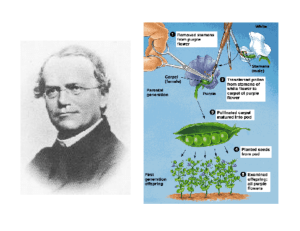
TRASMISSIONE
MENDELIANA
TRASMISSIONE MENDELIANA
OMIM
Sino ad ora sono stati catalogati più di 5000 fenotipi umani, ereditati in maniera mendeliana.
Più del 50% sono caratteri autosomici dominanti, il 36% sono autosomici recessivi e
meno del 10% sono X-linked.
Dei 5000 caratteri, circa 4000 sono
associati a malattie.
Di circa 600 di questi sono state
identificate una o più mutazioni
responsabili della malattia.
NCBI: Natl Center of
Biotechnology Information
TRASMISSIONE MENDELIANA
Le malattie mendeliane sono causate dalla mutazione di un singolo gene che ha un
notevole effetto sul fenotipo e sono ereditate in modo semplice simile o identico a
quello descritto da Mendel.
Le malattie mendeliane sono:
1. autosomiche: se i geni coinvolti si trovano su una delle 22 coppie di cromosomi
autosomi.
2. X-linked: se i geni coinvolti si trovano sul cromosoma X
Malattie dominanti: si manifestano negli eterozigoti (una copia di un allele normale)
Malattie recessive: si manifestano in individui omozigoti per l’allele mutato (doppia
dose del gene anormale).
PCR:
Polymerase Chain Reaction
PCR:
Polymerase
Chain
Reaction
Ricerca di sequenze virali, batteriche ecc.
PCR:
applicazioni
transilluminatore
Il sequenziamento
secondo Sanger
primer radioattivo
sintesi di DNA in presenza di
dideossinucleotidi che terminano
la catena
Elettroforesi
autoradiografia
Migrazione
dei
frammenti
2’,3’ dideossinucleotide
Sequenza del filamento
complementare
Mappatura e sequenziamento di genomi completi: il genoma umano
Filamento singolo di DNA
Sequenziamento
automatico del DNA
Primer fluorescenti diversi con ciascun
dideossinucleotide
ddA
ddG
ddC
ddT
Mescolamento dei prodotti
Elettroforesi
Fotomoltiplicatore
Migrazione dei frammenti
Raggio laser
Fluorescenza
Computer
Il sequenziamento automatico del DNA
Ibridazione di acidi nucleici
Miscela di molecole a
doppio filamento di DNA
cellulare totale
Denaturazione a
95°C
Si usa per rivelare
sequenze specifiche di
acidi nucleici
accoppiandoli alle basi di
filamenti complementari
di DNA o RNA.
Separazione dei filamenti
Aggiunta di una sonda
radioattiva di DNA
complementare ad una
sequenza specifica di
DNA cellulare
Ibridazione della sonda
radioattiva con sequenze
complementari presenti nel
DNA cellulari
Rinaturazione a 65°C
La marcatura radioattiva delle sonde
La sonda è uno specifico frammento di DNA, a sequenza nota,
che si utilizza per individuare un frammento di DNA o gene di
interesse in un pool di DNA totale.
La sonda per essere utile allo scopo deve essere marcata ad
esempio con il fosforo radioattivo P32.
Le sonde possono essere:
corte (20-30 oligonucleotidi)
oppure
lunghe (es. 2-3 kb)
La marcature delle due sonde avviene metodi diversi
La marcatura radioattiva delle sonde: sonde corte
22 oligo
5’ - ACTTAGTAAGGCTTGGAACCTC - 3’
+
g32ATP = A aP bP g32P
+
enzima: polinucleotide chinasi
=
5’ - g32PACTTAGTAAGGCTTGGAACCTC - 3’ La sonda è marcata
La marcatura radioattiva delle sonde: sonde lunghe
La nick translation
Ibridazione di acidi nucleici
Il DNA è digerito
con una endonucleasi
di restrizione
Migrazione
Utilizzando una sonda
marcata con radioattivo è
possibile individuare DNA o
geni di interesse in un pool di
DNA totale attraverso
l’ibridazione.
I frammenti di restrizione
di dimensione diverse
sono separati mediante
elettroforesi su gel.
Carta assorbente
Il DNA viene denaturato e trasferito su di
Filtro
un filtro mediante passaggio di una
Gel
Spugna
soluzione attraverso il gel
Soluzione salina
Frammenti
di DNA
Filtro
Il filtro viene ibridato
ad una sonda
radioattiva, che si
lega a sequenze
complementari di DNA
La sonda legata
al filtro è rivelata
in autoradiografia
Lastra autoradiografica
Identifica delezioni nella sequenza
1 2 C
EcoRI
EcoRI
C
EcoRI
EcoRI
EcoRI
EcoRI
1
2
Gli enzimi di restrizione
Tabella. Alcuni enzimi di restrizione e i loro siti di taglio sul DNA
Enzima
Batterio
Sito di taglio
___________________________________________________________________________________________
AluI
Arthrobacter luteus
5’-A-G-C-T-3’
3’-T-C-G-A-5’
BamHI
Bacillus amyloliquefaciens H
5’-G-G-A-T-C-C-3’
3’-C-C-T-A-G-G-5’
EcoRI
Escherichia coli RY13
5’-G-A-A-C-C-T-3’
3’-C-T-T-G-G-A-5’
EcoRII
Escherichia coli RY13
5’-C-C-T-G-G-3’
3’-G-G-A-C-C-5’
HaeIII
Aemophilus aegyptius
5’-G-G-C-C-3’
3’-C-C-G-G-5’
HindIII
Aemophilus influenzae b
5’-A-A-G-C-T-T-3’
3’-T-T-C-G-A-A-5’
PstI
Providencia stuartii
5’-C-T-G-C-A-G-3’
3’-G-A-C-G-T-C-5’
5’-G-T-C-G-A-C-3’
3’-C-A-G-C-T-G-5’
___________________________________________________________________________________________
SalI
Streptomyces albus
Tecnologia DNA Ricombinante
Enzimi di restrizione
TRASMISSIONE MENDELIANA
ALBERO GENEALOGICO
Coniugazionale
Generazionale
Fratria
TRASMISSIONE MENDELIANA
AUTOSOMICA DOMINANTE
MATRIMONIO PROTOTIPO
Trasmissione verticale
I
aa
Aa
1
2
II
III
1
2
3
4
5
6
7
8
9
10
1
2
3
4
5
6
7
8
9
10
11
11
1. OGNI INDIVIDUO AFFETTO HA UN GENITORE AFFETTO
12
12
13
13
♂
♀
14
15
16
14
15
16
A
a
2. GLI INDIVIDUI AFFETTI ETEROZIGOTI SE SI UNISCONO IN
MATRIMONIO CON INDIVIDUI SANI - OMOZIGOTI RECESSIVI (condizione più frequente) HANNO IL 50% DI PROBABILITA’ DI
TRASMETTERE LA MALATTIA AL FIGLIO
a
Aa aa
a
Aa aa
3. MASCHI E FEMMINE EREDITANO CON LA STESSA FREQUENZA LA
MALATTIA CONGENITA
50% Aa affetto
50% aa sani
TRASMISSIONE MENDELIANA
AUTOSOMICA DOMINANTE
MATRIMONIO MENO FREQUENTE MA POSSIBILE
Trasmissione verticale
Aa
Aa
1
2
I
II
1
2
3
4
5
6
7
8
9
10
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
14
15
16
III
11
12
13
♂
4. DUE INDIVIDUI ETEROZIGOTI (meno frequente) CHE HANNO
LA MALATTIA HANNO IL 75% DI PROBABILITA’ DI TRASMETTERE
LA MALATTIA AL FIGLIO
75%
♀
A
a
A
AA Aa
a
Aa aa
25% AA affetti gravi
50% Aa affetti
25% aa sani
TRASMISSIONE MENDELIANA
AUTOSOMICA DOMINANTE
MATRIMONIO RARO
Trasmissione verticale
I
AA
Aa
1
2
II
III
1
2
3
4
5
6
7
8
9
10
1
2
3
4
5
6
7
8
9
10
11
11
12
13
12
14
15
16
13
14
15
16
♀
A
♂
5. DUE INDIVIDUI, UNO OMOZIGOTE DOMINANTE (raro) E L’ALTRO
ETEROZIGOTE, ENTRAMBI EFFETTI HANNO IL 100% DI PROBABILITA’
DI TRASMETTERE LA MALATTIA AI FIGLI
100%
a
A
AA Aa
A
AA Aa
50% AA affetti
50% Aa affetti
TRASMISSIONE MENDELIANA
AUTOSOMICA DOMINANTE
Esempi di caratteri a trasmissione autosomica dominante
• Fossetta sul mento
• Mascella asburgica o prognatismo
• Capacità di arrotolare la lingua
• Capelli lanosi
TRASMISSIONE MENDELIANA
Capelli lanosi
Autosomico
dominante
TRASMISSIONE MENDELIANA
MALATTIE AUTOSOMICHE DOMINANTI
IPERCOLESTEROLEMIA FAMILIARE
Trasmissione: autosomica dominante
Frequenza: 1 malato: 500 nati sani
Patogenesi: mancata eliminazione delle LDL a livello epatico che determina un aumento dei livelli di LDL (e
quindi colesterolo) in circolo e conseguenti gravi manifestazioni cliniche
Causa: gli individui malati sono portatori di mutazioni sul gene che codifica per il recettore delle LDL
(cromosoma 19). Il difetto genico si fa risentire soprattutto a livello degli epatociti, preposti
all’endocitosi e degradazione delle LDL circolanti.
Aa
AA
N:220 mg/dl
P:300-600 mg/dl
P:600-1200 mg/dl
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
NH2
HOOC
Gene: 45 Kb
839 aa
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
apolipoproteina
core
(esteri del
colesterolo)
LDL
La complessazione con l'LDL determina il "clustering" dei recettori che
prendono contatto con la clatrina. Quest'interazione è necessaria alla
formazione della fossetta rivestita e quindi all'internalizzazione delle LDL
fossetta rivestita
recettore per le LDL,
riconosce la
apolipoproteina
presente su LDL
clatrina
vescicola
rivestita
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
recettore per LDL ri-esposto
Vescicola
rivestita
immagazzinamento
(sotto forma di esteri del
colesterolo)
vescicola
di ricircolo
endosoma
sintesi di
componenti
della
membrana
cellulare,
ormoni
steroidei,
acidi biliari
ACAT
Aminoacidi
(metabolismo)
lisosoma
Colesterolo libero
Acil-coenzima A-colesterolo aciltransferasi: acil-CoA + colesterolo ⇄ CoA + colesterolo estere
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
esterificazione
(immagazzinamento)
del colesterolo
enzima
ACAT
apparato del
Golgi
+
enzima HMG-Coa
reduttasi
sintesi endogena
del colesterolo
disponibilità
eccessiva di
colesterolo
ribosomi
RER
-
RNA
sintesi del recettore per le LDL
DNA
La idrossimetilglutaril-CoA reduttasi (o HMG-CoA reduttasi) è un enzima appartenente alla classe delle ossidoreduttasi che risiede nel reticolo endoplasmatico
liscio degli epatociti (cellule del fegato)
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
5'
Sequenze
promotore
1
trascrizione
Dominio di
legame per
LDL (292 aa)
NH2
2
Dominio di
omologia per
l'EGF (400 aa)
3
4
5
6
7
8
9
10
11
12
13
14
15
splicing
Dominio di
glicosilazione
(58 aa)
traduzione
Modificazioni
posttraduzionali
16
17
HOOC
18
3'
Dominio transmembrana (58
aa)
Dominio
citoplasmatico
(50 aa)
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
Sono state finora evidenziate più di 400 mutazioni diverse sul gene che
codifica per il recettore delle LDL che a livello fenotipico danno origine ad
effetti raggruppabili in 5 classi
inserzione
Cromosoma 19
delezione
G>A
nonsenso
missenso
+ 2 kb
+ 18 nt
+ 4 nt
D splicing
1
2
3
+ 7 kb
(duplicazione)
4 5 6
7 8 9 10 11 12 1314 15
-1 kb -4 kb
-10 nt
-5 kb
16 17
-5 kb
-7 kb
-20 kb
-25 kb
18
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
I soggetti affetti da FH in condizioni di omozigosi, rappresentano un modello sperimentale prezioso
per lo studio della funzionalità del gene, in quanto permettono di analizzare le alterazioni a carico
del recettore LDLR, causate dall’allele mutante, in assenza completa degli effetti di fondo prodotti
dall’allele normale. Questi studi sono effettuati mediante un modello in vitro nel quale si valuta
l’interazione tra fibroblasti in coltura (ottenuti da biopsie cutanee di soggetti omozigoti per
mutazioni perfettamente caratterizzate dal punto di vista molecolare) e LDL marcate. Si è così
stabilito che esistono 5 diverse classi di fenotipi difettivi, secondo la fase del meccanismo di
funzionamento del recettore LDLR che è compromessa.
Precisamente:
– Classe 1: fenotipo “allele nullo”
– Classe 2: fenotipo “trasporto difettivo”
– Classe 3: fenotipo “legame difettivo”
– Classe 4: fenotipo “internalizzazione difettiva”
– Classe 5: fenotipo “riciclo difettivo”
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
CLASSE 1: FENOTIPO «ALLELE NULLO»
Legame ligando EGF omology O-gli trans citoplasm
Mancata sintesi
(recettore negativo)
1
2
3
4 5 6
7 8 9 10 11 12 1314 15
16 17
18
DELEZIONE
-20 kb
No mRNA
No recettore
•
•
Mancanza del promotore che impedisce la trascrizione
dell'allele mutato (allele nullo)
Deficit quantitativo del recettore LDL sulla superficie delle
cellule
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
CLASSE 1: FENOTIPO «ALLELE NULLO»
Mancata sintesi
(recettore negativo)
Legame ligando EGF omology O-gli trans citoplasm
1
2
3
4 5 6
7 8 9 10 11 12 1314 15
16 17
18
NONSENSO
Sì mRNA
No recettore
•
•
mRNA contiene codoni di stop (allele nullo)
Deficit quantitativo del recettore LDL sulla superficie delle
cellule
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
O-gli
CLASSE 2: FENOTIPO «TRASPORTO DIFETTIVO»
b.
mancata
N- o O-glicosilazione
1
2
3
4 5 6
7 8 9 10 1112 1314
15
18
missenso
MSSENSO
Mutazione missenso nell’esone 15 che
comporta la mancata modificazione posttraduzionale nel Golgi. La mancata
glicosilazione O-linked impedisce ai recettori
LDL di raggiungere la membrana
citoplasmatica.
Oppure mutazioni che non consentono la
glicosilazione N-linked nel RER (no
trasporto nel Golgi)
16 17
Alterazione
delle
modificazioni
post-traduzionali
HOOC
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
CLASSE 3: FENOTIPO «LEGAME DIFETTIVO»
a. anomala interazione
recettore-ligando
1
2
3 456
DUPLICAZIONE
Duplicazione degli esoni 3-6 che determina
la duplicazione del dominio di legame del
recettore LDL. Nel RER la proteina è
altamente instabile ed è frequentemente
degradata o inserita sulle membrane del
RER con molta difficoltà. Il recettore è
trasportato alla membrana cellulare ma ha
poca affinità con LDL (30% del wild type)
7 8 9 10 1112 1314 15
16 17
18
8 kb
NH2
Duplicazione
del dominio
di legame
HOOC
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
missenso
CLASSE 3: FENOTIPO «LEGAME DIFETTIVO»
b. anomala interazione
recettore-ligando
DELEZIONI
MISSENSO
Delezioni negli esoni 3, 4 e 7,8
che eliminano rispettivamente i
repeats 2, 3, 4, 5, e A e B nel
recettore LDL. La mancanza di
questi repeats riduce l’affinità
del recettore con le LDL.
Mutazioni missenso negli stessi
esoni che eliminano dei reapet.
1
2
3 4
5 6
7 8 9 10 1112 1314 15
16 17
delezioni
7repeat
3
r
e
p
e
a
t
18
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
Trans Cit
CLASSE 4: FENOTIPO «INTERNALIZZAZIONE DIFETTIVA»
a. Deficit recettoriale
MISSENSO
1
2
3
4 5 6
7 8 9 10 1112 131415
16
17
18
Mutazione missenso (Tyr790 in Cys) nella
sequenza del dominio citoplasmatico
Il recettore ha una lunghezza corretta ma perde la
struttura 3D e non può interagire con la clatrina.
No internalizzazione
Tyr
Cys
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
Trans Cit
CLASSE 4: FENOTIPO «INTERNALIZZAZIONE DIFETTIVA»
b. Assenza recettoriale
NONSENSO
1
2
3
4 56
7 8 9 10 1112 1314 15
16
17
18
• Mutazione nonsenso nella sequenza del
dominio intracellulare
Stop
• Il recettore non reagisce correttamente
con la clatrina e non consente il clustering
dei recettori. No internalizzazione
RECETTORE LDL
IPERCOLESTEROLEMIA FAMILIARE
CLASSE 5: FENOTIPO «RICICLO DIFETTIVO»
EGF omology
missenso
Deficit recettoriale
DELEZIONI
MISSENSO
1
Mutazioni missenso o delezioni
nei repeats della sequenza del
dominio di omologia per l’EGF
Il recettore dopo internalizzazione
non ritorna sulla membrana.
No riciclaggio
2
3
4 5 6
7 8 9 10 1112 1314 15
16 17
delezioni
7repeat
3
r
e
p
e
a
t
18
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
1. Allele
nullo
3. Assenza di
legame
5. Assenza di
2. Trasporto
riutilizzazione
assente
4. Assenza di
internalizzazione
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
D200g (FH Padova-1): la più frequente in
Italia.
Mutazione missenso esone 4: A:G =
D200g= acido aspartico in glicina
FH Ferrara-1: Ex 4 c.557 del G
IPERCOLESTEROLEMIA FAMILIARE
RECETTORE LDL
- Classe 1: fenotipo “allele nullo”
– Classe 2: fenotipo “trasporto difettivo”
– Classe 3: fenotipo “legame difettivo”
– Classe 4: fenotipo “internalizzazione difettiva”
– Classe 5: fenotipo “riciclo difettivo”
IPERCOLESTEROLEMIA FAMILIARE
•
•
•
Il deposito di colesterolo a livello della
valvola aortica può provocare una
stenosi aortica sintomatica con
conseguente infarto del miocardio.
I sintomi si sviluppano nella seconda
(omozigoti) o terza/quarta decade di vita
(etrozigoti).
All'età di sessant'anni più dell' 85% degli
eterozigoti ha avuto un infarto
miocardico.
IPERCOLESTEROLEMIA FAMILIARE
DIAGNOSI
SANGUE: 1. analisi della concentrazione del colesterolo;
2. elettroforesi delle LDL
MOLECOLARE: è importante conoscere il tipo di mutazione nell’ambito delle etnie (effetto del
fondatore). PCR o IBRIDAZIONE IN SOUTHERN BLOT (previa digestione
enzimatica del DNA con enzimi di restrizione)
DIAGNOSI NEONATALE
SANGUE DAL CORDONE OMBELICALE: 1. analisi della concentrazione del colesterolo (risulta
doppio o triplo negli eterozigoti e 6-8 volte > negli
omozigoti);
2. elettroforesi delle LDL
MOLECOLARE: è importante conoscere il tipo di mutazione nell’ambito delle etnie (effetto del
fondatore). PCR o IBRIDAZIONE IN SOUTHERN BLOT (previa digestione
enzimatica del DNA con enzimi di restrizione)
DIAGNOSI PRENATALE
CELLULE DEL LIQUIDO AMNIOTICO E CARATTERIZZAZIONE MOLECOLARE
COREA DI HUNTINGTON
còrea (greco) = danza
Descritta da George Huntington nel 1872
Trasmissione: autosomica dominante
Frequenza: 1 malato: 10.000 nati sani
Patogenesi: malattia neurodegenerativa; manifestazioni cliniche a livello neurologico e motorio.
Causa: gli individui malati sono portatori di mutazioni sul gene che codifica per la proteina huntingtina
(cromosoma 4). Il difetto genico si fa risentire a livello dei neuroni.
Gene HTT o IT-15. Scoperto nel 1993; codifica per l’huntingtina (3144aa)
CAG repeat
glutammine
La proteina è necessaria per la sopravvivenza dei neuroni.
COREA DI HUNTINGTON
gene normale
huntingtina normale
10-35 CAG repeat
10-35 glutammine
> 35 CAG repeat
poliglutammina
gene mutato
huntingtina mutata
neurone sano
neurone malato
COREA DI HUNTINGTON
HUNTINGTINA
COREA DI HUNTINGTON
HUNTINGTINA
COREA DI HUNTINGTON
HUNTINGTINA
BDNF
BDNF: brain-derived neurotrophic factor
COREA DI HUNTINGTON
HUNTINGTINA
COREA DI HUNTINGTON
ANTICIPAZIONE
COREA DI HUNTINGTON
DIAGNOSTICA MOLECOLARE
• PCR
• SEQUENZIAMENTO AUTOMATICO
OSTEOGENESI IMPERFETTA
Trasmissione: autosomica dominante
Frequenza: 1 malato: 15.000 nati sani
Patogenesi: malattia apparato scheletrico (fragilità ossea).
Causa: gli individui malati sono portatori di mutazioni sul gene che codifica per il collagene, tipo 1, alfa 1
(cromosoma 17). Il difetto genico si fa risentire a livello degli osteoblasti.
Two RNA of 5,8 kb and 4,8 kb differing by their Ô3 terminus non coding sequence and giving rise to a single 140 kDa protein. The COL1A1
gene is 18 kb in size and is composed of 52 exons. Exons 6 to 49 encode the alpha helical domain.
1464 aa
Type I collagen is the most abundant protein in vertebrates and a
constituent of the extra cellular matrix in connective tissue of bone, skin,
tendon, ligament and dentine. It is mostly produced and secreted by
fibroblasts and osteoblasts. Member of the collagen family.
OSTEOGENESI IMPERFETTA
OSTEOGENESI IMPERFETTA
Collagene: Proteina a struttura quaternaria
Col1alfa1 sul cromosoma 17
Col1alfa2 sul cromosoma 7
OSTEOGENESI IMPERFETTA
Sintesi del collagene
OSTEOGENESI IMPERFETTA
2 fenotipi patologici
OI tipo 1: allele null. Le mutazioni sono
diverse: delezioni e inserzioni in più esoni,
mutazioni nonsenso e di splicing; viene
prodotto il 50% in meno di collagene.
Compatibile con la vita. Gli individui affetti
hanno fragilità ossea e sono suscettibili a
fratture multiple delle ossa lunga delle
gambe, dello sterno e delle ossa piccole
delle mani e piedi. In 50% dei casi perdita
di udito a partire dalla seconda decade di
vita. Sclera blu.
OI tipo 2: allele mutato. E’ una malattia
molto grave che porta alla morte nelle
prime settimane o mesi di vita (ancora
prima della nascita numerose fratture
delle ossa in tutto il corpo). Il difetto
dipende da mutazioni puntiformi de novo
(sostituzioni di glicine) o riarrangiamenti
negli alleli alfa1 o alfa2. Le mutazioni in
COOH più gravi che in NH2. Le fibre non si
assemblano correttamente e vengono
degradate, oppure se si assemblano non
funzionano correttamente
Allele null: delezioni,
inserzioni, mutazioni
splicing, nonsenso
Allele mutato: mutazioni puntiformi, riarrangiamenti
OSTEOGENESI IMPERFETTA
OI1
OI2
TRASMISSIONE MENDELIANA
MALATTIE AUTOSOMICHE RECESSIVE
1. Le malattie autosomiche recessive sono caratterizzate da manifestazioni cliniche soltanto
negli individui omozigoti per il gene mutante.
2. Il pedigree tipico è orizzontale in quanto gli individui affetti tendono a rimanere limitati ad
una singola fratria e la malattia non si presenta nelle generazioni successive.
3. I maschi e le femmine sono affetti con uguale probabilità.
GENITORI
MEIOSI
GAMETI
FECONDAZIONE
Portatori
eterozigoti
obbligati
PROLE
ETEROZIGOTE
ETEROZIGOTI AFFETTI
GAMETI
PATERNI
GAMETI
PATERNI
GAMETI
MATERNI
GAMETI
MATERNI
A.
Ereditarietà autosomica recessiva altre possibili combinazioni
NORMALE
AFFETTO
B. ETEROZIGOTE AFFETTO
Tutti
A = NORMALE, a = MUTANTE
A = NORMALE, a = MUTANTE
Ereditarietà autosomica recessiva
ETEROZIGOTI
GENITORI
MEIOSI
GAMETI
FECONDAZIONE
PROLE
NORMALE
ETEROZIGOTI
AFFETTA
GAMETI
PATERNI
GAMETI
MATERNI
Fibrosi cistica,
Anemia falciforme, Talassemie,
A = NORMALE, a = MUTANTE
TRASMISSIONE MENDELIANA
MALATTIE AUTOSOMICHE RECESSIVE
La fibrosi cistica è un carattere recessivo 1/2.000
La fibrosi cistica (CF, Cystic Fibrosis) (MIM/OMIM 219700) è una
patologia genetica disabilitante e letale che si eredita come carattere
autosomico recessivo; colpisce le ghiandole esocrine (mucose –muco- e
sierose-enzimi della digestione- e del sudore). Se le ghiandole del
sudore non funzionano bene rilasciano una quantità eccessiva di sale.
La malattia viene spesso diagnosticata analizzando la quantità di sale
nel sudore. In base ad alcune leggende popolari, le nutrici un tempo
leccavano la fronte dei neonati: se il sudore aveva un sapore troppo
salato, predicevano che il bambino sarebbe morto prematuramente di
congestione polmonare. La malattia ha effetti ad ampio spettro perché
ghiandola
le ghiandole colpite svolgono alcune importanti funzioni vitali. La CF
comporta la produzione di un muco spesso, che ostruisce i dotti che
sudoripara
portano gli enzimi della digestione dal pancreas all’intestino tenue,
riducendo gli effetti della digestione. Come conseguenza, i bambini
colpiti spesso soffrono di malnutrizione, nonostante un aumento di
ovaio
appetito e di quantità di cibo consumato. Col progredire della malattia,
si formano delle cisti nel pancreas e la ghiandola degenera in una
struttura fibrosa, che dà il nome alla malattia. Dal momento che il muco utero (il muco
blocca l’ingresso
spesso provocato dalla CF blocca anche le vie aeree dei polmoni, la
maggior parte dei pazienti colpiti da fibrosi cistica sviluppa patologie
dell’utero) cervice
ostruttive dei polmoni e infezioni, che portano a morte prematura.
Quasi tutti i casi di CF si registrano in figli di genitori fenotipicamente
normali, eterozigoti.
Ha una frequenza molto alta: 1/2000
Diversa incidenza tra le popolazioni: 1/25 portatori sani tra gli americani; 1/46
ispanici; 1/65 afroamericani; 1/150 asiatici. Molto grave, molto debilitante,
invalidante e porta alla morte.
parotide
polmone
fegato
pancreas
cistifellea
intestino
deferente
testicolo
(il muco
blocca i
dotti che
portano lo
sperma, e
solo circa il
2-3% dei
maschi
colpiti è
fertile).
FIBROSI CISTICA
Il prodotto del gene CF è una proteina che si inserisce
nella membrana plasmatica di alcune cellule ghiandolari.
La proteina è chiamata regolatore della conduttanza
transmembrana della fibrosi cistica o CFTR. Il CFTR
regola il flusso di ioni cloro attraverso la membrana
plasmatica della cellula.
Gene CFTR:
250.000 pb
1480 aa
FIBROSI CISTICA
Negli individui affetti da CF la proteina CFTR è assente o difettosa. Dal momento che i fluidi si muovono attraverso le membrane del plasma in
risposta a movimenti degli ioni, una proteina CFTR difettosa comporta una diminuzione dei fluidi che devono aggiungersi alla secrezione delle
ghiandole. Le secrezioni ispessite provocano i sintomi caratteristici della CF.
Esone 10
Mutazione delta F508: delezione
70% dei casi
Una delezione CTT nell’esone 10 causa la
perdita di una fenilalanina nella posizione 508
della proteina. La isoleucina nella posizione 507
rimane inalterata perché ATC e ATT entrambi
codificano per una isoleucina.
MALATTIE AUTOSOMICHE RECESSIVE
MALATTIE AUTOSOMICHE RECESSIVE
MALATTIE AUTOSOMICHE RECESSIVE
Terza, quarta settimana: sacco vitellino e fegato rispettivamente
Inizio secondo trimestre: milza
Fine secondo trimestre: ossa
(beta)
alfa
(beta)
(alfa)
Adulta: alfa/beta 97/98% - HbA1
alfa/delta 2/3% - HbA2
alfa/gamma <1% - HbF
MALATTIE AUTOSOMICHE RECESSIVE
MALATTIE AUTOSOMICHE RECESSIVE
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME: 1: 500 (afroamericani)
L'anemia falciforme è una malattia ereditaria, che si
esprime in forma conclamata solo negli omozigoti, vale a
dire nei figli che ricevono il gene anomalo da entrambi i
genitori. Negli omozigoti la Hb-S é la sola forma di
emoglobina presente negli eritrociti. Gli omozigoti non
vivono generalmente oltre l'età di 25 anni.
Sickle=falce
HbS
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
Effetto pleiotropico
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
ESONE 1, CODONE 6
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
HbA
MstII
MstII
364 pb
201 pb
90 pb
74 pb
HbS
291 pb
74 pb
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
CTC transizione TTC: mRNA=AAG=lisina
Malattia da emoglobina C: anemia lieve, l’emoglobina precipita solo quando è in alte concentrazioni
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
Gli eterozigoti, cioè i figli che
ricevono il gene anomalo da uno
solo dei genitori, posseggono il 50%
di Hb-S ed il 50% di Hb-A. In essi
l'anemia falciforme è asintomatica,
salvo crisi sporadiche che possono
verificarsi in condizioni di bassa
tensione di O2 (montagna,
sott’acqua, anestesia): maschio
falciforme (sickle cell trait).
Dominanza incompleta
MALATTIE AUTOSOMICHE RECESSIVE
ANEMIA FALCIFORME
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA
Comuni in tutto il mondo; ma soprattutto mediterraneo e sud est asiatico (20-30% affetti)
Talassa=mare Mediterraneo)
Malattie monofattoriali: ereditarietà autosomica recessiva. Talassemie
Sono un gruppo si malattie ereditarie determinate da anomalie nell’emoglobina, nelle quali il fenotipo deriva da
un’alterazione del rapporto reciproco tra globine alfa e beta. La sintesi di una delle globine può essere ridotta o anche
assente, causando la formazione di molecole di emoglobina aberranti con uno squilibrio tra le subunità. Questo tipo di
molecole non lega l’ossigeno efficientemente e determina effetti gravi se non addirittura fatali.
Le talassemie sono comuni in
diverse aree del mondo,
soprattutto nel Mediterraneo e
nel Sud Ovest asiatico, dove il 2030% della popolazione risulta
affetta. Il nome “talassemia”
deriva dal greco thalassa, ovvero
mare, quindi il nome della
malattia enfatizza una condizione
caratteristica di individui del
bacino del Mediterraneo.
Malattie monofattoriali: ereditarietà autosomica recessiva. Talassemie
Le talassemie sono anomalie ereditarie della produzione di emoglobina in cui il problema principale è un difetto
quantitativo della alfa-globina (alfa Talassemia) o della beta-globina (beta Talassemie). Come nel caso dell’anemia falciforme
la distribuzione della Talassemia coincide con quella della malaria.
L’alfa Talassemia è caratterizzata da un difetto relativo di sintesi delle catene alfa-globiniche, con normale produzione di
catene beta. Se alcune catene alfa possono ancora essere prodotte, si formerà una piccola quantità di tetrameri normali, ma
ci sarà un forte eccesso di catene beta. In queste circostanze beta è in grado di formare omotetrameri (beta4). Questa
emoglobina detta emoglobina H (HbH), può essere visualizzata sotto forma di corpi inclusi all’interno dei globuli rossi degli
individui affetti da alfa-Talassemia. La HbH ha una capacità marcatamente ridotta di trasportare ossigeno. La conseguenza di
questo eccesso di catene beta e del difetto di catene alfa è che i globuli rossi hanno dimensioni e numero ridotti, con
conseguente anemia.
Nella beta-Talassemia c’è un difetto delle catene beta. In queste circostanze l’alfa globina è in eccesso forma degli
omotetrameri che precipitano in aggregati moto insolubili. Gli eritrociti vengono distrutti precocemente distruzione nel
midollo osseo e milza.
Normale
tetrameri
ab
ba
ab
ba
alfa-Tal
ab
ba
ab
ba
bb
bb
bb
b
bb
b
beta-Tal
ab
ba
aa
aa
aa
aa
a
aa
a aa
globuli
rossi
Corpi inclusi di
beta4 (HbH)
Precipitazione di alfa4
(molto insolubile)
Malattie monofattoriali: ereditarietà autosomica recessiva. Talassemie
Ci sono due tipi di Talassemie: 1. Talassemia alfa, in cui la sintesi della globina alfa è
ridotta o assente;
2. Talassemia beta, che riguarda la sintesi ridotta o
assente della globina beta.
In entrambi i casi vi possono essere cause differenti e benché queste condizioni siano ereditabili
come caratteri autosomici recessivi, entrambe le forme di talassemia danno effetti fenotipici in
eterozigosi.
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA alfa
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA alfa
Malattie monofattoriali: ereditarietà autosomica recessiva. Beta-talassemie
Nelle beta Talassemie si riscontra un deficit delle catene beta.
Minor
Tal-minor
asintomatici
Sintomatica,
anemia
significativa ma
non richiede
trasfusioni
Major
Trasfusione dipendenti
Morbo di Cooley (precipitazione tetrameri)
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA beta
Mutazione comune in Sardegna e Sicilia T39 (codone 39)
CAG glutammina con TAG stop: talassemia beta 0
Solo 10% prodotta con mutazione
nel promotore
Almeno 100 mutazioni
Le mutazioni nella frazione COOH sono meno dannose
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA beta
Nel mediterraneo e in Italia la più frequente: Tal beta+
Mutazione puntiforme introne 1
MALATTIE AUTOSOMICHE RECESSIVE
TALASSEMIA beta
TRASMISSIONE X-LINKED
IL CROMOSOMA Y: 70 geni IL CROMOSOMA X: 1200 geni
I maschi sono emizogoti
IL CROMOSOMA X
LAYONIZZAZIONE
LYONIZZAZIONE
IL CROMOSOMA X
L’ipotesi di Lyon stabilisce che nelle cellule somatiche:
a) L’inattivazione del cromosoma X si verifica precocemente durante la vita embrionale
(stadio di blastocisti avanzata, nell’uomo).
b) L’inattivazione è casuale.
c) L’inattivazione dell’X è completa.
d) L’inattivazione è permanente e si propaga clonalmente.
LYONIZZAZIONE
IL CROMOSOMA X
XIST= X Inactivation Specific Transcript
RNA= 15 kb
Il fenomeno della lyonizzazione
Eccezioni all’ipotesi di Lyon
a) Sebbene l’inattivazione sia solitamente casuale, un cromosoma X
strutturalmente anomalo, ad es. delezione, viene inattivato preferenzialmente.
b) Sebbene l’inattivazione della X sia estensiva, tuttavia non è completa; sono
noti almeno 16 umani che sfuggono all’inattivazione.
Allele A= codifica l’enzima che converte eumelanina in feomelanina (rosso)
Allele a= manca enzima (nero)
Mosaicismo e chimere
I mosaici posseggono due o più linee cellulari geneticamente differenti, che derivano da un unico zigote. La mutazione
genetica indicata può essere una mutazione genica, una mutazione cromosomica numerica o strutturale o, nel caso particolare
della lyonizzazione, l’inattivazione dell’X.
Una chimera deriva da due zigoti, che solitamente sono entrambi normali, ma geneticamente differenti.
Mosaicismo della pelle
Mosaicismo dell’occhio
Ereditarietà X-linked
Le malattie X-linked sono provocate da geni mutati sul cromosoma X.
I geni mutati X-linked sono completamente espressi nei maschi che hanno un
solo cromosoma X e sono quindi emizigoti per i geni X-linked (eredità diaginica).
Una malattia prodotta da un gene mutato X-linked può essere o non essere
espressa clinicamente nelle femmine eterozigoti (fenomeno della lyonizzazione).
Le malattie che raramente hanno una espressione clinica nelle femmine
eterozigoti sono X-linked recessive, mentre le eterozigoti per malattie Xlinked dominanti solitamente sono affette in modo lieve e variabile dei maschi
Non esistono malattie legate all’Y
I geni legati all’Y codificano caratteri non essenziali o funzioni strettamente
maschili, ed è improbabile che loro difetti causino alterazioni patologiche, a parte
l’infertilità maschile. Trasmissione olandrica dei caratteri.
Ereditarietà X-linked dominante: es. Ipofosfatemia = bassi livelli di fosfato nel sangue = deformità dello sheletro: rachitismo
1. Gli individui maschi affetti generano solo figlie femmine affette e nessun figlio maschio affetto:
X
Y
X
XX
XY
X
XX
XY
1
2
2. Una femmina eterozigote affetta
trasmetterà il carattere a metà dei suoi figli e
maschi e femmine ne saranno ugualmente
affetti
X
Y
X
XX
XY
X
XX
XY
Ereditarietà X-linked dominante
1
3. Come atteso una femmina omozigote
trasmetterà il carattere a tutti i suoi figli
2
3
X
Y
X
XX
XY
X
XX
XY
Variazioni del modello di ereditarietà
La letalità dei maschi
Le condizioni legate all’X letali nei maschi
Per alcune condizioni dominanti legate all’X l’assenza dell’allele normale è
letale prime della nascita. Quindi non nascono maschi affetti e si osserva la
malattia solo nelle femmine, che la trasmettono a metà delle loro figlie
femmine.
2. Una femmina eterozigote affetta
trasmetterà il carattere a metà dei suoi figli e
maschi e femmine ne saranno ugualmente
affetti
X
Y
X
XX
XY
X
XX
XY
I maschi
affetti vengono
abortiti spontaneamente
Ereditarietà X-linked recessiva
Ereditarietà X-linked recessiva
L’ereditarietà X-linked può essere meglio compresa seguendo il cromosoma X.
L’incrocio meno comune è tra una donna
omozigote e un uomo normale
A. FEMMINA OMOZIGOTE
MASCHIO NORMALE
Un secondo incrocio tipico è tra un uomo
affetto e una donna omozigote normale.
B.
FEMMINA NORMALE
MASCHIO AFFETTO
GENITORI
MEIOSI
GAMETI
FECONDAZIONE
PROLE
FEMMINA
ETEROZIGOTE
M.
AFFETTO
FEMMINA
ETEROZIGOTE
MASCHIO
AFFETTO
FEMMINA
ETEROZIGOTE
GAMETI PATERNI
XaXA
XaXA
Le femmine
trasmettono l’X
a tutti i figli
GAMETI MATERNI
GAMETI MATERNI
GAMETI PATERNI
XA
MASCHIO
NORMALE
I maschi
trasmettono l’X
a tutte le figlie
femmine
A = NORMALE, a = MUTANTE
Ereditarietà X-linked recessiva
L’ereditarietà X-linked può essere meglio compresa seguendo il cromosoma X.
L’incrocio più comune è tra una donna
portatrice eterozigote e un uomo normale
C. FEMMINA ETEROZIGOTE
MASCHIO NORMALE
GENITORI
MEIOSI
GAMETI
FECONDAZIONE
PROLE
F
M.
NORMALE NORMALE
FEMMINA
ETEROZIGOTE
MASCHIO
AFFETTO
GAMETI MATERNI
GAMETI PATERNI
Le femmine
trasmettono l’X
al 50% dei figli.
Maschi e
femmine
Daltonismo
Cecità al rosso e al verde
Ereditarietà X-linked recessiva
Europa occidentale:
8% maschi, 0,7% femmine
Ereditarietà X-linked recessiva
Daltonismo
fotopsine
Ereditarietà X-linked recessiva
Daltonismo
Ereditarietà X-linked recessiva
Emofilia A
Ereditarietà X-linked recessiva
Emofilia A
Gene mutato: Fattore VIII della coagulazione
Manca la conversione del
fibrinogeno (solubile) in fibrina (insolubile)
Ereditarietà X-linked recessiva
Emofilia A
VIII
Ereditarietà X-linked recessiva
Emofilia A
Ereditarietà X-linked recessiva
Emofilia A
142 pb
99 pb
43 pb
Ereditarietà X-linked recessiva
Diastrofia muscolare
1/3500 maschi
Degenerazione muscolare
Esito infausto nell’arco dei primi 20 anni
Femmine eterozigoti normali (casi in
omozigosi)
Ereditarietà X-linked recessiva
Distrofina
Gene distrofina
Occupa l’1% del cromosoma X
Ereditarietà X-linked recessiva
60% delezioni
30% mutazioni puntiformi
10% duplicazioni intrageniche
Ereditarietà X-linked recessiva
Distrofina
Fibre muscolari
Normale
Becker
Duchenne
Fila 1-2: D. Becker
Fila 3: Normale
File 4-5: D. Duchenne
Western blot
Ereditarietà X-linked recessiva
Ereditarietà mitocondriale
matrilineare
matrilineare
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Riarrangiamenti del DNA mitocondriale
(delezioni)**
Sindrome di Kearns-Sayre (KSS)
Oftalmoplegia Esterna Progressiva (PEO)
Sindrome di Pearson (anemia sideroblastica e malassorbimento
connatali)
Mutazioni puntiformi*
Neuropatia ottica ereditaria di Leber (LHON)
Sindrome di NARP (neuropatia, atassia, retinite pigmentosa)
Sindrome MILS (Sindrome di Leigh ereditata per via matrilineare)
Encefalopatia mitocondriale con acidosi lattica e strokes (MELAS)
Mioclono-epilessia con fibre "ragged-red"(MERRF)
Miopatia e cardiomiopatia (MIMYCA)
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Ereditarietà mitocondriale
Soltanto le femmine possono trasmettere le malattie mitocondriali e
passano la mutazione a tutti i figli di entrambi i sessi.
Le malattie mitocondriali sono spesso caratterizzate da miopatie ed
encefalopatie (il cuore, i muscoli scheletrici e il sistema nervoso
centrale dipendono maggiormente dalla fosforilazione ossidativa).
Il pedigree di una famiglia con
una encefalopatia mitocondriale.
Si osservi la trasmissione materna
a tutti i figli maschi che femmine
e l’assenza della trasmissione per
via paterna.