervento chirurgico (funzione renale, epatica o polmonare gravemente compromessa).
Procedura: sostituzione valvolare aortica: viene eseguito in CEC e ipotermia moderata con
cardioplegia. Si può eseguire con ministernotomia. Si effettua cannulazione dell’aorta
ascendente (e cave) per la CEC, viene poi incisa l’aorta per poter esporre la valvola aortica
che deve essere asportata e poi sostituita con protesi valvolare. La protesi viene posizionata
così da poter essere suturata a livello dell’anulus artico, si sutura poi anche l’aorta
ascendente e si decannula.
Insufficienza aortica
Definizione: consiste nella chiusura incompleta delle cuspidi valvolari durante la diastole del
ventricolo sinistro, con relativo reflusso diastolico dall’aorta. Il sovraccarico di volume
comporta un’ipertrofia eccentrica che alla lunga deprime la funzione ventricolare causando
scompenso. È dilatata anche l’aorta e nelle fasi avanzate l’anello mitralico (insufficienza
mitralica secondaria funzionale)
Eziologia: un tempo le cause più comuni, ora in netta diminuzione, erano malattia reumatica
(tramite sclerosi e retrazione dei lembi) e sifilide. Altre cause sono malformazioni tipo aorta
bicuspide, vegetazioni da endocardite (che possono provocare perforazioni), connettivopatie
(tipo sindrome di Marfan), malfunzionamenti delle protesi.
Malattie della radice aortica possono causare insufficienza per dilatazione e distorsione
della radice (come nell’aortiti, ad esempio da lue o AR, e nelle aortopatie non
infiammatorie) o per perdita del supporto commissurale (come nella dissecazione o
58
aneurisma dissecante dell’aorta in cui l’anulus si può dilatare notevolmente).
Patogenesi: si distinguono una forma acuta ed una cronica.
Cronica: il reflusso cronico di sangue aumenta il riempimento tele diastolico del ventricolo
sinistro il quale, per mantenere una gittata normale aumenta di volume (si dilata) e inizia a
ipertrofizzarsi con ipertrofia eccentrica (relplicazione in serie dei sarcomeri). Il cuore può
arrivare a pesare anche più di 1kg. L’insufficienza è ben tollerata anche per decadi, perché il
compenso con dilatazione e ipertrofia riesce a mantenere adeguata la gittata. Alla lunga la
pressione tele diastolica ventricolare aumenta in quanto la riserva di precarico (capacità di
dilatazione e ipertrofia delle fibre) si esaurisce e non riesce ad affrontare l’aumento del post
carico.
Si va verso la disfunzione ventricolare sinistra (in genere dopo la 4° decade) che come primo
sintomo presenta la dispnea da sforzo (aumento della pressione polmonare). Un’altra
caratteristica è un aumento della pressione differenziale a causa dell’aumento della
pressione sistolica (maggiore volume di sangue) e diminuzione della diastolica (a causa del
rigurgito).
La minore pressione diastolica aortica corrisponde ad una minore pressione di perfusione
coronarica, che associata all’aumentata richiesta di ossigeno da parte del miocardio
ipertrofico può comportare episodi ischemici (per lo più in fase di sforzo), come succede
anche nella stenosi aortica. Si possono distinguere quattro fasi evolutive:
Prima fase: si ha un equilibrio stabile nonostante l’insufficienza, malgrado la cardiomegalia
la funzione ventricolare risulta normale .
Seconda fase: dopo anni, un po’ a causa di fibrosi e un po’ per l’aumento dello stress di
parete, l’ipertrofia diviene inadeguata e si ha depressione della contrattilità, ma ancora
senza causare sintomi.
Terza fase: aumenta lo stress, si deprime ancora la contrattilità, l’ipertrofia è sempre più
inadeguata per riduzione della riserva di miocardio contrattile e il paziente può avere
sintomi sotto sforzo.
Quarta fase: decisamente sintomatica, con frazione di eiezione ridotta anche a riposo e
scompenso sinistro come in una cardiomiopatia dilatativa. La chirurgia in questa fase
presenta alto rischio.
Acuta: se l’evento che causa insufficienza (endocardite acuta o dissezione aortica) è
improvviso si ha rigurgito acuto con il ventricolo che non può rispondere con l’ipertrofia
eccentrica. Si ha un aumento improvviso della pressione tele diastolica con contemporaneo
aumento della pressione atriale sinistra e quindi nel circolo polmonare (sintomi come
dispnea, ma anche edema polmonare). L’unica risposta possibile per il cuore non potendo
svuotarsi, è la tachicardia (si attiva infatti il sistema simpatico e si ha anche vasocostrizione e
sudorazione), si può avere shock cardiogeno ed edema polmonare acuto.
Clinica:
Cronica: in prima fase, ma anche in seconda e in terza il paziente è asintomatico, e si può
mantenere tale per moltissimi anni. In terza fase può lamentare astenia (ridotta gittata) e
più raramente dispnea da sforzo. Questa fase dura circa tre anni, poi si entra nella quarta, in
cui il paziente, a causa dell’insufficienza sinistra può lamentare angina pectoris (anche a
riposo e poco responsiva a nitrati) e anche dispnea parossistica notturna ed edema
polmonare da stasi. L’ipertensione arteriosa polmonare con associato scompenso sinistro è
59
un dato prognostico molto grave. La morte improvvisa è più frequente se c’è forte
dilatazione del ventricolo sinistro. Ci può essere coronaropatia associata nel 20% dei casi.
Acuta: si ha rapidamente ipotensione e sincope, il paziente suda ed è tachicardico, compare
edema polmonare.
Diagnosi:
Esame obiettivo: il polso appare spesso ampio e celere (polso scoccante di Corrigan) a causa
della forte grande pressione differenziale. La stessa provoca la cosiddetta danza della arterie
con vistose pulsazioni soprattutto delle arterie superficiali (carotidi e omerali) e che causa a
volte oscillazioni ritmiche del capo (segno di De Musset, poeta francese affetto) così come
quelle degli arti superiori (segno di Ascoli), o degli inferiori accavallati o della lingua profusa.
Ciò si può trasmettere anche ai capillari causando il polso capillare di Quincke (facendo
pressione sul letto ungueale si nota ritmico arrossamento e impallidimento) e hippus
circolatorio (ampliamento e restringimento della pupilla). Tutti questi segni sono dovuti alla
veloce e potente eiezione del ventricolo sinistro ipertrofico seguita dal rigurgito con relativo
calo di pressione.
L’itto è spostato in basso a sinistra e iperdinamico. Ci può essere un III tono, mentre la
precoce chiusura della mitrale può far ridurre il I tono.
All’auscultazione possono essere evidenti tre soffi:
Soffio diastolico da rigurgito in area aortica subito dopo il II tono(dura di più se è grave);
Soffio da eiezione aortico a causa della gittata sistolica elevata (quota anterograda +
rigurgito);
Soffio di Austin-Flint: telediastolico puntale, dovuto allo spostamento del lembo mitralico da
parte del rigurgito aortico.
Nel rilevare la pressione si nota naturalmente una sistolica elevata e diastolica bassa e
questa può anche sembrare 0 (anche se in realtà non scende mai sotto 30) perché i toni di
Korotkoff possono persistere a lungo.
Acuta: caratteristiche meno evidenti, il polso è piccolo ma non scoccante perché
l’attivazione simpatica limita l’aumento della pressione differenziale. Il soffio diastolico
anche è meno evidente perché si ha aumento precoce della pressione diastolica ventricolare
(ciclo cardiaco più rapido) e quindi gradiente trans valvolare ridotto. Può sfuggire.
ECG: il complesso QRS può apparire di ampiezza aumentata con onde R spesso elevate e/o a
salita rapida. Nelle fasi più gravi possono apparire onde T invertite simmetriche e BBS.
Radiografia del torace: può vedersi ventricolo sinistro dilatato e anche l’aorta ascendente
allargata (più che nella stenosi).
Ecocardiogramma: fondamentale per valutare i lembi valvolari e la presenza di rigurgito,
eventuale fluttuazione dei lembi, dimensioni e dinamica del ventricolo sinistro e suoi indici
funzionali. Con l’eco-doppler si valuta la gravità del rigurgito e si può calcolare il gradiente
trans aortico. Nel follow-up è utilizzata l’angioscintigrafia con tecnezio che valuta funzione
ventricolare e frazione di rigurgito.
Cateterismo cardiaco: attualmente meno indicato perché l’ecocardio è molto preciso.
Permette di rilevare la pressione diastolica ventricolare e la precisa entità del rigurgito
(tramite aortografia con m.d.c.) classificandolo da 1 a 4 (lieve, moderato, medio, massivo).
La frazione di rigurgito è calcolata con FR=volume rigurgitante per battito/gittata sistolica
totale (anterograda+rigurgito). Se FR>60% è massiva.
60
Prognosi: le forme lievi possono essere asintomatiche anche tutta la vita, anche se
peggiorano rapidamente con endocardite infettiva. Dopo la comparsa di disfunzione
ventricolare la sopravvivenza a 10 anni è del 50%. Sono però pericolosi emboli, aritmie e
l’insorgenza, per marcata dilatazione ventricolare, di insufficienza mitralica anche grave.
Nelle forme acuta la mortalità è molte elevata anche in ore o giorni.
Terapia medica: nell’acuta c’è risposta a diuretici e vasodilatatori, ma è indicata la chirurgia
urgente. Nella cronica oltre all’uso di anticoagulanti in pazienti con episodi embolici, sono
indicati diuretici e vasodilatatori se non per curare lo scompenso, almeno per mantenere la
pressione sistolica <140mmHg. Bisogna evitare esercizio fisico isometrico, possono essere
utili (non quanto nella coronaropatia) nitrati a lunga durata d’azione. Nel caso in cui sia
depressa la contrattilità, agenti inotropi positivi come la digitale sono utili.
Terapia chirurgica: Nelle forma acute l’indicazione è estensiva.
Nelle forme croniche vengono sicuramente indirizzati pazienti gravemente sintomatici, ma
anche i pazienti asintomatici devono essere valutati e seguiti possibilmente con un follow-up
che preveda un’ecocardiogramma ogni 6 mesi (o 3-12).
Questo perché bisogna ricorda che i sintomi compaiono solo dopo comparsa di una
disfunzione miocardica e che il trattamento chirurgico non ripristina la normale funzione
ventricolare se eseguito con troppo ritardo. Un controllo costante permette di eseguire
l’intervento nel momento migliore, quando è ancora utile e quando la mortalità è bassa.
La mortalità è inoltre di circa il 3% nei pazienti con funzione ventricolare buona, e di quasi il
20% negli scompensati (in cui però comunque la terapia medica è poco utile). Oltre ai
sintomatici (anche sotto sforzo, come sempre), l’intervento è indicato se
all’ecocardiogramma risulta FE<50%, diametro tele sistolico>55mm o diametro tele
diastolico >75mm. Nella maggior parte dei pazienti l’intervento consiste nella sostituzione
della valvola. In caso di dilatazione aneurismatica dell’aorta ascendente e
conseguentemente dell’anulus si potrebbe eliminare solo il tratto di aorta dilatato o
restringere l’annulus, nel 50% dei pazienti con dissecazione aortica (che hanno alto rischio di
mortalità), è possibile un intervento ripartivo. Risulta invece impossibile se c’è perforazione
dei lembi (endocardite) o distaccamento dell’anulus. I risultati a distanza degli interventi
dipendono dal recupero funzionale del ventricolo (si possono usare nel post-operatorio
digitale e vasodilatatori). A 15 anni una buona sopravvivenza si ha nel 70% dei casi.
I più comuni sono comunque gli interventi di sostituzione valvolare.
La sostituzione può avvenire utilizzando protesi valvolare (intervento di Wheat con
sostituzione separata dell’aorta scendente con protesi tubulare in dacron e della valvola con
protesi valvolare) o applicazione di tubi valvolati tipo Bentall (sostituzione di aorta
ascendente e valvola insieme, se come spesso accade, la patologia si associa a dilatazione e
patologia della radice aortica o se semplicemente si ritiene più semplice perché ad esempio
la radice aortica è troppo dilatata).
Si possono anche evitare le protesi meccaniche della valvola, maggiormente soggette a
complicanze e con minore durata nel tempo tramite: Interventi che prevedono l’uso di un
tubo protesico aortico: conservazione della valvola se tricuspide e anatomicamente normale
(interventi di David e Yacoub, c’è sempre bisogno di reimpiantare gli osti coronarici);
Intervento di Ross: trasposizione aortopolmonare. Consiste nella sostituzione della valvola
aortica e/o della radice aortica con la valvola polmonare dello stesso paziente (autograft) e
nella ricostruzione della via di efflusso polmonare mediante protesi presa da cadavere
(homograft). Può essere preferibile alla sostituzione con protesi perché: la valvola
61
polmonare ha struttura analoga all’aortica e sembra avere una minore tenuta e durata
emodinamica rispetto alle protesi biologiche o meccaniche, inoltre l’incidenza di endocardite
sembra bassa e non c’è bisogno di lunga anticoagulazione (meno episodi trombo embolici).
È indicato in pazienti giovani, per disfunzione di protesi, se c’è controindicazione
all’anticoagulazione, nelle donne che intendono avere gravidanze.
È controindicato se la polmonare è danneggiata, se c’è coronaropatia multi vasale, nei
pazienti con grave disfunzione ventricolare che non possono tollerare lunghe procedure
chirurgiche, nella malattia reumatica e FA cronica.
Si può eseguire con: tecnica d’inclusione: non possibile se l’aorta è molto dilatata, consiste
nell’inserire nella radice aortica la valvola polmonare con un tratto di arteria; sostituzione
completa della radice aortica: tecnica meno impegnativa, si adatta meglio e in modo più
semplice alle asimmetrie della radice aortica. Complicanze dell’intervento di Ross sono
sanguinamento, aritmie e IMA. Per quanto restino dei dubbi, l’autograft e (meno)
l’homograft vanno meno incontro a processi degenerativi rispetto alle protesi.
Procedura: si fa stereotomia longitudinale mediana, cannulazione arteriosa e venosa e avvio
della circolazione extracorporea. Si fa aortotomia e si escide la valvola aortica, si rimuove
l’autograft polmonare (dopo incisione dell’arteria polmonare) lo si impianta a livello aortico
con reimpianto dei osti coronarici (nei seni di Valsalva). Si utilizza l’homograft (preso da
cadavere, preparato e conservato a bassissima temperatura) per ricostruire la via d’efflusso
polmonare.
Sostituzione valvolare aortica con homograft: non necessita di trattamento anticoagulante,
si può utilizzare anche per ricostruire la radice aortica e ha maggiore resistenza
all’endocardite. Nei pazienti sopra i 35 anni si possono anche applicare valvole porcine
stentless (biologiche non montate su stent) che hanno una maggiore durata nel tempo delle
valvole biologiche trattate con glutaraldeide.
Stenosi tricuspidale
Definizione: ST, rara condizione clinica con ostacolo attraverso la tricuspide al flusso
dall’atrio al ventricolo destro durante il suo riempimento diastolico.
Eziologia: è raro che sia isolata, in buona parte dei casi è associata ad una vavulopatia
mitralica e/o anche aortica in quanto la causa più comune è la malattia reumatica. È più
comune nelle donne. Può essere causata anche da mixomi, LES, sindromi congenite,
sindrome da carcinoide (che di solito causa insufficienza).
Anatomia patologica: come nella stenosi mitralica, si ha fibrosi e fusione delle commessure
o dei lembi. Rara la calcificazione. L’atrio destro è spesso molto dilatato, ma non ipertrofico.
Fisiopatologia: l’area normale della tricuspide è di 5-7cm 2, un restringimento dell’ostio
inferiore a 1,5 crea un significativo ostacolo al riempimento del ventricolo destro.
Si ha aumento della pressione nell’atrio destro e riduzione della gittata cardiaca.
Si ha quindi uno stato congestizio del sistema venoso sistemico con turgore giugulare ed
epatosplenomegalia (vene del circolo splancnico sono spesso coinvolte), oltre ad un
progressivo scompenso cardiaco. L’onda presistolica atriale destra (onda a) è gigante. Nella
fibrillazione atriale la pressione può aumentare ancora.
La riduzione della portata spiega perché i livelli della pressione in atrio sinistro e circolo
62
polmonare sono normali o poco aumentati anche in presenza (molto comune) di una
valvulopatia mitralica. L’atrio destro in genere si dilata anche più precocemente del sinistro.
Clinica: sintomi modesti e atipici, con affaticabilità e astenia (ridotta portata) e dispnea da
sforzo. Sensazione di replezione per l’epatomegalia e la stasi venosa addominale (che può
anche causare anasarca). Nonostante possa coesistere molto spesso una stenosi mitralica i
sintomi di questa sono attenuati per un cosiddetto effetto “smorzante” o “protettivo” della
stenosi tricuspidale che evita i sintomi della congestione polmonare (si ha ridotta gittata del
ventricolo destro, così in genere DPN e ortopnea sono insoliti e rarissimi sono edema
polmonare ed emottisi). Si può avere evoluzione in cirrosi.
Diagnosi:
Esame obiettivo: Onda a prominente al polso giugulare, I tono può essere aumentato
(chiusura della valvola ispessita) mentre la componente polmonare del II è molto ridotta. Si
può avvertire un soffio diastolico in area tricuspidale (IV-V spazio sinistro) che aumenta alla
fine di un’inspirazione profonda (segno di Carvallo) per aumento del ritorno venoso all’atrio
destro e quindi del flusso attraverso la valvola ristretta. Si possono avvertire i segni di stenosi
mitralica e/o aortica.
ECG: ci sono onde P atriali elevate.
Radiografia del torace: dilatazione dell’atrio destro e possibile elevazione del diaframma per
epatomegalia.
Ecocardiogramma: esame fondamentale anche in questa valvulopatia perché anche se la
tricuspide è di difficile visualizzazione si possono vedere lembi ispessiti, mobilità ridotta e
aspetto “a cupola” in apertura. Il gradiente pressorio non supera 1mmHg in condizioni
fisiologiche, perciò già un gradiente di 2-3mmHg è indicativo di stenosi.
Terapia medica: si fa generalmente profilassi per l’endocardite e i diuretici attenuano i
sintomi della stasi venosa (associati a dieta povera di sodio).
Terapia chirurgica: Se l’orifizio è inferiore a 2cm2 e il gradiente supera i 4mmHg è indicato
l’intervento, in genere contemporaneamente a valvulotomia mitralica. Si può pensare ad
una commissurotomia che si esegue a cuore aperto aprendo le commessure fra i lembi
anteriore e settale e i lembi posteriore e settale. Se non è possibile si può fare anuloplastica
o sostituzione valvolare preferendo una protesi biologica (meno complicanze
tromboemboliche). La valvuloplastica con palloncino è fattibile, ma con efficacia tuttora
incerta.
Insufficienza tricuspidale
Definizione: IT, caratterizzata dal rigurgito di sangue dal ventricolo all’atrio destro durante la
sistole ventricolare, con sovraccarico di volume nelle due cavità e dilatazione.
L’insufficienza può essere dovuta a lesioni dell’apparato ventricolare destro (lembi anteriore
posteriore e settale, corde tendinee, muscoli papillari e muscolo di Lancisi per il lembo
settale) stesso (insufficienza organica, o organo-valvolare secondo Carral) o per dilatazione
ventricolare e dell’anulus (insufficienza funzionale o organo-muscolare secondo Carral).
La forma funzionale, mista, è più spesso associata a stenosi mitralica di origine reumatica.
Eziologia: la cardiopatia reumatica resta la prima causa di insufficienza tricuspidale cronica.
63
La forma cronica si accompagna più spesso a stenosi mitralica (e si ha IT sia per
coinvolgimento reumatico diretto che per motivi funzionali secondari a ipertensione
polmonare). Altre cause sono patologie che colpiscono il ventricolo destro, sindrome da
carcinoide, cardiopatia ischemica cronica. L’ischemia acuta è invece una delle cause di IT
acuta che può essere anche dovuta ad embolia polmonare oppure ad endocardite.
Patogenesi: IT cronica: è raramente un evento isolato. Si ha sovraccarico cronico di volume
con dilatazione di atrio e ventricolo destro, con pressione aumentata nell’atrio destro e nelle
vene cave. Aumenta principalmente la pressione in fase sistolica con flebogramma giugulare
che indica onde c+v fuse insieme. La pressione diastolica ventricolare destra resta a lungo
normale (intorno a 7mmHg) perché il ventricolo destro ha grande compliance. Questo è vero
se l’IT non è associato ad una stenosi mitralica che a causa di ipertensione polmonare ha
portato ad ipertrofia (e ridotta compliance) del ventricolo destro. In questo causo le
pressioni atriale destra e venosa sistemica salgono molto con evidente turgore giugulare ed
epatomegalia, anche ascite (scompenso congestizio). È frequente l’associazione (date il
notevole disturbo all’atrio destro) con fibrillazione atriale.
IT acuta: aumento della pressione atriale sistolica con valori pressori atriali medi però non
troppo elevati. Il sovraccarico di volume è spesso ben tollerato nel ventricolo sinistro, ma
l’aumento della pressione atriale destra e venosa sistemica è tollerato in modo assai
variabile.
Clinica: nelle forme croniche associate a valvulopatia mitralica il segno di insufficienza
tricuspidale compare lentamente con edemi declivi, epatomegalia anche notevole e
tensione al collo da turgore giugulare. Nell’acuta può esserci epatomegalia anche dolorosa.
Diagnosi
Esame obiettivo: si può rilevare epatomegalia anche pulsante (alta pressione venosa
sistolica). Si può notare una pulsazione al precordio da ipertrofia ventricolare destra.
Soprattutto se l’ipertrofia ventricolare destra è importante si apprezza un soffio olosistolico
in area tricuspidale (che si accentua in apnea postinspiratoria e si riduce in stazione eretta e
nella fase di compressione della manovra di Valsalva). Se coesiste ipertensione polmonare (e
quindi il gradiente pressorio ventricolo-atrio destro è maggiore) si possono notare pulsazioni
delle varici degli arti inferiori. Il polso venoso ed epatico positivi sono evidenti nelle forme
acute (come lo è anche il reflusso epatogiugulare).
ECG: Si può notare ipertrofia ventricolare destra e atriale destra, quest’ultima a volte
mascherata dalla non infrequente fibrillazione atriale.
Radiografia del torace: cardiomegalia da dilatazione ventricolare destra e segni di
distensione cavale superiore.
Ecocardiografia: evidenzia la dilatazione delle camere atriale e ventricolare, la dinamica del
ventricolo destro ed anche le caratteristiche morfologiche della tricuspide. Con l’eco-doppler
si può evidenziare il rigurgito e stimare la pressione sistolica nel ventricolo destro. Un esame
con eco contrasto con soluzione fisiologica o sostanze che formano micro cavità mostra un
movimento va e vieni delle micro cavità che è molto sensibile nell’insufficienza tricuspidale.
Cateterismo cardiaco: è da porre solo in pazienti con indicazioni chirurgiche in quanto gli
altri sistemi hanno una precisione sufficiente. Può misurare la pressione atriale destra e
cavale, oltre a registrare l’aumento anche della pressione tele diastolica ventricolare destra.
La curva di pressione atriale mostrerà onde c+v elevate, e si potrà valutare la presenza di
ipertensione polmonare con cateterismo in arteria polmonare. La ventricolografia permette
64
di valutare il grado (1-4) del rigurgito e la funzione ventricolare.
Diagnosi: spesso bisogna valutare la coesistenza di IT in corso di altre valvulopatie. Mentre
nelle forme acute ci si può confondere con un addome acuto, nelle forme croniche si rischia
di non comprendere se un’insufficienza tricuspidale sia primitiva o secondaria allo
scompenso stesso (influenza la terapia).
Prognosi: nelle forme croniche con disfunzione ventricolare destra il quadro procede verso
uno scompenso con rischio di complicazioni con ipertensione portale, ascite, cirrosi epatica,
malassorbimento intestinale (cachessia cardiaca). L’intervento sulla mitrale, nei casi
associati a valvulopatia mitralica,può risolvere l’IT o attenuarla, ma non tanto se c’è già
disfunzione ventricolare destra. Nelle forma acuta la trombo lisi per l’embolia o la
risoluzione dell’endocardite possono risolvere l’IT, magari con eventuale protesi valvolare.
Terapia: in sostanza l’IT senza ipertensione polmonare è ben tollerata, e, se l’ipertensione
manca, anche un eventuale rimozione senza sostituzione di tricuspide infetta per
endocardite può non dare problemi. Se associata a valvulopatia mitralica, come detto, può
bastare risolvere il problema mitralico. In generale le forme di IT lievi non vanno trattate.
L’intervento può essere considerato in caso di IT rilevante si può praticare anuloplastica con
inserzione dell’anello di Carpentier o secondo De Vega senza sostituzione dell’anello. È
possibile anche la sostituzione protesica (in genere biologica). La mortalità intraoperatoria,
più elevata nei casi di scompenso, è del 7% nelle tecniche conservative e del 15% nelle
sostituzioni valvolari.
Valvulopatie miste:
Steno-insufficienza mitralica: in genere da malattia reumatica. I sintomi dipendono dalla
prevalenza di un sintomo o dell’altro. In genere si associa a valvola molto calcificata e
compromessa. Il decorso è in genere più rapido rispetto alle forme pure, con sopravvivenza
più bassa.
Steno-insufficienza aortica: doppio vizio aortico se coesiste reflusso che aumente almeno
del 30% il volume ventricolare e gradiente pressorio trans aortico superiore a 30mmHg. Non
è infrequente. Si ha sempre ipertrofia (adeguata) ventricolare con ridotta compliance. In
genere la causa è una malattia reumatica.
Valvulopatie polmonari: a parte cause congenite (ad esempio stenosi polmonare congenita
o insufficienza polmonare dei pazienti sottoposti a intervento di ricostruzione del tratto di
efflusso polmonare nella tetralogia di Fallot) sono piuttosto rare. Una causa può essere la
sindrome da carcinoide, mentre la cardiopatia reumatica colpisce la polmonare con minore
frequenza rispetto a tutte le altre valvole. Causa di insufficienza polmonare può essere
notevole ipertensione polmonare a causa della dilatazione dell’anulus. In questo caso si
avverte soffio diastolico di Graham-Steel sul focolaio polmonare.
Sostituzione valvolare
Definizione: intervento che prevede l’eliminazione della valvola lesa o mal funzionante e
impianto di una protesi. I risultati della sostituzione dipendono da: 1) funzione miocardica e
condizioni cliniche del paziente. 2) capacità tecniche dell’equipe. 3) caratteristiche di durata
e trombogenicità della protesi. La mortalità aumenta con l’età e con le patologie
concomitanti. Complicanze tardive sono distacco della protesi, trombo embolie, emorragie
65
da terapia anticoagulante, deterioramento della protesi, endocardite infettiva. Oggi esistono
molti tipi di protesi valvolari, sia meccaniche che biologiche.
Indicazioni: La scelta tra protesi meccanica e biologica è condizionata da diversi aspetti. Le
protesi meccaniche sono a rischio di complicanze tromboemboliche. Il paziente è costretto
a fare un trattamento anticoagulante a vita, mentre il rischio trombo embolico delle protesi
biologiche è quasi nullo dopo tre mesi. Le protesi meccaniche possono però avere una
durata anche illimitata, a differenza di quelle biologiche che sono molto più sensibili al
deterioramento meccanico e per questo i pazienti necessitano di intervento risostitutivo nel
30% dei casi a 10 anni e nel 50% a 15 anni. Le protesi mitraliche si deteriorano ( a causa delle
maggiori pressioni a cui sono sottoposte) prima di quelle aortiche. In teoria in pazienti
giovani (<65 anni), data la migliore durata, si preferisce l’uso di protesi meccaniche a meno
che non ci siano controindicazioni alla terapia anticoagulante o il paziente si rifiuti di
assumerla. La protesi biologica è indicata in pazienti con età >65 anni che possono essere
coperti dalla protesi biologica per il resto della loro vita, in pazienti con controindicazioni
all’anticoagulazione o che hanno sviluppato endocardite su protesi meccanica
precedentemente installata e in donne desiderose di gravidanza.
Protesi meccaniche: ve ne sono di vari tipi, possono essere installate da sole o all’interno di
tubi valvolati (ad es. per sostituzione della radice aortica). Sono costituite da materiale
sintetico come dacron, teflon e titanio. La prima valvola utilizzata è stata la protesi di StarrEdwards di cospicue dimensioni con una pallina di silastic (rivestita di bario per renderla
radiopaca) che si muove all’interno di una gabbia di stellite (cromo+nichel) con una anello di
sutura rivestito in dacron. Il flusso era un po’ turbolento e il paziente poteva accusare fastidi.
È stata poi introdotta la protesi di Bjork-Shiley con un disco oscillante trattenuto da due
supporti che gli permettevano di ruotare di 60°. Ultimamente la scelta cade principalmente
sulla protesi di St.Jude (bi-leaflet) formata da due emidischi che aprendosi al centro
permettono un flusso centrale con un basso gradiente trans valvolare (indicata anche con
anulus di piccole dimensioni).
Protesi biologiche: sono costituite di materiale di origine valvolare (protesi di Hancock) o
non valvolare (come il pericardio). Questo può provenire dallo stesso paziente (autograft),
ma anche da cadavere (homograft) o da animale (xenograft, valvole aortiche porcine o
pericardio bovino criopreservato). L’homograft vengono prelevate da cadavere poi
sterilizzate a bassa concentrazione antibiotica (minore potere citotossico) e criopreservate.
Si possono anche asportare da cuori battenti da cuori non trapiantabili (homovital, con
vitalità cellulare preservata) e in tal caso non vengono né trattate né preservate, mantenute
a 4° con tempo tra espianto e impianto minore di 48 ore. L’homograft è molto utilizzato per
patologie aortiche soprattutto in caso di controindicazioni a terapia anticoagulante e
nell’endocardite batterica, con ottimi risultati a distanza. Normalmente comunque il tessuto
biologico è installato su supporto meccanico (stent) e le protesi vengono sterilizzate e
conservate in gliceraldeide. La trombogenicità è bassa anche perché il supporto meccanico è
ricoperto da strato pseudo endoteliale, il flusso è meno turbolento e i fastidi per il paziente
sono minori, tranne un maggiore deterioramento rispetto alle meccaniche. Le valvole
stentless sono delle valvole biologiche non montate su di uno stent meccanico. Queste
valvole hanno pertanto una maggiore area valvolare (non limitata dallo stent) e si aprono
con un minor gradiente. Sono più difficili da impiantare perché un non corretto allineamento
delle commessure o calibro inadeguato possono causare insufficienze.
66
Ipertensione arteriosa e rischio cardiovascolare
Ipertensione arteriosa
L’ipertensione non è propriamente una patologia, quanto piuttosto una condizione che
rappresenta un importante problema medico, soprattutto negli ultimi anni e soprattutto nei
paesi occidentali. La diagnosi è semplice in quanto esistono metodi rapidi per la misurazione
della pressione (sfigmomanometro, che può essere oltre che manuale anche elettronico,
comodo per il paziente). Di per sé non comporta una precisa sintomatologia, ma talvolta ha
complicanze (tipicamente sistemiche) che possono divenire anche mortali (danni vascolari,
renali, cardiaci, cerebrali, etc.).
Si definisce iperteso per l’OMS un adulto con pressione arteriosa superiore a 140/90 mmHg,
fermo restando che i valori ideali si pongono anche più in basso (120/70). Questo limite è del
tutto arbitrario e soprattutto non tiene conto di particolari condizioni e dell’età dell’individuo.
Infatti non ha un significato patologico il riscontro di una pressione più elevata della norma in
pazienti con età superiore ai 70 anni, in quanto la pressione tende ad aumentare con l’età.
Inoltre i bambini hanno normalmente una pressione più elevata, così come le donne in
gravidanza e condizioni come la febbre possono influenzarla (aumenta di 10 mmHg per ogni
grado sopra i 37) Fino ai 50 anni L’ipertensione ha una predilezione per il sesso maschile, ma
dopo i 50 anni aumenta il numero di donne ipertese (forse a causa degli squilibri ormonali
dovuti alla menopausa). Quando si parla di ipertensione arteriosa si intende comunque
principalmente l’ipertensione sistemica, risultando più rara l’ipertensione polmonare e
scatenata generalmente da condizioni diverse.
Si distinguono vari stadi: ipertensione lieve: Sistolica: 140-159, Diastolica: 90-99; ipertensione
moderata: S: 160-179, D: 100-109; ipertensione severa: S: 180-209, D: 110-119; ipertensione
grave: S: >210, D: >120.
L’ipertensione costituisce da sola un’importante fattore di rischio per le malattie cardiovascolari
(rischio che aumenta ad esempio di circa 2 volte per ogni aumento di 20mmHg della sistolica e
10 della diastolica). Il rischio dovrebbe essere basso nei pazienti con lieve ipertensione (circa
l’80% degli ipertesi), ma spesso questa si associa ad altri fattori di rischio quali fumo,
ipercolesterolemia, diabete, familiarità, cattive abitudini alimentari (abuso di sodio, di grassi, di
alcol, di caffè) cosicché il rischio aumenta in maniera esponenziale e pertanto il soggetto deve
essere curata adeguatamente.
Regolazione della pressione arteriosa: in senso generale: pressione arteriosa = gittata cardiaca x
resistenze periferiche. La gittata cardiaca, GC è a sua volta GC = gittata sistolica x frequenza
cardiaca e dipende quindi dal volume ematico, dalla quantità di sodio da contrattilità del
miocardio e frazione di eiezione (insomma dalla funzionalità cardiaca). Le resistenze periferiche
sono finemente regolate da ormoni e fattori (molto vari, influenzati anche da condizioni
particolari come ad esempio l’infiammazione) che possono portare vasocostrizione o
vasodilatazione. Nella regolazione della pressione a breve termine hanno un ruolo particolare i
chemocettori ed i barocettori che intervengono con aggiustamenti rapidi e principalmente
tramite il SNA fanno variare: attività e frequenza cardiaca, frequenza respiratoria, tono arterioso
e venoso. Per il controllo a lungo termine è determinante il rene che è deputato all’escrezione di
sodio (variazione volemia) , ma anche alla produzione della renina che stimola l’angiotensina
(vasocostrittore) che a sua volta stimola l’aldosterone (stimola il riassorbimento si sodio e acque
e l’escrezione di potassio). Il rene produce anche sostanze vasodilatatrici come NO, chinine e
67
prostaglandine. Altri fattori determinanti per la regolazione della pressione sono endoteline,
leucotrieni, istamina, etc.
Patogenesi: Nel 90-95% dei casi l’ipertensione non riconosce una singola causa ben definita e si
dice essenziale o primitiva o idiopatica. In tal caso più che una patologia possiamo considerarla
l’estremo della curva di distribuzione gaussiana della pressione nella popolazione.
È una patologia su base poligenica o meglio ancora multifattoriale dipendente da molteplici
fattori genetici e ambientali. Nel restante 5-10% dei casi è secondaria ad una patologia ben
definita.
Fattori genetici: presenta sicuramente una familiarità, ma su base poligenica. Sono coinvolti
geni che aumentano la sintesi epatica di angiotensinogeno, l’attività della renina, alterato
metabolismo dell’aldosterone. Inoltre alcune patologie che comportano ipertensione sono su
base familiare (ad esempio diabete, iperlipidemie, etc.).
Fattori ambientali: le abitudini alimentari influenzano notevolmente l’insorgenza di
ipertensione. Un eccessivo consumo di sodio (circa 10g/die) è comune nei paesi occidentali ed è
certamente correlata ad un incremento pressorio. Il sodio infatti è prima di tutto
osmoticamente attivo e pertanto trattiene liquidi aumentando la volemia, inoltre un aumento
del sodio extracellulare causa una maggiore attività della pompa Na +/Ca2+ con aumento del
calcio intracellulare (risposta vascolare aumentata agli stimoli vasocostrittori). Si pensa cha
anche la stessa quota di calcio fornito con la dieta sia implicata nella patogenesi. L’obesità è
sicuramente un fattore di rischio, con livelli di pressione dei pazienti obesi molto superiori ai
normopeso. Pare che il motivo sia dovuto all’attività ormonale del grasso (principalmente il
grasso viscerale) con aumento del riassorbimento di sodio e iperattivazione adrenergica. Si ha
infatti iperinsulinemia (il grasso produce fattori come IL-6 che è anti- insulinica) che ha un
effetto diretto sull’ipertensione e facilita l’ipertrofia vascolare (oltre ad essere diabetogena, per
aumento dell’insulino-resistenza). Inoltre comporta un sovraccarico cardiaco. Anche il fumo,
l’alcol ed il caffè sono implicati. Renina: l’attività del sistema RAA è fondamentale.
Negli ipertesi i valori di renina possono essere diversi, e questo si pensa sia associato a cause
diverse di ipertensione: ipertensione a bassa renina: (comune nei neri) forse dovuta a un
mineralcorticoide che aumenta il sodio e sopprime la renina; ipertensione ad alta renina:
causata dalla renina stessa anche se i sartani (ARB, bloccano i recettori dell’angiotensina) non
sempre bastano (forse c’è iperattivazione adrenergica); ipertensione con renina normale: non
modulabile, forse per troppa sensibilità al sale da cucina (da ridurre).
Pertanto l’ipertensione può essere dovuta a: ritenzione renale di sodio in eccesso: forze per
mutazioni a carico del rene (sempre con fattori ambientali) possono diminuire l’escrezione di
sodio e aumentando il VEC portano ipovolemia e forse ipertrofia vascolare e vasocostrizione di
adattamento; vasocostrizione ed ipertrofia vascolare: si ha aumento delle resistenze periferiche
per aumentato rapporto parete/lume e quindi vasocostrizione, forse mediata da fattori come
l’insulina.
Ipertensione secondaria: riconosce una causa precisa e si risolve una volta eliminata.
Può essere legata a:
Cause renali: ogni condizione che comporta una ridotta escrezione di sodio (aumento volemia):
glomerulo nefrite acuta, emangiopericitomi (aumento cellule iuxtaglomerulari e quindi renina),
ipertensione nefrovascolare (obliterazione arterie e arteriole renali che comporta un aumento
della produzione di renina, curabile con ACE-inibitori), insufficienza renale acuta e cronica
(talvolta associate anche a iperparatiroidismo).
Cause endocrine: acromegalia e iperparatiroidismo, aumento dei mineralcorticoidi (Cushing,
68
adenoma o iperplasia surrenalica, altre patologie del surrene, in genere associata a ipokaliemia e
potassiuria), ma anche feocromocitoma, che comporta per lo più episodi di ipertensione
parossistica dovuti all’iperproduzione di adrenalina e noradrenalina.
Altre cause: contraccettivi orali: se ad alto contenuto di estrogeni (che forse stimolano il sistema
RAA, ma ormai il contenuto di estrogeni è più basso), coartazione aortica, traumi, interventi
chirurgici, ipertensione psicogena, da trasfusioni, policitemia e altre.
Conseguenze: i principali danni sono a livello vascolare. L’ipertensione è un fattore di rischio
cardine per l’aterosclerosi che a sua volta può causare danni a molti organi.
Cuore: aumento del rischio di aterosclerosi delle arterie coronariche e dunque di cardiopatie
ischemiche (facilitate pure dal maggiore consumo di ossigeno da parte del miocardio sotto
sforzo). Il cuore cerca di compensare l’ipertensione con l’ipertrofia concentrica cioè aumento
dello spessore di parete (per rispondere all’aumento di pressione in base alla legge di Laplace),
pertanto c’è anche un maggiore rischio di scompenso.
SNC: aterosclerosi dei vasi cerebrali facilita l’ictus. Sono più comuni anche emorragie
conseguenti ad aneurismi facilitati dagli elevati livelli pressori (aneurismi di Charcot-Bouchard).
Encefalopatia ipertensiva: aumento pressione intracranica, confusione, convulsioni. È possibile
anche la retinopatia ipertensiva, con edema, scotomi, cataratte, glaucomi e alla lunga persino
cecità.
Rene: perdita progressiva dei nefroni per obliterazione alteriole, si ha microematuria e
albuminuria con progressiva insufficienza renale.
Valutazione del paziente iperteso: è una valutazione complessa che si compone di:
Anamnesi: per prima cosa bisogna escludere la possibilità di un’ipertensione secondaria (e
pertanto curabile risolvendo la patologia di base). Questa è più comune se il paziente è giovane
e senza fattori di rischio né familiarità. Un episodio di infezione urinaria o ematuria può far
pensare ad un problema renale, habitus tipico può far pensare al Cushing, parossismi al
feocromocitoma, etc. Successivamente bisogna indagare l’abuso di alcol, caffè, fumo, sale,
liquirizia, oppure l’uso di contraccettivi orali o cortisonici. Bisogna rilevare familiarità ed
eventuali fattori di rischio quali: obesità, diabete, dislipidemie, etc. Bisogna chiedere di sintomi
di cardiopatia ischemica o eventuali manifestazioni cardiache o cerebrali. Altri sintomi (in genere
aspecifici e comunque lievi) sono cefalea, vertigini, ronzii alle orecchie, disturbi visivi.
All’esame obiettivo bisogna cercare segni di eventuali patologie di base e valutare la presenza di
soffi (addominali, carotidei).
Diagnosi: le indagini diagnostiche devono essere ridotte (è inutile e dispendioso abusarne per
una patologia così comune), può essere utile un ECG (rileva un’ipertrofia, e lo fa ancora meglio
l’ecocardio che però è più costoso), screening primari (emocromo, creatininemia, kaliemia, etc.)
e opzionali (glicemia, lipidi, uremia e calcemia). Per valutare il danno cerebrale è utile la TAC,
l’esame del fondo oculare controlla lo stato di retinopatia (indicato in tutti gli ipertesi,
specialmente i diabetici).
Terapia: esistono misure generali e terapia farmacologica. Le misure generali che riguardano le
abitudini, la dieta e lo stile di vita devono essere applicate a tutti, i pazienti con ipertensione di
grado lieve senza fattori di rischio associati possono anche fare a meno della terapia
farmacologica.
69
Misure generali: 1) controllo degli stress fisici (evitare grande attività anaerobica) e psichici. 2)
Dieta: ridurre il consumo di sodio al massimo fino 5g/die, ridurre alcol e caffè oltre che di
colesterolo e grassi saturi. 3) Decremento di peso nei pazienti obesi con dieta ipocalorica e
attività fisica (preferibilmente anaerobica), anche se non bisogna puntare subito al pesoforma
(un decremento del 10-15% già riduce di molto la mortalità). 4) Smettere di fumare 5) Curare
fattori di rischio come ipercolesterolemia e diabete 6) Eliminare i contraccettivi estro
progestinici 7) Educare ad un controllo assiduo della pressione.
Terapia farmacologica: farmaci anti ipertensivi sono indicati per tutti i pazienti con diastolica
superiore a 90 e >65 con sistolica >160. La valutazione deve essere personale, vi sono più classi
di farmaci:
Diuretici: molto diffusi per il basso costo, assunzione per os e buona tollerabilità.
Si usano i tiazidici (idroclorotiazide) che favoriscono l’eliminazione di sodio agendo sul tubulo
contorto distale, inefficaci però nell’insufficienza renale. Possono causare ipokaliemia (rischio
aritmico), iperglicemia e ipercolesterolemia (da evitare in diabetici e gottosi).
I diuretici d’ansa (tipo furosemide) sono più potenti e più utili nelle emergenze ipertensive
(somministrabili pure per ev).
I diuretici risparmiatori di potassio (tipo spironolattone, bloccano l’aldosterone), spesso usati
insieme ai tiazidici per evitare l’ipokaliemia oppure da soli nell’iperaldosteronismo.
Controindicati nell’insufficienza renale.
Beta-bloccanti: (tipo propanololo, metoprololo) riducono gittata cardiaca (azione cronotopo e
inotropo negativa) e produzione di renina. Controindicati nello scompenso, blocchi AV, diabete,
asma etc.
Alpha-bloccanti: tipo fentolamina usata nel frocromocitoma, prazosina e doxazosina da usare
con cautela perché possono causare ipotensione (utili nell’iperlipidemia perché abbassano le
LDL e alzano le HDL).
Altri simpaticolitici: tipo clonidina, usati meno per vari effetti collaterali.
Calcio-antagonisti: utili nell’ipertensione lieve o moderata o grave ma in associazione ad altri
farmaci. Si usano diidropiridine (nifedipina), benzodiazepine (diltiazem), fenilalchilamine
(verapamil). Non sono da dare a pazienti con scompenso cardiaco o blocchi AV e da non
associare a beta-bloccanti.
Vasodilatatori: idralazina e minoxidil sono da associare ad un beta-bloccante e un diuretico in
quanto possono dare tachicardia (no idralazina nel LES). Diazossido e nitroprussiato nei casi di
ipertensione grave.
ACE-inibitori: per os, tipo captopril, enalapril, molto utilizzati. Da evitare nell’insufficienza
renale, stenosi arteria renale e gravidanza (teratogeni). Possono dare iperkaliemia, usati anche
per l’ipertensione grave, sono molto ben tollerati.
Sartani: ARB, agiscono sui recettori dell’angiotensina (tipo valsartan), non hanno gli efftti
collaterali degli ACE-inibitori (tipo tosse e angioedema) che derivano dall’accumulo di chinine
(bradichinina). Sono ben tollerati, ma non da dare in gravidanza. Si può fare uso di politerapie ad
esempio: beta-bloccanti + ACE-inibitore + diuretico; calcio antagonista + ACE-inibitore + sartano.
Ipertensione maligna: marcata ipertensione con sintomi di encefalopatia ipertensiva, vomito,
cefalea e disturbi visivi, insufficienza cardiaca e progressiva insufficienza renale. Si ha dilatazione
delle arterie cerbrali e necrosi fibrinoide delle arteriole. Coinvolge l’1% delle ipertensioni, è più
maschile (età media 40 anni), la prognosi ora è migliorata (5 anni). Trattata con urgenza con
nitro prussiato e diazossido per ev.
70
Rischio cardiovascolare
Rischio cardiovascolare: le malattie cardiovascolari sono la prima causa di morte al mondo (circa
il 30%) dei decessi e la prospettiva, considerando anche il progressivo invecchiamento della
popolazione, è di un incremento.
Fattori comportamentali di rischio: tabacco, dieta (particolarmente pericoloso e aterogeno il
consumo di grassi saturi), inattività fisica (l’attività raccomandata è di almeno 30 minuti
continuativi almeno cinque giorni a settimana)
Fattori metabolici di rischio: dislipidema, ipertensione (causa quasi il 50% e circa il 60% di
cardiopatie ischemiche e ictus), obesità, diabete mellito (colpisce il 5% della popolazione
mondiale).
Transizione epidemiologica: trasformazione delle cause di morbilità e mortalità avvenuta nel
corso del ventesimo secolo a causa dell’industrializzazione, dell’urbanizzazione e dell’associato
cambiamento dello stile di vita. Si distinguono quattro fasi, forse cinque (in alcuni paesi
attualmente):
Prima fase, l’era della pestilenza e della carestia; Seconda fase, l’era di recessione delle
pandemie; Terza fase, l’era delle malattie degenerative e causate dall’uomo; Quarta fase, l’era
delle malattie degenerative tardive ; Quinta fase, l’era dell’inattività e dell’obesità. Dal punto
di vista geografico invece, in Europa si nota un effetto protettivo della cosiddetta dieta
mediterranea, ricca in carboidrati e grassi insaturi.
Paradosso francese: i francesi hanno un’incidenza relativamente bassa di malattie
cardiovascolari perché, pur fumando molto e consumando grandi quantità di formaggi, bevono
molto vino rosso che contiene rosveratrolo antiossidante.
Lo studio intraheart è uno studio caso-controllo che ha individuato 9 fattori di rischio (concetto
epidemiologico e statistico, ma non fisiopatologico) ordinandoli per OR:
1) Rapporto Apo B/Apo A-1: indica dislipidemia e sostituisce il dosaggio LDL (tramite formula di
Freidwald: Col tot - HDL - trigliceridi/5= LDL). 2) Fumo 3) DM tipo 2 4) Ipertensione 5) Fattori
psicosociali (depressione e ansia) 6) Obesità 7) Frutta e verdura giornaliere 8) Esercizio fisico 9)
Consumo moderato di alcol. 1) è circa OR=5/1 6) OR=3/1 7), 8) e 9) sono fattori protettivi con
OR<1. Questi sono fattori di rischio modificabili. Da soli spiegano quasi il 90% degli eventi
cardiovascolari, ma conta anche il corredo genetico.
L’ipertensione costituisce un caso a sé stante. Mentre per il rischio di ictus è sicuramente un
fattore di rischio assoluto, per il rischio di infarto può non essere utile (ed anzi dannoso) un
eccessivo abbassamento della pressione. Questo è valido soprattutto per i pazienti anziani.
Certo non è valido il discorso che la PA debba essere =100+età, ma un’ottantenne non può avere
la sistolica di 110. Questo perché il rischio cardiovascolare associato ad ipertensione dà una
curva con aspetto a J (o curva U), ossia se c’è eccessiva riduzione di pressione il rischio aumenta
perché può diminuire la perfusione.
Sinergismo dei fattori di rischio: si calcola il rischio cardiovascolare globale facendo una
sommatoria pesata dei vari fattori di rischio. Si incasella quindi il paziente in una categoria di
rischio che può essere: Basso (<10% a 10 anni, <1% all’anno); Intermedio (<1-2% all’anno); Alto
(>2% all’anno). Questo si applica per pazienti tra 40 e 70 anni. Sono automaticamente
considerati ad alto rischio pazienti che hanno già avuto un evento ischemico, pazienti con
aneurisma aorta addominale, DM, vascolopatia, patologia carotidea. Infatti le categorie di
rischio servono soprattutto per distinguere gli asintomatici (per esempio per decidere se fare o
meno una coronarografia per es. paziente ad alto rischio, sintomatico per angina). Oggi la prima
causa di morte per patologia cv. è la morte improvvisa, 70% per cardiopatia ischemica (quasi nel
100% associata ad aritmie).
71
Cardiopatia ischemica
Circolo coronarico: formato dalle arterie coronariche di destra e di sinistra, riceve il 5% della
gittata cardiaca. Le arterie coronariche decorrono coperte dall’epicardio sulla superficie esterna
del cuore all’interno dei solchi coronario(atrioventricolare) e interventricolare, a volte
circondate da accumuli di grasso e coperte a ponte da esili fascetti di miocardio. Solo i piccoli
rami penetrano nel miocardio. La circolazione coronarica avviene prevalentemente durante la
diastole perché durante la sistole i vasi risultano compressi.
Le due arterie coronarie originano dall’inizio dell’aorta ascendente a livello dei seni aortici di
Valsalva e hanno un calibro simile (spesso, 80%, leggermente maggiore la sinistra, raramente
può sorgere una terza coronaria falsa). Tra di loro ci sono delle anastomosi, ma piccole e non
sufficienti a stabilire un circolo collaterale.
Coronaria destra: origina dal seno aortico (o coronario) destro e percorre il solco coronario fino
alla crux cordis (punto in cui il solco atrioventricolare si incrocia con il solco interventricolare
posteriore). Ora, nell’80-90% dei casi la coronaria destra prosegue oltre la crux cordis e percorre
il setto interventricolare posteriore con il nome di interventricolare posteriore. In questo caso si
parla di dominanza destra. Nei casi di dominanza sinistra la coronaria destra termina prima di
giungere alla crux cordis. Da numerosi collaterali, nell’ordine: arteria infundibolare: si
distribuisce al tratto di efflusso (cono) del ventricolo destro; rami atriali: tra cui l’arteria del
nodo seno-atriale (55% dei casi); rami ventricolari: tra cui l’arteria del margine acuto: che
vascolarizza la parete laterale del ventricolo destro fino quasi alla punta del cuore; ramo
discendente posteriore destro: che sarebbe l’arteria interventricolare posteriore (IVP), che
origina dalla divisione ad angolo retto della coronaria destra una volta giunta alla crux cordis.
Dall’IVP originano rami per le pareti posteriori dei ventricoli destro e sinistro e rami perforanti
per il setto interventricolare, mentre a livello della sua origine nella crux cordis può continuare
un piccolo ramo arterioso all’interno del solco atrioventricolare che ha leggere anastomosi con
l’arteria circonflessa della coronaria sinistra. A livello della crux si stacca anche l’arteria del nodo
atrioventricolare. La coronaria destra inoltre irrora il fascio di His e parte del ramo posteriore
della branca sinistra.
Coronaria sinistra: il tratto di origine detto tronco comune è lungo solo un cm e decorre coperto
dal tronco polmonare fino al solco coronario dove si divide in due rami (nel 30% anche un terzo,
l’arteria intermedia) di calibro simile: l’arteria interventricolare anteriore (IVA) e l’arteria
circonflessa. Il ramo discendente anteriore sinistro (o IVA appunto), decorre nel solco
interventricolare anteriore e fornisce rami per la parete anteriore del ventricolo destro, rami
perforanti per il setto interventricolare che si anastomizzano con quelli della coronaria destra e
arterie diagonali (come la prima arteria diagonale, grande) per la parete anteriore del
ventricolo sinistro. L’arteria circonflessa decorre invece nel solco atrioventricolare fino alla crux
cordis e fornisce: rami atriali: tra cui nel 45% il ramo per il nodo seno-atriale e un ramo per
l’atrio sinistro che può unirsi ad un ramo atriale della coronaria destra (circolo arterioso di
Kugel); rami ventricolari: importante è l’arteria del margine ottuso vascolarizza la parete
laterale sinistra sino all’apice del cuore. Circa nel 10% dei casi vi può essere dominanza sinistra e
pertanto è la coronaria sinistra a fornire il ramo interventricolare posteriore. Nell’1% dei casi vi è
una circolazione bilanciata con due arterie parallele fornite da entrambe le coronarie. La
coronaria sinistra, tramite il suo ramo interventricolare anteriore irrora parte del ramo
posteriore della branca sinistra, il ramo anteriore della branca sinistra e la branca destra.
72
Vene: Seno coronario (nell’omonimo solco), si apre nell’atrio destro vicino allo sbocco della cava
inferiore, tramite la valvola di Tebesio. Tra le tante raccoglie la vena cardiaca magna (anche la
media nel solco interventricolare posteriore, e la parva nel solco atrioventricolare) che segue
l’IVA e la vena del margine ottuso oltre a vene per atrio e ventricolo sinistro (tra cui la vena
obliqua dell’atrio sinistro e la vena posteriore del ventricolo sinistro). Oltre al seno coronario vi
sono le vene cardiache anteriori per la parte anteriore del ventricolo destro (come la piccola
coronaria, spesso tributaria della parva) e le vene minime di Tebesio (che sboccano
direttamente nelle camere cardiache, principalmente nell’atrio destro).
Fisiopatologia coronarica: i vasi coronarici possono essere suddivisi in vasi di conduttanza ossia
le grosse arterie epicardiche ed i loro rami e in vasi di resistenza ossia le per arteriole e i piccoli
rami intramiocardici che sono il principale determinante delle resistenze coronariche.
La circolazione coronarica è influenzata e controllata dalle richieste di ossigeno da parte del
cuore. Queste richieste aumentano con la frequenza cardiaca, la contrattilità e lo stress di
parete. Il livello di estrazione di ossigeno del miocardio è del 70%, molto elevato. Pertanto se vi
è un’aumentata richiesta metabolica, l’unico meccanismo che permette un adeguato apporto di
ossigeno è la vasodilatazione del vasi di resistenza con conseguente diminuzione della resistenza
al flusso.
La riserva coronarica è la massima capacità di vasodilatazione e conseguente flusso in risposta
ad uno stimolo. Il fabbisogno miocardico di ossigeno regola pertanto la vasodilatazione dei vasi
intramiocardici (che normalmente hanno grande capacità di dilatarsi) che costituiscono pertanto
un particolare microcircolo dotato di capacità di regolazione metabolica e di autoregolazione
dipendente più dal fabbisogno cardiaco che dal controllo sistemico.
L’aumento della richiesta miocardica pare si esplichi in un aumento dell’idrolisi di ATP con
conseguente rilascio nell’interstizio di adenosina, la quale vaso dilata i rami intramiocardici con
incremento del flusso in proporzione alla richiesta. Il flusso coronarico è generalmente fasico, in
quanto è possibile solo durante la diastole (nelle tachiaritmie pertanto può ridursi a causa della
diminuzione del tempo di diastole) perché in sistole i vasi intramiocardici sono occlusi dalla
contrazione. Gli strati sub endocardici sono quelli maggiormente esposti all’ischemia.
L’ischemia è una riduzione del flusso che si associa naturalmente a danno cellulare (di vario
grado). La principale causa di ischemia è l’aterosclerosi coronarica in quanto la placca
ateromatosa ostruisce il flusso con diminuzione del calibro delle arterie. All’inizio anche una
notevole ostruzione non causa sintomi proprio perché le arterie a vale compensano questa
situazione con una vasodilatazione, ma quando l’ostruzione raggiunge l’80% del vaso si ha una
riduzione del flusso che alla lunga può essere tale da far comparire ischemia. L’ischemia
miocardica può verificarsi anche, molto più raramente, da spasmo, da trombi o emboli, da
anomalie congenite come l’origine del ramo discendente anteriore della coronaria sinistra
dall’arteria polmonare (ischemia e infarto in età infantile), grave ipotrofia ventricolare da stenosi
aortica (ma anche ipetrofia ventricolare con associata maggiore richiesta), e inoltre la soglia
ischemica può essere ad esempio ridotta da una riduzione della capacità di trasporto di
ossigeno.
Aterosclerosi coronarica: l’aterosclerosi è la prima causa di ischemia e ha sede principalmente
nelle arterie coronarie epicardiche (il circolo coronarico è in generale una delle sedi preferite
dall’aterosclerosi). I fattori come l’aumento delle LDL e la diminuzione delle HDL, il fumo,
l’ipertensione e il diabete provocano (insieme a fattori di minore importanza, all’omicisteinemia,
all’età, all’obesità, al sesso) la cosiddetta disfunzione endoteliale (che non è per forza una
73
lesione) che favorisce l’inizio della formazione delle placche ateroma tose.
Le placche tendono a concentrarsi nei punti in cui il flusso passa da laminare a turbolento come
le diramazioni delle arterie epicardiche.
La prima lesione è la stria lipidica che comincia ad osservarsi già intorno ai vent’anni in molti
soggetti. È un’area giallastra con monociti ed esteri del colesterolo. Questa può evolvere in una
vera e propria placca ateromatosa costituita da lipidi, macrofagi, cellule muscolari lisce ,
fibroblasti e piastrine,la quale è frutto di un processo infiammatorio cronico mediato, oltre che
da vari mediatori di infiammazione, da LDL ossidate e macrofagi (cellule schiumose).
La placca tende ad ingrandirsi con ulteriore deposito di materiale grasso e fibroso e può
condurre ad una progressiva stenosi del vaso.
Quando la stenosi riduce il diametro dell’arteria epicardica del 50% non basta un pieno
incremento del flusso per rispondere agli aumenti delle esigenze metaboliche (ad esempio sotto
sforzo), se l’ostruzione è del 75-80% il flusso può essere ridotto anche a riposo.
All’inizio i vasi a valle provano a dilatarsi per compensare questo flusso ridotto, ma con il tempo
si giunge ad ischemia miocardica soprattutto quando un aumento delle richieste del miocardio
non riesce ad essere compensato da una vasodilatazione già a riposo molto elevata. Questo
spiega perché spesso risultano asintomatici anche pazienti con alto grado di stenosi delle
coronarie. La gravità e l’insorgenza dell’ischemia dipende anche dal punto in cui si verifica
l’ostruzione (più grave se prossimale, tipo nel tronco comune della coronaria di sinistra) e dalla
possibilità (in genere presente, soprattutto se l’ostruzione è graduale) di sviluppo di circoli
collaterali.
Le placche ateromatose possono andare incontro a varie lesioni come: calcificazione (arterie
rigide), rottura e ulcerazione (si liberano sostanze trombo geniche con possibile sviluppo di
trombi a valle).
Generalità sulla cardiopatia ischemica: la cardiopatia ischemica è una condizione di
insufficiente apporto di ossigeno al miocardio, spesso causato dall’ostruzione dei vasi a causa di
aterosclerosi. Quando si presenta ischemia si hanno alterazioni dell’attività biochimica, elettrica
e meccanica del miocardio. Il tessuto sub endocardico è più sensibile di quello subepicardico.
In genere l’ischemia da aterosclerosi delle coronarie è un processo focale che si accompagna a
blocco della normale contrazione e rilassamento delle fibre. Se coinvolge ampie porzioni di
ventricolo può causare insufficienza ventricolare .
Se l’ischemia è transitoria (da 0 a 20 minuti di occlusione totale, danno reversibile) si hanno
modificazioni reversibili della contrattilità miocardica e può associarsi ad angina pectoris,
mentre se è prolungata (oltre 20 minuti) provoca una necrosi tissutale con danno contrattile
permanente e può associarsi al quadro clinico di infarto acuto del miocardio.
In condizioni ischemiche cambia il metabolismo cardiaco con passaggio da un metabolismo di
tipo aerobico ad uno anaerobico con formazione ed immissione di acido lattico (un segno di
ischemia è la produzione di lattato).
L’ischemia si associa a modificazioni dell’ECG dovute ad anomalie della ripolarizzazione quali
inversione dell’onda T e slivellamento dell’ ST (sottoslivellamento=subendocardio,
sopraslivellamento=ischemia trans murale).
La cardiopatia ischemica può essere sintomatica o meno (e magari essere rilevabile solo con
coronarografia o ECG sotto sforzo) e persino l’IM pare restare non diagnosticato nel 25% dei
pazienti. La fase sintomatica è invece caratterizzata da dolore toracico dovuto ad angina pectoris
o infarto acuto del miocardio.
Le conseguenze dell’aterosclerosi coronaria e dell’ischemia possono essere:
1) Aritmie fatali: l’ischemia causa instabilità elettrica con eccitazione di focolai ectopici che
74
causano extrasistoli, tachicardie e fibrillazioni ventricolari con conseguente morte improvvisa.
2) Cardiomiopatia ischemica: condizione dovuta a ripetuti danni miocardici che si presenta con
cardiomegalia e scompenso cardiaco (ventricolo sinistro danneggiato) che non hanno
determinato alcun sintomo prima dell’insorgenza di insufficienza cardiaca.
3) Formazione di trombi: Nell’angina stabile l’endotelio che ricopre la placca è liscio, nell’angina
instabile la superficie tende a ulcerarsi con fenomeni di aggregazione piastrinica. A seguito di
rottura o ulcerazione delle placche (che sono instabili a causa di fattori come il trombossano
prodotto dalle piastrine) possono formare un trombo che può depositarsi a valle e causare
condizioni cliniche differenti: 1) Se non occlude il vaso coronarico si manifesta un incremento
della gravità del quadro anginoso. 2) Se la progressione è rapida ma comunque non occlude il
vaso si ha infarto sub endocardico (non Q) 3) Se l’occlusione è completa si ha infarto trans
murale (infarto Q).
Indagini diagnostiche per cardiopatia ischemica:
ECG: la patologia ischemica, modificando l’attività elettrica dei miociti, comporta anche della
alterazioni elettrocardiografiche (prevalentemente riguardanti il ventricolo sinistro, più
frequentemente sede di episodi ischemici) quali:
1) Onda T invertita simmetrica: è un segno più evidente nelle derivazioni toraciche. Indica
un’ischemia subepicardica (trans murale). Nella sindrome di Wellens c’è un’onda T invertita in
V2 e V3 (stenosi della coronaria anteriore discendente).
2) Sopraslivellamento del tratto ST: indica la presenza di una lesione ischemica (nell’infarto
indica un episodio acuto). È presente nell’angina variante (transitorio). Può essere di varia
entità. Una pericardite o un aneurisma ventricolare possono provocare innalzamenti del tratto
ST (nella sindrome di Brugada è soprasilvellato in V 1 e V3 e c’è BBD). Se non associato a onda Q
può indicare un infarto non Q.
3) Sottoslivellamento del tratto ST: segno di intossicazione da digitale, ma soprattutto di infarto
sub endocardico. L’ST appare sottoslivellato e appiattito. È un infarto non Q e non transmurale.
Può essere un’immagine indiretta di sopraslivellamento del tratto ST.
4) Onda Q: un’onda Q significativa (almeno 1mm di ampiezza o 1/3 dell’altezza del QRS) è indice
di necrosi e quindi di infarto. È un segno che in genere non scompare e permane per tutta la
vita.
ECG da sforzo: test provocativo in grado di indurre ischemia in soggetti con ostruzione delle
coronarie. La manifestazione ischemica appare in genere come un sottoslivellamento del tratto
ST.
Scintigrafia con tallio 201: il tallio 201 è un isotopo che si fissa al miocardio in una
concentrazione dipendente dal flusso coronarico regionale ed evidenzia quindi alterazioni della
perfusione coronarica in presenza di stenosi emodinamicamente significative (soprattutto di
IVA). Pertanto permette di vedere dove sono ostruzionie e se l’ischemia è reversibile (alterazioni
che scompaiono dopo circa quattro ore) o meno. È anche molto utile nella valutazione dei
pazienti dopo interventi di rivascolarizzazione miocardica quali angioplastica coronarica e bypass.
ECG secondo Holter: registrazione continua per 24-48 ore. Con la collaborazione del paziente
permette di evidenziare disturbi del ritmo e anomalie di ripolarizzazione ventricolare tipiche
delle fasi ischemiche e di metterle in relazione con la sintomatologia e la frequenza cardiaca del
paziente durante la giornata. Si possono così vedere anche eventuali attacchi ischemici
asintomatici ed aritmie verificatesi nel contesto o meno di tali attacchi.
Arteriografia coronarica: o coronarografia, è l’indagine più specifica per le patologie
coronariche. È un esame invasivo tramite un catetere inserito per via venosa (più femorale) che
75
giunge fino agli osti coronarici dove poi viene iniettato un mezzo di contrasto per l’esame
radiologico delle coronarie che vengono ben evidenziate. In genere si esegue anche una
ventricolo grafia per valutare contrattilità e valvole ventricolari. Trova indicazioni per valutare
una dubbia cardiopatia ischemica o per quantificare l’ostruzione delle coronarie in paziente con
malattia coronarica già avanzata. In genere si esegue quando la terapia medica non controlla
bene i sintomi o se l’angina è instabile. È un esame non privo di rischi quali l’infarto miocardico
(0,7%) e accidenti cerebrovascolari (0,07%).
Classificazione generale della cardiopatia ischemica: i pazienti con cardiopatia ischemica si
dividono in due grandi gruppi:
1) quelli con malattia coronarica cronica che in genere si presentano con angina pectoris
stabile;
2)quelli con sindromi coronariche acute (SCA) che possono essere pazienti con:
- infarto miocardico acuto (IMA) con sopraslivellamento del tratto ST all’ECG (STEMI)
- angina instabile o infarto miocardico acuto senza sopraslivellamento del tratto ST
(AI/NSTEMI).
Angina pectoris stabile
Definizione: sindrome clinica relativa ad un’ischemia miocardica transitoria. È una malattia
coronarica cronica.
Clinica: il tipico paziente è un uomo oltre 50 anni o una donna oltre 60 che lamenta disturbi al
torace in genere descritti come senso di pesantezza, pressione, oppressione, soffocamento e
solo più raramente come vero e proprio dolore (che comunque è un dolore toracico retro
sternale costrittivo). È buona abitudine farsi indicare dal paziente la sede del dolore in quanto
può avvenire che esso sia indicato con la mano aperta o il pugno chiuso sullo sterno (e non con
la punta del dito) per indicare un senso di schiacciamento retro sternale (segno di Levine). Il
dolore anginoso può avere un andamento crescente, in genere non supera i 20 minuti (l’angina
stabile anche molto meno) e cessa comunque in poco tempo con l’interruzione delle’attività
fisica. Il dolore può irradiarsi al collo, alla regione intrascapolare (se al trapezio è più
pericardite), alla mascella, ai denti, all’epigastrio e alle braccia (più sinistro). A volte il dolore può
non insorgere in sede retro sternale bensì solo nelle sedi secondarie.
Gli episodi sono tipicamente scatenati dallo sforzo fisico o dalle emozioni e cessano con il riposo,
ma è possibile anche che si verifichino durante il riposo specie in posizione supina (angina da
decubito o angina a riposo) e il paziente può persino essere svegliato di notte da dispnea e
dolore tipico.
Si distingue pertanto un’angina primaria con ischemia causata da una transitoria riduzione del
flusso e un’angina secondaria causata da un incremento delle richieste di ossigeno.
L’angina può anche essere silente, ossia asintomatica. La soglia di sforzo fisico per la comparsa
di angina può essere stabile (angina da sforzo stabile) o variare molto, anche in rapporto allo
stato emotivo del paziente.
L’impatto dell’angina da sforzo sulla capacità funzionale del paziente è descritto dalla
classificazione NYHA in quattro fasi (1=cardiopatico senza limitazioni nell’attività fisica 2=lieve
limitazioni 3=marcata limitazione 4=anche a riposo). Dolori taglienti e immediati o prolungati e
lievi sono angina solo molto raramente. Nausea e vomito sono meno comuni che nell’infarto.
Poiché l’aterosclerosi coronaria può essere associata da lesioni simili in altre arterie si deve
esaminare un’eventuale claudicatio intermittens (o ictus o altro) e fare un anamnesi di
76
familiarità per cardiopatia, oltre che valutare la presenza di fattori di rischio per aterosclerosi
coronarica. L’angina può anche essere associata ad anemia, tachicardie, anomalie delle
coronarie, ipertrofia ventricolare sinistra.
Diagnosi: Esame obiettivo: può essere presente un terzo o quarto tono se vi è compromissione
ventricolare causata da precedenti ischemie, ma l’angina può anche causare uno scompenso
ventricolare sinistro transitorio (nel corso di un attacco). Si possono avere soffi carotidei da
aterosclerosi e riduzione dei polsi arteriosi degli arti inferiori. La stenosi e l’insufficienza aortica,
l’ipertensione polmonare e la miocardiopatia ipertrofica possono causare angina anche senza
aterosclerosi coronarica e pertanto vanno escluse. Se la parete toracica è dolorabile alla
palpazione questo fa verosimilmente escludere l’angina.
Esami: oltre ad analisi di laboratorio per valutare fattori associati ad aterosclerosi (lipidi,
glucosio, rene, etc.) anche una radiografia al torace può indirizzare se presente cardiomegalia o
segni di scompenso. L’ECG può mostrare modificazione dell’ST, inversione dell’onda T e segni di
ipertrofia ventricolare che non sono specifici a meno che non siano concomitanti alla
sintomatologia.
Test da sforzo: con sforzo su tappeto rotante o cicloergometro (o dipiridamolo o adenosina,etc.
se il paziente non è in condizione) può permettere l’esecuzione di vari esami con maggiori
informazioni: ECG che indica sottoslivellamento del tratto ST o la comparsa di pericolose aritmie,
ecocardiografia che indica difetti di cinesi del ventricolo sinistro, scintigrafia con tallio 201 o
tecnezio 99, coronarografia.
Prognosi: dipende da vari fattori. 1) Maggiori sono il numero e la gravità dei fattori di rischio per
aterosclerosi coronarica (età, lipidi, etc.) peggiore è la prognosi. 2) Segni obiettivi di scompenso
cardiaco e segni di disfunzione ventricolare sinistra (riduzione della frazione di eiezione,
aumento della pressione telediastolica, discinesia evidente all’ecocardio o ventricolo grafia
sinistra ) sono prognostici negativi indipendentemente da grado e sede di ostruzione coronarica.
3) Vi è quasi una correlazione diretta tra la comparsa precoce di segni o sintomi nei test da
sforzo e la mortalità 4) La sede della lesione è importante: lesioni ostruttive del tronco comune
della coronaria sinistra o del discendente anteriore (IVA) sono più gravi perché finiscono per
riguardare porzioni più estese di miocardio.
Terapia: ciascun paziente deve essere valutato individualmente e controllato sia per quanto
riguarda la sintomatologia sia per quanto riguarda le complicanze gravi come l’IM e aritmie
fatali. La terapia si fonda su:
1) Misure generali e stile di vita: Il paziente deve prima di tutto essere rassicurato affinché
capisca che può condurre una vita lunga e attiva (magari portare qualche esempio di paziente).
Potrà condurre le normali attività lavorative ed extralavorative (tranne lavori manuali pesanti),
magari solo conducendo il tutto più lentamente ed evitando carichi di lavoro al mattino o dopo i
pasti. Un buon controllo dell’attività fisica migliora la resistenza all’esercizio pertanto è bene che
i pazienti capiscano che l’angina (l’ischemia in realtà) è dovuta ad una discrepanza tra la richiesta
d’ossigeno da parte del miocardio e l’effettiva perfusione. Ad alcuni pazienti bisogna far
comprendere che è necessario un controllo dello stress anche emotivo (ira e frustrazione).
2) Trattamento dei fattori di rischio: bisogna curare i problemi cardiaci associati come
cardiomiopatia ipertrofica o valvulopatie ed inoltre patologie quali obesità, ipertiroidismo ed
ipertensione che aumentano il lavoro cardiaco e la richiesta di ossigeno da parte del miocardio.
È necessario naturalmente curare anche il diabete ed eliminare il fumo di sigaretta. È
importante una dieta che limiti l’apporto di grassi saturi e di sale e bisogna sempre monitorare
77
un’eventuale dislipidemia che può richiedere un trattamento anche farmacologico con inibitori
della HMG-CoA reduttasi (statine).
3) Terapia farmacologica: ha lo scopo di ridurre gli eventi di angina e l’incremento improvviso di
frequenza cardiaca e pressione arteriosa che innalzano il lavoro cardiaco.
Nitroderivati: sono in uso da più di 125 anni, hanno azione benefica, anche immediata, nel
ridurre gli episodi di angina in quanto comportano vasodilatazione delle arterie coronariche
(aumentando il flusso miocardico) e di tutto il circolo sistemico. La loro azione si esplica sia sulle
arterie (riducendo il post-carico, la pressione arteriosa) che sulle vene (aumento della
capacitanza venosa e quindi riduzione del pre-carico). Vengono assorbiti più rapidamente e
completamente attraverso le mucose (pertanto, soprattutto nell’immediato, si predilige
l’assunzione sublinguale). Per un effetto a lungo termine sono possibile anche l’assunzione trans
dermica (unguento di nitroglicerina) o per os. Per minimizzare la tolleranza bisogna utilizzare la
dose minima efficace e interrompere la somministrazione per almeno 8 ore tutti i giorni. Gli
effetti collaterali sono tachicardia e aumento della contrattilità in risposta alla diminuita
pressione arteriosa, ma anche tolleranza al farmaco, cefalea pulsante e ipotensione ortostatica.
Il più utilizzato è la nitroglicerina, ma anche l’isosorbide dinitrato.
Beta-bloccanti: provocano una riduzione della richiesta di ossigeno, effetto inotropo e
cronotropo negativo (inoltre soprattutto durante l’esercizio fisico più che a riposo). Sono molto
utili in associazione con i nitrati, in quanto prevengono l’aumento della frequenza e della
contrattilità che provocano questi ultimi. Come effetti collaterali provocano bradicardia,
claudicatio intermittens, peggioramento dell’asma bronchiale e dell’ipoglicemia (ad esempio se
insieme ad antidiabetici). I più utilizzati sono atenololo e metoprololo oltre a propanololo e
nadololo.
Calcio antagonisti: vasodilatatori coronarici che determina riduzione della domanda di ossigeno,
di pressione arteriosa e di contrattilitàI più usati sono diltiazem e verapamil, ma anche
nifedipina e amlodipina. Il verapamil (soprattutto) e il diltiazem (negli scompensati) non sono da
associare ai beta-bloccanti per la loro azione inotropo-negativa che potrebbe peggiorare una
disfunzione ventricolare sinistra. L’angina di Prinzmetal risponde bene ai calcio-antagonisti
(soprattutto diidropiridinici). È importante in ogni caso individualizzare la terapia e il dosaggio
soprattutto se si scelgono associazioni di farmaci. Nell’approccio iniziale sono preferibili i betabloccanti perché rispetto ai calcio-antagonisti determinano un miglioramento dell’aspettativa di
vita. Se però i beta-bloccanti non funzionano, o sono controindicati (asma o BPCO) o hanno
comportato effetti collaterali, o il paziente è affetto da angina di Prinzmetal, si preferiscono i
calcio-antagonisti. Altri farmaci che possono essere utili sono gli ACE-inibitori (negli ipertesi
specie se diabetici), le statine, farmaci antipiastrinici come l’acido acetilsalicilico. Se il paziente è
affetto da scompenso cardiaco la somministrazione di ACE-inibitori, diuretici e digossina servono
per lo scompenso e deve essere fatto un tentativo di introduzione dei beta-bloccanti. Interventi
non farmacologici sono descritti in seguito.
Angina instabile/infarto miocardico senza sopraslivellamento del tratto ST: AI/NSTEMI.
L’angina instabile una volta detta angina preinfartuale o insufficienza coronarica acuta è una
forma di angina che ha alta probabilità di evolvere in infarto miocardico acuto.
La diagnosi e anche la definizione di AI è basata sulla presentazione clinica, è infatti un’angina
pectoris che ha una di queste caratteristiche: 1) compare a riposo (o con minimo sforzo) e di
solito dura più di 10 minuti (l’angina stabile dura meno ed è più associata allo sforzo), 2) è
intensa e di recente insorgenza (pazienti con angina insorta nelle ultime 3-4 settimane) 3) si
verifica seguendo un pattern in crescendo (nel tempo la sintomatologia anginosa è divenuta più
78
grave, prolungata o frequente).
La diagnosi di NSTEMI viene posta se alla clinica di AI si aggiungono segni di necrosi miocardica
evidenziata dall’aumento degli enzimi di danno miocardico.
Patogenesi: è ovviamente causata da un disequilibrio tra apporto e richiesta di ossigeno che
causa ischemia miocardica. Il processo alla base di quest’ischemia può essere: rottura o erosione
di placca con trombi non occlusivi, che sono la causa più comune, ostruzione dinamica (tipo
spasmo coronarico angina di Prinzmetal), ostruzione meccanica progressiva ma comunque non
completa (cosa che spesso causa infarto sub endocardico, non Q) e AI secondaria a maggiore
richiesta o altre cause di minore apporto (anemia o tachicardia che causa riduzione dei tempi di
diastole). A volte queste cause si presentano insieme. Il 40% dei pazienti ha patologia
monovasale (soprattutto IVA), il 5% ha ostruzione del tronco comune, il 10% nessuna ostruzione
critica (spesso questi hanno Prinzmetal). La lesione responsabile visibile all’angiografia è più
spesso un trombo bianco (piastrine) rispetto all’IM. Spesso ci sono placche multiple e può essere
utile valutarne il numero, la percentuale e la localizzazione delle ostruzioni.
Clinica: sintomi e segni sono come quelli dell’angina stabile, in questo caso però il dolore
toracico è abbastanza intenso da essere riconosciuto come doloroso. Se il paziente ha un’ampia
area necrotica di NSTEMI si possono avere segni e sintomi tipici dell’infarto STEMI. ECG: si ha in
genere un sottoslivellamento del tratto ST o anche un’onda T invertita. (le onde T sono meno
specifiche se non sono abbastanza profonde cioè >0,3mV). Nuove alterazioni dell’ECG in pazienti
con sintomi di AI sono segni prognostici negativi.
Enzimi di danno miocardico: livelli elevati di enzimi come CK-MB e troponina comportano
rischio maggiore di morte o recidiva infartuale (cioè però vale solo per pazienti con chiara storia
clinica di ischemia, per cui c’è relazione diretta tra aumento troponina e mortalità).
Diagnosi: bisogna prima di tutto determinare la probabilità che i sintomi siano dovuti a
sindrome coronaria. Dolore ischemico, precedente infarto, segni chiari all’angiografia, nuove
alterazioni ischemiche all’ECG, scompenso cardiaco sono molto indicativi. In secondo luogo
contano i fattori di rischio per patologia coronarica. Gli strumenti diagnostici principali sono
anamnesi, ECG, enzimi e test da sforzo. Bisogna verficare se c’è infarto (ECG ed enziimi), valutare
se c’è ischemia a riposo o grave malattia coronarica (dopo test da sforzo). Incrementi di enzimi o
nuove alterazioni dell’ECG mentre il paziente è ricoverato sono segno di AI/NSTEMI.
Prognosi: mortalità tra 1-10% in 30 giorni e rischio o recidiva d’infarto tra 3-10%. Fattori di
rischio per malattia coronarica, età superiore a 65 anni, sviluppo di sintomi nonostante terapia
con acido acetilsalicilico, slivellamento del tratto ST maggiore o uguale a 0,5mm peggiorano la
prognosi.
Terapia: il paziente deve essere messo a riposo (ricoverato) con monitoraggio ECG continuo. La
terapia prevede l’uso di nitrati e beta-bloccanti oltre ad un trattamento con antitrombotici quali
acido acetilsalicilico o anche eparina non frazionata o enoxaparina (eparina a basso peso
molecolare). Il trattamento a lungo termine è in tutto e per tutto uguale a quello dei pazienti
con angina, anche per quanto riguarda le misure generali.
Angina variante di Prinzmetal
Definizione: è una forma di angina che compare prevalentemente a riposo associata ad un
79
transitorio sopraslivellamento del tratto ST. L’evidenza all’ECG è presente solo durante l’attacco
e dura in genere, a differenza dell’IM, 5-20 minuti, risultando invece un ECG normale nel resto
della giornata. Gli attacchi tendono a comparire nelle stesse ore e dissociati dallo sforzo.
La sindrome è dovuta ad uno spasmo focale di un’arteria epicardica (più la destra, ma spesso
varie) cui consegue un’ischemia miocardica. In alcuni pazienti lo spasmo arterioso è sistemico
(sincope, fenomeno di Raynaud, emicrania). La causa dello spasmo è forse un’ipercontrattilità
della muscolatura liscia vascolare dovuta al rilascio di fattori vasocostrittori, leucotrieni,
serotonina, etc. In genere lo spasmo si verifica entro 1 cm da una placca aterosclerotica (che
quindi comunque è presente anche in questi pazienti, che però in genere, a parte il fumo, hanno
meno fattori di rischio per patologia coronarica e sono più giovani). Lo spasmo coronarico
transitorio visto all’angiografia è l’elemento diagnostico fondamentale. A volte possono aversi
episodi di ischemia silente (asintomatica) o anche lievi aumenti di CK-MB e troponina.
Prognosi: dopo un periodo acuto di 3-6 mesi in cui c’è anche un maggiore rischio di infarto
miocardico non fatale (che in 5 anni è del 20%) la sopravvivenza a 5 anni è eccellente (90-95%).
La prognosi è peggiore in pazienti con gravi ostruzioni o che sviluppano importanti aritmie
durante gli episodi.
Terapia: nitrati e soprattutto calcio-antagonisti.
Ischemia asintomatica
Definizione: anche detta silente. I pazienti con IMA, ischemia transitoria e coronaropatia
ostruttiva sono spesso asintomatici anche se hanno segni evidenti all’ECG. Questi episodi
comportano comunque un rischio elevato di sviluppo di infarto miocardico (e talvolta morte) e
in base a test da sforzo, all’ECG e all’età deve essere scelta comunque una terapia adeguata.
Infarto miocardico con sopraslivellamento del tratto ST
Definizione: L’infarto miocardico costituisce la necrosi tissutale del muscolo cardiaco che si
verifica in seguito ad un’ostruzione delle coronarie. STEMI è una diagnosi associata ad alta (30%)
mortalità precoce (ossia nei 30 giorni successivi) con metà dei decessi avvenuti prima dell’arrivo
in ospedale. La mortalità resta alta (1/25) nell’anno successivo all’episodio e dipende molto
dall’età. Una volta posta diagnosi di sindrome coronarica acuta (sintomatologia tipica) è
principalmente l’ECG che permette di distinguere il tipo di patologia.
La sindrome coronarica acuta può infatti essere senza sopraslivellamento del tratto ST o con
sopraslivellamento del tratto ST. Nel primo caso in un’alta percentuale dei casi non vi sono
proprio enzimi di danno e pertanto non vi è necrosi, perciò non è infarto bensì angina instabile.
In una percentuale comunque alta vi sono enzimi di danno, dunque vi è infarto, ma è un infarto
non Q (se il tratto di ST non è normale ma è sottoslivellato potrebbe essere un infarto sub
endocardico) ossia non vi sono le tipiche onde Q che indicano necrosi all’ECG (sono ben pochi i
casi di sindrome coronarica acuta con assenza di sopraslivellamento del tratto ST, ma presenza
di onde Q -questo è un quadro comune se l’infarto magari è pregresso-).
Nel caso di sopraslivellamento del tratto ST nella maggior parte dei casi l’infarto è con onde Q e
solo in una minoranza dei casi è un infarto non Q.
Patogenesi: è ben noto che la causa principale di IHD (cardiopatia ischemica) sia l’aterosclerosi
coronarica. Questa può causare ostruzione più o meno grave dei vasi coronarici (per lo più
epicardici) e può coinvolgere uno o più spesso diversi vasi. Mentre generalmente le stenosi fisse
causano patologie come l’angina e in genere tanto gravemente quanto grave è la lesione (75%
80
occlusione ischemia da sforzo, 90% ischemia anche a riposo), le sindromi coronariche acute
dipendono generalmente da impreviste e brusche trasformazioni della placca aterosclerotica
quali rottura o fissurazione, ulcerazione, erosione. In breve, generalmente:
1) Angina stabile: è causata da un’ostruzione fissa (senza rottura della placca) e da un’ischemia
da aumentata richiesta del miocardio (raro che ci sia necrosi perché si riesce in genere a formare
una rete di vasi collaterali, dato che l’ostruzione si ingrossa lentamente).
2)Angina instabile: si ha rottura della placca, ma il trombo è solo parzialmente occlusivo.
3) Infarto miocardico: a causa dell’alterazione della placca si forma un trombo che occlude
totalmente il vaso, e rapidamente, in modo che non si riesce a formare un circolo collaterale e si
ha pertanto necrosi.
In pratica la sequenza di eventi risulta questa:
Le placche aterosclerotiche hanno una composizione dinamica (per esempio il cappuccio fibroso
va incontro a rimodellamento) e in particolare risultano “vulnerabili” quelle con core lipidico
ricco di cellule schiumose, quelle con molte cellule infiammatorie o con cappucci fibrosi sottili.
Si ha pertanto un’improvvisa modificazione della placca ateromatosa (ulcerazione, emorragia
intramurale, erosione, rottura o fessurazione) con relativa esposizione delle componenti
altamente trombo gene (come il collagene subendoteliale) della placca che causano
un’attivazione piastrinica anche a seguito del rilascio di agonisti. Si ha rilascio di trombossano
A2 e ulteriore attivazione piastrinica e d esposizione del fattore tissutale con relativa attivazione
della cascata della coagulazione. Il risultato è la formazione di un trombo murale che occlude
rapidamente il vaso.
Altre cause come emboli, anomalie congenite, vasospasmo possono causare infarti, ma molto
più raramente.
L’ostruzione arteriosa causa ischemia nell’area irrorata dall’arteria occlusa, definita area a
rischio. Il miocardio perde la sua funzione contrattile, avvia un metabolismo anaerobico (di acido
lattico, potenzialmente dannoso), ma soltanto a seguito di un’ischemia severa (meno del 10%
del flusso) e duratura (20-30 minuti almeno) si ha necrosi dei cardiomiociti (ossia danno
irreversibile) con rottura della membrana e fuoriuscita di proteine miocardiche. Un danno
permanente di notevole entità si ha dopo un periodo di 2-6 ore dall’inizio dell’ischemia (dipende
anche dal livello di circolo collaterale) e pertanto risulta fondamentale una rapida diagnosi e
intervento.
Anatomia patologica: la forma di infarto più frequente è quella completa, ossia l’infarto trans
murale che coinvolge appunto l’intero spessore della parete ventricolare.
Soprattutto se il trombo si è sciolto rapidamente per trombo lisi può verificarsi un
esclusivamente un infarto sub endocardico (terzo o metà interna della parete) ossia
concentrato solo nella sezione più sensibile all’ischemia (in quanto peggio irrorata).
Quasi tutti gli infarti sono localizzati nella parete del ventricolo sinistro e risparmiano solo una
sottile rima (0,1mm) di miocardio sub endocardico direttamente irrorata dal sangue contenuto
nel lume ventricolare. La sede dell’infarto dipende naturalmente dalla coronaria occlusa:
1) Coronaria discendente anteriore sinistra (40-50%): infarto “anteriore” che coinvolge la
parete anteriore del ventricolo sinistro, l’apice e la parte anteriore del setto interventricolare.
2) Coronaria destra: “infarto posteriore” (ma solo in caso di dominanza destra) che coinvolge la
parete inferiore/posteriore del ventricolo sinistro, la parte posteriore del setto interventricolare
e, nel 15-30% dei casi, parte del ventricolo destro.
3) Coronaria circonflessa sinistra: infarto “laterale” che coinvolge la parete laterale del
ventricolo sinistro, ma non l’apice.
Vi sono anche sedi meno consuete come il tronco comune della sinistra oppure rami secondari.
81
Le aree miocardiche danneggiate vanno incontro ad una serie di modificazioni che partono dalla
necrosi coagulativa e continuano on processi di infiammazione e riparazione (anche se l’aspetto
dipende naturalmente dalla sopravvivenza del paziente). Macroscopia: Nelle prime 12 ore gli
infarti non sono in genere affatto riconoscibili macroscopicamente. Successivamente è prima
visibile come un’area rossastra ( a causa del ristagno ematico), poi è apprezzabile un colorito
giallo a causa dell’infiltrato infiammatorio. Lo spessore della parete cardiaca appare ridotto
perché il tessuto necrotico è progressivamente rimosso. In seguito il centro giallognolo è
circondato da una zona iperemica di tessuto di granulazione (entro 2 settimane). Dopo circa un
mese l’area diviene grigiastra sino alla formazione di una cicatrice dura successivamente.
Microscopia: le cellule non mostrano segni prima delle 12 ore. Dopo un giorno si nota la necrosi
cellulare e la perdita delle fibre e successivamente infiltrato infiammatorio che tramite la
liberazione di enzimi da parte dei neutrofili e la fagocitosi dei cardiomiociti morti da parte dei
macrofagi eliminano il tessuto necrotico, che già dopo una settimana viene sostituito da
abbondante tessuto di granulazione. Si ha poi lentamente, nel corso del primo mese, la
sostituzione del tessuto di granulazione con tessuto fibroso e almeno entro sei mesi la
formazione finale della cicatrice. Una volta guarita una lesione non è più databile. Nell’infarto
trans murale queste modificazioni sono molto più evidenti e nette che nell’infarto sub
endocardico.
Clinica: Il sintomo più caratteristico è il dolore. Il dolore è profondo e viscerale, in genere
descritto come pesante, opprimente o a morsa. Coinvolge la parte centrale del torace e/o
l’epigastrio e si può irradiare alle braccia (più il sinistro) a collo, mandibola, addome e schiena,
anche fino all’occipitale ma mai sotto l’ombelico. A differenza di quella della pericardite acuta
non si irradia al trapezio. Può assomigliare al dolore dell’angina pectoris, ma, anche se in circa
della metà dei casi è preceduto da un fattore precipitante (stress fisico o emotivo, patologia), in
genere si presenta a riposo e comunque non è alleviato dal riposo e dura più di venti minuti. Il
dolore può essere accompagnato da sintomi diversi quali astenia, nausea, vomito, ansia e senso
incombente di morte.
Un quarto dei casi di infarto anteriore è associato ad un ipertono adrenergico con tachicardia,
ipertensione, agitazione e tremore; mentre la metà dei casi di infarto inferiore si associa ad un
ipertono vagale con bradicardia, nausea, vomito e collasso. L’infarto può anche essere silente
(asintomatico), più comunemente nei pazienti anziani e soprattutto nei diabetici (minore
sensibilità dolorifica e funzionalità del SNA). Può esserci dispnea soprattutto se è presente
insufficienza ventricolare sinistra. In qualche caso possono sopraggiungere pericolose aritmie.
Diagnosi: fondamentali l’ECG e gli enzimi cardiaci sierici.
Esame obiettivo: dolore toracico retro sternale di durata superiore a 30 minuti, pallore e
sdorazione sono suggestivi di STEMI. Ci possono essere segni di disfunzione ventricolare: l’itto
così come il polso carotideo sono ridotti, ci possono essere un terzo e un quarto tono,
indebolimento del primo tono e sdoppiamento paradosso del secondo, soffio sistolico mitralico,
pressione arteriosa a volte ridotta di 10-15mmHg.
ECG: costituisce un mezzo fondamentale nella diagnosi di infarto, soprattutto in fase acuta. È
inoltre in grado di individuare un infarto pregresso. Tramite l’ECG si distinguono infarti con o
senza sopraslivellamento del tratto ST, e infarti Q e non Q (suddivisioni che combaciano spesso,
ma non sempre). L’area miocardica colpita presenta delle alterazioni nella conduzione degli
impulsi che vengono rilevate.
I principali segni di cardiopatia ischemica e infarto sono:
82
Onda T invertita: segno di ischemia.
Tratto ST sottoslivellato: segno di ischemia in ECG da sforzo, comune nell’infarto sub
endocardico, ma può anche essere un’immagine indiretta di lesione.
Tratto ST sopraslivellato: segno di lesione (immagine diretta), è presente anche in altre
condizioni. In un ECG da sforzo in paziente con pregresso infarto può essere un segno di
discinesia ventricolare (vedi ECG cardiopatia ischemica).
Onda Q: è un segno di necrosi. Il tessuto necrotico è del tutto incapace di condurre impulsi e
pertanto crea un vuoto o finestra elettrica che l’ECG è in grado di rilevare. L’elettrodo positivo
più vicino alla zona infartuata rileva infatti l’assenza di vettori nella propria direzione mentre,
attraverso questa specie di finestra elettrica, vede i vettori che si propagano dalla parte opposta
e dunque nella direzione opposta. Il risultato è una profonda deflessione negativa, un’onda Q
appunto. Questa è significativa (e non una normale onda q del complesso QRS dovuta alla
propagazione dell’impulso attraverso il setto interventricolare da sinistra a destra) solo quando
ha un’ampiezza maggiore di 1mm o un’altezza almeno di 1/3 del QRS.
Solitamente le lesioni causate dall’infarto vengono distinte in tre o quattro stadi.
Inizialmente, nella fase acuta, compaiono i segni di lesione come il sopraslivellamento del tratto
ST. Della lesione si può avere, come anche per l’ischemia, un’immagine diretta (da parte degli
elettrodi che guardano l’area lesa) e un’immagine indiretta (da parte degli elettrodi che
guardano l’area opposta).
Successivamente, dopo ore, ma anche alcuni giorni, possono comparire inversione dell’onda T
(segno di ischemia, che in realtà può anche essere presente prima, magari in pazienti con angina
o comunque con episodi ischemici) e onda Q, segno di necrosi.
Successivamente le onde T e le variazioni del tratto ST tendono a scomparire lasciando come
unico segno l’onda Q o meglio il complesso QS. È possibile localizzare l’infarto valutando in quali
derivazioni si presentano queste alterazioni. Si può fare il discorso pensando alle sole onde Q,
ma naturalmente è valido anche per il tratto ST e per le onde T invertite.
Infarto anteriore: le onde Q appaiono nelle derivazioni “anteriori” ossia le toraciche: da V1 a V4.
Alcuni dividono ulteriormente l’infarto anteriore in antero-settale (onde Q solo in V1 e V2) e
antero-laterale (onde Q solo in V3 e V4).
Infarto laterale: onde Q nelle derivazioni “laterali” I e aVL.
Infarto inferiore: onde Q nelle derivazioni “inferiori” II, III e aVF.
Infarto posteriore: poiché non possiamo averne immagini dirette ne abbiamo solo immagini
indirette e quindi opposte a quelle visibili nell’infarto anteriore. Se nell’anteriore si avrà nelle
derivazioni toraciche onde Q e sopraslivellamento del tratto ST nel posteriore, nelle stesse
derivazioni e principalmente V1 e V2 si vedrà un’onda R alta, un sottoslivellamento del tratto ST
nelle stesse derivazioni e una possibile Q in V6. Per facilitare la diagnosi di infarto posteriore si
può far uso del test dello specchio o della lettura in trasparenza che, dopo aver capovolto il
tracciato, ci faranno vedere un infarto anteriore (opposto).
In presenza di blocco di branca sinistra la diagnosi di infarto con ECG non è facile.
Gli infarti si associano spesso ad emiblocchi (ischemia branche) e quindi anche a deviazioni
dell’asse cardiaco.
Esami di laboratorio: a parte una lieve leucocitosi polimorfo nucleata con lieve aumento della
conta dei bianchi e anche lieve aumento della VES, il segno più specifico di infarto sono gli
enzimi cardiaci sierici.
Normalmente infatti, gli enzimi miocardici hanno una bassa concentrazione in circolo, quando
però vi è un danno miocardico le proteine contenute all’interno dei cardiomiociti vengono
liberate nel sangue, raggiungendo concentrazioni molto superiori alla norma e costituendo un
83
importante indice diagnostico.
Ogni marker cardiaco sierico ha una sua cinetica, ossia un suo picco di concentrazione e un suo
tempo di comparsa e di scomparsa. Interventi di rivascolarizzazione possono modificare questa
cinetica. Valutando gli enzimi cardiaci si può integrare l’esame clinico e l’ECG per confermare la
diagnosi di infarto, soprattutto nei casi di ECG non diagnostici (assenza di onde Q, da ciò si
deduce che sono un valido supporto in caso di infarti non trans murali).
È stata dimostrata la correlazione diretta tra estensione dell’infarto ed entità del rilascio degli
enzimi (come CK-MB o troponine), mentre è meno definito il rapporto con il picco di
concentrazione. I principali enzimi sono:
Creatin fosfochinasi, CK: aumenta entro 4-6 ore dall’inizio dei sintomi, raggiunge il picco nelle
24-36 ore e scompare dopo circa 4 giorni. Ha una specificità limitata perché può aumentare in
ogni danno muscolare e in altre patologie. L’isoforma CK-MB è invece tipicamente cardiaca
anche se aumenta anche in interventi di cardiochirurgia e miocarditi oltre alla cardioversione
elettrica (che causa spesso aumento anche di altri enzimi) che è un comune intervento in caso di
aritmie pericolose come la fibrillazione ventricolare.
Troponine miocardio-specifiche T e I, TnT e TnI: insieme alla troponina C hanno il compito di
regolare lo spostamento della tropo miosina e in generale l’interazione tra actina e miosina
tramite un meccanismo di risposta calcio-dipendente (la troponina C rappresenta il sito di
legame con il calcio). Le troponine I e T miocardiche hanno (a differenza della C), una
composizione aminoacidica diversa rispetto a quelle di altri tessuti muscolari e pertanto sono
specifiche di danno miocardico. Il dosaggio si effettua tramite specifici anticorpi monoclonali, e
in caso di infarto possono aumentare anche di 20 volte. TnI e TnT sono i marcatori sierici
preferiti per IM, soprattutto quando ci sono valori al limite di CK-MB. Hanno un picco più tardo
rispetto alla CK, ma possono restare elevate anche per una settimana o poco più.
Altri enzimi sono: transaminasi glutammico-ossalacetica (GOT, compare con la CK e torna a
valori normali in 5-7 gg) e lattato-deidrogenasi (LDH, torna alla normalità in 10 giorni). I pazienti
che fanno terapia trombo litica o interventi di rivascolarizzazione hanno, a causa della maggiore
perfusione, un rilascio degli enzimi molto più rapido, con picco di concentrazione e ritorno alla
normalità in meno tempo. In questo modo la quantità di enzima rilasciata è minore, e questo
indica pertanto anche un minore danno miocardico.
Imaging cardiaco: Ecocardiogramma: permette di valutare le conseguenze di un infarto
miocardico. Non è in grado di distinguere un infarto miocardico pregresso da un evento acuto,
né è in grado di definire bene le dimensioni dell’infarto in quanto non distingue un tessuto
necrotico da un tessuto ischemico. È però semplice e rapido e permette di valutare le anomalie
della cinesi parietale (100% deigli infarti Q e 80% dei non Q) e il diminuito spessore delle zone
miocardiche colpite, potendo in tal modo ipotizzare anche quale vaso sia occluso in base alla
localizzazione dell’infarto. Permette in generale la rilevazione di complicanze come l’aneurismo
ventricolare, la preforazione del setto interventricolare e l’insufficienza mitralica ed inoltre
permetta la valutazione della funzionalità del ventricolo sinistro (eventuali disfunzioni
ventricolari, diminuzione della frazione di eiezione). È inoltre in grado di valutare l’efficacia di
interventi di riperfusione coronarica.
Scintigrafia miocardica: con tallio o tecnezio, rileva segni di ischemia residua postinfartuale.
Ventricolografia con tecnezio: serve a localizzare la necrosie valutare la frazione di eiezione e
quindi la funzione ventricolare. Le tecniche radioisotopiche sono in genere cmq meno usate
perché indaginose e spesso poco specifiche.
La risonanza magnetica con gadolinio è un’alternativa.
Tomografia a emissione di positroni: (PET) permette tramite l’impiego di FDG
84
(fluorodesossiglucosio) di valutare il metabolismo di glucosio da parte del tessuto miocardico e
di considerare la presenza o meno di zone miocardiche vitali. Possono esservi zone che, pur
restando vitali, hanno caratteristiche particolari. Si distinguono:
Miocardio ibernato: Tessuto temporaneamente addormentato, forse a causa di episodi ripetuti
di stordimento) che può ancora ritornare allo stato normale nel momento in cui il flusso
coronarico viene completamente ripristinato. I cardiomiociti sono ipocinetici, hanno ancora la
capacità di estrarre glucosio (come si può vedere alla PET), ma hanno un metabolismo ed una
funzionalità ridotti. In genere è dovuto ad una coronaropatia che magari fa mantenere un flusso
normale a riposo, ma che (ad esempio per sforzi) causi episodi ricorrenti di ischemia che lasciano
il miocardio addormentato. Questo tessuto può ancora essere recuperato tramite
rivascolarizzazione (molto più raro che capiti con tessuto che ha anche una ridotta capacità di
estrarre glucosio).
Miocardio stordito: Condizione caratterizzata dalla presenza di una prolungata disfunzione
miocardica post-ischemica (quindi dopo che il flusso è tornato normale) con ritorno finale ad
una normale attività contrattile. La riperfusione infatti, se fatta in tempo, è in grado di
recuperare il tessuto ischemico, ma ancora vitale (l’ischemia non causa subito morte cellulare). Il
miocardio può però rimanere stordito anche dopo rivascolarizzazione. Se si associa uno studio
funzionale (come scintigrafia o PET) ad uno studio morfologico delle coronarie (per verificare se
il flusso è effettivamente presente o no) si può diagnosticare uno stato di stordimento (ossia di
compromissione reversibile delle capacità contrattili del miocardio). Lo stordimento può
verificarsi in numerose situazioni cliniche come in seguito ad un episodio ischemico da sforzo
(angina da sforzo) o dopo trombo lisi o PTCA con riperfusione coronarica o anche dopo
cardioplegia. Pare che alla base dello stordimento vi siano i meccanismi biologici dell’aumento di
calcio nel citoplasma e la formazione di radicali liberi a seguito della riperfusione (addirittura, nel
cosiddetto precondizionamento brevi attacchi di ischemia possono proteggere da un
susseguente reale infarto per meccanismi ancora non ben chiari).
La riperfusione (segue dopo, con le varie tecniche farmacologiche, interventistiche, chirurgiche)
può anche causare una danno da riperfusione pur non essendo ancora chiara la rilevanza clinica
di quest’ultimo.
Complicanze: quadri clinici che possono presentarsi durante l’infarto o in tempi successivi:
1) Disfunzione ventricolare e scompenso cardiaco congestizio: a seguito di un infarto si hanno
dei fenomeni di rimodellamento ventricolare con ingrandimento del ventricolo a seguito
dell’allungamento delle fibre nell’area infartuata e successivamente anche intorno. La
dilatazione è associata ad una disfunzione ventricolare che può evolvere (anche in anni) in uno
scompenso cardiaco (sistolico da ridotta gittata o diastolico da ridotta compliance). Il deficit di
pompa è la principale causa di morte dopo IMA, e ha come segni un terzo o quarto tono e
congestione polmonare (radiografia al torace). I pazienti con scompenso presentano
un’aumentata pressione di riempimento ventricolare e dell’arteria polmonare (verificate con
inserimento di un catetere a palloncino). Una funzione ventricolare anomala è in genere
associata ad un deficit di contrattilità di almeno il 20-25% del ventricolo (oltre il 40% si può
avere shock cardiogeno ossia un deficit di pompa con volumi ventricolari di riempimento che
risultano aumentati e forte ipotensione che può portare anossia cerebrale, nell’80% dei casi
risulta mortale). La terapia per lo scompenso cardiaco congestizio dopo IMA è la stessa del
normale con diuretici, vasodilatatori (nitrati) e ACE-inibitori e/o ARB. I diuretici possono
peggiorare la situazione causando ipovolemia.
2) Infarto ventricolare destro: più che una complicanza è una condizione che può avvenire in
concomitanza all’infarto del ventricolo sinistro (30% si infarti laterali). Si può associare a segni di
85
scompenso cardiaco destro e si può notare in genere un sopraslivellamento del tratto ST nelle
derivazioni precordiali destre dell’ECG.
3) Aritmie: l’incidenza delle aritmie in pazienti con IMA è molto alta in quanto queste possono
essere causate da squilibri del SNA, disturbi elettrolitici, ischemia e rallentamento di conduzione
nelle aree necrotiche. Le aritmie possono aggravare la necrosi diminuendo la perfusione
coronarica e incrementando il consumo di ossigeno (le tachiaritmie riducono il tempo di diastole
e aumentano il lavoro cardiaco) o aggravando il deficit di pompa (bradi aritmie).
Tachiaritmie:
- Extrasistoli ventricolari: i battiti ectopici ventricolari sono comuni e sono dovute all’ipossia e
aggravate da eventuali squilibri idroelettrici quali ipopotassiemia e ipomagnesemia (che sono
aggravanti di molte forme aritmiche e devono essere corretti!). Di per sé non rappresentano un
fenomeno preoccupante perché non compromettono la funzione emodinamica, ma se molto
precoci possono favori l’insorgenza di TV e FV (fenomeno R/T). I beta-bloccanti le riducono
notevolmente.
- Tachicardia ventricolare e fibrillazione ventricolare: sono potenzialmente molto pericolose (si
aggiunge a queste anche la torsione di punta) in quanto possono compromettere la condizione
emodinamica. Anche se la terapia profilattica antiaritmica con lidocaina non è più raccomandata
si usano i beta-bloccanti per prevenire queste condizioni. Si trattano con amiodarone, o, se le
condizioni del paziente sono gravi, con cardioversione elettrica (scarica non sincronizzata di
200-300J, se non funziona si associa ad adrenalina). L’impianto di un ICD è indicato in pazienti
con frazione di eiezione ventricolare inferiore al 40% con episodi di TV e FV post-STEMI.
- Ritmo idioventricolare accelerato: il ritmo idioventricolare può avvenire solo se la funzione del
NSA come pacemaker dominante viene a mancare. È una forma di tachicardia ventricolare in
genere quindi associata a bradiacardia sinusale. Non è tendenzialmente pericolosa perché il
ritmo è di 90-100bpm e non è un preludio a FV, può essere trattata con atropina per favorire un
aumento dell’attività del NSA (tachicardia sinusale).
- Aritmie sopraventricolari: la più comune è la tachicardia sinusale che a meno che non sia
sostenuta (100-120 bpm) e duratura (2 ore, e in tal caso si usa defibrillatore) non è
particolarmente preoccupante. Si può trattare con beta-bloccanti perché in genere è associata a
ipertono simpatico. Ritmi idiogiunzionali accelerati sono possibili.
Bradicardie:
- Bradicardia sinusale: è comune nell’infarto inferiore ed è in genere associata ad ipertono
vagale. Il trattamento con atropina è indicato se c’è compromissione emodinamica. Se persiste
si pratica pacing.
- Blocchi di conduzione atrioventricolare e intraventricolare: possono avere cause differenti.
Negli infarti inferiori i blocchi AV sono dovuti in genere a ipertono vagale, negli anteriori
principalmente alla necrosi e ostruzione delle coronarie che vascolarizzano anche le branche del
sistema di conduzione. Il malfunzionamento su base ischemica del sistema di conduzione è alla
base anche dei frequenti emiblocchi o blocchi di branca che si verificano comunemente in caso
di IMA. Il BAV è la condizione più grave e deve essere trattato in caso di compromissione
emodinamica con pacing (magari preceduto da elettrostimolazione temporanea esterna non
invasiva). Comunque è indicato in tutti i casi di blocco bi fascicolare, Mobitz II, BAV di terzo
grado, se non rispondono a terapia medica e presentano bradicardia.
4) Aneurisma del ventricolo sinistro: è una discinesia o espansione parietale del ventricolo
sinistro. In genere è associato ad infarti anteriori e coinvolge la parete antero-settale . La parete
ventricolare perde il tessuto muscolare che viene sostituito da tessuto fibroso acinetico e sottile
(ben visibile all’esame ecocardiografico). Può associarsi a disfunzione ventricolare o scompenso
e ad aritmie ventricolari. Con il tempo la zona può dilatarsi e diventare sede di ancoraggio di
86
trombi (con un rischio, raro, di fenomeni embolici sistemici).
La terapia è chirurgica e consiste nell’aneurismectomia ventricolare sinistra. Quest’intervento è
eseguito in ipotermia moderata (28°) e circolazione extracorporea con infusione cardioplegica
potassica a 4°C. L’aneurisma deve essere inciso e i asportato lasciando però dei margini di
tessuto necrotico sui quali poi dovranno essere passati dei punti con sutura in Tevdek 2-0 a
punti staccati su quadratini di Teflon. Se nella parete ventricolare ci sono dei trombi questi
dovranno essere rimossi con cautela. Dopo l’asportazione dell’aneurisma bisogna eseguire
interventi di rivascolarizzazione miocardia mediante applicazione di by-pass.
5) Insufficienza mitralica acuta secondaria: può aversi per rottura dei muscoli papillari e delle
corde tendinee a causa di un grosso infarto antero-laterale, ma può essere anche conseguenza
di una progressiva ischemia dei muscoli papillari. Si può avere dispnea da sforzo fino anche
scompenso cardiaco (e anche shock cardiogeno). La terapia è chirurgica con sostituzione della
valvola con una protesi.
6) Perforazione del setto interventricolare: in genere si manifesta 3-5 giorni dopo l’infarto e può
essere localizzato nella parete inferiore del setto (infarto antero-settale inferiore per ostruzione
IVP) o all’apice del setto (infarto antero-settale apicale per ostruzione di IVA). La conseguenza è
uno scompenso cardiaco grave e il trattamento è chirurgico con intervento eseguito in ipotermia
moderata dopo clampaggio aortico e protezione miocardica con cardioplegia potassica a 4°C.
Viene incisa la zona infartuarta e riparata la perforazione con innesto di un patch in Dacron
suturato con punti staccati ad U di Tevdek 2-0 su Teflon.
7) Dolore toracico ricorrente: nel 25% dei pazienti si sviluppa un’angina residua.
8) Pericardite: il dolore acuto che si estende al trapezio può facilitarne la distinizione dal dolore
toracico ricorrente. La pericardite, definita epistenocardica, si può verificare nella regione
sovrastante la necrosi tra il 2° e il 4° giorno post-infarto. Si può trattare con acido acetilsalicilico.
9) Tromboembolia: possono esserci segni evidenti nel 10% dei pazienti, anche se almeno un
altro 10% presenta fenomeni silenti. Episodi trombo embolici a livello polmonare e sistemico
sono più comuni in infarti estesi, per lo più anteriori, soprattutto se complicati da scompenso e
ancor più in caso di aneurima ventricolare, in cui i trombi murali nel ventricolo sinistro sono una
complicanza comune.
Terapia: la prognosi di STEMI è in gran parte condizionata dal verificarsi di complicanze
meccaniche (deficit di pompa) ed elettriche. La maggior parte dei decessi extraospedalieri
avvengono per fibrillazione ventricolare che quasi nella metà dei casi si verifica nella prima ora.
È essenziale quindi che il paziente venga portato il prima possibile in ospedale (conta molto che
sia educato a dare il giusto peso ai sintomi e che chiami presto soccorso).
Lo scopo principale della terapia è, soprattutto nella fase iniziale, evitare l’insorgenza di
complicanze meccaniche ed elettriche e di iniziare al più presto un adeguato trattamento
(medico o chirurgico) che permetta riperfusione e dunque una limitazione dell’area infartuata.
Una volta giunti in ospedale i pazienti vengono ammessi nell’unità di terapia intensiva
coronarica dove vi è un monitoraggio continuo delle funzioni vitali del paziente, della frequenza,
del ritmo e della pressione arteriosa. Vengono eseguiti ECG ed esami di laboratorio urgenti per
verificare IMA e sono comunque disponibili defibrillatori, respiratori, pacemaker trans toracici e
altre apparecchiature. Il trattamento nella fase ospedaliera consiste prima di tutto in alcune
misure generali:
1) Attività: bisogna evitare i fattori che aumentano il lavoro e quindi il consumo di ossigeno da
parte del miocardio, pertanto nelle prime 12 ore almeno il paziente deve restare a letto. I
pazienti devono essere incoraggiati ad assumere posizione eretta nelle prime 24 ore in modo da
ridurre la pressione capillare polmonare ma soprattutto per dare incoraggiamento psicologico.
87
Già dal secondo-terzo giorno i pazienti possono camminare per la stanza e provvedere da soli
al’igiene personale, successivamente possono fare anche 200 metri di cammino due o tre volte
al giorno.
2) Dieta e alvo: nelle prime (12) ore è indicato digiuno e dieta idrica. Successivamente la dieta
deve essere equilibrata con adeguato introito soprattutto di potassio e magnesio oltre che di
fibre per evitare la stipsi che spesso i narcotici possono causare (si può anche aggiungere un
emolliente fecale, il dioctil-sulfosuccinato sodico, e se non basta somministrare lassativi). Le
restrizioni riguardano i grassi ed il colesterolo, oltre che i carboidrati (normali per un paziente
sano, da evitare nel diabete).
3) Terapia del dolore e sedazione: si può somministrare sin da subito nitroglicerina sublinguale
per diminuire il dolore toracico (oltre a favorire la vasodilatazione coronarica e un minore sforzo
cardiaco). Sono da evitare in pazienti ipotesi, o con infarto del ventricolo destro o che hanno
assunto sildenafil. La morfina a piccole dosi e in piccoli boli per ev è molto efficace, ma può
causare, soprattutto all’inizio, stipsi, nausea e vomito (per la sua azione anticolinergica, per la
quale potrebbe causare anche blocco AV e bradicardia). I beta bloccanti sono utili perché
riducono la richiesta di ossigeno e l’ischemia (e quindi il dolore ad essa associato). Possono
essere somministrate benzodiazepine (tranquillanti) per facilitare il riposo del paziente, il cui
sonno può essere disturbato da ansia e da fastidio per il monitoraggio continuo soprattutto il
primo giorno.
4) Ossigeno: la somministrazione di ossigeno è indicata nel caso in cui la saturazione sia
diminuita (spesso).
Terapia farmacologica:
Antitrombotici: la fibrinolisi è una strategia fondamentale, e la terapia sarebbe bene che fosse
iniziata entro 30 minuti dalla presentazione dei sintomi. Lo scopo è quello si eliminare il trombo
nella coronaria occlusa riassicurando la perfusione e di evitare episodi trombotici a livello
sistemico. La terapia fibrinolitica può ridurre il rischio di morte ospedaliera del 50% riducendo
l’area infartuata, limitando la disfunzione ventricolare sinistra e l’incidenza di complicanze gravi
come perforazione del setto interventricolare, shock cardiogeno ed aritmie fatali. I benefici sono
notevoli se iniziata entro le preme 1-3 ore e forse anche fino a 6-12. È preferita la trombo lisi
rispetto all’intervento di rivascolarizzazione percutanea in tutti i pazienti che si presentano nella
prima ora, o nei vari casi in cui l’interveto non sia possibile.
Gli agenti utilizzati sono: t-PA (attivatore tissutale del plasminogeno), streptokinasi, e il
complesso attivatore streptokinasi plasminogeno anisoilato (APSAC). Questi farmaci
favoriscono l’attivazione del plasminogeno in plasmina che lisa i trombi. Il farmaco più utilizzato
è il t-PA con 15mg di bolo iniziale, 50mg ev nella prima mezz’ora, 35mg ev nella successiva ora.
Controindicazioni assolute: pregresso ictus cerebrale emorragico nell’ultimo anno, emorragia
interna o dissezione aortica in atto, ipertensione grave (sistolica >180mmHg), neoplasie
intracraniche.
Controindicazioni relative: età avanzata, intervento chirurgico recente, ulcera peptica attiva.
Complicanze: le principali sono reazioni allergiche (principalmente alla streptokinasi) ed
emorragia (da banali sanguinamenti a ingenti che richiedono trasfusioni, a emorragie gravi
gastrointestinali o cerebrali come l’ictus emorragico). In aggiunta alla fibrinolisi si usano farmaci
anticoagulanti (antitrombinici ed antipiastrinici) sempre per assicurare una perfusione adeguata.
Già in pronto soccorso viene somministrato acido acetilsalicilico (160-325mg) che comporta
inibizione della COX2 con riduzione dei livelli di trombossano. Dovrebbe poi essere seguita da
somministrazione giornaliera.
Anticoagulanti: servono perché può capitare che una placca residua possa con l’attivazione della
cascata coagulativa rioccludere precocemente il vaso. Il farmaco di scelta è l’eparina non
88
frazionata (ENF) per ev (bolo da 5000 unità poi per ev continua di 1000 unità). La sinergia tra
acido acetilsalicilico, eparina e un trombo litico comporta diminuzione del rischio di mortalità
senza un grande aumento del rischio emorragico.
Farmaci per la protezione del miocardio: Beta-bloccanti: sia in fase acuta che cronica,
comportano diminuzione della richiesta di ossigeno, blocco dell’ischemia e dunque
miglioramento anche del dolore ed hanno un effetto antiaritmico. ACE-inibitori: bloccano il
sistema renina-angiotensina-aldosterone che si attiva durante la fase acuta dell’IMA. Nitrati: la
nitroglicerina per ev è poco utile in caso si diano ACE-inibitori + beta-bloccanti. Calcioantagonisti: non vi è indicazione precisa e sicura.
Rivascolarizzazione coronarica: viene essenzialmente eseguita mediante due modalità:
l’angioplastica coronarica transluminale percutanea (PTCA, o più in generale interventi
coronarici percutanei, PCI) e bypass coronarico.
La PTCA, introdotta nel 1977 ma notevolmente rimodernata fino ai giorni nostri, ha ormai un
ruolo dominante nel trattamento della coronaropatia, e ha portato alla creazione di una nuova
disciplina nota come cardiologia interventistica.
Indicazioni terapeutiche e criteri di scelta: Scegliere quando effettuare una rivascolarizzazione
piuttosto che semplicemente una terapia medica e, soprattutto, scegliere tra la PTCA ed il
bypass non è facile. La terapia medica nel trattamento della cardiopatia ischemica risulta
estremamente rilevante, ma molti pazienti traggono giovamento dalla rivascolarizzazione
coronarica in associazione con la terapia medica. La rivascolarizzazione è indicata sia in pazienti
con sindrome coronarica acuta, sia in alcuni casi di cardiopatia ischemica non ben controllati
dalla terapia. La rivascolarizzazione deve essere pensata per:
1) Pazienti con cardiopatia ischemica e bassa capacità di svolgere esercizio, con un’ampia area di
ischemia miocardica e frazione di eiezione ventricolare <40% o con sintomi non controllabili in
corso di esercizio fisico.
2) Pazienti con presentazione di sindrome coronarica acuta che allo screening con ECG al
momento del triage (trasporto in ospedale) che mostrano sopraslivellamento del tratto ST di
almeno 2mm in due derivazioni precordiali contigue e di 1mm in due derivazioni periferiche.
3) Pazienti che hanno eseguito terapia fibrinolitica ma hanno ancora segni di mancata
riperfusione dopo 90 minuti (dolore toracico e sopraslivellamento ST) e si parla di PCI di
salvataggio
4) Pazienti con riocclusione della coronaria o sviluppo di un’angina ricorrente.
6) Pazienti che presentano complicanze gravi come lo shock cardiogeno, che hanno diagnosi
dubbia, che hanno rischio di sanguinamento aumentato o che hanno sintomi presenti da almeno
2-3 ore e pertanto coaguli più maturi e meno facilmente lisati da farmaci.
In tutti questi casi naturalmente prima dell’indicazione all’intervento è necessaria
coronarografia e cateterismo cardiaco per valutare la possibilità di eseguire l’intervento.
La scelta tra bypass e PTCA è ancora più complessa. Con il tempo il numero di rivascolarizzazioni
eseguite con PTCA ha raggiunto e poi anche superato il numero di rivascolarizzazioni eseguite
con bypass grazie soprattutto alle innovazioni tecniche che hanno migliorato di molto l’efficacia
di questa procedura. Un tempo la PTCA presentava grosse limitazioni in condizioni anatomiche,
valutabili con coronarografia, quali lesioni eccentriche calcifiche, occlusioni croniche totali delle
coronarie o ad esempio lesioni localizzate in biforcazioni. Adesso queste limitazioni sono meno
nette. La valutazione deve comunque essere individuale e molto dipendente dall’anatomia
coronarica del paziente, dall’età, dall’insufficienza ventricolare e da malattie concomitanti.
In generale la PCI è consigliata in pazienti con malattia monovasale (o anche bivasale) con lesioni
anatomiche idonee, mentre il bypass aortocoronarico è consigliato in pazienti con malattia
89
trivasale, lesioni complicate o ostruzioni totali, malattia bivasale che include la coronaria sinistra
discendente prossimale o il tronco comune, alterata frazione di eiezione ventricolare sinistra e
pazienti affetti da diabete mellito.
La PCI presenta un maggiore rischio di ristenosi rispetto al bypass in quanto mentre la PCI si
focalizza sulla risoluzione delle lesioni responsabili, il bypass pone rimedio non solo al tratto
interessato, ma fornisce una via di perfusione anche qualora si sviluppino lesioni native
prossimali all’anastomosi dell’impianto del vaso nativo.
La PCI ha un minore rischio di mortalità a breve termine (intervento meno invasivo e meno
rischioso), ma ha una maggiore mortalità a lungo termine (es. 5 anni).
Rivascolarizzazione coronarica percutanea: è un intervento, nato semplicemente come
angioplastica coronarica transluminale percutanea (poi rimodernato e complicato da nuove
tecniche) che deriva in pratica dal cateterismo cardiaco diagnostico. L’indicazione principale è la
presenza di stenosi coronariche ritenute responsabili della sindrome clinica e che necessitano di
rivascolarizzazione. Le attuali indicazioni di PCI coprono l’intero spettro della cardiopatia
ischemica, dai pazienti con angina instabile o anche con ischemia silente a pazienti con STEMI
(nei quali è necessario che il tempo tra la presentazione dei sintomi e l’intervento sia
possibilmente inferiore ai 90 minuti). L’intervento di rivascolarizzazione coronarica per via
percutanea anche primario (non preceduto da terapia fibrinolitica) se eseguito da operatori
esperti e in centri medici specializzati si associa ad una migliore prognosi sia a breve che a lungo
termine rispetto alla semplice terapia medica.
Procedura: La PTCA può essere eseguita durante l’indagine coronarografica o in una fase
successiva. Con l’angiografia che precede l’intervento vengono identificate le lesioni bersaglio e
pianificata la strategia di intervento. In genere l’anticoagulazione si ottiene mediante iniezione
di eparina non frazionata e in si pretratta con acido acetilsalicilico. Si utilizzano due cateteri, un
catetere guida in genere con diametro esterno 6F (2mm) uguale a quello usato per la
coronarografia (ma diametro interno maggiore) si inserisce, come nel cateterismo cardiaco
diagnostico, attraverso l’arteria femorale e viene introdotto fino all’ostio dell’arteria coronarica
da trattare (in realtà il catetere è inserito attraverso una guaina di accesso vascolare). Al suo
interno si introduce un catetere dilatatore che all’estremità monta un palloncino il quale,
gonfiandosi in prossimità della placca ateromatosa, causa frantumazione della placca intimale e
dilatazione di media e avventizia del vaso rendendolo pervio. Dopo lo “sgonfiaggio” del
palloncino ci può essere un ritorno elastico dell’arteria che lascia un certo grado di ostruzione. Il
palloncino è in genere gonfiato a 4-10 atm (anche di più in condizioni particolari) per un tempo
di 5-15 secondi. Si possono effettuare più dilatazioni dopo le quali è opportuno eseguire una
coronarografia di controllo. La guaina in genere viene rimossa quando il tempo di coagulazione
torna normale (2-4 ore). Il tempo totale dell’intervento può essere di 60-90 minuti e la
procedura, per quanto poco invasiva, non è esente da complicanze come chiusura di un vaso o
perforazione di una coronaria che comunque sono rare (mortalità di circa l’1% nonostante i
pazienti siano gravi e con funzioni cardiache alterate). Controindicazione all’intervento è di
solito la presenza di una stenosi del tronco comune della coronaria sinistra. La percentuale di
successo è quasi del 95% con una possibilità di ristenosi nei 6 mesi del anche superiore al 20%
(maggiore ad esempio in pazienti diabetici).
Lo sviluppo di nuove tecniche ha migliorato notevolmente però l’efficacia della procedura che
ora si può avvalere ad esempio di:
Stent: sono protesi metalliche tubulari che vengono inserite nel vaso non dilatato e poi dilatate
dal palloncino (superando due dei principali problemi ossia la dissezione intimale e la retrazione
elastica del vaso). Attualmente gli stent sono anche usati come piattaforme per il rilascio di
90
farmaci che bloccano la proliferazione intimale e antinfiammatori che migliorano la durata
dell’intervento e riducono il rischio di ristenosi.
Cateteri da aterectomia: con lame rotanti che possono rimuovere la massa della lesione
trattata allargando così il lume. Durante un intervento di angioplastica è comunque sempre
bene avere una sala operatoria cardiochirurgia pronta ad intervenire nel caso in cui (ad esempio
per occlusione improvvisa della coronaria o dissecazione intimale) ci fosse bisogno di un bypass
d’urgenza. L’angioplastica può anche essere utilizzata per dilatare un bypass in caso di eventuale
ristenosi a distanza di tempo.
Bypass aorto coronarico: (CABG) l’intervento ha una mortalità intorno all’1% che, in pazienti
molto debilitati come quelli con gravi cardiopatie ischemiche, è più che accettabile, si ha un
minore rischio di ristenosi rispetto alla PCI, si ha risoluzione completa o molto buona dell’angina
nel 90% dei casi. Inoltre l’intervento è adatto anche in pazienti con stenosi complete bivasali o
trivasali (e anche del tronco comune della coronaria sinistra) con compromissione ventricolare
sinistra(FE<50%) e con lesioni anatomicamente complesse, tutte condizioni in cui la PCI è poco
indicata. Chiaramente l’età avanzata e il diabete peggiorano la prognosi.
Procedura: l’intervento è eseguito a torace aperto e consiste nell’utilizzare tratti venosi o
arteriosi che saltano le ostruzioni coronariche permettendo così di nuovo l’apporto ematico.
L’intervento in genere si esegue in circolazione extracorporea e ipotermia moderata con arresto
diastolico del cuore tramite soluzione cardioplegica potassica a 4°C.
[Cardioplegia: protezione del cuore. Oggi si tende a preferire una soluzione di KCL in quanto il
potassio permette un arresto cardiaco rapido e con il cuore in diastole (rispetto al calcio che
pure è rapido, ma blocca in sistole, condizione meno manipolabile). Si associa a circolazione
extracorporea (CEC) e clampaggio aortico (ossia blocco della aorta, in questo caso a livello dei
seni coronarici, per sgomberare il campo operatorio dal sangue) che causerebbero danno
anossico al cuore se questo non fosse protetto e “metabolicamente rallentato”. Si inietta la
soluzione in genere nel bulbo aortico (o osti coronarici se c’è insufficienza aortica) ogni 20
minuti durante l’intervento.]
Soprattutto un tempo si potevano (ma ancora ora si possono) utilizzare condotti venosi, in
genere costituiti dalla vena safena autologa adeguatamente preparata per evitare lesioni
intimali la quale andrà quindi a costituire la nuova coronaria. Viene prelevata dalla gamba, ne
vengono chiusi i collaterali e suturati eventuali punti di perdita e dopo essere stata capovolta
viene suturata a monte e a valle dell’ostruzione coronarica. L’anastomosi a valle è di tipo
termino-laterale perché distale all’ostruzione, mentra l’anastomosi a monte è confezionata sulla
radice dell’aorta ascendente (bypass aortocoronarico). Nel caso in cui bisogna rivascolarizzare
più rami (ostruzione bi o trivasale) si può utilizzare un unico tratto lungo di vena safena che
viene anastomizzato normalmente a monte e a valle, ma anche lateralmente per vascolarizzare
gli altri rami vicini (bypass sequenziale).
Oggi si utilizzano anche dei rami arteriosi, ossia tratti di arteria mammaria interna offrono un
migliore rendimento a distanza. L’arteria mammaria interna, AMI, nasce dalla parte inferiore
della prima porzione di succlavia ed è incrociata dietro l’articolazione sternoclaveare dal frenico.
Poi scende lateralmente allo sterno tra le cartilagini costali e la pleura parietale accompagnata
da due vene satelliti e da linfonodi. Termina a livello del sesto spazio dando origine ad
epigastrica superiore e muscolo frenica (ha anche rami intercostali, sternali, mediastinici). Viene
preparata scheletrizzata (cioè senza i tessuti molli e la fascia che la circondano) o all’interno del
peduncolo che contiene anche le vene satelliti (in questo caso si fa stereotomia longitudinale
medinana e scollamento della parte mediale della pleura parietale). Il peduncolo è isolato con
l’elettrobisturi (a 1cm di distanza dalle vene satelliti per evitare danni da calore all’arteria),
91
vengono poi occluse tramite clip metalliche i rami collaterali emergenti. Viene poi mobilizzato il
peduncolo mammario (AMI, vene satelliti e tessuto adiposo periavventiziale) che viene suturato
sulla coronaria da trattare. In genere l’impiego dalla mammaria richiede un tempo operatorio
maggiore perché il peduncolo mammario deve essere adeguatamente scollato e i collaterali
chiusi, ed inoltre si procede prima all’anastomosi termino-laterale a valle dell’ostruzione e poi
alla fissazione del peduncolo sulla parete epicardica. (in sostanza la mammaria viene lasciata in
genere in situ, ossia resta legata alla sua origine naturale sulla succlavia). Si può però anche
ricorrere al free-graft di mammaria ossia un segmento di arteria a cui sono sezionate l’estremità
prossimale e distale (soprattutto in condizioni anatomiche in cui la mammaria è corta). In genere
la mammaria viene confezionata sull’interventricolare anteriore (IVA).
Mentre si è sempre utilizzata un’unica arteria mammaria (più la sinistra), negli ultimi anni si è
assistito al sempre maggiore utilizzo di entrambe le arterie mammarie. Le due mammarie
possono essere suturate a Y, tecnica in grado di favorire un’ottima e diffusa vascolarizzazione.
Persino la gastroepiploica destra (sempre più usata) può contribuire anche insieme alla Y creata
dalle due mammarie. La tendenza dunque ad utilizzare graft arteriosi multipli si è sempre più
diffusa (funzionano meglio).
L’uso della mammaria è controindicato nella sindrome da furto della succlavia. L’uso della
mammaria (o delle mammarie) è vantaggioso a lungo termine perche il calibro dell’AMI è simile
a quello della coronaria. Altri condotti vascolari utilizzabili, specie nei re interventi, sono:
epigastrica inferiore e gastroepiploica sinistra.
La circolazione extracorporea presenta, come è noto, delle complicanze, pertanto un’alternativa
può essere la rivascolarizzazione miocardica a cuore battente: si fa bypass senza arresto
dell’attività cardiaca tramite soluzione cardioplegica.
In base al numero di bypass da eseguire si può scegliere poi la stereotomia longitudinale
mediana o altrimenti la minitoracotomia. Per bloccare la contrazione del miocardio sottostante
la coronaria da trattare (ed evitare la fuoriuscita di sangue) si fa uso di stabilizzatori. Questi non
danno problemi sui vasi anteriori come l’IVA, mentre la difficoltà aumenta ad esempio per la
circonflessa. Per garantire un flusso coronarico distale ed evitare traumatismi alle coronarie si
può fare uso di shunt che vengono posizionati all’interno delle arterie tramite arteriotomia e
vengono asportati subito dopo aver completato l’anastomosi. La chirurgia a cuore battente ha
maggiori difficoltà tecniche e non rende possibile estendere o modificare l’incisione
anastomotica. Quando si pratica insieme ad una sternotomia mediana è possibile un migliore
approccio a tutti i vasi coronarici e se insorgessero complicazioni sarebbe possibile avviare
subito la circolazione extracorporea.
Bypass a cuore battente in minitoracotomia si può praticare per: rivascolarizzazione di IVA non
trattabile con PCI; notevoli fattori di rischio per circolazione extracorporea; re intervento per
bypass singolo su IVA, ristenosi dopo PCI; bypass singolo su IVA in pazienti già sottoposti ad un
intervento di chirurgia valvolare. È controindicato se: presenza di aritmie pericolose, ischemia
miocardica acuta, coronari non favorevole a tale procedura. Bisogna anche considerare che
comunque nella chirurgia a cuore battente possono aversi danni cerebrali da perfusione o da
emboli (formatisi dopo il clampaggio dell’aorta ascendente durante l’esecuzione delle
anastomosi a monte o per immissione in circolo di trombi localizzati nel ventricolo sinistro).
Bypass coronarico in minitoracotomia con approccio port-access: vengono cannulate arteria e
vena femorale per avviare la circolazione extracorporea, poi si introduce un catetere occludente
nella femorale che, posizionato nell’aorta ascendente la occlude e permette l’infusione della
soluzione cardioplegica. Si pratica quindi la circolazione extracorporea dopo arresto cardiaco,
ma si esegue l’intervento con minitoracotomia.
92
Malattie del miocardio
Miocarditi
Definizione: sono processi infiammatori che coinvolgono il miocardio.
Eziologia: la causa è principalmente infettiva, per lo più virale. I patogeni più comuni sono
Coxsackie virus (ma anche altri virus, come ecovirus). Può però esserci invasione diretta anche
da parte di batteri, rickettsie e parassiti, oltre che indiretta, come ad esempio accade per azione
delle tossine prodotte da Corynebacterium dyfteriae. Le cause non infettive possono essere
metaboliche (ad esempio in corso di uremia), da malattie sistemiche infiltrative o di origine
immunologica, ma anche da ipersensibilità (soprattutto a farmaci).
Anatomia patologica: l’aspetto dipende molto dalla durata e dalla gravità della patologia.
L’interessamento miocardico può essere diffuso o focale. L’infiltrato infiammatorio interstiziale
si associa spesso a focolai di necrosi miocardica. A causa dell’interessamento spesso focale le
biopsie miocardiche possono non essere diagnostiche. In fase acuta possono essere evidenti
segni di infiltrazione leucocitaria ed edema, successivamente può aversi fibrosi che può ridurre
la compliance e la contrattilità miocardica. Il cuor può apparire normale, dilatato, ipertrofico o
anche raggrinzito. In forme particolari di miocardite il quadro può essere più specifico.
Clinica: In corso di molte patologie infiammatorie si può avere una miocardite a decorso
asintomatico. Alcune miocarditi presentano solo alterazioni transitorie e fugaci dell’ECG.
I sintomi sono piuttosto aspecifici con segni di malessere generale, febbre, astenia, mialgie,
artalgie, a volte nausea vomito o diarrea. Questo può dipendere dal tipo di infezione (digestiva o
delle vie respiratorie) che precede la miocardite (in genere di 15-20 giorni).
I segni di interessamento cardiaco sono affaticabilità, astenia, dispnea da sforzo, ma anche
possibili aritmie, cardiomegalia, soffi cardiaci e addirittura insufficienza cardiaca conclamata.
Il decorso è assai vario, la patologia può essere silente, può avere solo sintomi generali o può
essere dominata dall’insufficienza cardiaca (simile ad una miocardiopatia dilatativa) o
dall’ostacolo al riempimento diastolico (miocardiopatia restrittiva).
Meno spesso l’evoluzione è cronica (può condurre ad una miocardiopatia dilatativa
“idiopatica”), comunque la maggior parte delle volte guarisce spontaneamente in 4-6 settimane.
Ad ogni modo l’insufficienza cardiaca e le di aritmie fatali sono la principale causa di morte
(improvvisa). In genere la clinica può portarci ad eseguire un ECG che potrà essere approfondito
con un ecocardio. La conferma di alterazioni e la clinica ci aiuteranno a ricercare con esami di
laboratorio la causa scatenante.
Diagnosi:
ECG: alterazioni varie di onda T e tratto ST, aritmie e possibili anche onde Q (sono tutte
alterazioni transitorie).
Ecocardiogramma: alterazioni anche qui aspecifiche della cinetica e a volte dello spessore
(edema). Scintigrafi isotopica, biopsia endomiocardica (per i motivi suddetti) e cateterismo
cardiaco sono raramente diagnostici perché poco sensibili.
Esami di laboratorio: frequente l’anemia, possono essere aumentati enzimi come la CPK, ma
soprattutto bisogna cercare anticorpi relativi alla possibile eziologia.
93
Terapia: misure causali e generali, associate se serve ad una terapia per l’insufficienza cardiaca
soprattutto con vasodilatatori e diuretici. Le aritmie, che possono essere gravi, sono trattate con
stimolazione elettrica.
Forme particolari di miocardite:
Miocardite in corso di difterite: causata dalla tossina prodotta da Corynebacterium dyfteriae
che può causare insufficienza cardiaca e anche blocco AV (che una volta rappresentava la prima
causa di morte in questi pazienti).
Malattia di Chagas: o tripanosomiasi. È quasi del tutto sconosciuta in Italia, perché causato da
Trypanosoma cruzi, protozoo trasmesso all’uomo tramite la cimice “del bacio” (morde, spesso
sul viso). È endemica in America latina (30% delle cardiopatie). Vi è una forma acuta che può
risolvere (se non si muore) in qualche mese, caratterizzata da insufficienza cardiaca e aritmie,
un’altra forma invece, cronica, che si presenta con 20 anni di latenza nel 30% dei soggetti colpiti
dall’acuta che causa una progressiva miocardiopatia dilatativa. Terapia con antiparassitari. Vi
sono anche miocarditi in corso di toxoplasmosi e in corso di infezioni parassitarie come la
trichinosi e l’echinococcosi. Vi sono anche miocardiopatie da ipersensibilità (per lo più a farmaci
come antibiotici e antracicline contro alcune neoplasie).
Miocardite a cellule giganti: caratterizzata da un infiltrato infiammatorio nel miocardio con
cellule giganti multinucleate. Comporta aritmie e insufficienza congestizia, spesso rapidamente
fatali.
Cardiomiopatie
Definizione: sono processi morbosi che colpiscono elettivamente il muscolo cardiaco e non sono
la conseguenze di altre malattie cardiovascolari. Provocano generalmente alterazioni dello
spessore e delle dimensioni delle camere cardiache con eventuali disfunzioni meccaniche ed
elettriche (e alterazione della contrattilità o della distensibilità).
Si distinguono in primitive in cui non si distingue un agente eziologico e la malattia non fa parte
di un processo a carattere sistemico e in secondarie in cui la causa è nota o la malattia fa parte
di un processo sistemico.
Un’ulteriore distinzione, fisiopatologica, comprende tre forme: cardiomiopatia dilatativa (90%
dei casi), ipertrofica e restrittiva (meno frequente).
Cardiomiopatia dilatativa
Definizione: CMPD, è una forma di cardiomiopatia caratterizzata da una progressiva dilatazione
cardiaca (in particolare ventricolo sinistro, ma anche tutte le camere) e da una disfunzione della
contrattilità (disfunzione sistolica) con frazione di eiezione sinistra che passa dal normale 60% a
<40%. È CMPD quando c’è assenza di altre cardiopatie oppure (secondo l’OMS) le alterazioni
delle condizioni di carico, a danno ischemico, o delle coronarie, non sono tali da spiegarne la
presenza.
Eziologia: nella maggior parte dei casi la causa precisa è ignota e pertanto si definisce CMPD
idiopatica. Sono stati riscontrati diversi possibili fattori eziologici:
Fattori genetici: nel 20-50% dei casi vi è una certa familiarità, con la presenza di anomalie
94
genetiche soprattutto a carico di proteine citoscheletriche dei miociti. La trasmissione appare
essere varia, anche se spesso autosomica dominante e più raramente legata all’X o ancora di
altro genere. A volte c’è mutazione della distrofina (forme X-linked), che è mutata anche in
miopatie scheletriche genetiche (come la distrofia di Duchenne) nelle quali ci può essere franca
CMPD.
Miocarditi: grazie a biopsie endomiocardiche sequenziali si è visto che a volte (circa 15% delle
miocarditi) ci può essere evoluzione dalla miocardite in cardiomiopatia dilatativa. In questi casi
sono stati spesso ritrovati frammenti di genoma virale (Coxsackie virus) nelle cellule
miocardiche, facendo immaginare come in alcuni casi di miocardite, magari solo silente, ci possa
essere un’attivazione del sistema immunitario (soprattutto se il virus rimane nei miociti senza
compiere il ciclo litico) a seguito dell’infezione virale, in soggetti predisposti.
Alcool: l’alcool e i suoi metaboliti (principalmente l’acetaldeide), hanno un effetto tossico sul
miocardio. Mentre un consumo moderato pare possa essere addirittura protettivo, un abuso
causa certamente danno miocardico, ma in due modi: uno diretto, l’altro indiretto attraverso la
carenza di tiamina che può causare il beri-beri (fra l’altro causa di scompenso ad alta gittata). La
CMPD alcolica è indistinguibile da quella idiopatica, ma a volte (non sempre, e forse in questi
casi l’associazione è casuale o l’alcool è solo una concausa) l’astensione dall’alcool è in grado di
far regredire la patologia.
Farmaci: antineoplastici, in particolare le antraci cline, hanno effetto tossico sul miocardio.
Parto: cardiomiopatia peripartum può insorgere nell’ultimo mese di gravidanza o nei primi mesi
dopo il parto, per cause non ben definite, forse per ipertensione, sovraccarico di volume,
carenze nutrizionali, ma più probabilmente attraverso un processo autoimmune. Nella maggior
parte dei casi regredisce dopo pochi mesi, ma a volte può causare danni persistenti (molto
evidenti soprattutto in caso di seconda gravidanza). Altre cause sono virali, malattie
immunologiche, deficit metabolici e cause nutrizionali, agenti fisici e cause endocrine.
Anatomia patologica: il cuore appare globalmente ingrandito (2 o 3 volte) con dilatazione anche
di tutte le camere. Valvole e coronarie sono solitamente normali e comunque prive di danni in
grado di spiegare la cardiomiopatia. È frequente la presenza di trombi intracavitari che possono
causare emboli. I miociti paiono spesso ipertrofici, a volte allungati e irregolari. C’è fibrosi
interstiziale ed endocardica ed anche lesioni e cicatrici forse derivate da necrosi a causa di
processi di ischemia dovuti a squilibri da ipetrofia e perfusione. La parete infatti può essere
ispessita anche se comunque non è proporzionata alla cavità (inadeguata).
Patogenesi: il primum movens è la riduzione di contrattilità del miocardio e della funzione
sistolica principalmente del ventricolo sinistro.
La dilatazione che ne consegue mantiene all’inizio normale la gittata sistolica, ma con un
conseguente aumento della pressione tele diastolica ventricolare. Il grado di ipertrofia
(inadeguata) non è sufficiente a mantenere nella norma lo stress di parete, che pertanto cresce
e si determina un aumentato consumo di ossigeno (aumentato lavoro cardiaco) e
deterioramento della funzione ventricolare con riduzione della frazione di eiezione (<40%) e
della gittata cardiaca. La dilatazione ventricolare sinistra causa insufficienza mitralica.
L’aumento della pressione tele diastolica e l’insufficienza mitralica determinano un’ipertensione
dapprima atriale sinistra, poi anche del circolo polmonare (prima vene e capillari) con comparsa
di scompenso sinistro. Con l’instaurarsi dell’ipertensione arteriosa polmonare si deteriora anche
la funzione del ventricolo destro e quindi si ha insufficienza tricuspidale, la quale, con l’aumento
ora della pressione tele diastolica del ventricolo destro determina aumento della pressione
anche in atrio destro e circolo sistemico, con un quadro di scompenso globale.
95
Clinica: è molto varia, con possibili sintomi di scompenso sinistro e/o destro. Compare anche
molto dopo l’inizio della patologia. Vi sono pazienti quasi asintomatici e pazienti fino alla classe
IV NYHA. I sintomi più comuni sono dispnea, affaticamento, astenia (ridotta portata),
cardiopalmo e segni di scompenso destro come epatomegalia, turgore delle giugulari ed edemi.
All’esame obiettivo è possibile rilevare un III tono ed un soffio sistolico all’itto, segno di
insufficienza mitralica oltre ai segni dello scompenso destro o sinistro (rantoli alla base dei
polmoni) e a segni di cardiomegalia (itto più all’esterno, ingrandimento aia cardiaca).
Diagnosi: l’anamnesi ci può indicare familiarità, una recente infezione, un elevato consumo di
alcol.
Radiografia del torace: cardiomegalia e stasi polmonare possono essere evidenti.
ECG: sia normale che Holter, possono trovare fibrillazione atriale e aritmie ventricolari (anche
solo extrasistoli) con possibili segni di BBS o di ipertrofia sinistra.
Ecocardiogramma bidimensionale: è l’esame più importante, usato anche come screening sui
familiari dei portatori di CMPD. Permette di rilevare l’aumento del volume tele diastolico, la
ridotta frazione di eiezione, la ridotta contrattilità (pressione tele sistolica/volume tele sistolico),
l’ipertrofia inadeguata (massa/volume). Con il color doppler si vedono anche eventuali
insufficienze valvolari e si può stimare la pressione arteriosa polmonare.
Cateterismo cardiaco: ridimensionato dall’eco, permette di misurare con precisione la pressione
nella cavità cardiache e di valutare la portata cardiaca.
Biopsia endomiocardica: distingue la forma idiopatica da forme più specifiche, molto utile in
caso di miocardite.
Coronarografia: permette di distinguere la cardiomiopatia da sindromi coronariche (che pure
possono portare disfunzione ventricolare), ed è molto utile se il paziente ha angina.
Prognosi: l’evoluzione è verso una sempre maggiore disfunzione ventricolare (con frazione di
eiezione che può scendere anche sotto il 25%) e insufficienza cardiaca. La sopravvivenza a 5 anni
è del 25%, anche se attualmente vi è un miglioramento con i nuovi farmaci che si usano per lo
scompenso.
Terapia: i farmaci sono quelli dello scompenso. Principalmente si usano ACE-inibitori che
comportano anche un rallentamento dell’evoluzione della malattia. Se la frazione di eiezione
scende sotto il 40% si usano anche beta-bloccanti (da iniziare sempre a basse dosi), e se vi sono
segni di ritenzione idrica si fa uso di diuretici (in particolare furosemide e spironolattone che
forse rallenta la disfunzione ventricolare). La terapia definitiva resta però il trapianto, tant’è che
la CPMD è la causa di quasi la metà dei trapianti.
Cardiomiopatia aritmogena del ventricolo destro
Definizione: è una malattia ereditaria del miocardio che si associa ad aritmie che possono essere
il solo sintomo o comunque il sintomo dominante e che possono causare morte improvvisa in
pazienti giovani. Scoperta praticamente in Italia, perché ha una particolare frequenza in veneto
(studi a Padova a partire proprio da Dalla Volta) ed è infatti cause del 12% di morti improvvise
giovanili nel Veneto.
Anatomia patologica: caratteristica essenziale della patologia è la trasformazione parziale o
totale del miocardio del ventricolo destro in tessuto fibrolipomatoso con alterazioni che si
concentrano alla parte sottotricuspidale del setto interventricolare destro, all’apice del
96
ventricolo destro, alla parte di deflusso sottopolmonare. Può riguardare in qualche caso anche il
ventricolo sinistro. Si associa a cardiomegalia nel 30% dei casi, ma non si sa se questa sia una
conseguenza.
Eziologia: le noxae patogene ipotizzabili sono meccanismi che hanno agito in vita fetale. La
patologia è trasmessa per via autosomica dominante.
Patogenesi: la trasformazione del ventricolo destro genera un anello che crea un circuito di
rientro che causa diverse aritmie ventricolari, principalmente extrasistoli ventricolari, ma anche
tachicardie ventricolari che, siccome si verificano a causa di un circuito di rientro nel ventricolo
destro, hanno spesso la forma del BBS. Non si sa se la cardiomegalia sia una conseguenza a
lungo termine nei pazienti che non muoiono di aritmie.
Clinica: i pazienti sono all’inizio asintomatici e la malattia è scoperta o quando avviene l’episodio
aritmico di morte improvvisa, o durante visite casuali, o per ricerca sistematica in parenti di
individui affetti. Il malato a volte si rivolge al medico per episodi di palpitazioni e per le
extrasistoli.
Diagnosi: ECG: possono notarsi onde T invertite, anomalie del QRS, aritmie ventricolari con
aspetto del blocco di branca sinistro (Holter).
Ecocardiogramma: si possono vedere ispessimenti e diltazioni nel ventricolo destro (zona di
efflusso polmonare e zona sottotricusipdale), buona specificità e sensibilità. Il cateterismo
cardiaco può poi permettere una ventricolo grafia destra e la biopsia endomiocardica, che
risulta spesso importante. La diagnosi è però difficile, poiché la patologia è sottostimata e spesso
la prima presentazione è l’episodio di aritmia, spesso fatale.
Prognosi: difficile definirla, in pazienti in terapia c’è un maggiore riscontro di cardiomegalia in
quanto le aritmie (che in genere uccidono prima) possono essere controllate da una
Terapia: basata essenzialmente su beta-bloccanti.
Malattia di UHL
Definizione: è una miocardiopatia del ventricolo destro, con quasi totale assenza del miocardio
ventricolare e “atrializzazione del ventricolo destro” con insufficienza cardiaca destra molto
precoce (dipende tutto dall’atrio).
Cardiomiopatia ipertrofica
Definizione: CMPI, è un’ipertrofia del miocardio del ventricolo sinistro non associata a
dilatazione e non riconducibile ad altre cause cliniche o ad altre patologie. È un malattia
genetica con incidenza di 1:500 e questo fa di essa la più comune patologia cardiovascolare
causata dalla mutazione di un singolo gene.
È sempre associata ad un difetto di riempimento diastolico, dovuto alla minore compliance, e si
distingue in ostruttiva e non ostruttiva a seconda della presenza o meno di un gradiente
pressorio dinamico che impedisce il flusso ventricolare. Bisogna distinguerla dalle forme di
ipertrofia secondaria, quali comunemente si verificano nella cardiopatia ipertensiva e nella
stenosi aortica.
97
Eziologia: è trasmessa in modo autosomico dominante con penetranza ed espressività variabili,
dipendenti molto probabilmente dal tipo di mutazione. Nella maggior parte dei casi la
mutazione è missenso (ma ne esistono 400 tipi diversi) e sono concentrate principalmente sul
gene che codifica per la catena pesante della beta-miosina (quasi 50%), troponina T (alta
frequenza di aritmie), proteina C legante la miosina (con queste ultime due si arriva all’80%),
alpha-tropomiosina ed altri ancora. È considerata una malattia del sarcomero.
Anatomia patologica: è presente ipertrofia massiva, non associata a dilatazione. Il cuore appare
ipercontratto al contrario della CMPD in cui è ipocontratto, dilatato e flaccido.
Ciò che la distingue dalle altre forme di ipertrofia (secondarie) è che nella maggior parte dei casi
vi è ipertrofia settale asimmetrica in quanto l’ispessimento del setto interventricolare è
sproporzionato rispetto alle altre pareti (tranne in un 10% di casi di ipertrofia simmetrica).
A causa della protrusione del setto interventricolare nella cavità ventricolare, la cavità perde la
forma rotondeggiante-ovalare, a risulta quasi essere “a banana”. L’ipertrofia può coinvolgere
l’intero setto o essere concentrata specialmente nella regione subaortica e questa sporgenza è
quella principalmente in grado di ostacolare l’efflusso durante la sistole.
Oltre a questo è comune un movimento anteriore sistolico della valvola mitrale, SAM il quale
porta a contatto il lembo mitralico anteriore con il setto ispessito causando spesso la formazione
di placche fibrose dovute al trauma.
Dal punto di vista istologico si rileva 1) un’estesa ipertrofia dei cardiomiociti, con diametro
trasversale che supera anche in 40micrometri (contro i normali 15); 2) un disarrangiamento
(disarray) o disorganizzazione strutturale delle fibre miocardiche (interessa oltre il 5% delle fibre,
con a volte anche miociti bizzarri), soprattutto nel setto, con un grado molto maggiore rispetto
ad altre ipertrofie 3) fibrosi interstiziale.
Patogenesi: Si ha ipertrofia ventricolare sinistra con associata disfunzione diastolica per ridotta
compliance (distensibilità) e stato di ipercontrattilità con relativo aumento della frazione di
eiezione (anche 70-90%). La ridotta distensibilità è causata dall’aumentato spessore, dal
disarrangiamento delle fibre e dalla fibrosi.
Pertanto si ha un’aumentata pressione di riempimento ventricolare sinistra (ventricolo sinistro
piccolo e poco distendibile) con aumento della pressione atriale sinistra e conseguentemente
del circolo polmonare.
Oltre a questa anomalia diastolica si ha anche un’anomalia sistolica, ossia un’ostruzione
dinamica (non fissa, varia tra battito e battito, e può scomparire o accentuarsi con manovre
provocatorie) all’efflusso ventricolare. Si ha infatti un gradiente pressorio intraventricolare
sinistro tra apice e tratto di efflusso a causa del SAM e della sporgenza del setto
interventricolare (che stringe il tratto di efflusso).
Il SAM è causa ed effetto dell’ostruzione (perché la valvola è tirata in avanti per suzione dal
flusso ematico rapido attraverso un tratto di efflusso stretto, effetto Venturi).
L’ostruzione può essere aggravata da condizioni che aumentano la contrattilità (esercizio fisico,
beta-agonisti, digitale) e anche da manovre o farmaci che riducono il precarico o il post carico
(manovra di Valsalva in fase di pressione, nitrati, anche ortostatismo prolungato).
Clinica: molto vario. Il sintomo più frequente è la dispnea (causata da pressione atriale sinistra e
quindi delle vene polmonari), maggiormente sotto sforzo. Nel 40% si può avere angina da sforzo
(il muscolo ipertrofico chiede più ossigeno, che può rendere difficile la distinzione da ostruzioni
delle coronarie. Nel 20% ci sono sincopi, lipotimie a volte da sforzo o da posizione erette causate
da aritmie o da un aumento dell’ostruzione.
98
La morte improvvisa per fattori emodinamici, ischemici, ma soprattutto aritmici (fibrillazione
atriale e tachicardia e fibrillazione ventricolare) è l’evento più temuto (più comune in giovani e
in mutazioni della troponina T).
All’esame obiettivo, in modo incostante in dipendenza del gradiente variabile, è possibile
avvertire un polso celere, e all’ascoltazione un soffio sistolico al mesocardio (che aumenta ad
esempio con la manovra di valsalva che riduce il ritorno venoso e il postcarico), ma anche
sistolico puntale (insufficienza mitralica). Itto a volte doppio per la forte contrazione atriale
sinistra.
Diagnosi: oltre all’esame clinico può essere d’aiuto:
ECG: raramente è normale, presenta: onde Q profonde (pseudonecrosi); ipertrofia ventricolare
sinistra; onde T negative grandi; ma anche segni di ipertrofia atriale sinistra e alterazioni
aspecifiche del tratto ST (segni aspecifici). All’Holter è possibile notare tachicardia ventricolare
non sostenuta (ma anche sostenuta) e fibrillazione atriale.
Radiografia del torace: ingrandimento ombra cardiaca e soprattutto dell’atrio sinistro, non
sempre evidente.
Ecocardiografia: è la metodica di scelta per diagnosi e follow up. Rileva l’alterato rapporto
setto/parete e in generale l’ipertrofia asimmetrica. Nei 2/3 dei casi è visibile il SAM (tanto grave
quanto grave è l’occlusione). Nel 65% dei casi l’ipertrofia è nel setto e parete anterolaterale
(tipo III di Maron), 25% setto anteriore (tipo I), e nel resto a tutto il setto (II) e solo all’apice (IV).
Con il color-doppler è possibile identificare l’insufficienza mitralica, e l’entità della frazione di
eiezione e del gradiente. Il cateterismo cardiaco al giorno d’oggi non è più necessario
abitualmente.
Prognosi: è generalmente buona, con mortalità annua anche inferiore all’1%. Fattori prognostici
negativi sono morti improvvise in famiglia, evidenza di tachicardia ventricolare all’Holter e
sintomatologia sincopale.
Terapia: bisogna evitare attività faticose e sport agonistici. L’uso di beta-bloccanti è
storicamente il più comune (riduzione contrattilità e richiesta di ossigeno, diminuzione del
gradiente pressorio dinamico). Calcio-antagonisti e in particolare il verapamil sono stati pensati
perché in questa patologia si ha un aumento del calcio intracellulare (e quindi maggiore
contrattilità). Possono causare forme di blocco della conduzione, e bisogna comunque evitare
quelli con prevalente azione vasodilatatrice. Negli adulti con tachicardia ventricolare non
sostenuta all’Holter si impiega amiodarone. Assolutamente da evitare digitale, beta-agonisti e
diuretici. Se adulti sintomatici, con FV o TV è obbligatorio un defibrillatore impiantabile.
Terapia chirurgica: l’intervento è di miomectomia settale che ha però rischio operatorio
variabile e pertanto si indica solo a pazienti sintomatici con ostruzione del flusso importante. La
sostituzione della valvola mitrale è possibile in caso di gravi insufficienza mitrale. A volte è
indicato il trapianto. L’uso di pacemaker bilaterale può essere utile principalmente in casi di
blocco AV o blocco seno atriale. Nei bambini ci sono schemi terapeutici meno rigidi e si tende a
valutare caso per caso
Cardiomiopatia restrittiva
Definizione: è la più rara della cardiomiopatia, caratterizzata da una riduzione primitiva della
compliance ventricolare con alterato riempimento diastolico (disfunzione diastolica). La
99
funzione contrattile del ventricolo e la frazione di eiezione sono generalmente inalterate (o poco
alterate). Può essere idiopatia o associata a diverse patologie e processi patologici che
colpiscono il miocardio.
Eziologia: nell’idiopatica o primaria si riscontra una certa familiarità che fa pensare ad un’origine
genetica. A volte nella stessa famiglia è presente anche CMPI.
Anatomia patologica: i ventricoli sono in genere di dimensione normale, con pareti normali o
solo di poco ispessite, endocardio, pericardio e valvole sono normali, mentre gli atri sono
dilatati. Può essere evidente al microscopio una fibrosi interstiziale e un certo grado di
disarrangiamento dei miociti, principali cause della minore distensibilità.
Patogenesi: si ha aumentata pressione tele diastolica (aumentata pressione di rendimento) con
conseguente aumento della pressione atriale e nei distretti venosi sistemico e polmonare.
Clinica: più comune nei giovani adulti, sintomi frequenti sono astenia e dispnea da sforzo e
soprattutto segni di scompenso destro come edemi declivi, turgore delle giugulari (con segno
venoso di Kussmaul ossia pressione nelle giugulari che non diminusisce, anzi può aumentare,
con linspirazione), epatomegalia. Possono esserci soffi mitralici e tricuspidali e segni di stasi
venosa polmonare.
Diagnosi: può essere molto difficile la diagnosi differenziale con pericardite costrittiva. All’ECG ci
sono segni di ingrandimento biatriale, volte fibrillazione atriale, alla radiografia del torace
talvolta cardiomegalia , principalmente da ingrandimento biatriale che è poi il segno più
evidente anche all’ecocardiogramma (che evidenzia in genere ventricoli normali). Al cateterismo
cardiaco si rileva aumento della pressione tele diastolica ventricolare. L’esecuzione della biopsia
endomiocardica, tramite le caratteristiche istologiche, può permettere la differenziazione con la
pericardite.
Terapia: il decorso della patologia è lentamente progressivo, con un peggioramento dello
scompenso. I farmaci più utilizzati sono i diuretici, meno utili ACE-inibitori e digitale. L’unica
terapia possibile se manca il controllo con diuretici è il trapianto cardiaco.
Altre patologie con cardiomiopatia restrittiva
Il quadro clinico della cardiomiopatia restrittiva si riscontra anche sia in altre malattie primitive
del miocardio (anche una forma particolare di cardiomiopatia dilatativa senza dilatazione
ventricolare), sia in alcune patologie secondarie del miocardio:
Fibrosi endomiocardica: malattia tipica di climi tropicali (in Africa), interessa più bambini e
giovani. È caratterizzata da una progressiva fibrosi a livello del ventricolo destro e sinistro che
provoca un difetto di tipo restrittivo. Si possono sviluppare trombi murali.
Per alcuni autori costituisce la stessa patologia dell’Endocardite di Loeffler: è un processo che
comincia con una mio endocardite, probabilmente causata dalla sindrome ipereosinofilica
(forte aumento degli eosinofili che con la loro proteina basica maggiore sono tossici per il
miocardio).
Negli stadi più avanzati si ha trombosi a volte delle coronarie, a volte trombi murali che si
organizzano e vanno ad obliterare le regioni apicali e di afflusso dei ventricoli (processo in
100
comune con la endomiocardiofibrosi), a volte con interessamento della valvole atrioventricolari.
Si ha riduzione della distensibilità ventricolare con aumento delle pressioni tele diastoliche
ventiricolari, con progressivo scompenso, soprattutto destro. All’ecografia e all’angiografia sono
visibili i trombi. La terapia all’inizio può essere con corticosteroidi. Successivamente si è rivelato
utile l’intervento chirurgico con rimozione del materiale fibroso e trombotico e, se serve,
sostituzione delle valvole AV.
Amiloidosi: è una patologia sistemica con infiltrazione dell’amiloide in vari tessuti. A livello
cardiaco l’amiloide (principalmente le proteine immunoglobuliniche in corso di mieloma) infiltra
l’interstizio miocardico. Si può avere un certo gradi di cardiomegalia (visibile alla radiografia del
torace), in alcuni casi i ventricoli sono dilatati, in altri la forma della patologia è soprattutto di
tipo restrittivo con progressivo quadro di scompenso. All’ECG il QRS ha voltaggio diminuito
(possono esserci disturbi di conduzione e onde Q), all’ecocardiogramma si nota iperiflettenza del
miocardio dovuta ai depositi di amiloide. La certezza si ottiene però con biopsia endomiocardica
che (tramite colorazione rosso congo) identifica l’amiloide.
Altre forme sono l’emocromatosi, le malattie metaboliche di Fabry e Gaucher,
mucopolisaccaridosi (Hurler), glicogenosi (Pompe), e la rara fibroelastosi endocardica
(ispessimento fibroelastico a eziologia ignota).
101
Malattie del pericardio ed endocarditi
Versamento pericardico
Definizione: normalmente nel pericardio è presente un liquido limpido e paglierino di circa 3050 ml. In alcune condizioni il pericardio parietale si distende per l’accumulo di liquido. Questo
può essere sieroso con basso contenuto proteico, ossia un trasudato (idropericardio, o vero e
proprio versamento pericardico), sangue (emopericardio), un essudato, dovuto
all’infiammazione del pericardio, (pericardite vera e propria o purulenta). Il pericardio può
dilatarsi in seguito all’aumento di pressione o di volume di lunga durata, pertanto versamenti
cronici fino a 500ml non portano nessun sintomo o conseguenza evidente poiché il pericardio
può divenire ampio e permettere al cuore di dilatarsi. L’unica evidenza clinica in questo caso è
l’aumento dell’ottusità cardiaca e dell’ombra cardiaca alla radiografia. Un aumento rapido può
generare condizioni più pericolose come IM, dissezione aortica, e, per compressione delle pareti
di atri grossi vasi e anche ventricoli può ostacolare il riempimento cardiaco determinando un
tamponamento cardiaco.
Pericarditi
Pericarditi: sono malattie infiammatorie del pericardio caratterizzate da: ispessimento del
connettivo del foglietto parietale e/o viscerale; essudato infiammatorio che si versa nella cavità
pericardica.
Le pericarditi vengono distinte in tre varianti (per l’anatomia patologica le varianti sono diverse):
Pericardite acuta: scarso versamento, sono visibili segni di flogosi: dolore, sfregamenti, febbre,
ECG tipico. Si risolve in poche settimane (per definizione meno di sei).
Pericardite cronica essudativa: pericardite che dura più di sei mesi. Versamento abbondante,
che può non dare segni clinici o provocare una serie di segni e sintomi.
Pericardite cronica costrittiva: dopo un periodo infiammatorio di lunga durata, provoca
coartazione del pericardio per cui il cuore resta “strozzato” e non si dilata sufficientemente
durante la diastole. Una pericardite che dura dalle sei settimane ai sei mesi è detta pericardite
subacuta che può essere l’evoluzione di un’acuta verso una cronica o una pericardite acuta
prolungata.
Eziologia: è molto varia, tutte le pericarditi possono esordire in forma acuta ed alcune evolvere
in essudative croniche. Le forme più comuni sono:
1) Pericardite virale: in genere è una pericardite acuta che si risolve in tre-quattro settimane. I
virus coinvolti sono il virus della parotite, Cocksackievirus A e B, Echovirus e altri, spesso però
poiché la ricerca del patogeno nel pericardio non è facile, l’eziologia resta indeterminata
(pericardite virale idiopatica).
2) Pericardite batterica: in genere provocata da Streptococchi, Stafilococchi, Neisserie,
Haemophilus ed altri. La più comune è provocata dal Micobacterium tuberculosis (pericardite
tubercolare) e in genere si presenta in forma cronica essudativa che evolve in costrittiva. Da
tenere presente in soggetti esposti o comunque positivi al test alla tubercolina. L’esame del
liquido pericardico è spesso negativo (anche perché micobacterium è di difficile coltura, anche 1
mese). In genere non consegue ad una tubercolosi, ma è associata alla diffusione per via ematica
del micobatterio da un focolaio primario infetto (ad esempio un linfonodo mediastinico).
102
Vi sono anche pericarditi da funghi o da altri parassiti (Candida, blastomiceti, aspergilli).
3) Pericardite uremica: l’uremia può provocare direttamente versamento pericardico, infatti
spesso nella fase finale dell’insufficienza renale cronica si ha pericardite (complicanza frequente
anche negli emodializzati). La patogenesi è ignota, ma si pensa che l’acido urico possa
comportare flogosi (in generale è noto che è in grado di attivare l’infiammazione) a livello del
pericardio.
4) Pericardite da mixedema: il mixedema nel 30% dei casi è associato a pericardite. La
patogenesi è anche qui ignota, ma è noto che il liquido pericardico risulta ricco di colesterolo e
si ipotizza che questo (che nel mixedema può essere liberato dalle cellule danneggiate) possa
scatenare una risposta infiammatoria che può evolvere in pericardite costrittiva. Infatti anche in
caso di ipercolesterolemia può verificarsi pericardite.
5) Pericarditi neoplastiche: sono causate o da rari tumori primitivi del pericardio (mesoteliomi,
linfangiomi ed emangiomi) oppure più probabilmente un versamento pericardico può formarsi a
causa di metastasi di un tumore che interessa il pericardio per continuità (o per via linfatica)
come il carcinoma polmonare, il carcinoma della mammella, il linfoma o per via ematogena
come il melanoma. In questi casi in genere si ha un emopericardio (pericardite emorragica) e si
può avere tamponamento cardiaco.
6) Pericarditi da traumi: sono in genere forme di pericardite emorragica con emopericardio
dovuti a traumi con ferite penetranti o toracici. Un trauma violento può causare tamponamento
cardiaco rapido e fatale oppure con il tempo una pericardite costrittiva.
7) Pericarditi da farmaci: di tipo essudativo provocata da procainamide (antiaritmico) o
idralazina (vasodilatatore).
8) Malattie reumatiche: le collagenopatie possono causare pericarditi, soprattutto LES e AR
(prevalenza di pericardite tra25-50%, in genere essudativa).
9) Pericardite epistenocardica: successiva ad un episodio di infarto miocardico complicatosi, in
genere dovuta al rilascio di enzimi proteolitici in seguito alla necrosi miocardica che agiscono sul
pericardio. Può comparire dopo 2-4 giorni (pericardite precoce) oppure può presentarsi dopo
qualche settimana (sindrome di Dressler, associata a febbre, leucocitosi e dolore).
Pericarditi acute
Definizione: tipicamente insorgono per cause virali, quadro clinico uniforme.
Clinica: Dolore: sintomo caratteristico, ma molto variabile. In genere è improvviso e trafittivo
con sede retro sternale, ma può irradiarsi a collo e spalla sinistra, persino al braccio sinistro
(simulando un infarto). Una differenza rispetto al dolore da IM è che il dolore pericardico si
accentua con il respiro, la rotazione del tronco, deglutizione e tosse (aumentata pressione). A
volte è localizzato all’epicardio perché solo la parte inferiore del pericardio riceve innervazione
dolorifica (dal frenico). Il dolore è accentuato nella tosse e nella respirazione profonda. S
intomi di accompagnamento: soprattutto se è infettiva si ha febbre, mialgia diffusa, malessere,
astenia, dispnea (tachipnea).
Esame obiettivo: poiché l’infiammazione determina una formazione costante di essudato che si
versa nella cavità pericardica si crea un attrito tra i foglietti pericardici durante i movimenti del
cuore che genera un rumore ruvido (come sfregamento del cuoio) che può coincidere con
sistole, presistole o proto diastole. I rumori sono transitori, diventare più netti se si preme con lo
stetoscopio o nell’ispirazione (il diaframma in basso fa avvicinare i due foglietti). Se il
versamento diviene abbondante i foglietti viscerale e parietale si allontanano ed i rumori
103
svaniscono (si può però rischiare un tamponamento cardiaco).
Alla percussione si nota il segno di Ebstein (ingrandimento ottusità cardiaca con angolo epatocardiaco ottuso), segno di Gendrin (se l’itto è apprezzabile batte all’interno dell’ottusità). I toni
cardiaci si sentono meno. Il miocardio sottile degli atri può risentire della pressione esercitata
dal liquido e c’è un maggiore rischio di aritmie atriali.
Diagnosi: Ecocardiogramma: esame indispensabile, permette di accertare la presenza di un
versamento superiore ai 100 cc. Si vedrà uno spazio chiaro ampio dietro il ventricolo sinistro
associato ad uno spazio chiare davanti al ventricolo destro. Un versamento abbondante può
anche provocare movimenti ampi delle cavità cardiache (cuore oscillante).
Pericardocentesi: il prelievo del liquido pericardico si esegue con una puntura in sede
sottoxifoidea, dopo anestesia locale. È un esame non scevro di rischi e pertanto da praticare con
cura per il rischio di rottura di una coronaria. Va eseguito se gli altri esami non permettono una
diagnosi e se il paziente non beneficia della terapia.
Il liquido ha caratteristiche diverse a seconda dell’eziologia: liquido giallo denso: sospetta
pericardite batterica; liquido giallo chiaro: virale; emopericardio: origine neoplastica o uremica.
La successiva analisi di laboratorio, oltre a permettere una distinzione tra un trasudato ed un
essudato permette analisi fisico-chimiche più complesse nonché la ricerca (magari con coltura)
dei germi presenti. La pericardocentesi è terapeutica nel tamponamento cardiaco.
ECG: si hanno modificazioni della ripolarizzazione ventricolare, che però si distinguono in vari
stadi: 1) inizia con il dolore e si ha sopraslivellamento del tratto ST in tutte le derivazioni tranne
aVR e V1, con QRS che però resta normale (a differenza dell’IM) 2) ritorno dell’ST all’isoelettrica,
ma con appiattimento dell’onda T. 3) inversione dell’onda T 4) tracciato di nuovo normale. In
genere il sopraslivellamento deell’onda T appare appiattito o concavo (derivazioni con QRS
negativo) o coinvolge anche tutta l’onda T (derivazioni con QRS positivo).
Analisi del sangue: segni di infiammazione acuta come aumento VES, PCR e globuline e talvolta
modesto aumento degli enzimi cardiaci (nell’IM è molto più netto).
Prognosi: in genere buona se la patologia è infettiva (virale o batterica), anche se tende a
recidivare anche molte volte prima di risolversi completamente. Negli altri casi la gravità
dipende dalla patologia di base. Nei pazienti con insufficienza renale cronica è un segno di grave
peggioramento (un tempo precedeva la morte di circa tre settimane).
Terapia: nelle forme infettive è indicata, con FANS preferiti ai corticosteroidi. Nel
tamponamento cardiaco è d’obbligo l’intervento.
Tamponamento cardiaco
Definizione: è una delle complicanze più pericolose del versamento pericardico e della
pericardite. È una riduzione del riempimento diastolico del cuore provocata da un aumento
eccessivo della pressione intrapericardica (dovuto all’aumento di liquido). Altre cause di
tamponamento sono traumi e interventi chirurgici, dissecazioni aortiche, etc.
La variabile determinante perché avvenga tamponamento è la compliance del pericardio, molto
di più dell’entità del versamento: se il liquido si accumula rapidamente e il pericardio non riesce
a far fronte all’accumulo anche poche centinaia di ml possono provocare tamponamento; al
contrario anche 1-2 litri di liquido possono non provocarlo se il pericardio riesce a dilatarsi
(magari perché sfiancato da una pericardite essudativa) oppure se il liquido si accumula
lentamente e dà modo al pericardio di “abituarsi”.
104
Un’altra discriminante è il volume intracardiaco: se è diminuito (ipovolemia) si può avere
tamponamento anche per pressioni intrapericardiche basse, se è aumentato (come nello
scompenso cardiaco congestizio) si può avere anche un’alta pressione intrapericardica senza che
si verifichi tamponamento (forte pressione intracardiaca contrastante).
Patogenesi: la pressione intrapericardica influenza la funzione cardiaca a due livelli: quando la
pressione intrapericardica eguaglia la pressione diastolica del ventricolo destro (prima del
sinistro ovviamente, perché più bassa), il ventricolo destro non potrà riempirsi di sangue perché
compresso; questo effetto si trasmette al circolo venoso periferico (tamponamento) e si
determinerà stasi venosa.
Si potrà notare aumentata PVC e turgore delle giugulari, inoltre il polso giugulare diventa più
turgido con l’inspirazione (segno venoso di Kussmaul) Quando, nei casi avanzati, la pressione
intrapericardica raggiunge valori più alti influenzerà anche la funzione sistolica dei ventricoli
(pressione tale da poter intaccare la pressione sistolica).
Si avrà una riduzione della gittata del ventricolo destro che comporta anche un minore afflusso
di sangue al ventricolo sinistro il quale a sua volta è anche direttamente compresso (anche se
più resistente).
Si avrà una riduzione della gittata cardiaca e della pressione arteriosa che in parte sono
compensate da un aumento della frequenza cardiaca e da un aumento delle resistenze
periferiche. Quando però la pressione intrapericardica raggiunge valori ancora più elevati questi
meccanismi di compenso non funzionano più e quindi si riducono sia la pressione arteriosa che
la frequenza cardiaca.
Clinica: Ipotensione arteriosa: tale da rendere impalpabili i polsi, cute fredda e sudata.
Tachicardia: riflessa, ma un brusco versamento come da trauma o da perforazione cardiaca, può
causare bradicardia.
Aumento della pressione venosa centrale: può superare i 15mmHg o i 20.
Polso paradosso: nei soggetti normali è comune una caduta della pressione lieve durante
l’inspirazione (5-10mmHg). Nei pazienti con tamponamento si ha una caduta abnorme della
pressione in inspirazione (più di 10mmHg): i polsi periferici possono anche sparire del tutto. Una
possibile spiegazione è che anche nel soggetto normale durante l’inspirazione si ha un
intrappolamento del cuore nel torace per depressione intratoracica, però si ha di rimando anche
un maggiore richiamo di sangue nelle cavità cardiache e pertanto l’aumento del pre-carico (con
conseguente aumento della forza di contrazione per la legge di Frank-Starling) evita il crollo
pressorio (comunque arriva un po’ meno sangue al ventricolo sinistro). Questo avviene invece
nel paziente con tamponamento perché non essendo il ventricolo destro in grado di dilatarsi
(perché compresso) non si può avere il normale aumento del pre-carico. Il polso paradosso si
riscontra anche nelle gravi patologie respiratorie ostruttive.
Diagnosi: gli esami sono quelli della pericardite, fermo restando che l’ecocardiogramma resta il
mezzo diagnostico più sicuro. Terapia: d’urgenza, pericardocentesi.
Pericardite cronica essudativa
Definizione: la causa è difficilmente virale, molto più spesso autoimmunitaria. Il volume del
versamento può anche essere notevole senza però causare tamponamento cardiaco (l’essudato
si forma lentamente e il pericardio si dilata di conseguenza). I pazienti possono essere anche
asintomatici e scoprire la malattia per caso con una radiografia (o con un esame obiettivo ben
105
fatto…eh sì magari!). All’inizio l’unico sintomo può essere un senso di pienezza e oppressione del
torace. Quando il volume raggiunge i 2-3 litri si può avere dispnea per compressione dei
polmoni. Continuano tuttavia a non esserci segni di compressione cardiaca a meno che non
coesista pregressa cardiopatia. Il liquido appare citrino, limpido, senza germi ma ricco di cellule.
La diagnosi richiede ecocardiogramma.
Pericardite cronica costrittiva
Definizione: mentre il tamponamento cardiaco è un evento acuto, in questo caso si ha una
compressione cronica del cuore. Può avere delle gravi conseguenze emodinamiche .
L’eziologia era un tempo prevalentemente tubercolare, oggi è per lo più dovuta a tumori,
collagenopatie come l’AR, e terapia radiante (anche se ogni tipo di pericardite può causarla).
È associata alla formazione di uno spesso rivestimento fibro-calcifico che trasforma il pericardio
in un involucro rigido riducendone la compliance.
Clinica: il pericardio rigido esercita un’azione costrittiva sul cuore, la quale interessa però
soprattutto la parte finale della diastole (perché a differenza del versamento pericardico, il
pericardio rigido non esercita una pressione durante tutte le fasi, ma è una resistenza invincibile
quando il cuore si espande). I sintomi sono dispnea da sforzo, astenia (per riduzione della
gittata) e talvolta dispnea parossistica notturna.
Esame obiettivo: per la congestione venosa si hanno edemi declivi, ascite (con addome
globoso), epatomegalia con fegato da stasi, mentre la parte superiore del corpo appare
ipotrofica. Ci possono essere ittero e cianosi che conferiscono un colore olivastro. Le vene del
collo sono turgide senza riduzione nell’inspirazione profonda (segno di Kussmaul).
Diagnosi: All’ascoltazione del cuore si sente uno schiocco pericardico, knock pericardico,
rumore intenso dovuto all’urto delle pareti ventricolari in diastole contro il pericardio rigido. Si
distingue dal III tono perché compare subito dopo il II. Si può anche avere uno sdoppiamento del
II tono (per anticipo della componente aortica).
Radiografia del torace: l’ombra cardiaca non è ingrandita e nel 50% dei casi mostra calcificazioni
all’interno, campi polmonari senza strie, chiari a differenza dello scompenso.
Si può notare profilo incrociato di Wenckebach (nell’inspirazione si proietta in avanti solo la
parte superiore dello sterno mentre la parte inferiore e l’epigastrio restano fissi, tipico di
aderenze o accretio pericardica)
ECG: mostra la triade: onda P mitralica, basso voltaggio delle derivazioni periferiche, ischemia
subepicardica diffusa.
Ecocardiogramma: ispessimento del pericardio e ridotta espansione diastolica.
La diagnosi in realtà presenta non pochi problemi in quanto i sintomi sono spesso sovrapponibili
a: cirrosi epatica, scompenso cardiaco destro e cardiomiopatia restrittiva.
DD con cirrosi: (pseudo cirrosi pericardica o morbo di Pick) turgore delle giugulari e segno di
Kussmaul possono escludere un problema epatico (insieme ad assenza di enzimi epatici alterati).
DD con scompenso destro: non vi è cardiomegalia, ma questo è possibile nello scompenso
destro da cuore polmonare cronico (ma in questo caso sono in genere presenti pneumopatie
croniche associate). In ogni caso l’ECG mostrerebbe ipertrofia atriale e ventricolare destra.
DD con cardiomiopatia restrittiva: è la più simile, mancano solo le calcificazioni pericardiche alla
radiografia (peraltro non costanti). In alternativa biopsia miocardica.
106
Terapia: i casi lievi devono solo essere sorvegliati, se il paziente si aggrava si può ricorre a
terapia con diuretici (per diminuire il ritorno venoso e non sforzare il cuore che comunque ha
una diastole limitata) e digitale (inotropo-positivo, per favorire una buona contrazione anche
senza il vantaggio della legge di Starling). Se la sintomatologia è importante è indicata
pericardectomia che libera il cuore dal guscio costrittivo normalizzando il riempimento. Risultati
buoni e rischio operatorio accettabile.
Endocarditi
Endocardite infettiva: EI, è un processo infettivo dovuto alla colonizzazione e all’inasione da
parte di alcuni microorganismi (per lo più batteri e poi miceti) dell’endocardio, delle valvole
soprattutto (anche protesi), e della parete arteriosa (aorta, endoaortite). Nettamente più
comuni sono le endocarditi batteriche. L’infezione causa in genere la formazione di vegetazioni
costituite da detriti trombotici e microrganismi associate a distruzione dei tessuti cardiaci
sottostanti. Possono essere distinte in endocarditi delle valvole naturali, delle protesi valvolare e
dei tossicodipendenti. Storicamente la distinzione è sempre stata in:
Endocarditi acute: causate da patogeni molto virulenti, tipo S. Aureus (10-20% delle
endocarditi, causa lesioni necrotizzanti e ulcere), in genere attaccano valvole sane e possono
causare morte in 1-2 mesi, anche poche settimane, spesso nonostante il trattamento (antibiotici
poco efficaci, meglio l’intervento).
Endocarditi subacute: causate da germi a bassa virulenza, tipo Streptococcus viridans (50-60%,
sono parte della normale flora batterica orale), in genere attaccano valvole già anatomicamente
alterate, con infezioni insidiose e a decorso lungo, ma che in genere guariscono dopo terapia
antibiotica.
[Morfologia: il segno distintivo dell’EI è la presenza di vegetazioni destruenti, voluminose e
friabili contenenti fibrina, cellule infiammatorie e microrganismi. Le valvole più coinvolte sono
l’aortica e la mitralica. Le vegetazioni possono essere singole o multiple, anche a più valvole. A
volta causano erosioni del miocardio sottostante creando una cavità ascessuale (ascesso
anulare). Emboli possono essere liberati in ogni momento dalle vegetazioni, e poiché vi sono
numerosi microrganismi si possono formare infarti settici laddove si depositano.
Nell’endocardite subacuta le vegetazioni sono meno distruttive e hanno tessuto di granulazione,
indice di riparazione, alla base. Si può avere fibrosi ed infiltrato infiammatorio cronico.]
Nel 10-15% dei casi non si scopre l’eziologia dell’endocardite (endocardite a coltura negativa).
Oggi si preferisce una classificazione eziologica in quanto lo stesso germe può causare sia
endocardite acuta che subacuta ed è associato ad un decorso ed una terapia specifica.
Endocarditi su valvole naturali: possono essere coinvolti stafilococchi, streptococchi ed
enterococchi. Vengono colpiti più i maschi di età superiore ai 50 anni (rare nei bambini). Spesso
si associano a condizioni predisponenti (lesioni endocardiche) quali cardiopatia reumatica,
prolasso mitralico, stenosi valvolare calcifica degenerativa, cardiopatie congenite.
Endocardite su protesi valvolari: le protesi valvolari predispongono all’endocardite (come anche
le suture vascolari o i fili del pacemaker). Spesso è dovuta a stafilococchi coagulasi negativi (tipo
S.Epidermidis). Sono per lo più interessate le valvole aortiche. Può esordire precocemente
(entro 2 mesi dall’intervento) oppure tardivamente (dopo i 2 mesi). Sia stafilococchi che
streptococchi (più nella tardiva).
Endocardite nei tossicodipendenti: colpisce nel 50% dei casi la tricuspide, poi aortica e mitrale.
Il principale patogeno è S.Aureus e la sorgente principale dell’infezione è la cute(aghi infetti).
107
Patogenesi: la via di ingresso dei patogeni può essere: cavo orale (specie dopo interventi
chirurgici o odontoiatrici), lesioni cutanee contaminate, apparato uro-genitale, intestino, etc. I
germi entrano nel sistema vascolare spesso a seguito di interventi chirurgici condotti con poca
attenzione alla sterilità e arrivano all’endocardio. Spesso in germi si impiantano in aree in cui è
presente un’elevata pressione (cuore sinistro) a valle di valvole attraverso cui c’è un forte
gradiente pressorio (situazione comune nelle cardiopatie congenite). Nei tossicodipendenti
finiscono spesso nel cuore destro a causa delle continue iniezioni endovenose. Si possono avere:
vegetazioni estese che possono occludere l’orifizio valvolare oppure si possono determinare, per
risposta immunitaria, delle cicatrici con conseguente stenosi o insufficienza valvolare (anche
cavità ascessuali). Si possono avere anomalie della conduzione e emboli settici (coronarie, reni,
cervello).
Clinica: la febbre è il segno più costante di endocardite. Nell’endocardite acuta esordisce in
maniera improvvisa e violenta, con febbre alta, brividi spossatezza e debolezza. Nell’endocardite
subacuta la febbre può essere lieve o assente (specie negli anziani) e gli unici sintomi possono
essere astenia, calo ponderale e sintomi simil-influenzali. Senza trattamento le embolie sono
frequenti. Nel 90% dei casi sono presenti soffi, spesso sono presenti altralgie e mialgie,
splenomegalia e petecchie (forse da microtromboembolie) soprattutto nel cavo orale e
congiuntiva. Si possono riscontrare macchie di Roth (emorragie ovali retiniche e con un chiaro
centro pallido), noduli di Osler (noduli sottocutanei sui polpastrelli delle dita delle mani o pianta
dei piedi che possono persistere per giorni), lesioni di Janeway (lesioni dure ed eritematose su
palmo della mano e pianta del piede). Le complicanze delle endocarditi compaiono a poche
settimane dall’esordio e sono: a livello cardiaco (insufficienza e stenosi valvolare, pericardite
suppurativa), a livello extracardiaco (emboli polmonari se l’infezione è a destra e cerebrali o
renali se è a sinistra). Queste manifestazioni sono meno comuni con il trattamento.
Diagnosi: Criteri di Duke: Patologici: esame colturale o istologico che evidenzia microrganismi in
una vegetazione o embolo o ascesso, conferma istologica su una vegetazione o ascesso.
Clinici: Maggiori: emocoltura positiva per microrganismi tipici o inusuali, identificazione di
massa o ascesso correlati ad una valvola con l’ecocardio, insufficienza valvolare appena insorta.
Minori: lesioni cardiache predisponenti o uso di farmaci per ev, febbre, lesioni vascolari
emorragie e petecchie (lesioni di Janeway), moduli do Osler e macchie di Roth, coltura positiva,
reperti ecocardio compatibili, ma non diagnostici. Oltre ad un accurata visita, si basa
sull’emocoltura, che risulta essere positiva nel 90% dei casi (negativa in caso di germi di difficile
coltura come aspergillo, candida, haemophilus). Alle analisi risulterà un’anemia normocromica
normocitica, VES elevata, FR positivo. L’indagine più accurata resta l’ecografia trans toracica.
Terapia: con antibiotici, la guarigione richiede l’eradicazione di tutti i microrganismi dalle
vegetazioni, dunque dosi sufficientemente elevate per temi prolungati. Per streptococchi si usa:
penicillina+gentamicina per 2-4 settimane, o ceftriaxone per 4 settimane o vancomicina per 4
settimane. Per stafilococchi si usa: nafcillina o oxacillina per 4-6 settimane, cefazolina +
gentamicina per 4-6 settimane, vancomicina per 4-6 settimane. Nel caso non sia possibile una
terapia antibiotica come nell’endocardite fungina oppure se le emocolture rimangono positive si
può pensare di sostituire la valvola con una protesi. La malattia guarisce in genere 3-7 giorni
dopo l’inizio della terapia. La profilassi antibiotica assume un ruolo fondamentale per i pazienti
con anomalie cardiache, protesi valvolari, interventi chirurgici o dentari (soprattutto se
determinano significativi traumi o sanguinamento).
108
Circolazione extracorporea, CEC
Tecnica di assistenza cardiocircolatoria che permette di vicariare le funzioni di cuore e polmoni
in interventi che richiedono l’arresto dell’attività cardiaca. Prima bisogna somministrare eparina
con 300 UI/kg per evitare la coagulazione del sangue e monitorare sempre pT e piastrine (rischio
emorragie). Il sangue viene raccolto da una cannula nell’atrio destro o da due nelle cave, passa
poi all’ossigenatore che lo ossigena e depura da scorie e anidride carbonica, poi viene ponmpato
attraverso cannulazione all’aorta ascendente (cannulazione centrale) o arteria e vena femorale
(periferica). Nel frattempo una centralina prevede a riscaldarlo o raffreddarlo secondo
necessità. Gli aspiratori aspirano il sangue dal campo operatorio e sono collegati a delle pompe
roller che spingono il sangue in un filtro (cardiotomo) e poi va all’ossigenatore (anch’esso con
una pompa roller). Prima dell’intervento il circuto estracorporeo deve però essere riempito di
una soluzione priming (un tempo era sangue fresco, ora si usano soluzioni isotoniche che
possono sovraccaricare di liquidi e dare aumento di peso, reversibile comunque con diuretici)
per migliorare la circolazione ed evitare l’immissione di aria in circolo.
Ossigenatori: 3 tipi: A bolle: scambio gassoso ottenuto facendo gorgoliare ossigeno in una
camera di ossigenazione, il sangue ossigenato ricade attraverso un sistema antischiuma per
esere pompato privo di bolle. A membrana: scambio attraverso una membrana
sempipermeabile che separa sangue e gas (azzerando il rischio di embolia e di emolisi, spesso
usati per i periodi lunghi). Ossigenatore a fibre cave: fasci di fibre cave permeabili ai gas che vi
scorrono dentro, il sangue intorno. Privo di bolle, stessi vantaggi.
Pompe: Pompa roller: due rulli schiacciano un tubo su di un supporto, alternandosi nella
compressione (per non dare un flusso retrogrado e discontinuo). Pompe centrifughe: un rotore
pompa il sangue contro la parete di un contenitore che lo raccoglie in un foro di uscita. Il
vantaggio sono i minori traumatismi.
Ipotermia: Nei lunghi interventi chirurgici si provoca l’ipotermia perchè comporta una
diminuzione del consumo di ossigeno (così si può ridurre il flusso ematico in circolazione
extracorporea); ritarda il riscaldamento del cuore da parte del mediastino; aumenta la quantità
di ossigeno disciolto e l’affinità dell’emoglobina per esso (con relativo minor volume di
ossigeno). Può però provocare aumento della viscosità, vasocostrizione, aritmie, ridotta
secrezione di insulina. Si devono monitorare il parametri vitali.
Può essere: Lieve: 32-37; Moderata: 26-32; Profonda: 18-25 (interventi che compromettono la
vascolarizzazione cerebrale o prevedono arresto cardiaco).
Il raffreddamento si pratica con borse e materassini termici ( di superficie) o con la macchina
cuore-polmone (con uno scambiatore di calore).
Viscosità: influenza le resistenze periferiche. All’inizio a causa dell’infusione del priming, per
emodiluizione, è minore, poi con l’abbassamento della temperatura aumenta.
Pressione oncotica: si abbassa con l’emodiluizione. Una complicanza può essere l’edema
polmonare, e, se si ha forte alterazione del microcircolo, si può avere sindrome da distress
respiratorio (ARDS, anche fatale).
Coagulazione: Si altera con la CEC e torna normale solo dopo 4-5 giorni. Si dà infatti una dose
più bassa di solfato di protamina per non neutralizzare del tutto l’eparina. Il rischio di trombi è
109
dovuto all’aumento del fibrinopeptide A, della betatromboglobulina, e diminuzione di
antitrombina III e del numero di piastrine.
Monitoraggio del paziente: fondamentale prima e dopo l’intervento chirurgico. Si valutano:
1) Pressione arteriosa, PA: tramite incannulamento dell’arteria radiale (accessibile e a basso
rischio di trombosi) o dell’arteria femorale (non risente della vasocostrizione periferica). Si
valuta sistolica, diastolica, pressione media e curva di pressione. Nei bambini si fa isolamento
chirurgico (difficile incannulare).
2) ECG
3) Pressione venosa centrale, PVC: uno o più cateteri in atrio destro tamite la giugulare.
4) Diuresi: durante il CEC si controlla il bilancio di entrata (liquidi somministrati per os e ev) e
uscita (diuresi, drenaggi, perspiratio) di liquidi. In genere si ha un sovraccarico di 4-7kg che
favoriscono la funzione cardiaca e evitano l’abbassamento di qualche elettrolita , e vengono
eliminati nei successivi tre giorni in modo non forzato. Durante l’intervento, tramite catetere
vescicale si raccolgono normalmente 50-100ml di urina. Riduzione di tale valore può indicare
ipovolemia o insufficienza renale.
5) Gittata cardiaca e resistenze periferiche: la gittata si misura con il metodo della diluizione
tramite catetere di Swan-Ganz (dalla succlavia o giugulare viene fatto penetrare lentamente fino
alle cavità destre del cuore o areteria polmonare, poi ad un ramo arterioso periferico per
misurare pressione capillare polmonere, volume telediastolico, gittata). Le resistenze vascolari
sistemiche (RVS) e polmonari (RVP) si calcolano tramite le formule RVS=(PAP-PAS)*80/GC. [con
PAP pressione arteria polmonare e PAS pressione atriale sinistra] RVP=(PA-PVC)*80/GC.
6) Funzione polmonare: bisogna ripetere regolarmente l’emogasanalisi. Anche radiografia del
torace per visualizzare complicanze respiratorie e cardiache.
7) Drenaggi mediastinici e pleurici: alla fine dell’intervento sono posti due tubi di drenaggio per
lavare dalle perdite ematiche verificatesi (nella sostituzione valvolare aortica c’è abbondante
sanguinamento).
8) Parametri ematochimici: analisi di laboratorio sono: azotemia e cretinemica, elettroliti,
ematocrito, glicemia, emogasanalisi, enzimi sierici, parametri di coagulazione.
110
Indagini strumentali cardiache
Radiologia: Esame radioscopico: immagini ingrandite rispetto alla realtà perché le radiazioni
sono a proiezione conica e a distanza ravvicinata. Esame radiografico: immagini a grandezza
naturale. È possibile uno studio dell’ombra cardiaca e delle sue anomalie. Si fa anche uno studio
volumetrico del cuore (vari diametri). Eventuali modificazioni riguardano sporgenze e riduzioni
dei grossi vasi (tipo arco aortico), pulsazioni, dilatazione delle camere cardiache. È possibile poi
notare ingrandimenti dell’ombra cardiaca (cardiomegalia, versamento pericardico), diminuzione
di volume del cuore (micro cardia), alterazione dei contorni cardiaci (accretio pericardica), etc. Si
possono poi notare eventuali calcificazioni (aortiche, cardiache, valvolari), modificazione della
trama nei campi polmonari (iper e ipoafflusso, segni di edema, strie di Kerley, etc.).
ECG: esame fondamentale, basato sulla registrazione dell’attività elettrica cardiaca.
Normale: Normalmente è registrato su carta millimetrata, con velocità della carta=25mm/s. Le
divisioni più piccole corrispondono ad 1mm e sono percorse in un tempo di 0,04 s (40ms), le
linee più marcate ne comprendono 5 e sono quindi da 0,2s. Un secondo sono 5 quadrati grandi.
In genere nel valutare l’ampiezza si considera che 10mm (2 quadrati grandi) sono 1mV.
Gli intervalli principali tra le onde sono RR, PR, QRS, QT. Il punto di ricongiungimento tra QRS e
ST è il punto J, inizio della ripolarizzazione ventricolare.
La frequenza può essere calcolata dividendo 300 per il numero di quadrati grandi o 1500 per il
numero di quadrati piccoli. Un modo più rapido è mettere sui primi quadrati grandi i numeri
300-150-100-75-60-50.
Intervallo PR=120-200ms; tempo tra depolarizzazione atriale e ventricolare, comprende tutta
l’onda P e arriva all’inizio della R.
QRS: 100-110ms o meno, durata della depolarizzazione ventricolare.
QT: durata di depolarizzazione e ripolarizzazione ventricolare, comprende tutta l’onda Q e tutta
l’onda T. Dovrebbe essere meno della metà di RR, ed è inversamente proporzionale alla
frequenza. Nella sua valutazione deve essere corretto per la frequenza (QTc) tramite la formula
QT/radice quadrata di R-R. QTc=440ms o meglio 430 ms per l’uomo e 450 per la donna. Nel QRS
la prima deflessione negativa è l’onda Q, la prima positiva è l’onda R, la negativa che segue la R
è la S. Se vi sono successive onde positive o negative sono dette R’ e S’. Si usano le lettere
minuscole qrs per dire che le onde hanno scarsa ampiezza. Un complesso tutto negativo è QS.
Derivazioni: L’ECG completo è costituito di 12 derivazioni, divise in due gruppi, sei localizzate
sugli arti, che valutano potenziali trasmessi sul piano frontale, e sei toraciche o precordiali che
valutano il piano orizzontale. Le derivazioni degli arti sono 3 unipolari e 3 bipolari.
Le derivazioni unipolari vanno in direzione di un unico elettrodo come se venissero da un punto
elettricamente nullo e sono poi aumentate (“a” piccola) elettricamente del 50%. Sono aVF
(verso la gamba sinistra), aVL (braccio sinistro), aVR (braccio destro).
Le derivazioni bipolari usano due elettrodi di cui uno negativo ed uno positivo: D1 (o I)=braccio
sinistro (+) braccio destro (-); D2=gamba sinistra (+) braccio destro (-); D3= gamba sinistra (+)
braccio sinistro (-). Insieme, le sei derivazioni degli arti, sono disposte su di un piano esassiale
equidistanti tra loro e alternate tra unipolari e bipolari.
Le sei derivazioni toraciche sono registrate da sei elettrodi posti in sei punti della zona
precordiale: V1: 4° spazio sulla margino sternale dx; V2: 4° spazio sulla margino sternale sx; V3:
intermedia tra V2 e V4; V4: 5° spazio sull’emiclaveare sx (itto); V5: ascellare anteriore allo stesso
livello di V4; V6: ascellare media allo stesso livello di V4 e V5.
Regola fondamentale: In generale, se un’onda depolarizzante (positiva) va verso l’elettrodo
positivo si rileverà una deflessione positiva. Se l’orientamento medio del vettore di
111
depolarizzazione è ad angolo retto rispetto una derivazione si avrà una lieve deflessione bifasica.
Onda P: il vettore di depolarizzazione atriale è normalmente orientato verso il basso verso il
fianco sinistro. (propagazione dal NSA prima verso destra e poi verso sinistra nell’atrio). È diretto
verso il polo positivo di D2, negativo di aVR, l’onda P normale è positiva in D2 e negativa in aVR.
In V1 è bifasica (così come in V2-aVL, perché ad angolo retto). È positiva in D1, D3
(leggermente), aVF (molto vicina a D2!), V3-V6 (leggermente).
Onda T: normalmente ha la stessa direzione del complesso QRS, in quanto è una
ripolarizzazione, ma che avviene in direzione opposta.
Onda U: spesso non visibile, dovrebbe essere la ripolarizzazione dei muscoli papillari. È una lieve
deflessione con la stessa direzione dell’onda T, che risulta aumentata spesso in caso di
ipokaliemia o farmaci. Una sua inversione può essere un subdolo sintomo di ischemia.
Complesso QRS: Due fasi di depolarizzazione ventricolare: Fase 1: depolarizzazione del setto
interventricolare da sinistra verso destra e anteriormente. Fase 2: depolarizzazione
contemporanea di entrambi i ventricoli in cui quello sinistro prevale per cui il vettore è diretto
posteriormente a sinistra. V1 è anteriore destra per cui vedrà prima un’onda r (piccola e
positiva, no onda Q!) e poi un’onda S (negativa grande). V6 vedrà un’onda q e poi R. Le
derivazioni intermedie mostrano un aumento dell’ampiezza dell’onda R e diminuzione dell’onda
S, con V3 e V4 con onde di dimensioni simili e che sono definite appunto zona di transizione. La
morfologia del QRS nei sani varia a seconda dell’asse elettrico cardiaco, il quale è normalmente
compreso tra -30° e +100°. C’è pertanto deviazione assiale oltre questi valori.
Deviazione dell’asse elettrico: è uno spostamento dell’asse sul piano frontale. Per valutare
l’asse cardiaco bisogna considerare il vettore medio, molto più influenzato dal ventricolo sinistro
perché più spesso. Origina dal nodo AV, in quanto corrisponde al vettore di depolarizzazione
ventricolare. Normalmente l’asse cardiaco è tra -30 e +100 quindi per lo più nel quadrante tra 0
e +90, ossia nella maggior parte dei casi il vettore è rivolto in basso a sinistra. Nei soggetti
longitipi può esserci un cuore verticale (più a destra), nei brachitipi orizzontale (più a sinistra).
L’ipertrofia di un ventricolo fa deviare l’asse verso la zona ipertrofica, l’infarto fa deviare l’asse
nella parte opposta rispetto alla zona infartuata.
Per valutare in quale quadrante è rivolto l’asse basta guardare il QRS nelle derivazioni I e AVF,
orizzontale e verticale. La I divide il quadrante (ossia la proiezione frontale del cuore), in sopra
(negativo) e sotto (positivo), AVF divide il quadrante in destra (negativo) e sinistra (positivo).
Perciò bisogna guardare se il QRS in queste derivazioni è positivo o negativo. I+ aVF+ = in basso a
sinistra (normale). I+ aVF- = deviazione assiale sinistra (alto a sinistra) I- aVF+ = deviazione
assiale destra (basso a destra) I- aVF- = deviazione assiale sinistra estrema (alto a destra). In
realtà anche se l’asse non è tra 0 è +90 è ancora normale se deviato a sinistra fino a -30. In tal
caso basterà guardare il QRS in II: se positivo è sotto -30, se negativo è oltre, e pertanto è
deviato a sinistra.
Per determinare precisamente il grado di deviazione bisogna valutare quale derivazione è più
isoelettrica poiché la derivazione isoelettrica è esattamente quella perpendicolare al vettore.
Pertanto se, per esempio, dopo aver determinato che c’è I- aVF+, ossia deviazione assiale destra,
si nota che la derivazione più isoelettrica è la II, sapremo che la deviazione è di -150°.
Rotazione dell’asse elettrico: è uno spostamento dell’asse su un piano orizzontale. Si valuta
controllando le derivazioni toraciche. Normalmente la zona di transizione, isoelettrica e quindi
perpendicolare all’asse, corrisponde a V3 e V4. Se l’asse ruota la zona di transizione si sposta
verso destra (V1 e V2) o verso sinistra (V5 e V6).
Ipertrofia: consiste in una aumento dello spessore della parete di una cavità cardiaca. Quando
una cavità è ipertrofica, i vettori che rappresentano la depolarizzazione delle pareti (e quindi del
miocardio) della cavità hanno un modulo maggiore.
112
Ipertrofia atriale: indica più precisamente una “dilatazione” degli atri. Comporta, specialmente
in V1, la derivazione posta più in corrispondenza degli atri, un’onda P difasica (sia positiva che
negativa). Se la componente iniziale dell’onda difasica è quella più ampia si tratta di
ingrossamento atriale destro, se la componente terminale è quella più ampia si tratta
ingrossamento atriale sinistro. Un’onda P che supera i 2,5mm in tutte le derivazioni, anche non
difasica, è sospetta di ingrossamento atriale destro.
Ipertrofia ventricolare: il complesso QRS in V1 è normalmente più negativo che positivo, con
onda S più ampia dell’onda R. Nell’ipertrofia ventricolare destra si ha un’onda R ampia in V 1 con
complesso QRS che appare più positivo del normale, l’onda R si fa inoltre man mano più piccola
da V1 a V4. In caso di ipertrofia atriale destra può notarsi una deviazione assiale destra ed una
rotazione assiale destra. Nell’ipertrofia ventricolare sinistra si ha un’onda S in V 1 ancora più
sottoslivellata ed un’onda R in V5 molto ampia. Se la somma delle due è >35mm si ha ipertrofia
ventricolare sinistra. In questo caso si verificherà anche deviazione assiale sinistra e rotazione
assiale sinistra, inoltre è facile che l’onda T sia invertita e asimmetrica con discesa lenta verso il
basso e salita ripida verso l’alto. In caso di blocco di branca, i parametri usati per valutare
l’ipertrofia ventricolare non sono del totalmente affidabili.
ECG secondo Holter: registrazione continua di almeno 24 ore con elettrodi cutanei collegati ad
un apparecchio di registrazione portatile. Vi è anche l’ECG ad alta risoluzione, e ECGrafia
endocavitaria.
Test provocativi nella diagnostica della cardiopatia ischemica: sono test usati per valutare la
presenza di cardiopatia ischemica e coronaropatia. Con differenti meccanismi d’azione si induce
ischemia del miocardio (entro certi limiti) e nel frattempo si monitora la funzione cardiaca con
tecniche di imaging, ECG e valutazione dei markers.
I meccanismi utilizzati sono l’esercizio fisico (in assoluto l’ECG da sforzo è il test più utilizzato)
tramite tappeto rotante o cicloergometro, atrial pacing (aumento frequenza atriale),
dobutamina (che ad alti dosaggi ha effetto cronotropo positivo), dipiridamolo (induce
maldistribuzione di flusso), adenosina (effetto vasodilatatorio). Il monitoraggio consiste in
genere nell’ECG e nel valutare i parametri come il lavoro muscolare e quello cardiaco (ottenuto
valutando frequenza, pressione arteriosa).
È possibile fare un’ecocardiografia da stress che permette di valutare anomalie nella
contrazione. Il test deve essere eseguito solo se strettamente necessario (alcuni rischi, elevati
costi), solo dopo attenta valutazione clinica del paziente, possibilmente in sospensione dei
farmaci cardiaci, e bisogna controllare con frequenza lo stato del paziente durante l’esecuzione
del test.
Ecocardiografia: è un test semplice, non costoso, non invasivo, ripetibile e applicabile in ogni
paziente. Si basa sull’impiego di ultrasuoni (frequenza superiore a 20000Hz) e sulla capacità di
rilevare l’eco, ossia la quota di essi che viene riflessa dai tessuti. Gli ultrasuoni infatti
nell’attraversare i tessuti vengono per una parte riflessi, per un’altra proseguono e per un’altra
sono assorbiti (ossia attenuati). La profondità che possono raggiungere è inversamente
proporzionale alla frequenza che però aumenta con la definizione dell’immagine. I tessuti più
riflettenti sono quelli che hanno maggiore impedenza acustica. A produrre le onde è un
materiale piezoelettrico (modifica la propria forma se investito da una scarica elettrica). Il
cristallo piezoelettrico in genere funziona per un decimo di secondo da trasmittente e per nove
decimi da ricevente. Infatti è in grado di rilevare gli echi e di trasmetterli al tubo catodico. La
registrazione può essere in M-mode (unidimensionale) o in B-mode (bidimensionale).
113
Ecocardiografia tridimensionale: si preferisce l’analisi tramite vie trans esofagea. Permette una
visione tridimensionale e pertanto è utilissima per valutare la morfologia e le dimensioni dei
difetti settali. Esiste anche l’ecografia intravascolare se il trasduttore è montato su cateteri. Si
può fare inoltre ecocontrastografia tramite iniezione intravasale di una sostanza liquida
arricchita con pochissimo gas che produce micro bolle d’aria che fungono da mezzo di contrasto.
Ecocardiografia Doppler: utilizza l’effetto doppler per valutare il flusso all’interno dei vasi e
delle camere cardiache (molto utile ad esempio per le insufficienze valvolari). La versione
ecocolordoppler, con colori diversi (rosso e blu) per distinguere flusso anterogrado e retrogrado
è molto precisa. Èanche possibile distinguere il moto laminare dal turbolento e valutare
eventuali danni o difetti che comportano shunt. Attualmente con questa metodica è possibile
anche valutare le pressioni vigenti in vasi e cavità (una stima).
Cateterismo cardiaco: per valutare le pressioni vigenti in vasi e cavità cardiache con precisione si
utilizzano particolari cateteri. Procedura non priva di rischi (es. aritmie,) anche se la mortalità
effettiva è circa di un caso su diecimila. Un intervento di cateterismo che preveda coronarografia
può trasformarsi senza interruzione se ve ne è indicazione in un intervento coronario
percutaneo. Mentre una volta l’indicazione al cateterismo risultava automatica in tutti i pazienti
candidati all’intervento cardochirurgico, attualmente si riserva angiografia coronarica solo a
pazienti anziani e con alti fattori di rischio.
La coronarografia risulta in effetti la principale procedura eseguita tramite cateterismo cardiaco.
L’intervento viene normalmente eseguito con paziente cosciente, ma in genere sedato con
diazepam. La terapia anticoagulante deve essere sospesa 48 ore prima per evitare
sanguinamento (INR deve essere <2). Nella stragrande maggioranza dei cateterismi l’accesso è
percutaneo femorale (arteria o vena). Attraverso questo ago viene inserita una guida flessibile
che permette l’inserimento di una guaina di accesso vascolare attraverso la quale vengono fatti
avanzare i cateteri. La stessa tecnica può adattarsi ad arteria brachiale o radiale e a giugulare
interna. Il cateterismo dura in media 30 minuti e utilizza 60 ml di mezzo di contrasto, 90 se si
effettua una ventricolo grafia sinistra. Le guaine vengono rimosse a fine procedura, e l’emostasi
può essere ottenuta con compressione locale manuale sul sito di accesso vascolare.
Cuore destro: catetere introdotto nella vena periferica, e (mentre c’è controllo ecgrafico) è
guidato con attenzione fino all’atrio destro e magari arteria polmonare o ventricolo destro. Si
determinano ad esempio O2 e pressione.
Cuore sinistro: per via retrograda da un’arteria o per via percutanea si raggiungono cuore
sinistro e vasi.
Coronarografia: costituisce l’essenza del moderno cateterismo. È un metodo contrastografico
per valutare la circolazione coronarica. Il catetere è introdotto nell’arteria omerale o femorale e
serve ad iniettare il mezzo di contrasto. Si filmano immagini in diverse proiezioni per visualizzare
le coronarie in tutta la loro lunghezza. Si valutano prima la coronaria sinistra, poi la destra ed
eventuali bypass. Ogni arteria è visualizzata in varie proiezioni per poter localizzare e misurare al
meglio le lesioni. Una stima visiva permette di valutare il grado (in %) di stenosi.
L’ecografia intravascolare offre un’alternativa interessante soprattutto per poter valutara la
presenza di eventuali ostruzioni.
Misurazione della portata cardiaca: si usa la termo diluizione. Un bolo di liquido freddo viene
iniettato nell’atrio destro e poi si valuta la variazione di temperatura nell’arteria polmonare.
Esistono anche metodiche radioisotopiche incruente.
114
Angiocardiografia: studio radiologico praticato dopo iniezione di un mezzo di contrasto in una
vena del braccio (venosa) o direttamente in una cavità cardiaca (selettiva) mediante cateteri. Si
valutano anatomia della cavità cardiaca e dei grossi vasi. Riprese radiografiche in serie. Vi è una
variante radio isotopica. Una recente alternativa è la scintigrafia miocardica che fa uso di
radioisotopi che dimostrano un particolare tropismo ad esempio per tessuti ischemici o necrotici
oppure per tessuti sani (anche più utile e preciso).
Altre tecniche: è possibile l’utilizzo della tomografia ad emissione di positroni (PET, per
indagare biochimica e metabolismo), risonanza magnetica nucleare (RMN, valutazione
dell’apparato), etc.
Metodiche per lo studio del polso venoso sono il flebogramma giugulare (rivela le cinque onde
venose) e la misurazione mediante catetere in atrio destro o nella cava superiore della pressione
venosa centrale (PVC). Lo studio delle lesioni arteriose nel modo più preciso è invece possibile
tramite l’angiografia, tecnica radiologica effettuata dopo iniezione di un mezzo di contrasto.
115
Indice Analitico
Scompenso cardiaco
3
Ipertensione polmonare e cuore polmonare
11
Cardiopatie congenite
18
Elettrofisiologia cardiaca e aritmologia
25
Infezione reumatica
43
Malattie valvolari
46
Ipertensione arteriosa e rischio cardiovascolare
67
Cardiopatia ischemica
72
Malattie del miocardio
93
Malattie del pericardio ed endocarditi
102
Circolazione extracorporea
109
Indagini strumentali cardiache
111
116