CHIMICA FARMACEUTICA 2
12 Ottobre
Per raggiungere il SNC i farmaci nicotinici devono superare la barriera ematoencefalica.
Attraverso studi di attività si è osservato che la porzione di ACh che interagisce con il sito attivo dei
recettori è la testa ammonica quaternaria, ma questa struttura difficilmente oltrepassa la BEE perciò si
preferisce utilizzare analoghi sottoforma di ammine terziarie o secondarie: la molecola supera la BEE come
neutra e, per equilibrio acido/base, una piccola percentuale si protona riuscendo ad interagire con nAChR.
Per quanto riguarda i recettori muscarinici (mAChR), invece, sono attivati quasi esclusivamente dalla testa
ammonica quaternaria e solo in alcuni casi da ammine terziarie.
Fino a pochi anni fa non si conosceva la correlazione tra i recettori nicotinici neuronali (Nn) ed il dolore, era
però noto che alcune secrezioni cutanee di rane tropicali (Epipedobates) avevano proprietà analgesiche: tra
le secrezioni sono stati infatti ritrovati sia peptidi con azione endorfinica (recettori morfinici), sia sostanze
con attività nicotinica.
In particolare è stata ricavata l’Epibatidina che si è rivelato essere un agonista Nn α4β2.
La sostanza ho molti effetti collaterali soprattutto a livello cardiaco perciò non è
utilizzabile come tale, ma ha dato il via alle ricerche su questa struttura.
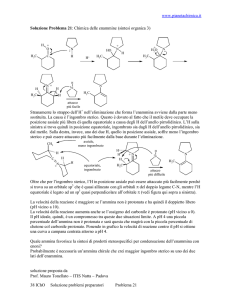
Dal Maggiociondolo (pianta), invece, è stata estratta la
Citisina avente un sistema 2-Pirrolidonico; è anch’esso un
agonista Nn α4β2 e ne hanno derivato un farmaco
attualmente utilizzato per la disassuefazione da Nicotina:
Vareniclina (CHANTIX).
STUDI DI SAR
Partendo dalla struttura base della nor-Nicotina, tramite approfonditi studi di SAR, sono stati
identificati dei derivati attivi:
1) Il legame singolo tra la Piridina e la Pirrolidina permette la rotazione della molecola che quindi può
assumere sia una conformazione coplanare, sia una conformazione non-coplanare.
Sono stati generati dei derivati con struttura ciclica “incatenata” in modo da
valutare l’attività:
- Forzando la coplanarietà sono stati ottenuti derivati inattivi.
N.B.: I derivati potrebbero essere inattivi non a causa della coplanarietà, ma a
causa dell’ingombro sterico provocato dal ponte etilenico.
Bloccando la coplanarietà verso l’alto si ottengono addirittura potenti
antagonisti α4β2.
- Forzando la struttura non-coplanare (ponte etilenico tra C4 e C5’) sono stati ottenuti derivati
con attività maggiore: questo dimostra che la struttura privilegiata è non-coplanare.
2) Si è poi osservato che la struttura ottenuta è molto simile ad una tossina animale (Anatossina) che
si distingue per la presenza di un carbonile α,β instaturo al posto della Piridina.
L’elevata attività dell’Anatossina può essere spiegata col fatto che il carbonile α,β instaturo (che
sostituisce la Piridina della nicotina) ha la funzione di instaurare un legame H con una porzione del
recettore. La Ferroginina è un altro derivato molto simile ma che ha un ponte metilenico anziché
etilica.
3) Con approcci di semplificazione della molecola è stato ottenuto l’Arecolone che ha dimostrato
avere un’attività paragonabile alla Nicotina per i recettori centrali neuronali (Nn).
Utilizzando lo stesso approccio di semplificazione a partire dall’Epibatidina si ottiene un derivato
nicotinico inattivo perché è stata alterata la distanza tra i due gruppi funzionali. Date queste
osservazioni è stata fatta una prima descrizione della struttura del farmacoforo nicotinico.
Sono poi stati eseguiti molti altri studi di SAR da cui derivano strutture con anello amminico a 4 termini
particolarmente attive che sono tuttora in fase preclinica o clinica (solo la Vareniclina è già arrivata sul
mercato).
FARMACOFORO NICOTINICO
Dagli studi di SAR è emersa la struttura attiva principale del farmacoforo nicotinico:
a è un sito anionico del recettore che da
interazione ionica con N protonato
b (recettore) è un donatore di legame H che
interagisce con un atomo elettronricco
c è una porzione del ligando (sistema π di
carbonile o eterociclo) capace di dare interazione
con un sistema elettronico π o un residuo
cationico localizzato nel binding-site recettoriale
Per quanto riguarda la differenza di interazione specifica coi sottotipi recettoriali, essa dipende da
differenze di carattere sterico del ligando.
Per ottenere nuove molecole attive sul recettore nAChR (nicotinico) si può operare con due approcci:
-
Screening sistematico di librerie virtuali
Studi di SAR su molecole attive note (ACh)
Partendo dalla struttura di ACh sono stati infatti eseguiti studi di SAR:
A: Gruppo Estereo
B: Ponte etilenico
C: Testa ammonica quaternaria
A – Gruppo Estereo:
Può essere sostituito da un etere etilico (-OCH2CH3) senza perdita significativa di attività
B – Ponte Etilenico:
Sostituendo gli H dei gruppi metilenici si osservano delle variazioni di attività:
-
CH3 in α: Attività nicotinica dimezzata; attività muscarinica 180 volte minore
CH3 in β (Metacolina): Attività nicotinica 180 volte minore; attività muscarinica dimezzata
Significa che introducendo CH3 in α si ottengono composti ad attività quasi unicamente
nicotinica, introducendolo in β, invece, si ottengono composti ad attività muscarinica.
Inoltre l’introduzione del gruppo metilico crea ingombro sterico riducendo la velocità di idrolisi.
I parametri si misurano tramite EPRM (EquiPotent Molar Ratio), ovvero la quantità del farmaco necessaria
ad avere lo stesso effetto (maggiore è EPRM, minore è la potenza).
C – Testa ammonica quaternaria:
1) Modifica di N con altri eteroatomi (P, S, As): Perdita di attività
2) Sostituzione dei gruppi metilici quaternari con H: Si ha una riduzione di attività, le ammine 2° e 3°
rimangono comunque sufficientemente potenti.
N.B.: Le ammine 2° e 3° sono più resistenti all’idrolisi rispetto al gruppo ammonico quaternario!
3) Omologazione dei gruppi metilici quaternari a gruppi etilici: Perdita di attività per ingombro sterico
ANTAGONISTI NICOTINICI
A differenza degli agonisti, che sono rappresentati solo da poche molecole, gli antagonisti sono vari:
-
Neuronali (Nn): Ganglioplegici (attività principalmente simpaticolitica, molti effetti collaterali)
Le molecole di base sono il Pentametonio e l’Esametonio ( Me3N–(CH2)6–NMe3 ) che possiedono le
due teste ammoniche quaternarie poste alla distanza corretta (5,3 Å) per poter interagire con i due
siti anionici: il primo è il sito di legame fisiologico per ACh, il secondo è un sito accessorio.
-
Placca neuromuscolare (Nm): Il recettore è leggermente diverso e la distanza tra i due siti anionici
(principale ed accessorio) è di circa 10,6 Å. Sono composti curarosimili che possono distinguersi in:
1) Leptocurari: Molecola a catena lunga e lineare, si ottiene un attività massima quando i
gruppi ammonici quaternari sono separati da 10-16 unità metileniche (es.: Decametonio).
Riescono a legare il recettore bloccandolo però in conformazione attiva (paralisi spastica).
La Succinilcolina (Suxametonio) è un derivato del decametonio avente però due gruppi
esterei; per questo motivo ha un’emivita decisamente minore il che può essere utile per
poter gestire al meglio le tempistiche durante gli interventi chirurgici.
2) Pachicurari: Sono molecole molto più grosse e complesse.
Il notevole ingombro sterico riesce ad impedire il legame di ACh perciò bloccano il recettore
in forma inattiva (paralisi flaccida) ed hanno un impiego come coadiuvanti miorilassanti agli
anestetici generali.
Il capostipite è la D-Tubocurarina (derivato vegetale del Chondrodendron), attiva per e.v.
I derivati più interessanti sono Gallamina (scaffold benzenico), attiva per e.v. ad alte dosi ed
il Pancuronio (scaffold ciclopentanoperidrofenantrenico).
Le CONOTOSSINE sono peptidi complessi prodotte da alcune specie di lumache di mare australiane.
I peptidi presentano almeno 2 ponti disolfuro importanti per determinare la struttura tridimensionale.
-
-
Conantochine: Antagonisti NMDA
α-Conopeptidi: Antagonisti Nicotinici
Essi si distinguono, a seconda della distanza amminoacidica tra i ponti disolfuro, in:
α4/7: Antagonisti Nn
α3/5: Antagonisti Nm
ω-Conopeptidi: Bloccanti Ca-N (mediano il dolore); la Ziconotide (derivato) è usato nelle neuropatie
SINTESI PANCURONIO (Pachicuraro):
1)
Reazione di Transesterificazione: l’Ossigeno enolico (per tautomeria del gruppo chetonico) del derivato steroideo attacca
il carbonio estereo dell’Enolestere che si scinde liberando Acetone (infatti esso era formato da Acetone+Ac.Acetico).
La reazione di transesterificazione di per sé è una reazione all’equilibrio. Per ottenere una resa quantitativa è necessario
spostare l’equilibrio a destra utilizzando un forte eccesso di reagente enolestere (esso è utilizzato addirittura come
solvente).Con una successiva distillazione è possibile allontanare l’eccesso di enolestere e l’acetone generato.
2)
Si utilizza MCPBA (Acido m-Cloro Perbenzoico), un peracido, per ossidare i doppi legami generando epossidi. Gli
epossidi si formano sotto il piano della molecola perché l’attacco è favorito per ragioni di ingombro sterico dei
sostituenti sopra al piano.
3)
L’Azoto nucleofilo attacca il Carbonio δ+ con una SN2. L’attacco avviene sopra al piano per motivi di ingombro sterico
dei gruppi epossidici. Sul Carbonio 17 si forma quindi un glicole che rigenera la funzione chetonica. Per quanto riguarda
l’altro epossido, probabilmente l’attacco del nucleofilo può avvenire sia sul carbonio 2 (come in figura), sia sul C3.
4)
La riduzione del chetone è
stereoselettiva: H- (idruro)
attacca da sotto per
ingombro e si genera OH
sopra il piano.
5)
La Piridina (Py) è uno scavenger di AcOH (generatosi dalla reazione). Uno Scavenger di questo tipo deve essere basico
per neutralizzare l’acido, ma non deve avere carattere nucleofilo altrimenti potrebbe reagire con la sostanza.
In alternativa alla Piridina si può usare la Trietilammina (NEt3) che dato l’ingombro attorno ad N è poco nucleofilo.
6)
Siccome CH3Br è
volatile, in
laboratorio si
preferisce usare
CH3I (più costoso).