
UNITA’ DI RICERCA DI ANCONA
Direttore: Prof. Paolo Bruni
Studio FT-IR sull’effetto di ioni metallici bivalenti su complessi DNA-liposomi
Il lavoro condotto nel corso dell‟anno 2000 rappresenta il punto d‟arrivo sul contributo che la
spettroscopia IR-FT può fornire allo studio del tema: sono stati infatti definiti i pattern spettrali
completi di ogni singolo componente (DNA, liposoma), delle singole miscele binarie (metallo
catione/DNA, metallo catione/liposoma, DNA/liposoma) nonché dei vari complessi ternari
Me2+/DNA/liposoma. Come liposoma è stata usata la dimiristoilfosfatidilcolina (DMPC) al posto
della fosfatidilcolina (egg-yolk) allo scopo di semplificare gli spettri; gli ioni metallici studiati sono
Ca2+, Mg2+, Mn2+, Cu2+, Cu2+. Lo studio ha permesso di valutare l‟influenza sia del liposoma che
dei vari ioni metallici sulle interazioni di “base pairing” e “base stacking” del polinucleotide,
nonché sulle transizioni tra le sue forme A, B ,Z; nonché sulle modifiche indotte dai vari ioni
metallici sul complesso DNA/DMPC. I principali risultati ottenuti sono di seguito descritti.
Complessi DNA-ioni metallici.
Mentre nel semplice DNA si ha prevalenza della conformazione B, in presenza di tutti gli ioni
metallici aumentano le forme A e Z (fosfati e carbonili), ciò che contraddice quanto da altri
affermato e cioè che gli ioni metallici inibiscono la transizione verso la forma A. Tutti i metalli
aumentano il grado di polarizzazione del legame P=O.
Complessi DMPC-ioni metallici.
Si nota un aumento nell‟ordine delle catene alifatiche, in particolare con Ca2+ e Mg2+ ed una
riduzione dei legami idrogeno.
Complessi DMPC-DNA.
Si nota un aumento della conformazione B del DNA, mentre quest‟ultimo agisce sui modi
vibrazionali dei CH2 e del C=O allo stesso modo degli ioni metallici.
Complessi tripi.
Mentre l‟azione indipendente sia del DNA che degli ioni metallici favorisce una geometria
esagonale del DMPC il loro effetto congiunto non porta a nessun mutamento nell‟organizzazione
della catena idrocarbonica del liposoma. Solo il Mg2+ non annulla l‟effetto del DNA e mantiene la
geometria esagonale della DMPC. Mentre gli ioni Ca2+ ed Mg2+ non intervengono a modificare il
maggio ordine indotto dal DNA sulle catene idrocarboniche della DMPC, Co2+, Cu2+ e Mg2+, ne
riducono fortemente l‟effetto.
In generale valgono le seguenti conclusioni. L‟analisi dei modi vibrazionali dei gruppi CH,
carbonile e fosfato del DNA e del liposoma dimostra che l‟andamento spettrale dipende dal
rapporto molare tra i due componenti (complesso DNA/DMPC) e dalla natura dello ione metallico
(complessi binari DNA/Me2+ e DMPC/Me2+): gli ioni Ca2+ ed Mg2+ mostrano il maggior effetto sul
CH, gli ioni Co2+ e Cu2+ sui gruppi polari, con lo ione Mn2+ che viene a collocarsi in una posizione
intermedia. Nei tripli complessi tutti i cationi eccetto Cu2+ aumentano l‟ordine lungo le catene
lipidiche e tutti eccetto Mg2+, preservano la conformazione originale delle catene idrocarboniche
della DMPC. Questo risultato è di una certa importanza in quanto sembra confermare
un‟osservazione fatta da altri autori e non dimostrata e cioè che esista una correlazione long-range
tridimensionale nel corso della formazione dei complessi tra liposomi e DNA. Un altro importante
risultato ottenuto con la tecnica IR-FT è che, a differenza di Ca2+ e Mg2+, che non hanno
praticamente effetto, Co2+ e Cu2+ favoriscono la forma Z del DNA nel triplo complesso. Ciò
significa che questi ioni debbono essere evitati in eventuali applicazioni terapeutiche di complessi
DNA-liposomi (processi di DNA transfection).
In conclusione la tecnica IR-FT è in grado di caratterizzare in modo soddisfacente i complessi
ternari distinguendoli da quelli binari DNA-DMPC.
Pubblicazioni del gruppo
UNITÀ DI RICERCA DI BARI
Direttore: Prof. Giovanni Natile
L'attività di Ricerca dell'Unità Locale di Bari ha riguardato lo sviluppo di farmaci
antitumorali a basi di Platino "non convenzionali" in quanto caratterizzati dall'avere geometria trans
e non geometria cis come il capostipite cisplatino, cis-[PtCl2(NH2)2], il farmaco antitumorale più
venduto nei paesi avanzati. La diversa struttura geometrica comporta una diversa interazione con il
recettore (il DNA cellulare) e quindi il superamento di alcune limitazioni riscontrate nell'uso del
cisplatino quali la resistenza di alcune linee tumorali sia intrinseca che acquisita a seguito del
trattamento. L'attivazione della geometria trans é stata ottenuta mediante l'uso di leganti carrier
diversi dall'ammoniaca ed, in particolare, mediante l'uso di leganti iminoeteri di formula generale
HN = C(OR')R. Questi leganti sono caratterizzati dall'avere carattere intermedio tra amine alifatiche
ed amine aromatiche. Infatti come le ammine aromatiche hanno struttura planare e presenza di
orbitali molecolari di tipo p-greco, mentre come le ammine alifatiche recano un atomo di idrogeno
legato all'azoto donatore ed un ridotto ingombro sterico. Composti di Platino con ammine
aromatiche avevano mostrato possedere attività citotossica nei confronti di linee tumorali, in vitro,
ma non attività antitumorale in vivo. Al contrario i composti di platino con iminoeteri hanno
mostrato possedere due caratteristiche peculiari e cioé: ii) citotossicità degli isomeri a geometria
trans maggiore di quella dei corrispondenti isomeri a geometria cis, (ii) attività antitumorale in vivo
sia nei confronti di Leucemie che di tumori solidi. La sintesi di questi composti per alcolisi dei
corrispondenti composti nitrilici e il loro potenziale uso come farmaci antitumorali è stata oggetto di
brevetto esteso alle nazioni della Comunità Europea e del Nord America.
L'unità di Bari ha quindi approfondito le indagini di tipo sia chimico che farmacologico
necessarie perché uno o alcuni di questi composti possano entrare in fase clinica. In particolare dal
punto di vista chimico é stata valutata la stabilità dei composti in soluzione acquosa per ottenere
informazioni necessarie alla formulazione del composto ed alle modalità di somministrazione. E'
stata valutata la velocità di solvolisi in assenza di eccesso di ioni clururo e la reattività nei confronti
di substrati solforati, quali il glutatione, e di basi azotate del DNA.
Da un punto di vista farmacologico é stato effettuato uno screening dell'attività di campioni
rappresentativi di questa classe di composti nei confronti di una ampia gamma di linee tumorali
umane selezionate in base alla vastità della loro incidenza ed alla resistenza al trattamento col
cisplatino. Inoltre é stata studiata la distribuzione intracellulare, l'alterazione del ciclo cellulare e la
morte cellulare per apoptosi. E' Stato siglato un accordo di collaborazione con l'industria
farmaceutica Sigma-Tau, la quale valuterà la possibilità di sviluppo industriale di alcuni di questi
composti. Parallelamente é stato intrapreso un intenso studio di caratterizzazione degli addotti col
DNA, delle distorsioni conformazionali che ne conseguono e del loro presumibile effetto sulla
biologia cellulare. I risultati di questa indagine, riportati in tre lavori su riviste internazionali hanno
permesso di accertare che: i) il farmaco interagisce preferenzialmente con guanine in sequenza pyG-py; ii) gli addotti monofunzionali evolvono ad addotti bifunzionali nel caso di singolo filamento
mentre risultano stabili nel caso di doppio filamento; iii) le maggiori distorsioni della doppia elica
sono localizzate sulla coppia di base adiacente in posizione 5'. Infine lo studio NMR di un
decanucleotide 5'-d(CCTCGCTCTC) platinato alla singola guanina ed ibridizzato con il filamento
complementare 5'-d(GAGAGCGAGG) ha mostrato, contro ogni aspettativa, come l'addotto
monofunzionale sia in grado di provocare un bending della doppia elica di circa 45° confrontabile
con quello provocato dagli addotti bifunzionali del cisplatino. Inoltre il bending é diretto verso il
solco minore (come negli addotti crociati intercatena) e non verso il solco maggiore (come negli
addotti crociati intracatena).
Uno studio dettagliato (HPLC, NMR) della reazione del trans-iminoetere complesso, trans[PtCl2{HN=C(OMe)Me}2], con il dinucleotide ApG ha permesso di evidenziare per la prima volta,
dopo 30 anni di ricerca in questo settore, la possibilità anche per i composti di platino a geometria
trans di formare addotti intracatena con basi puriniche adiacenti dello stesso filamento. Tale tipo di
interazione era stata sempre ritenuta impossibile.
L‟acquisito, in collaborazione con altri gruppi di ricerca del dipartimento Farmaco-Chimico,
di uno spettropolarimetro CD-ORD della JASCO S.p.A., ha permesso lo studio delle alterazioni
conformazionali del DNA conseguente alla interazione con farmaci a base di platino. In particolare
è stata messa in evidenza una “comunicazione di seconda sfera” tra i leganti “carrier” e le basi
nucleiche coordinate al platino e tra basi nucleiche coordinate al platino in posizione cis. Queste
interazioni di “seconda sfera” determinano la stereochimica e la diversa stabilità di diversi
conformeri. Per la prima volta è stato messo in evidenza che il legame crociato del cisplatino con
due purine adiacenti dello stesso filamento (questo rappresenta da solo circa il 90% degli addotti del
cisplatino con il DNA) non è affatto statico ma dinamico e può assumere, nel tempo, diverse
conformazioni. L‟uso sapiente di “retromodels”, cioè di modelli più complessi del sistema che si
vuole studiare, ha permesso la caratterizzazione strutturale delle diverse conformazioni. Una volta
acquisite le caratteristiche spettroscopiche (NMR e CD) dei singoli conformeri, è stato possibile
calcolare il contributo delle singole conformazioni nel sistema naturale. Inoltre un'altra quota di
£.25.000.000 é stata utilizzata per il cofinanziamento di un assegno di ricerca biennale. Il tema di
ricerca di tale assegno di ricerca é esclusivamente quello indicato in questo progetto di ricerca e
permetterà al vincitore di acquisire competenze nell'ambito della sintesi dei composti di platino,
dell'uso di tecniche di biologia molecolare e di biofisica strutturale.
UNITÀ LOCALE DI BOLOGNA
Direttore: Prof. Alberto Ripamonti
Descrizione attività svolta
L‟attività di ricerca si inquadra nella tematica del Consorzio: Biomineralizzazione e
Biocristallografia.
a) Biomineralizzazione
Il progetto di ricerca è indirizzato allo studio degli aspetti chimici e strutturali dei processi di
biomineralizzazione come sistemi modello per la progettazione di materiali con particolari proprietà
tecnologiche.
In questo ambito è stata proseguita l‟indagine sull‟effetto di alcuni polielettroliti sulla stabilità
relativa di fosfati di calcio. In particolare è stato eseguito uno studio strutturale e morfologico
sull‟idrolisi dell‟ottacalcio fosfato in presenza di poliacrilato di sodio. L‟ottacalcio fosfato,
Ca8H2(PO4)6 5H2O (OCP), appare spesso come intermedio durante la precipitazione della fase
termodinamicamente più stabile, l‟idrossiapatite (HA), ed è considerato un probabile precursore
dell‟apatite che si deposita durante la biomineralizzazione dei tessuti duri dei vertebrati. La struttura
dell‟OCP è molto simile a quella dell‟HA: la cella elementare è costituita da uno strato praticamente
identico a quello della cella elementare dell‟HA, e da uno strato in cui gli ioni calcio e i gruppi
fosfato sono più distanziati a causa della presenza di molecole di acqua di idratazione. E‟ stato
suggerito che la trasformazione dell‟OCP in HA in soluzione acquosa avviene secondo un
meccanismo di idrolisi in situ e/o attraverso la dissoluzione dell‟OCP e successiva
ricristallizzazione dell‟HA.
Dal momento che la deposizione dei fosfati di calcio nei tessuti biologici avviene in un
ambiente ricco di macromolecole acide che possono influenzarne la nucleazione e la crescita,
l‟idrolisi dell‟OCP è stata studiata in presenza di un polielettrolita contenente numerosi gruppi
carbossilato quale il sodio poliacrilato (NaPA, peso molecolare 2100). Generalmente i polielettroliti
in soluzione inibiscono la cristallizzazione dei fosfati di calcio. Inoltre vengono facilmente ed
irreversibilmente adsorbiti su cristalli di HA attraverso interazioni di tipo elettrostatico e/o legame a
idrogeno. I prodotti ottenuti dopo permanenza in soluzioni a diversa concentrazione di
polielettrolita sono stati analizzati con tecniche di diffrazione di raggi X, di spettroscopia FTIR, di
microscopia elettronica a scansione e a trasmissione. Il sodio poliacrilato mostra una notevole
affinità per la struttura dell‟OCP, su cui viene adsorbito in quantità nettamente maggiore rispetto
all‟HA. L‟adsorbimento avviene in modo preferenziale sullo strato idrato della faccia (100)
dell‟OCP, dove i gruppi carbossilato possono possono interagire sia con gli ioni calcio, attraverso
interazioni di tipo elettrostatico, sia con le molecole d‟acqua attraverso legami a idrogeno. Il
polielettrolita adsorbito sui cristalli di OCP ne inibisce la trasformazione in HA, e controlla la
morfologia dei cristalli.
Nell‟ambito delle ricerche indirizzate a chiarire e dettagliare il ruolo della struttura del collageno
sulla nucleazione e crescita di fasi inorganiche sono stati analizzati tessuti calcificati di pesci ossei
in cui la deposizione della fase inorganica avviene su fibre collagene con struttura molecolare
leggermente diversa da quella tipica del collagene di tipo I presente nei vertebrati superiori. I
risultati delle indagini condotte prevalentemente con tecniche di diffrazione di raggi X ad alto e
basso angolo indicano che, sebbene il processo di calcificazione avvenga su fibrille collagene con
un impaccamento molecolare leggermente modificato, le relazioni strutturali fra cristalli apatitici e
fibrille collagene sono del tutto simili a quelle riscontrate nei tessuti calcificati dei vertebrati
superiori. In particolare, i cristalli apatitici crescono con il loro asse c preferenzialmente orientato in
direzione parallela a quella dell‟asse lungo delle fibrille di collageno, che fungono da templato per
la deposizione ordinata della fase minerale.
E‟ stato studiato il ruolo della struttura della matrice organica sulla cristallizzazione di fosfato di
calcio, utilizzando approcci di tipo biomimetico. A tal fine sono stati preparati films di gelatina,
componente che simula la macromolecola strutturale, contenenti sodio poliacrilato, quale analogo
sintetico delle macromolecole acide, che sono quindi stati sottoposti ad interazione con SBF
(Simulated Body Fluid). Le variabili prese in esame, oltre alla quantità relativa di polielettrolita
contenuto nei films di gelatina, sono il tempo di permanenza nella soluzione di SBF e
l‟organizzazione strutturale dei films di gelatina. Infatti parte dei campioni è stata sottoposta a
deformazione uniassiale prima di essere utilizzata per le prove di calcificazione, al fine di allineare
le porzioni di molecole di collageno lungo una direzione preferenziale.
I risultati ottenuti indicano che l‟orientamento della matrice organica rappresenta un fattore
determinante per la precipitazione della fase inorganica. Tuttavia, in assenza del polielettrolita, la
deposizione è solo superficiale e non interagisce con la struttura intima dei films. L‟arricchimento
della matrice con gruppi carbossilato, favorisce la crescita di cristalli apatitici all‟interno dei film
orientati di gelatina. I cristalli si depongono tra gli strati di gelatina e crescono con il loro asse
cristallografico c preferenzialmente orientato parallelamente alle porzioni di molecole di collageno,
mimando la deposizione preferenziale dei cristalli apatitici biologici rispetto all‟asse delle fibre
collagene.
Analogamente, sono stati utilizzati Xerogel di gelatina come templanti per la crescita ordinata di
cristalli di fosfato di calcio. La precipitazione è stata indotta facendo diffondere soluzioni di ioni
calcio e ioni fosfato dalle parti opposte dei film di gelatina. La morfologia e l‟orientamento dei
cristalli sulla superficie ed all‟interno dei film sono state analizzate con tecniche di microscopia
ottica ed elettronica e con diffrazione di raggi X. La fase cristallina è stata identificata come OCP. I
cristalli crescono con morfologia a piattina allungata lungo l‟asse cristallografico c, con le facce
(100) parallele agli strati di gelatina. Nei film di gelatina sottoposti ad allungamento per
deformazione uniassiale, la direzione preferenziale di allineamento dell‟asse c dei cristalli di OCP
risulta pressochè parallela alla direzione di deformazione.
Matrici collagenose contenenti poli-L-aspartato (poly-Asp) e poli-L-glutammato (poly-Glu)
sono state utilizzate anche per studiare la deposizione orientata dei polimorfi del carbonato di
calcio. I risultati ottenuti indicano che il controllo del polimorfismo e dell‟organizzazione strutturale
dei cristalli sono correlati alla struttura dei siti di nucleazione in termini di geometria,
concentrazione dei polipeptidi, della sovrasaturazione locale. Sono stati osservati effetti di
orientamento dei cristalli di calcite solo nelle matrici contenenti poly-Asp, mentre l‟orientamento
dei cristalli di aragonite e vaterite può essere controllato attraverso la deformazione dei substrati.
L‟insieme dei risultati ottenuti suggerisce cha la sovrasaturazione locale nel microambiente in cui
avviene la nucleazione e la crescita può rappresentare un importante fattore di controllo della
deposizione dei polimorfi del carbonato di calcio e che la struttura dello stampo organico può
condizionare la morfologia dei depositi cristallini.
b) Biocristallografia
E‟ stato ideato e messo a punto un nuovo metodo di cristallizzazione di proteine ed è stata
determinata la struttura cristallina della forma A
della gliceraldeide –3-fosfato deidrogenasi
plastidiale.
La crescita di cristalli singoli di proteine rappresenta una delle maggiori difficoltà, che si
incontrano nello studio della struttura di proteine mediante diffrazione di raggi X. La nucleazone
dei cristalli di macromolecole biologiche è il risultato di un bilancio delicato di parecchi fattori ed è
un processo lento, che può avvenire in periodi di giorni o settimane. L‟importanza di fattori quali la
forza ionica, il pH, la natura del precipitante, la temperatura, varia da caso a caso ed è difficile fare
delle generalizzazioni. L‟alta supersaturazione necessaria per aumentare la probabilità di
formazione di un nucleo cristallino può portare con facilità alla formazione di un precipitato
amorfo. Si può tuttavia avere la formazione di nuclei ad una supersaturazione più bassa sfruttando
un processo di nucleazione eterogenea. E‟ stata riportata la nucleazione eterogenea di proteine su
facce di cristalli di minerali e su strati lipidici. Strati lipidici carichi inducono la nucleazione
concentrando le molecole di proteina sulla superficie mediante interazioni elettrostatiche non
specifiche. E‟ stato dimostrato che un meccanismo simile opera nelle cristallizzazioni biomimetiche
di carbonato di calcio su superfici di polistirensolfonato, su films di -chitina e di fibroina della seta
con assorbite macromolecole acide estratte da conchiglie di mollusco e su matrici collagenose
contenenti poli-L-aspartato.
La similarità del meccanismo, che opera quando le cariche sono localizzate su monostrati o su
films polimerici, suggerisce che le superfici di films polimerici possono essere considerate adatte
per la nucleazione di cristalli proteici. Sono state pertanto utilizzate per la nucleazione eterogenea di
proteine
superfici
di
films
polimerici
contenenti
gruppi
ionizzabili
ed
in
specifico
polistirensolfonato a diversi gradi di solfonazione, gelatina reticolata e fibroina della seta contenenti
poli-L-lisina o poli-L-aspartato. Come proteine modello sono state usate la concanavalina A e il
lisozima. E‟ stato trovato che la cristallizzazione della concanavalina A mediante la tecnica della
diffusione di vapore è fortemente influenzata dalla presenza di gruppi ionizzabili sulla superficie del
film. Il tempo di induzione e la concentrazione della proteina necessaria per la nucleazione
diminuiscono entrambi, mentre la densità di nucleazione aumenta passando dal vetrino siliconizzato
di riferimento alla superficie polimerica ed ancora di più alla superficie polimerica carica.
Interazioni non specifiche e locali fra la proteina e la superficie del film sono responsabili delle
collisioni molecolari e dell‟assemblaggio con la dovuta simmetria per al formazione del nucleo
cristallino. I risultati ottenuti indicano che l‟impiego di superfici polimeriche cariche può essere
particolarmente utile per la cristallizzazione di proteine da soluzioni a bassa concentrazione, usando
quindi minori quantità di sostanza, e per la riduzione dei tempi di induzione.
La gliceraldeide-3-fosfato deidrogenasi plastidiale (GAPDHp) è stata estratta e purificata dalle
foglie di spinacio. La GAPDHp è un enzima chiave nel ciclo di Calvin ed è presente nella forma
(A2B2)4 con peso molecolare di 600 kDa. L‟enzima presenta un meccanismo regolativo, un‟alta
attività NAD+ ed una bassa attività NADP+-dipendente. Due forme diverse dell‟enzima A4 e A2B2
sono state cristallizzate in presenza di NADP* ed i corrispondenti dati di diffrazione sono stati
raccolti utilizzando la luce di sincrotrone (Elettra, Trieste).
Le strutture di entrambe le forme sono state determinate con il metodo “Molecular
Replacement”, utilizzando come modello la struttura molecolare della gliceraldeide-3-fosfato
deidrogenasi citosolica dal Bacillus stearothermophilis, che presenta un‟omologia di sequenza del
59% con l‟enzima plastidiale. I risultati hanno mostrato la presenza nell‟unità asimmetrica di un
monomero e di un dimero per l‟isoforma A4 e di un tetramero e di un dimero per la forma A2B2.
L‟affinamento della forma A4 ha permesso di localizzare il coenzima NADP+ in tutte le subunità e
di eseguire un‟analisi dettagliata delle interazioni enzima-NADP+ allo scopo di dare una risposta in
termini strutturali al problema della selettività NAD+/NADP+. La struttura tridimensionale rivela
una grande omologia strutturale con le note GAPDH citosoliche. Si deve però notare che nella
isoforma A4 dell‟enzima plastidiale, a differenza che nelle GAPDH da eucarioti e batteri, si forma
un ponte disolfuro fra la cisteina 203 di un monomero con con la corrispondente cisteina del
monomero riferito per simmetria. Questo ponte non si forma neanche nell‟isoforma B in cui la
cisteina 203 è sostituita da un‟alanina.
Il programma di ricerca del gruppo CIURLI, qui riportato, si propone di studiare le
proprieta' chimico-fisiche e bio-fisiche dei sistemi biologici contenenti ioni metallici.
Le metallo-proteine studiate sono: (a) rame-proteine (b) citocromi; (c) proteine ferro-zolfo
(d) ureasi.
(a)
La plastocianina da Synechocystis PCC6803 è stata clonata, overespressa e purificata
in grandi quantità (30-50 mg), arricchita con 15N. La struttura in soluzione di questa proteina allo
stato ossidato paramagnetico è stata determinata. Tale determinazione strutturale è stata possibile
grazie allo sviluppo ed all'utilizzo di metodologie NMR per segnali molto paramagnetici. La
struttura della proteina ridotta, diamagnetica, è stata determinata in soluzione. Le proprietà
dinamiche di questa proteina sono state determinate su una scala di tempi molto ampia (da 10-12 a
105 secondi) sia per la forma ridotta che per la forma ossidata, utilizzando misure di 15N T1, 15N
T2, 1H-15N NOE eteronucleari, esperimenti di 15N T2 spin-echo, scambio con il solvente
deuterato. Questo lavoro rappresenta il primo studio integrato di struttura e mobilità in soluzione
sulla stessa rame-proteina in due forme redox diverse, di cui una altamente paramagnetica.
La struttura della plastocianina da spinaci nella forma ridotta è stata determinata in soluzione
mediante spettroscopia NMR, utilizzando un nuovo approccio per l‟assegnamento automatico dei
cross-peaks NMR con il programma GARANT. La struttura della plastocianina da piante superiori
così ottenuta è stata quindi confrontata con quella dell'alga unicellulare Synechocystis PCC6803.
Uno studio computazionale sul pathway di trasferimento elettronico tra queste due plastocianine da
organismi diversi con il loro partner redox fisiologico, il citocromo f, è in corso di svolgimento
utilizzando il programma AMBER-6 e GREENPATH. Uno studio ibrido QM/MM è stato iniziato
utilizzando il metodo Carr-Parrinello, allo scopo di individuare i parametri strutturali che regolano il
potenziale di riduzione, le proprietà magnetiche, e le proprietà cinetiche di trasferimento elettronico
nelle proteine blu di rame.
Il mutante C84S della plastocianina da Synechocystis PCC6803 è stato ottenuto e purificato
in grandi quantità (100-150 mg). Studi di binding con metalli diversi (Cu, Co, Ni, Fe) sono in corso
di svolgimento. La caratterizzazione strutturale e biofisica di tali mutanti proteici renderà più chiaro
il ruolo dei suddetti amminoacidi nel determinare le proprietà funzionali delle plastocianine.
(b)
Il citocromo c6 da Cladofora glomerata è stato isolato e purificato in grandi quantità
(10-20 mg). Sono stati svolti studi elettrochimici che hanno permesso di determinare sia il
potenziale standard di riduzione che i parametri termodinamici (entropia ed entalpia) della reazione.
La proteina è stata cristallizzata e dati di diffrazione di raggi X sono stati raccolti con una
risoluzione di 2.0 Å. Lo studio strutturale verrà proseguito dopo avere raccolto dati a risoluzione
maggiore utilizzando luci di sincrotrone.
Il citocromo c553 da Bacillus pasteurii è stato sequenziato, e la struttura cristallografica
determinata alla risoluzione di 0.97 Å. La struttura del citocromo c553 ossidato paramagnetico da
B. pasteurii è stata determinata in soluzione mediante tecniche NMR. Lo studio della mobilità in
soluzione è stato svolto sulla proteina clonata, overespressa e arricchita con 15N.
(c)
La HiPIP (High Potential Iron-sulphur Protein) da Rhodoferax fermentans è stata
cristallizzata e la sua struttura è stata determinata utilizzando tecniche MAD e luce di sincrotrone.
(d)
La struttura ai raggi X di due complessi tra l'ureasi isolata da B. pasteurii e l'acido
acetoidrossammico, il fosfato, e la mercaptoetanolammina sono state determinate. Studi teorici sui
meccanismi catalitico e di inibizione e per il design razionale di nuovi inibitori sono stati condotti
utilizzando tecniche di docking e di molecular modeling accoppiate con studi quanto-meccanici
(HF-SCF/DFT), in un approccio nuovo che potrebbe rappresentare un modello di studio più
generale per lo sviluppo di inibitori di metallo-enzimi. Questo studio è in corso di applicazione su
altri enzimi idrolitici contenenti un centro bimetallico, come nel caso delle fosfatasi acide
purpureee.
Dei quattro geni accessori dell‟ureasi da B. pasteurii, ureD, ureE, ureF e ureG, due (ureE e
ureG) sono stati amplificati e separatamente clonati in un vettore di espressione (pET3d). Il ceppo
BLR(DE3) di E. coli è stato trasformato con tali costrutti e la e proteine sono state overespresse. La
purificazione e la caratterizzazione di queste due proteine sono state effettuate, e le loro
caratteristiche chimico-fisiche sono state determinate anche utilizzando tecniche di spettrometria di
massa (MALDI-TOF). Studi di binding di nichel (per UreE) e di GTP (per UreG) sono in corso di
svolgimento. Si prevede di effettuare studi spettroscopici per determinare la struttura del centro
metallico nella UreE, così come di effettuare tentativi di cristallizzazione di entrambe le proteine.
Uno studio analogo sulle altre due proteine accessorie UreF e UreD è in corso di svolgimento.
UNITA’ LOCALE DI CAMERINO
Direttore: Prof. Bianca Rosa Pietroni
Gli obiettivi della Chimica Inorganica stanno subendo un'evoluzione e il campo di ricerca
non è più principalmente focalizzato sulla sintesi di composti aventi numeri di coordinazione,
strutture e stereochimiche interessanti bensì sulla preparazione di composti aventi potenzialmente
utili applicazioni pratiche. Due di tali applicazioni sono nell'area dell'ottica e dell'elettronica. A tal
proposito, in accordo alle linee guida del Programma Nazionale di Ricerca, l'unità operativa
dell'Università di Camerino nel corso dell'anno 2000 si è dedicata alla sintesi di nuovi materiali che
potrebbero avere applicazioni come sensori o come materiali per l'optoelettronica. Questi nuovi
materiali potrebbero avere un impatto su una pluralità di settori tra cui la biomedicina.
In considerazione di quanto detto l'u.o. di Camerino ha continuato a studiare il
comportamento come base di Lewis mostrato da alcuni composti trinucleari ciclici di Au(I) (Chem.
Comm. 1998). Sfruttando tale proprietà, si è messa a punto una via di sintesi innovativa di composti
supramolecolari. Infatti si è visto che substrati trinucleari ciclici di Au(I) aventi un intorno C-Au-N
si comportano da basi di Lewis verso ioni metallici aventi guscio chiuso d10 o s2 (Ag(I), Tl(I)).
L'analisi strutturale ai raggi-X effettuata su cristallo singolo di alcuni dei derivati ottenuti, ha messo
in evidenza la natura supramolecolare di tali composti (Inorg. Chem. 2000). Gli ioni Ag(I) o Tl(I) si
intercalano tra le specie trimeriche formando delle unità a sandwich che attraverso interazioni
Au...Au intermolecolari formano catene polimeriche inorganiche con interessanti proprietà
luminescenti. Un preliminare studio di queste proprietà condotte su cristallo singolo ha messo in
evidenza che questi composti mostrano una luminescenza termocromica. Infatti, quando i cristalli
vengono raffreddati a 77 K, gli spettri di luminescenza mostrano degli shifts nel rosso dei massimi
di emissione. Gli spettri di emissione mostrano una fosforescenza a bassa energia che è consistente
con le estese strutture a catena osservate aventi interazioni intermolecolari Au---Au. Ulteriori
indagini sulle proprietà luminescenti di questi composti sono attualmente in corso. Le proprietà
luminescenti dei nostri derivati fanno si che essi possano essere usati come congegni molecolari, ad
esempio nella misura della pressione parziale dell'O2 in fluidi biologici (diagnostica, industrie
biotecnologiche) (A. Mills, A. Lepre, B. R. C. Theobald, E. Slade, B. A. Murrer, Gold Bull 31
(1998) 68). Inoltre, dato che la luce fluorescente emessa cambia colore in funzione del tipo di ioni
intercalati, i trimeri di oro potrebbero essere utilizzati come sensori per rivelare ioni o composti
presenti in una miscela o in un essere vivente.
Il nostro studio è poi continuato partendo dal presupposto che un derivato perfluorurato
trinucleare ciclico di Hg(II) si comporta in modo opposto ai derivati trinucleari ciclici di oro(I),
strutturalmente analoghi. Infatti è noto in letteratura che il derivato di mercurio agisce come acido
di Lewis verso ioni alogenuro (anti-crown) formando strutture polimeriche intercalate da anioni.
Facendo reagire un trimero di oro con un trimero di mercurio, rispettivamente in un rapporto molare
2:1, si sono ottenute per assemblaggio catene supramolecolari neutre in cui il derivato di mercurio si
trova intercalato tra due trimeri di oro. Anche questi derivati sono luminescenti (J.A.C.S. in press).
Studi teorici (calcoli DFT) hanno mostrato che i trimeri di oro mostrano valori negativi di
potenziale elettrostatico al di sopra e sotto del piano del ciclo mentre valori positivi di potenziale
sono osservati in regioni simili del ciclo di mercurio. Questi risultati dimostrano chiaramente la
natura elettrostatica dell'interazione fra i cicli di mercurio e di oro e quindi anche tra i trimeri di oro
e gli ioni metallici (Ag+ e Tl+). Queste interazioni ricordano quelle catione-
in molecole
aromatiche descritte da Dougherty (Science 1996, 271, 163).
Un altro tema di ricerca svolto dall'unità operativa di Camerino ha riguardato lo studio delle
reazioni di equilibrio che si hanno quando la TFP (tri-2-furilfosfina) è messa a reagire con sali di
argento. Lo studio è stato effettuato analizzando gli spettri 31P NMR dei derivati solidi cristallini
isolati e caratterizzati strutturalmente. L'analisi spettroscopica ai raggi X di uno dei prodotti mostra
una struttura ad elica che si ottiene per self-assemblaggio delle unità [Ag(TFP)]+ in presenza di ioni
nitrato (manoscritto in preparazione); è importante segnalare che recentemente un altro derivato di
argento avente struttura elicoidale è risultato avere attività antibatterica e antifungina (Nomiya,
K. et al. Inorg. Chem. 2000, 39, 3301).
Infine allo scopo di ottenere modelli per sistemi bioinorganici, sono stati sintetizzati diversi
derivati dell'argento con leganti polidentati, derivati del pirazolo e dell'imidazolo, opportunamente
funzionalizzati. Inoltre in collaborazione con i biochimici presenti nell'unità operativa è iniziato uno
studio che si prefigge come obiettivo quello di capire il meccanismo di azione di alcuni farmaci di
oro utilizzati nel trattamento dell'artrite reumatoide. Lo studio è iniziato individuando alcuni target
biologici coinvolti nell'artrite reumatoide che sono inibiti da derivati di oro(I).
UNITA’ LOCALE DI CATANIA
Direttore: Prof. Enrico Rizzarelli
Complessi di rame(II) con oligopeptidi di interesse biologico.
E' noto che l'ottapeptide della regione N-terminale della proteina del prione (PrPc) è capace
di legare lo ione rameico. Per capire quali sono gli atomi donanti e, quindi, il tipo di coordinazione
che si ottiene, l'ottapeptide Ac-PHGGGWGQ-NH3, che corrisponde nella proteina ad una sequenza
che si ripete otto volte, e stato sintetizzato e purificato insieme al tetrapeptide HGGG-NH2.I
complessi di rame(II) formati da questi oligopeptidi sono stati studiati mediante tecniche
spettroscopiche che hanno coinvolto misure di dicroismo circolare, spettroscopia UV-Vis e misure
di risonanza di spin elettronico. Entrambi i peptidi formano con il rame(II) complessi di
stechiometria 1:1 sia a pH intorno alla neutralità che a pH basici.
Le misure spettroscopiche danno evidenza di una coordinazione in una geometria quadrato
planare a pH neutro, con il coinvolgimento di tre atomi di azoto di tipo peptidico ed uno derivante
da un immidazolo del residuo istidinico, mentre a pH basici, si ha un'espansione della
coordinazione con il coinvolgimento di un altro atomo di azoto di tipo peptidico. Le misure di
voltammetria ciclica effettuate agli stessi valori di pH indicativi del formarsi delle specie suddette,
ha confermato l'esistenza di due geometrie sostanzialmente diverse quando si passa da valori di pH
introno alla neutralità a quelli basici. L'osservazione di valori negativi del potenziale redox sono
stati ritenuti in buon accordo con le geometrie molecolari proposte, in particolare che il potenziale
della specie a pH più basico sia stato trovato più negativo porta a concludere che è proprio avvenuta
un'espansione della coordinazione. Tali potenziali di valore negativo fanno escludere la possibilità
che questi complessi possiedano attività antiossidante. Inoltre, simulazioni delle geometrie
molecolari più probabili con programmi di calcolo, che partono da dati strutturali di tipo
cristallografico per complessi simili, hanno confermato che la coordinazione di questi peptidi al
rame(II) coinvolge sia atomi di azoto peptidico, immidazolico, ed eventualmente o qualche
molecola d'acqua oppure qualche atomo di ossigeno derivante da gruppi carbonilici.. Tutti questi
risultati suggeriscono che l'ottapeptide Ac-PHGGGWGQ-NH3 possiede un singolo sito di
coordinazione disponibile per il rame(II) e che questo coinvolge in particolare la regione contenente
la sequenza HGGG. Infatti esperimenti condotti sia in eccesso di rame(II) che in eccesso di
legante(per entrambi i peptidi) non hanno condotto a specie complessate diverse.
Processi di riconoscimento molecolare.
E' stata sintetizzata una nuova -ciclodestrina funzionalizzata in posizione 6 mediante Boccarcinina. La struttura cristallina, risolta ai raggi X, rivela una forma a "sleeping swan", nella quale
il gruppo Boc della carcinina è inserito nella cavità idrofobica della ciclodestrina, che viene
stabilizzata dalla formazione di legame idrogeno. La cavità ciclodestrinica differisce poco da una
ciclodestrina non modificata e mantiene la sua caratteristica simmetria. La struttura in soluzione,
determinata mediante uno studio accoppiato mediante spettroscopia NMR e CD, ha dimostrato che
l'inclusione del gruppo Boc nella cavità della ciclodestrina è mantenuta in soluzione acquosa. Per
determinare l'entità delle forze deboli coinvolte nel processo d'inclusione, sono stati fatti tutta una
serie di esperimenti di competizione, usando come molecola competitiva l'1-adamantanolo, di cui
sono note le capacità di inclusione in cavità idrofobiche, seguiti mediante la tecnica spettroscopica
CD.
L'aggregazione di porfirine, campo di vasto interesse per possibili applicazioni biomediche e
tecnologiche, attraverso la possibilità di guidare i processi di "assembly" riveste un ruolo centrale
per ottenere strutture e funzioni specifiche. Recentemente abbiamo dimostrato come l'autoaggregazione di porfirine solubili in acqua su matrici polimeriche di carica opposta non è
semplicemente legata alla tendenza delle porfirine ad aggregare, ma è guidata da specifici processi
di riconoscimento molecolare. Si sono anche studiate interazioni tra porfirine aventi cariche opposte
(si parla di eteroaggregazione).Comunque, nessuno studio sistematico è stato rivolto alla
comprensione di come le specifiche caratteristiche steriche ed elettroniche delle porfirine possono
condurre alla formazione di eteroaggregati aventi una struttura predeterminata. I nostri studi hanno
portato alla conclusione che anche nel caso dell'eteroaggregazione tra porfirine di segno opposto
sono coinvolti processi di riconoscimento molecolare. Abbiamo usato come porfirina anionica la
meso-tetrakis(4-sulfonatophenyl)porfirina (H2TPPS), la cui forma protonata (H4TPPS ha un pK =
4.8) da luogo ad aggregati di tipo J-(edge-to-edge) che di tipo H(face-to-face). La formazione di
aggregati sef-assembly dipende dalla concentrazione, dalla forza ionica e dal pH. Generalmente si
formano a pH<1 ed alte forze ioniche. Nei nostri lavori abbiamo dimostrato come questi aggregati
possono essere ottenuti in condizioni molto più blande, inducendo l'aggregazione con porfirine
cationiche. Le più efficaci si sono dimostrate le porfirine che contengono un metallo con una o due
molecole di acqua, oppure presentano una carica positiva nel "core" della porfirina.
Purificazione e caratterizzazione di enzimi ad attività degradativa.
Sono state estratte e purificatele varie isoforme di laccasi dal fungo Rigidoporus Lignosus, e
si sono studiate le sue caratteristiche in soluzione in paragone ad altre laccasi estratte da funghi
diversi (Pleurotus Ostreatus). In particolare, si è voluto saggiare l'entità delle variazioni
conformazionali di questi enzimi al variare di condizioni esterne come variazioni di pH, di forza
ionica e di temperatura. Si sono usate le tecniche spettroscopiche di Dicroismo circolare e di
Risonanza magnetica di Spin Elettronico, l'una perché permette nella regione dell'UV (si possono
usare piccole quantità di proteina) di monitorare eventuali variazioni di conformazione rispetto alla
conformazione della proteina nativa (generalmente queste proteina si ritrovano in conformazioni di
-sheet), l'altra perché dà informazioni su variazioni geometriche che potrebbero essere subite dai
siti metallici. Le misure CD indicano che la struttura secondaria delle proteine è poco dipendente
dalla forza ionica e dal pH, sebbene forti incrementi di forza ionica possono svolgere un ruolo
indiretto nei processi di riconoscimento molecolare tra l'enzima e l'eventuale substrato. Questi
enzimi sono però altamente destabilizzati quando vengono esposti per molto tempo (questo dato è
interessante per eventuali applicazioni biotecnologiche) a bassi valori di pH o ad alte temperature.
Si osserva "unfolding" delle proteine che coincide con la loro inattivazione ed in qualche caso
anche con la loro precipitazione. E' interessante osservare che anche nelle condizioni più estreme,
anche quando la proteina ha perso le sue capacità catalitiche, i siti metallici sono conservativi, nel
senso che non si evidenziano drammatiche variazioni nelle geometrie di coordinazione dei siti a
rame(II) T1 e T2. Ciò suggerisce, come d'altronde già visto nella letteratura chimica, che la regione
che include i siti metallici è la regione più stabile a variazioni di conformazione di queste proteine.
In particolare, si è anche visto che la forza ionica non influenza il riconoscimento molecolare delle
proteine verso i substrati allo stesso modo, nel senso che ci sono substrati per i quali si ha un crollo
dell'attività catalitica ed altri per i quali tale attività si mantiene pressoché costante. Questa evidenza
sperimentale suggerisce la possibilità di modulare la specificità di questi enzimi (è noto infatti che
le laccasi sono degli enzimi altamente aspecifici) attraverso una modulazione della forza ionica e
questo potrebbe essere un fattore importante da tenere conto nell'eventuale applicazione
biotecnologica di questa classe di enzimi.
UNITA’ DI RICERCA DI FERRARA
Direttore: Prof. Roberto Rossi
Design e sintesi di complessi di Re, Tc e Pt come potenziali radiodiagnostici e chemioterapici,
loro reattività chimica e attività biologica.
I composti del Tc-99m e del platino hanno un ruolo ben consolidato nell‟ambito della farmaceutica
inorganica. Il tecnezio infatti è largamente usato come radiofarmaco diagnostico in medicina
nucleare, mentre i composti del platino (cisplatino, carboplatino) trovano una estesa applicazione
come farmaci antitumorali. A differenza del platino l‟affermarsi di altri metalli di transizione in
campo clinico come antitumorali è stato particolarmente lento.
L‟attività antimetastatica manifestata da alcuni complessi ottaedrici di rutenio rappresenta
un importante sviluppo nel campo dei chemioterapici ed ha stimolato la ricerca a tutto campo
all‟interno della famiglia dei metalli di transizione, in considerazione anche del fatto che nonostante
il successo del cisplatino, esso presenta diversi svantaggi che includono principalmente l‟elevata
tossicità e l‟applicabilità ad una relativamente ristretta casistica di tumori.
Per quanto riguarda il renio, i suoi isotopi -186, -188 promettono interessanti sviluppi come
radiofarmaci terapeutici dei tumori, ed inoltre i suoi complessi “freddi” possono essere valutati
come potenziali farmaci chemioterapici in analogia a quelli di rutenio precedentemente menzionati.
La biodistribuzione e l‟attività biologica dei complessi degli elementi citati è spesso
determinata sia dalla natura del metallo considerato, che dal tipo di leganti in essi presenti.
Per quanto riguarda il renio ed il tecnezio, la ricerca attuale è prevalentemente rivolta alla
loro chimica nei bassi stati di ossidazione poiché in essi il loro comportamento chimico è
particolarmente simile. Ciò consente di indagare la chimica del tecnezio utilizzando l‟elemento non
radioattivo renio. Quindi se si riesce ad ottenere un composto con proprietà radiofarmacologiche
sarà possibile sviluppare un kit farmaceutico identico per entrambi gli usi, diagnostico (Tc) e
terapeutico (Re). Inoltre i complessi di Renio(I) sono molto importanti nel campo organometallico e
recentemente è stato suggerito che questa classe di composti possa avere interessanti sviluppi nel
settore dei radiofarmaci.
Per queste ragioni si è avviato uno studio metodico delle potenzialità coordinative espresse
da specifici leganti atti a stabilizzare il metallo in bassi stati di ossidazione, con l‟intento anche di
pervenire alla messa a punto di metodiche sintetiche innovative di complessi di renio(I) e
tecnezio(I) privi di gruppi carbonilici. Questa necessità deriva dal fatto che in Medicina Nucleare
l‟utilizzo di ossido di carbonio limiterebbe pesantemente l‟eventualità applicativa a livello di Tc99m.
Poiché le proprietà biodistributive dei complessi, come inizialmente ricordato, possono
essere determinate dal sistema legante (sovente di tipo organico), si stanno studiando sistemi
coordinativi dotati di caratteristiche bifunzionali che li rendono idonei sia a coordinare stabilmente
il metallo, che a legarsi a molecole di rilevanza biologica in modo da mantenere il più possibile
inalterate le loro proprietà biodistributive.
Tecnezio
Negli ultimi anni si è osservato un crescente interesse verso complessi di Re(I) e Tc(I), sia
per studi di metallorganica (Re) che per lo sviluppo di nuove procedure di sintesi che possano
essere applicate alla Medicina Nucleare (Tc, Re). Un nostro obiettivo era quello di realizzare nuovi
complessi di Tc(I) da utilizzare come precursori in reazioni di scambio con leganti di interesse per
la M. N. E‟ stato iniziato uno studio per trovare procedure sintetiche per trasferire al Tc la sintesi
dell‟analogo complesso di renio [ReV(N2COPh)(PPh3)2Cl2], il quale può essere facilmente ridotto a
Re(I). Il pertecnetato è stato fatto reagire con benzoilidrazina in presenza di acidi alogenidrici (HCl,
HBr) a temperatura ambiente per 5-10 min. L‟addizione di PPh3 in rapporto stechiometrico favoriva
la precipitazione di un solido arancione (HBr) o giallo (HCl) che, da un‟indagine cristallografica è
stato identificato come un esa-alogenuro di Tc(IV) avente la fosfina protonata come contro-ione
[HPPh3]2[Tc(X)6] (X=Cl, Br), rivelando quindi il ruolo di riducente della benzoilidrazina e non di
legante. Questi composti potevano essere facilmente ridotti a complessi carbonilici di Tc(I) se ad
una loro sospensione in toluene mantenuta a riflusso, veniva aggiunto goccia a goccia BH3.THF e
facendo gorgogliare monossido di carbonio per un tempo complessivo di 45 min.
L‟analisi strutturale condotta sul complesso isolato come sale di tetrafenilarsonio mostrava
essere il complesso dinucleare [AsPPh4][Tc2(CO)6Br3]. Facendo reagire questo con la trifosfina MeC(CH2PPh2)3 (triphos), o aggiungendola direttamente alla miscela di reazione, si è ottenuto il
complesso [Tc(triphos)(CO)2Cl] del tutto simile a quello di renio. In prospettiva per un futuro
impiego in M. N., è importante sottolineare che è stato possibile isolare complessi di Tc(I) in un
unico step partendo da [TcO4]- via la formazione intermedia di esa-alogenuri di Tc(IV), e con tempi
di reazione sufficientemente brevi, ca. 1h e 30 min.
Renio
Gli studi condotti con amminoacidi funzionalizzati con 2-amminotiazolo hanno dimostrato,
anche mediante indagini strutturali, che la coordinazione coinvolge sempre l‟atomo di azoto
dell‟anello tiazolico e l‟ossigeno del gruppo ammidico deprotonato dell‟amminoacido. Nel
procedimento di sintesi dei diazoto-derivati del renio(I) a partire da perrenato di potassio,
l‟intermedio chiave è rappresentato dal composto di renio(V) [Re(N2COPh)(PPh3)2(Cl)2], dove è
presente il gruppo chelante diazobenzoile. La classe di leganti precedentemente menzionati
coordinano su questo formando composti mono- e bisostituiti e favorendo l‟apertura dell‟anello a
livello del dente ad ossigeno del frammento diazobenzoile, il quale assume una disposizione Re-NN lineare, con conseguente riduzione del centro metallico. In tutti questi composti il biolegante è
direttamente coinvolto nella coordinazione; volendo evitare ciò, si è sintetizzato l‟analogo
complesso di Re(V) con p-ammino-benzoilidrazina [Re(N2COPh-NH2)(PPh3)2Cl2] per poi in una
seconda fase condensare al gruppo amminico il biolegante in questione.
Per tale operazione si sono utilizzati i cloruri acilici dei leganti onde consentire la
condensazione al gruppo p-amminico. Tra gli acidi organici scelti, oltre all‟acido benzoico, è stato
usato il cloruro acilico dell‟Ibuprofen HOOCCH(CH3)C6H4CH2CH(CH3)2 noto antinfiammatorio
non steroideo. I complessi ottenuti, trattati con piridina o acetonitrile hanno portato all‟apertura del
dente labile ad ossigeno del legante benzoilidrazinico con conseguente riduzione del renio ed
ulteriore allontanamento da esso del frammento biologico. Queste procedure consentono quindi di:
avere il biolegante non direttamente coinvolto nella coordinazione al metallo, aumentare la
probabilità di una sua interazione con il recettore, ed in ultimo, di rendere disponibile sul metallo un
sito coordinativo suscettibile di attacco nucleofilo da parte di proteine recettoriali o di membrana.
Nell‟ambito della chimica del renio stiamo anche indagando l‟eventuale utilizzo dei suoi
complessi “freddi” come potenziali farmaci chemioterapici. Le indagini in corso in collaborazione
con il dipartimento di Biochimica su cellule in vitro per complessi di Pt(II), sono state recentemente
estese ad alcuni complessi di renio, ed i risultati fino ad ora ottenuti sembrano incoraggianti.
Platino.
Poichè il complesso [PtOTf(triphos)]OTf, [triphos = bis(2-difenilfosfinoetil)fenilfosfina],
caratterizzato dalla presenza di una fosfina tripodale e di un legante labile, è un sistema
particolarmente adatto allo studio delle sostituzioni nucleofile al platino, ne è stata migliorata la
sintesi. La via sintetica più conveniente prevede due soli step: essi possono essere eseguiti in
sequenza, senza necessità di isolare il prodotto intermedio. La reazione tra [PtMe2(COD)] e triphos
produce la specie [PtMe2(triphos-P,P‟)], in cui la trifosfina agisce da legante bidentato; il
trattamento di questa specie con due equivalenti di acido triflico produce infine il complesso
[PtOTf(triphos)]OTf.
Il complesso [PtOTf(triphos)]OTf è stato completamente caratterizzato mediante analisi
elementare, spettroscopia IR e
31
P{1H} NMR, spettrometria di massa ed anche mediante analisi ai
raggi X.
Lo spettro
31
P{1H} NMR di [PtOTf(triphos)]OTf in CDCl3 denota la presenza di fenomeni
di flussionalità in soluzione, che dipendono da alcuni fattori, quali la polarità del mezzo, la
concentrazione e la temperatura; l'osservazione NMR del complesso al variare delle condizioni
sopra citate sembra indicare che il legante triflato labilmente coordinato sia soggetto ad uno
scambio continuo con il triflato esterno alla sfera di coordinazione, dovuto all'attacco nucleofilo di
quest'ultimo al platino. Abbiamo interpretato questo fenomeno come una manifestazione
dell'estrema suscettibilità del complesso all'attacco nucleofilo al metallo.
Il complesso è stato utilizzato per ottenere un‟ampia serie di analoghi e per indagini
meccanicistiche sulla sostituzione nucleofila al platino. Il legante labile OTf è infatti sostituito da
diversi anioni e molecole elettron-donatrici, quali SMe-, N3-, PhCC-, PPh3, SMe2, CO e solventi,
come acetonitrile e acetone, incluse specie con scarsa tendenza a coordinare al platino. Alla luce di
questa caratteristica è ragionevole aspettarsi che il complesso [PtOTf(triphos)]OTf reagirà con
diversi gruppi funzionali di biomolecole legandosi per esempio a peptidi e acidi nucleici. Sono state
eseguite prove preliminari di attività antiproliferativa in vitro di questo complesso, con risultati
incoraggianti sia sulla linea cisplatino sensibile T2, che sulla linea cisplatino resistente SKOV3.
Catalisi biomimetica mediante l'uso di ferroporfirine fotoeccitate.
Uno dei principali obiettivi di questa ricerca è quello di ottenere nuovi sistemi catalitici di
interesse nella ossidazione selettiva di idrocarburi scarsamente reattivi in condizioni blande di
temperatura e di pressione. La via da seguire è quella di "mimare" il comportamento catalitico di
alcune ossigenasi naturali (citocromo P450, perossidasi) utilizzando modelli attivati attraverso
processi fotoossidoriduttivi. Oltre che dal punto di vista catalitico, questa indagine è interessante per
le informazioni che può dare sul meccanismo d'azione in vivo di queste stesse emoproteine. Ferro
porfirine fotoeccitate possono funzionare come modelli dei sitemi naturali NO sintetasi (NOS). In
particolare, l'eccitazione fotochimica di soluzioni acquose di ferro tetrafenil porfirina tetrasolfonata
provoca l'ossidazione di arginina a citrullina e NO. Il meccanismo di reazione che coinvolge
intermedi di contenenti il metallo ad alto stato di ossidazione, analoghi a quelli caratteristici del
ciclo catalitico del citocromo P450.
E‟ stata realizzata la funzionalizzazione del TiO2 con una ferro porfirina seguendo una
nuova procedura secondo la quale il complesso porfirinico viene preventivamente silanizzato. La
caratterizzazione
mediante
laser
flash
fotolisi,
spettroscopia
uv-visibile
e
tecniche
fotoelettrochimiche ha messo in evidenza che la natura del mezzo disperdente controlla i processi
redox che coinvolgono la ferro porfirina. In particolare, si sono osservati effetti marcati sulla
stabilità del complesso ridotto, formato per cattura dell'elettrone promosso nella banda di
conduzione in seguito a fotoeccitazione del TiO2. L'attività fotocatalitica del sistema è stata studiata
nei processi di monoossigenazione del cicloesano e del cicloesene da parte dell'O2. E' stato visto che
la ferro porfirina provoca un aumento delle quantità di prodotti di monoossigenazione rispetto alla
totale degradazione a CO2 per entrambi i substrati esaminati. Su questa base, si può affermare che la
presenza della porfirina ha la funzione di modulare l'elevato potere ossidante del TiO2 e
contemporaneamente di aumentare l'efficienza fotocatalitica. Inoltre, nel caso del cicloesano, il
complesso cambia la selettività del processo, aumentando il rapporto alcol/chetone.
La fotoeccitazione di (nBu4N)4W10O32 in presenza di O2 permette di ossidare cicloesene e
cicloottene a temperatura ambiente e a pressione atmosferica. Questo processo può essere realizzato
sia in fase omogenea sia usando il decatungstato in fase dispersa dopo la sua eterogeneizzazione su
silice. I due cicloalcheni sono principalmente ossidati ai corrispondenti idroperossidi in
conseguenza del processo fotochimico primario che porta alla formazione di radicali allilici. La
presenza di ferro tetrakis 2,6-diclorofenil porfirina (FeTDCPP), modifica le proprietà fotocatalitiche
del decatungstato, giocando un ruolo importante soprattutto per quanto riguarda i meccanismi di
ossidazione dei due cicloalcheni, mediate dagli idroperossidi allilici. Nel caso del cicloesene, la
porfirina aumenta l'efficienza catalitica in termini di turnover totale e catalizza la decomposizione
dell'idroperossido con formazione selettiva dell'alcool allilico. Nel caso del cicloottene, favorisce la
formazione del corrispondente epossido per addizione di radicali ROO. sul doppio legame.
L'N,N dimetil tetradecil N-ossido (DTAO) si è rivelato un buon tensioattivo per formare un
sistema micellare capace di ospitare FeTDCPP. Il catalizzatore microeterogeneo così ottenuto può
indurre processi biomimetici su substrati organici in mezzo acquoso, utilizzando luce solare e
ossigeno molecolare come reagenti. In particolare, l'ambiente micellare può controllare alcuni
parametri importanti da cui dipende la reattività di alcuni intermedi e quindi anche la selettività nel
processo di ossidazione. Al contrario di quanto osservato in precedenza in fase omogenea, sia il
cicloesene che il cicloottene sono ossidati ai corrispondenti epossidi; nel caso del cicloottene la
selettività è maggiore del 90%. Questo sistema composito è risultato anche molto promettente nella
conversione di piccole quantità di alcani alogenati presenti nelle acque in prodotti meno tossici. In
particolare, CCl4 può essere ridotto sia da etanolo sia da un alcol non miscibile con l'acqua, come il
cicloesanolo, con buone rese e buoni valori di turnover.
Biocatalisi per l’ottenimento di molecole chirali
Negli ultimi decenni microrganismi ed enzimi sono stati ampiamente usati nella sintesi di
molecole otticamente attive sfruttando reazioni di ossido-riduzione e di idrolisi. In particolare le
riduzioni enantioselettive di chetoni prochirali con lievito di birra e altri microrganismi, quali lieviti
e muffe, hanno permesso la sintesi di alcoli omochirali con prevalente stereochimica S, in accordo
con la “regola di Prelog,” mentre nelle reazioni di idrolisi/esterificazione con lipasi si risolvono
cineticamente esteri/alcoli racemici ottenendo alcoli con stereochimica R.
Nella nostra ricerca ci siamo interessati ,in primo luogo, di riduzioni di chetoni prochirali con lieviti
e muffe, focalizzando il nostro interesse su ceppi di Yarrowia lipolytica che hanno mostrato in molti
casi stereospecificità “anti-Prelog”. Gli stessi ceppi di Yarrowia lipolytica hanno dato risultati
soddisfacenti in reazioni di idrolisi di esteri racemici.
Nel nostro gruppo di ricerca per la prima volta sono state utilizzate le reazioni di ossidazione
microbiologica mediate da batteri per risolvere alcoli chirali e sempre nell‟ambito delle reazioni di
ossidazione sono staiti ossidati stereoselettivamente (reazioni di Bayer-Villiger) chetoni per ottenere
esteri ed alcani per ottenere alcoli (reazioni di idrossilazione).
UNITA’ DI RICERCA DI FIRENZE
Direttore: Prof. Pierluigi Orioli
L‟attività dell‟Unità operativa di Firenze si è concentrata essenzialmente su tre linee di ricerca:
1) complessi di rutenio(III) come farmaci antitumorali;
2) sviluppo di complessi di oro(III) come agenti citotossici ed antitumorali,
3) studio di antracicline e loro interazione con complessi metallici.
Complessi di rutenio(III)
Per quanto riguarda il primo tema di ricerca la nostra attenzione si è rivolta all‟analisi delle
interazioni in vitro di alcuni promettenti complessi di rutenio(III) con possibili target
biomacromolecolari (DNA, proteine). Le interazioni con DNA in vitro sono state descritte
utilizzando tecniche quali il dicroismo circolare, la spettroscopia elettronica, l‟analisi delle curve di
fusione del DNA, la dialisi di equilibrio, la mobilità elettroforetica, l‟inibizione di enzimi di
restrizione. Globalmente i risultati ottenuti permettono di dimostrare che i complessi di rutenio(III)
legano (debolmente) il DNA ed inducono piccole variazioni strutturali e funzionali. Gli effetti
descritti sono comunque assai più piccoli di quelli prodotti da concentrazioni equimolari di
cisplatino. Le interazioni con l‟albumina serica hanno rappresentato l‟oggetto di un altro studio.
Interazione con proteine
La scelta di studiare l‟interazione con le proteine plasmatiche deriva principalmente dal fatto
che i complessi di rutenio(III) vengono somministrati per via intravenosa, ed è quindi importante
considerare le interazioni che si producono con queste biomolecole, le quali possono svolgere
funzioni di trasporto, ma che possono anche costituire una modalità di inattivazione dei complessi
stessi. Si può inoltre considerare l‟ipotesi che i complessi di rutenio(III) esplichino parte della loro
attività biologica danneggiando direttamente alcune proteine di membrana o della matrice
extracellulare, fondamentali nei processi di proliferazione cellulare o di invasione metastatica
I modelli biologici presi in esame nel nostro laboratorio sono principalmente l‟Albumina
Serica e la Transferrina. I complessi di rutenio studiati sono NAMI, [Na][trans(Im)2RuCl4] (ICR),
Ru(III)Cl2(EDTA) e Ru(III)Cl2(PDTA) (RAP).
Caratterizzazione chimica mediante spettroscopia elettronica UV-VIS, Assorbimento atomico ed
IR
Il processo d‟idrolisi dei complessi di rutenio in presenza di quantità crescenti di proteina è
stato analizzato spettrofotometricamente. Il legame con le proteine si stabilisce dopo l‟idrolisi dei
gruppi cloruro coordinati equatorialmente (l‟aggiunta di proteina accelera il processo d‟idrolisi). Gli
spettri sono caratterizzati da intense bande nel visibile assegnabili a transizioni LMCT dei centri di
rutenio; inoltre si notano significative variazioni legate alla stechiometria crescente di rutenio,
indicando la presenza sulla proteina di siti multipli non equivalenti di legame per il rutenio. Dopo
incubazione ed estesa ultradialisi, la stechiometria dell‟interazione rutenio/proteina è stata valutata
tramite misure di assorbimento atomico. Sono stati condotti inoltre esperimenti sull‟interazione tra i
complessi di Ru(III) e proteine modificate con Dietilpirocarbonato (DEPC), il quale
preferenzialmente si lega alle Istidine poste in superficie.
Caratterizzazione chimica tramite Dicroismo Circolare (CD)
Tramite spettroscopia CD si possono ottenere informazioni più dettagliate sulle
caratteristiche spettrali degli addotti di rutenio/proteine. Il dicroismo circolare è infatti una tecnica
specifica per analizzare l‟intorno dei centri metallici legati a proteine. Dai dati ottenuti si nota che le
caratteristiche spettroscopiche dei singoli centri di legame rutenio-proteina sono abbastanza
differenti e ciò può implicare che sulle proteine esistono siti di legame distinti. In particolare il
primo equivalente di rutenio dà luogo ad un particolare spettro CD (significativo della presenza di
un sito di legame preformato), mentre gli altri centri di rutenio danno luogo a segnali meno
caratteristici.
Interazione con acidi nucleici
Il motivo che ci ha spinto a studiare l‟interazione dei complessi di rutenio(III) con il DNA è
il fatto che esso rappresenta il bersaglio presunto di vari complessi metallici ad attività antitumorale
nota, come avviene anche per le antracicline e la bleomicina. In particolare il DNA è il target
riconosciuto dei complessi di platino che sono strutturalmente e chimicamente simili ai complessi di
rutenio(III). E‟ pertanto ragionevole considerare il DNA come possibile bersaglio per i complessi di
rutenio anche se non vi è ancora evidenza definitiva.
Il modello biologico utilizzato è il DNA calf thymus, gli addotti sono stati studiati
principalmente mediante spettroscopia UV-VIS, CD ed Assorbimento Atomico.
Caratterizzazione chimica mediante spettroscopia elettronica UV-VIS, CD ed Assorbimento
atomico (AAS)
I risultati ottenuti mediante spettroscopia UV-VIS e CD hanno mostrato che i complessi di
rutenio(III) reagiscono con il DNA dopo che sono andati incontro ad idrolisi i due gruppi cloruro
coordinati equatorialmente. Non c‟è evidenza di riduzione del rutenio ed inoltre il legame al DNA
ha piccoli effetti sulla sua struttura a doppia elica. La quantità di rutenio legata verrà determinata
mediante misure di assorbimento atomico.
Esperimenti di Melting
Sono stati eseguiti esperimenti di melting del DNA in collaborazione con la Prof. E. B.
Dalian dell‟Università di Yerevan (Armenia). Questo tipo di tecnica è molto utile e sensibile per
determinare piccoli cambiamenti nella conformazione del DNA indotti da complessi metallici o da
altri leganti durante il processo di denaturazione termica (si determina la Tm che è la temperatura di
fusione corrispondente alla transizione doppia elica singolo filamento). Al fine di determinare il tipo
d‟interazione si può seguire il processo inverso (rinaturazione). Dati ottenuti forniscono prova del
fatto che i complessi di rutenio danno luogo ad un‟interazione “bidentata” interstrand.
I complessi di rutenio come scavengers di NO
Negli ultimi anni è chiaramente emerso il ruolo centrale svolto dall‟ossido di azoto (NO) in
molti processi biologici tra i quali la vasodilatazione, la neuromodulazione, e l‟angiogenesi.
Variazioni nella concentrazione di NO sono associate a differenti stati patologici. La possibilità di
intervenire sulle concentrazioni locali di NO apre nuove aspettative terapeutiche nel trattamento di
alcune malattie neoplastiche. Sotto quest‟ottica si può considerare la possibilità di rimuovere
l‟ossido di azoto a livello intracellulare o intravasale mediante appositi complessi di rutenio(III) che
possano fungere da “scavengers” ad elevata selettività.
Si è cercato di valutare se i complessi di rutenio(III) in analisi possono comportarsi da
scavenger di NO. Alcune indicazioni in tale senso sono disponibili in letteratura. La formazione di
addotti NO-Ru è stata analizzata mediante spettroscopia IR. Abbiamo preliminare evidenza che
alcuni dei nostri complessi di rutenio(III) sono i grado di fissare NO e le conseguenze biologiche di
tale interazione sono confermate da studi condotti presso il Dipartimento di Farmacologia della
Prof. Ziche.
Studi farmacologici di Biologia Molecolare e
E‟ possibile valutare l‟attività antitumorale in vitro e in vivo dei complessi di rutenio in
collaborazione con il Laboratorio di Chemioterapia dell‟Università di Firenze e con il gruppo del
Prof. Sava della Fondazione Callerio di Trieste.
Mediante tecniche di Biologia Molecolare e Genetica è possibile caratterizzare in maggior
dettaglio l‟interazione dei complessi metallici con DNA in sistemi in vitro, mediante misure di
mobilità elettroforetica, analisi di inibizione di endonucleasi di restrizione e applicazioni della
tecnica Polymerase Chain Reaction (PCR).
Complessi di oro(III)
Presso il Dipartimento di Chimica dell‟Università di Firenze sono stati presi in esame complessi
di oro(III), isoelettronici ed isostrutturali a quelli del platino(II), sui quali i dati di letteratura sono
scarsi. La stretta somiglianza che intercorre tra Au(III) e Pt(II) rende i complessi di oro(III) dei
buoni candidati per uno studio più approfondito come agenti antitumorali, nonostante l‟elevata
labilità cinetica e gli alti potenziali redox li rendano instabili in ambiente fisiologico. I complessi di
Au(III) sono interessanti sia perché potrebbero costituire una nuova classe di farmaci antineoplastici
con un nuovo profilo di attività antitumorale, sia perché rappresentano un valido modello per
determinare il meccanismo d‟azione dei complessi metallici d8 con geometria quadrata planare.
Caratterizzazione chimica dei complessi mediante spettroscopia FTIR, UV-VIS, NMR,
Potenziometria e Spettrometria di massa
Le caratteristiche chimiche dei complessi sintetizzati sono state definite mediante spettroscopia
infrarossa (FTIR), mentre la stabilità dei complessi in ambiente fisiologico è stata analizzata
mediante spettroscopia, risonanza magnetica e misure potenziometriche; dall‟analisi degli spettri di
massa dei campioni si è determinata la specie presente in maggior quantità in soluzione acquosa.
Prove farmacologiche
Presso il Laboratorio di Chemioterapia dell‟Università di Firenze, è stato studiato l‟effetto
inibente dei complessi di oro(III) sulla crescita delle linee di carcinoma ovarico A2780/S, A2780/R
(con resistenza acquisita al cisplatino) ed SKOV3 (con resistenza acquisita ad esso) mediante il test
della sulforodamina-B. Per individuare possibili specificità dei complessi, la loro attività è stata
testata anche nei confronti di linee leucemiche (CEM-CH/S e CEM-CH/R). I complessi analizzati
risultano avere citotossicità comparabile a quella del cisplatino; essi sono inoltre capaci di superare
la resistenza ad esso nelle linee A2780/R,SKOV3 e CEM-CH/R.
Interazione con il DNA: determinazione del meccanismo d’azione dei complessi e di eventuali
danni molecolari al DNA
I vari complessi metallici reagiscono con le diverse biomolecole con meccanismo dipendente
dalla loro chimica (la labilità cinetica, l‟idro- e la lipofilicità, il comportamento redox e la carica
elettrica), dando luogo a lesioni biologiche differenti. In prima approssimazione possiamo
ipotizzare che il target principale di complessi antitumorali che siano simili chimicamente e
strutturalmente al cisplatino sia il DNA, anche se non possiamo escludere altri meccanismi
d‟azione. E‟ noto che il cisplatino, legandosi al DNA, lo modifica creando un danno localizzato non
riconosciuto dai sistemi di riparazione, che porta all‟inibizione dei processi di replicazione e
trascrizione.
Il primo passaggio consiste nel determinare se il complesso di Au(III) si leghi
covalentemente o meno al DNA. Il test COMET (microgel-elettroforesi su singole cellule) è in
grado di evidenziare rotture dirette o alterazioni convertibili in rotture delle catene del DNA, sia che
interessino un singolo o il doppio filamento dello scheletro fosfodiesterico. Esso permette quindi di
valutare immediatamente il danno al DNA prodotto ad esempio dalla formazione di legami
covalenti tra il complesso ed il DNA stesso, confrontando il percorso elettroforetico del DNA
cellulare trattato con i complessi con quello di DNA cellulare non trattato.
Con un‟analisi di tipo chimico è inoltre possibile misurare la quantità di complesso legata
covalentemente al DNA, dopo aver eliminato dall‟ambiente di reazione tutto il complesso libero. Si
utilizzano misure dirette di assorbimento atomico (AAS) su campioni preparati seguendo varie
procedure (DNA analizzato sia di origine cellulare che acellulare).
Per definire le modificazioni conformazionali prodotte sulla doppia elica del DNA, dopo
aver fatto reagire in vitro i complessi di oro(III) con DNA calf thymus, abbiamo analizzato le
variazioni indotte sullo spettro CD del DNA-B.
Ulteriori informazioni sulla stabilità del DNA si sono ottenute mediante misure di
denaturazione termica sul DNA, dopo incubazione con il complesso in tampone fisiologico.
Variazioni nella Tm indicano che il complesso si è legato covalentemente al DNA, stabilizzandolo
o destabilizzandolo. E‟ possibile anche quantificare lo srotolamento della doppia elica mediante
misure elettroforetiche (l‟angolo
dipende dalla densità della superelica e dal valore rb al quale la
forma superavvolta e rilassata comigrano).
I risultati ottenuti indicano la presenza di interazione, più o meno marcata, con il DNA: il
meccanismo con cui i complessi di oro(III) interagiscono con esso sono differenziati a causa delle
diverse caratteristiche chimiche dei composti scelti per lo studio.
Antracicline
Le antracicline sono ancor oggi tra i farmaci antitumorali più comunemente utilizzati nei
trattamenti clinici; in particolare la Doxorubicina è il maggior agente anticancro introdotto su larga
scala nell‟uso clinico a partire dagli anni 70. Chimicamente le antracicline sono amminoglicosidi
contenenti il cromoforo tetraciclico chinone. Inizialmente esse venivano ottenute dalla
fermentazione aerobica di differenti microorganismi del ceppo Streptomyces, oggi sono in larga
parte prodotti di sintesi. E‟ stato osservato che il maggior sito di azione per le antracicline è la
doppia elica del DNA dove tali farmaci si intercalano in modo tale da inibire l‟azione dell‟enzima
Topoisomerasi II e quindi la duplicazione del DNA stesso.
Le antracicline sono attive contro un‟ampia varietà di tumori maligni solidi e del sangue.
Tuttavia questi farmaci non mancano di numerosi effetti collaterali, fra questi il più preoccupante e
dannoso è la cardiotossicità. Per far fronte a questo problema è stato necessario sviluppare nuovi
derivati che abbiano minore tossicità e maggiore efficacia. E‟ stato dimostrato che anche piccole
variazioni chimiche delle strutture classiche, quali per esempio la sostituzione o modificazione di
uno o più gruppi funzionali, producono importanti effetti sull‟attività biologica e farmacologica di
questi composti.
La nostra ricerca consiste in un‟indagine delle proprietà chimiche e strutturali di nuovi
derivati delle antracicline per mezzo di tecniche spettroscopiche e cristallografiche. Di particolare
interesse è lo studio dell‟interazione di questi potenziali farmaci con il DNA e con le proteine
plasmatiche. Attualmente è in corso anche lo studio sull‟interazione di antracicline e complessi
metallici.
Questo progetto di ricerca è condotto in collaborazione con il gruppo MENARINI RICERCHE
di Firenze.
UNITÀ DI RICERCA DI NAPOLI
Direttore: Prof. Carlo Pedone
1.
Sviluppo di Peptidi Funzionalizzati con Metallo-Macrocicli aventi Alta Affinita’ Recettoriale
per il Targetting di Cellule Tumorali
Negli ultimi anni un crescente interesse è stato mostrato per lo sviluppo di sonde biospecifiche
in grado di riconoscere con alta affinità recettori espressi in cellule tumorali, con lo scopo di
ottenere agenti targetting per la diagnosi e per la terapia del cancro.
A seconda dell‟azione, terapeutica o diagnostica, che si intende conferire alla sonda
biospecifica, essa trasporta due diversi tipi di metalli: isotopi radioattivi quali
99m
Tc e
111
In per
utilizzo in medicina nucleare, oppure metalli paramagnetici quali Gd(III), Mn(II) e Mn(III) per
impiego di tecniche di risonanza magnetica imaging.
A causa della elevata tossicità mostrata tanto dagli ioni metallici paramagnetici quanto dai
radioisotopi, è necessario che questi siano trasportati in vivo irreversibilmente legati alla sonda
biospecifica. A tale scopo vengono utilizzati agenti chelanti bifunzionali, ossia in grado di legarsi
alla sonda trasportatrice e di formare complessi stabili con lo ione in esame.
Durante lo scorso anno sono state messe a punto le sintesi di derivati del peptide CCK8, con
macrocicli porfirina e DO3A. Il peptide CCK8 funge da sonda biospecifica ed interagisce con alta
affinità con i due recettori della colicistochinina CCK-A e CCK-B che sono stati dimostrati
recentemente essere sovraespressi in una serie di cellule tumorali.
CONHAspTyrNleGlyTrpNleAspPheNH2
HOOC
N
N
Gd
N
HOOC
N
COOH
Cl
Cl
Cl
Cl
N
N
In
N
N
Cl
Cl
Cl
Cl
CONH
(CH2)4
CH
CONHAspTyrNleGlyTrpNleAspPheNH2
NH2
Quest‟anno i due composti sono stati accuaratamente caratterizzati sia dal punto di vista
strutturale che per le loro proprietà chimico-fisiche.
In particolare il derivato con la porfirina di indio è stato caratterizzato mediante spettroscopia
NMR. Lo studio NMR condotto in DMSO ed in sistemi misti DMSO/acqua ha confermato che il
peptide assume una conformazione molto simile a quella esibita da CCK8 nel complesso con un
modello del recettore CCKA costituito dall‟estremità N-terminale di 47 residui che è ritenuta
responsabile dell‟interazione con il peptide. Lo studio NMR inoltre conferma che l‟introduzione sul
residuo di Asp N-terminale di CCK-8 dell‟amminoacido di Lysine legato covalentemente con il
gruppo porfirinico, non modifica la struttura di CCK8, ed in particolare non modifica la
conformazione delle due catene laterali di Met e Tyr che sono ritenute responsabili del binding.
Inoltre il gruppo porfirinico è rivolto in una zona lontana dalla zona di binding e quindi non
dovrebbe influenzare le proprietà di binding del coniugato da noi ottenuto con il recettore.
Sempre nell‟ambito di questa linea di ricerca si è provveduto a mettere a punto la sintesi in fase
solida di un nuovo derivato peptidico del CCK8, in particolare un decapeptide funzionalizzato con
una molecola di fosfina sulla posizione N-terminale. Il nuovo derivato peptidico contiene dunque un
set coordinativo costituito da un atomo di fosforo (della fosfina), uno di zolfo (di un residuo di
cisteina) e due azoti ammidici (appartenenti al backbone peptidico) che dovrebbe essere
particolarmente adatto a complessare metalli come Renio e Tecnezio.
La caratterizzazione ei derivati con il Renio è tuttora in corso.
2. "Molecular Tools" per il "Design" di Peptidi Bioattivi
L'inserimento di residui C ,
dialchilati nella sequenza di peptidi bioattivi è stato di grande
aiuto nello studio delle relazioni struttura-attività di queste molecole, anche in considerazione del
numero sempre più crescente di residui -amminoacidici C , dialchilati finora caratterizzati, per
la loro capacità di indurre nelle molecole bioattive variazioni strutturali e conformazionali
specifiche che modulano o mimano particolari effetti biologici. Numerosi studi in questo campo
sono stati condotti presso il Gruppo negli ultimi anni. Proseguendo le ricerche sulle preferenze
conformazionali di residui, sono stati analizzati peptidi contenenti residui quali l'Ac10c (acido 1ammino-1-ciclodecan-carbossilico) ed è iniziata la caratterizzazione di residui Ac11c(acido 1ammino-1-cicloundecan-carbossilico) che presenta diverso ingombro sterico e flessibilità sulle
catene laterali. Lo studio conformazionale di questi residui è stato condotto su omopeptidi (dal
monomero fino al pentamero) e su peptidi (tripeptidi, tetrapeptidi e pentapeptidi) contenenti
ciascuno di questi residui , -dialchilati posizionati all'inizio, al centro ed alla fine di una catena
polipeptidica contenente residui codificati (quale ad esempio la L-alanina) alla scopo di misurare la
capacita' di questi residui nell'indurre particolari strutture secondarie.
Sono inoltre continuti gli studi strutturali di peptidi sintetici lineari e/o ciclici contenenti residui
homo- -amminoacidi per valutare l'effetto strutturale impartito di residui non proteinogenici sul
"backbone" peptidico. A tale riguardo è stato condotto uno studio preliminare di calcolo di energia
conformazionale su sistemi modello di homoimpartita dai residui homo-
e
ammino acidi che ha mostrano la flessibilità
allo scheletro peptidico evidenziato dalle nette prevalenze
conformazionali per le regioni elicoidali della mappa.
3. Studio delle Proprietà Conformazionali di Sistemi -Ciclodestrinici Mono e Difunzionalizzati
I sistemi ciclodestrinici rappresentano un‟interessante classe di oligosaccaridi ciclici costituiti da
6-12 unità D-glucopiranosidiche, legate da legami
(1-4)-glicosidici. Tra questi composti, le -
ciclodestrine (cicloeptaamilosio) sono quelle maggiormente studiate. Gli studi cristallografici hanno
dimostrato come questi composti, formati da 7 unità D-glucopiranosidiche, presentano una forma a
tronco di cono, con le unità di glucosio in conformazione a sedia di tipo 4C1. La presenza della
cavità idrofobica e dei gruppi idrossilici primari permette di poter utilizzare le -ciclodestrine per
costruire modelli molecolari con proprietà peculiari. In particolare, questi sistemi possono formare
composti di inclusione ed essere utilizzati come modelli di “carrier” di composti idrofobici in
ambienti polari, modificando le caratteristiche chimico-fisiche dei composti inclusi, e come modelli
di enzimi naturali e recettori. Inoltre questa classe di composti può essere utilizzata per il
riconoscimento enantioselettivo di composti amminoacidici chirali, attraverso la formazione di
complessi con metalli di transizione.
In quest'ambito sono stati caratterizzati in quest‟anno allo stato solido, presso l'unità operativa di
Napoli una -ciclodestrinica difunzionalizzati, con due punti di possibile coordimazione per ioni
metallici.
UNITA’ LOCALE DI PADOVA
Direttore Prof. Ulderico Mazzi
Sezione Radiofarmaci
Sintesi e Caratterizzazione di Complessi di Renio, Tecnezio-99 e
Tecnezio-99m quali Potenziali Radiofarmaci
INTRODUZIONE
I radiofarmaci sono sonde radioattive impiegabili in vivo a scopo diagnostico o terapeutico.
La loro applicazione medica è legata alle proprietà nucleari del radionuclide utilizzato e alle
proprietà biologiche della molecola marcata. Di recente sviluppo è la ricerca di prodotti ottenuti per
marcatura di molecole biologicamenente attive con 99mTc e 186/188Re impiegabili, rispettivamente, in
diagnostica clinica e in terapia dei tumori.
Una possibilità nella marcatura di peptidi biologicamente attivi è rappresentata
dall‟approccio del chelante bifunzionale (Bifunctional Chelating Approach) che prevede la
coniugazione della biomolecola con un sistema chelante in grado di fissare stabilmente il metallo.
La marcatura può essere effettuata dopo la coniugazione (Preformed Chelate Approach) oppure si
può inizialmente marcare il chelante e successivamente coniugare il complesso ottenuto alla
biomolecola (Chelate Conjugate Approach).
DISCUSSIONE
Questo lavoro è stato incentrato sulla sintesi di chelanti bifunzionali in grado di legare
stabilmente Renio e Tecnezio e nei quali fosse presente un gruppo funzionale utile per l‟ancoraggio
di molecole bioattive. In figura 1 è riportata la struttura generale di tali leganti derivati dalla
coniugazione dell‟acido (R1)2fospinopropionico con il dipeptide glicina-cisteina che presentano un
set coordinativo di tipo PN2S e un gruppo carbossilico disponibile per la coniugazione con molecole
bioattive.
O
R1
P
R1
O
NH
R1 = Ph, CH3, CH3O(CH2)3
R3
NH
O
R2 = Bzl, Trt
S
R2
R3 = OH, OCH3, NH-biomolecules
Fig. 1
I leganti con R1= Ph, R2= Bzl e R3= OCH3, OH (L-SBzl) sono stati in precedenza
ampiamente studiati: da questi sono stati ottenuti i complessi di Renio ed i corrispondenti analoghi
del
99m
Tc. I complessi aventi come gruppo centrale il Re(V)oxo mostrano una configurazione
ottaedrica nella quale il gruppo tioetereo della cisteina, i due gruppi ammidici deprotonati e il
gruppo fosfinico sono coordinati al metallo e disposti sul piano equatoriale, mentre la sesta
posizione trans all‟[Re=O] è occupata da un atomo di cloro.
Studi di spettrometria di massa hanno dimostrato come il [Re=O]3+ (V) formi complessi
stabili con chelanti del tipo PN2S; si è dedotto, inoltre, che il complesso maggiormente stabile è
quello nel quale l‟atomo di zolfo risulta coordinato e deprotetto.
Difficoltà incontrate nella rimozione del benzile utilizzato come gruppo protettore dello
zolfo cisteinico ha orientato la ricerca verso la sintesi di un legante, contenente la stessa sequenza
amminoacidica, nel quale fosse presente, però, un gruppo protettore rimovibile in condizioni più
blande rispetto al benzile. Si è optato per l‟utilizzo del trifenilmetile o tritile, che può essere
facilmente rimosso in ambiente acido o in presenza di sali di metalli pesanti, ottenendo il legante N[N-(3-difenilfosfinopropionil) glicinil]-S-trifenilmetilcisteina secondo lo schema in Fig.2.
O
O
Trt
O
N
OH
+
H2 N
DCCI / HOBt
CH2 Cl2
OH
AcOH / H2O
NH
Trt
OH
NH
O
+
AcO- H3 N
NH
O
S
S
S
Trt
Trt
Trt
+
O
O
CH2 Cl2
OH
Steam bath
O
O
TEA
O
NH
P
OH
NH
RT
P
O
N
O
S
O
Trt
Fig. 2
Sono stati successivamente condotti gli studi complessazione con il Renio sul legante
tritilato (L-STrt) e detritilato (L-SH). La detritilazione più conveniente ha previsto l‟impiego di
acido trifluoroacetico in presenza di uno scavengers donatore di idruro rappresentato dal
trietilsilano.
Per la complessazione sono stati impiegati diversi exchange ligands e diverse condizioni di
reazione al fine di valutare quelle che in grado di garantire l‟ottenimento del complesso con rese
elevate.
Come riportato in figura 3 il complesso è stato ottenuto dal legante tritilato e detritilato con
l‟impiego del trans-oxotriclorobistrifenilfosfina renio (V) e di tetrafenilfosfonio oxotetraclororenio
(V) in etanolo assoluto.
Negli spettri di massa, ottenuti con la tecnica dell‟Electro Spray Ionisation, è presente lo
ione con m/z 617 ([M-H]-) corrisponde al complesso in cui il Renio risulta coordinato dal set PN2S
e il frammento con m/z 573 ([M-COOH]-) che corrisponde al complesso dopo la perdita del gruppo
carbossilico: questo dimostra che nei composti isolati questo gruppo non risulta coinvolto nella
coordinazione del metallo.
Il riscaldamento a riflusso, necessario per favorire la solubilizzazione del transoxotriclorobistrifenilfosfina renio (V) e l‟eventuale rimozione del tritile ad opera del metallo, ha
comportato un abbassamento della resa per la concomitante comparsa di reazioni collaterali dovute
probabilmente all‟indebolimento della struttura del legante ad alte temperature.
Si è inoltre osservata una certa difficoltà nella cristallizzazione del prodotto imputabile, in
parte, alla presenza del gruppo carbossilico libero; quest‟ultimo può inoltre interferire anche durante
la reazione di complessazione in quanto può competere con il tiolato nella coordinazione del
metallo, soprattutto nel caso del legante non detritilato.
O
EtOH abs, reflux 30 min, N2
ReOCl3(PPh3)2 + L-SH
N
ReOCl3(PPh3)2 + L-STrt
(Ph4P)ReOCl4
O
O
+ L-STrt
EtOH abs, reflux 1h, N2
ReO(V)Gluconate* + L-STrt
(Ph4P)ReOCl4
+ L-SH
OH
Re
EtOH abs, reflux 4h, N2
P
Ph
ReO(V)Gluconate* + L-SH
O
N
S
Ph
MeOH, RT, N 2
MeOH, RT, N 2
UNKNOWN PRODUCTS
EtOH abs, reflux 15 min, N2
* in the reaction mixture
(NH4)ReO4 + NaGluconate + SnCl2 + TFA
MeOH, RT, N2
Fig. 3
ReO(V)Gluconate
Allo scopo di eliminare le possibili interferenze del gruppo carbossilico libero nella reazione
di complessazione, è stato sintetizzato il corrispondente legante metilestere N-[N-(3difenilfosfinopropionil) glicinil]-S-trifenilmetilcisteina metilestere (L‟-STrt). La sintesi può essere
condotta, secondo quanto riportato nello schema in figura 4, in due diversi modi: o per sintesi del
dipeptide
metilestere
glicina-S-tritilcisteina
difenilfosfinopropionico,
oppure
per
e
successiva
esterificazione
coniugazione
diretta
del
con
l‟acido
legante
N-[N-(3-
difenilfosfinopropionil) glicinil]-S-trifenilmetilcisteina.
O
O
Trt
O
N
OH
+
H2 N
DCCI / HOBt
Trt
CH2 Cl2
OH
OH
NH
O
+
AcO- H3 N
AcOH / H2O
NH
NH
OH
Steam bath
O
O
S
S
S
Trt
Trt
PyBOP
DIEA
Trt
+
MeOH
0°C
O
O
O
Trt
O
N
OH
+ H2N
O
N
NH
EDAC
Trt
OCH 3
O
P
OCH 3
NH
CH2 Cl2
O
O
S
S
Trt
CH2 Cl2
TEA
Trt
Steam
bath
AcOH / H2O
O
P
O
O
O
N
+
+ AcO- H3 N
NH
O
O
NH
OCH 3
P
OH
NH
O
O
RT
O
S
S
Trt
RT
Trt
CH2Cl2
TEA
PyBOP
DIEA
O
O
NH
P
MeOH
0°C
OCH3
NH
O
S
Trt
Fig. 4
Vantaggiosa risulta l‟esterificazione diretta del legante N-[N-(3-difenilfosfinopropionil)
glicinil]-S-trifenilmetilcisteina “via Py-BOP” con rese superiori al 90% operando a basse
temperature (0-4° C) e impiegando il metanolo sia come reagente che come solvente.
Il legante metilestere è stato quindi impiegato per la complessazione con il Renio dopo
detritilazione (fig. 5): in questo caso aggiungendo alla soluzione di tetrafenilfosfonio
oxotetraclororenio (V) in etanolo assoluto il legante metilestere, solubilizzato anch‟esso in etanolo
assoluto, si ottiene a temperatura ambiente un
precipitato verde che per successiva alcalinizzazione con trietilammina porta al complesso rosso
mattone ReO(L‟-S) con il gruppo metilestere esterno alla sfera di coordinazione.
O
O
NH
P
OCH 3
NH
O
S
Trt
Et3SiH
TFA
O
O
NH
P
OCH 3
NH
O
SH
Ph4 PReOCl4
EtOH Abs.
EtOH Abs
TEA
RT
O
O
O
N
O
N
OCH 3
Re
P
S
Fig. 5
La reazione procede senza formazione di sottoprodotti di degradazione e con rese
quantitative. Vantaggiosa risulta, inoltre, la possibilità di recuperare il prodotto intermedio con la
filtrazione che consente la sua purificazione da tutte le altre specie presenti nell‟ambiente di
reazione.
Il passo successivo è stata la modifica strutturale del legante al fine di ottimizzare la stabilità
dei complessi, la loro solubilità e idrofilicità e le loro interazioni a livello fisiologico. A tale scopo
l‟attenzione
è
stata
focalizzata
sulla
sostituzione
dei
gruppi
fenilici
dell‟acido
difenilfosfinopropionico con gruppi metilici o metossipropilici che conferiscono una maggiore
solubilità in acqua: sono stati quindi sintetizzati i corrispondenti leganti con il dipeptide glicina-Stritilcisteina secondo lo schema in figura 6.
O
Trt
O
O
N
OH
+
H 2N
DCCI / HOBt
OH
CH2 Cl2
AcOH / H2O
NH
Trt NH
OH
O
O
Trt
P
EDAC, TEA, 4-DMAP
OH
NH
CH2 Cl2, RT
O
R1
Trt
O
O
NH
R1
OH
S
+
Trt
O
NH
Steam bath
S
S
O
+
AcO- H 3N
R1
OH
R1
S
Trt
P
R1 = CH3, CH3O(CH2)3
Fig. 6
Tali leganti dimostrano un‟elevata idrofilia rispetto al precedente legante difenilfosfinico
che risulta ulteriormente marcata in seguito a detritilazione.
Studi preliminari di complessazione hanno dimostrato la formazione di un complesso
analogo
a
quello
ottenuto
con
il
legante
N-[N-(3-difenilfosfinopropionil)
glicinil]-S-
trifenilmetilcisteina.
CONCLUSIONI
Il set coordinativo PN2S mostra un comportamento diverso verso il gruppo ReO3+ a seconda
che il gruppo tiolico della cisteina sia protetto oppure libero.
Il L-SBzl stabilizza il gruppo ReO3+ in una configurazione ottaedrica con il legante disposto
sul piano equatoriale rispetto al gruppo oxo. Un atomo di cloro in trans all‟ossigeno del gruppo oxo
completa la esacoordinazione.
Il L-STrt reagisce con l‟oxorenio con perdita del gruppo protettore dello zolfo cisteinico
grazie alle proprietà coordinanti del metallo, formando un complesso oxorenio-PN2S con il set
coordinativo disposto sul piano equatoriale in una configurazione a piramide a base quadrata.
Il L-SH reagisce con il ReOCl3(PPh3)2 producendo lo stesso complesso ottenuto con la
specie L-Strt, mentre reagisce con il tetrafenilfosfonio oxotetraclororenio (V) portando alla
formazione di un composto insolubile non ben definito.
Per ovviare ad eventuali interferenze durante la complessazione da parte del gruppo
carbossilico libero presente nei composti L-STrt e L-SH, è stato sintetizzato il corrispondente
legante metilestere: il L‟-SH reagisce con il tetrafenilfosfonio oxotetraclororenio (V) portando
ancora una volta al complesso pentacoordinato, ma attraverso una reazione più vantaggiosa sia dal
punto di vista qualitativo (prodotto più pulito) che quantitativo (resa più elevata).
La sostituzione dei gruppi fenilici nel L-Strt con gruppi metilici e metossipropilici al fine di
aumentarne l‟idrofilia ha effettivamente portato a composti con una elevata solubilità in acqua. Gli
studi preliminari di complessazione confermano le proprietà coordinanti del set PN2S e fanno
supporre la possibilità di ottenere complessi marcati con 99mTc e 186/188Re altrettanto idrofili che non
alterino la solubilità, in condizioni fisiologiche, delle molecole biologicamente attive ad essi
coniugate.
PARTE SPERIMENTALE
1) N- -Tritil-glicinil-S-tritil-L-cisteina (1).
Ad una soluzione di tritilglicina (1.50 g, 4.7 mmoli) e idrossibenzotriazolo (0.71 g, 5.2 mmoli)
in cloroformio anidro (100 ml) raffreddata in bagno di ghiaccio è stata aggiunta N,N‟dicicloesilcarbodiimmide (0.98, 4.7 mmoli): la miscela è stata mantenuta per 15 minuti a 0° C,
quindi portata a temperatura ambiente e agitata per 3 ore. Il precipitato di dicicloesilurea formatosi è
stato filtrato e alla soluzione risultante sono state aggiunte S-tritil-L-cisteina (2.06 g, 5.6 mmoli) e
trietilammina (0.79 ml, 5.6 mmoli) in cloroformio (50 ml). Dopo 5 ore il solvente è stato rimosso
per evaporazione a pressione ridotta; il residuo è stato quindi ripreso con etile acetato, lavato con
2% KHSO4 (2 100 ml), 5% Na2CO3 (2 100 ml) e acqua (1 100 ml), quindi anidrificato su Na2SO4
ed evaporato a secchezza. L‟olio residuo è stato ricristallizzato da etere/etere di petrolio. Resa
97.60%1H NMR (CDCl3): 2.72-2.78 (m, 2, CH2 Cys), 2.89 (d, 2, CH2 Gly,), 4.28 (q, 1, CH Cys),
7.14-7.99 (m, 30, ArH), 7.96 (d, 2, NH,). 13C NMR (CDCl3): 35.10 C Cys, 49.04 C Gly, 61.53 C
Cys, 67.45 CPh3, 72.04 CPh3, 127.72, 127.82, 129.05, 129.20, 129.82, 130.64, 145.64, 146.22 CArom,
172.59 CONH.
2) Glicinil-S-tritil-L-cisteina acetato (2).
Il composto 1 (1.00 g, 1.50 mmoli) è stato sciolto in una miscela di acido acetico (8 ml) e acqua
(2 ml) e riscaldato per 50 minuti in bagno di vapore. Per raffreddamento è precipitato il
trifenilcarbinolo: per aggiunta di etere il carbinolo è passato in soluzione con contemporanea
precipitazione del pordotto 2. Resa 98.72%. 1H NMR (DMSO-d6): 1.91 (s, 3H, COCH3), 2.40-2.42
(m, 2H, CH2 Cys), 3.47 (dd, 2H, CH2 Gly), 4.15 (q, 1H, CH Cys), 7.20-7.33 (m, 15H, ArH), 8.28 (b,
1H, NH)
3) N- N-(3-Difenilfosfinopropionil)glicinil -S-tritil-L-cisteina (3).
Il composto 2 (0.50 g, 1.04 mmoli) è stato sospeso in atmosfera inerte in diclorometano anidro e
degasato (50 ml) ed è stata quindi aggiunta trietilammina (0.14 ml, 1.04 mmoli). Alla soluzione
risultante è stata addizionato l‟estere succinimmidico dell‟acido difenilfosfinopropionico (0.34 g,
0.95 mmoli) ed il pH portato a 8.5-9 con trietilammina. Dopo 6 ore di agitazione in atmosfera inerte
il solvente è stato rimosso per evaporazione a pressione ridotta; il residuo oleoso è stato quindi
ripreso con etile acetato, lavato con 2% KHSO4 (2 50 ml), 5% Na2CO3 (2 50 ml) e acqua (1 100
ml), quindi anidrificato su Na2SO4 ed evaporato a secchezza.. L‟olio residuo è stato ricristallizzato
da etere/etere di petrolio degasati. Resa: 88.76%. Anal.: calcolato: C39H37N2PSO4: C, 70.89; H,
5.64; N, 4.24; trovato: C, 69.98; H, 5.67; N, 4.22. 1H NMR (CDCl3): 2.23-2.37 (m, 2H, PCH2),
2.61-2.76 (m, 4H, PCH2CH2, CH2 Cys), 3.75-3.94 (ddd, 2H, CH2 Gly), 4.40 (“q”, 1H, CH Cys),
6.51 (b, 1H, NH), 6.91 (d, 1H, NH), 7.16-7.49 (m, 25H, ArH). 13C NMR (CDCl3): 23.34 PCH2,
32.22 PCH2CH2, 32.40 C Cys, 42.74 C Gly, 52.15 C Cys, 66.37 CPh3, 126.61, 127.84, 128.44,
128.51, 128.68, 129.51, 132.61, 132.79, 137.87, 144.60 CArom, 172.55 CONH, 174.68 CONH.
4) N,S-Ditritil-L-cisteina metilestere (4).
Ad una sospensione di S-tritilcisteina metilestere hydrochloride (0.50 g, 1.2 mmoli) in
diclorometano anidro e degasato (100 ml) è stata aggiunta trietilammina (0.67 ml, 4.8 mmoli)
seguita da trifenilclorometano (0.67 g, 2.4 mmoli). Dopo 20 ore a temperatura ambiente la
soluzione è stata lavata con 2% KHSO4 (2 100 ml) e acqua (1 100 ml), quindi anidrificata su
Na2SO4 solvente è stato rimosso per evaporazione a pressione ridotta. Il prodotto è stato
ricristallizzato per lenta evaporazione di una soluzione in etere/metanolo (1:4). Resa: 68.45%. 1H
NMR (CDCl3): 2.50-2.60 (m, 2, CH2), 3.15 (s, 3, OCH3,), 3.40-3.45 (m, 1, CH), 7.20-7.45 (m, ArH,
NH).
5) S-Trityl-L-cysteine metilestere (5).
Il composto 4 (0.25 g, 0.4 mmoli) è stato sospeso in acido acetico (1.6 ml) e acqua (0.4 ml) e
scaldato per 40 minuti in bagno di vapore. Dopo raffreddamento la miscela è stata diluita con
diclorometano (50 ml) e aggiunta di trietilammina, quindi lavata con 2% KHSO4 (2 50 ml) e acqua
(2 50 ml). Il solvente è stato quindi evaporato a pressione ridotta e il composto 5 è stato purificato
con flash column chromatography (CHCl3:MeOH 9:1). Resa 91%. 1H NMR (CDCl3): 2.40-2.60 (m,
2, CH2), 3.20-3.25 (m, 1), 3.65 (s, 1, OCH3), 7.20-7.40 (m, ArH).
6) N- -Tritil-glicinil-S-tritil-L-cisteina metilestere (6).
Metodo A. Ad una soluzione del composto 5 (0.66 g, 1.7 mmoli) in diclorometano anidro
(100 ml) è stata aggiunta 1-etil-3-(3-dimetilamminopropil)carbodiimide (0.40 g, 2.04 mmoli) e Ntritilglicina (0.50 g, 2.04 mmoli): la miscela è stata mantenuta per 15 minuti a 0° C, quindi portata a
temperatura ambiente e addizionata di trietilammina fino a pH 8.5-9. La soluzione mantenuta sotto
agitazione per 1 ora è stata quindi lavata con 2% KHSO4 (2 100 ml), 5% Na2CO3 (2 100 ml) e
acqua (1 100 ml), quindi anidrificato su Na2SO4. Il solvente è stato rimosso a pressione ridotta
ottenendo un residuo oleoso ed il prodotto è stato ricristallizzato da etere/etere di petrolio. Resa
72.05%.
Metodo B. Una soluzione del composto 1 (0.30 g, 0.45 mmoli) in metanolo anidro è stata portata
a 0° C e aggiunta di Py-BOP (0.23g, 0.45 mmoli) e diisopropiletilammina (0.12 ml, 0.45 mmoli).
La soluzione è stata mantenuta a 0° C per 8 ore. Il solvente è stato quindi rimosso a pressione
ridotta ed il residuo ripreso con acetato di etile, lavato con 2% KHSO4 (2 50 ml), 5% Na2CO3
(2 50 ml) e acqua (1 100 ml), quindi anidrificato su Na2SO4. Il solvente è stato rimosso a pressione
ridotta ed il prodotto cristallizzato da etere/etere di petrolio. Resa 85.36%. 1H NMR (CDCl3): 2.702.80 (m, 2, CH2), 2.92 (t, 2, CH2), 3.76 (s, 3, OCH3), 4.68 (q, 1, CH), 7.20-7.45 (m, ArH), 8.07 (d,
1, NH).
7) Glicinil-S-tritil-L-cisteina metilestere acetato (7).
Una soluzione del composto 6 (0.25 g) in acido acetico e acqua è stata scaldata in bagno di
vapore per 2 ore. Dopo raffreddamento è stato aggiunto diclorometano e la soluzione è stata lavata
con 5% Na2CO3 (2 50 ml) e acqua (1 100 ml), quindi anidrificata e il solvente rimosso a
pressione ridotta. Il composto 7 è stato separato con flash column chromatography. Il prodotto è
stato quindi ricristallizzato per triturazione con etere di petrolio. Resa 65%. 1H NMR (CDCl3): 1.98
(s, 3H, COCH3), 2.44-2.47 (m, 2H, CH2 Cys), 3.54 (dd, 2H, CH2 Gly), 3.77 (s, 3, OCH3), 4.17 (q,
1H, CH Cys), 7.22-7.40 (m, 15H, ArH), 8.11 (b, 1H, NH).
8) N- N-(3-Difenilfosfinopropionil)glicinil -S-tritil-L-cisteina metilestere (8)
Metodo A. Il composto 7 (0.50 g, 1.04 mmoli) è stato solubilizzato in atmosfera inerte in
diclorometano anidro e degasato (50 ml) ed è stata quindi aggiunta trietilammina (0.14 ml, 1.04
mmoli). Alla soluzione risultante è stata addizionato l‟estere succinimmidico dell‟acido
difenilfosfinopropionico (0.34 g, 0.95 mmoli) ed il pH portato a 8.5-9 con trietilammina. Dopo 10
ore di agitazione in atmosfera inerte il solvente è stato rimosso per evaporazione a pressione ridotta;
il residuo oleoso è stato quindi ripreso con etile acetato, lavato con 2% KHSO4 (2 50 ml), 5%
Na2CO3 (2 50 ml) e acqua (1 100 ml), quindi anidrificato su Na2SO4 ed evaporato a secchezza.
L‟olio residuo è stato ricristallizzato da etere di petrolio degasato. Resa: 88.76%.
Metodo B. Una soluzione del composto 3 (0.40 g, 0.60 mmoli) in metanolo anidro e degasato è stata
portata a 0° C e aggiunta di benzotriazoliloxi-tris(pirrolidino)-fosfonio esafluorofosfato (PyBOP,
0.31 g, 0.60 mmoli) e diisopropiletilammina (0.20 ml, 0.60 mmoli). La soluzione è stata mantenuta
a 0° C per 6 ore. Il solvente è stato quindi rimosso a pressione ridotta ed il residuo ripreso con
acetato di etile, lavato con 2% KHSO4 (2 50 ml), 5% Na2CO3 (2 50 ml) e acqua (1 100 ml),
quindi anidrificato su Na2SO4. Il solvente è stato rimosso a pressione ridotta ed il prodotto
cristallizzato da etere di petrolio. Resa 92.00% 1H NMR (CDCl3): 2.30-2.47 (m, 2H, PCH2), 2.602.75 (m, 4H, PCH2CH2, CH2 Cys), 3.63 (s, 3H, OCH3), 3.87 (d, 2H, CH2 Gly), 4.70 (“q”, 1H, CH
Cys), 6.49 (t, 1H, NH), 7.22 (d, 1H, NH), 7.21-7.43 (m, 25H, ArH).
Oxo N- N-(3-Difenilfosfinopropionil)glicinil -L-cisteina metilestere Renio (V).
Il composto 8 (0.30 g, 0.43 mmoli) è stato solubilizzato in acido trifluoroacetico: è stato
quindi aggiunto treitilsilano fino a decolorazione della soluzione. Dopo 1 ora l‟acido
trifluoroacetico è stato rimosso in corrente di azoto e per evaporazione a pressione ridotta. Il residuo
è stato quindi solubilizzato in etanolo assoluto degasato ed è stato aggiunto ad una soluzione in
etanolo assoluto di tetrafenilfosfonio oxotetraclororenio (V) (0.16 g, 0.43 mmoli): si è formato un
precipitato verde che è stato filtrato, risospeso in etanolo assoluto e addizionato di trietilammina
fino a pH 8.5-9 ottenendo una soluzione rosso mattone. Il prodotto è stato cristallizzato per lenta
evaporazione del solvente. ESI MS: m/z 617 M-CH3 -; m/z 573 M-CO2CH3 -.
UNITA’ LOCALE DI PALERMO
Direttore: Prof. Lorenzo Pellerito
Indagini pH-metriche e spettroscopiche (1H,13C,119Sn NMR;
119
Sn Mössbauer) sono state
condotte per caratterizzare l'interazione dello ione dimetilstagno2+ con amino acidi e dipeptidi quali
glicina (gly), glicilglicina(gly-gly) acido imidazolo-4-acetico, istamina, istidina, glicil-istamina,
glicil-istidina(gly-his) e beta-alanil-istidina (carnosina). La istamina e la glicil-istamina, che
posseggono un solo atomo di azoto donatore, non si è osservata formazione di complessi. Le specie
idrolizzate del catione dimetilstagno(IV) sono sempre dominanti rispetto ai complessi formati con
gli altri leganti, ad eccezione dei complessi formati con gly-gly e gly-his. Per questi due leganti,
nell'intervallo di pH neutro si ottengono complessi COO-, N- e NH2 coordinati, con struttura
trigonale bipiramidale. Questi complessi rappresentano i primi esempi di complessi in cui cationi
diorganostagno sono capaci di provocare la deprotonazione dell'azoto peptidico in soluzione
acquosa, a pH inaspettatamente basso. In questo processo lo ione carbossilato si comporta da
gruppo chelante, in contrasto con gli altri ioni metallici che invece coordinano l'azoto amidico.
Sono stati sintetizzati complessi di diorgano e triorganostagno(IV) con l'acido orotico, (2,6diidrossipirimidin-carbossilico)= H3Or, in cui l'acido orotico si comporta sia da mono che da
dianionico nei confronti della metà diorganostagno(IV), originando complessi di formula
R2Sn(H2Or)2 e R2Sn(HOr), mentre nei confronti dei triorganostagno(IV)orotati avviene soltanto la
monodeprotonazione dell'acido orotico dando R3SnH2Or. La struttura allo stato solido è stata
proposta sulle basi di dati spettroscopici IR e Mossbauer, mentre la natura dei complessi in
soluzione è stata indagata mediante 1H e 13C NMR. Infine, i complessi ottenuti hanno mostrato, in
vivo, proprietà inibenti nei confronti di embrioni di Ciona intestinalis. I risultati mostrano che gli
orotato complessi interferiscono con la polimerizzazione della tubulina durante il processo di
divisione cellulare durante lo sviluppo embrionale.
Derivati dialchil e trialchilstagno(IV)tiamminapirofosfato hanno mostrato avere formule
generali del tipo R2Sn(HTPP)2·nH2O [n = 2 per il metil derivato ed n = 4 per il butilderivato] e
R3SnHTPP·nH2O [n = 2 per il metil derivato ed n = 1 per il butil derivato]. I dati infrarossi
suggeriscono un coinvolgimento del solo ossigeno del gruppo fosfato nella coordinazione dello
stagno sia nei diorgano che nei triorganostagno derivati originando così strutture ottaedriche e
trigonale bipiramidale, strutture confermate mediante arazionalizzazione dello splitting di
quadrupolo nucleare, determinato con spettroscopia Mossbauer. Dati 1H e
13
C NMR suggeriscono
che le strutture dei complessi ottenuti vengono mantenute in soluzione di D2O. Per tutti i complessi
stabili in soluzione è stata studiata l'attività citotossica nei confronti di fibroblasti embrionali di topo
immortalizzati (NIH-3TP, verso i quali mostrano un chiaro effetto inibitore della loro crescita.
Inoltre la citometria di flusso è stata utilizzata per indagare l'attività citotossica di diorgano e
triorganostagno derivati della [meso-tetra(4-carbossifenil)porfina], (=TPPC), con stechiometria
(R2Sn)2TPPC e (R3Sn)4TPPC nei confronti del ciclo cellulare di cellule renali umane per cercare di
capire l'origine della inibizione della crescita cellulare da parte dei complessi precedentemente
menzionati. La distribuzione del contenuto di DNA in funzione del ciclo cellulare è stata investigata
mediante la citometria a flusso, una tecnica potente capace di evidenziare alcuni aspetti della
citotossicità indotta. Un approccio ANN (artificial neural network) è stato utilizzato per valutare la
geometria di coordinazione attorno all'atomo di stagno(IV) per complessi pentacoordinati di
diorganostagno con dipeptidi. L'applicazione di tale metodo fornisce un rapido ed accurato metodo
per la corretta classificazione di spettri Mössbauer.
La spettroscopia Mössbauer è stata utilizzata per investigare la natura delle interazioni tra sali e
complessi di organostagno(IV) con molecole biologiche. In particolare è stata riportata l'analisi e la
speciazione di organostagno derivati presenti nell'ambiente circostante e l'interazione di composti
organostagno(IV) con emoglobina, acido desossiribonucleico, e di sistemi ternari quali
organostagno-amino acidi-acidi nucleici. La struttura delle specie ottenute e la natura degli atomi
coordinati è stata investigata mediante tecniche spettroscopiche, sia allo stato solido che in
soluzione.
Gli
effetti
di
alcuni
organostagno(IV)[meso-tetra(4-carboxyporphinate)]
con
formula
(R2Sn)2TPPC e (R3Sn)4TPPC (R= Me, n-Bu, Ph)sono stati utilizzati in vivo sullo sviluppo
embrionale di ascidiacee. Il composto più tossico è il (Bu3Sn)4TPPC, il quale già a concentrazione
dell'ordine di 10-7 M impedisce la suddivisione in due cellule. Per correlare questo arresto di
sviluppo embrionale con il percorso metabolico, e per capire perchè mai le organelle cellulari per
primi subiscono danni chimici, le uova fecondate in soluzioni 10-5 e 10–7 M di (Bu3Sn)4TPPC sono
stati analizzati per il loro contenuto in DNA, RNA, proteine, glucosio, lipidi ed ATP, confrontando i
valori ottenuti con quelli delle uova fecondate di controllo.
Gli effetti pleiotropici dell‟esposizione a derivati organostagno ed in particolare ai
tributilstagno(IV) derivati sono stati studiati in funzione della concentrazione della metà
tributilstagno. In particolare sono stati indagati I valori dei livelli di ATP, lipidi ed acidi nucleici,
glucosio ed ioni Ca2+ e l‟influenza che I tributilstagno derivati hanno sulla sintesi di queste
molecole essenziali per lo sviluppo embrionale. I risultati ottenuti mentre mostrano una
diminuizione di queste sintesi, indicano un marcato aumento della concentrazione in ioni Ca2+ e di
glucosio. Inoltre viene inibita l‟attività enzimatica della Creatina chinasi e viene stimolata l‟attività
della Fosfatasi alcalina e della Colinesterasi (a concentrazioni dell‟ordine di 10-5 M in soluzione di
acqua di mare sterile). Gli effetti sulla riproduzione e sullo sviluppo embrionale sono causati da un
meccanismo di estrema difesa cellulare che si instaura per evitare deleterie conseguenze sulla
sopravvuivenza cellulare. Sono state determinate le geometrie molecolari, parametri termodinamici
e distribuzioni di carica elettronica di diorgano e triorganostagno(IV) derivati di [protoporfirina-IX]
e di [meso-tetra(4-carbocarbossifenil)porfina] con metodi semi-empirici ed ab initio. Per studiare i
dettagli molecolari dei complessi, sono stati calcolati anche modelli moleoclari più semplici con il
metodo ab initio pseudo-potential. I risultati teorici estratti dai calcoli sono in perfetto accordo con i
dati sperimentali ottenuti con spettroscopia Mossbauer.
L‟idrolisi di metà organostagno e le interazioni con amminoacici, peptidi, carboidrati e loro
derivati, con acidi nucleici ed altri bioleganti sono stato oggetto di particolare attenzione, ciò che è
sfociato in una articolo-rivista accettato per la pubblicazione su Coordination Chemistry Review.
Infine, sono stati investigati gli effetti di tributilstagno(IV)derivati sulla metamorfosi di larve di
Ciona intestinalis. Le Ascidie sono Protocordati che non hanno follicoli tiroidei, ma che posseggono
ormoni tiroidei (THs) ed i loro precursori, 3-monoiodo-tirosina (= MIT) e 3,5-diiodo-tirosina (=
DIT) nel loro endostilo. Questi ormoni sono presenti anche allo stato larvale, localizzati nelle
cellule mesenchimali e la loro funzione sembra essere prevalentemente correlata alle trasformazioni
larvali. In conseguenza di ciò sono stati investigati gli effetti di tributilstagno derivati sul contenuto
e localizzazione della Tirosina (T4) dopo esposizione di larve di C. intestinalis per tempi e
concentrazioni differenti di tributilstagno(IV) derivati, i quali bloccano la metamorfosi, il che porta
al blocco della coda ed alla immobilità larvale. Come rivelato da indagini immmunoistochimiche,
nelle larve normali, T4 si trova in tutte le cellule mesenchimali, diffusa nella cavità del corpo, sotto
le papille adesive e intorno all‟intestino. In contrasto, nelle larve esposte a soluzioni 10 -5 e 10-7 M di
tributilstagno derivati, T4 viene rilevato rispettivamente solo nel 5 e 25% di tutte le cellule
mesenchimali. Indagine Radio-immunologiche (RIA) mostrano una diminuizione del 70% del
contenuto di T4 nelle larve esposte ai composti, rispetto a quello non esposte.
Inoltre viene inibita la neosintesi delle TH. I risultati indicano che gli organostagno derivati si
comportano da distruttori endocrini (= ED) nei confronti delle Ascidie e scompensano il
metabolismo di T4. Questi risultati implicano che l‟attività ED si conserva dagli invertebrati agli
esseri umani.
Sono stati analizzati dal punto di vista termodinamico le problematiche riguardanti:
(a) le interazioni tensioattivo-ciclodestrina
(b) le interazioni sostanze macromolecolari-tensioattivi
a) Sono state determinate le entalpie di trasferimento di ciclodestrine modificate da acqua a
soluzioni acquose di alchilcarbossilati di sodio. L‟analisi quantitativa dei risultati sperimentali è in
accordo con l‟idea che nella regione pre-micellare abbia luogo la formazione del complesso di
inclusione tra ciclodestrina ed il tensioattivo e che la ciclodestrina interagisca con le micelle. Per
una data ciclodestrina, la termodinamica di complessazione è influenzata notevolmente dalla natura
del tensioattivo.
b) Le interazioni tensioattivo-macromolecola hanno riguardato classici tensioattivi e polimeri
ed additivi di varia natura e copolimeri a blocchi del tipo poli(etilenossido)a-poli(propilenossido)bpoli(etilenossido)a, dove a e b sono le unità ripetitive. Le proprietà termodinamiche del tipo
poli(etilenossido)13-poli(propilenossido)30-poli(etilenossido)13 (L64) in presenza di perclorato di
sodio evidenziano che l‟elettrolita gioca lo stesso ruolo della temperatura mentre quelle in presenza
di elettroliti idrofobi evidenziano essenzialmente interazioni idrofile elettrolita-L64. Le interazioni
tra L64 (sia in forma dispersa che micellizzata) e micelle di sodio decilsolfato portano alla
formazione di micelle miste con un grado di dissociazione elevato.
Le entalpie di trasferimento del poli(etileneglicole) 400 e 35000 da acqua a soluzioni acquose
di perfluoroalcanoati di sodio hanno messo in evidenza la presenza di un processo cooperativo sia
nella regione pre- che in quella post-micellare. L‟energia libera standard di formazione del
complesso polimero-tensioattivo di stechiometria1:n indica che la driving force del processo è di
natura idrofobica e che clusters di tensioattivo sono associati al polimero. L‟entropia governa detto
processo. L‟energia libera, l‟entalpia e l‟entropia standard di formazione del complesso polimerotensioattivo micellizzato di stechiometria 1:q indicano che le micelle siano avvolte dalle catene
polimeriche.
Nel quadro degli studi di speciazione sono state effettuate indagini sistematiche sulle
interazioni di leganti carbossilici nei confronti di ioni metallici e organometallici. In particolare è
stata definita la speciazione di leganti policarbossilici in acqua di mare artificiale, studiando anche il
comportamento di poliacrilati e polimetacrilati (P.M. 2000, 5000, 2000 u.m.a.) come molecole
modello rappresentative delle frazioni solubili degli acidi fulvici nelle acque naturali.
Sono stati approfonditi gli studi sulla chimica in soluzione acquosa dei cationi di
organostagno(IV), definendo in particolare i processi di idrolisi e la speciazione in acqua di mare di
mono-, di- e trimetilstagno(IV).
E‟ stato pubblicato il libro “Chemical Processes in Marine Environments” quale risultato della
2^ edizione della International School on Marine Chemistry, organizzata in Ustica (Settembre 1998)
in collaborazione con la Riserva Marina e con il patrocinio della SCI e del CNR.
UNITA’ DI RICERCA DI PARMA
Direttore: Prof. Marisa Ferrari Belicchi
L’unità di ricerca di Parma si compone di 3 gruppi che si distinguono per le proprie diverse
competenze nel campo della chimica dei sistemi biologici. L’attività di ricerca di ciascun gruppo
verrà pertanto illustrata separatamente.
1. Gruppo di ricerca della Prof. A. M. Lanfredi Manotti
(F. Ugozzoli e C. Massera)
Studio di complessi di rutenio(II) e rutenio(III) ad attività antitumorale
Il gruppo, che ha una pluriennale attività di ricerca nel campo della Strutturistica
Diffrattometrica, si occupa sia della caratterizzazione strutturale allo stato solido mediante
diffrazione di raggi-X sia dello studio delle correlazioni proprietà-struttura di complessi metallici
aventi potenziale attività antitumorale.
Negli ultimi anni, in collaborazione con il Department of Chemistry dell‟ Università di
Leiden (Olanda) sono state sviluppate ricerche su complessi metallici con frammenti s-triazolici
variamente sostituiti. In particolare l‟attenzione è stata rivolta a complessi di rutenio con purine
modificate, simili a derivati di rutenio ad attività anticancerogena per i quali, come riporta la
letteratura, sembrano svolgere un ruolo determinante ai fini dell‟attività antiproliferativa le loro
reazioni idrolitiche di acquazione. In questa ottica la caratterizzazione di due monoacqua tricloro
complessi di rutenio(III) con leganti purinici modificati ci ha permesso di evidenziare che tali
leganti eterociclici sono in grado di finalizzare le proprietà fisiche e chimiche dei complessi
attraverso un sistema intramolecolare multiplo di legami ad idrogeno.
Essendo d‟altra parte importante studiare il binding dei rutenio-complessi con basi modello
del DNA sono state studiate le proprietà del complesso di rutenio(II)
(azpy=2-(fenilazo)piridina;
-[Ru(azpy)2(NO3)2],
indica l‟ isomero in cui i gruppi coordinanti ONO2, N(py), ed N(azo)
sono cis, trans e cis rispettivamente) ed il suo modo di binding a basi modello del DNA quali 9etilguanina (9egua) e guanosina (guo). I risultati sono stati anche confrontati con quelli ottenuti dal
binding di basi modello al complesso bis(bipiridil)rutenio(II). Lo studio strutturale del complesso [Ru(azpy)2(NO3)2] ha permesso di interpretare gli spettri NOESY NMR dei suoi complessi con le
basi modello e quindi di determinare le orientazioni del 9egua e guo mostrando come i derivati della
guanina in complessi di azpy possono avere maggiori libertà stereospaziali che non nei complessi
[Ru(bpy)2Cl2] e che tale flussionalità e‟ importante nel binding del complesso -[Ru(azpy)2(NO3)2]
al DNA.
2. Gruppi di ricerca della Prof. Marina Cingi Biagini
a) (M. Lanfranchi, L. Marchiò, M. A. Pellinghelli, M. Tegoni)
Nuovi complessi con leganti triazolici, triazolinici e triazolidinici
I derivati ciclici della tiocarbonoidrazide rappresentano una classe di composti relativamente
poco studiata. I derivati 4-ammino-5-tiosso-s-triazolinici sono conosciuti in letteratura per la loro
capacità di complessare numerosi ioni metallici con caratteristiche soft e sono da considerarsi acidi
monoprotici. In soluzione mostrano tautomeria tione-tiolo e hanno mostrato un interessante
meccanismo di desolforazione promosso dal rame(II).
I complessi di Werner del rutenio con derivati ciclici della tiocarbonoidrazide sono una
nuova classe di composti che possono essere interessanti nello sviluppo della chimica di
coordinazione dei metalli di transizione. In tempi recenti alcuni ricercatori hanno studiato le
proprietà antitumorali di alcuni complessi di Ru(II) e Ru(III), in particolare i derivati con dmso. Dal
nostro gruppo di ricerca sono stati sintetizzati e caratterizzati, spettroscopicamente e tramite
diffrazione ai raggi X su cristallo singolo, alcuni leganti triazolici solforati e alcuni loro complessi
di Ru(II) e Ru(III). E‟ stato studiato il comportamento chimico di derivati triazolici, tiodiazolici e
triazolinici della tiocarbonoidrazide verso il complesso cis-[RuCl2(dmso)4] (1). La reazione di 4ammino-5-metiltio-3-(2-piridil)-1,2,4-triazolo (L1) con 1, forma due complessi diastereomerici,
trans,cis-[RuCl2(dmso)2(L1)] (2) e il complesso chirale cis,cis-[RuCl2(dmso)2(L1)] (3).
La reazione di 1 con 2-(2-formilidrazino)-1,3,4-tiodiazolo in metanolo ha formato il
complesso trans,cis-[RuCl2(dmso)2(L2)] (4) in cui il legante L2 risulta deformilato. Il processo è
stato catalizzato da Ru(II) ma con resa molto bassa. La sintesi di complessi di Ru con 4-ammino-3metil-1,2,4- 2-triazolina-5-tione (L3) e 4-ammino-3-etil-1,2,4- 2-triazolina-5-tione (L4) ha rivelato
un interessante processo di ossidazione del metallo promosso da dmso. La reazione di 1 con questi
leganti in soluzione acquosa di HCl 6 M ha portato alla formazione dei complessi mer[RuCl3(dmso)(L3)] ½H2O
(5)
e
mer-[RuCl3(dmso)(L4)]·½H2O
(6).
Il
complesso
mer-
[RuCl3(dmso)(L4)]·CH3COOH·H2O (7) è stato ottenuto ricristallizando 6 da etilacetato. A nostra
conoscenza questi complessi rappresentano il primo caso di chelazione di un sistema di tipo N-N-CS.
L3 è stato trovato agire come monodentato tramite l‟atomo di zolfo o bidentato chelante
tramite lo zolfo e l‟azoto amminico o tridentato chelante N,S e a ponte con l‟azoto endociclico ed
infine tridentato
3.
Data la grande versatilità di L3 è stato studiato il comportamento legante di
composti simili come 4-ammino-1,2,4- 2-triazolina-5-tione (L5) e 4-ammino-3-etil-1,2,4- 2triazolina-5-tione (L4) rispettivamente verso Ag(I) e Cu(I). Tramite analisi di diffrazione ai raggi X
su cristallo singolo sono state determinate le strutture molecolari di [Ag(HL5)2]NO3 e
[CuCl(HL4)3]. Nel composto di argento sono presenti (evento non frequente) contemporaneamente
specie dimeriche e polimeriche e il legante mostra un nuovo modo di coordinarsi chelando N,S e
disponendosi a ponte tramite l‟atomo di zolfo. Il complesso tetraedrico di rame(I) (simmetria C3) è
chirale e questo è dovuto alla disposizione elicoidale delle 3 molecole triazoliniche S-coordinate
che interagiscono tramite legami d‟idrogeno con l‟atomo di cloro coordinato e con disposizione
testa-coda con altri complessi formando catene che scorrono lungo c. E‟ stata osservata una
risoluzione spontanea e nel cristallo analizzato sono presenti solo conformeri .
I derivati ciclici della carbonoidrazide sono oggetto di studio nel campo biologico,
tecnologico e analitico, a tal fine sono state studiate le capacità complessanti di 4-ammino-1,2,4triazolidina-3,5-dione (urazina) sia come legante neutro (Hur) che monoanionico (ur) verso cationi
divalenti (Co, Ni, Cu, Zn). Sono stati sintetizzati e caratterizzati i complessi CoCl 2(Hur)2.4H2O,
NiCl2(Hur)2.4H2O e Zn(SO4)(Hur).2H2O nei quali l‟urazina agisce come legante neutro. Il
complesso polimerico di zinco ha dato luogo a risoluzione spontanea e nel cristallo esaminato è
presente solo l‟isomero A. Partendo dal sale sodico dell‟urazina sono stati sintetizzati i complessi
Co(ur)2.4H2O, NiCl(ur).H2O e CuCl(ur).2H2O. Partendo da soluzioni acquose di urazina sono stati
sintetizzati i complessi Cu3(NO)3(ur)4.6H2O, Cu(ur)2.4H2O e Cu2(SO4)(ur)2.2H2O. Di questi ultimi
tre complessi e di CoCl2(Hur)2.4H2O e Zn(SO4)(Hur).2H2O sono state determinate le strutture
cristalline ai raggi X. In questi composti Hur agisce solo come chelante N,O mentre ur nei
complessi di rame esaminati agisce o come monodentato tramite l‟azoto endociclico deprotonato o
come tridentato o come tetradentato mostrando una grande versatilità.
b) (F. Dallavalle, M. Tegoni)
Equilibri di formazione di complessi ternari di Cu(II) con acidi (S)-amminoidrossammici e D- o
L-amminoacidi in soluzione
Gli acidi amminoidrossammici e più in generale leganti idrossammici variamente
funzionalizzati sono oggetto di studio sia in campo chimico che biologico, specialmente per il ruolo
che rivestono nell‟accumulo e nel trasporto di ioni metallici negli organismi viventi. E‟ ben noto il
caso del sideroforo naturale Desferossiammina B, impiegato nel trattamento di pazienti beta-
talassemici come sequestrante specifico per ferro(III). Inoltre, alcuni acidi idrossammici e
amminoidrossammici presentano la capacità di funzionare come inibitori enzimatici mediante un
meccanismo che sembra coinvolgere la chelazione di questi leganti con gli ioni metallici dei
metalloenzimi, anche se sono stati scarsamente studiati i complessi ternari che questi leganti
formano con ioni metallici di transizione e amminoacidi.
Nell‟ambito della ricerca riguardante la stereoselettività termodinamica nella formazione di
complessi ternari di ioni bi- e trivalenti con leganti chirali ammino-idrossammici e D- o Lamminoacidi in soluzione è stato preso in esame il sistema Cu2+/ acido (S)-triptofanoidrossammico /
D- o L-amminoacido (Pro, Phe, Trp).
Gli equilibri di protonazione e di complesso-formazione sono stati studiati in soluzione
acquosa mediante potenziometria con elettrodo a vetro e spettrofotometria di assorbimento nel
visibile.
E‟ noto che le specie formate da Cu(II) con acidi -amminoidrossammici (HL) sono [CuL]+,
[Cu2L2H-1]+, [CuL2], [CuL2H-1]-. Lo studio degli equilibri del sistema Cu(II) / acido (S)triptofanoidrossammico (Trpha, HL) è risultato complicato a causa della insolubilità della specie
[Cu2L2H-1]+ che precipita nella zona di pH 4-6 per poi trasformarsi completamente nella specie
solubile [CuL2] e quest‟ultima nella specie [CuL2H-1]-.
Per superare questa difficoltà si è scelto un legante competitore di Trpha abbastanza forte,
quale EDTA, in grado di ridurre la concentrazione del complesso insolubile così da evitarne la
precipitazione anche a concentrazioni di metallo non troppo basse, ma con una scarsa tendenza a
formare complessi ternari con Trpha.
I valori delle costanti globali di protonazione e di complessazione (log
,
pqr
=
[CupLqHr]/[Cu]p[L]q[H]r a T = 25 ° C e I = 0.1 M (KCl)) sono risultati:
HL
9.13(1)
H2 L +
[CuL]+
[Cu2L2H-1]+
[CuL2]
16.16(1)
10.62(4)
20.68(6)
20.53(2)
[CuL2H-1]10.37(2)
I complessi sono stati caratterizzati mediante spettroscopia di assorbimento nel visibile
calcolando i parametri di
max/
che sono risultati in buon accordo con quelli già noti per altri
amminoidrossammati. In particolare è stato possibile confermare che la specie [CuL2H-1]- non è un
idrossocomplesso ([CuL2(OH)]-), ma contiene il gruppo idrossammico deprotonato anche
all‟ossigeno.
Nell‟ipotesi che in questi complessi lo ione Cu2+ presenti sempre una geometria ottaedrica
distorta, con due molecole di acqua assiali, si sono calcolati i contributi al campo dei leganti da
parte degli atomi di azoto del gruppo idrossammato (0.479(5) m-1) e idrossammato deprotonato
anche all‟ossigeno (0.623(12) m-1) secondo il modello di Billo.
Per valutare l‟eventuale presenza di stereoselettività termodinamica nella formazione di
specie ternarie con D- o L-amminoacidi, sono stati scelti prolina, fenilalanina e triptofano, cioè
quegli amminoacidi che avevano in precedenza mostrato le maggiori stereoselettività in analoghi
sistemi con leganti (S)-ammino-ammidici.
Rapporti favorevoli alla formazione della massima quantità delle specie miste (Cu:L:A =
1:1:1 o 1:1:2, HA = amminoacido) non sono risultati praticabili a causa della insolubilità del
complesso dinucleare [Cu2L2H-1]+, quindi per evitarne la precipitazione si è dovuto operare in
presenza di un forte eccesso di amminoacido (Cu/A da 1:15 a 1:30). Le specie ternarie individuate
sono [CuLA] e [CuLH-1A]- e i corrispondenti valori di log
Pro
D
sono risultati:
Phe
L
D
Trp
L
D
L
[CuLA]
18.67(2)
18.80(1)
18.01(3)
17.94(2)
18.46(4)
18.35(3)
[CuLH-1A]
9.22(1)
9.12(1)
8.02(5)
8.30(3)
8.25(5)
8.07(4)
La stereoselettività nella formazione dei complessi ternari è stata espressa come
log
SR
– log
SS
log
=
. I valori ottenuti sono i seguenti:
Pro
Phe
Trp
-0.13(2)
+0.07(4)
+0.11(5)
[CuLH-1A]- +0.10(1)
-0.28(6)
+0.18(6)
[CuLA]
Si può considerare significativa la stereoselettività presentata da entrambi i complessi
contenenti prolina e quella del complesso [CuLAH-1]- con fenilalanina. Comunque, l‟entità di
questa stereoselettvità è minore di quella ottenuta in precedenza con l‟analogo legante (S)triptofanammide.
La stereoselettività osservata è attribuibile a interazioni steriche tra i due residui amminoacidici
sulla base di modelli molecolari.
3. Gruppo di ricerca della Prof. M. Ferrari Belicchi
(F.Bisceglie, G.Pelosi, P.Tarasconi, R.Albertini, S.Pinelli)
Relazione struttura-attività biologica di nuovi complessi con tiosemicarbazoni aromatici
E' proseguita la nostra attività di ricerca basata su sintesi, caratterizzazione e valutazione di
alcune proprietà biologiche di complessi di interesse farmacologico, non analoghi al cis-platino,
contenenti il frammento tiosemicarbazonico con sostituenti di diverso ingombro e natura sugli
atomi di azoto amminico ed idrazinico, con lo scopo di confrontare le caratteristiche geometriche e
gli effetti biologici con quelli dei corrispondenti composti senza sostituenti sulla catena
tiosemicarbazonica.
In particolare i leganti aromatici precedentemente caratterizzati, Me-H3ut e Me2-H3ut (4metil e 4,4-dimetiltiosemicarbazide), ottenuti condensando il 5-formiluracile con tiosemicarbazidi
sostituite, sono stati fatti reagire con sali di rame variando anche il controione inorganico. Sono stati
isolati in forma cristallina e caratterizzati anche mediante diffrazione ai raggi X i complessi
[Cu(Me-H3ut)Cl2].H2O 1 [Cu(Me2-H3ut)Cl2].H2O 2 e [Cu(Me-H3ut)(NO3)(OH2)2]NO3 3. Si è
potuto osservare che nè il differente tipo di controione nè il numero diverso di sostituenti ha
influenzato la deprotonazione del legante che in tutti i complessi è presente in forma neutra. In
entrambi i complessi derivati dal cloruro rameico la coordinazione risulta quadrata piramidale (4+1)
ed implica tre atomi donatori SNO del legante ed un atomo di cloro nel piano di base. L'apice della
piramide è occupato dal secondo atomo di cloro con distanze molto più lunghe rispetto a quelle nel
piano di base in analogia con quanto rilevato in analoghi complessi di rame con il tiosemicarbazone
dell'uracile non sostituito studiati in precedenza. La struttura del composto 3 è costituita da cationi
complessi [Cu(Me-H3ut)(NO3)(OH2)2]+ e anioni nitrato. La geometria di coordinazione (4+2) è
una bipiramide allungata con gli atomi SNO del tiosemicarbazone neutro tridentato e una molecola
d'acqua nel piano di base, mentre le posizioni apicali sono occupate dall'ossigno dell'altra molecola
di acqua e dall'atomo O3 del gruppo nitrato monodentato più debolmente legati all'atomo di rame.
Legami di idrogeno tra O1W e O2W e gli atomi di ossigeno O4 ed O5 del gruppo nitrato coordinato
di una molecola in posizione x, y, z+1 creano catene di poliedri che si sviluppano nella direzione
dell'asse
z.
Il
corrispondente
complesso
con
il
legante
non
sostituito
di
formula
[Cu(H3ut)(OH2)2](NO3)2.H2O presenta invece geometria di coordinazione piramidale quadrata
(4+1) e i nitrati sono entrambi ionici. Legami di idrogeno tra le molecole d'acqua coordinate
appartenenti a poliedri centrosimmetrici determinano la formazione di dimeri.
Sui leganti ed i relativi complessi sono stati effettuati saggi biologici, in particolare prove di
inibizione della proliferazione cellulare, su linee cellulari leucemiche umane U937 in
collaborazione con l'Istituto di Patologia Speciale Medica dell'Università di Parma. Il legante libero
H3ut, sciolto in DMSO e addizionato al mezzo di cultura, immediatamente precipita, pertanto non è
stato possibile valutare la sua attività biologica. Entrambi i leganti, con uno o due gruppi metile
sull'azoto amminico Me-H3ut e Me2-H3ut, non inibiscono la proliferazione cellulare. Per quanto
riguarda i complessi è stato fatto un confronto della loro attività di inibizione con quella dei
corrispondenti
complessi
con
il
legante
non
[Cu(H3ut)Cl2].2H2O
sostituito
4
e
[Cu(H3ut)(OH2)2](NO3)2.H2O 5. Mentre i complessi 1, 2 e 3 non inibiscono la proliferazione,
entrambi i complessi con il legante H3ut presentano una inibizione maggiore del 50% della crescita
cellulare ad una concentrazione rispettivamente di 40
g/ml per il cloruro e di 35
g/ml per il
nitrato.
INIBIZIONE DELLA PROLIFERAZIONE CELLULARE SULLA LINEA LEUCEM ICA UM ANA U937
4
x 10 cells/ml
300
250
200
150
100
50
0
CTR
Me-H3ut
Me 2-H3ut
1
2
3
4
5
Per questi composti attivi si è eseguito anche il saggio sulla linea cellulare leucemica CEM e
si è osservata una analoga attività, per il nitrato complesso già alla concentrazione di 30 g/ml.
Questi risultati suggeriscono che il gruppo amminico terminale non sostituito sembra avere un ruolo
rilevante nel determinare un effetto di inibizione che però non si verifica attraverso un meccanismo
apoptotico.
In parallelo seguendo sempre un procedimento di screening, per valutare una possibile
attività biologica anche di leganti potenzialmente SN bidentati, sono stati sintetizzati diversi
tiosemicarbazoni di aldeidi naturali tra i quali il tiosemicarbazone del citronellale che, alla
concentrazione di 10 g /ml inibisce del 75% la proliferazione cellulare ed è anche in grado di
indurre apoptosi sulla linea cellulare leucemica umana U937. Sulla base dei risultati ottenuti sono
stati studiati nuovi tiosemicarbazoni SN bidentati, aventi differente carattere idrofilo-lipofilo,
derivati dalla condensazione della p-fluorobenzaldeide con tiosemicarbazidi variamente sostituite
con gruppi alifatici ed aromatici e sono stati caratterizzati per mezzo di spettroscopia IR ed NMR. Il
tiosemicarbazone della p-fluorobenzaldeide (Hfbt) ed il 4-feniltiosemicarbazone della pfluorobenzaldeide (Ph-Hfbt) sono stati anche caratterizzati mediante diffrazione ai raggi X. Per
verificare l'effetto della chelazione metallica sulle proprietà biologiche è stato pure studiato il
complesso di nichelio [Ni(fbt)2], tenendo presente che, pur essendo nota l'essenziale presenza di
questo metallo in enzimi batterici, non è ancora chiaro il ruolo che esso ha come elemento in tracce
negli esseri umani. I saggi biologici sulla linea cellulare U937 hanno evidenziato che il legante Hfbt
inibisce la proliferazione cellulare del 50% ad una concentrazione di 50 g/ml, il legante, Ph-Hfbt
più attivo, inibisce del 50% già ad una concentrazione di 10 g/ml, il complesso [Ni(fbt)2] a 30
g/ml inibisce del 75%. Queste concentrazioni corrispondono alla massima inibizione senza
osservare effetto citotossico sulle cellule. Nessuno di questi composti è in grado di indurre apoptosi.
Anche il complesso di nichelio, che è costituito da molecole neutre [Ni(fbt)2], con il metallo su un
centro di simmetria, e presenta una coordinazione quadrata planare con i due leganti SN bidentati in
posizione trans non induce apoptosi. Questo indica che l'incompleta saturazione delle posizioni di
coordinazione osservata in complessi di rame studiati in precedenza è una condizione necessaria,
ma non sufficiente per rendere attivo il composto riguardo il meccanismo di apoptosi.
UNITA’ DI RICERCA DI PAVIA
Direttore: Prof. Luigi Casella
Gruppo di Ricerca del Prof. Luigi Casella
Studi su Metalloproteine e loro Modelli
Sono stati studiati vari sistemi modello di rame enzimi, sia mononucleari sia binucleari. Per i
sistemi mononucleari sono stati in particolare studiati l‟attività ed il meccanismo di riduzione del
nitrito a monossido di azoto ai centri rame(I). Questa reazione riproduce l‟attività dei rame enzimi
nitrito riduttasici attivi nella denitrificazione batterica, il cui meccanismo catalitico non è noto, ma
potrebbe avere rilevanza anche nel campo delle patologie degli organismi superiori, nelle quali si ha
sovraproduzione di specie azotate reattive. E‟ stata impiegata in questi studi una serie di complessi
di rame con leganti tridentati con donatori azotati correlati all‟imidazolo. Gli studi cinetici hanno
evidenziato per la reazione nitrito riduttasica una dipendenza di tipo iperbolico dalla concentrazione
del nitrito ed una dipendenza lineare dalla concentrazione del protone. Ciò indica che lo stadio
chiave della reazione è costituito dalla protonazione di una specie rame(I)-nitrito. Tra i sistemi
binucleari si è studiata l‟ossigenazione reversibile a bassa temperatura (-80° C) di un complesso con
un legante esadentato poliazotato e si è potuto dimostrare seguendo le reazioni a –60° C che questo
addotto è la specie competente per realizzare l‟ossigenazione del nucleo fenolico di un substrato
esterno. Si tratta dell‟unico sistema modello per il quale sia stata dimostrata un‟attività tirosinasica
dell‟addotto rame-ossigeno su un substrato fenolico esterno. Altri sistemi modello correlati al
precedente esibiscono attività tirosinasica ma non danno addotti sufficientemente stabili con
l‟ossigeno. La rilevanza biologica dei sistemi modello è confermata da studi sul meccanismo di
inibizione esercitato nelle reazioni di ossidazione dei substrati fenolici dall‟acido kojico, uno dei più
potenti inibitori della tirosinasi.
Sono stati inoltre iniziati degli studi sulla possibilità di convertire la mioglobina in una
proteina con attività catalitica. Essi si sviluppano secondo due linee parallele. Da un lato è stata
presa in considerazione la possibilità di incrementare l‟attività catalitica sostituendo il gruppo eme
nativo con un derivato eme modificato chimicamente. Dall‟altro si è proceduto all‟espressione e
purificazione di mutanti sito specifici della proteina nei quali sono stati introdotti nella cavità distale
dell‟eme dei residui analoghi a quelli che promuovono l‟attività di eme enzimi quali le perossidasi.
Sono stati infine iniziati degli studi di ricostituzione della mioglobina con cofattori eme modificati.
La modifica iniziale è stata l‟aggancio al complesso protoeminico di un residuo di arginina, il quale
avrebbe la funzione di simulare l‟effetto di un residuo di arginina presente nella tasca distale delle
perossidasi. La mioglobina ricostituita con questo gruppo eme presenta in effetti un‟attività
perossidasica più marcata rispetto alla proteina nativa, anche se questo incremento di attività è
dovuto più alla facilità con cui reagisce il substrato con la proteina che alla vera e propria
attivazione della reazione col perossido di idrogeno. La ricostituzione della mioglobina con un
gruppo eme modificato con un‟istidina produce una proteina eterogenea, con una frazione
minoritaria contenente ferro ad alto spin (pentacoordinato), come nella proteina nativa, ed una
frazione maggioritaria contenente ferro a basso spin (esacoordinato). Il sesto legante è
probabilmente rappresentato dall‟istidina distale e può essere scalzato dal legame di buoni donatori
per il ferro quali gli ioni azide o cianuro. Per questo motivo la mioglobina ricostituita esibisce
un‟attività perossidasica significativa, nonostante la ridotta accessibilità del ferro.
Questo approccio viene seguito anche negli studi su complessi ferroporfirinici modello, nei
quali la porfirina viene modificata chimicamente con l‟aggancio di gruppi sostituenti identici o
analoghi ai residui distali delle eme perossidasi. I modelli studiati in questa fase sono dei derivati
eme-peptidici, denominati microperossidasi, i quali sono stati ottenuti per modificazione covalente
del residuo eme-octapeptidico risultante dalla digestione del citocromo c. La modificazione è
consistita nell‟aggancio di residui di prolina all‟estremo N-terminale della catena, che corrisponde
al residuo che lega il gruppo eme. Le proline ingombrano lo spazio “distale” dell‟eme e aumentano
notevolmente la stabilità chimica del complesso durante la catalisi ossidativa, perché ne riducono la
velocità di degradazione ossidativa, in competizione alla reazione che avviene sul substrato.
Gruppo di Ricerca del Prof. Luigi Fabbrizzi
Studi di Chimica Supramolecolare
Nel corso dell‟anno 2000, le ricerche sono proseguite secondo le due linee principali di
seguito descritte:
a) Sensori molecolari fluorescenti per anioni e analiti di rilevanza biologica
Sono stati sintetizzati sistemi a due componenti nei quali un recettore selettivo per l‟analita
di interesse viene legato covalentemente ad una subunità segnalante capace di comunicare
all'esterno in processo di riconoscimento. Si è scelto come segnale l'emissione di fluorescenza, in
quanto (i) tale proprietà viene percepita sia visualmente che strumentalmente anche a livelli di
concentrazione molto bassi, (ii) la sua determinazione può essere effettuata con una strumentazione
relativamente poco sofisticata e costosa, (iii) essa può essere infine controllata attraverso due
meccanismi ben definiti: il trasferimento elettronico e il trasferimento di energia. Tali sistemi
consentono in particolare la determinazione di analiti all‟interno della cellula, con risoluzione nel
tempo e nello spazio. I sensori fluorescenti sviluppati in questo Laboratorio vengono studiati per
l‟applicazione da fisiologi cellulari dell‟Ospedale S. Raffaele di Milano. Risultati di particolare
interesse sono stati ottenuti nella determinazione dell‟acido glutammico. Il riconoscimento
dell‟analita si basa sulle interazioni metallo-legante, che presentano maggiore intensità e
direzionalità delle interazioni elettrostatiche (legame di idrogeno incluso). In questo senso, la
subunità recettore contiene un centro metallico (es. ZnII) coordinativamente insaturo, capace di
interagire con il residuo carbossilato dell‟amminoacido.
b) Interruttori molecolari
Sono stati sviluppati interruttori molecolari capaci di attivare-disattivare la luminescenza di
frammenti fotoattivi attraverso processi di natura sia redox che acido-base. Tale funzione, di
interesse nel campo della generazione di segnali a livello molecolare, viene realizzata per mezzo di
sistemi a due componenti, nei quali il frammento fotoattivo è connesso covalentemente ad una
subunità bistabile. Quando l‟input ha una natura redox, la subunità di controllo è costituita da un
metallo incorporato in una cavità macrociclica e dà luogo a due stati di ossidazione adiacenti di
stabilità comparabile. Lo switching della luminescenza è assicurato se uno dei due stati di
ossidazione del centro metallico interagisce con il lumoforo adiacente con meccanismo fotoindotto
e l'altro stato di ossidazione no. Nel caso di input acido-base, uno ione metallico viene traslocato
reversibilmente tra due compartimenti coordinativamente non equivalenti di un legante ditopico, a
seguito di una moderata variazione del pH. La posizione del metallo viene segnalata attraverso
l‟accensione-spegnimento di un frammento fluorescente covalententemente legato allo scheletro del
legante. In questo ambito è stato riportato il primo esempio di interruttore meccanico della
fluorescenza.
UNITA’ LOCALE DI ROMA
“LA SAPIENZA”
Direttore: Prof. Elena Borghi
L'attivita' di ricerca di questa Unita' afferisce alla tematica "Metalloproteine come catalizzatori
biologici" e nell'anno 2000 si e' articolata come segue.
A) STUDIO DELLA STRUTTURA DEL SITO ATTIVO METALLICO DI CITOCROMI
MEDIANTE SPETTROSCOPIA EXAFS E SIMULAZIONI DI DINAMICA MOLECOLARE.
I citocromi sono una famiglia di metallo proteine contenenti ferro, largamente distribuiti in
moltissime specie animali e nei microrganismi. Tra i citocromi esiste una subclasse che prende il
nome di citocromo c in cui il gruppo prostetico è la ferro porfirina detto anche eme. Queste metalloproteine hanno la funzione specifica di trasportare elettroni all‟interno del complesso ciclo di
reazioni redox che è noto con il nome di fosforilazione ossidativa. In particolare nella fosforilazione
ossidativa le cellule realizzano la sintesi di ATP guidata dal trasferimento di elettroni all‟ossigeno.
Nei microrganismi, la sintesi dell‟ATP è guidata dalla luce e prende il nome di fosforilazione.
Entrambi i due meccanismi, sono stati largamente studiati in quanto, senza dubbio, sono i due
sistemi di trasduzione dell‟energia più importanti della biosfera. Il processo di trasporto di elettroni
da parte dei citocromi, è realizzato mediante il cambio reversibile dello stato di ossidazione del Fe
del gruppo prostetico
Gli spettri sperimentali EXAFS dei campioni di citocromo C di cuore di cavallo in forma
ossidata Fe(III) e in forma ridotta Fe(II) sono stati registrati utilizzando campioni di proteina con
una concentrazione molare di ferro (atomo assorbitore ) 10 Mm. Il pH delle soluzioni è stato
mantenuto costante a pH=7.5 con tampone fosfato 0.01 M e i campioni di citocromo sono stati
preparati introducendo una quantità di ditionito 5 volte in eccesso rispetto alla concentrazione di
ioni ferro.
Anche se il potenziale redox del citocromo c (250 mV) non consente la sua ossidazione da
parte dell‟aria, i campioni sono stati preparati subito prima della misura.
L‟eccesso di ditionito avrebbe dovuto garantire che il Fe fosse rigorosamente mantenuto allo
stato di ossidazione più basso (Fe(II)).
Questa condizione sperimentale normalmente realizzabile con estrema facilità si è rivelata
invece più complessa di quanto ragionevolmente ipotizzabile.
Infatti, un eccesso di ditionito permette di realizzare soluzioni di citocromo C ridotto a pH
controllato stabili per un periodo di tempo sufficiente alla realizzazione delle misure chimico fisiche
di laboratorio.
B) CARATTERIZZAZIONE STRUTTURALE DI FORME OSSIDATE DI EMOCIANINE.
Ci stiamo interessando allo studio delle relazioni struttura-funzione catalitica (reattivita'
chimica) di forme ossidate di Hcs di mollusco e di artropodo. Il sito binucleare delle Hcs subisce
una complessa ed estesa chimica di ossidazione che porta ad una serie di derivati ossidati in cui sia
lo stato di ossidazione sia la geometria di coordinazione per lo ione metallico nel sito attivo sono
differenti. La reattivita' chimica del centro a rame binucleare di tipo 3 delle Hcs avviene con piccoli
anioni e molecole neutre con effetti differenti.
Le reazioni dei leganti esogeni sulle forme funzionali ossidate (ossi-Hc, centro
[Cu(II)O2CU(II)] chimicamente sono interazioni di scambio legante-legante, passano attraverso lo
stato funzionale deossi-Hc [Cu(I) Cu(I)] e sono favorite da una diminuzione di ossigeno nel mezzo,
dipendono dalla natura dei leganti, sono pH dipendenti ed avvengono generalmente con affinita'
maggiore nei molluschi rispetto agli artropodi.
La ricerca e' focalizzata sulla caratterizzazione del sito attivo di forme met-Hc (centro [Cu(II)
Cu(II)] EPR-silente) e dei loro derivati coordinanti legante(i) esogeno(i) in differenti condizioni di
pH e temperatura con uno studio di tipo XANES ed EXAFS in approccio di multiplo scattering
(MS).
Per l'analisi dei dati EXAFS con un approccio di MS usiamo il pacchetto di programma
GNXAS. La regione XANES viene simulata con i programmi G4XANES e CONTINUUM che
calcolano gli spettri con l'approccio MS.
Le forme ossidate che abbiamo gia' considerato sono le forme met-Hc ed i loro derivati
coordinanti azide come legante esogeno a pH 7.5.(Hc da mollusco Octopus Vulgaris e Hc da
artropodo Carcinus maenas). Questo studio, che e' stato approvato dal comitato scientifico di ESRF
a Grenoble (Francia), e' focalizzato sulla caratterizzazione del sistema di leganti del sito attivo della
proteina e di eventuali leganti esogeni a ponte tra i due ioni metallici responsabili
dell'accoppiamento antiferromagnetico. Il legante azide permette di perturbare le proprieta'
strutturali del sito binucleare in modo controllato e di valutare l'effetto di modificazioni strutturali e
conformazionali responsabili dell'accoppiamento antiferromagnetico tra i due ioni rameici osservato
in questi derivati.
I composti modello considerati sono i complessi a rame(II), mono e dinucleari con leganti a
poli(benzoimidazolo) che modellano il centro Cu2 nel sito attivo proteico e le cui caratteristiche
sono rilevanti per la chimica biomimetica delle emocianine [L. Casella, Unita' CIRCMSB di
Pavia].
Abbiamo
caratterizzato
con
esperimento
XAS
i
sei
composti
binucleari
[Cu(II)2(L)(X)2]+n(ClO4)n con L=L-5,5 e L-6,6 e X=OH-, OH2, N3-. I leganti L-5,5 e L-6,6 sono
di tipo poli(benzoimidazolo) con identici gruppi azoto-donatori e formano chelati con anelli di
differente grandezza (5-T per L-5,5 e 6-T per L-6,6). Questi composti hanno caratteristiche
spettroscopiche essenziali per comprendere la struttura del sito attivo delle forme met-Hcs e per
differenziare il modo di coordinazione del legante azide ( -1,1 per L-5,5 e
-3,3 per L-6,6).
Tuttavia nessuno dei sei modelli binucleari e' stato caratterizzato cristallograficamente. Soltanto
l'analogo complesso [Cu(II)2(L-5,5)(OMe)2](ClO4)2 ha struttura ai raggi X risolta. Abbiamo
considerato anche i complessi mononucleari [Cu(II)(L)(X)]+n(ClO4)n con X=OH2, N3- e L=2-BB
perché sono caratterizzati cristallograficamente. Inoltre il legante 2-BB e' correlato al legante L-6,6,
mima il motivo tris(imidazolo) con differenti numeri di coordinazione e differenti stereochimiche e
mostra una coordinazione "end-on" del legante azide con due legami N-N statisticamente uguali.
I risultati dello studio XAS sui derivati dei due phyla della proteina a pH 7.5 e sui complessi
considerati sono in corso di pubblicazione.
L'analisi EXAFS di prima shell, accompagnata da un'analisi XANES qualitativa, e i primi
risultati dell'analisi di MS con il doppio approccio (EXAFS e XANES) sui quattro derivati di Hcs a
pH 7.5 e su gli otto composti modello sono stati presentati in diversi Congressi Internazionali.
C) SINTESI E CARATTERIZZAZIONE DI AZAPORFIRINE SOSTITUITE. STRUTTURA E
REATTIVITA' CON METALLI DI TRANSIZIONE.
Macrocicli, quali le porfirine, le ftalocianine e loro derivati, hanno ricevuto molta attenzione
a causa del loro ruolo nei processi di ossido-riduzione che avvengono nei sistemi biologici e,
relativamente a questi macrocicli, esiste una vastissima letteratura.
Noi siamo interessati a studiare le proprietà, anche comparativamente a questi macrocicli più
noti, di composti metallo-porfirino-simili ed in particolare dell'emiporfirazina e dei suoi derivati
metallici. La sostituzione rispetto alle ftalocianine di due gruppi iso-indolici con due piridine in
posizione trans porta all'interessante situazione di quattro azoti coordinanti non equivalenti ed a una
struttura elettronica particolare. In generale, i metallo derivati dell'emiporfirazina mostrano una
spiccata reattività in posizione assiale cosicché derivati metallici sono stati ottenuti sia sotto forma
di monomeri che di polimeri. Rispetto alle porfirine, l'emiporfirazina presenta allo stato
fondamentale una coniugazione dei legami p greco più localizzata, ma calcoli teorici prevedono una
coniugazione estesa a tutto il macrociclo per il I∞ stato eccitato. Da qui nascono interessanti
proprietà spettroscopiche.
Allo stato solido l'emiporfirazina si trova in due forme, una rossa e una gialla (probabilmente idrata)
e non sono chiare le ragioni per le grandi differenze negli spettri di riflettanza (uv-vis) e di
assorbimento(ir).
La ricerca si e' sviluppata lungo le seguenti linee:
1) Studio dell'influenza del solvente e in particolare dell'acqua disciolta in solventi organici
sulle proprietà spettroscopiche (assorbimento uv-vis-nir e fluorescenza statica e dinamica) e sulla
struttura dell'emiporfirazina in soluzione.
2) Sintesi di omologhi dell'emiporfirazina con sostituenti atti a promuovere la coniugazione
dell'intero macrociclo e caratterizzazione spettroscopica in soluzione e allo stato solido.
Questi studi, ancora in corso, sono volti anche a dimostrare la possibile esistenza di una
forma tautomerica dell'emiporfirazina in cui i due H, notoriamente legati agli N-isoindolici, si
trovano invece sugli N-piridinici presenti nel macrociclo e a verificare l'esistenza di tale forma
tautomerica nel I∞ stato eccitato prevista dai calcoli teorici.
D)
COMPLESSI
MACROCICLICI
DI
LEGANTI
TETRAPIRROLICI
(PORFIRINE,
PORFIRAZINE, FTALOCIANINE)
Nell‟anno 2000 le linee principali di lavoro riguardanti la sintesi e caratterizzazione chimicofisica di nuovi complessi macrociclici tetrapirrolici appartenenti alle classi di composti del tipo delle
porfirine e delle porfirazine (tetrabenzoporfirazine) hanno abbracciato aspetti riguardanti un
argomento di lavoro da lungo tempo portato avanti ed imperniato sulla preparazione di composti
stabili contenenti ferro in alto stato di ossidazione (> +3). Questa tematica, mentre si collega a
processi chimici strettamente attinenti al mondo biologico, quali quelli che concernono il ruolo
svolto dal gruppo ferroporfirinico presente nel citocromo P 450 (attivazione di O2 e trasferimento
selettivo di atomi di ossigeno su legami C-H), ha permesso di isolare une larga serie di composti
contenenti Fe(IV), in forma stabile, all‟interno di sistemi bimetallici a ponte di singolo atomo,
prevalentemente di tipo nitrurico di formule generali (L)M-N-M‟(L‟) con L ed L‟ leganti
macrociclici uguali o diversi, ed M ed M‟ a costituire ponti di tipo Fe-N-Fe, o Fe-N-M, od ancora
M-N-M con M un metallo diverso quale Ru, Mn, e più recentemente Cr. Infatti, per l‟anno appena
trascorso l‟attenzione è stata accentrata sull‟ulteriore studio della specie (TPP)Mn-N-Cr(Pc)N3,
della quale è nota la struttura da lavoro cristallografico su cristallo singolo, ma della quale non erano
precedentemente state definite con precisione linee di sintesi, e reattività, anche in termini di
proprietà redox, e di associati composti complementari quali l‟azide PcCrN3 e la corrispondente
specie nitrurica PcCr-N-CrPc. Lavoro ulteriore è stato compiuto anche con tentativi di sintesi di
specie quali (TPP)Fe-N-CrPc, composto a valenza mista nel quale dovrebbe risultare la presenza
della coppia bimetallica Fe(IV)-N-Cr(III).
La seconda linea di lavoro riguarda l‟impostazione preliminare della sintesi di un sistema
cromoforico di tipo porfirazinico combinato con un sito di coordinazione che richiama il cis-platino.
E‟ stato al momento individuato un percorso di tale tipo attraverso la sintesi della
ottamminoporfirazina, già precedentemente caratterizzata nell‟ambito del gruppo di lavoro di cui
alla presente relazione (JPP, 3, 371-379, 1999).
E) REATTIVITA' DI COMPLESSI FTALOCIANINICI DI METALLI DI TRANSIZIONE.
Il proponente è da tempo impegnato nello studio della reattività di complessi metallo
ftalocianinici e, in particolare, dei derivati ftalocianinici del ferro.
In questo ambito sono state sviluppate due linee di ricerca parallele che riguardano (a) la
reattività assiale di complessi nei quali il metallo mantiene il proprio numero di ossidazione, e (b) le
reazioni redox, nelle quali il metallo subisce una variazione del numero di ossidazione.
Sono state finora studiate, sia cineticamente che dal punto di vista termodinamico, le
reazioni di interscambio dei leganti che occupano le posizioni assiali nel complesso
(dmso)2PcFe(II) (dmso = dimetil solfossido; Pc = anione ftalocianinato(2-)) con leganti come CO,
O2, NO, o basi azotate del tipo piridina, piperidina, imidazolo e imidazoli sostituiti.
Per quanto riguarda le reazioni redox, l‟attenzione è stata rivolta essenzialmente allo studio
della reattività del complesso a ponte di ossigeno (PcFe(III))2O nel quale il metallo viene ridotto a
Fe(II) da riducenti appropriati, in soluzione di piridina. Lo stesso complesso tende spontaneamente
a ridursi nello stesso solvente ed in presenza di tracce di acqua, apparentemente con il
coinvolgimento dell‟anello ftalocianinico che agirebbe da riducente interno. Questa reazione è
molto complessa e presenta aspetti ancora da chiarire.
Le due linee di ricerca ricordate (a) e (b) sono reciprocamente complementari in quanto si
ammette generalmente che i processi di ossidoriduzione a carico di questi complessi siano preceduti
dalla coordinazione del partner redox in una posizione assiale. L‟importanza di questo studio è
evidente se si considera che le metallo ftalocianine sono spesso considerate, assieme alle metallo
porfirine, come modelli artificiali di siti attivi enzimatici come l'eme, nei quali, sia la coordinazione
assiale, sia gli eventuali processi redox coinvolgenti il metallo, rappresentano le fasi più
significative del loro meccanismo di azione.
UNITA’ LOCALE DI ROMA
“TOR VERGATA”
Direttore: Prof. Massimiliano COLETTA
Nel corso del 2000 l’Unità Operativa dell’Università Tor Vergata di Roma ha operato secondo le
seguenti linee di ricerca:
1) Emoproteine
2) Metalloenzimi
Emoproteine
In questo ambito lo studio si è prevalentemente rivolto all studio funzionale e strutturale di
a) Citocromi
b) Perossidasi
c) Emoglobine
a) Citocromi
Per quanto riguarda i citocromi l‟indagine si è incentrata prevalentemente su studi di tipo
strutturale concernenti processi di folding/unfolding in citocromi sia eucariotici che procariotici. In
particolare:
1) si sono caratterizzate delle transizioni conformazionali nella forma ferrica del citocromo
batterico da Methylophilus methylotrophus in condizioni ambientali estreme, evidenziando
l‟importanza dei leganti assiali dell‟eme nel regolare e nello stabilire l‟energia associata a tali
transizioni;
2) si è studiato il ruolo delle interazioni eme-globina nel citocromo c, caratterizzando le proprietà
strutturali e funzionali sia in solventi misti aquo-organici che mediante l‟utilizzo di sali.
Tali studi hanno permesso di evidenziare come a pH acidi il citocromo c” da Methylophilus
methylotrophus mostri una serie di transizioni conformazionali perfettamente reversibili una volta
ricondotto il campione a pH neutro. Tale caratteristica ha permesso un‟indagine accurata di tali
processi fino a pH 0.3, una situazione mai affrontata in precedenza con altri sistemi. Tale studio,
che è oggetto di una pubblicazione sulla rivista Biochemistry, è stata condotta in parallelo ad una
analoga sul citocromo c da cuore di cavallo, il quale presenta una differente coordinazione assiale
del eme (metionina ed istidina come leganti assiali). Si è evidenziato come il legame Fe-metionina
sia molto più debole che quello Fe-istidina; inoltre, nel citocromo c” da M. methylotrophus si è
osservato che la rottura di uno dei due legami assiali dell‟istidina facilita la rottura del legame in
posizione trans, mettendo in evidenza un ruolo concertato dei due legandi assiali nel regolare
l‟energia associata ai due legami assiali. Tali transizioni a livello dell‟eme sono accompagnate da
modificazioni strutturali dell‟intera molecola, che le fanno assumere una struttura assimilabile a
quella dello stato A osservata nel citocromo c a pH più elevati.
Nel caso del citocromo c da cuore di cavallo lo studio delle proprietà strutturali e funzionali in
solventi non convenzionali ha permesso di osservare come le variazioni strutturali indotte dal
solvente influenzino la funzionalità del biosistema, anche nella prospettiva di potenziali
applicazioni in area biotecnologica.
Inoltre, l‟utilizzo solventi misti aquo-organici, come anche di sali in particolari condizioni
sperimentali, permette di stabilizzare forme con struttura intermedia tra quella tipica dello stato
nativo e dello stato completamente denaturato. Ciò ha reso possibile studiare le caratteristiche
strutturali di intermedi di „equilibrio‟ del citocromo c, non sempre rivelabili a pH fisiologico perché
transienti, e di ipotizzare possibili meccanismi che regolano il processo di ripiegamento della catena
polipetidica dal momento della biosintesi alla formazione della struttura proteica biologicamente
attiva. Infine, si sono prodotti dei frammenti della catena polipeptidica, di cui si sono studiate le
proprietà strutturali in condizioni parzialmente denaturanti.
Si sono anche caratterizzate proteine immobilizzate in membrane polimeriche (da applicarsi
successivamente su superfici elettrodiche) nella prospettiva di una loro utilizzazione in area
biosensoristica (costruzione di biosensori elettrochimici della terza generazione).
b) Perossidasi
Nel caso delle perossidasi lo studio si è indirizzato alla correlazione fra variazioni strutturali e
conseguenze funzionali, sia utilizzando variazioni strutturali indotte dall‟ambiente (es., pH) che da
mutazioni sito- specifiche. In particolare:
1) si è caratterizzata dal punto di vista spettroscopico e funzionale una transizione conformazionale
indotta dal pH nella perossidasi di rafano, evidenziando anche in questo caso l‟importanza dei
leganti coordinanti il ferro dell‟eme nel determinare l‟energia e la dinamica di tale transizione;
2) si è completata l‟indagine funzionale sugli equilibri ossido-riduttivi della manganese
perossidasi, sia per quanto riguarda la forma ricombinante immodificata che alcuni mutanti sitospecifici di aminoacidi localizzati nelle immediate vicinanze dell‟eme, in particolare della zona
prossimale della tasca dell‟eme;
3) si è iniziato uno studio sulla lattoperossidasi recombinante, volta alla caratterizzazione in
funzione del pH dei potenziali di ossido-riduzione e di cinetica con leganti sia della forma
ferrica (es. Sodio Azide) che ferrosa (es., CO).
Tali studi hanno permesso di evidenziare una correlazione fra alcuni determinanti strutturali
della regolazione dei potenziali di ossido-riduzione delle perossidasi e la reattività verso leganti sia
della forma ferrica che della forma ferrosa.
In particolare, nel caso della perossidasi di rafano è stato caratterizzato sia l‟aspetto funzionale
che strutturale di una transizione conformazionale indotta dal pH. In particolare, è stato evidenziato
come tale transizione sia indotta, oltre che dal pH, anche dalla presenza di fosfato inorganico, e che
la dinamica di tale transizione è influenzata dallo ione cloruro. Tale aspetto è stato oggetto di una
pubblicazione su Journal of Inorganic Biochemistry. E‟ stato inoltre portata a termine un‟indagine
più quantitativa di tale variazione strutturale, studiando sia l‟andamento dei potenziali di ossidoriduzione nel corso di tale transizione, sia una descrizione quantitativa dell‟effetto del cloruro sulla
dinamica. Inoltre, tali effetti sono stati caratterizzati anche mediante spettroscopia di risonanza
Raman. Tale secondo approccio sarà oggetto nell‟immediato futuro di un‟altra pubblicazione.
Per quanto riguarda la manganese perossidasi da Phanerochaetes chrysosporium abbiamo
studiato il comportamento in funzione del pH dei potenziali di ossido-riduzione nella specie
“selvaggia” e in due tipi di mutanti, che interessavano due residui aminoacidici situati
nell‟immediata vicinanza della zona prossimale dell‟eme. La comparazione dei risultati ha
permesso di dimostrare come un elemento fondamentale nel determinare il potenziale di osiidoriduzione sia la distribuzione elettronica sull‟imidazolo dell‟istidina coordinante il ferro in
posizione prossimale. Infatti, allorchè la sostituzione aminoacidica non permetteva la formazione di
un legame idrogeno fra il residuo e l‟azoto in posizione
dell‟imidazolo il pKa della transizione
veniva alterato, variando anche i potenziali corrispondenti. Inoltre, si è osservato che la presenza di
Mn2+ nel sito specifico non altera i potenziali di ossido-riduzione, ma ha un effetto soltanto sulla
velocità di scambio elettronico intramolecolare. Tale studio è stato oggetto di una pubblicazione su
Biochemical Journal.
Per quanto riguarda le perossidasi animali, si è iniziato uno studio sulla lattoperossidasi di latte
bovino. Su questa proteina abbiamo iniziato a determinare la misura dei potenziali di ossidoriduzione in funzione del pH, al fine di determinare il pKa dei residui modulanti i potenziali sia
nella forma ossidata che in quella ridotta. Inoltre si sono cominciate da effettuare misure della
cinetica di interazione con il CO, per il quale la lattoperossidasi mostra delle costanti estremamente
basse (simili a quelle osservate nel caso della perossidasi di rafano).
c) Emoglobina
L‟indagine si è incentrata sulo studio dell'organizzazione genomica dei geni A e B della
emoglobina tetramerica del mollusco Scapharca inaequivalvis, con lo scopo di verificare
l'esistenza, in questi geni, di un "minigene"ancestrale, corrispondente all'esone centrale,
funzionalmente attivo, in grado di trascrivere una "miniglobina", cioè un"unità funzionale" capace
di legare il gruppo eme e quindi l'ossigeno in modo reversibile.
In particolare, va sottolineato che i geni globinici di Scapharca inaequivalvis hanno
l'organizzaziuone a tre esoni/due introni, tipica delle globine dei vertebrati e di molti vertebrati, con
gli introni in posizioni molto conservate. Studi di sequenza dei geni A e B dell'emoglobina
tetramerica di tale mollusco avevano dimostrato l'esistenza di "minigeni" comprendenti l'esone
centrale codificante il dominio legante l'eme e, in "frame", corte sequenze fiancheggianti, che si
estendono nel primo e nel secondo introne. Inoltre, l'analisi delle sequenze aveva permesso di
identificare, nella regione 3' del primo introne, potenziali sequenze regolatorie, corrispondenti a
promotori e a sequenze in grado di legare fattori di trascrizione. Abbiamo quindi clonato e
analizzato funzionalmente queste sequenze, di circa 400 bp, e abbiamo potuto stabilire, attraverso
esperimenti di trasfezione transiente in cellule eritroidi K562, la loro attività come promotori,
potenzialmente in grado di esprimere i geni delle miniglobine A e B. Inoltre esperimenti di RT-PCR
(reazione con la trascrittasi inversa e reazione a catena della polimerasi) sull'RNA citoplasmatico
totale del mollusco hanno permesso di ottenere molecole di cDNA, la cui sequenza è risultata in
accordo con l'esistenza, negli eritrociti di S. inaequivalvis, di minigeni A e B trascrizionalmente
attivi in vivo. Quindi i risultati ottenuti hanno mostrato che nell'organizzazione attuale dei geni
globinici di questo invertebrato, è possibile riconoscere ancora oggi l'esistenza di un gene globinico
ancestrale, in accordo con la teoria dell'evoluzione genica, nota come "exon shuffling".
Metalloenzimi
In questo ambito ci si è rivolti verso due argomenti prevalenti:
a) Metalloproteinasi
b) Superossido dismutasi
a) Metalloproteinasi
Nel caso delle MetalloProteinasi l‟indagine si è rivolta prevalentemente alla collagenasi da
neutrofilo (MMP-8) e alle gelatinasi A (MMP-2) e B (MMP-9), le quali ultime sono pesantemente
coinvolte nei processi di diffusione metastatica attraverso la matrice extracellulare.
Abbiamo inizialmente diretto la nostra attenzione alla comprensione delle differenze di
meccanismo fra le due classi di MetalloProteinasi, effettuando uno studio sistematico in funzione
del pH del meccanismo catalitico di questi tre enzimi con lo stesso substrato sintetico. Tale studio,
che è oggetto di una pubblicazione sulla rivista Biophysical Journal, ha permesso di evidenziare
come le differenze maggiori fra gelatinasi e collagenasi risiedano prevalentemente nel meccanismo
di riconoscimento del substrato, in quanto è questo il parametro che più differisce in funzione del
pH nei tre enzimi, presupponendo il coinvolgimento di residui diversi, o con diversi valori di pKa
(che rispecchierebbero un diverso ambiente).
Si è inoltre approfondita l‟analisi del meccanismo catalitico della MMP-8 con il substrato
naturale, il collagene di tipo I (presente nei tendini). Questo studio, che è oggetto di una
pubblicazione sulla rivista Journal of Biological Chemistry , ha permesso di caratterizzare le
proprietà catalitiche del processo enzimatico sul collagene, evidenziando come il processo di
riconoscimento di una macromolecola è assai più complesso di quello di un substrato sintetico,
mostrando il coinvolgimento anche di residui piuttosto lontani dal sito attivo dell‟enzima. Questa
osservazione, che è stata fatta per la prima volta, rende conto del fatto che inibitori specifici basati
su un‟indagine utilizzando substrati sintetici, si sono sempre mostrati fallimentari “in vivo”.
b) Superosido dismutasi
Numerosi funzioni cellulari ed eventi patologici sono modulati da processi redox. Il nostro
gruppo è coinvolto in attività di ricerca che riguardano l'attività redox di metalloproteine con
particolare interesse per la funzione dell'enzima antiossidante superossido dismutasi.
Una delle principali linee di ricerca del nostro gruppo riguarda i meccanismi molecolari ramedipendenti in due diverse malattie neurodegenerative: la sclerosi laterale amiotrofica (SLA) e la
malattia di Menkes. La SLA è una patologia caratterizzata dalla progressiva degenerazione dei
neuroni motori corticali e del midollo spinale, che nelle sue forme familiari è associata a mutazioni
nel gene codificante per l'enzima superossido dismutasi a rame e zinco (Cu,ZnSOD), mentre la
malattia di Menkes è caratterizzata da un'alterazione dell'assorbimento del rame, anch'essa dovuta
ad un'alterazione genetica, che porta ad una diminuzione dei livelli di questo metallo e degli enzimi
rame-dipendenti nel sistema nervoso centrale. Negli ultimi anni abbiamo sviluppato modelli
cellulari e utilizzato modelli animali di queste malattie. Attraverso l'uso di questi modelli abbiamo
recentemente dimostrato che l'esposizione a effettori noti della regolazione redox, quali l'ossido
nitrico o radicali dell'ossigeno, induce l'apoptosi in cellule neuronali e che la resistenza all'apoptosi
indotta da NO è strettamente correlato ai livelli e alla integrità della Cu,ZnSOD.
Recentemente abbiamo esteso i nostri studi alla ricerca di bersagli molecolari alterati dalla
espressione di Cu,ZnSOD mutanti associate alla SLA. Siamo stati in grado di dimostrare che la
calcineurina (CN), una Ser/Thr protein-fosfatasi Ca++- e calmodulina-dipendente, implicata nella
regolazione di processi neuronali cruciali quali il metabolismo del calcio, l‟assemblaggio di
neurofilamenti e la morte apoptotica, non solo presenta una modulazione ossidoriduttiva della sua
attività, ma risulta anche significativamente inibita in due diversi modelli di SLA (cellule di
neuroblastoma umano transfettate per l‟espressione di enzima mutante G93A o H46R e corteccia
motoria di cervelli di topi topi transgenici per la G93A). Tale inibizione e‟ di tipo ossidativo, in
quanto e‟ prevenuta e revertita dalla somministrazione di antiossidanti (ad es. ascorbato). Sono in
corso esperimenti volti a valutare se le Cu,ZnSOD mutanti esercitano la loro azione pro-ossidante
direttamente sulla CN (una ferro proteina) o indirettamente (ad es. attraverso inibizione ossidativa
della calmodulina o mediante attivazione di fattori di trascrizione redox-regolati).
Siamo inoltre coinvolti nello studio delle proprietà strutturali e funzionali della Cu,ZnSOD, con
particolare interesse per le varianti espresse da batteri patogeni. Questo specifico interesse è
riconducibile all'osservazione che nei
batteri la Cu,ZnSOD
è
esportata negli
spazi
extracitoplasmatici (periplasma, membrane) dove potrebbe svolgere un ruolo di primo piano nella
protezione della cellula batterica dai radicali dell'ossigeno prodotti dai fagociti attivati. A questo
proposito abbiamo dimostrato che questo enzima, oltre ad offrire una significativa protezione nei
confronti del burst ossidativo dei macrofagi, è capace di modulare la sopravvivenza batterica anche
in cellule di tipo epiteliale. Questa osservazione suggerisce che anche in questo tipo di cellule siano
operative dei meccanismi antimicrobici basati sulla produzione di specie radicaliche dell'ossigeno.
Le Cu,ZnSOD batteriche, inoltre, presentano proprietà strutturali ben distinte rispetto alle
varianti isolate da organismi eucariotici. Ad esempio, abbiamo recentemente dimostrato che nella
Cu,ZnSOD da Escherichia coli, unica Cu,ZnSOD monomerica finora isolata, è assente la molecola
d'acqua che coordina il rame nello stato ossidato in tutte le altre Cu,ZnSOD note. Nonostante questa
differenza l'attività enzimatica è ancora alta, suggerendo quindi che tale molecola non sia necessaria
per la reazione di dismutazione.
Abbiamo anche caratterizzato le proprietà funzionali e strutturali di una delle Cu,ZnSOD
espresse dai ceppi più virulenti di Salmonella. Misure di attività enzimatica hanno dimostrato che
questo enzima è la superossido dismutasi più efficiente mai trovata e che tale caratteristica è
associata ad un incremento dell'accessibilità del rame catalitico. La struttura tridimensionale di
questa proteina è simile a quella di altre Cu,ZnSOD batteriche dimeriche, anche se significative
differenze possono essere messe in evidenza nella regione di contatto tra le subunità. In
considerazione della recente dimostrazione dell'esistenza di una comunicazione tra l'interfaccia del
dimero ed il sito attivo, è possibile ipotizzare che mutazioni a livello della superficie di contatto tra
le subunità possano essere responsabili dell'iperattività di tale enzima.
Infine, abbiamo studiato la stabilità delle due Cu,ZnSOD espresse dall'anfibio Xenopus laevis
(XSODA e XSODB). Queste due Cu,ZnSOD mostrano una diversa stabilità al calore, la XSODA
essendo molto più termolabile della XSODB. Tramite la costruzione di mutanti sito specifici ed
esperimenti di microcalorimetria abbiamo dimostrato che la minore stabilità di una delle due
varianti è essenzialmente dovuta alla presenza di un residuo di cisteina all'interfaccia del dimero. I
nostri risultati suggeriscono che la rimozione di cisteine non impegnate in ponti disolfuro
rappresenti una valida strategia per aumentare la resistenza delle proteine nei confronti
dell'inattivazione termica.
UNITA’ LOCALE DI SIENA
Direttore: Prof. Piero Zanello
L‟Unità di Ricerca di Siena è composta da quattro distinti Gruppi di ricerca, ciascuno con proprie
competenze nel settore di interesse di questo Consorzio. Verrà pertanto brevemente illustrata
l‟attività scientifica svolta nel corso del 2000 da ciascun Gruppo.
1. Gruppo di ricerca del Prof. Piero Zanello
Questo Gruppo si occupa della caratterizzazione ossido-riduttiva di molecole di potenziale
interesse biomedico o mimetiche di funzioni biologiche.
Per quanto concerne l‟attività svolta nel settore di metallo-complessi come potenziali agenti
antitumorali: (a) in collaborazione con l‟Unità di Firenze, è stato completato lo studio di complessi
di oro(III) con una serie di leganti azotati, che si sono mostrati possedere attività citotossiche; (b) in
collaborazione con il locale Dipartimento di Dipartimento di Fisiopatologia e Medicina
Sperimentale (di cui al punto 2) e l‟Unità del Piemonte Orientale è stato completato lo studio del
meccanismo d‟azione antitumorale dei sali di ferricinio.
Per quanto riguarda le indagini su complessi metallici biomimetici, in collaborazione con le
Unità di Pavia e Trieste è stato completato lo studio su complessi di rame capaci di mimare
l‟attivazione dell‟ossigeno molecolare tipica delle catecolasi.
Si è iniziata una nuova linea di ricerca su biosensori elettrochimici. Si sono studiati una serie
di stannaferrocenofani capaci di riconoscere selettivamente anioni del tipo Cl-, F-, H2PO42-.
2. Gruppo di Ricerca del Prof. Marco Ferrali
Il gruppo si occupa del ruolo patogenetico del ferro sia nei casi di mobilizzazione “in vivo”
dai suoi carriers fisiologici, sia di quello somministrato artificialmente a colture cellulari “in vitro”
Per quanto riguarda il primo punto il nostro sforzo si è di recente concentrato sull‟ideazione
e sintesi di chelanti del ferro da usarsi “in vivo” in sostituzione di quelli a tutt‟oggi in uso, che non
si sono dimostrati del tutto adatti allo scopo. Sono stati sintetizzati, ed attualmente sotto studio, due
differenti cromanoli, uno dei quali chela il ferro come bidentato ed un altro come tridentato.
Per quanto riguarda il secondo punto abbiamo potuto vedere che il complesso ferro-citrato
stimola la produzione di collageno in cellule epatiche di Ito in coltura. Tale effetto è annullato dalla
presenza nella coltura stessa di chelanti del ferro. Ciò suggerisce che il processo di fibrosi epatica
che accompagna spesso processi di sovraccarico epatico del metallo in oggetto potrebbe essere
prevenuto dall‟impiego farmacologico di adatti chelanti.
3. Gruppo di ricerca del Prof. Renzo Cini
Questo Gruppo di ricerca si occupa di sintesi e caratterizzazione strutturale allo stato solido
ed in soluzione di composti di coordinazione ed organometallici con leganti di interesse biologico
(frammenti di acidi nucleici ed amminoacidi) e farmacologico (farmaci anti-infiammatori ed
antitumorali).
Sono stati continuati e completati gli studi sulla caratterizzazione strutturale di una nuova
forma di un composto di coordinazione Platino(II)-acyclovir (farmaco antivirale) in cooperazione
con il Dipartimento Farmaco-Chimico dell‟Università di Bari e con il Dipartimento di Chimica
dell‟Università di Ljubljana, Slovenia. Il composto è di potenziale interesse antivirale ed
antitumorale.
È stato completato un articolo monografico di analisi e critica di tutte le strutture (e dei
relativi metodi di sintesi) di complessi con farmaci anti-infiammatori non steroidei, determinate
mediante metodi diffrattometrici. Il lavoro ha mostrato delle interessanti correlazioni strutturali, ha
suggerito nuovi sviluppi in questo campo della chimica di coordinazione ed ha messo in risalto le
potenziali attività in campo farmacologico di questa classe di composti contenenti metalli.
È stata effettuata la sintesi, la caratterizzazione strutturale mediante diffrazione di raggi X e
la analisi mediante DFT del complesso [VO(terpiridina)(SO4)] che contiene l‟inusuale legante
chelante SO42-. Il composto è un modello dei sistemi biologici contenenti vanadio e capaci di
concentrare solfati ed acido solforico in alcuni organismi marini della famiglia delle Ascidie.
È stata effettuata la sintesi, la caratterizzazione strutturale mediante diffrazione di raggi X e
la analisi mediante DFT del complesso trans(C,N7), trans(S,S), trans(P,N7)-[Rh(C6H5)(H1,H9H2tp)2(PPh3)][Rh(C6H5)(H1,H9-H2tp)(H9-Htp)(PPh3)]Cl3•HCl•6H2O, contenente il farmaco
anti-leucemico tiopurina. Il complesso è di potenziale interesse anti-tumorale e contiene un
appaiamento delle basi tiopuriniche mai descritto prima, che coninvolge legami ad idrogeno NH…N(-) ed interazioni del tipo C-H…S.
4. Gruppo di ricerca del Prof. Gianni Valensin
Il gruppo si occupa principalmente del ruolo di ioni metallici biologicamente rilevanti nel
processo di selezione conformazionale nell‟interazione tra molecole biologicamente attive (in
prevalenza di natura peptidica) e proteine. Lo studio è stato condotto mediante spettroscopia NMR,
supportata da UV-Vis, CD e EPR.
L‟attività di ricerca si è articolata come segue:
(i)
in collaborazione con il gruppo di ricerca del Prof. Henryk Kozlowski (Faculty of
Chemistry, University of Wroclaw, Polonia) è stato condotto uno studio su alcuni leganti specifici
degli ioni Cu(II) e Ni(II) e sono stati ottenuti dati interessanti sulla stabilità del complesso tra CMTI
I (un peptide di 29 aminoacidi) e gli ioni Cu(II);
(ii)
è stato definito l‟effetto degli ioni rame sulle probabilità di esistenza dei quattro isomeri del
tetrapeptide Tyr-Pro-Phe-Pro ( -casomorfina 4): la stabilizzazione di determinate conformazioni,
osservata nel corso della ricerca, può risultare rilevante per il ruolo biologico del rame stesso;
(iii)
particolare attenzione è stata rivolta alla definizione del ruolo svolto dagli ioni calcio
nell‟interazione tra peptidi e proteine;
(iv)
è stato studiato il complesso generato nel corso della reazione in ambiente acquoso tra
cromato e glutatione in assenza ed in presenza di aspartato e glutammato, particolarmente
interessante ai fini di una più esatta comprensione di una forma biologicamente attiva di cromo
(Natural Chromium Factor – LMWCr) e dei meccanismi di tossicità del cromo stesso;
(v)
infine, in collaborazione con i gruppi di Ricerca dei Proff. Ivano Bertini (Università di
Firenze) e Silvio Aime (Università di Torino), è stata iniziata una nuova linea di ricerca volta alla
caratterizzazione della superficie proteica di calcioproteine: i primi studi hanno riguardato la
calbindina, la cui superficie proteica è stata accuratamente caratterizzata sfruttando le proprietà del
complesso Gd3+-DO3A.
UNITA’ DI RICERCA DI TORINO
Direttore: Prof. Silvio Aime
Attività scientifiche del gruppo S. Aime
L‟attività di ricerca relativa all‟anno 2000, nel campo dello studio di nuovi agenti di contrasto per
MRI, si è svolta seguendo diverse linee di ricerca:
Studio di sistemi ad alta relassività per Imaging Angiografico
Nell‟ottica di trovare sistemi dotati di relassività sempre maggiori, con lo scopo di trovarne
un‟applicazione in angiografia, si è proseguita la ricerca di substrati macromolecolari la cui
interazione con i complessi di Gd(III) ne assicurassero elevata relassività e lungo tempo di
residenza nei vasi sanguigni. Dopo averne testato l‟affinità si sono caratterizzati gli addotti con
substrati macromolecolari di diverso tipo ( HSA, Poli- -Ciclodestrine, polimeri misti di Acido
Ialuronico e -Ciclodestrine e Poliamminoacidi). Si è inoltre studiato un sistema alternativo che
consiste nell‟inglobazione di un certo numero (ca. 10) di complessi paramagnetici all‟interno della
proteina di stoccaggio del ferro: la Ferritina. Per l‟ottimizzazione di sistemi di questo tipo è stato
necessario studiare in modo approfondito il processo di scambio della molecola di acqua coordinata
al metallo in modo da limitare il più possibile l‟effetto limitante sulla relassività di un lungo valore
di
M,
una volta stabilita l‟interazione con il substrato macromolecolare.
Messa a punto di sistemi responsivi
Un‟altra linea di ricerca è stata rappresentata dallo studio di MdC la cui relassività sia
dipendente da uno specifico parametro del micro-ambiente in cui vengono a distribuirsi (pH,
temperatura, pO2, attività enzimatica, …). Per esempio nel settore dei MdC che agiscono come
reporter del pH sono stati messi a punto sistemi di diverso tipo per i quali la relassività può essere
resa dipendente dal pH attraverso approcci diversi: i) la variazione del pH può causare variazioni
strutturali di sistemi macromolecolari contenenti molti complessi di Gd(III) con una conseguente
variazione del tempo di reorientazione del sistema e quindi della relassività; ii) il cambiamento di
pH può influenzare la forza dell‟interazione di un complesso di Gd(III) con un particolare substrato
macromolecolare; iii) in complessi contenenti un gruppo protonabile nel range di pH di interesse, la
variazione del pH è in grado di far variare la denticità del legante e quindi l‟idratazione del centro
paramagnetico.
Si è inoltre affrontato il problema dello sviluppo di un MdC che agisca come reporter della
pressione parziale di Ossigeno (pO2), che è un parametro di rilevante interesse in molte patologie.
L‟idea di un MdC responsivo alla pO2 si basa sulla realizzazione di un complesso il cui ione
metallico possa trovarsi in due stati redox caratterizzati da relassività molto diverse.
L‟abilità del complesso [MnII(TPPS)]4- di agire come MdC responsivo alla pO2 è stato testato
“in vitro” misurando le velocità di rilassamento di soluzioni contenenti una quantità fissa di
complesso e quantità variabili di O2 in un tubo NMR ermeticamente chiuso.
Tra i vari sistemi di tipo responsivo particolare importanza hanno acquistato sistemi sensibili
alla concentrazione di un dato metabolita. Tra questi il lattato è sicuramente uno degli anioni di
maggior rilevanza, in quanto nello sviluppo di numerose patologie si verificano variazioni della sua
concentrazione. Queste variazioni di concentrazione possono essere convenientemente seguite
tramite la spettroscopia di risonanza magnetica, tramite cui è possibile visualizzare il picco di
assorbimento del metile del lattato. Il potenziale diagnostico di questa tecnica è stato notevolmente
incrementato progettando un sistema in grado di ottenere due segnali distinti dal lattato presente nei
due compartimenti extracellulare ed intracellulare.
Imaging Molecolare
Lo sviluppo di sistemi in grado di visualizzare recettori sovraespressi da cellule tumorali
sarebbe di estremo interesse poiché, rispetto agli analoghi approcci utilizzati in Medicina Nucleare,
l‟approccio MRI si avvale della grande superiorità in termini di risoluzione di immagine. Purtroppo
il numero relativamente basso di questi recettori rende problematico questo approccio . Si rende
quindi necessario mettere a punto sistemi in grado di accumulare un gran numero di complessi
paramagnetici in prossimità di ciascun sito recettoriale. Si è proceduto alla sintesi di complessi
dotati di un particolare gruppo di riconoscimento specifico verso determinati target e alla loro
caratterizzazione rilassometrica.
Ferro e Neuromelanina nel Morbo di Parkinson
Spettroscopia 13C-NMR
Lo spettro 13C-NMR della NM mostra i segnali tipici delle melanine nella regione aromatica
compresa tra 110 e 160 ppm e nella regione relativa ai gruppi carbonilici (160-180 ppm). Tale
“impronta digitale”, comune a tutti i tipi di melanine esistenti, naturali e sintetiche, non risulta del
tutto conservata nel pigmento patologico, il cui spettro
13
C-NMR mostra solo il segnale relativo ai
gruppi carbonilici riconducibile anche ai residui amminoacidici delle proteine. Questo indizio ha
suggerito la preparazione di un modello sintetico di melanoproteina, che, nonostante il colore nero,
mostra uno spettro mancante dei segnali aromatici (110-160 ppm), così come appare anche lo
spettro dell‟albumina serica liofilizzata.
Spettroscopia EPR in banda Q e magnetometria
Entrambe le tecniche sono state applicate ai due pigmenti, neuromelaninico e patologico.
Nel caso della NM la spettroscopia EPR in banda Q, così come lo studio magnetometrico, ha fornito
dei risultati più dettagliati, tuttavia sovrapponibili a quelli ottenuti in banda X: sono presenti più
domini ossi-idrossilici di ferro tipo ferritina, ed è inoltre presente un contributo paramagnetico del
ferro romboedrico meno evidente nella ferritina. Anche per quanto riguarda il pigmento patologico
entrambe le tecniche forniscono risulati coerenti: almeno uno dei domini ossi-ossidrilici del ferro
scompare, ed è assente il segnale relativo al contributo paramagnetico del Fe(III).
Rilassometria a ciclo di campo
Inizialmente l‟indagine rilassometrica è stata finalizzata alla valutazione del Fe(III) (in
forma di piccoli complessi) quale possibile agente di contrasto endogeno a livello cerebrale. E‟
infatti risaputo che la SNpc è particolarmente ricca di Fe(III) e che questo metallo paramagnetico è
in grado di aumentare la relassività protonica . Confrontando i profili rilassometrici delle SNpc,
patologiche e di controllo, con i profili di soluzioni di Fe(III)-EDTA e di modelli di proteine
reticolate non sono, però, comparse differenze significative imputabili al decorso della malattia.
Uno studio più approfondito della forma funzionale dei profili rilassometrici mesencefalici ha
invece indicato una maggiore presenza di proteine aggregate nelle SNpc dei soggetti affetti da MP.
Come già detto precedentemente, i profili rilassometrici dei tessuti possono essere studiati tramite
dei modelli in vitro costituiti da proteine reticolate, essendo entrambi contraddistinti da alti valori di
relassività a basso campo e della comparsa di due picchi nella regione 1.5-4 MHz. Tali profili
rilassometrici, dalla tipica forma funzionale, sono studiabili tramite un‟equazione parametrica detta
di Hallenga-Koenig. Poichè l‟ampiezza dei profili dei modelli in vitro è riconducibile al contenuto
di proteine reticolate, ne deriva che è possibile studiare il contributo dell‟aggregazione proteica nei
tessuti.
Attività scientifiche del gruppo B. Fubini
L‟attività svolta nel campo della tossicità dei materiali inorganici finemente suddivisi si è
svolta nelle seguenti direzioni:
Tossicità e carcinogenicità della silice cristallina:
Uso di solidi modello cristallini a porosità controllata (porosils) per evidenziare i
meccanismi di citotossicità: caratterizzazione chimico fisica e studio della citotossicità su linee
cellulari macrophage –like (coll Università di Modena)
Preparazione e caratterizzazione di silici modificate ad hoc per evidenziare effetti
trasformanti su cellule embrionali (coll INRS Nancy)
Studio dell‟azione dell‟ascorbato sulla silice anche in relazione all‟attività di organismi
viventi nei confronti di silice (spugne, diatomee) in vista di uno studio filogenetico di relazione
silice-organismi viventi.
Meccanismi molecolari di carcinogenesi degli amianti
Studio della mobilità del ferro, della generazione di radicali liberi da parte di amianti
variamente modificati.
Studio dell‟attivazione del complemento in siero umano da parte di amianti modificati in
seguito a incubazione in chelanti (collaborazione Univ Ancona).
Studio del ruolo del ferro nella attivazione di INOS da parte degli amianti.
Altro
Studio delle caratteristiche superficiali di materiali di interesse tossicologico: fibre di vetro,
silici porose
Attività scientifiche del gruppo C. Nervi
Il marcamento di ormoni può essere utile sia per scopi diagnostici che terapeutici. In
recettorologia per dosare il livello di specifici recettori (per esempio nei tumori ormone-dipendenti),
mentre nei metodi di radioimmunoassay (RIA), largamente usati per l‟analisi in tracce di molecole
di importanza biologica, per dosare l‟ormone nei fluidi biologici.
Un nuovo approccio del problema viene offerto dalla complessazione della biomolecola
con un frammento organometallico che può essere facilmente rilevato tramite diversi metodi
analitici, come l‟assorbimento atomico o tecniche spettroscopiche ed elettrochimiche. La serie di
ormoni steroidei (estradiolo) da noi marcati con diversi frammenti organometallici in posizione 17o 11- esemplifica questo approccio, in quanto la coordinazione del frammento organometallico
permette il mantenimento di un alto livello di riconoscimento dell‟estradiolo da parte dei suoi
recettori specifici.
Dopo il successo del cisplatino come uno dei più importanti agenti antitumorali, l'interesse
dell'uso dei complessi dei metalli di transizione in medicina ed in altre aree biologiche è cresciuto
rapidamente.
In questa classe di composti i metalloceni hanno dimostrato di essere particolarmente attivi
verso svariate tipologie di tumore, ma con un meccanismo d'azione diverso da quello del cisplatino.
Abbiamo testato l'attività antitumorale di alcuni sali di ferrocinio su linee cellulari di
adenocarcinoma mammario (MCF-7).
Consistenti dati sperimentali dimostrano il coinvolgimento dell‟enzima telomerasi nel
processo oncogenetico e nella proliferazione delle cellule tumorali. Questa caratteristica permette
alle cellule tumorali di eludere la senescenza replicativa e di proliferare in modo indefinito. Queste
caratteristiche fanno della telomerasi un target biochimico ideale per una lotta ai tumori mirata.
Sono stati sintetizzati e testati come inibitori dell'enzima telomerasi alcuni derivati del cisdicloroplatino(II) con leganti aromatici contenenti azoto come atomo donatore.
UNITA' DI RICERCA DI TRIESTE
Direttore: Prof. Lucio Randaccio
Sono di seguito descritti i risultati ottenuti nell‟anno 2000 dall‟ Unità di Trieste
relativamente alle varie tematiche di ricerca. Gran parte dei risultati sono stati oggetto di
pubblicazioni scientifiche, come da elenco allegato a questa relazione, e di comunicazioni a
convegni nazionali ed internazionali.
1. Biocristallografia e struttura di modelli di sistemi biologici
Uno accurato studio EXAFS di tipo metodologico su cinque cobalossime, di cui era nota la
struttura XRD, ha permesso di stabilire i limiti e le potenzialità dell'analisi di spettri EXAFS per
la determinazione del numero e delle distanze di coordinazione. In particolare si è messo in
evidenza che non di rado le analisi EXAFS nel passato sono state condotte con una metodologia
non adeguata e i relativi risultati strutturali vanno considerati con molta attenzione.
Sono stati preparati e analizzati mediante diffrazione di raggi X con radiazione di
sincrotrone i cristalli singoli di diverse cobalammine, incluse le forme cristalline di cobalammine
contenenti elettroliti del tipo LiCl e KCl, che hanno fornito un modello strutturale di interazione
delle catene ammidiche delle cobalammine con gli ioni. E‟ stato anche possibile analizzare in
modo quantitativo le similarità e le differenze, sia strutturali che chimico-fisiche, tra cobalammine
e cobalossime.
Proseguendo i precedenti studi sui modelli di proteine con centri di-rame, in collaborazione
con il Prof. L. Casella (Università di Pavia), sono state determinate le strutture di complessi
mononucleari, modelli del modo di legarsi dello ione nitrito e della sua attivazione riduttiva a
ossido di azoto su centri biomimetici di rame.
Sono stati ottenuti cristalli singoli del ferricitocromo c2 da Rhodopseudomonas palustris
(114 amminoacidi, PM 12783), in ambiente acido (forma A, monoclina) e in ambiente basico
(forma B, trigonale), dei quali sono stati raccolti dati di diffrazione con luce di sincrotrone a 100
K. Le strutture sono state risolte e affinate ad un fattore di disaccordo R = 0.178 (A) e 0.173 (B).
L'analisi strutturale della forma A del citocromo c2 e il suo confronto con analoghe strutture di
citocromi c2 da batterio e di citocromi c mitocondriali ha permesso di ipotizzare che il maggior
potenziale redox di questa proteina procariotica, rispetto all‟omologo citocromo c eucariotico,
deriva da un parziale allentamento della forza dei legami ad idrogeno che "circondano" il gruppo
prostetico. La forma B, ottenuta in ambiente alcalino, si è rivelata estremamente interessante in
quanto la metionina 93 che coordina il ferro dell' eme è stata sostituita da una molecola di
ammoniaca con una variazione conformazionale che coinvolge i residui Lys92-Met93-Thr94Phe95. Sono stati inoltre raccolti i dati di diffrazione da luce di sincrotrone, a 2.0 Å di risoluzione,
della forma ridotta del citocromo c2 da Rhodopseudomonas palustris. Questa struttura è in corso
di affinamento. Sono in corso tentativi di cristallizzazione dell‟omologo citocromo c eucariotico
da spinacio, fornito in forma purificata dall‟ Unità di Ricerca di Modena.
Di particolare rilievo è stata la risoluzione della prima struttura di un modello
miniaturizzato di proteine con centri dimetallici, come la ferritina, sintetizzata dall‟ Unità di
Napoli sulla base di un nuovo approccio di de novo design. Questa proteina, la cui unità
monomerica è composta da 50 amminoacidi, in presenza di metalli dà origine ad una struttura
quaternaria dimerica in cui è presente il motivo "four-helix bundle". L‟analisi dei dati
diffrattometrici della metallo proteina contenente ioni zinco, raccolti con luce di sincrotrone a 100
K, ha permesso la risoluzione a 2.5 Å della struttura cristallina. Nell‟ unità asimmetrica della cella
elementare un “four-helix bundle” è disposto su un asse binario cristallografico e un altro
possiede una pseudo simmetria binaria. In ciascuna unità due ioni Zn sono ingabbiati all'interno di
questi dimeri proteici. In particolare i due ioni zinco sono coordinati da quattro gruppi
carbossilato di residui glutammato, due a ponte e due chelati, e da due -N donatori di residui
istidinici, cosicché ogni ione Zn ha un contorno di leganti con geometria tetredrica distorta.
2. Verso una nuova generazione di modelli della Vitamina B12
La difficoltà di studiare direttamente i complicati sistemi naturali che costituiscono la
famiglia degli enzimi B12 ha incoraggiato nel corso degli anni la sintesi e lo studio di un gran
numero di complessi che potessero in qualche modo essere considerati dei modelli del composto
naturale. Oltre alle ben note cobalossime, sono stati utilizzati anche dei composti con leganti
imino-ossimici e basi di Schiff. E‟ stato quindi studiato l‟effetto sulla struttura e sulla reattività
variando sistematicamente i leganti assiali e modificando il chelante equatoriale. Ciò che risultava
chiaramente impossibile da studiare su questi modelli era l‟effetto dell‟interazione con la proteina
nel composto naturale. Ci è quindi sembrata interessante la possibilità di sintetizzare e studiare dei
complessi organometallici di Co comprensivi di catene peptidiche laterali in grado di interagire
con il frammento assiale R-Co-L. Lo spunto è stato offerto da un lavoro apparso recentemente,
[S.Ranganathan et al Inorg.Chem. 38 (1999) 1019] nel quale veniva descritta la sintesi di un
complesso di Co(II) con una base di Schiff derivante dalla 3-acetil tirosina per condensazione con
etilendiamina. La reazione porta alla formazione del complesso di Co(II) e implica l‟acetilazione
della tirosina, la protezione del gruppo carbossilico e del gruppo amminico, la condensazione con
etilendiammina per dare la base di Schiff, e infine la metallazione del chelante. Il complesso di
Co(II) che si forma è relativamente stabile nei confronti dell‟ossidazione all‟aria e viene
rapidamente ridotto da NaBH4 in presenza di PdCl2. L‟aggiunta di un opportuno alogenuro
alchilico porta alla formazione del corrispondente derivato organometallico. E‟ stata così
sintetizzata una serie di complessi organometallici (R = Me, Et, n-Bu, i-Pr, CH2Cl, CH2CF3), che
sono stati caratterizzati tramite analisi elementare, NMR ed Electrospray Mass Spectrometry. Il
gruppo
amminoacidico della 3 acetiltirosina può costituire il punto di partenza per la sintesi di
complessi contenenti catene laterali di- o poli-peptidiche. Un primo tentativo è stato fatto
sintetizzando il dipeptide che deriva dalla condensazione tra acetiltirosina protetta al gruppo
amminico e istidina metilestere e condensandolo con etilendiammina per formare la base di
Schiff. Per reazione del legante con un sale di cobalto e piridina, in soluzione metanolica e in
presenza di ossigeno, si ottiene il complesso [(py)2Co(III)(chel)]+, che può essere ridotto a Co(I) e
trasformato nel corrispondente derivato organometallico per addizione ossidativa dell‟opportuno
alogenuro alchilico. Questa nuova serie di complessi è attualmente in fase di sintesi e di
caratterizzazione.
3. Nuovi complessi polinucleari derivanti da organocobalossime assemblate mediante acidi
boronici
Questa linea di ricerca è volta all‟assemblaggio di sistemi polinucleari sfruttando legami di
coordinazione e interazioni deboli del tipo - . La relativa facilità di reazione degli acidi borinici
con i ponti O-H-O delle organocobalossime, che ricorda da vicino quella degli acidi boronici con i
dioli, ci ha suggerito che degli acidi borinici o boronici contenenti gruppi -donatori nell'anello
potrebbero essere validamente utilizzati come templati per assemblare diverse unità di
cobalossime, sfruttando le interazioni del boro con l'ossigeno e del gruppo donatore con il centro
metallico. La geometria del prodotto finale è determinata sia dalla posizione del gruppo donatore
nell'anello aromatico che dalla conformazione del complesso di partenza. Gli studi compiuti negli
anni scorsi hanno dimostrato che quest‟ultima può essere efficacemente modulata sfruttando le
interazioni steriche ed elettroniche, essenzialmente di tipo
- , dei leganti assiali delle
organocobalossime con gli anelli aromatici degli acidi fenilborinici. Un primo tentativo di
ottenere complessi polinucleari è stato compiuto facendo reagire il complesso MeCo(DH)2H2O
con gli acidi 3- e 4-piridinilboronico. In entrambi i casi si ottiene un complesso dinucleare a
struttura chiusa, nel quale la piridina coordina assialmente il cobalto dell'altra unità, ma la
determinazione strutturale ai raggi X ha dimostrato che nei due casi il complesso assume la forma
di “rettangolo molecolare” e di “scatola molecolare”. L'esterificazione del gruppo OH legato
all'acido boronico in presenza di metanolo suggerisce che questo residuo della molecola
costituisce un sito molto reattivo, che potrebbe essere sfruttato per un ulteriore accrescimento del
complesso.
4. Nuovi Alchilderivati del Cobalto(III) con Leganti Tridentati Amminoossimici
La chimica del legame Cobalto-Carbonio si articola su vari settori di indagine, quali:
i) l'identificazione dei fattori che determinano la stabilità di tale legame; ii) lo studio dei
meccanismi di formazione e di rottura del legame mediante termolisi o fotoscissione in condizioni
aerobiche ed anaerobiche; iii) lo studio dei processi di trasferimento di gruppi alchilici su substrati
organici ed inorganici; iv) la reattività di gruppi alchilici legati al metallo che non implichi
scissione del legame Co-C.
In relazione a quest'ultima tematica abbiamo osservato che i complessi del tipo [XCH2Co(III)(LNHPy)(HLNHPy)]+ (X = Cl, Br, I, HLNHPy = 2-(2-piridiletil)-3-butanoneossima e
LNHPy = la sua base coniugata) per trattamento con NaOH dil. a temperatura ambiente porta alla
formazione di un interessante derivato caratterizzato dalla presenza del metallociclo a tre termini
Co-CH2-N (equatoriale). Il processo di ciclizzazione intramolecolare (Figura 1) implica la
deprotonazione dell'azoto equatoriale recante il gruppo piridile libero seguito dallo stadio di Nalchilazione.
Cl
Cl
CH2
CH2
H
H3 C
H
O
O
N
N
CH3
H3 C
HN
CH3
Co
CH3
H3 C
H
N
N
N
-H
N
O
O
+
Co
H3 C
H2O
CH3
N
HN
N
N
N
O
H
Cl
H2
C
CH2
H
H
H3 C
N
N
CH3
-Cl -
H3 C
CH3
N
HN
N
N
Co
Co
H3 C
O
O
O
O
H3 C
CH3
N
CH3
HN
N
N
N
N
Figura 1
I dati cinetici ottenuti per le reazioni a partire dal cloro-, bromo- e iodometilderivato
mostrano che le velocità di formazione del metallociclo aumentano nell'ordine Cl < Br < I in
accordo con l'attesa diminuzione della forza di legame carbonio-alogeno e con il trend delle
velocità osservate nei processi di solvolisi degli alogenuri alchilici. Ciò suggerisce che lo stadio
lento del processo sia costituito dalla rottura del legame carbonio-alogeno e la concomitante Nalchilazione e piuttosto che dallo stadio di deprotonazione dell‟azoto oppure dal processo di
distorsione dell'anello equatoriale nell'avvicinamento dell'atomo di azoto al carbonio assiale.
Il composto è stato caratterizzato mediante diffrattometria RX che evidenzia la presenza
del metallociclo avente un angolo di legame CH2-Co-N di 40°. La reazione, che costituisce un
raro esempio di N.alchilazione nell'ambito della chimica dei complessi del Cobalto, può esser
considerata come la fase iniziale per il trasferimento di un‟ unità monocarboniosa da un atomo
metallico ad un substrato organico.
5. Sviluppo di complessi di Ru(III) dotati di attività antitumorale.
Da più di un anno (Ottobre 1999), il complesso di
rutenio(III) sviluppato a Trieste e denominato NAMI-A
([ImH][trans-RuCl4(Me2SO)(Im)]), viene sperimentato in fase
clinica I su pazienti come potenziale agente antimetastatico
N
Cl
Cl
presso il Netherland Cancer Institute di Amsterdam. Esso è
uno dei pochissimi composti antitumorali non-platino ad
entrare in fase di sperimentazione clinica e, in assoluto, il
primo composto a base di rutenio. La fase clinica I, tuttora in
-
HN
NH+
Ru
Cl
Cl
O
S
NH
CH3
CH3
corso, sta dando risultati incoraggianti e si prevede che nei primi mesi del 2001 inizierà la fase
clinica II. Lo scopo degli studi di fase clinica I è quello di individuare la dose massima tollerata
dai pazienti, correlata alla tossicità del composto, e di stabilire le migliori modalità di
somministrazione.
Il raggiungimento di questo primo traguardo ha richiesto un approfondimento delle
indagini sul NAMI-A sia di tipo chimico che farmacologico. In particolare, dal punto di vista
chimico è stata studiata la stabilità del NAMI-A in soluzione acquosa, per ottenere informazioni
necessarie alla sua formulazione ed alle successive modalità di somministrazione ai pazienti.
Inoltre è stato approfondito lo studio del comportamento del complesso in soluzione fisiologica
tamponata in seguito a riduzione del nucleo metallico da Ru(III) a Ru(II) con riducenti biologici
quali acido ascorbico e cisteina. In collaborazione con l‟ unità di Firenze è stata studiata
l‟interazione del NAMI-A e di altri composti di rutenio con l‟albumina e con il DNA.
La sintesi chimica si è inoltre indirizzata, come programmato, verso nuovi addotti di rutenio
che potessero presentare attività antineoplastica, i cosiddetti “composti di seconda generazione”.
Gli studi di fase clinica sul NAMI-A hanno evidenziato l‟importanza della stabilità dei composti
in soluzione acquosa; infatti, i processi di sterilizzazione e di formulazione del composto e,
soprattutto, la somministrazione per infusione lenta implicano che il complesso di rutenio deve
essere il più possibile stabile in soluzione acquosa. Sono quindi stati sintetizzati composti
analoghi al NAMI-A, di formula generale (LH)[trans-RuCl4(Me2SO)(L)], e ne è stata studiato il
comportamento in soluzione acquosa ed in soluzione fisiologica. Si è osservato che complessi con
leganti azotati meno basici dell‟imidazolo, quali ad esempio pirazina, pirazolo, ossazolo e
tiossazolo, sono più inerti del NAMI-A in soluzione acquosa, mentre presentano cinetiche di
idrolisi paragonabili a quelle del NAMI-A in soluzione fisologica. Quindi, se tali composti
mantenessero l‟attività antimetastatica del NAMI-A (prove in vivo attualmente in corso), essi
presenterebbero degli indubbi vantaggi dal punto di vista della formulazione e della
somministrazione rispetto al composto attualmente in fase clinica.
Inoltre è proseguita la sintesi di composti dinucleari di rutenio utilizzando leganti azotati
con due siti di coordinazione, facendo così seguito alla messa a punto della sintesi di due nuove
classi di composti dimerici simmetrici, sia dianionici che neutri, recanti il legante azotato a ponte
di
formula
rispettivamente
[Na]2[{trans-RuCl4(Me2SO)}( -L)]
e
[{mer-RuCl3(Me2SO)
(Me2SO)}( -L)] con L = pirazina, pirimidina, 4,4'bipiridile e derivati, ad esempio il dimero non
simmetrico
monoanionico
di
formula
[NH4]
Cl6
C6
C5
S2
Cl7
O2
C8
Cl5
[ trans-RuCl4(Me2SO-S) ( -pyz) mer-RuCl3 (Me2
SO-S)(Me2SO-O) ] (vedi Figura). Sono in corso
S3
Ru2
Cl3
C7
N2
O3
N1
Cl4
C9
C4
C10
studi per valutare l‟attività citotossica (in vitro) ed
Cl2
Ru1
S1
C3
antitumorale (in vivo) dei nuovi dimeri; inoltre, in
O1
Cl1
C2
C1
collaborazione con le unità di Bari e di Firenze,
vengono investigate le interazioni dei composti con DNA e proteine in vitro.
Sono infine proseguiti gli studi sulle caratteristiche strutturali dei complessi metallici con
solfossidi. Dal punto di vista farmacologico è stato osservato che il NAMI-A risulta privo di
effetti citotossici diretti contro le cellule tumorali sia in vitro sia in vivo, a concentrazioni
confrontabili ed equivalenti nei due sistemi. L‟ordine di grandezza, di 10-4 M, risulta da dati
ottenuti mediante studi di spettroscopia ad assorbimento atomico, sia su cellule trattate in vitro sia
su cellule ottenute ex vivo da tumori prelevati da animali trattati i.p. con NAMI-A alla dose
convenzionale di 35 mg/kg/die per 6 giorni consecutivi. In particolare, a livello polmonare la
concentrazione di 10-4 M sembra mantenersi costante anche dopo sospensione del trattamento per
tempi relativamente lunghi suggerendo che il NAMI-A mantenga la capacità di esercitare gli
effetti farmacologici sulle metastasi polmonari anche dopo interruzione di un ciclo di
somministrazione.
Confrontato con il cisplatino, il NAMI-A mostra un comportamento marcatamente diverso;
la tossicità sistemica del NAMI-A risulta inferiore, anche se va sottolineato che la manifestazione
tossica più evidente, analogamente al cisplatino, è a carico dell‟apparato renale; studi preliminari
indicano comunque che l‟effetto tossico del NAMI-A sul nefrone è almeno in parte reversibile.
Infine, ma non meno importante, il NAMI-A è almeno altrettanto o anche più attivo del cisplatino
in esperimenti di confronto usando tumori murini che metastatizzano nel polmone. Infatti, sia
considerando gli effetti sul prolungamento del tempo di sopravvivenza, sia considerando l‟effetto
di riduzione delle metastasi polmonari, il NAMI-A, al contrario del cisplatino che mostrava una
dipendenza dal tumore trattato, ha causato effetti molto marcati sia utilizzando il Lewis lung
carcinoma, sia impiegando il carcinoma mammario MCa del topo CBA o l‟adenocarcinoma
mammario TS/A nel topo Balb/c. Questi risultati assumono ulteriore importanza se si considera
che in due dei tre modelli utilizzati, il trattamento è stato effettuato su tumori in fase di crescita
avanzata, anche dopo rimozione chirurgica del tumore primario.
6. Studi di chimica supramolecolare
In anni recenti l‟ Unità di Trieste ha anche dato importanti contributi nel settore della
chimica supramolecolare inorganica, in particolare nella costruzione e caratterizzazione, sia in
N
trans-DPyP
Figura 1. Trans- e cis-dipiridil
porfirine e loro rappresentazione
schematica come building blocks.
cis-DPyP
NH
N
NH
N
N
NH
N
NH
N
N
N
soluzione che allo stato solido, di sistemi supramolecolari formati da porfirine e centri
metallici. La presenza di porfirine o metallo-porfirine nei sistemi supramolecolari è di primario
interesse, in quanto queste molecole sono in grado di introdurre utili proprietà fotochimiche e
redox nel sistema. Gli addotti porfirinici supramolecolari sono utilizzati come modelli di sistemi
biologici nello studio dei meccanismi di cattura della luce solare (“light-harvesting devices”) e di
reazioni di trasferimento elettronico fotoindotto
(sistemi fotosintetici), oltre che in settori quali:
riconoscimento molecolare, sensori, catalisi,
polimeri conduttori, composti con proprietà
ottiche non lineari.
La
metodologia
di
sintesi
per
autoassemblaggio, che sfrutta la formazione di
legami di coordinazione tra siti basici periferici
sull‟anello porfirinico e centri metallici, ha
recentemente fornito numerosi esempi di
aggregati di porfirine di grosse dimensioni
ottenuti con buona resa.
Figura 2.
Quadrato molecolare omometallico di porfirine
Recentemente sono state messe a punto
delle procedure di sintesi efficienti per la costruzione di quadrati molecolari di porfirine e
composti di coordinazione.
2+
Y
Ru
X
Cl
Cl
N
Y
N
N
M
N
N
N
Ph 2
P
TfO
Cl
Ru
X
N
N
M
N
N
N
Cl
N
Pd
TfO
N
N
-
P
Ph 2
2TfO
N
N
N
N
N
M
N
N
N
M
N
N
Ph
Pd P 2
P
Ph2
Figura 3. Procedura di sintesi di un quadrato di porfirine etero-bimetallico.
Tali addotti vengono costruiti tramite auto-assemblaggio di bis-4'piridilporfirine, cioè
porfirine che presentano due siti basici periferici a 90° o a 180° fra di loro, e opportuni composti
di coordinazione ottaedrici o planari quadrati con due siti labili in geometria cis o trans.
Dall‟opportuna combinazione di questi building blocks è possibile ottenere quadrati molecolari di
tipo 2:2 o 4:4 in cui le porfirine occupano rispettivamente gli angoli o i lati del metallaciclo. In
generale, i centri metallici che costituiscono gli angoli (o che giacciono lungo i lati) del quadrato
molecolare possono essere tutti uguali fra loro (quadrati omo-metallici), oppure di natura diversa
(quadrati etero-metallici). Sono state sviluppate procedure di sintesi per la costruzione di quadrati
molecolari di porfirine sia omo- che etero-metallici (Figure 2 e 3).
Una scelta opportuna dei centri metallici e dei leganti ancillari (non aventi cioè funzione
strutturale) permette di variare la carica complessiva (e quindi la solubilità) dell‟addotto e di
introdurre opportune funzionalità (e.g. magnetiche, fotofisiche, centri chirali). Inoltre, è stata
messa a punto la sintesi di quadrati molecolari in cui le piridilporfirine, oltre ad essere coordinate
ai centri metallici con funzione strutturale (angoli del quadrato), sono anche coordinate ad altri
centri metallici periferici. L‟importanza di questo approccio sintetico è facilmente comprensibile:
la scelta opportuna dei centri metallici periferici (spettatori) e di quelli strutturali potrebbe
favorire i trasferimenti di elettroni e/o di energia, eventualmente fotoindotti, tra i diversi siti della
supramolecola.

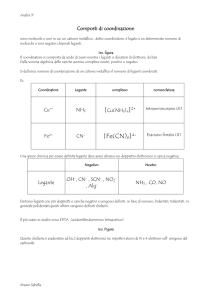










![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)