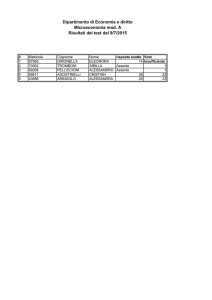
Copyright “Tutti i diritti sono riservati”
Difetti del recettore per LH (Ipoplasia delle cellule di Leydig)
Gianni Russo G, Silvia Meroni
UO Pediatria e Medicina dell’adolescenza, Università “Vita e Salute”, Ospedale San Raffaele,
Milano.
LH e hCG si legano allo stesso recettore (LH-R) situato sulla membrana cellulare delle
cellule bersaglio. Il gene codificante per il recettore è situato sul braccio corto del cromosoma 2
(2p21) ed è formato da undici esoni. Il recettore è costituito da un’ampia porzione extracellulare, responsabile del legame con l’ormone, sette domini trans-membrana e una coda Cterminale intra-cellulare. La sua azione è mediata da una proteina G e, quindi, dalle variazioni
dei livelli intra-cellulari di cAMP. Tale recettore è essenziale per una corretta crescita e
differenziazione delle cellule di Leydig fetali e per la produzione di androgeni.
Le mutazioni inattivanti del LH-R (tipicamente situate nel dominio trans-membrana)
determinano un’assente responsività delle cellule di Leydig a hCG e LH. I soggetti 46,XY con
omozigosi o doppia eterozigosi per mutazioni del LH-R (autosomica recessiva) presentano
ipoplasia/agenesia delle cellule di Leydig (evidenziabile all’esame istologico) e difetto di
virilizzazione in utero e alla pubertà (1).
La forma completa presenta le seguenti caratteristiche:
·
fenotipo femminile con assegnazione del sesso femminile;
·
assente sviluppo dei caratteri sessuali secondari alla pubertà;
·
testicoli non discesi di dimensioni leggermente inferiori rispetto alla norma, con conservazione
dei tubuli seminiferi, ma assenza di cellule di Leydig mature;
·
presenza di strutture Wolffiane ipoplasiche e assenza di strutture mülleriane;
·
ridotta/assente concentrazione di T, nonostante elevati livelli di gonadotropine (LH > FSH);
·
assente risposta al test da stimolo con hCG, in assenza di deficit nella sintesi del testosterone
(non accumulo di precursori).
Se presente una parziale responsività del LH-R, con conseguenti livelli sub-ottimali di
testosterone, il fenotipo è prevalentemente maschile con deficit di virilizzazione, quali micropene
e/o ipospadia; i testicoli possono essere criptorchidi o localizzati nello scroto. Alla pubertà si
assiste a una parziale virilizzazione con dimensioni testicolari normali o solo lievemente ridotte,
ma con evidente compromissione della crescita peniena (1).
Bibliografia
Copyright It.DSD Gruppo di Studio – “Tutti i diritti sono riservati”
www.gruppodistudio-it-dsd.org
Copyright “Tutti i diritti sono riservati”
1. Segaloff DL. Diseases associated with mutations of the human lutropin receptor. Prog Mol Biol
Transl Sci 2009, 89: 97-114.
Copyright It.DSD Gruppo di Studio – “Tutti i diritti sono riservati”
www.gruppodistudio-it-dsd.org