Parte 3
cinetica chimica
147
La cinetica chimica studia proprietà macroscopiche di non equilibrio: come
evolvono prodotti e reagenti durante una reazione chimica. Richiede una
descrizione più difficile a causa della dipendenza temporale e spaziale
delle variabili. In questo senso, è un caso particolare della termodinamica
di non-equilibrio.
Semplificazioni:
1) mescolamento completo (trascuriamo dipendenza spaziale)
2) T e V costanti: possiamo lavorare con concentrazioni che semplificano
le leggi cinetiche!
definiamo velocità di reazione per reazione generica irreversibile:
(−vA ) A + (−vB ) B → (vC ) C + (vD ) D
1 d [J ]
v ≡
vJ dt
148
esempio:
A + B → 2C
d [ A]
d [ B]
1 d [C ]
v =
=−
=−
2 dt
dt
dt
il problema fondamentale della cinetica è descrivere la dipendenza di v
dalle proprietà macroscopiche.
ipotesi: per un fissato ambiente di reazione (T, V, solvente,…) v dipende
solo dalla concentrazioni istantanee delle specie (tipicamente solo dei
reagenti):
v = f ( [ A ], [ B ])
legge cinetica
questa assunzione significa supporre che il sistema non abbia memoria
degli stati precedenti.
149
Costanti cinetiche e Ordini di reazione
da misure sperimentali si trova che spesso:
v = k [ A]
mA
[ B]
mB
dove:
k = costante cinetica
mA, mB = ordini di reazione parziali
(mA+mB) = ordine di reazione totale
gli ordini di reazione NON coincidono con coefficienti stechiometrici.
la legge cinetica e gli ordini di reazione sono determinati
sperimentalmente ad esempio con il metodo di isolamento.
Non sempre gli ordini di reazione sono definiti:
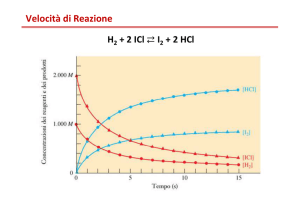
H 2 (g) + Br2 (g) → 2HBr(g)
3/2
k [ H 2 ] [ Br2 ]
v =
[ Br2 ] + k ' [ HBr ]
150
Leggi Cinetiche Integrate
Nota la legge cinetica, la dipendenza temporale delle concentrazioni è
ottenibile come soluzione di equazione differenziale. Il risultato è la legge
cinetica integrata.
Esempio: reazione del primo ordine
A→P
d [ A]
v = k [ A]
v =−
!
#"#
$ !#
dt$
#
"
##
legge cinetica
definizione di velocità
d [ A]
−
= k [ A]
dt
A]
[
ln
= −kt ⇒
[ A ]0
[ A] = [ A]0 e−kt
151
reazione del secondo ordine con un reagente:
A→P
v = k [ A]
d [ A]
2
−
= k [ A]
dt
1
1
=
+ kt ⇒
[ A ] [ A ]0
2
A ]0
[
[ A] =
1+ [ A ]0 kt
152
Dipendenza della costanti k dalla temperatura: legge di Arrhenius
legge empirica:
k = A exp {−Ea / RT }
ln k = ln A −
Ea
RT
parametri di Arrhenius:
A fattore pre-esponenziale
Ea energia di attivazione
153
energia di attivazione e stato di transizione
Energia di attivazione: energia dello stato di transizione.
Lo stato di transizione è una fase intermedia della reazione chimica che si
ha quando i reagenti si stanno per trasformare nei prodotti di reazione.
A livello molecolare, allo stato di transizione corrisponde il complesso
attivato che è un complesso altamente instabile nel quale i legami tra i
reagenti si stanno rompendo e se ne stanno formando di nuovi.
teoria delle collisioni: la reazione avviene quando le molecole dei
reagenti collidono. L’efficacia degli scontri dipende principalmente
dall’orientamento reciproco delle molecole che si accingono all’urto e dalla
quantità di energia in gioco, che deve essere sufficiente affinché nuovi
legami chimici possano formarsi. L’energia di attivazione è quindi il minimo
di energia specifico per ogni reazione.
All’aumentare di T, si accresce l’energia cinetica disponibile e di
conseguenza aumenta la anche frequenza degli urti, tra i quali quelli
efficaci. Ci sono quindi più molecole che hanno un’energia cinetica
superiore all’energia di attivazione minima, e di conseguenza la reazione
154
avverrà più velocemente.
Giustificazione delle leggi cinetiche
leggi cinetiche anche semplicissime possono in realtà nascondere
meccanismi complicati che passano per esempio, attraverso un alto
numero di intermedi. Non solo, se la reazione non è a completamento,
allora si raggiunge un equilibrio tra reagenti e prodotti e insieme ala
reazione diretta va considerata anche la reazione inversa.
A !k!
→B
'
B !k!
→A
la velocità netta di formazione di B è: v = k [ A ] − k ' [ B ]
all’equilibrio (la reazione si è fermata!): v = 0
⇒
k [ A ]eq = k ' [ B ]eq
la costante termodinamica di equilibrio:
[ B]eq k
K=
=
[ A]eq k '
supponendo che l’attività di reagenti e prodotti all’equilibrio sia
approssimabile con la loro concentrazione
155
Nesso fondamentale tra termodinamica e cinetica
[ B]eq k
K=
=
[ A]eq k '
K >> 1 ⇒ k >> k '
la reazione diretta prevale e quindi
spontaneamente i reagenti si convertono in
prodotti
K << 1 ⇒ k << k '
prevale la reazione inversa
156
reazioni consecutive (o con intermedi)
Ipotesi: ogni stadio del meccanismo dia un contributo indipendente alla
comparsa/scomparsa di una specie
A
B
A !k!
→ B !k!
→C
d [ A]
= −k A [ A ]
dt
d [ B]
= k A [ A ] − kB [ B ]
dt
d [C ]
= kB [ B ]
dt
velocità con cui A si consuma per dare B
velocità con cui B viene prodotto dal consumo di A meno
la velocità con cui B si consuma per dare C
velocità con cui C si produce da B
sistema di equazioni differenziali, non sempre di facile soluzione. In questo
caso specifico vale:
[ A] = [ A]0 e−k t
A
kA
e−kAt − e−kBt ) [ A ]0
(
kB − k A
" k A e−kBt − kB e−kAt %
' [ A ]0
[C ] = $1+
k
−
k
#
&
B
A
[ B] =
157
Derivazione della legge cinetica dal meccanismo di reazione
si applica dunque approssimazione dello stato stazionario che suppone
che la concentrazione degli intermedi resti costante e piccola durante la
reazione
Esempio:
reazione sequenza di due stadi unimolecolari, in cui B è intermedio labile
A
B
A !k!
→ B !k!
→C
e di cui sono note condizioni iniziali:
[ A ]0 ,
e
[ B ] 0 = [C ] 0 = 0
158
se B è intermedio labile, allora:
[ B] ≈ 0
e
d [ B]
≈0
dt
d [ A]
= −k A [ A ] ⇒ [ A ] = [ A ]0 exp (−k A t )
dt
d [ B]
= k A [ A ] − kB [ B ] ≈ 0 ⇒ kB [ B ] ≈ k A [ A ]
dt
d [C ]
d [ A]
= kB [ B ] ≈ k A [ A ] = −
dt
dt
159
Catalisi
Un catalizzatore è una sostanza che accelera una reazione senza subire
trasformazioni chimiche. Il catalizzatore abbassa l’energia di attivazione della
reazione attraverso l’introduzione di un cammino di reazione più efficiente.
Gli enzimi sono i catalizzatori biologici.
160
Catalisi enzimatica: meccanismo di Michaelis-Menten
valido per reazioni in ambiente acquoso. tutte le reazioni supposte del primo
ordine nei reagenti.
S=substrato(reagenti); E=enzima; P=prodotti; ES=complesso enzima+substrato
a
S + E !k!
→ ES
formazione del complesso substrato-enzima
'a
ES !k!
→E + S
decomposizione del complesso
b
ES !k!
→P
formazione dei prodotti
a basse concentrazioni di enzima si può applicare ipotesi dello stato
stazionario. si trova che la velocità di formazione del prodotto è:
v = k [ E ]0
kb [ S ]
k=
[S ] + K M
k ' a + kb
KM =
ka
costante di Michaelis-Menten
161
Catalisi enzimatica: meccanismo di Michaelis-Menten
a
1) S + E !k!
→ ES
'a
2) ES !k!
→E + S
b
3) ES !k!
→P
v = ka [ E ] [ S ]
velocità di decomposizione di ES in (2): v = −k 'a [ ES ]
velocità di formazione di ES in (1):
velocità di consumo di ES in (3)* :
v = kb [ ES ]
*uguale alla velocità di produzione di P ma con segno opposto
velocità netta di formazione dell’intermedio nell’ipotesi dello stato stazionario:
v = ka [ E ] [ S ] − k 'a [ ES ] − kb [ ES ] = 0
ka [ E ] [ S ]
[ ES ] =
k ' a + kb
162
[ E ] = [ E ]0 − [ ES ]
[S ]
dove [ E ]0 è la concentrazione totale di enzima
si può ritenere costante perché la porzione di S che va a formare
il complesso ES è trascurabile rispetto alla concentrazione totale
quindi:
ka [ E ] [ S ] ka ([ E ]0 − [ ES ]) [ S ]
=
[ ES ] =
k ' a + kb
k ' a + kb
k 'a [ ES ] + kb [ ES ] = ka [ E ]0 [ S ] − ka [ ES ] [ S ]
(k ' + k
a
b
− ka [ S ]) [ ES ] = ka [ E ]0 [ S ]
" k 'a + k
%
+ [ S ] ' [ ES ] = [ E ]0 [ S ]
$
# ka
&
[ E ]0 [ S ]
E ]0 [ S ]
[
=
[ ES ] = k ' + k
a
+ [S ] K M + [S ]
ka
163
v = kb [ ES ] =
kb [ S ]
[ E ]0 = k [ E ]0
K M + [S ]
!#
"#
$
k
kb [ S ]
[ E ]0
KM
[ S ] << K M
⇒ v ≈
[ S ] >> K M
⇒ v ≈ v MAX = kb [ E ]0
[S ] = K M
kb
v MAX
⇒ v = v1/2 = [ E ]0 =
2
2
164
v = kb [ E ] 0
0.35
Reaction rate
0.30
Vmax
0.25
½Vmax
0.20
0.15
kb
v =
[ E ]0
KM
Km
0.10
0.05
0.00
0
1000
2000
3000
4000
Substrate concentration
[S ] = K M
165