La carica elettrica è una grandezza scalare (numero reale)
che caratterizza lo stato di elettrizzazione di un corpo.
In particolare la più piccola carica elettrica
misurata è quella dell’elettrone, che viene detta
carica elementare:
e = –1,610–19 C ,
dove 1 C = 1 Coulomb è l’unità di misura della carica
elettrica.
q1q2
F
2
4 0 r
0 8,9 1012
Legge di Coulomb
2
C2
C
12
8
,
9
10
2
N m
J m
Costante
dielettrica nel
vuoto o
Permittività
elettrica nel vuoto.
In elettrostatica (cariche elettriche ferme) la forza
elettrica ed il campo elettrico sono conservativi.
q1q2
U
4 0 r
Energia Potenziale di
Coulomb
Descrive l’interazione
coulombiana, cioè
elettrostatica
In sostanza è legata al lavoro necessario per
portare una carica q1 dall’infinito a distanza r
dalla carica q2. Infatti il lavoro che il Campo
Elettrico esercita per spostare una carica positiva
q+ da un punto A verso un punto B è uguale alla
differenza di energia potenziale:
W = DU = U2 –U1
Se la forza elettrica è conservativa allora esiste una funzione
scalare U(r), detta energia potenziale elettrica, ed una funzione
scalare V(r), detta potenziale elettrico, tali che
dU
dV
F q
dr
dr
dove U(r) = q V(r).
L’unità di misura dell’energia potenziale elettrica è il Joule, mentre l’unità di
misura del potenziale elettrico è il Volt (V),
dove 1Volt = 1 Joule/Coulomb.
-Due cariche elettriche uguali, per esempio positive, si
respingono. Per avvicinarle si compie un lavoro.
-Le due cariche vicine hanno un’energia potenziale maggiore di
due cariche lontane.
-Nel caso di due cariche elettriche opposte, ossia di una carica
positiva (+) e una carica negativa (-), la situazione si rovescia.
Per separare una carica positiva da una carica negativa, si
deve compiere un lavoro, necessario per vincere la forza di
attrazione. Una volta separate, le due cariche si troveranno, a
causa della loro posizione, in uno stato con energia potenziale
più elevata.
-Se le due cariche opposte vengono a contatto, il lavoro fatto
per allontanarle verrà restituito interamente e le due cariche
avranno un’energia potenziale più bassa.
Il lavoro della forza elettrica conservativa per andare da un
punto A ad un punto B risulta allora
B
B
dV
F dr q E dr q dr
dr
A
A
A
B
LAB
q(V (rB ) V (rA )) (U (rB ) U (rA ))
Quindi il lavoro della forza elettrica è pari alla variazione di
energia potenziale elettrica cambiata di segno. Ed anche, il
lavoro del campo elettrico è pari alla variazione del potenziale
elettrico cambiata di segno.
-
+
+
+
-
+
+
Vuoto
H2O
q1q2
F
2
4 0 r
q1q2
F
2
4 0r
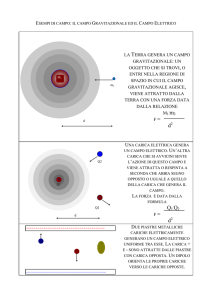
Intorno a qualsiasi oggetto, corpo o sistema carico, positivamente o
negativamente, esiste una zona in cui si possono avvertire gli
effetti ossia le interazioni dovute alla presenza del corpo carico.
Intorno a un corpo carico elettricamente, insomma, si crea una
speciale zona di influenza detta campo elettrico. Il campo di forza,
prodotto da una o più cariche elettriche (sorgente), è di tipo
vettoriale e si rappresenta con frecce, che si allontanano dalla
carica positiva e convergono verso la carica negativa.
In che modo possiamo verificare la presenza o meno del campo
elettrico? Immaginiamo di portare una seconda carica elettrica, che
chiameremo di prova, nella zona dove sospettiamo vi sia un campo
elettrico. Per carica di prova intendiamo un corpo con una piccolissima
carica elettrica positiva. Effettuato l’esperimento, possono accadere
tre cose:
◊ la carica di prova sarà respinta, se il campo è generato da una
carica positiva;
◊ la carica di prova sarà attratta, se il campo è generato da una
carica negativa;
◊ la carica di prova rimarrà ferma.
Il concetto di campo elettrico fu inventato da Michael Faraday nel XIX
secolo, per fare chiarezza fra tante teorie, più o meno corrette, sulla
natura dell’elettricità.
Si chiama campo elettrico la regione di spazio in cui una carica
esercita una azione di repulsione o di attrazione su una piccola carica
elettrica positiva di prova q0.
Ma chimicamente come funziona?
Un cristallo di un solido ionico contiene cariche positive e negative
ordinatamente alternate. Se questo solido viene riscaldato fino alla
temperatura di fusione, essendo già presenti delle cariche, allo stato
liquido si avrà un sistema conduttore e si parla di IONOFORO.
soluto ionoforo: solidi ionici (come il cloruro di sodio)
costituiti da ioni aventi carica opposta; quando un soluto
ionoforo è immerso in un solvente gli ioni che lo costituiscono
vanno in soluzione solvatandosi, cioè si ha la formazione
attorno allo ione del soluto di un guscio di solvente orientato
in base alla carica dello ione centrale; ad esempio il cloruro di
sodio (NaCl) in acqua si dissocia in ioni Na+ (circondati da ioni
OH-) e ioni Cl- (circondati da ioni H3O+);
soluto ionogeno: composti con legame covalente (come
l'acido cloridrico); quando un soluto ionogeno è immerso in un
solvente hanno luogo reazioni chimiche che producono ioni di
segno opposto. Ad esempio l'acido cloridrico (HCl) in acqua
(H2O) dà luogo a ioni H3O+ e ioni Cl-.
I punti principali della teoria di Arrhenius
sulla dissociazione elettrolitica possono essere
riassunti come segue:
1) Una soluzione elettrolitica contiene ioni liberi –
in altre parole, essi sono dissociati anche se
non passa alcuna corrente;
2) la conduzione della corrente elettrica
attraverso la soluzione dipende dal numero e
dalla velocità di migrazione degli ioni che si
trovano in essa;
3) nel caso di un elettrolita debole, il grado di
dissociazione degli ioni cresce al crescere della
diluizione;
4) a diluizione infinita, la dissociazione degli ioni in
un elettrolita debole è sempre completa;
5) in un elettrolita forte la ionizzazione è sempre
incompleta, perché gli ioni si ostacolano
reciprocamente nella migrazione, anche se
questa interferenza è minore in soluzioni
diluite.
Le interazioni intermolecolari nella solubilizzazione possono essere
così classificate:
• molecole polari:
- Ione-dipolo permanente
- dipolo-dipolo (Forze di Keesom)
• molecole non polari
- dipolo-dipolo indotto (Forze di Debye)
- dipolo istantaneo-dipolo indotto (forze di dispersione di
London)
• Legame ad idrogeno
Interazioni ione - dipolo permanente
(SOLVATAZIONE)
Si instaura tra uno ione e la parte della
molecola polare che possiede
carica opposta. Sono le interazioni
intermolecolari più forti (ma meno
di quelle interioniche ione-ione).
L’energia è valutata dalla legge di
Coulomb e varia con 1/r2
Interazioni
ione-dipolo
12 6
4
r
r
Dipendono da:
- distanza tra ione e
dipolo (r)
- carica dello ione (q)
- valore del dipolo (μ)
Potenziale di Lennard-Jones
Vi sono, tuttavia, anche forze di
natura repulsiva che derivano dalla
repulsione nucleo-nucleo e
soprattutto da quella fra le nuvole
elettroniche . Tali repulsioni
diventano molto importanti a
distanze interatomiche corte.
Per solubilizzare
un composto
ionico è
necessario un
solvente polare o
polare indotto.
Ruolo del solvente nelle sostanze ionogene:
determinare la natura delle specie cariche
ottenere la separazione degli ioni
Chimicamente parlando, particolarmente importante risulta la
valutazione della costante dielettrica che caratterizza un dato
solvente, in quanto questa grandezza permette di stabilire quale
solvente sia più opportuno per portare in soluzione un dato
soluto. Infatti per sciogliere un cristallo ionico bisogna superare
le intense interazioni elettrostatiche tra gli ioni e generalmente
più elevata è la costante dielettrica del solvente, più efficace è
questo processo.
Ad esempio, l'acqua possiede costante dielettrica relativa molto
elevata (78,5) ed è perciò in grado di solubilizzare i composti
ionici o fortemente polari.
Di contro il benzene, con valore della costante dielettrica
relativa 2,30, risulta un solvente molto utile per ottenere
soluzioni di composti organici poco polari (o apolari) e non
idrosolubili.
Nel liquido esiste un equilibrio tra molecole di H2O strutturate in
clusters stabilizzati da legami idrogeno e molecole libere di ruotare nella
fase liquida (rotameri liberi).
nH2O ⇄ (H2O)n
Struttura tetraedrica
lacunare dell’acqua
I dipoli dell’ H2O interagiscono con gli ioni, in modo tale che risultino
idratati, cioè circondati da gusci di molecole di H2O
Numero di solvatazione = Numero di idratazione =
Numero di molecole di solvente legate al singolo ione.
Le molecole di solvente nella sfera di solvatazione
hanno proprietà diverse dalle molecole di bulk.
Sono ioni strutturanti (structure makers) ioni piccoli
con carica elevata, che presentano campi elettrici intensi
in grado di indurre un ordine aggiuntivo oltre la prima
sfera di solvatazione.
Sono ioni destrutturanti (structure breakers) ioni con
carica meno intensa che presentano campi elettrici meno
intensi che non polarizzano le molecole di acqua al di
fuori della prima sfera di solvatazione.
Ioni strutturanti
(structure making)
Li+, Na+,Mg2+, Al3+, OH-, F-
Ioni destrutturanti
(structure breaking)
K+, Rb+, Cs+, NH4+, Cl-, NO3-
Solvation of ion
Coordination number: Li+: 4, K+: 6
Primary solvation shell: 4-9
6 is the most common number
Secondary solvation shell: 6-8
for Al3+ and Cr3+: 10-20
a
lim 0 1
c
a a a
1
lim 0 1
1
A B A z B z
A B
z
KD
z
AB
A B
z
K
'
D
KD
z
AB
'
A B
KD
A B
A B
aA aB
A B
a A B
FORZE ELETTROSTATICHE
DEVIAZIONE DALLA IDEALITA’
Coefficiente di attività
dipenderà dalla carica
di tutti gli ioni
Lewis
Randall
Brønsted
lg Az z I
Legge limite di Debye-Hückel
Approfondiamo il concetto di attività.
Soluzione ideale di un
A B A z elettrolita:
B z
NaCl Na Cl
0
RT ln mNa
0
0
0
RT
ln
m
RT ln mCl
m
NaCl
RT
ln
Na
Cl
0
Na
RT
ln m
Cl
Cl
Cl
0
0
RT ln m
NaCl
RTln mNaCl
Na
Na
m m
m m
RT ln m RT ln m
0
0
RT ln m m
0
0
m m m
1
Molalità ionica
media
Questo comporta…
RT ln m
0
0
Ma la maggior parte delle soluzioni elettrolitiche non si comporta idealmente:
in soluzioni di specie non cariche le interazioni dipendono da r-7,
mentre la legge di Coulomb dipende da r-2 quindi anche in soluzioni
diluite le forze elettrostatiche tra ioni sono in grado di indurre
deviazioni dalla idealità
a a a
a m
1
1
Il potenziale chimico di una soluzione elettrolitica non
ideale diventa:
0 0 RT ln a 0 0 RT ln a
-
• Ideal solution: cations and
anions do not interact.
-
+
+
-
-
+
+
ideal
-
+
+
+
+
+
Shaded region:
Solvation cage
+
+
-
• Real solution: Electrostatic
interactions.
– Ions of opposite charge are
more likely to be near each
other.
-
+
-
real
Una soluzione di elettroliti, pur essendo complessivamente un
corpo elettricamente neutro, è sede di interazioni tra cariche
elettriche localizzate sugli ioni prodotto della dissociazione
degli elettroliti stessi.
Gli ioni in soluzione sono solvatati, per cui le forze
elettrostatiche attrattive e repulsive tra ioni sono
"schermate" parzialmente dalle molecole di solvente
interposte tra ione e ione.
È ragionevole tuttavia attendersi che ogni ione
carico positivamente sia prevalentemente
circondato da ioni con carica elettrica negativa e
viceversa.
Naturalmente gli ioni possono interagire anche con
molecole di soluto non dissociate (è quanto si
verifica nel caso di soluzioni di elettroliti deboli).
Potenziale su uno ione dovuto a tutti gli altri ioni che lo circondano
Si considera il tipico insieme medio
Potenziale elettrostatico totale del sistema.
Da cui si può calcolare….
Effetti delle interazioni elettrostatiche su molte proprietà termodinamiche
Energia libera di Helmholtz
Potenziale chimico
Energia libera di
Gibbs
+
-
-
+
+
+
Atmosfera ionica o nube ionica
+
+
+
+
-
-
-
-
+
+
+
-
+
+
-
+
-
-
-
+
+
+
-
Tutti gli ioni sono
uguali
Ciascuno è ione
centrale ed è
circondato da una
nube ionica
Ma a sua volta è
anche parte della
nube ionica di un
altro ione centrale
Secondo D-H: la nube ionica distingue le soluzioni
elettrolitiche reali da quelle ideali
Approssimazioni utilizzate:
dielettrico
ad ioniche,
un sistema
1.Unsicontinuo
considerano
solo equivale
interazioni
noncon
si 0 basso:
considerano interazioni con il solvente per cui si ha un
continuo dielettrico;
qq
F
2
4 0r
qq
W
4 0r
2. si considerano
1 2
1 2 solo interazioni elettrostatiche
molto basso F o W sono molto
grandi. Quindi il
soluto si scioglie poco
avendo una grande
forza di attrazione o
una grande energia
potenziale di
attrazione.
molto grande F o W sono
molto piccoli.
Quindi esiste una
debole interazione
tra gli ioni ed il
soluto si scioglie
facilmente.
Vale sempre la condizione di elettroneutralità della
soluzione
1) Condizione a diluizione
la distribuzione
di carica attorno
allo ione centrale
è sferosimmetrica
infinita = condizione
vicina all’idealità
r
+
2) Aumentando la
concentrazione,
diminuisce la distanza
media tra gli ioni che si
attraggono.
3) Ad una certa distanza si
ione J
centrale
sentiranno sia le forze di
repulsione, che l’agitazione
termica.
4) Si avrà un bilancio di forze
unidirezionali (sia attrattive
che repulsive) in competizione
con l’agitazione termica.
La soluzione viene idealizzata come mezzo continuo e isotropo "visto"
dalla posizione di uno "ione centrale"; questo è soggetto alle forze di
un campo elettrostatico dovuto alla sua stessa carica e a quella di una
"atmosfera ionica" che lo circonda. Il potenziale chimico di questo
ione viene espresso come quello di un qualunque composto,
Il termine che contiene il coefficiente di attività viene inteso come
misura della deviazione dal comportamento ideale; questa, a sua
volta, viene attribuita alla presenza della carica elettrica sullo ione,
cioè alle interazioni elettrostatiche che sono direttamente
correlate col valore di questa e con la densità di carica elettrica
dell'atmosfera ionica circostante, cioè con la concentrazione ionica
complessiva della soluzione.
Il potenziale chimico dello ione viene scomposto in un contributo
ideale, pari al potenziale chimico in assenza di interazione, e in un
contributo elettrostatico, pari al lavoro di trasferimento di una
carica elettrica, Zi e, uguale alla carica ionica, sulla superficie di una
sfera di raggio uguale al raggio dello ione:
Zi e
i iideale RT ln i iideale N y r 0dq
0
dove y(r=0) è il potenziale elettrostatico nel punto in cui è localizzato lo ione
centrale e N è il numero di Avogadro. Tutto lo sviluppo della teoria di Debye
Hückel mira alla valutazione di y(r=0).
Il valore di questo potenziale dipende dal contributo di tutte le
cariche elettriche presenti nella soluzione, ivi compresa quella
dello ione centrale. Tuttavia, mentre la posizione di quest'ultima
è ben definita, ad esempio come origine di un sistema di assi
cartesiani o polari, non altrettanto si può dire delle cariche di
tutti gli altri ioni. Per superare questa difficoltà, si tratta la
soluzione come un mezzo continuo, le cui proprietà sono isotrope
e pari, in ogni punto, alla risultante dei contributi di tutti gli ioni
della soluzione stessa.
L’elettroneutralità vale per qualunque elemento di
volume sufficientemente grande rispetto agli ioni.
La carica di un elemento di volume dV nelle vicinanze
di uno ione centrale che si muove attraverso la
soluzione durante l’agitazione termica, è diversa da
zero per il principio di elettroneutralità.
Valgono le seguenti relazioni:
q1q2
U
4 0 r
DU q1 y zi e y j
W
y
q1
DU
q2
DU W y
q1
4 0 r
Sia y il potenziale elettrostatico alla distanza r dallo ione centrale. Uno ione «j»
che si trovi in questa posizione avrà energia potenziale elettrostatica
U j q j y zi e y j
Se consideriamo Uj come una soglia di energia che permette di distinguere la
frazione di ioni «j» posti a distanza r dallo ione centrale secondo la relazione di
Boltzmann, cioè
nj nj e
'
Uj
kBT
legge di distribuzione di Boltzmann:
nj’ è il numero di particelle per unità di volume che
risentono dello ione centrale
nj sono le particelle nel bulk per unità di volume dove
yj=0
si potrà esprimere la densità di carica elettrica a distanza r dallo ione centrale
come:
n j z j e
'
j
Densità di carica dell’elemento di
volume, con nj’ numero di ioni
nell’elemento dV a distanza r dallo ione
centrale.
n j Z j e e
j
Uj
kBT
n j Z j e e
j
Z j e
kBT
Poiché Uj<<kBT (eZj<< kBT) cioè le interazioni
elettrostatiche prevalgono sull'agitazione termica
l’espressione di ρ può essere linearizzata utilizzando
Espansione in serie di Taylor della funzione e-x:
2
23
4
3
eziy
1 xeziy
eziy 1x eziy x
x) 11
...
exp
x
...
exp(
k BT 2! k BT 3! k BT
k BT
2! 3! 4!
ez jy
exp
kBT
n
ez jy
1
kBT
z j e nj e
kBT
j 1
Z e n j y
eZ jy
Z j e n j
Z j e n j 1
k
T
kBT
j 1
j 1
B
j 1
n
n
n
2
j
2
Poichè la soluzione è
complessivamente neutra, cioè
n
Z j e nj
j
1
0
Uj
Ricapitolando:
U j z j ey j
nj nj e
'
Uj
kBT
Uj grande nj’<<nj
Uj=0 nj’=nj
n
n
j 1
j 1
Z j e nj
per
l’elettroneutralità
va a zero
Z j e nj
2
2
kBT
va a zero
solo nel bulk
per yj=0
yj
Dalla teoria di D-H la distribuzione di carica dipende solo dalla
distanza r, per cui si hanno superfici equipotenziali attorno allo
ione centrale, a cui si può applicare l’equazione di Poisson che
correla densità di carica e potenziale elettrostatico:
y
2
4
Definisce il potenziale generato da una
distribuzione di carica
2 2 2
x
y
z
2
2
2
2
2
n
Z
e
4
4
e
j
j
2
y
y
j
k T
j 1 kBT
B
n
2
2
n
nj Z j y j
j
2
1
Data la distribuzione sferosimmetrica, si preferisce
esprimere in coordinate polari:
1 2 y
y 2
r
r r r
2
è la costante
dielettrica della
soluzione
Conoscendo i valori di ψ in ogni punto, nel caso di un distribuzione di
carica sferosimmetrica, che implica un r uniforme in tutte le
direzioni, si ottiene una relazione in cui ψj è direttamente correlato
alla carica e alla distanza:
n n Z 2e 2
1 d 2 dy
j
j
yj
r
2
r dr dr
j 1 kBT
2
e
2
k
kBT
n
2
1
e
k T
B
n
2
n
Z
j jy j
j 1
nj z j
j
2
1
Equazione differenziale
risolvibile integrando su tutta
la sfera (volume nube
ionica).
1 d 2 dy
2
r
k
yj
2
r dr dr
La risoluzione è del tipo:
e
kr
kr
e
y A1
A2
r
r
Bisogna
determinare le
costanti A1 e A2
Condizioni al contorno per la valutazione delle costanti:
1 yj=0 per r=∞
2) In prima approssimazione gli ioni possono
essere considerati punti materiali con una
certa carica per cui a distanza nulla dallo ione
centrale non vi è che la carica dello ione
centrale, e quindi è facile dedurre che : per
r→0 il potenziale yj è
y r 0 y j
eZ j
4r
e kr
La prima condizione è soddisfatta solo se A2=0, altrimenti si avrebbe:
y per r
L’equazione diventa:
y A1
e
kr
r
Per valutare A1 si considera la seconda condizione espandendo in serie il
termine e-kr:
kr
1
1
2
y A1
A1 1 kr kr ...
r
r
2!
e
Dove per r→0 tutti i termini sono trascurabili rispetto all’unità, ottenendo:
y r 0
1
y i A1
r
eZ j
da cui: A
con D 4
1
D
Quindi:
eZ j e kr eZ j kr
y
e
r r
4r
Poiché è necessaria l’elettroneutralità del sistema
ione centrale-atmosfera ionica, la carica spaziale
attorno allo ione nell’intero volume di soluzione sarà
della stessa intensità, ma di segno opposto.
Il potenziale ne deve tenere conto per cui integrando r
sull’intero volume in coordinate polari si ha:
z je
z je 1
yJ
4r 4r L
z je
z je 1
yJ
4 0r 4 0 L
a
b
yJ è il potenziale nella posizione dello ione
centrale (di carica Zje) e dovuto all’atmosfera
ionica.
Equivale al potenziale generato da una carica
individuale (-Zje) posta a distanza L dallo ione
centrale.
Che significato ha L?
L è detta lunghezza di Debye = raggio della nube
Dipende dalla concentrazione
Tra la costante k e L esiste una relazione:
L
1
k
Rappresenta la
condizione di diluizione
infinita (soluzioni ideali)
a
2
e
2
k
kBT
n
nj Z j
j
2
1
Introducendo il concetto di forza ionica:
1 n
I n j Z j2
2 j 1
e
k
2I
kBT
2
2
Più grande è la I, più grande è k2 e quindi
k 1/k sarà piccolo.
Viceversa più piccola è la I, più piccolo è
k2 e quindi k 1/k sarà grande.
Come influenza la L?
Se 1/k è piccolo (alti valori di I), la distanza L
k piccolo 1/k grande 1/k>>a L dipende
dipende solo da a
dalla forza ionica yJ=f(I)
Se 1/k è grande (bassi valori di I), si ha 1/k>>a,
k grande 1/k piccolo 1/k<<a L dipende
quindi L dipenderà dalla forza ionica.
solo da a
q1q2
DU
4 0r
Uj
Zj e
2
Energia elettrostatica
2
1
4 a
k
Energia di interazione ione-nube
Più grande è i
contenuta in k più
alta è l’interazione
ione centrale-nube
Aumentando la concentrazione Uj sarà
molto grande perché la forza ionica aumenta
e quindi 1/k sarà piccolo come L.
Soluzioni ideali Du=0 ioni a distanza tale da non
interagire
ideale
j
RT ln
0
j
cj
concentrazione stechiometrica
(c=a per idealità)
c 0j
concentrazione stato standard
soluzioni concentrate Du=DG perché l’unica
interazione è di tipo elettrostatico
ideale
reale
Delettrico
j
j
0
0
0
reale
RT
ln
a
RT
ln
c
j
j
j
j
j j
j RT ln j RT ln c j
Il lavoro necessario per addizionare una quantità di carica allo
ione centrale di potenziale yj è:
DU
Z j2e 2
1
4 a
k
DG Delettrico jreale ideale
j
ideale
0
0
reale
RT
ln
RT
ln
c
j
j
j
j
j
j RT ln c j RT ln j
D j RT ln j
Z j2e 2
1
4 a
k
D j RT ln j
Z j2e 2
1
4 a
k
Si ricorda che k:
2
e
k2
kBT
k
2
e
2
n
z
2I
j j
kBT
j 1
2I
n
kBT
e
2
kBT
I
Se I è molto piccola (diluizione infinita) allora 1/k>>a per cui:
RT ln j
Z j2e 2
4
1
k
le uniche variabili sono la carica Zj e la forza ionica contenuta in k
Zj e
2
4
2
k AZ j I
2
con A costante dipendente
solo dal solvente:
e2
2
e3
A
e
4
kBT 4
2
kBT
Quindi:
RT ln j AZ j I
2
ln j A'Z j I
2
non può essere associato ad un singolo ione perché c’è sempre
associato un controione, per cui si parla di medio:
Quindi la legge limite di D-H diventa:
ln A' z z I
Sperimentalmente si ha:
Questo dipende dalle approssimazioni
fatte:
uso della permettività della soluzione
in massa
ioni considerati puntiformi
combinazione di due equazioni (eq. di
Poisson ed eq. di Boltzmann) in
approssimazione lineare
attribuzione della non idealità solo ad
interazioni coulombiane
Una legge più corretta, indicata come legge estesa è:
ln A' z z I BI
in cui si cerca di modificare l’approssimazione di non considerare
le interazioni tra gli ioni.
Fattori che influenzano la conducibilità
(1) Dipendenza
della
conducibilità
dalla
concentrazione
1.Acidi e basi
hanno
conducibilità
maggiore
2. C < 5 mol dm-3,
aumenta con C
3. Per CH3COOH la conducibilità non
dipende da C
(2) Dipendenza
dalla
temperatura
1.38
%
H2SO4
viene usato nelle
batterie
al
piombo.
2. Elettroliti caldi
vengono utilizzati
per elettrolisi e
elettrodeposizione
A
temperatura
ambiente
la
conducibilità delle soluzioni aumenta
del 2% per grado centigrado.
(T ) 25[1 ' (T 25) ' (T 25)]
T P T P
Dependenza della conducibilità molare
dalla concentrazione
m diminuisce con la
concentrazione.
A causa delle
interazioni tra gli
ioni: attrazione
interionica.
Kohlrausch ha
riportato m in
funzione di C1/2
Per un
elettrolita 1:1
C < 0.002~
0.003 mol dm-3
Relazione
Lineare tra m
e C1/2.
formula empirica di Kohlrausch
m
m A c
Estrapolando la parte lineare di m ~
C1/2 a basse concentrazioni fino a C=0,
si può ottenere m.
m è il valore limite di m a
diluizione infinita: conducibilità
molare limite