Laura Ferrucci, Valerio Alfieri, Raimondo Sollazzo
Brain glycogen in health and disease
Il glicogeno è un polimero ramificato del glucosio immagazzinato principalmente nel fegato e nei
muscoli scheletrici: in questi è utilizzato come rifornimento energetico durante la contrazione
muscolare, mentre è impiegato dal fegato come materiale di riserva a ridosso dei periodi di digiuno.
Il glicogeno è stato identificato anche in altri tessuti dove ricopre ruoli fino a poco tempo fa
sconosciuti. Tuttavia recenti studi riportano la loro attenzione sulla presenza di glicogeno all’interno
del sistema nervoso, in particolar modo in relazione alle sue funzionalità ed alle conseguenze del
malfunzionamento nel suo metabolismo.
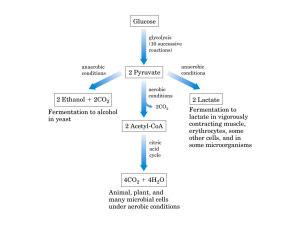
Il glicogeno è un omopolimero costituito da una catena di residui glicosidici uniti linearmente tra
loro da legami α (1-4) e da ramificazioni laterali connesse da legami α (1-6) presenti ogni 8-10
residui. La sintesi del glicogeno inizia con l’entrata del glucosio all’interno della cellula mediante
trasportatori specifici: qui avverrà la fosforilazione del glucosio in glucosio6-P, il quale subirà
l’azione di un’isomerasi a glucosio1-P ed in fine andrà a formare una molecola di uridina-glucosio5’difosfato, il quale è un donatore diretto di glucosio atto alla sintesi del glicogeno.
La glicogenina catalizza la formazione di un
corto polimero di glucosio, il quale subirà un
allungamento grazie all’azione della
glicogeno sintasi. L’enzima ramificante del
glicogeno, a sua volta, aggiungerà dei legami
α (1-6) per permettere la formazione delle
ramificazioni laterali; l’azione coordinata di
questi enzimi contribuirà alla corretta sintesi
di una molecola di glicogeno, il quale è
altamente solubile e pronto a subire
degradazione (quando richiesta) da parte
dell’enzima glicogeno fosforilasi (GP) e
Immagine 1. Sintesi del glicogeno
dell’enzima deramificante.
La glicogeno sintasi possiede due isoforme: una muscolare presente nella maggior parte dei tessuti
(MGS, codificata dal gene GYS1) ed una specifica per il fegato (LGS). Per quanto invece riguarda
l’enzima glicogeno fosforilasi, questo esiste in tre isoforme: una muscolare (MGP), una specifica
per il fegato (LGP) ed una terza specifica per il cervello (BGP), sebbene ivi sia stata riscontrata
anche la presenza di MGS. La sintesi e la degradazione del glicogeno sono due processi altamente
regolati i quali permettono l’omeostasi di questo metabolita all’interno dei tessuti. Per assicurare ciò
la cellula possiede numerosi meccanismi di controllo dell’attività della glicogeno sintasi: la proteina
PTG, la quale è una sub-unità regolatoria della protein-fosfatasi PP1, gioca un ruolo chiave
nell’attivazione della glicogeno sintasi mediante defosforilazione. È stato stimato che la quantità di
glicogeno presente all’interno del cervello sia intorno ad 1g (0.1% del peso del tessuto), e che
questa concentrazione sia 10 volte inferiore rispetto a quella presente nel muscolo scheletrico e 100
volte inferiore a quella presente nel fegato. Fino a poco tempo fa si riteneva che, solamente durante
la fase di sviluppo embrionale, il glicogeno cerebrale fosse presente sia nelle cellule gliali che
quelle neuronali; e che invece negli adulti questo polisaccaride si potesse ritrovare solamente
all’interno degli astrociti. Alla luce di queste motivazioni, il ruolo del glicogeno come riserva di
energia cerebrale è stato poco considerato, complice anche il fatto che il cervello è energeticamente
dipendente dal glucosio derivante dalla circolazione sistemica.
Ciononostante, è stato ipotizzato che il contenuto di glicogeno cerebrale possa rappresentare una
risorsa a breve termine di vitale importanza per specifiche attività neurali: come ad esempio la
formazione di memorie, i processi di stimolazione sensoriale ed i cicli di sonno-veglia.
Inoltre il glicogeno cerebrale rappresenta una fondamentale protezione durante l’esercizio intenso e
le situazioni di stress cellulare o condizioni patologiche come l’ipoglicemia, l’ischemia o le crisi
epilettiche. È largamente accettato anche il fatto che, attraverso l’azione di neuromodulatori e
neurotrasmettitori, le cellule neuronali siano in grado di stimolare la mobilizzazione di riserve
glicogeniche all’interno degli astrociti, i quali a loro volta saranno in grado di convertirlo in lattato:
quest’ultimo sarà infine internalizzato ed utilizzato dai neuroni. In uno studio del 2013 di Duran et
al., al fine di poter determinare in maniera inequivocabile il ruolo del glicogeno nel cervello, è stato
generato un modello animale GYS1nestin KO in cui si è stati in grado di ottenere un KO condizionale
dell’enzima glicogeno sintasi specificatamente nel sistema nervoso. In seguito sono state analizzate
le capacità di apprendimento di questi animali e sono state identificate differenze delle proprietà
elettrofisiologiche delle sinapsi dei sottocampi CA3-CA1 dell’ippocampo. In particolar modo sono
stati analizzati i livelli di LTP (long-term potentiation) delle sinapsi CA3-CA1 nel gruppo di
animali GYS1nestin KO rispetto ad un gruppo di controllo: dopo una sessione di stimolazioni ad alta
frequenza, solo il gruppo di controllo presentava un livello significativo di LTP nel primo giorno di
registrazioni, mentre i topi GYS1nestin KO non presentavano alcun segno evidente di LTP; addirittura
questi ultimi hanno mostrato dei fenomeni di depotenziamento nei giorni successivi di registrazione.
Oltre a questo sono stati effettuati dei test di tipo comportamentale mediante l’utilizzo di una
Skinner box al fine di monitorare i livelli di capacità di apprendimento delle cavie: i topi GYS1nestin
KO
sono risultati carenti in maniera significativa per quanto riguarda il consolidamento dei vari task
di apprendimento associativi rispetto al gruppo di controllo.
Queste osservazioni indicano che il glicogeno cerebrale gioca un ruolo chiave nella giusta
acquisizione e nel consolidamento di memorie, nonché nella capacità di compiere lavori di
apprendimento complessi.
Immagine 2: A. alterata performance comportamentale nella skinner box.
B.modificazioni elettrofisiologiche delle sinapsi ippocampali in topi
GYS1nestin-KO
Nonostante il fatto generalmente accettato che il
glicogeno non sia ritrovabile all’interno dei neuroni,
queste cellule presentano al loro interno MGS,
nonché tutti i vari meccanismi molecolari finalizzati
alla sintesi glicogenica. L’esistenza di glicogeno
fosforilasi all’interno dei neuroni è stata lungamente
dibattuta, ma un esperimento del 2003 ne dimostra la
presenza della sola isoforma BGP, mentre invece è
stata riscontrata all’interno degli astrociti anche
l’esistenza di MGP. Per determinare in maniera
definitiva la presenza di un metabolismo glicogenico
autonomo nei neuroni, sono stati condotti
esperimenti sia in vitro che in modelli di Drosophila,
nei quali sono stati appurati gli effetti protettivi nei
confronti dei danni causati da stati di ipossia.
È bene tenere presente però che i livelli glicogenici all’interno dei neuroni è mantenuto a livelli
estremamente bassi, in quanto un eccessivo accumulo di glicogeno induce l’apoptosi della cellula.
Mediante l’utilizzo un modello animale che sovra-esprime condizionatamente una forma di
glicogeno sintasi resistente all’inattivazione, è stato possibile dimostrare che l’accumulo di
glicogeno nei neuroni in topi e Drosophila portano ad una perdita di questo tipo cellulare, difetti di
locomozione, e ridotta sopravvivenza. Questi risultati suggeriscono che i livelli di glicogeno
neuronali devono essere strettamente regolati e mantenuti ad un livello basso ma funzionale, in
quanto il sovra-accumulo può portare all’insorgenza di patologie neurodegenerative.
La malattia di Lafora è una forma rara di epilessia mioclonica progressiva che ha effetti invalidanti
a livello motorio ed intellettivo: colpisce soprattutto gli adolescenti con un’incidenza di 1 caso ogni
milione e, purtroppo, attualmente non esistono terapie. La diagnosi può essere fatta in base all’età in
cui compaiono i primi sintomi, ai precedenti familiari, al rapido deterioramento della funzione
cognitiva e anche in base ad una biospia. Infatti, la caratteristica principale di questa patologia è
l’accumulo di una tipologia di glicogeno poco ramificato all’interno di neuroni, fegato, muscoli e
ghiandole sudoripare, sottoforma di aggregati chiamati poliglucosani, in inclusioni denominate
Lafora Bodies.
Immagine3:Accumulo di corpi di
Lafora osservati in una sezione di
cervello(a), di muscolo
scheletrico(b), di ghiandole
sudoripare(c)
Brain (a)
Skeletal muscle (b)
Sweat glands (c)
L'evoluzione della malattia di Lafora è caratterizzata da una progressiva degenerazione del sistema
nervoso, dovuta alla crescita di queste inclusioni che, nel giro di pochi anni, riduce i malati in uno
stato vegetativo terminale portando così alla morte a causa dell’apoptosi dei neuroni stessi. La
Malattia di Lafora è una patologia autosomica recessiva dovuta a mutazioni o delezioni che possono
colpire due geni: EPM2A, che codifica per la laforina (una protein-fostatasi) e EPM2B, che codifica
per la malina (una ubiquitina E3 ligasi). Gli individui con mutazioni nel gene EPM2A o nel gene
EPM2B sono neurologicamente e istologicamente indistinguibili. Queste due proteine formano un
complesso che regola l’accumulo del glicogeno; l’azione fosfatasica della laforina, inoltre, assicura
la qualità del glicogeno prevenendo la sua iperfosforilazione, che è considerata uno dei motivi
principali della formazione dei Lafora Bodies.
Immagine4:Mutazioni nel gene
EPM2A portano ad un’alterazione
dell’attività della Laforina che non
sarà in grado di impedire
l’iperfosforilazione del glicogeno.
Per studiare su modelli animali la malattia di Lafora, i ricercatori hanno creato un topo KO per il
gene malina, il quale ha confermato la possibilità dell’utilizzo di questo modello per lo studio della
patologia dopo aver visto che quest’ultimo presentava accumuli di poliglucosani in numerosi
tessuti, cervello compreso. Il contenuto di glicogeno nel cervello di questi animali era più del
doppio rispetto ai rispettivi wild type. La formazione di questi
aggregati era accompagnata da una progressiva
Immagine5: fEPSP
perdita di neuroni, da alterazioni
misurato nel
neurofisiologiche, in particolare da cambiamenti
sottocampo CA1 in
KO
delle proprietà elettrofisiologiche a livello delle
topi WT e Malina in
seguito a stimolazioni
sinapsi ippocampali e da alterazioni del
di intensità crescente
comportamento. I topi KO per la malina, infatti,
a livello della
risultavano essere iperattivi e avevano un tasso
collaterale di Schaffer
d’ansia ridotto, nonché un’elevata suscettibilità ad epilessia indotta
dall’acido kainico. I cambiamenti delle proprietà elettrofisiologiche a livello delle sinapsi
ippocampali sono stati confermati attraverso metodi in vivo che hanno permesso di studiare tali
sinapsi in topi svegli. E’ stato misurato il potenziale eccitatorio postsinaptico nel sottocampo CA1
in seguito ad impulsi di intensità crescente al livello della collaterale di Schaffer. I topi KO per la
malina presentano ampiezze maggiori rispetto agli animali WT, suggerendo una maggiore
eccitabilità sinaptica.
Tuttavia non c’era una chiara evidenza della correlazione tra accumulo di glicogeno e causa della
malattia. Per capire se quest’ultimo è direttamente responsabile della neurodegenerazione osservata
nei topi KO per la malina, i ricercatori hanno creato un topo KO per la malina e KO anche per
l’isoforma muscolare della glicogeno-sintasi (MGS), responsabile della sintesi di glicogeno nel
cervello. Come ci si aspettava, il cervello di questi animali non accumulava Lafora Bodies,
dimostrando quindi che l’accumulo di glicogeno è direttamente responsabile dei fenomeni
neurodegenerativi, dal momento che i topi KO per la malina e per la MGS non mostravano
marcatori di neurodegenerazione, cambiamenti nelle proprietà elettrofisiologiche o cambiamenti
alla suscettibilità per epilessia indotta
da acido kainico. I ricercatori, inoltre,
hanno creato anche modelli animali
KO per la malina, con una ridotta
attività della MGS (hanno ottenuto ciò
eliminando un solo allele del gene).
Come conseguenza si è verificata la
riduzione del numero di Lafora Bodies
presenti nel cervello e una
diminuzione della suscettibilità ad
epilessia indotta da acido kainico. Ciò
è stato confermato dalle registrazioni
effettuate a livello ippocampale dopo
che ai topi è stato iniettato acido
kainico e sono stati stimolati. I topi
KO per la malina avevano una
Immagine6:Registrazioni effettuate in seguito ad iniezione di
suscettibilità maggiore rispetto ai topi
acido kainico, prima e dopo stimolazione ippocampale in topi
MalinKO + MGShet. Queste
KO
KO
KO
sani, Malin ,Malin +MGS
considerazioni dimostrano che la
malattia di Lafora può essere prevenuta
semplicemente inibendo parzialmente la sintesi del glicogeno; in linea di massima, una riduzione
del 50% è sufficiente. Laforina e malina sono coinvolti in processi di autofagia in cui i componenti
danneggiati della cellula sono sequestrati e degradati all’interno di lisosomi. Si è notato che modelli
animali KO per la malina presentano un indebolimento del meccanismo di autofagia, mentre i
modelli animali double KO sia per la malina che per la MGS presentano un meccanismo autofagico
normale. Questo comportamento è stato attribuito all’eccessiva produzione di glicogeno che porta
quindi ad un indebolimento del meccanismo autofagico..
I corpi amilacei sono strutture rotondeggianti con un diametro tra i 10-50
μm, basofile e composte da poliglucosani. Essi sono presenti a livello della
ghiandola prostatica, del sistema nervoso e degli alveoli polmonari. In
principio si pensava che queste si trovassero nel sistema nervoso soltanto a
livello delle cellule gliali, ma questa ipotesi è stata successivamente
smentita dal ritrovamento di tracce di corpi amilacei anche nei neuroni. I
corpi amilacei sono presenti in cervelli di umani sani durante la vecchiaia e
condividono molte caratteristiche istologiche e biochimiche con i Corpi di
Lafora.
Immagine 7: corpi amilacei
È stato dimostrato infatti che topi in età matura presentano accumuli di poliglucosani
principalmente a livello dell’ippocampo e del cervelletto. La presenza dei corpi amilacei è però
strettamente dipendente dalla presenza della glicogeno sintasi. Topi knock out per questo enzima
infatti non mostravano presenza di questi aggregati, indicando uno stretto legame tra metabolismo
del glicogeno e insorgere di accumuli
di poliglucosani. Altri esperimenti
eseguiti sulla Drosophila mostravano
che la riduzione dell’espressione della
glicogeno sintasi aumenta la durata di
vita di questi animali e ne migliora le
capacità motorie. Alcuni ricercatori
paragonarono la distribuzione di
aggregati di poliglucosani di topi
maturi con topi knock out per la malina
e osservarono che questi aggregati sono
del tutto simili, sia per la loro
localizzazione che per la loro
immunopositività ad un array di
proteine legate allo stress cellulare. Il
Immagine 8: ruolo della GS nell’inibizione della formazione dei corpi amilacei
tasso di aggregati di poliglucosani era
però minore nei topi in età di vecchiaia rispetto a quelli affetti dalla malattia di Lafora e l’accumulo
di corpi amilacei assomigliava infatti ai primi stadi di questa malattia.
La quantità di corpi amilacei aumenta in seguito a condizioni di stress cellulare e, come vedremo
successivamente, in condizioni neurodegenerative. Nei corpi amilacei è stata infatti rilevata la
presenza di proteine caratteristiche di stati di stress citologico, come l’ubiquitina, le heat-shock
proteins e i cosiddetti prodotti finali della glicosilazione, macromolecole glicosilate correlate a stati
di invecchiamento e a patologie neurodegenerative.
Nei corpi amilacei sono state trovate anche tracce di α-sinucleina. L’α-sinucleina è una cosiddetta
“proteina incline all’aggregazione”, cioè una proteina che tende a formare dei grossi aggregati a
livello intra ed extracellulare in casi di patologie neurodegenerative, soprattutto nella malattia di
Parkinson.
La presenza dei corpi amilacei è associata infatti anche a numerose malattie neurodegenerative,
come la malattia di Parkinson, di Alzheimer e la
demenza vascolare. Tramite uso di anticorpi
specifici per proteine come ubiquitina, tau e le
heat-shock proteins è possibile osservare come il
numero di corpi amilacei sia molto più elevato in
malati di Alzheimer rispetto a individui sani nel
periodo della vecchiaia. Nella sclerosi multipla
invece, alcuni ricercatori hanno ipotizzato che i
corpi amilacei potessero rappresentare resti di
cellule neuronali aggregate e degenerate.
Immagine 9: A) corpi amilacei nel cervello in casi di demenzia vascolare. Il ruolo dei corpi amilacei nelle malattie
B) Proteine dei copri amilacei nel SNC
neurodegenerative non è stato ancora del tutto compreso. Alcuni studi recenti hanno suggerito che
questi possano fungere da indicatori di neurodegenerazione. Nei corpi amilacei sono infatti presenti
proteine ubiquitinate e proteine del complemento. La funzione dei corpi amilacei potrebbe essere
quindi quella di proteggere queste proteine dal riconoscimento da parte delle cellule immunitarie
del Sistema Nervoso Centrale e quindi di bloccare o limitare i danni infiammatori.
Altri ricercatori hanno invece proposto che i corpi amilacei possano essere coinvolti nel
sequestramento di proteine che potrebbero essere deleterie per le cellule del Sistema Nervoso. In
ogni caso la presenza di corpi amilacei è legata a stati di stress cellulare. Una prova di ciò è stato il
ritrovamento di transaglutaminasi-1, enzimi che in seguito a situazioni di stress provocano il crosslinking di proteine che vanno a formare il core dei corpi amilacei.
L’unione di questi dati suggerisce che i corpi amilacei, in quantità fisiologiche, possano servire
come “protettori” delle cellule del Sistema Nervoso. In quantità eccessive però i corpi amilacei e in
generale l’accumulo di glicogeno, sono causa di gravi malattie neurodegenerative. È importante
capire quindi come il glicogeno nel sistema nervoso non abbia un ruolo di secondo piano ma che la
sua visione sia cambiata molto negli ultimi decenni, acquistando un ruolo importante in diversi
processi fisiologici e patologici. Il suo metabolismo deve essere minuziosamente regolato perché il
suo accumulo causa neurodegenerazione. A sua volta lo studio del metabolismo del glicogeno è di
grande interesse perché l’inibizione della sua sintesi potrebbe avere un importante impiego
terapeutico.
Bibliografia:
-Jordi Duran, Joan J. Guinovart. Brain glycogen in health and disease Molecular Aspects of Medicine 46 (2015)
http://dx.doi.org/10.1016/j.mam.2015.08.007
-Jordi Duran, Isabel Saez, Agne`s Gruart, Joan J Guinovart and Jose´ M Delgado-Garcia. Impairment in long-term
memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain Journal of
Cerebral Blood Flow & Metabolism (2013) doi:10.1038/jcbfm.2012.200
-Cissé S, Perry G, Lacoste-Royal G, Cabana T, Gauvreau D. Immunochemical identification of ubiquitin and heatshock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains. Acta Neuropathol. 1993;
85:233–240. [PubMed: 7681614]
-Rohn T. T. (2015). Corpora amylacea in neurodegenerative diseases: cause or effect? Int. J. Neurol. Neurother. 2:031.
-Sinadinos, C., Valles-Ortega, J., Boulan, L., Solsona, E., Tevy, M.F., Marquez, M., et al., 2014. Neuronal glycogen
synthesis contributes to physiological aging. Aging Cell 13 (5), 935–945.
-Singhrao SK, Neal JW, Piddlesden SJ, Newman GR. New immunocytochemical evidence for a
neuronal/oligodendroglial origin for corpora amylacea. Neuropathol Appl Neurobiol. 1994; 20:66–73. [PubMed:
8208342]
-Wilhelmus MM, Verhaar R, Bol JG, van Dam AM, Hoozemans JJ, et al. Novel role of transglutaminase 1 in corpora
amylacea formation? Neurobiol Aging. 2011; 32:845–856. [PubMed: 19464759]
Immagini:
-Immagine 1: http://www.webalice.it/r.taddei/neoglucogenesi.gif
-Immagine 2: Jordi Duran, Joan J. Guinovart. Brain glycogen in health and disease Molecular Aspects of Medicine 46
(2015) http://dx.doi.org/10.1016/j.mam.2015.08.007
-Immagine 3: Lafora's disease. The role of skin biopsy. Newton GA1, Sanchez RL, Swedo J, Smith EB.1987
1
-Immagine 4: Glycogen hyperphosphorylation underlies lafora body formation. Turnbull J , Wang P, Girard
JM, Ruggieri A, Wang TJ, Draginov AG, Kameka AP, Pencea N, Zhao X, Ackerley CA, Minassian BA. 2010
-Immagine 5: Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a
mouse model of Lafora disease. Valles-Ortega J1, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin
A, Cañas X, Soriano E, Delgado-García JM, Gruart A, Guinovart JJ. 2011
-Immagine 7: http://www.pathologicalbodies.com/bodies-a-b.html
-Immagine 8: Jordi Duran, Joan J. Guinovart. Brain glycogen in health and disease Molecular Aspects of Medicine 46
(2015) http://dx.doi.org/10.1016/j.mam.2015.08.007
-Immagine 9: Rohn T. T. (2015). Corpora amylacea in neurodegenerative diseases: cause or effect? Int. J. Neurol.
Neurother. 2:031.