Disordini Linfoproliferativi
Patologie dei globuli bianchi
Disordini Linfoproliferativi
•Esistono due grandi categorie di patologie dei GB:
•leucopenie (riduzione del numero)
•disordini proliferativi
•Le proliferazioni possono essere di due tipi:
•Reattive
•Neoplastiche
Le cellule del sistema immunitario proliferano in maniera altamente
controllata come risposta normale a infezioni o infiammazioni o nel loro
processo maturativo.
La proliferazione maligna incontrollata delle cellule del sistema immunitario
puo’ determinare lo sviluppo di leucemia, linfomi e mielomi.
Complessivamente leucemie, linfomi e mielomi sono noti come disordini
linfoproliferativi
Cause di leucocitosi
Downloaded from: StudentConsult (on 30 January 2011 10:47 AM)
© 2005 Elsevier
Disordini Linfoproliferativi
Definizioni
Vanno sotto il nome di leucemie, le proliferazioni maligne delle
cellule del midollo osseo che si trovano circolanti nel sangue o
nel midollo stesso. Le cellule maligne proliferanti delle leucemie
spesso infiltrano altri organi e possono determinare
linfoadenopatie, lesioni meningee etc.
Le neoplasie di cellule linfoidi non ricircolanti vanno a costituire
i linfomi. Comunque la continue fuoriuscita delle cellule maligne
dall’organo linfoide colpito risulta in uno stato “leucemico” che
puo’ essere confuso con le leucemia
Le discrasie delle plasmacellule prendono il nome di mielomi
Differenziamento delle cellule ematopoietiche
Downloaded from: StudentConsult (on 1 February 2011 11:44 AM)
(Modified from Wyngaarden JB, et al [eds]: Cecil Textbook of Medicine, 19th ed. Philadelphia, WB Saunders, 1992, p. 820.)
Disordini Linfoproliferativi
Considerazioni generali
A ciascuno step della differenziazione dalla cellula staminale pluripotente del midollo
osseo si puo’ determinare una trasformazione neoplastica della cellula e lo sviluppo di
un disordine linfoproliferativo
Disordini Linfoproliferativi
Identificazione dei tipi cellulari coinvolti
E’ estremamente importante la determinazione del tipo cellulare che determina il
disordine linfoproliferativo. Infatti dal tipo e dalle caratteristiche delle cellule coinvolte
dipende l’approccio terapeutico.
Una serie di tecniche vengono impiegate per l’esatta identificazione dei tumori
linfoidi e mieloidi:
-esame morfologico dell’aspetto delle cellule e dei nuclei all’esame microscopico
-colorazioni citochimiche speciali al fine di evidenziare caratteristici enzimi,
carboidarati o lipidi
-immunofenotipizzazione delle cellule tramite citometria di flusso per identificare
la linea cellulare mieloide o linfoide coinvolta ed il suo grado di differenziamento
-analisi citogenetica per identificare eventuali caratterstiche alterazioni
cromosomiche (traslocazioni/delezioni)
-studi di riarrangiamento genico per identificare o confermare la monoclonalita’
dei disordini linfoproliferativi soprattuto in linfociti T
Disordini Linfoproliferativi
Eziopatogenesi
Eziopatogenesi dei disordini linfoproliferativi (1)
Si pensa che ciascun disordine linfoproliferatico (leucemia, linfoma o
mieloma) derivi da una singola cellula che va in trasformazione maligna che
determina la sua incontrollata divisione.
I meccanismi con cui si determina la trasformazione neoplastica nelle cellule
derivate dal midollo osseo sono fondamentalmente gli stessi chiamati in causa
nella tumorogenesi di altri tessuti
La maggior parte dei casi e’ determinata dall’accumulo di mutazioni in uno o
piu’ geni coinvolti nella regolazione della proliferazione cellulare o del
differenziamento. Ad esempio si puo’ avere attivazione di protoncogeni o
l’alterazione del controllo da parte di geni tumore soppressori.
In alcune leucemia il materiale genetico e’ scambiato tra due geni
(translocazione), determinando lo sviluppo di un nuovo gene di fusione che
agisce da oncogene.
Eziopatogenesi dei disordini linfoproliferativi (2)
Cause innesco dei processi
Virus possono innescare il meccanismi di trasformazione neoplastica (tipo
EBV, HHV-8 HTLV-1), comunque non rappresentano la sola causa in quanto
solo l’1% circa dei soggetti infettati in aree endemiche sviluppa disordini
linfoproliferativi.
Altri fattori noti innescare la trasformazione in leucemie/linfomi sono
Radiazioni
Farmaci (agenti alchilanti)
Sostanza chimiche (es. espozione al benzene)
Fattori genetici (es. incrementata incidenza in alcune malattie genetiche
come la sindrome di Down).
Traslocazioni nella eziopatogenesi dei disordini linfoproliferativi
Il cromosoma Philadelphia
Il miglior esempio di cancerogenesi delle cellule del midollo osseo determinato da traslocazioni è lo
spostamento di una parte del cromosoma 9 sul cromosoma 22 [t(9;22)].
Trovata in forme leggerente diversa sia nella leucemia cronica mieloide che nella leucemia acuta
linfoblastica, genera un gene di fusione con funzione tirosin-kinasica BCR-ABL, dovuta alla apposizione del
gene abl del cromosoma 9 al gene bcr del cromosoma 22.
Dalla traslocazione si genera un nuovo cromosoma piu’ piccolo del 22 che prende il nome di cromosoma
Philadelphia
p210 nella maggiro parte di
leucemie mieloidi croniche
p190 nella maggiro parte di
leucemie linfoidi croniche
Traslocazioni nella eziopatogenesi dei disordini linfoproliferativi
Il cromosoma Philadelphia
Il miglior esempio di cancerogenesi delle cellule del midollo osseo determinato da
traslocazioni e’ lo spostamento di una parte del cromosoma 9 sul cromosoma 22 [t(9;22)].
Dalla traslocazione si genera un nuovo cromosoma piu’ piccolo del 22 che prende il
nome di cromosoma Philadelphia
Trovata in forme leggerente diversa sia nella leucemia cronica mieloide che nella
leucemia acuta linfoblastica, genera un gene di fusione con funzione tirosin-kinasica BCRABL, dovuta alla apposizione del gene abl del cromosoma 9 al gene bcr del cromosoma 22.
L’attività della tirosin-chinasi è normalmente regolata dalla dimerizzazione ligandomediata seguita dall’attivazione di molteplici vie a valle quali quelle del controllo della
sopravvivenza e della proliferazione cellulare
L’auto associazione della proteina di fusione BCR-ABL porta all’autofosforilazione
costitutiva di BCR-ABL e all’attivazione delle vie a valle.
L’effetto è la divisione cellulare e l’inibizione dell’apoptosi indipendentemente dal
legame con il ligando.
Individuazione di un gene di fusione BCR-ABL per ibridazione fluorescente in
situ
A cromosomi 9 e 22 e la posizione dei geni ABL e BCR. Il cromosoma Philadelphia si crea mediante la
traslocazione cromosomica bilanciata e uhe sostituisce la porzione telomerica di 22q con quella di 9q. A
livello molecolare la rottura e il ricongiungimento del DNA provocano la formazione del gene di fusione
sul cromosoma Ph derivante dall’estremità 5’ di BCR e 3’ di ABL e quindi porta le due sequenze in stretta
vicinanza fisica. Questa anomala collocazione di BCR e ABL può essere rilevata per ibridazione in situ con
una coppia di sonde fluorescenti di DNA, complementari alla sequenza genomica di DNA vicina ai punti
di rottura di BCR e ABL.
(Courtesy of Dr. Cynthia Morton and Ms. Debbie Sandstrom, Department of Pathology, Brigham and Women's Hospital, Boston, MA.)
Downloaded from: StudentConsult (on 30 January 2011 10:48 AM) © 2005 Elsevier
Individuazione di un gene di fusione BCR-ABL per ibridazione fluorescente in
situ
B. Una sonda verde per ABL e una rossa per
BCR sono state ibridate in su cromosomi
metafasici e nuclei interfasici preparati da
sangue periferico di individuo normale. Due
segnali rossi e due segnali verdi stanno ad
indicare la presenza di copie normali,
spazialmente distanziate di ABL e BCR.
C. Al contrario cromosomi metafasici e un
nucleo interfasico preparati sa midolleo osseo
di un soggetto con leucemia mieoloide cronica
mostrano un segnale per ABL (verde), uno per
BCR (rosso) e il segnale giallo che è il risultato
della sovrapposizione di un segnale BCR rosso
e ABL verde. Questo indica la presenza di un
gene di fusione BCR-ABL
(Courtesy of Dr. Cynthia Morton and Ms. Debbie Sandstrom, Department of Pathology, Brigham and Women's Hospital, Boston, MA.)
Downloaded from: StudentConsult (on 30 January 2011 10:48 AM) © 2005 Elsevier
Traslocazioni nella eziopatogenesi dei disordini linfoproliferativi
Altre traslocazioni
A parte la ben caratterizzata traslocazione t(9;22) altre traslocazioni identificate con
una certa frequenza in varie forme di leucemie sono la t(15;17) e la t(8;21)
L’applicazione di tecniche molecolari per la caratterizzazione fine di queste
mutazioni/traslocazioni (FISH, Gene array etc) ha chiaramente dimostrato che tutte le
leucemie hanno geni alterati.
L’importanza della identificazione di queste alterazioni genetiche e’ fondamentale
perche’ rappresentano nuovi specifici target terapeutici o prognostici, ad esempio:
L’imatinib mesilate (Glivec) nella leucemia cronica mieloide
Pazienti con t(8;21) nella leucemia acuta mieloide hanno una prognosi migliore
e non necessitano di trapianto di cellule staminali.
Classificazione WHO delle neoplasie del midollo osseo
Diverse classificazioni sono state impiegate nelle neoplasie del midollo osseo
in base alla istopatogenesi (mieloide o linfoide)
in base alla presentazione acuta o cronica,
con o senza leucemia,
Il WHO ha recentemente ridefinito la classificazione che tiene in
considerazione sia la morfologia cellulare, le caratteristiche immunologiche, e la
specifica abberrazione genetica osservata.
Questo approccio permette una migliore classificazione del paziente, la
definizione della prognosi e i diversi approcci terapeutici che si possono
impiegare come anche il piu’ corretto arruolamento in trial terapeutici.
Proliferazione neoplastiche dei GB (1)
Definizioni
Neoplasie linfoidi
Neoplasie mieloidi
• Derivano da cellule staminali
emopoietiche da cui originano le
cellule della linea linfoide
• Comprendono entità patologiche
distinte
• Il fenotipo è per lo più simile a
quello di un linfocita normale in
un particolare stadio
differenziativo.
• Il fenotipo è un marcatore
diagnostico che permette di
classificare tali patologie
• La caratteristica comune di
questo gruppo eterogeneo è
un’origine comune da cellule
progenitrici ematopoietiche
capaci di dar luogo a cellule
terminalmente differenziate della
serie mileoide
• Interessano principalmente il
midollo osseo e in minor grado gli
organi ematopoietici secondari.
• Si presentano con alterazioni
dell’ematopoiesi
Proliferazione
neoplastiche
dei
GB
(2)
Proliferazione neoplastiche dei GB (2)
Classificazione WHO
Neoplasie linfoidi
Neoplasie mieloidi
• Neoplasie dei precursori Bcellulari (neoplasie a cellule B
immature)
• Neoplasie delle cellule B
periferiche (neoplasie a cellule B
mature)
• Neoplasie dei precursori T
cellulari (neoplasie a cellule T
immature)
• Neoplasie delle cellule T/NK
periferiche (neoplasie a cellule T
e NK mature)
• Linfoma di Hodgkin
• Leucemie mieloidi acute
(caratterizzate da accumulo
forme mieloidi immature nel
midollo osseo e soppressione
normale ematopoiesi)
• Sindromi mielodisplastiche
(associate con ematopoiesi
inefficace e citopenie correlate)
• Sindromi mieloproliferative
croniche (associate con
aumentata produzione delle
cellule mieloidi terminalmente
differenziate)
Sommario dei maggiori tipi e sottotipi di disordini
linfoproliferativi in base alla classificazione WHO
Malattie mieloidi croniche linfoproliferative
Sindromi Mielodisplastiche/mieloproliferative-(sindromi crossover)
Sindromi Mielodisplastiche
Leucemia Mieloide Acuta (AML, Acute myeloid leukaemia)
Con ricorrenti anormalita’ citogenetiche
Con displasia multilinea cellulare
Associata a terapia
Con ambiguita’ di linea cellulare
Non altrimenti caratterizzata
Neoplasie dei linfociti B
Leucemie/linfomi dei precursori B-linfoblastici
Neoplasie delle cellule B mature (linfomi B-non-Hodgkin's e neoplasie delle plasmacellule)
Neoplasie dei linfociti T ed NK (natural killer)
Leucemie/linfomi dei precursori T-linfoblastici
Linfomi dei blasti delle cellule NK
Neoplasie dei linfociti T e cellule NK mature
Linfomi di Hodgkin‘s
Neoplasie degli istiociti e delle cellule dendritiche
Mastocitosi maligne
Leucemie
Neoplasie linfoidi
Neoplasie mieloidi
ACUTA
CRONICA
LINFOIDE
MIELOIDE
LINFOIDE
MIELOIDE
Leucemie
Le proliferazioni maligne delle cellule del midollo osseo che si trovano circolanti nel
sangue o nel midollo stesso interessano tutti gli stadi di differenziazione della cellula
staminale emopoietica.
Leucemie
Distinguiamo in prima analisi due forme Leucemie Acute e Leucemie Croniche.
LEUCEMIA
ACUTA
CRONICA
• Sebbene le forme acute siano a
carico di tipi cellulari a tutti i gradi
di differenziamento, le forme piu’
comuni sono tipiche delle cellule
meno differenziate della cellula
staminale emopoietica.
• Per contro, le forme di leucemia
cronica sono normalmente a
carico delle forme piu’ mature
delle varie linee cellulari linfoidi e
mieloidi
LEUCEMIA
capacità differenziativa delle cellule trasformate
ACUTA
Nelle forme acute le cellule
neoplastiche perdono ulteriore
capacita’ differenziativa
In entrambe le forme la
fenotipizzazione delle cellule
coinvolte e’ fondamentale
per il miglior approccio
terapeutico possibile
CRONICA
Nelle forme croniche la capacita’
differenziativa e’ normalmente
mantenuta
LEUCEMIA (andamento epidemiologico ed età)
ACUTA
Le forme acute hanno un andamento
bimodale con la massima incidenza
nei primi anni di vita che torna a
risalire in vecchiaia (questo
sottolinea anche le diverse linee
cellulari coinvolte
CRONICA
Le forme croniche sono tipiche
dell’eta’ adulta
Leucemie - sommario
Acuta vs Cronica
Acuta
Stadio
maturativo
Cronica
Puo’ essere a carico di cellule a
Tipica delle forme mature
diverso stato di differenziamento,
derivate dalle cellule staminali
prevalentemente meno differenziate
Differenziamento Le cellule leucemiche non
differenziano ulteriormente
Le cellule leucemiche
mantengono la capacita’ di
differenziare
Andamento
Andamento bi-modale con forme
tipiche del bambino e dell’adulto
Tipica dell’adulto
Danno
Danno midollare
Proliferazione normalmente
senza danno midollare
Prognosi
Rapidamente fatale se non trattata
Sopravvivenza di qualche anno
Terapia
Potenzialmente curabile
Al momento non curabile senza
trapianto del midollo
Leucemie acute
Leucemia Linfoblastica Acuta
(ALL, Acute Lymphoblastic Leukemia
a precursori B e T)
Leucemia Mieloide Acuta
(AML, Acute Myeloid Leukaemia)
Tipica del bambino
Tipica dell’adulto
Leucemie acute
ALL
(Leucemia Linfoblastica Acuta)
AML
(Leucemia Mieloide Acuta)
Tipica del bambino (4 anni)
Si osserva con una certa
frequenza la traslocazione t(9;22)
con presenza del cromosoma
Philadelphia,
Comunque nei piu’ piccoli la
presenza del cromosoma
Philadelphia e’ meno frequente e
sono presenti difetti genetici
complessi che si pensa si
sviluppino durante la vita fetale
Tipica dell’adulto (15-39 anni)
Anomalie genetiche di diverso tipo
anche in base alla linea mieloide
coinvolta con traslocazioni t(15;17) e
t(8;21) con una prognosi migliore
che altre anomalie come la perdita
completa del cromosoma 5 o 7 o piu’
complesse alterazioni genetiche
Altre caratteristiche delle leucemie acute
Cambi a livello ematico e di midollo osseo
Nel sangue periferico i leucociti aumentano
Si possono osservare anche conte superiori a 100 × 103/µl.
La maggioranza delle cellule nucleate sono blasti leucemici.
E’ presente normalmente una anemia normocitica normocromica.
La trobocitopenia e’ marcata, particularmente nella AML.
La cellularita’ midollare e’ marcatamente aumentata.
I blasti devono costituire almeno il 20% delle cellule nucleate, ma
spesso superano l’80%.
Normalmente non si ha erosione dell’osso con fratture conseguenti
Cambi a carico di altri organi
Linfonodi, fegato e milza possono essere infiltrati da blasti leucemici
L’ingrossamento linfonodale non e’ marcato
L’ingrossamento splenico, quando presente, e’ minore delle leucemie
croniche.
Si ha una aumentata suscettibilita’ ad infezioni
Possono essere presenti emorragie secondarie a trombocitopenia
Markers per la caratterizzazione delle forme di
leucemia acuta
Pannello di anticorpi per la diagnosi differenziale di leucemia acuta
B linfoide
T linfoide
Mieloide
Non linea ristretti
Prima linea
CD19, CD10 (earlyB),
CD79a (BCR), CD22
CD3, CD2
CD34, CD64, CD33, CD15,
CD117, CD13, antimieloperossidasi
Tdt
Seconda linea
Ig superficie ,
Ig citoplasmatiche, CD138
CD7
CD33, CD41, CD42, CD61
CD45
Bain et al. 2002 (modified)
Esempio di identificazione della forma di
leucemia linfoblastica acuta
Lo striscio di sangue periferico
evidenzia linfoblasti con nucleo
condensato
L’analisi del fenotipo tramite
citometria di flusso evidenzia che I
linfoblasti esprimono TdT e marker
dei linfociti B CD22, e che le stesse
cellule sono positivi per altri due
markers tipici delle cellule pre-B,
CD10 e CD19.
La diagnosi e’ quindi di un ALL pre-B.
Esempio di identificazione della forma di
leucemia mieloide acuta
Lo striscio di sangue periferico
evidenzia mieloblasti con delicata
cromatina
nucleare,
nucleoli
sporgenti e fini granuli azzurofili nel
citoplasma
L’analisi del fenotipo tramite
citometria di flusso evidenzia che B. I
mieloblasti (in rosso) esprimono
CD34 (marker delle cellule staminali
multipotenti) ma non il CD64 tipico
marker delle cellule mieloidi mature.
Inoltre, in C gli stessi mieloblasti
esprimono CD33 marker delle
mieloidi immature e un sottogruppo
il CD15 , un marker delle mieloidi più
mature.
Leucemie - sommario
Acuta vs Cronica
Acuta
Cronica
Stadio
Puo’ essere a carico di cellule a Tipica delle forme mature derivate
differenziamento diverso stato di differenziamento dalle cellule staminali
cellulare
Effetti
Le cellule leucemiche non Le cellule leucemiche mantengono
differenziano ulteriormente
la capacita’ di differenziare
Andamento
Andamento bi-modale con forme Tipica dell’adulto
tipiche del bambino e dell’adulto
Danno
Danno midollare
Prognosi
Rapidamente
trattata
Terapia
Potenzialmente curabile
fatale
Proliferazione
senza
midollare normalmente
se
danno
non Sopravvivenza di qualche anno
Al momento non curabile senza
trapianto del midollo
Leucemie Croniche
Le forme di leucemia cronica sono normalmente a carico delle forme piu’ mature
delle varie linee cellulari linfoidi e mieloidi
Tipica dell’eta’ adulta
La capacita’ differenziativa e’ normalmente mantenuta
Anche per le forme croniche distinguiamo le forme mieloidi (CML, Chronic Myeloid
Leukemia) da quelle linfoidi (CLL, Chronic Lymphocytic Leukemia)
Leucemia Linfoide Cronica (1)
La CLL, è relativamente comune nell’anziano
Normalmente presenta un decorso benigno
Il reperto tipico è la presenza di un numero eccesivo di piccoli linfociti nel sangue
periferico che è più contenuto delle ALL ed altre leucemie
Nel 90% dei casi di CLL le cellule neoplastiche sono linfociti B con le caratteristiche
di linfociti B immaturi circolanti
Striscio di sangue di CLL.
Numerosi piccoli linfociti e
cellule “smear” (frecce) sono
caratteristici.
Leucemia Linfoide Cronica (2)
La trasformazione e’ normalmente monoclonale ma i linfociti non sono
sufficientemente maturi da secernere l’immunoglobulina monoclonale o da essere
rilevata la sua presenza intracellulare
La CLL e’ di fatto un disordine linfoproliferativo con caratteristiche simili ad un
linfoma di basso grado ma con un coinvolgimento predominante del sangue e del
midollo
Si ha un lieve ma progressivo interessamento linfonodale, puo’ esserci una
splenomegalia a cui si associa anemia e trombocitopenia, ed interessamento epatico
Alcuni soggetti possono presentare aumentata suscettibilita’ alle infezioni
Il trattamento e’ normalmente solo sintomatico
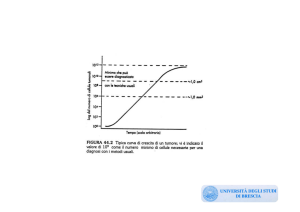
Decorso clinico e staging della CLL.
Nella classificazione di 'Rai' si riconoscono cinque fasi in con
interessamento progressivo dei linfonodi, milza e fegato
Markers per la caratterizzazione delle forme di leucemia
linfocitica cronica da altre forme di leucemie delle cellule mature
Linfocita B
Linfocita T
Linfocita B e T
Prima linea
CD19, CD33, CD23, CD79b
(BCR-bchain), surface Ig, k;l
CD2
CD5
Seconda linea per
determinare particolari
disordini
Disordini delle cellule B
immature
Sindrome di Sezary ed altri
disordini del lifocita T
“Linfoma
mantellare”
CD11C, CD103, HC2
Ig citoplasmatiche, CD79a,
CD138
CD3, CD7, CD4, CD8
Ciclina D
Leucemia Mieloide Cronica (1)
Sebbene sia definita cronica, la CML ha una sopravvivenza media di 5 anni in
pazienti non elegibili a trapianto di midollo
Il midollo osseo è sostituito progressivamente dal clone mieloide
Nella maggior parte dei casi si ha anomalia cariotipica, con presenza del
cromosoma Philadelphia con i precursori sia della linea eritroide, megacariocitica
granulocitica che dei linfociti B che portano il difetto
Leucocitosi è tipica con la conta cellulare che può occasionalmente eccedere i 300
elementi × 103/µl.
Lo striscio di sangue presenta
superficialmente le stesse caratteristiche
dell’aspirato midollare con mielociti,
promieolociti, mieloblasti e normoblasti
presenti in gran numero
Una anemia normocromica e’ spesso
presente.
Nel midollo vi e’ una riduzione dello
spazio grasso
Nelle fasi terminali il numero di blasti nel
sangue aumenta
Striscio di CML. Mielociti (frecce) e
metamieolciti (teste di frecce) entrano
nel circolo
Leucemia Mieloide Cronica (2)
Cambi a carico di altri organi
La milza e’ allargata per infiltrazione delle cellule CML con aree di infarto a
causa del rapido allargamento e mancanza di sangue
Epatomegalia e’ anche frequente
L’infiltrazione di altri organi occasionale.
Infezioni e sanguinamento non sono comuni nella
fase cronica
Decorso Clinico
I sintomi sono minimi nella fase cronica ed essenzialmene dovuti all’anemia ed
alla splenomegalia. Nella fase terminale in cui si sviluppa la crisi di blasti simile
alla leucemia acuta le caratteristiche cliniche somigliano a quella della leucemia
acuta.
Trattamento
Il trattamento delle forme BCR-ABL positive (cromosoma Philadelphia) sono
ora ben controllate con anti-metaboliti orali quali la idrossiurea. Il trapianto di
midollo è la terapia preferita nei soggetti elegibili a questo trattamento.
Chi non puo’ essere sottoposto a trapianto di midollo, la terapia con αinterferone ha aumentato la media di sopravvivenza da 3 a 5 anni.
Linfomi
LEUCEMIA LINFOCITICA
vs
LINFOMA
Leucemia linfocitica
•
Neoplasia linfoide che si presenta con
diffuso interessamento del midollo
osseo e che è accompagnata da un
alto numero di cellule neoplastiche
nel sangue periferico
Linfoma
•
Proliferazioni linfoidi che si
presentano come masse tissutali
distinte
I linfomi possono presentarsi con un quadro leucemico nel sangue periferico,
accompaganto da interessamento del midollo osseo. L’evoluzione a leucemia è
frequente nei linfomi “incurabili”
Al contrario tumori “identici” a leucemie insorgono a volte come masse dei tessuti
molli senza interessamento midollare.
Quindi quando sono applicati a particolari neoplasie, i termini “LEUCEMIA” e
“LINFOMA” descrivono puramente la tipica distribuzione tissutale della malattia al
momento della manifestazione clinica
Linfomi
Le neoplasie di cellule linfoidi non ricircolanti, che prendono origine
nei linfonodi, o dai tessuti linfatici extranodali
Rappresentano un’espansione clonale dei linfociti B, T o N
Vengono comunemente suddivisi in linfomi Hodgkin e non-Hodgkin
Leucemie vs Linfomi
Leucemie
Linfomi
Tumori delle cellule ematopoietiche
Tumori delle cellule ematopoietiche
Originano nel midollo, si diffondono nel sangue e nei
linfonodi
Originano nel linfonodo, si diffondono nel sangue e nel
midollo
Mieloidi e linfoidi
Solo linfoidi
Acute o croniche
Hodgkin o non-Hodgkin
Linfomi – caratteristiche generali
Rappresentano il 3% di tutti i tumori
Hanno una incidenza relativamente alta 100 nuovi casi/100.000
abitanti/anno
L’incidenza aumenta con l’età e la presentazione, decorso e
prognosi e’ spesso diversa
Linfomi indolenti: più frequenti nell’adulto-anziano (età media
55-60 anni)
Linfomi aggressivi: tutte le età con maggiore frequenza nel
giovane-adulto (età media 30-40 anni)
Presentano normalmente una linfoadenopatia a cui si associa
dolore, febbre, sudorazione notturna, perdita di peso, prurito,
aumentata frequenza di infezioni opportunistiche e di riattivazioni di
infezioni (infezioni da herpes simplex, herpes zoster etc)
Linfomi – linfoadenopatia
Distribuzione dei linfonodi e loro interessamento
nella linfoadenopatia conseguente a linfoma
L’interessamento linfonodale persistente viene
investigato tramite biopsia
La biopsia linfonodale si esegue
quando il paziente presenta una massa
linfonodale persistente da oltre 4 settimane o
quando nel sangue periferico o nel midollo
osseo si osserva un numero aumentato di linfociti
con fenotipo suggestivo per diagnosi di linfoma.
Oggi è suggerito di processare il linfonodo a fresco o
dopo congelamento in azoto liquido per favorire la
tipizzazione con anticorpi monoclonali di superficie
che permettono di identificare le cellule mantenendo
l’architettura del tessuto.
Sul linfonodo è possibile eseguire tecniche di
colorazione citochimica, caratterizzazione
immunofenotipica e molecolare
Linfomi
Follicolare e diffuso
Linfoma follicolare (milza) noduli
prominenti rappresentano follicoli di
polpa bianca espansi.
(Courtesy of Dr. Jeffrey Jorgenson, Department of
Pathology, Brigham and Women's Hospital, Boston,
MA.)
Linfoma diffuso a grandi cellule B
(milza). Presenza di una unica grande
massa
(Courtesy of Dr. Mark Fleming, Department of
Pathology, Brigham and Women's Hospital, Boston,
MA.)
Linfomi – coinvolgimento di virus e batteri nella
eziopatogenesi di alcuni sottotipi
Distribuzione geografica del linfoma e degli agenti infettivi
Tipo di Linfoma
Agente
infettivo
Distribuzione
geografica
Evidenza del ruolo
etiologico
Linfoma di Burkitt
Virus di
Forma endemica ristretta
Epstein-Barr
essenzialmente alle aree
(EBV) e malaria di endemia malarica
Correlazione con prevalenza
malarica. Costante presenza
di EBV nelle cellule tumorali
Linfoma nasal-type
(angiocentrico)
EBV
Aree del sud-est asiatico
e del Sud America
Presenza del virus nelle cellule
tumorali
Evidenza sierologica
dell’infezione attiva
Linfoma associato al
tessuto linfoide della
mucosa gastrica
(MALT)
Helicobacter
pylori
Associata a condizioni
socioeconomiche povere
Linfoma associato a gastrite da
H. pylori. Può rispondere al
trattamento antibiotico
Leucemia/linfoma
dell’adulto a cellule T
HTLV-1
Sud-est del Giappone
e Caraibi
Neoplasie trovate solo
nei portatori del virus
Linfomi –ruolo dei virus e batteri nel
meccanismo eziopatogenetico
L’infezione o la normale proliferazione in risposta all’agente infettivo possono
determinare traslocazioni geniche come quelle che portano il gene enhancer
delle Ig contiguo a bcl-2 che determina la costante produzione della proteina
che inibisce l’apoptosi con conseguente proliferazione incontrollata del clone
Linfoma di Hodgkin
E’ un linfoma delle cellule B
Caratterizzato dalla presenza di cellule binucleate di Reed-Sternberg
L’epidemiologia e’ bimodale, con un largo picco tra i giovano adulti ed un picco minore
dopo i 60 anni
La linfoadenopatia del collo e’ abbastanza tipica
Puo’ presentarsi in diverse varianti di cui 4 principali:
Predominanza linfocitaria
Sclerosi nodulare
Cellularità mista
Deplezione linfocitaria
La radioterapia viene impiegata nelle forme localizzate, mentre in quelle piu’ diffuse si
impiega una chemioterapia aggressiva o combinata a radioterapia
Il linfoma di Hodgkin si associa a marcata depressione della immunita’ cellulo-mediata.
Quindi il paziente e’ prone ad infezioni batteriche, virali e fungine, sia prima che durante
la terapia.
Altre complicazioni includono la comparsa di tumori secondari associati alla terapia ed
alterazioni della tiroide dopo irradiazione del collo
Linfoma di Hodgkin
Biopsia linfonodale nella malattia di Hodgkin. Le frecce mostrano cellule di notevoli dimensioni,
con nuclei pallidi ed evidenti nucleoli (Cellule di Reed-Sternberg), nell’ambito di una popolazione
cellulare costituita prevalentemente da piccoli lnfociti con occasionali istiociti.
Downloaded from: StudentConsult (on 30 January 2011 10:47 AM)
© 2005 Elsevier
Linfoma di Hodgkin
Linfoma Hodgkin tipo sclerosi nodulare. Basso ingrandimento mostra bande ben definite di
collagene acellulare rosa che suddividono le cellule tumorali e l’infiltrato reattivo descrivendo dei
noduli
(Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, TX.)
Downloaded from: StudentConsult (on 30 January 2011 10:47 AM)
© 2005 Elsevier
Linfoma di Hodgkin
EBV e NF-κB
Frequente è la presenza degli episomi di EBV nelle cellule di Reed-Sternberg di molti casi di LH
a cellularità mista.
Le cellule EBV-positive esprimono in modo latente la proteina latente di membrana (LMP-1) che
ha attività trasformante.
LMP-1 trasmette segnali che up-regolano NF-κB, un fattore di trascrizione importante per
l’attivazione linfocitaria.
L’inappropriata attivazione di NF-κB
κ sembra essere un evento comune nei LH classici
(Tale attivazione si verifica anche nei tumori EBV-negativi e dovuta a mutazioni acquisite in un
regolatore negativo di NF-κB, I κB).
In alcuni casi di LH i geni delle Ig delle cellule di Reed-Sternberg contengono mutazioni
somatiche “crippling” che prevengono l’espressione delle Ig di superficie necessaria per la
produzione di segnali che permettono la sopravvivenza delle cellule B nei centi germinativi.
L’attivazione di NF-κB da parte di EBV (o altri meccanismi) salverebbe queste cellule destinate
all’apoptosi ponendole in uno stadio adatto all’acquisizione di ulteriori mutazioni
sconosciute che collaborano per produrre le cellule di Reed-sternberg
Linfomi non-Hodgkin
Numerosi altri linfomi classificati come non-Hodgkin sono descritti:
Linfoma/leucemia linfoblastici (T e B)
Linfoma a piccoli linfociti/leucemia linfatica cronica (B)
Linfomi follicolari (B)
Linfoma mantellare (B)
Linfoma extranodale (B)
Lnfoma diffuso a grandi cellule (B)
Linfoma di Burkitt (B)
Micosi fungoide/sindrome di Sézary (T)
Linfoma dei linfociti periferici (T)
La presentazione e la varieta’ dei sintomi e’ simile al linfoma di Hodgkin
La caratterizzazione cellulare e della alterazione cromosomica/danno
molecolare permette il miglior trattamento delle varie forme
LH vs LNH
Linfoma di Hodgkin
Linfoma non-Hodgkin
• Generalmente circoscritto a
un singolo gruppo assiale di
linfonodi
• Diffusione sviluppata per
contiguità
• Noduli mesenterici e anello
di Waldeyer raramente
coinvolti
• Interessamento extranodale
raro
• Più frequente
interessamento di
molteplici linfonodi
peeriferici
• Diffusione non sviluppata
per contiguità
• Anello di Waldeyer e noduli
mesenterici
frequentemente coinvolti
• Interessamento extranodale
comune
Discrasie delle Plasmacellule
Mieloma Multiplo
Mieloma Multiplo
Il mieloma multiplo e’ la proliferazione maligna di plasmacellule, probabilmente
provocata da un eccesso di produzione di IL-6
Le cellule del mieloma overproducono il loro specifo monoclone di Ig
I criteri richiesti per la diagnosi di mieloma multiplo sono (almeno 2 dei 3):
Presenza di paraproteine (banda monoclonale) nel siero e/o urine
Aumento del numero di plasmacellule anormali nel midollo (>20%) o con provata
monoclonalita’ delle plasmacellule (>12% con un unico tipo di catena leggera)
Lesioni osteolitiche delle ossa
I soggetti con mieloma multiplo presentano comunemente:
infezioni ricorrenti a causa dello stato immunosoppressivo (diminuzione delle
immunoglobuline normali)
Ipercalcemia
Danno renale per la ipercalcemia e/o la deposizione di paraproteine
Rottura patologica delle ossa
Neutropenia ed anemia
Si può riscontrare aumento della viscosità del sangue per aumento della quantità
di proteine con agglomerati di emazie, alterazioni piastriniche ed aumento della
VES
Elettroforesi delle proteine sieriche
Elettroforesi delle sieroproteine normale
Presenza di banda monoclonale nel siero
Mieloma Multiplo
Striscio midollare di mieloma multipo da puntato
sternale o della cresta iliaca.
L’infiltrazione del midollo osseo da parte dei plasmociti
giovani di grandi dimensioni con anomalie nucleari o
citoplasmatiche. Questi impediscono lo sviluppo di altri
precursori ematici, con la conseguenza di anemia,
neutropenia, piastrinopenia
I plasmociti producono un fattore stimolante gli
osteoclasti
con
conseguenti
lesioni
litiche,
osteoporosi, ipercalcemia, danno renale
Elettroforesi ed immunoelettroforesi di siero ed urine.
Si riscontra banda monoclonale data da Ig
monoclonale isolata, IgG k (caso piu’ frequente,
seguito da IgA, IgM raro, IgD rarissimo, IgE
eccezionale).
Le catene leggere delle Ig monoclonali possono
ritrovarsi nelle urine come proteine di Bence-Jones
La catena leggera isolata è presente nel 20% dei
mielomi
Downloaded from: StudentConsult (on 30 January 2011 10:47 AM)
© 2005 Elsevier
Mieloma Multiplo - Terapia
• L’approccio terapeutico prevede radio e chemioterapia
• La sopravvivenza a 5 anni con la sola chemioterapia e del 20%
• Il trapianto di midollo sta diventando la terapia di elezione in
particolare il trapianto autologo
• Altre chemioterapie basata su fattori che potrebbero controllare la
crescita delle plasmacellule (anti-IL-6 etc) sono ancora in fase
sperimentale