Corso di laurea in SCIENZA DEI MATERIALI
Insegnamento di Applicazione del Computer in Scienza dei Materiali
1. Docenza
Docente: dr.Bartolomeo Civalleri
Dipartimento di Chimica IFM
Tel.: +39-011-6707564 ; Fax: +39-011-6707855
e-mail: [email protected] ; WEB: http://www.theochem.unito.it/didattica
2. Finalità ed obiettivi dell’insegnamento
Finalità
Il corso si propone di fornire agli studenti un’introduzione agli strumenti di calcolo quanto-meccanico utilizzati
nella moderna chimica computazionale molecolare e dello stato solido. L'obiettivo principale e' mostrare
l'utilita' dei programmi di calcolo quanto-meccanici nello studio modellistico di materiali
Obiettivi
L’allievo dovrà essere in grado di
a) conoscere che cosa si intende con approccio computazionale nella scienza dei materiali e come questo stia
diventando uno strumento importante nella ricerca scientifica e un utile complemento all’attività sperimentale;
b) apprendere l’utilizzo base di programmi di calcolo quanto-meccanici per lo studio di sistemi molecolari e
cristallini
3. Pre-requisiti in ingresso e competenze minime in uscita
Pre-requisiti (in ingresso)
Insegnamenti fornitori
Fondamenti di meccanica quantistica
Meccanica quantistica
Principi di fisica dello stato solido
Fisica dello stato solido
Elementi di cristallografia
Cristallografia
Competenze minime (in uscita)
Conoscere come si possano studiare le proprietà di materiali
attraverso tecniche di modellizzazione
Conoscere gli elementi di base per l’uso di programmi di calcolo
molecolare come Gaussian
Insegnamenti fruitori
4. Metodologia didattica
La metodologia didattica impiegata consiste in:
a) 6 ore di lezioni in aula
b) 12 ore di esercitazioni nel laboratorio informatico
5. Programma, articolazione e carico didattico
Argomento
Ore Lez.
Ore Eserc.
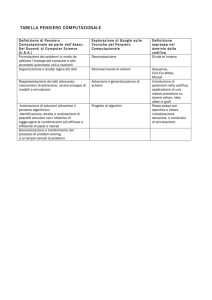
Approccio computazionale nella Scienza dei Materiali
2
Accenni alla simulazione multiscala
Definizione di scienza dei materiali computazionale
Tecniche per la definizione di un modello strutturale per la
simulazione di materiali
Fasi della progettazione di un esperimento al calcolatore
Accenni sui principali metodi quanto-meccanici per lo studio 2
di molecole e solidi:
a) principali approssimazioni dell’equazione di
Schrodinger
b) metodi ab-initio molecolari (HF e DFT)
c) estensione al trattamento di sistemi cristallini
Presentazione e caratteristiche dei principali programmi di
2
calcolo ab-initio molecolari e per sistemi cristallini (in
Totale Ore
2
2
2
particolare Gaussian e CRYSTAL)
Descrizione dell’input e analisi dell’output dei programmi
Gaussian e CRYSTAL
Accenni sull’ottimizzazione della struttura di molecole e
cristalli
Modulo1:
Introduzione all’uso di Gaussian98 per Windows
Introduzione ai programmi di visuallizzazione grafica
GaussView e Moldraw
La molecola d’acqua: ottimizzazione della geometria e
calcolo delle frequenze vibrazionali (Gaussian98)
Analisi dei risultati e discussione
Modulo 2:
Il dimero dell’acqua: definizione del problema,
ottimizzazione della geometria e calcolo delle frequenze
vibrazionali (Gaussian98)
Visualizzazione e analisi dei dati calcolati con discussione sul
cambiamento delle caratteristiche strutturali e vibrazionali
della molecola d’acqua isolata e nel dimero
Modulo 3:
Uso dell’approccio a cluster nella simulazione di materiali
Simulazione degli ossidrili superficiali di materiali silicei
Definizione del problema e creazione di un semplice modello
strutturale con l’ausilio di un programma di grafica
molecolare (Moldraw)
Calcolo delle proprietà strutturali e vibrazionali del modello
isolato e in interazione con ammoniaca
Analisi/discussione dei risultati e confronto con i dati
sperimentali
Modulo 4:
Introduzione all’uso di CRYSTAL
Esempi di uso dell’approccio periodico nella simulazione di
materiali (crustalli, superfici, difetti, ...)
Totale 6
2
2
2
4
6
6
2
2
12
18
6. Materiale didattico
I testi base consigliati per il corso sono:
lucidi delle lezioni e materiale usato per le esercitazioni (forniti dal docente e disponibili sul sito)
E’ consigliato l’utilizzo del seguente materiale per approfondimenti e integrazioni:
F. Jensen, Introduction to Computational Chemistry, Wiley, 1999.
Infine sono di seguito indicati siti internet di interesse:
http://www.gaussian.com
http://www.crystal.unito.it/tutojan2004/tutorials/index.html
7. Modalità di verifica/esame
L'esame si svolge , di norma, come segue:(dettagliare il più possibile:scritto,orale,prove in itinere,criteri di
valutazione ecc.)
L’esame consiste in una prova pratica e in una serie di domande scritte.
a) La prova pratica riprende il modulo 1 delle esercitazioni e rappresenta la parte più importante
dell’esame. Riguarda l’utilizzo del programma Gaussian e viene richiesto di saper calcolare le
proprietà strutturali e vibrazionali di un semplice addotto molecolare (es. FH…NH3) valutando la
variazione nelle caratteristiche dei singoli componenti in seguito alla formazione dell’addotto. Gli
input necessari vengono forniti nel testo dell’esame. Il testo comprende anche una serie di tabelle
che gli studenti devono riempire con i dati estratti dall’analisi dei dati prodotti dal programma. Gli
studenti devono quindi dimostrare di saper usare il programma e di essere in grado di analizzare gli
output estraendo le informazioni richieste.
b) Le domande scritte (solitamente tre), di cui è richiesto uno sviluppo molto sintetico, riguardano
alcuni punti generali discussi nella parte di lezioni in aula e sono mirate a valutare la comprensione
degli argomenti trattati.
Il voto finale (in trentesimi) è determinato in larga parte dall’esito della prova pratica (5/6), il resto (1/6) è
valutato sulla base delle risposte alle domande.