Società Italiana di Endocrinologia
Le informazioni presenti sul sito non intendono sostituire il programma diagnostico, il
parere ed il trattamento consigliato dal vostro medico. In caso di condizioni di
interesse medico o quesiti medici consultate sempre il vostro medico o lo specialista.
Acromegalia
L’Acromegalia è la crescita eccessiva
dei tessuti corporei che causa
accentuazione ed ingrossamento dei
tratti del viso ed ingrossamento delle
mani e dei piedi. Essa è causata da una
produzione continua ed eccessiva di
ormone della crescita (GH). In
condizioni nomali GH, prodotto dalle
cellule somatotrope dell’ipofisi, viene
veicolato dal sangue a tutti i tessuti fra
cui le ossa ed i muscoli dove esercita la
propria azione. Nel bambino GH stimola
la crescita ed è importante per lo
sviluppo corporeo, nell’adulto influenza
il livello di energia, la forza muscolare,
la salute dell’osso ed il senso di
benessere. L’eccessiva produzione di
GH nel bambino causa il gigantismo che
è una condizione estremamente rara;
nell’adulto l’acromegalia è una
condizione che colpisce uomini e donne
di mezza età. La causa più comune della
malattia è un tumore benigno (adenoma)
delle cellule somatotrope ipofisarie, le
cellule produttrici di questo ormone.
L’Acromegalia non è una malattia
comune: risulta che siano colpite non più
di 3 o 4 persone su un milione ogni anno.
Il decorso è generalmente così lento
che la malattia viene riconosciuta solo
quando è presente da molti anni.
L’acromegalia è una malattia che può
essere trattata con successo ma, se
non trattata, può causare gravi
complicanze ed anche la morte.
Sintomi
Complicanze
Diagnosi
Trattamento
Monitoraggio
Informazione sui farmaci in uso
Informazioni di carattere
amministrativo
Sintomi
L’eccessiva produzione di ormone della
crescita stimola la produzione, pure
eccessiva, di un altro ormone, chiamato
insulin-like growth factor-1 (IGF-1).
IGF1 stimola la crescita del tessuto
cutaneo, del connettivo, della
cartilagine, degli organi e diversi altri
tessuti nel corpo. Anche le ossa sono
stimolate a crescere, ma dopo lo
sviluppo puberale la crescita non è più in
lunghezza ma in larghezza. Possono
essere presenti anche sintomi causati
dalla presenza dell’adenoma ipofisario
che, crescendo, comprime le strutture
circostanti.
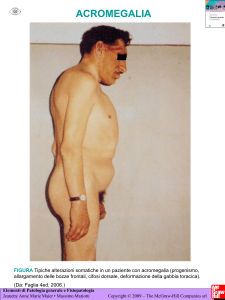
Le manifestazioni più visibili e
caratteristiche dell’acromegalia
derivano dalla crescita di tessuti molli,
delle cartilagini e ossa del viso, delle
mani e dei piedi.
Viso – naso, orecchie, labbra e fronte
diventano più larghi e accentuati, anche
la lingua si ingrossa; la mandibola
s’ingrossa e si estende causando
aumento dello spazio fra i denti e
difetto di opposizione. Aumenta anche
la crescita dei peli del viso (un problema
che sarà sentito particolarmente dalle
donne).
L’eccessiva crescita dei tessuti molli
della gola e del laringe (l’organo della
voce) causa un abbassamento ed
arrochimento della voce e può essere
causa di sleep apnea (una condizione di
arresto temporaneo del respiro durante
il sonno, che causa abbassamento del
tenore di ossigeno nel corpo e disturbo
del sonno). Può essere presente un
gozzo (ingrossamento della tiroide).
La crescita di mani e piedi causa la
necessità di cambiare la dimensione
dell’anello, dei guanti e delle scarpe. La
crescita dei tessuti ai polsi comprime i
nervi diretti alle mani causando
formicolii e dolore alle dita (la sindrome
del tunnel carpale).
La pelle si ispessisce, compaiono dei
“fibromi” (polipi fibroepiteliali, skin
tags), la sudorazione è eccessiva e
presente anche a riposo.
La crescita eccessiva dell’estremità
delle ossa causa danni alle cartilagini
vicine ed è causa di artrosi.
Complicanze
I pazienti con acromegalia presentano
un rischio aumentato di avere tumori
benigni, soprattutto se non ottengono il
controllo della concentrazione di
ormone della crescita: le donne sono più
spesso colpite da fibromi uterini ed
entrambi i sessi sono esposti a maggior
rischio di polipi del colon, che possono
degenerare se non asportati.
La acromegalia comporta un aumento
delle malattie cardiache,
verosimilmente come conseguenza
dell’aumento di volume del muscolo
cardiaco che ne ostacola il normale
funzionamento (la cosiddetta
cardiomiopatia). E’ comune
l’ipertensione arteriosa e qualche
paziente soffre di problemi alle valvole
cardiache. Se la acromegalia non è
curata si può arrivare allo scompenso
cardiaco.
L’eccessiva produzione di ormone della
crescita può causare aumento della
concentrazione di zucchero nel sangue
al di sopra dei valori di norma (diabete)
perché determina resistenza all’azione
dell’insulina (l’ormone che riduce il
livello di zucchero nel sangue). Infatti il
diabete è più comune fra gli
acromegalici.
Si è valutato che la prospettiva di vita
è ridotta di circa 10 anni nei pazienti
acromegalici che non ottengono il
controllo della malattia e soffrono di
diabete e cardiopatia. Se la malattia è
invece controllata la prospettiva di vita
è normale.
Se l’adenoma ipofisario diventa
voluminoso (più di 2 cm) può causare
sintomi dovuti all’aumento di pressione
sulle strutture circostanti; se l’adenoma
cresce verso l’alto, può causare uno
stiramento dei nervi ottici (chiasma
ottico) ed un difetto visivo,
prevalentemente nella visione
periferica (ai lati).
Diagnosi
Nel caso di sospetto di acromegalia
basato sull’aspetto di un soggetto, è
indispensabile confermare la diagnosi
mediante determinazione della
concentrazione di IGF1 e/o GH. La
determinazione di IGF1 può essere
effettuata in ogni momento della
giornata mentre la concentrazione di
GH deve essere determinata su diversi
campioni di sangue prelevati sia prima
che dopo aver bevuto una soluzione di
glucosio (carico orale di glucosio). In
condizioni normali il carico di zucchero
induce l’ipofisi ad interrompere la
secrezione di GH e la concentrazione di
ormone nel sangue si riduce; al
contrario un adenoma che produce GH
non interrompe la produzione di ormone
e quindi il livello di GH nel sangue non si
riduce.
Se la diagnosi di eccesso di GH viene
confermata è necessario effettuare la
risonanza magnetica della regione
ipofisaria per determinare se è
rilevabile un adenoma nell’ipofisi.
Trattamento
Il trattamento della acromegalia è volto
al controllo dell’eccessiva secrezione di
GH per evitare le complicanze della
malattia, anche in assenza di
particolari sintomi. L’obiettivo è quello
di riportare le concentrazioni di GH ed
IGF1 all’interno del range di normalità;
in questo modo le modificazioni dei
tessuti molli regrediranno nel corso di
diversi mesi ed il rischio di morte
anticipata regredirà. Talora un solo tipo
di trattamento non è sufficiente a
controllare la malattia e ne sono
necessari altri.
Ci sono tre tipi di terapia possibile
dell’acromegalia: chirurgica, medica e
radioterapia.
Terapia chirurgica
La chirurgia è completamente curativa
se è possible la completa rimozione
dell’adenoma ipofisario: questo è
possibile in genere nei pazienti il cui
adenoma non si estende al di fuori dei
limiti dell’ipofisi. La chirurgia è anche la
terapia di prima scelta nei casi di
adenoma che, essendo molto grosso,
compromette o minaccia la vista.
L’intervento chirurgico viene
effettuato microchirurgicamente con
una via di accesso attraverso il naso e
passaggio attraverso il seno sferoidale
(cavità ossea posta al di sotto
dell’ipofisi).
L’intervento può essere effettuato
utilizzando un endoscopio (un tubo
molto sottile dotato di illuminazione e
fotocamera) durante tutte le fasi
dell’intervento.
Nella chirurgia ipofisaria è dimostrata
una relazione fra l’esperienza del
neurochirurgo ed il successo degli
interventi effettuati; i pazienti che
devono essere sottoposti ad intervento
dovrebbero rivolgersi quindi ad un
chirurgo esperto in questo tipo di
chirurgia.
Dopo chirurgia la concentrazione di GH
ed IGF1 si riduce ma non sempre
normalizza; la probabilità di
normalizzazione è correlata alle
dimensioni dell’adenoma ipofisario. Nei
casi di microadenoma (inferiore ad 1
cm) la normalizzazione della
concentrazione di ormoni si ottiene
all’incirca nell’80% dei casi; al contrario
nei casi di macroadenoma, adenoma di
dimensione superiore al centimetro con
estensione al di fuori dell’ipofisi, la
probabilità di normalizzazione dei livelli
ormonali è solo del 30% circa. Quando
l’adenoma viene completamente rimosso,
la concentrazione di GH si normalizza
entro poche ore mentre la
normalizzazione di IGF1 richiede
settimane o mesi. Le possibili gravi
complicazioni dell’intervento
microchirurgico, quali il peggioramento
della vista, la meningite o la
rinoliquorrea (perdita di liquido spinale
attraverso la breccia chirurgica) sono
rare se il chirurgo è esperto (inferiori
al 5%), la comparsa di deficit ormonali
ipofisari che conducono a difetto della
funzione tiroidea, surrenalica, ovarica e
testicolare si verifica in meno del 10%
dei casi.
Terapia medica
Attualmente sono disponibili tre classi
di farmaci per il trattamento
dell’acromegalia
Analoghi di somatostatina
(octreotide, lanreotide)
Farmaci dopaminergici
(bromocriptina, cabergolina)
Antagonista del recettore
dell’ormone di crescita
(pegvisomant)
Analoghi di somatostatina – questi
farmaci inibiscono la secrezione di GH
da parte delle cellule ipofisarie che
producono l’ormone (cellule
somatotrope). Octreotide esiste in
forma a breve durata d’azione che deve
essere somministrato tre volte al
giorno ed a lunga durata con
somministrazione di una dose ogni 4
settimane. Lanreotide è disponibile in
forme a lunga durata d’azione che sono
somministrate ogni 2- 4 settimane.
Questi farmaci sono utilizzati sia come
trattamento iniziale (terapia primaria),
soprattutto nei casi in cui si debba
trattare un grosso adenoma con bassa
probabilità di asportazione chirurgica
completa, che dopo un intervento
chirurgico (terapia secondaria) nei
pazienti in cui persistono tessuto
adenomatoso ed elevata concentrazione
di GH dopo intervento chirurgico. Gli
analoghi di somatostatina riducono in
qualche misura i livelli di GH e IGF1
nella maggior parte dei pazienti, mentre
la normalizzazione dei livelli ormonali si
ottiene in circa il 40-50% dei pazienti.
Gli analoghi di somatostatina possono
ridurre il volume del tumore.
La terapia è generalmente ben tollerata
se si escludono disturbi intestinali, quali
dolori addominali, senso di gonfiore e
diarrea che in genere si risolvono dopo
le prime settimane di trattamento. Nel
20% circa dei pazienti trattati si
sviluppano calcoli alla colecisti, ma
raramente causano sintomi o rendono
ncessaria l’interruzione della terapia.
Antagonista del recettore dell’ormone
di crescita (pegvisomant) – blocca gli
effetti dell’ormone di crescita
legandosi al recettore dell’ormone,
diminuendo la produzione di IGF1 e
quindi diminuendo gli effetti di
crescita. Deve essere somministrato
quotidianamente mediante iniezioni
sottocutanee. Questo trattamento,
riservato in Europa a pazienti già
sottoposti a trattamento chirurgico e/o
radiante, ma con persistenza di valori
sopranormali di GH/IGF1, si è
dimostrato efficace nel normalizzare la
concentrazione di IGF1 in oltre il 70%
dei pazienti trattati.
Tra gli effetti indesiderati del farmaco
vanno ricordate le alterazioni
reversibili di alcuni indici di funzione
del fegato, che si verificano
occasionalmente e che ne rendono
necessario il periodico monitoraggio.
L’adenoma ipofisario, che è causa della
secrezione eccessiva di GH, può
continuare ad accrescersi durante
terapia con Pegvisomant per cui è
necessario controllarne annualmente lo
stato attraverso risonanza magnetica.
Farmaci dopaminergici – I farmaci
dopamino agonisti o dopaminergici
possono inibire la produzione di GH e
quindi diminuire secondariamente la
produzione di IGF1, ma non sono
altrettanto efficaci come le altre classi
di farmaci prima elencate. Alcuni
endocrinologi hanno rilevato efficacia di
cabergolina (farmaco dopamino
agonista) nel normalizzare le
concentrazioni di GH e IGF1 in un terzo
dei loro pazienti, ma la maggior parte
non concorda su una tale efficacia. Nei
pazienti che si dimostrano responsivi,
questa forma di trattamento è molto
vantaggiosa perché può essere assunta
per via orale ed è molto ben tollerata.
La Bromocriptina è un altro farmaco
dopaminergico, meno efficace e meno
ben tollerato di cabergolina e quindi non
più utilizzato.
Gli effetti indesiderati più comuni
legati all’uso di questi farmaci sono la
nausea (più frequente con
bromocriptina), abbassamento della
pressione arteriosa nel passaggio alla
stazione eretta con senso di
stordimento, talora disturbi del tono
dell’umore, stanchezza, congestione
nasale. Molti effetti indesiderati
possono essere controllati assumendo il
farmaco con i pasti o al momento di
coricarsi ed assumendo dosi iniziali
basse per poi aumentare gradualmente.
Radioterapia – E’ stata molto utilizzata
nel passato per il trattamento degli
adenomi ipofisari, inclusi quelli
secernenti ormone della crescita. La
terapia radiante può essere generata
da diverse sorgenti:
Un acceleratore lineare
Una sorgente di cobalto (raggi
gamma)
Un ciclotrone (radiazione
protonica)
La radioterapia può essere
somministrata in un’unica grossa dose o
in dosi più piccole frazionate; in ogni
caso viene diretta al bersaglio
attraverso un programma
computerizzato in guida tridimensionale
(stereotassica).
La radioterapia è in genere efficace sia
nel frenare la crescita del tumore
ipofisario che la eccessiva produzione
di GH e di IGF1; la riduzione però si
manifesta molto lentamente nel tempo
e così pure il miglioramento clinico.
Pertanto anche 10 anni dopo la
radioterapia solo una piccola
percentuale di pazienti ha raggiunto
valori ormonali normali.
Tra gli effetti collaterali della
radioterapia alcuni, che si manifestano
durante o subito dopo il trattamento
quali nausea, stanchezza, caduta dei
capelli e riduzione del senso del gusto e
dell’olfatto regrediscono nel corso di
settimane o qualche mese. La neurite
ottica che deriva da un danno al nervo
ottico (che trasmette la vista) può
esitare in perdita della vista nell’occhio
colpito, ma è un evento estremamente
raro.
Prima che la terapia radiante venisse
effettuata con la guida stereotassica
che si utilizza attualmente, era
possibile un danno alla parete dei vasi
cerebrali che si manifestava anni dopo
la radioterapia.
L’effetto collaterale più comune dopo
radioterapia è la comparsa di deficit di
uno o più ormoni ipofisari (quelli che
controllano la tiroide, i surreni, le ovaie
ed i testicoli) che si manifesta entro 10
anni dal trattamento in circa il 50% dei
pazienti.
Monitoraggio
I pazienti affetti da acromegalia
devono essere seguiti per tutto il corso
della vita per controllare l’efficacia dei
trattamenti e gli eventuali effetti
collaterali e per trattare le complicanze
della malattia.
Il paziente per primo deve rilevare se
la terapia è efficace attraverso
l’evidenza della riduzione della cefalea,
della sudorazione eccessiva, dello
spessore dei tessuti molli del viso, delle
mani e dei piedi (avvertita dal paziente
ma evidente anche agli altri).
Le concentrazioni di GH e IGF1 devono
essere monitorate con regolarità per
verificare l’efficacia del trattamento
prescelto.
Il paziente deve vigilare sulla eventuale
presenza e ricevere trattamento per la
sleep apnea (che può essere presente
per tutto il tempo che la
concentrazione di GH e IGF1 è
superiore alla norma)
Il paziente acromegalico dovrebbe
sottoporsi dopo i 50 anni a colonscopia,
da ripetere ogni 3-4 anni, per il rischio
aumentato di polipi al colon
(particolarmente i pazienti con
numerosi skin tags).
I pazienti trattati con chirurgia o
radioterapia sono a rischio di sviluppare
deficit ormonali ipofisari per cui le
concentrazioni di ormoni tiroidei,
surrenalici e testicolari dovrebbero
essere periodicamente monitorate, e la
presenza di regolari cicli mestruali
accertata, ed eventuali deficit corretti
mediante terapia sostitutiva.
In corso di terapia è importante
valutare (mediante risonanza
magnetica) se il trattamento in corso ha
indotto la riduzione di volume
dell’adenoma ipofisario.
Se il trattamento iniziale non è stato
sufficiente a controllare la malattia il
paziente deve essere consapevole che
sarà necessario fare trattamenti
aggiuntivi perché solo con il controllo
completo della malattia si ottiene la
riduzione del rischio di complicazioni ed
una normale sopravvivenza.
Gravidanza
Non vi sono molti dati sulla gravidanza
nella acromegalia pur essendo chiaro
che una donna acromegalia può condurre
a termine una normale gravidanza.
Se la paziente ha desiderio di
gravidanza dovrebbe discutere questo
progetto con l’endocrinologo curante
per essere informata sulla necessità di
interrompere alcuni farmaci che stesse
eventualmente assumendo.
Se la paziente è portatrice di un
adenoma ipofisario di volume superiore
ad 1 cm, dovrebbe controllare il campo
visivo in corso di gravidanza e
sottoporsi a risonanza magnetica in
caso di comparsa o peggioramento del
difetto del campo visivo. La risonanza
magnetica è considerata sicura per la
gestante e per il feto.
patologia (acromegalia) della quale il
paziente è affetto.
Somavert è un farmaco di classe H che
viene fornito gratuitamente ai pazienti;
a seconda dei casi la fornitura dei
farmaci può avvenire tramite il Presidio
Ospedaliero o tramite la ASL di
appartenenza del paziente.
Informazione sui farmaci in uso
Lanreotide ed octreotide (nelle varie
forme presenti in commercio) vengono
prescritti dal medico specialista su
apposito piano terapeutico che ha
validità massima di un anno ed è
rinnovabile.
Ai fini dell’erogazione dei farmaci in
regime di esenzione, copie del piano
terapeutico dovranno essere fatte
pervenire al medico curante (Medico di
Medicina Generale o Pediatra di Libera
Scelta) e alla ASL di residenza
dell’assistito.
A seconda dei casi la fornitura dei
farmaci può avvenire tramite il Presidio
Ospedaliero, tramite la ASL di
appartenenza del paziente o tramite le
farmacie aperte al pubblico. Il medico
curante può prescrivere i farmaci
indicati nella Scheda per la Prescrizione
dei Farmaci sul ricettario regionale,
indicando sulla ricetta il codice della
La diagnosi di Acromeglia da diritto
all’esenzione dalla partecipazione al
costo delle prestazioni sanitarie
correlate (Decreto 28 maggio 1999 n.
329, aggiornato dal d.m. 21 maggio
2001, e dal regolamento delle malattie
rare d.m. 18 maggio 2001). Il codice di
esenzione ticket è 001.
Per ottenere il rilascio del tesserino di
esenzione il paziente deve disporre di
certificazione attestante la patologia
invalidante rilasciata da una struttura
ospedaliera pubblica (ASL o Aziende
Ospedaliere) o da un medico specialista
ambulatoriale dell’Azienda Sanitaria.
Non è ammessa la certificazione
rilasciata da specialisti di struttura
privata ambulatoriale, anche se
accreditata. Il rilascio del tesserino è
immediato
Per chiarimenti visita:
http://www.ministerosalute.it
Informazioni di carattere
amministrativo