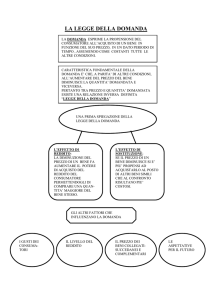
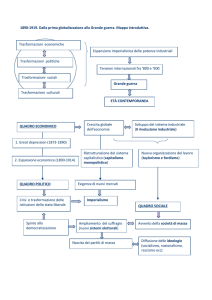
G. Moro
Ott. 99
2. ENERGIA INTERNA ED ENTALPIA.
Nel seguito si considerano trasformazioni di un sistema chiuso tra due ben
definiti stati di equilibrio. Solo con questo vincolo le variazioni delle proprietà
tra stato iniziale e finale sono descrivibili con le grandezze di stato. Gli stati
iniziali e finali sono quindi rappresentabili come punti in un diagramma di
stato T, p.
Catalogazione delle trasformazioni termodinamiche come reversibili/irreversibili.
• Trasformazioni reversibili: il sistema passa attraverso stati molto prossimi a quelli di equilibrio.
In questo caso non solo gli stati iniziali e finali, ma anche gli stati intermedi sono rappresentabili termodinamicamente, ottenendo così una traiettoria nel diagramma di stato. La natura reversibile di tali trasformazioni deriva
dalla necessità di operare con piccoli (infinitesimali) incrementi dei parametri
esterni rispetto alla condizione di equilibrio, cosicchè il senso della trasformazione può essere facilmente invertita cambiando il segno di tali incrementi.
Esempio delle compressioni/espansioni (un gas in un cilindro con pistone): l’equilibrio meccanico richiede l’uguaglianza della pressione esterna
pext (operante sul pistone) e la pressione p del sistema (supposto in equilibrio
termodinamico)
p = pext
Per comprimere reversibilmente il sistema si dovrà incrementare la pressione
esterna di una quantità molto piccola δp (tendenzialmente infinitesima)
pext = p + δp
in modo tale da non distruggere lo stato di equilibrio del sistema. Invertendo
il segno di δp, si ha la trasformazione opposta (espansione). Volendo in tutta
generalità comprimere il sistema dalla pressione iniziale p1 alla pressione finale p2 , si dovrà operare con una pressione esterna dipendente dal tempo, ad
esempio
p1
(t ≤ t1 )
=
pext (t) = p1 + r(t − t1 ) (t1 < t < t2 )
=
p2
(t ≥ t2 )
1
dove r ≡ (p2 − p1 )/(t2 − t1 ) è la velocità di compressione. Affinchè la trasformazione sia reversibile, la variazione della pressione deve essere asintoticamente lenta (r → 0). Compressioni a velocità r finita sono irreversibili
nella misura in cui si generano stati di non-equilibrio non rappresentabili
nel diagramma di stato (o, più precisamente, rappresentabili solamente in
un ipotetico diagramma multidimensionale che includa anche gradi di libertà
descrittivi degli stati di non-equilibrio).
• Le trasformazioni reversibili sono un caso limite delle trasformazioni reali.
Analogamente si possono descrivere i processi di riscaldamento/raffreddamento, immaginando il sistema a contatto con un corpo esterno la cui temperatura Text possa essere modificata a piacimento. La condizione di equilibrio
termico equivale all’uguaglianza con la temperatura T del sistema
Text = T
In un riscaldamento reversibile bisogna incrementare di una quantità infinitesima la temperatura del corpo esterno
Text = T + δT
in modo tale da conservare la condizione di equilibrio del sistema. Per variazioni brusche di Text si hanno trasformazioni irreversibili attraverso stati
di non-equilibrio del sistema (ad esempio si vengono a stabilire gradienti di
temperatura che determinano moti convettivi).
Dati due stati di equilibrio, ad esempio determinati dalle coppie (T1 , p1 )
e (T2 , p2 ) di temperatura e pressione, esiste una infinità di trasformazioni (sia
reversibili che irreversibili) che li connettono. Il I0 principio della termodinamica determina i vincoli per tali trasformazioni sotto forma di conservazione
dell’energia. Un aspetto non ovvio della questione deriva dall’esistenza dei
fenomeni di dissipazione (di energia macroscopicamente misurabile in energia distribuita a livello microscopico/molecolare). Si consideri come esempio
la caduta nel vuoto di un peso di massa m inizialmente a riposo ad un altezza h0 dal suolo. Calcoliamo l’energia del sistema come somma dell’energia
potenziale e cinetica
E = mv 2 /2 + mgh
dove g e l’accelerazione di gravità (g = 9.81 m/sec2 ), con E0 = mgh0 come
valore iniziale. Immediamente prima dell’urto con il suolo, tutta l’energia
2
potenziale si è trasformata in energia cinetica in corrispondenza della velocità
√
v0 = 2gh0 . Dopo un tempo sufficiente il peso è in uno stato di immobilità
sul suolo, a cui va apparentemente attribuita una energia nulla, così violando
il principio della conservazione dell’energia. Per ripristinarlo bisogna tener
conto della dissipazione dell’energia cinetica mv02 /2 in “energia termica” del
peso (assumendo che non ci sia dissipazione verso il sistema suolo), che si
manifesta in un suo incremento di temperatura. Nel bilancio dell’energia
bisogna includere anche tale contributo descritto dall’energia interna U , per
cui a conservarsi è l’energia totale
E = mv 2 /2 + mgh + U
Nella trasformazione in questione si avrà allora una variazione di energia interna ∆U = mgh0 .
La termodinamica statistica fornisce una interpretazione microscopica
dell’energia interna come valore medio dell’energia di interazione delle molecole e della loro energia cinetica rispetto al baricentro del sistema (ovviamente
ci sono fluttuazioni di tale energia attorno al valore medio quando si consideri
il sistema in equilibrio con l’ambiente).
Enunciazione del Io principio della termodinamica:
• per ogni sistema chiuso esiste una funzione di stato detta energia interna
U la cui variazione in una trasformazione è data da
∆U = q + w
dove q e w sono rispettivamente il calore assorbito dal sistema ed il lavoro
fatto sul sistema.
Da notarsi la convenzione sui segni di q e w, con valori positivi corrispondenti
ad incrementi energetici del sistema. Inoltre
• se il sistema non è immobile allora il principio va riferito all’energia totale
∆E = q + w con E inclusiva sia dell’energia interna che dell’energia
cinetica e potenziale.
• Ha senso considerare solo trasformazioni tra stati di equilibrio in cui
sono definite le grandezze di stato, quale l’energia interna che può essere
pensata come funzione di stato di variabili indipendenti, ad esempio U =
U (T, p).
3
• L’energia interna è una proprietà estensiva.
• L’energia interna è determinabile solamente in base al Io principio. Ne
consegue che sono misurabili solo variazioni di energia interna. Si può
attribuire un valore assoluto ad U solo se si fissa un qualche stato di
riferimento (come per altre forme di energia).
L’affermazione che U è una grandezza di stato costituisce il postulato del Io
principio.
Il calore non è una grandezza di stato caratterizzante lo stato di equilibrio di un sistema, ma il flusso di energia (interna) che si stabilisce tra
1) due corpi a temperatura differente e
2) separati da una superficie diatermica (cioè conduttrice di calore).
Le superfici adiabatiche sono per definizione quelle che non consentono il flusso
di calore.
• Definizione di processi adiabatici come trasformazioni in assenza di flusso
di calore: q = 0.
o
Il I principio fornisce anche il criterio di misura della quantità di calore. Si
consideri una generica trasformazione tra due stati di equilibrio
∆U = q + w
Introducendo una trasformazione adiabatica tra gli stessi due stati e quindi
con la stessa variazione di energia interna (essendo una grandezza di stato)
∆U = wad
si può derivare la quantità di calore
q = wad − w
• La quantità di calore è definita sulla base di misure di lavoro.
Da questo punto di vista si può analizzare l’esperimento di Joule per
la determinazione dell’equivalente meccanico della kilocaloria, definita come
quantità di calore nel processo:
1 kg H2 O (p = 1Atm, T = 287.65K) → 1 kg H2 O (p = 1Atm, T = 288.65K)
cioè per riscaldare un kg di acqua da 14.5◦ C a 15.5◦ C alla pressione atmosferica. Una trasformazione alternativa tra gli stessi due stati si può realizzare
4
in maniera adiabatica, utilizzando un peso di massa m che per caduta da una
altezza ∆h mette in rotazione un agitatore. Per opportuna scelta (empiricamente verificabile) di m e ∆h, la dissipazione dell’energia meccanica da parte
dell’agitatore assicura la trasformazione adiabatica tra i due precedenti stati.
Applicando il primo principio si ha
∆U = mg∆h + wvol
dove wvol è il lavori di volume fatto dalla pressione atmosferica (essendoci
comunque variazioni di volume dell’acqua). Se la stessa trasformazione viene
effettuata con una una quantità di calore q fornita da un corpo esterno, si
ottiene
∆U = q + wvol
con lo stesso lavoro di volume. Per confronto si deriva
q = mg∆h
e la realizzazione dell’esperimento fornisce il seguente valore numerico della
kilocaloria
1 kcal = 4.184 kJ
Da notarsi che in questo esperimento ambedue le trasformazioni sono irriversibili.
Es. Quale dovrebbe essere l’altezza ∆h se m = 1 kg? Come si deve realizzare
in pratica l’esperimento?
Normalmente le misure di quantità di calore si effettuano nei calorimetri
utilizzando una sostanza di riferimento. Il principio basilare è che la quantità
di calore assorbita da un corpo deve essere esattamente uguale a quella ceduta
dal corpo con cui è a contatto. Applichiamo il primo principio singolarmente
a ciascun corpo
∆U1 = q1 + w1
∆U2 = q2 + w2
ed al sistema costituito dal loro insieme tenuto conto del carattere estensivo
dell’energia interna
∆U = ∆U1 + ∆U2 = q + w
5
dove
q = q1 + q2
w = w1 + w2
se il sistema globale è mantenuto in condizioni adiabatiche, allora
q = q1 + q2 = 0
cioè le quantità di calore assorbite dai due corpi sono uguali in modulo e di
segno opposto.
Anche il lavoro è descrittivo di una trasformazione e non dello stato di
equilibrio di un sistema. Se ne possono distinguere diversi tipi a seconda della
forma di interazione con il sistema, in particolare
1) lavoro meccanico di una forza meccanica che agisce sul sistema producendo il moto di una superficie di separazione,
2) lavoro elettrico prodotto da una differenza di potenziale imposta dall’esterno che genera una corrente nel sistema.
Forme di lavoro associate ad altri tipi di campi esterni, sebbene possibili, non
verrano esaminate.
Per il lavoro meccanico si distinguono due tipologie fondamentali a seconda delle modalità della sua esecuzione
a) lavoro puramente dissipativo (sfregamento, agitazione) come nell’esperimento di Joule, che non provocano variazioni di volume se non per effetto
termico,
b) lavoro di volume in cui la forza esterna è utilizzata per modificare il volume del sistema e che sotto opportune condizioni può essere restituito
dal sistema (cicli di compressione/espansione).
Il primo tipo essendo stato precedentemente esaminato, consideriamo ora il
lavoro di volume ed i metodi per la sua determinazione. Il caso più semplice si
ha quando su un sistema inizialmente all’equilibrio con dati (p1 , V1 ) si esercita
una pressione esterna costante pext ̸= p1 . Dopo un tempo sufficiente il sistema
raggiunge il nuovo stato di equilibrio (p2 , V2 ) con p2 = pext . Immaginando
un cilindro di superficie S in cui il pistone esegua uno spostamento ∆x nella
direzione della forza esterna Fext , si ottiene per il lavoro di volume
wvol = Fext ∆x = pext S∆x = −pext ∆V
dove ∆V = V2 − V1 .
6
• La precedente relazione determina il lavoro fatto sul sistema solo in assenza di attrito tra pistone e cilindro, altrimenti si avrebbe parziale dissipazione del lavoro meccanico sul contenitore.
Affinchè tale lavoro sia anche calcolabile, oltre che misurabile da differenze
di volume ∆V , occorre che lo stato finale sia determinato univocamente. Ma
ciò dipende dalla temperatura raggiunta dal sistema in relazione allo scambio
termico con l’ambiente.
• Definizione di termostato: sistema in grado di scambiare velocemente
grandi quantità di calore senza cambiare la sua temperatura (esempio:
miscela di acqua e ghiaccio).
Per i processi isotermi nei quali il sistema è a contatto con un termostato a
temperatura T fissata, il lavoro di volume è deducibile dall’equazione di stato
V = V (T, p)
Es. Dato lo stato iniziale (p1 , T ), esprimere il lavoro di volume come funzione
della pressione esterna costante pext = p2 nel caso di i) un gas ideale, ii)
un liquido nell’approssimazione a compressibilità costante.
Nel caso generale in cui la pressione esterna non è costante ma cambia
con il tempo t in una qualche prefissata maniera, pext = pext (t), si dovrebbe
misurare il volume del sistema V (t) in funzione del tempo. L’esperimento è
rappresentabile come due grafici per la pressione esterna ed il volume come
funzioni del tempo. Se quest’ultimo ha un andamento monotono, la funzione
può essere invertita come t = t(V ) che sostituita in pext (t) genera la rappresentazione
pext = pext (V )
Dalla quantità di lavoro infinitesima dwvol per uno spostamento del pistone
dx con variazione di volume dV
dw = Fext (V )dx = −pext (V )dV
si deriva il lavoro per integrazione
∫
wvol = −
V2
pext (V )dV
V1
che (a parte il segno) coincide con l’area sottesa dalla curva pext (V ). In
presenza di una pressione esterna variabile, il lavoro non è calcolabile se non
7
si conosce la risposta del sistema, cioè il suo volume in funzione del tempo.
Si consideri allora il caso particolare in cui la trasformazione sia isoterma e
reversibile, cosicchè la pressione esterna è deducibile dall’equazione di stato
per la pressione pext = p(T, V ):
∫
wvol = −
V2
p(T, V )dV
V1
Nel caso particolare dei gas ideali si ottiene
∫
wvol = −
V2
nRT dV /V = −nRT ln(V2 /V1 ) = −nRT ln(p1 /p2 )
V1
Es. Integrare wvol per compressioni isoterme reversibili del gas di van der
Waals.
Es. Integrare wvol per compressioni isoterme reversibili di un liquido nell’approssimazione a kT costante.
Es. Confrontare il lavoro di compressione isotermo reversibile a 25◦ C da p1 =
1bar a p2 = 2bar per una mole di gas ideale ed una mole di acqua (kT =
5.01 × 10−5 /bar).
Es. Confrontare il lavoro di volume per la compressione isoterma di una mole
di gas ideale a 25◦ C da p1 = 1bar a p2 =2bar, condotta i) reversibilmente
o ii) a pressione esterna costante.
Es. Dimostrare che in tutta generalità (wvol )rev. ≤ (wvol )irr. per trasformazione isoterme (sia compressioni che espansioni o loro insiemi).
Es. Può il lavoro di volume fatto da una forza esterna avere un esito puramente dissipativo? Considerare come esempio una compressione seguita
da una espansione (ambedue isoterme) tra gli stessi due stati, ed eseguite
i) con pressione esterna costante o ii) reversibilmente.
Per il lavoro elettrico consideriamo solamente quello derivante dal passaggio nel sistema di una corrente. Quindi il sistema deve presentare due capi
metallici attraverso cui si possa stabilire un flusso di carica. Si consideri prima
il caso in cui i capi siano connessi da una semplice resistenza R a cui si possa
applicare la legge di Ohm, ∆V = Ri. Imponendo una differenza di potenziale
∆V costante per un tempo ∆t si effettua un lavoro elettrico
wel = q∆V
8
dove q = i∆t è la carica trasportata. Nel caso di potenziali applicati dipendenti dal tempo, si dovrà integrare la forma differenziale corrispondente
∆V 2
dwel = ∆V dq = ∆V idt =
dt
R
È da sottolineare che nel caso di una resistenza, il lavoro elettrico è puramente
dissipativo e non può essere operato in maniera reversibile.
Es. Calcolare il lavoro fatto su una resistenza R nel tempo ∆t da una differenza di potenziale dipendente dal tempo come i) ∆V (t) = at, ii)
∆V (t) = a sin(ωt). Sotto quali condizioni il lavoro elettrico è all’incirca
proporzionale a ∆t nel caso ii)?
La cella galvanica (pila) costituisce l’altro importante sistema. La generazione di una corrente per semplice cortocuitazione dei suoi capi evidenzia
la differenza rispetto al caso della resistenza. Altrimenti si può misurare la
corrente come funzione della differenza di potenziale applicato ∆V ai suoi
capi, così determinando la differenza di potenziale a corrente nulla.
• Definizione di forza elettromotrice E (f.e.m) di una pila: differenza di
potenziale ai capi della pila in assenza di passaggio di corrente
E ≡ (∆V )i=0
• Solo per ∆V = E (o a circuito esterno aperto) la cella galvanica può
essere in uno stato di equilibrio. Altrimenti si ha passaggio di corrente
che produce una trasformazione chimica dei suoi costituenti.
La forza elettromotrice è una proprietà di stato: due pile identicamente costituite e nelle stesse condizioni termodinamiche hanno la stessa forza elettromotrice. Uno degli obiettivi della termodinamica è di stabilire l’equazione
di stato per la forza elettromotrice, vale a dire la relazione con le grandezze
termodinamiche dei costituenti della cella galvanica. A tale scopo è indispensabile attribuire il segno delle grandezze elettriche in modo non ambiguo.
Si supponga di avere comunque scelto l’elettrodo di destra (e l’elettrodo di
sinistra), allora per definizione
∆V ≡ Vdestra − Vsinistra
ed il lavoro elettrico è dato da wel = q∆V se viene attribuito il segno positivo
alla carica q che circola esternamente dall’elettrodo di sinistra a quella di
9
destra. Un lavoro elettrico positivo per ∆V > 0, corrisponde al trasferimento
di carica positiva dall’elettrodo di sinistra a potenziale più basso, all’elettrodo
a potenziale più elevato. Se la pila è cortocircuitata (resistenza esterna nulla
e quindi ∆V = 0), il lavoro elettrico è nullo.
Es. Il lavoro elettrico dipende dalla scelta dell’elettrodo di destra/sinistra?
Con le pile il lavoro elettrico può essere effettuato in maniera reversibile,
applicando una differenza di potenziale che differisce dalla f.e.m a meno di
una quantità δV piccola a piacere
∆V = E + δV
La direzione del flusso di carica, e quindi anche il lavoro elettrico esprimibile
in forma differenziale come
dwel = Edq
viene a dipendere dal segno di dq (e quindi di δV ). L’integrazione della
precedente forma differenziale non è banale poichè il flusso di carica (che
produce modificazioni chimiche nei costituenti) in generale modifica la forza
elettromotrice della pila.
Es. Può una pila essere assimilata ad una pura resistenza se la sua forza
elettromotrice è nulla?
In molti contesti risulta importante predire gli effetti termici di un processo (termochimica). Quindi si pone la questione: si può determinare la
quantità di calore assorbita in un processo sulla base della sola conoscenza
delle grandezze di stato? Poichè
q = ∆U − w
una risposta affermativa è possibile se è assente ogni forma di lavoro, w = 0,
ed il volume è costante (altrimenti ci sarebbe lavoro di volume), nel qual caso
la quantità di calore è data dalla variazione della grandezza di stato energia
interna indipendentemente dal carattere reversibile o irreversibile del processo
(q)w=0,V =cost. = ∆U = U (T2 , V ) − U (T1 , V )
In questo caso è conveniente considerare l’energia interna come funzione di
stato della temperatura e del volume. Per caratterizzarne la dipendenza dalla
10
temperatura a volume fissato, si introduce il corrispondente coefficiente differenziale chiamato calore specifico a volume costante
(
)
∂U
CV ≡
∂T V
che è una grandezza estensiva. Per sostanze pure è sufficiente la conoscenza
del calore specifico molare
cV ≡ CV /n
che normalmente si esprime in unità J/ mol K. In generale il calore specifico
dipende dalle variabili di stato indipendenti, e la sua integrazione è richiesta
nel calcolo delle variazioni di energia interna
∫
T
U (T, V ) − U (T0 , V ) =
dT ′ CV (T ′ , V ) ≃ (T − T0 )CV
T0
dove l’approssimazione è applicabile per intervalli di temperatura ristretti.
Es. Una pallina di piombo del peso di 0.1 kg è lasciata cadere da una altezza di
10 m dal suolo. Assumendo che durante l’urto tutta l’energia cinetica sia
dissipata sotto forma di energia interna della sola pallina, determinare
l’incremento della sua temperatura, noto il calore specifico del piombo
0.128 J/g K. Come dipende l’incremento della temperatura dalla massa
della pallina?
Normalmente però le trasformazioni termodinamiche vengono eseguite a
pressione costante (spesso quella atmosferica) invece che a volume costante.
È quindi necessario introdurre la grandezza di stato estensiva entalpia
H ≡ U + PV
per descrivere gli effetti termici a pressione costante. Si dimostra infatti che
(q)w=wvol ,pext =cost. = ∆H
in una trasformazione con solo lavoro di volume e con pressione esterna pext
rigorosamente costante. In tal caso la pressione iniziale p1 e finale p2 del
sistema uguagliano la pressione esterna
pext = p1 = p2 ≡ p
11
e conviene considerare l’entalpia come funzione di stato della pressione e della
temperatura: H = H(T, p). Per applicazione del primo principio si dimostra
il precedente teorema
q = ∆U − wvol = U2 − U1 + pext (V2 − V1 ) = H2 − H1
indipendentemente dalla natura reversibile o irreversibile della trasformazione
tra i due stati di equilibrio del sistema chiuso. Per caratterizzare la dipendenza
dell’entalpia dalla temperatura a pressione costante, si definisce il calore specifico a pressione costante
(
)
∂H
Cp ≡
∂T p
con la quantità molare cp ≡ Cp /n per sostanze pure. La variazione di entalpia
è deducibile via integrazione del calore specifico
∫
T
H(T, p) − H(T0 , p) =
dT ′ Cp (T ′ , p) ≃ (T − T0 )Cp
T0
con l’ultima approssimazione valida per intervalli di temperatura ristretti.
Come applicazione si consideri la miscelazione di n1 moli di un liquido puro
alla temperatura T1 con n2 moli dello stesso sostanza alla temperatura T2 .
Nell’ipotesi che il processo avvenga a pressione costante, si può esplicitare la
variazione entalpica considerando (T, p, n) come variabili indipendenti:
∆H =[H(T, p, n1 ) − H(T1 , p, n1 )] + [H(T, p, n2 ) − H(T2 , p, n2 )] =
=n1 cp (T − T1 ) + n2 cp (T − T2 )
dove si è utilizzata l’approssimazione a cp costante e la proprietà addittività
H(T, p, n1 +n2 ) = H(T, p, n1 )+H(T, p, n2 )] della grandezza estensiva entalpia.
Se il processo avviene adiabaticamente, ∆H = 0, la precedente relazione
permette di calcolare la temperatura finale T
T =
n1 T1 + n2 T2
n1 + n2
Es. Quant’è la variazione di entalpia dell’acqua nell’esperimento di Joule utilizzando un peso di 1 kg che scende di un’altezza di 3 m?
Es. Stimare il tempo richiesto per portare all’ebollizione l’acqua per la pasta
se si utilizza un fornello elettrico con una potenza di 1 kw (calore specifico
12
dell’acqua: cp = 89.1 J/mol K). Quanto viene a costare se l’Enel fa pagare
100 lire per 1 kwh?
Ad una data pressione le trasformazioni di fase avvengono a temperatura
costante ed il calore assorbito in presenza di solo lavoro di volume definisce la
variazione entalpica del processo o calore latente. Ad esempio per la fusione
del ghiaccio
H2 O (s, p = 1bar, T = 273.15K) → H2 O (l, p = 1bar, T = 273.15K)
si denota con ∆Hf il calore di fusione per mole di acqua. Analogamente si
definiscono i calori latenti di ebollizione ∆Hv , di sublimazione ∆Hs , o per la
trasformazione tra due fasi solide (ad esempio Srombico → Smonoclinico ), che in
generale dipendono dalla pressione a cui il processo avviene.
Es. È definito il calore specifico Cp in corrispondenza di una transizione di
fase?
Considerando in una miscela la grandezza estensiva entalpia come funzione di T , p e del numero di moli dei costituenti, si possono definire le entropie
parziali molari
(
)
∂H
Hi ≡
∂ni T,p,nj
Esse determinano l’entalpia di mescolamento tra due liquidi puri
∆Hmix =
∑
ni (Hi − Hi∗ )
i
e quindi il calore assorbito nel mescolamento se si opera a (p, T ) costanti.
13