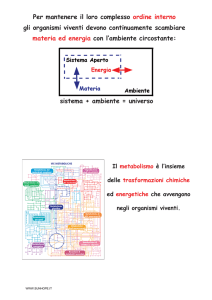
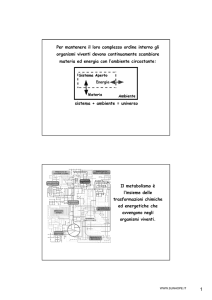
SBOBINATURA della PRIMA LEZIONE di EMATOLOGIA
Prof. Guastafierro – 07 marzo 2014
Voi sapete che l’insegnamento dell’ Ematologia, assieme a quello dell’ Oncologia Clinica,
fa parte della Patologia Integrata V, quindi sapete bene che esistono questi esami di
patologia integrata, in cui non è possibile prendere 30 in uno di questi esami e meno di 18
nell’altro, altrimenti poi bisogna ripetere tutto. Quindi, vi prego, venite a fare l’esame
quando vi sentite veramente sicuri, non venite a tentare! Allora, vediamo come
l’ematologia, come le altre branche della medicina, è una disciplina che comprende diverse
patologie, ma noi dobbiamo considerare le malattie dei pazienti. Quindi, il nostro primo
obiettivo è quello di saperci approcciare al paziente; quindi dobbiamo sapere come costruire
un iter diagnostico che ci possa portare, con un’alta probabilità, ad una diagnosi
possibilmente precisa. Quindi, i vari step di questo iter, sono quelli che voi già conoscete,
così come è per tutte le altre branche della medicina. In primo punto, l’ ANAMNESI :
l’anamnesi fatta per bene ci dice tante cose ; l’ ESAME OBIETTIVO ; gli ESAMI
SEMPLICI DI LABORATORIO ( è inutile che noi chiediamo esami complessi, di
secondo/terzo livello, che ci portano fuori strada e non ci consentono nemmeno di fare la
diagnosi); il quarto punto è un altro punto molto importante: vedete, quando voi venite agli
esami, una delle domande che io pongo è sempre “ Ma il paziente che ha tale malattia,
perché potrebbe essere spinto ad andare dal medico?” . Cioè, questo che cosa vuole dire?
Che voi abbiate la conoscenza della modalità di presentazione della malattia, perché una
cosa è che voi vi avviciniate per la prima volta a un quadro clinico e lo studiate
sistematicamente nella sua interezza ( troverete una serie di segni, sintomi, etc.etc.)… però,
nella realtà clinica, voi ne troverete soltanto alcuni, e allora dovete sapere, innanzitutto, la
malattia come si presenta, che cos’è che è importante, che cos’è che vedete ! Allora,
secondo questi 4 steps, cominciamo proprio dalle particolarità dell’ ANAMNESI.
Innanzitutto l’anamnesi è familiare, deve essere precisa, soprattutto se pensiamo alla
possibilità di una malattia ereditaria – quanti di voi avranno avuto modo di sentire “ Bè,
qualcuno mi ha detto che nella mia famiglia c’è l’anemia mediterranea”… guardate, ragazzi,
WWW.SUNHOPE.IT
che noi in Italia abbiamo 3 milioni di talassemici, per fortuna la stragrande maggioranza
sono eterozigoti, ma è importante già sentirsi dire questo. Oppure, se ci troviamo davanti a
un paziente che ha un subittero sclerale - un giovane che, peraltro, sta bene - bene, pensiamo
anche alla possibilità di una sferocitosi ereditaria. Oppure, nel caso in cui un soggetto
giovane, di sesso maschile, ha una crisi emolitica violenta dopo aver mangiato delle fave o
dei piselli , poi scopriamo che la madre era portatrice di un’eredità di tipo diaginico ( madre
portatrice e figlio maschio affetto). Quindi vedete come l’anamnesi familiare già può
indirizzarci su una determinata strada, e gli esempi sono vari… Anamnesi patologica
remota: è importante sapere se il paziente è stato sottoposto ad interventi chirurgici e se ci
sono stati dei problemi; a volte i pazienti che non sono al corrente, arrivano in pronto
soccorso (ad esempio, in seguito ad un incidente, a un evento traumatico) e devono essere
operati d’urgenza e gli esami di primo livello non ci danno sufficienti informazioni ( sembra
tutto normale) ; oppure ancora, un paziente che ha un’ emofilia c, per cui è deficitario del
fattore XI della coagulazione ( trasmissione, in questo caso, autosomica recessiva) ma ha un
quadro clinico dubbio, non si capisce da dove viene fuori questo sanguinamento
prolungato… c’è qualche dubbio e ciò ci invita a chiedere al nostro paziente se ha subito
interventi chirurgici e se ci sono state eventuali complicanze. E’importante sapere, in caso
di tumore, se è stato trattato con chemioterapia oppure con che mio/radioterapia e se vi sono
state neoplasie precedenti. Facciamo un esempio: un giovane affetto da linfoma di Hodgkin
( voi sapete, naturalmente, che il linfoma di Hodgkin ha una prognosi buona, l’80 % dei
pazienti guarisce). Ora si è visto che questi pazienti, trattati con protocolli di chemioterapia
oppure chemio + radioterapia, rispetto alla popolazione generale, hanno un rischio maggiore
di andare incontro ad una leucemia mieloide acuta, oppure a una sindrome mielodisplastica.
Malattie infettive pregresse: vedete, voi potreste visitare un paziente e trovate una
splenomegalia. Non disdegnate la possibilità di una splenomegalia, ma dovendo procedere
in ordine razionale, cominciamo a valutare la possibilità di malattie infettive. Ne sanno
qualcosa i pazienti che hanno avuto una leishmaniosi, pazienti che oggi ( ma è una cosa che
vedremo di più nei decenni successivi) che vengono da aree geografiche in cui è ancora
endemica la malaria… pazienti che anche essendo guariti possono aver residuato una
splenomegalia. Anamnesi patologica prossima: delle patologie microproliferative è
importante sapere se il paziente che viene alla nostra osservazione, nel caso in cui ha avuto
un’ epatomegalia, se ha una febbre - per caso di sera, di pomeriggio – superiore a 38 gradi,
poi scompare autonomamente senza prendere nessun antipiretico ; se il paziente ha profuse
sudorazioni notturne ; se ha un calo ponderale. Guardate, anche nel caso del calo ponderale,
bisogna stare molto attenti. Non dobbiamo pensare al calo ponderale come valore assoluto,
e vi dico perché in maniera molto semplice: se viene da noi una paziente che pesa 50 kg (
qualcuno in sottofondo comincia a fischiettare) che ha perso ,negli ultimi 6 mesi, 6 kg, è un
discorso; se invece viene un uomo di 100 kg che ha perso 6 kg ( sempre 6 kg sono !), ha un
significato totalmente diverso perché nell’ultimo caso sarebbe il 6% dell’intero peso
corporeo, mentre invece 6 kg su 50 sarebbero il 12 % e, guardate, noi dobbiamo porre
un’attenzione particolare a quel calo ponderale che è superiore o uguale al 10 % del peso
corporeo in un intervallo soprattutto limitato agli ultimi 6 mesi. Il paziente o la paziente che
vi vengono a raccontare “ Sì, io sono dimagrito”. “Quanto ha perso?” “Ho perso 2 kg” , bè,
insomma, voi non dovete interpretare in senso assoluto. Ridotta capacità di esercizio,
dispnea, cardiopalmo : cosa vi richiamano? Ah, un’altra cosa volevo dirvi… Tenete conto
che ogni paziente è un vuoto (?!?) a sé: ogni paziente ha un suo modo di esprimersi
variabilissimo, a seconda del proprio livello culturale. Noi dobbiamo avere la capacità di
WWW.SUNHOPE.IT
interpretare quello che il paziente vuole dirci. Vedete, tutto questo comporta una cosa: che
noi non dobbiamo essere frettolosi, dobbiamo discutere, colloquiare con il nostro paziente,
perdere del tempo con lui ! Allora, ridotta capacità di esercizio/dispnea/cardiopalmo: c’è la
donna che vi racconterà lei “ Appena faccio qualche servizio in più in casa, mi sento subito
stanca” oppure un uomo che vi dirà “ Io per tutta la vita ho fatto l’autotrasportatore, adesso
non ce la faccio più a scaricare nemmeno pesi superiori a 10 kg circa” (che a noi sembrano
chissà che, però, in rapporto alle sue abitudini, il paziente avvertiva una limitazione);
dispnea: nessun paziente ci verrà a dire “ Dottore, ho la dispnea!”… Il paziente ci dirà “ Ho
il sovraffiato, mi manca il respiro” ; oppure il cardiopalmo: cioè, in effetti, quella che
obiettivamente è una tachicardia, il paziente me la esprimerà come “ Ho il cuore in gola, mi
sento sbattere in petto” e la nostra bravura sarà quella di capire ciò che il paziente ci sta
dicendo. Vedete, ridotta capacità di esercizio, dispnea e cardiopalmo fanno parte di che
cosa? Di un insieme di segni e sintomi che ci richiamano che cosa? ( Si deve accendere una
lampadina nella nostra mente…) Uno stato anemico! Benissimo!! Stomatiti ricorrenti e
infezioni ricorrenti: il paziente ci verrà a raccontare “ Bè io ho notato che da diverso tempo
ho delle afte, delle stomatiti etc.etc.” e questo è importante perche potrebbe suggerirci che
cosa? Un immunodeficit, molto spesso acquisito ; Emorragie spontanee o provocate: il
paziente ci potrà raccontare una storia di epistassi oppure sanguinamenti abbondanti dopo
un taglio superficiale. State tranquilli che mai nessun paziente entrerà nel vostro studio
dicendo “ Dottore, ho una splenomegalia (o un’ epatomegalia)”. Ognuno racconterà la cosa
in rapporto al suo vissuto; c’è il paziente che vi dirà “ Ho un senso di peso” riferendosi
ovviamente all’ipocondrio sinistro oppure al destro; qualcuno vi dirà “ Io appena comincio a
mangiare subito sono pieno”. E’ chiaro: una sensazione di sazietà precoce che è legata ad un
ingrossamento della milza che comprime lo stomaco e quindi dà questa sensazione di
sazietà precoce. Sintomatologia neurologica: c’è il paziente che vorrà raccontarci di
un’alterazione della sensibilità, ci farà un racconto di una storia di parestesie, di quella che
noi chiamiamo ipostenia e che il paziente ci dice “ Ho una riduzione della forza muscolare
nel braccio sinistro, nel braccio destro etc.etc.” Guardate, allora, noi abbiamo delle malattie
patologiche in cui la sintomatologia neurologica è importante (basta pensare al deficit di
vitamina B12, o anche, per esempio al mielosa, in cui per effetto del crollo vertebrale e delle
lesioni osteolitiche , viene fuori una sintomatologia neurologica che può essere talora
drammatica, con dei dolori violenti). E’ importante conoscere se il paziente ha allergie
(allergie a farmaci, ad alimenti). Quindi questa è una cosa importante anche in rapporto ad
un’eventuale terapia, che dobbiamo instaurare. Il paziente che ha un’allergia alle betalattamine ce lo dice , e questa è una cosa importante che dobbiamo tenere presente;
dobbiamo cercare di capire cosa è successo… Il paziente ci racconterà “ Ho fatto
un’iniezione” oppure “ho preso dei farmaci” “Che farmaci?” Tenete presente che il paziente
non ricorderà mai il principio attivo del farmaco che ha preso, ricorderà il nome
commerciale, e spetterà a voi capire di cosa si tratta. Trasfusioni: il paziente che ha una
epato-splenomegalia, è un paziente che potrebbe anche aver, in passato, avuto delle
trasfusioni ed è diventato poi un paziente cronico per, per esempio, un’epatite cronica da
virus C ; oppure un paziente politrasfuso, che a casa (o a caso…boh !!!) viene nuovamente
trasfuso, e in occasione dell’ultima trasfusione, dopo alcuni giorni, presenta un subittero
sclerale. Valutiamo il paziente, gli facciamo fare un esame emocromocitometrico, non
abbiamo avuto l’incremento di emoglobina atteso, e, alla fine, andando avanti nell’iter
diagnostico, scopriamo che il paziente ha avuto “una reazione trasfusionale emolitica
ritardata” da incompatibilità di sistemi gruppo-ematici diversi dall’ AB0 e dall’ Rh e di cui
WWW.SUNHOPE.IT
poi parleremo a suo tempo. Etilismo: bene, se voi vedete l’emocromo di un etilista, il
sospetto vi potrebbe anche venire perché il paziente etilista ha una tendenza alla
macrocitosi, cioè tende ad avere delle emazie un poco più grandi della norma. Questo
perché ? Perché l’etilista è il paziente che ha una riduzione delle scorte di acido folico. Se
un paziente ha uno stato anemico con emolisi dopo che è tornato da un viaggio, dopo un
soggiorno all’estero in zone in cui alcune malattie sono endemiche, allora questo per noi è
importante per farci collegare questo soggiorno in quella determinata area geografica e quali
sono le malattie tipiche di quella zona. Passiamo al terzo step del nostro iter diagnostico,
cioè l’ ESAME OBIETTIVO. Cominciamo dalla cute : la cute, ovviamente , può essere
pallida, cianotica, può essere itterica e ciascuno di questi aspetti ci indica e ci indirizza in
modo diverso ; oppure, se sono presenti delle petecchie. Sapete cosa sono le petecchie?
Ragazzi, ditelo, perché noi qui non stiamo facendo gli esami, non abbiate timore di
sbagliare!!! Su, le petecchie cosa sono? (Risposta di sottofondo da parte di qc. delle prime
file : “delle macchioline”) Obiettivamente, si notano, come tu hai detto, come delle
macchioline di colorito rossiccio, ma sono espressione di che cosa? Sì, esatto! Sono delle
emorragie capillari! Queste petecchie hanno un diametro di 1-2 mm. E dove le dobbiamo
ricercare? Essenzialmente nelle parti declivi, quindi a livello delle caviglie o sotto gli occhi.
Ma dobbiamo però esaminare il paziente nella sua interezza; potrebbe avere delle petecchie
anche a livello della superficie addominale o sulla superficie volare degli avambracci.
Teleangectasie, oppure lesioni da grattamento: il paziente può avere prurito che è
giustificato da determinate lesioni cutanee. Guardando il paziente negli occhi cosa possiamo
vedere? La prima cosa, a livello sclerale – per valutare se un paziente è anemico o meno, la
prima cosa che facevano i vecchi medici era guardare le mucose, soprattutto quindi le
sclere, e rendersi conto se quel colorito era pallido oppure se più che pallore vi era una
sfumatura giallastra, allora, ovviamente, ci indirizza verso una iperbilirubinemia, voi sapete
benissimo che c’è un rapporto tra il livello di bilirubina, la concentrazione di bilirubina
ematica e il subittero sclerale o, addirittura, la colorazione itterica della (mucosa ?!?) e
quindi ittero franco. Emorragie congiuntivali o retiniche: vedete, in determinate malattie
l’esame del fondo oculare ci può dimostrare la presenza di emorragie, per esempio in caso
di una severissima piastrinopenia o nel corso di una leucemia acuta. Ectasia dei vasi retinici:
un paziente con macroglobulinemia di Waldenstrom presenta una particolare (controttusità
?!?) dei vasi retinici . Una stomatite: un paziente, dopo che ha fatto una chemioterapia ,
soprattutto dopo 7-8 giorni, 10 giorni, può presentare delle lesioni, delle ulcerazioni
necrotiche; oppure un bambino che ha una porpora trombocitopenica idiopatica ( Morbo di
White ?!?), cioè una patologia autommune che può presentarsi, in età pediatrica, dopo 2-4
settimane da un’infezione perlopiù virale. Bene, questi bambini hanno dei tempi di piastrina
così bassi da avere non solo delle manifestazioni cutanee ma addirittura possono formarsi
delle bolle emorragiche, cioè delle bolle ripiene di sangue. Infiltrazioni gengivali: in
particolari tipi di leucemie, in particolare la leucemia mieloide acuta M5 cioè la leucemia
monoblastica, è frequente l’infiltrazione gengivale, cioè i blasti di questa variante di
leucemia mieloide acuta vanno ad infiltrarsi anche a livello delle gengive. Adesso, un punto
molto importante nella valutazione dell’esame obiettivo del paziente, è la valutazione delle
stazioni linfonodali. Ragazzi, la stragrande maggioranza dei pazienti che verrà alla vostra
osservazione per una linfadenomegalia, ha patologie gravi, e solo una percentuale
bassissima avrà invece una malattia linfoproliferativa . Allora, di fronte ad un paziente con
linfoadenomegalia, dobbiamo escludere determinate malattie, in primis determinate malattie
infettive. Allora, vediamo insieme quali malattie infettive possono accompagnarsi ad una
WWW.SUNHOPE.IT
linfoadenomegalia. Cominciamo dalle malattie virali: (la parotite? Eh no, la parotite no,
perché in realtà vi è un ingrossamento della parotide che crea turgore ) la mononucleosi,
infezione da virus di Epstein Barr, oppure, altra malattia virale, il Citomegalovirus… bene!!
(Studente: “ l’ Herpesvirus) …l’ Herpesvirus, bè , sì, può essere! (Studentessa: “ la
Rosolia”) … la rosolia, benissimo!! (Studente senza farsi però sentire dal prof. : “ il
Morbillo”)… Ragazzi, ma come… l’ HIV. Poi vediamo un po’ quelle di origine non virale:
( lo stesso insospettabile studente di prima : “ la Leishmaniosi”) Ecco, la linfadenopatia
tubercolare… Ragazzi, non pensate che la tubercolosi sia scomparsa, eh! Questo era
l’obiettivo che si era posta l’ OMS all’inizio degli anni ’80, cioè appunto di eradicare la
tubercolosi dall’Europa Occidentale e dal Nord America. Però all’inizio degli anni ’80 non
si erano ancora scoperti due fattori: la (prevenzione?!?) di massa in zone in cui la
tubercolosi era ancora frequente e contemporaneamente lo smantellamento della nostra rete
di sorveglianza e, ovviamente, il persistere poi dell’impossibilità di (guardare?!?) le lesioni
tubercolari con lo sviluppo di resistenze verso i classici criteri di prevenzione delle infezioni
tubercolari. Quindi considerate che, ecco, voi vi troverete, quando sarete ,tra qualche anno,
sul campo, anche di fronte a pazienti che non sono regolari, che non sono registrati, che non
si rivolgono ai medici per paura e quindi sfuggono ad ogni perizia. Quindi la tubercolosi è
una malattia che c’è ancora. Poi ancora, dicevamo, ancora un’altra linfoadenomegalia da
non dimenticare è quella da Toxoplasma gondii, la toxoplasmosi. Ora vabbè, una volta che
abbiamo tenuto conto più o meno di queste situazioni valutiamo obiettivamente questi
linfonodi. Voi, naturalmente, capite che non possiamo valutare le stazioni linfonodali
profonde, ma soltanto le stazioni linfonodali superficiali. Quali sono le stazioni linfonodali
superficiali che noi possiamo valutare? ( Allora, qui è partito un coro di voci all’unisono
protrattosi per qualche minuto, riassumo un po’ la cosa sinnò facimm nott !) In senso
cranio-caudale: postoccipitali, retroauricolari, cervicali posteriori, angolomandibolari,
sottomandibolari, laterocervicali ( situate sulla superficie dello sterno-cleido-mastoideo),
sovraclaveari, ascellari, linfonodo epitrocleare, inguinali, poplitei. Bene, che cosa dobbiamo
valutare di questi linfonodi? Ragazzi, un poco di silenzio per favore!! ( Fine del vocìo
iniziato qualche minuto addietro…). Guardate un paziente che ha la linfoadenomegalia; la
prima cosa, prima di andarlo a visitare, che facciamo cos’è ? Guardiamo se la cute è
arrossata o meno, se vi è una flogosi sovrapposta. La seconda cosa, cosa chiedete al
paziente? Se il paziente vi dice “ Non mi fa male” oppure “ Mi fa male” , voi chiedete anche
“ Ma spontaneamente fa un po’ male?” . Poi a questo punto cosa dovete stabilire ? La
mobilità, benissimo ! Dobbiamo valutare se il nostro linfonodo è spostato rispetto ai piani
superficiali oppure no; ma poi dobbiamo valutare anche la consistenza, noi possiamo avere
un linfonodo leggermente aumentato di consistenza. Un’altra cosa è se abbiamo un
linfonodo duro, ligneo, che ha un significato totalmente diverso. Ci rimane poi da valutare
un’ultima cosa: se questi linfonodi sono isolati o hanno la tendenza a riunirsi insieme, a
formare, tecnicamente, gruppi. Valutiamo poi la “ simmetricità” (voci di sottofondo: “ si
dice ‘simmetria’ !!”) . Queste informazioni che ci vengono fuori dall’esame obiettivo
devono indurci a pensare. Bene, questa è un’immagine schematica (cfr. slides) che ci fa
vedere , nel collo, come si dispongono le varie stazioni linfonodali che abbiamo già detto.
All’esame obiettivo del torace che cosa dobbiamo chiedere? Cosa dobbiamo vedere?
Innanzitutto se vi sono dei versamenti pleurici. Non è la prima volta che un linfoma può
associarsi anche a versamenti pleurici. Inoltre, la digitopressione sullo sterno, oppure la
digitopressione delle costole, o anche delle vertebre dorsali; il paziente che ha un mielosa, in
cui vi è una proliferazione del midollo osseo soprattutto delle ossa “corte e chiatte”, una
WWW.SUNHOPE.IT
proliferazione di plasmacellule atipiche, si hanno delle erosioni a stampo dello scheletro, di
queste strutture scheletriche. E quindi, la digitopressione, in prossimità di una di esse, può
provocare dolore. Fegato e milza: dobbiamo precisare non solo di quanto è aumentato
volumetricamente uno di questi organi, ma precisarne più o meno le caratteristiche.
Ragazzi, vedete che in ematologia abbiamo alcune malattie in cui la splenomegalia è
veramente notevole, il polo inferiore della milza può arrivare addirittura nella fossa iliaca
sinistra, cioè va quasi a occupare metà dell’addome. Questo è quello che si può osservare in
pazienti con mielofibrosi idiomatica, per esempio. Apparato osteo-articolare e
malformazioni scheletriche: bè, non so se qualcuno di voi ha avuto modo di vedere, o
almeno da qualche immagine, qualche bambino affetto da Morbo di Cooley. Il Morbo di
Cooley sapete che è una talassemia, la beta-talassemia maior e che è caratterizzato da che
cosa? Da un’anemia severa, ma allo stesso tempo questi bambini sviluppano delle deformità
ossee per una espansione del midollo eritropoietico in aree ossee dove non vi dovrebbe
essere midollo eritropoietico, quindi con assottigliamento dei tavolati. Allora questi bambini
presenteranno una sporgenza degli zigomi, un appiattimento della radice del naso,
prognatismo. Come pure alterazioni scheletriche sono presenti nei bambini e nei pazienti
che hanno una eritroblastopenia congenita. L’esame neurologico: l’esame obiettivo
neurologico è importante e focalizzeremo una maggiore attenzione quando parleremo del
paziente con deficit di vitamina B12 oppure del paziente con mielosa.Anche, per esempio, il
paziente con una leucemia acuta linfoblastica in cui possiamo avere una infiltrazione
neoplastica del liquor o del sistema nervoso centrale, delle meningi. Quindi, bisogna
conoscere e saper eseguire le manovre fondamentali dell’esame obiettivo neurologico.
Vediamo un poco, adesso, gli esami di laboratorio di base, che hanno però perso gran parte
della loro importanza, ma tuttavia, anche nelle malattie ematologiche, qualche informazione
ce la potrebbero dare. Innanzitutto, a prescindere da condizioni che non dipendono dallo
stato patologico, una VES eccessivamente bassa (3-4) viene riscontrata in pazienti che
hanno una poliglobulia; mentre invece, all’opposto, un paziente anemico, ha una VES
particolarmente alta. Ma la VES è influenzata, quindi, non soltanto dalla massa eritrocitaria,
ma è influenzata anche dal rapporto tra albumina e globulina. Più si abbassa questo rapporto
( e più aumentano le globuline), maggiore sarà la velocità di eritrosedimentazione, quello
che per esempio noi osserviamo in un paziente con mielosa in cui è presente una
componente monoclonale vistosa che è alla base di questo valore particolarmente alto.
Veniamo un attimo all’esame emocromocitometrico: allora, oggi, con i contaglobuli
automatici noi abbiamo una serie di informazioni notevoli. La prima cosa è che nessuno
giudicherà uno stato anemico sulla base del numero dei globuli rossi. Prima abbiamo detto
che la maggioranza dei pazienti talassemici, in Italia, la stragrande maggioranza, è
eterozigote, cioè significa soggetti che hanno una forma (falciata ?!?), più frequentemente la
beta-2 (??) . Bene, questi pazienti possono essere dei pazienti totalmente asintomatici che
addirittura possono non essere anemici o avere un’anemia di grado lieve. Quindi questo
perché ? Perché i globuli rossi sono più piccoli. O una donna che ha un’anemia sideropenica
che può essere anche importante, 8 grammi di emoglobina per decilitro, oppure 7 grammi e
poi avete un numero di globuli rossi di 4.000.000 / 3.700.000… Il numero non ci dice
niente, non ci consente di giudicare uno stato anemico. Vedete, quello che vi voglio
raccontare è non solo di non dirlo all’esame, ma , nella vostra pratica, è il parametro che,
all’emocromo, trascurate, giudicate con minore attendibilità. Lo potete trascurare perché vi
dice poco o niente. Invece, che cos’è che ci dà informazioni? La concentrazione di
emoglobina. Noi solitamente esprimiamo la concentrazione di emoglobina in grammi per
WWW.SUNHOPE.IT
decilitro ( g/dl). La concentrazione dell’emoglobina è un fedele indicatore dello stato
anemico in tutte le condizioni cliniche tranne una, anzi due : la prima condizione è la donna
in stato di gravidanza. Nella donna in stato di gravidanza, per un effetto di retroinibizione, il
valore è più basso. Per esempio se noi valutiamo l’esame emocromocitometrico di una
donna in stato di gravidanza, al settimo mese, e vediamo che la sua emoglobina è 9,5 g/dl,
noi dobbiamo che, per effetto della retroinibizione, la sua condizione reale è migliore di
quello che può esprimere quel 9.5 . Condizione opposta: arriva da voi in pronto soccorso un
politraumatizzato; dopo un incidente, ha avuto un’emorragia. Fatto subito l’emocromo,
supponiamo che il paziente ( di sesso maschile) ha 9 g/dl di emoglobina. Viene operato
d’urgenza; l’intervento va bene, il paziente esce dalla sala operatoria, gli viene fatto un
emocromo di controllo e il paziente scende a 8 g/dl di emoglobina. Se , dopo 3-4 ore, gli
fate di nuovo un esame emocromocitometrico, vedrete che l’emoglobina non sarà più 8, ma
sarà , se è il caso, 5-6 g/dl. Eppure il paziente non ha perso sangue, ma per quale motivo?
Perché quando si ha una perdita acuta, si ha una emoconcentrazione. Quindi questa è una
situazione diametralmente opposta a quella di prima. Quindi, la vera astrazione di queste
due condizioni è che possiamo fidarci sufficientemente della concentrazione
dell’emoglobina che ci darà il nostro valore. Però, una cosa importante: allora, quali sono i
limiti al di sotto dei quali possiamo parlare di anemia? Nel 1968 l’ OMS aveva stabilito che
questi limiti fossero di 13 g/dl per il sesso maschile e 12 per il sesso femminile.Sono passati
da allora più di quarant’anni, quasi cinquant’anni e questi valori, bene o male, sono ancora
accettabili.Tenendo conto però di che cosa? Di alcune differenze razziali e tenendo conto
anche che, grosso modo, sono valori che possono essere utilizzati per un confronto nelle
varie parti del mondo. Quindi, è un riferimento. E’ vero che oggi alcuni autori parlano anche
di anemia al di sotto di 11,5-12 g/dl ma, grosso modo, tra 11,5 e 12 non è che la cosa cambi
notevolmente.Quello che è importante è comunque che un’anemia è un’anemia di grado
lieve se l’emoglobina è compresa fra tali valori e 10 g/dl; tra 8 e 10 è di grado intermedio o
moderato; al di sotto di 8 g/dl è invece di grado severo. Una precisazione che voglio farvi a
questo riguardo è di non confondere l’anemia severa con l’anemia acuta.Cioè, cosa voglio
dire? Il termine “anemia acuta” che cosa sta a indicare ? Bravo ! ( riferito non si capisce a
chi…) Un concetto temporale ! Cioè, io posso avere un’anemia acuta però di grado
moderato, cioè un paziente che ha avuto un’emorragia e passa nel giro di poche ore da 14
g/dl di emoglobina a 9 g/dl di emoglobina (??). Questo paziente è un paziente che ha
un’anemia intermedia, però quella è un’anemia acuta. Se invece consideriamo una donna
che , per effetto di mestruazioni, flussi mestruali abbondanti, si è anemizzata lentamente nel
tempo, questa donna può arrivare nel nostro ambulatorio con 7 g/dl di emoglobina, quindi
avere un’anemia di grado severo… ma quella non è un’anemia acuta ! E se noi non
bolliamo il paziente che ha avuto un’anemia acuta rispetto a quello che ha avuto un’anemia
cronica, anche se di grado maggiore, guardate che quel paziente che ha avuto un’anemia
acuta clinicamente sta peggio, perché non c’è stato il tempo che i meccanismi omeostatici si
siano messi in moto. Una volta che noi abbiamo stabilito che è presente uno stato anemico,
abbiamo stabilito il grado dell’anemia, noi dobbiamo valutare, fra i parametri eritrocitari (
che non sono dei numeri che il contaglobuli ci dà semplicemente per caso, ma per noi sono
importanti…) , il volume corpuscolare medio. Che cosa rappresenta ? Rappresenta
sostanzialmente la media dei volumi della nostra popolazione eritrocitaria.Cioè noi non
abbiamo che il nostro midollo produce dei globuli rossi, degli eritrociti che sono standard,
cioè tutti della stessa grandezza, ma alcuni un po’ più piccoli, altri un po’ più grandi. Cioè il
contaglobuli che fa? Ci fornisce un valore che è la media di questi valori. Noi sappiamo che
WWW.SUNHOPE.IT
i volumi globulari di riferimento sono compresi più o meno tra 80 e 100 ( o secondo altri
autori tra 82 e 98) femtolitri, o micron cubi , che è la stessa cosa. Allora è chiaro che voi, se
avete un’anemia e avete stabilito che è un’anemia di grado intermedio, e poi vedete che il
volume corpuscolare medio è piccolo, vedete che quell’anemia di grado intermedio è
microcitica. Se invece il volume corpuscolare medio è 110 femtolitri, vedete che quella è
un’anemia macrocitica. E se invece è di 87 micron cubi, è normocitica. Ma sapere questo
già ci indirizza verso un determinato ambito, già vi fa sfogliare fra un diverso numero di
anemie, vi indirizza già, vi consente di costruire il vostro iter diagnostico. Allora voi già
sapete che se è presente un volume corpuscolare ridotto, dove dovete focalizzare la vostra
attenzione e la vostra possibilità ? Per esempio, l’anemia microcitica qual è ? ( Tutti gli
studenti in coro: “ l’ anemia sideropenica”) L’ anemia sideropenica, o anche il paziente
portatore di tratto talassemico. Allora già avete cominciato la vostra diagnosi ! L’ MCH: l’
MCH che cosa rappresenta? E’ il parametro eritrocitario che ci dice quanta emoglobina si
trova mediamente in un globulo rosso come quantità espressa in peso. In realtà noi
sappiamo che l’intervallo di riferimento è tra 27 e 31-32 picogrammi. Bè in realtà è come se
noi avessimo contato tutta l’emoglobina presente in ciascun globulo rosso, avessimo fatto la
media ed è venuto fuori questo valore, quindi “contenuto emoglobinico medio” . Quando
non avevamo i contaglobuli automatici, l’unica nostra possibilità era guardarci questi
globuli rossi al microscopio… e il globulo rosso al microscopio come appare? Appare di
forma rotondeggiante, con una parte più rosa perifericamente e una parte chiara
centralmente. Questo perché? Perché, voi sapete, che il globulo rosso tridimensionalmente
ha una forma biconcava, più stretto al centro, quindi perciò passa più luce e noi lo vediamo
più chiaro. Quindi quando i medici di una volta vedevano al microscopio le emazie di un
soggetto con, per esempio, un’anemia sideropenica, vedevano questi globuli rossi come ?
Meno colorati , con quella zona chiara più ampia, e parlavano di “ ipocromia”.Noi sappiamo
oggi che la ipocromia corrisponde ad un MCH al di sotto dei parametri, dei valori che noi
abbiamo detto.Se invece l’intervallo dei valori di riferimento del nostro paziente è del valore
di 28-29 picogrammi, allora la nostra anemia è normocromica. Allora, vedete, a questo
punto abbiamo aggiunto un altro tassello : se abbiamo un’anemia di grado severo,
ipocromica e microcitica, già possiamo orientarci in una certa maniera; se invece l’anemia è
normocromica, normocitica etc.etc. , ci direzioniamo in un’altra maniera. L’ MCHC che
cosa sta ad indicare? E’ un altro parametro, che è l’ emoglobina corpuscolare media
espressa percentualmente. Cioè la quantità di emoglobina, all’interno del globulo rosso,
espressa percentualmente. Vedete, questo parametro ha perso un poco il suo valore, e oggi i
risultati vengono espressi in percento, ma anche in g/dl. Ma quello che è importante è sapere
che valori oltre 36 % , 37 % massimo, li troviamo soltanto in un numero limitato di
patologie, soprattutto nell’anemia sferocitica. Ma , in tutti gli altri casi, guardiamo
l’emocromo e troviamo un MCHC di 40; abbiamo escluso che il paziente abbia un’anemia
emolitica. Ragazzi, o il contaglobuli non è settato bene, oppure c’è qualcosa che interferisce.
Per esempio il paziente ha una crioalbumina o delle crioglobuline che alterano la
determinazione. L’indice di distribuzione volumetrica dei globuli rossi (RDW) : in realtà,
che cosa sta ad indicare? Io vi ho detto , quando abbiamo parlato del volume corpuscolare
medio, che il nostro midollo non produce globuli rossi di dimensioni standard, ma vi sono
globuli rossi un poco più piccoli e globuli rossi un poco più grandi. Quando non avevamo i
contaglobuli automatici, al microscopio vedevamo questi globuli rossi in due dimensioni e
potevamo valutare un aspetto bidimensionale dei globuli rossi. Allora in base a a che cosa
potevamo valutare se la nostra popolazione era (autogenica ??) ? In base al diametro dei
WWW.SUNHOPE.IT
globuli rossi, e se i diametri erano molto diversi, dicevamo “ anisocitosi”. Oggi quando
diciamo “ anisocitosi” , vogliamo dire “ un’alterazione dell’ RDW” cioè dell’indice di
distribuzione volumetrica, perché i contaglobuli ci indicano la variabilità di volume dei
globuli rossi. Allora noi sappiamo che se troviamo dei globuli rossi che anche se
volumetricamente sono più piccoli, ma che hanno una variabilità notevole tra di loro, questo
ci indirizza ad una determinata malattia. Per esempio, l’anemia microcitica ipocromica
legata ad una iposideremia, ha un RDW ampliato, oltre quel valore di 14-15 che abbiamo
ritenuto normale. Questo ci dice l’espressione di una anisocitosi. Molto spesso poi la
valutazione morfologica microscopica , soprattutto se sono presenti forme strane di globuli
rossi, allora accanto al termine di anisocitosi, ci consente di porre il termine di “
poichilocitosi” . Molto spesso voi troverete nel commento ad un esame
emocromocitometrico “ spiccata, lieve aniso-poichilocitosi” . Il contaglobuli cosa ci dà,
quali informazioni ci dà ? Allora, voi penso che conosciate il numero normale, in un
soggetto, di globuli bianchi. Allò ? Queste sò cose che dovreste sapere!! ( Studente
sicuramente non in prima fila : “ Sono 4.000-10.000 per metro cubo”) 4.000-10.000 per
metro cubo. Ovviamente, voi sapete, che si tratta di almeno 5 popolazioni diverse. In realtà
abbiamo i polimorfonucleati, i neutrofili, i basofili, gli eosinofili e poi abbiamo i monociti e
i linfociti . Voi conoscete i valori di una formula leucocitaria normale ? ( Qualcuno nelle
prime file ha detto qualcosa, ma a voce bassa, sì da non essere percepita dal mio modesto
apparecchio registratore…). Allora, tu hai detto i valori percentuali : 50-70% sono i
neutrofili, gli eosinofili dall’ 1 al 4-5 %, basofili 0-1%, monociti e linfociti ( Guagliù, qua
non si è capito nulla, avete cominciato tutti a parlare all’unisono… comunque, fonti
wikipediche affermano: monociti 2-8 %, linfociti 20-50 %). Non è la prima volta che in
ambulatorio vengono inviati dei pazienti etichettati come “ pazienti con linfocitosi” , che
hanno un 60% di linfociti. Se un paziente ha 3000 globuli bianchi in totale, il 60%
rappresenta una quota di linfociti pari a 1800, quindi un valore perfettamente normale.
Quindi quel nostro paziente non ha una linfocitosi; guardando bene , avrà una mielopenia.
Allora, non vi fidate dei valori percentuali, fate riferimento ai valori assoluti. Molti
contaglobuli mostrano, esprimono anche i valori interni assoluti. Laddove questi valori non
siano forniti, voi, mentalmente, calcolate il percento rispetto al totale. E così eviterete di
incombere in errori che potranno portarvi fuori strada. Il numero totale delle piastrine:
150.000- 400.000 . Ovviamente, non ci fideremo in senso assoluto dei parametri piastrinici,
che sono una nullità diagnostica, hanno una bassa sensibilità quindi non possiamo utilizzarli
così come utilizziamo i parametri eritrocitari . L’esame emocromocitometrico fatto con il
contaglobuli automatico ci dà, ma noi possiamo fidarci sempre ? Allora, se si tratta di un
paziente normale e nei “frames”, nelle segnalazioni che ci vengono fornite dal contaglobuli
non c’è nulla, noi possiamo accettare quel (valore numerico ??); ma , se c’è qualcosa di
alterato nella formula leucocitaria, abbiamo l’obbligo della valutazione microscopica. Quali
sono gli esami che possono essere importanti per la valutazione del paziente ematologico?
In particolare la lattico-deidrogenasi ( LDH) , che voi conoscete in molti isoenzimi. In molte
malattie cardiologiche l’ LDH può essere abnormemente alto. Quando dovete valutare un’
eventuale ipotetica anemia sideropenica, non vi accontentate mai, anche se l’anemia è
un’anemia microcitica ipocromica , non vi accontentate di valutare solo la sideremia. Il vero
stato carenziale è caratterizzato da una iposideremia, una ipertransferrinemia ( e in
particolare aumenta la quota insatura). Vedete, noi possiamo avere dei pazienti che hanno
un’anemia leggermente microcitica, leggermente ipocromica, con una iposideremia, ma che
non hanno un’anemia ferro-carenziale. Andiamo a valutare la transferrina e troviamo una
WWW.SUNHOPE.IT
transferrinemia normale o bassa, una ferritinemia addirittura elevata. Questa è l’anemia “ da
disordine cronico” , cioè da malattia cronica associata; vuol dire che vi è un’altra condizione
patologica associata, di cui la nostra anemia è solo un corollario. Se noi ci accontentiamo di
valutare soltanto la sideremia, al paziente cosa facciamo ? Gli prescriviamo l’eparina e non
abbiamo agito in modo corretto. Voi sapete che il rilascio del ferro è regolato ; la cellula
epatica produce un peptide di 25 amminoacidi che si chiama “ epcidina” . L’ epcidina cosa
fa? Va a legarsi con la transportina ( o ferroportina) provocando una parziale degradazione.
La ferroportina che cos’è ? E’ l’unica proteina in grado di regolare l’export del ferro dalla
cellula digiunale verso il sangue. Se questa proteina è degradata dalla epcidina in eccesso, è
chiaro che il ferro non passa nel sangue e quindi viene eliminato con lo sfaldamento della
cellula intestinale. Ma, ovviamente, come agisce anche l’epcidina ? Agisce anche mediante
una collaborazione con le cellule del reticolo istiocitario. Nelle malattie croniche abbiamo
un aumento della produzione di determinate citochine. Queste citochine possono provocare
un aumento della produzione dell’epcidina. Quindi, da un lato viene bloccato
l’assorbimento a livello della cellula ileale, d’altro lato viene bloccata la liberazione a livello
del reticolo istiocitario. Quindi, nella valutazione dello stato anemico , consideriamo 3
parametri: SIDEREMIA, TRANSFERRINEMIA e URICEMIA. Uricemia: voi sapete
benissimo che l’acido urico è il catabolita ultimo del metabolismo delle purine e sapete
benissimo che dopo aver fatto una chemioterapia , in cui abbiamo prodotto una distruzione
cellulare, abbiamo fatto in modo che vi sia un aumento del catabolismo purinico e quindi
aumenta la produzione di acido urico. Noi dobbiamo evitare che l’acido urico raggiunga dei
valori elevati ( e poi tende a precipitare). Allora, quindi, noi dobbiamo evitare la formazione
dell’acido urico. Noi sappiamo che l’acido urico da cosa deriva? Deriva dalla xantina che a
sua volta deriva dall’ ipoxantina . Questi due ultimi passaggi ( ipoxantina-xantina; xantinaacido urico) vengono tutti e due catalizzati dalla xantina ossidasi . Ora noi, utilizzando
l’allopurinolo, che è il farmaco che inibisce la xantina ossidasi, preveniamo la formazione
dell’acido urico. Ma nel caso in cui l’acido urico è già alto, dobbiamo invece iniziare
l’uricasi che consente di “spaccare” la molecola di acido urico e trasformarla in allantoina
per cui poi i livelli di acido urico alla fine si abbassano e quindi non abbiamo più il pericolo
di una precipitazione.Considerate che l’acido urico elevato è una delle condizioni che si
associa ad altre alterazioni metaboliche che si possono defilare in una sindrome (ipomolare
??), quindi condizioni per le quali bisogna intervenire perché è a rischio la vita del paziente.
Allora, ancora, in un paziente che fa determinate terapie, dobbiamo valutare che cosa? Nel
paziente che fa le chemioterapie ci possono essere malattie onco-ematologiche. Bene, urea e
creatininemia. Urea: qua dovrei fare una precisazione: noi abbiamo, molto spesso abbiamo,
abbiamo molto spesso ancora abbiamo ( sì, è proprio come pensate: il prof. si è incantato, a
mò di cd pezzotto !) un’azione azotemica. Azotemia che cosa significa ? Azoto proteico !
Mentre invece con le valutazioni meccaniche che ci sono oggi , voi cosa dosate? Le ureasi
in realtà vanno a demolire soltanto la molecola dell’urea. Ora, i risultati però potrebbero
essere forniti come urea oppure come azoto proteico, e non significano la stessa cosa. Per
esempio, se io ho un valore di 25 mg/dl di urea, questo è un valore che è normale, ma non lo
è se è di azoto proteico, perché ? Voi vi ricordate la formula di struttura dell’ urea ? Un
carbonio, il doppio legame con l’ossigeno, poi 2 legami con 2 gruppi NH2. Se voi andate a
vedere il peso molecolare dell’intera molecola, è di 60. I due azoto ( 14 + 14) sono 28,
quindi l’azoto vero è soltanto 28/60 dell’intera molecola di urea (fattore di conversione =
2,14). Ancora, l’iperbilirubinemia
WWW.SUNHOPE.IT
: l’iperbilirubinemia coniugata e quella non coniugata.A noi, per la valutazione ematologica,
interessa cosa, fondamentalmente? L’anemia emolitica che cos’è ? Un’anemia
normocromica . Questa bilirubina non coniugata…allora…voi vi ricordate qual è il destino
dei globuli rossi una volta che diventano senescenti, no? La stragrande maggioranza viene
captata dal reticolo istiocitario del torrente splenico e che succede? Il globulo rosso viene
distrutto, l’eme viene separata dalla parte proteica, il gruppo eme viene privato del ferro; la
molecola che rimane è quella della protoporfirina IX che diventa prima, come composto
intermedio, la biliverdina e poi bilirubina . La bilirubina viene veicolata dove ? Al fegato.
Dove? A livello del polo vascolare dell’epatocita. A livello del polo vascolare dell’epatocita
vi sono dei sistemi specifici che fanno in modo che questa bilirubina libera possa passare
nella cellula epatica. Attraverso due step successivi, viene prima legata a una prima
molecola di acido glicuronico e poi a una seconda molecola di acido glicuronico. A questo
punro si è formata la nostra bilirubina che però non ritorna nel sangue, ma viene espulsa
dall’epatocita . Allora, vedete un poco come conoscere queste cose ci può far capire una
serie di condizioni diversissime. Nel caso di anemie extravascolari, quindi distruzione
abnorme di questi globuli rossi a livello del sistema reticolo-istiocitario, si forma
quest’eccesso di bilirubina non coniugata. Ora, la cellula epatica ha la capacità di aumentare
il trasporto attivo verso il polo vascolare di altre 4 volte. Ma quando questa capacità viene
superata che cosa succede ? Che questa bilirubina non coniugata si accumula. Se invece è
presente un ostacolo delle vie biliari, la bilirubina ha un ostacolo, che può essere
rappresentato da una calcolosi oppure una massa che comprime “ab estrinseco” , una
neoplasia della testa del pancreas, che succede? Che la bilirubina coniugata torna nella
cellula epatica dopodichè si riversa attraverso il polo vascolare nel sangue periferico. Quindi
avere una molecola di bilirubina coniugata vuol dire avere che il problema è delle vie biliari.
E nel caso in cui il problema è a livello della cellula epatica, è compromessa la
microcircolazione pertanto possiamo avere tanto un aumento della bilirubina coniugata
quanto di quella non coniugata.Quindi quello che è importante è conoscere il valore della
bilirubina. Ci vediamo la prossima volta!
(Sbobba Made By D.G.C.)
WWW.SUNHOPE.IT
EMATOLOGIA
II LEZIONE 12/03/2014
ANEMIE
Nella scorsa lezione abbiamo introdotto il concetto di anemia. Ora cerchiamo di inquadrare le varie forme di anemia.
Il voler classificare qualunque cosa, schematizzare, incasellare è un elemento quasi distintivo dell’uomo. Anche per le
anemie noi potremmo seguire criteri diversi per fare una classificazione (criteri clinici, criteri epidemiologici);
tuttavia, uno dei criteri più facile da ricordare è il criterio patogenetico, quello che tiene conto del momento
patogenetico più importante di una determinata anemia. È vero che esistono anemie che hanno più momenti
patogenetici, però in tal caso teniamo conto del momento patogenetico più importante.
ANEMIE DEL I GRUPPO
Sulla base di questo criterio noi possiamo distinguere 4 gruppi. Il primo gruppo è caratterizzato da quelle anemie in
cui è difettiva la ERITROBLASTOGENESI, cioè vi è un ridotto apporto di eritroblasti a partire dai progenitori. È chiaro
che se gli eritroblasti sono pochi, anche i globuli rossi saranno scarsi,saranno scarsi anche i globuli rossi giovani,
quindi è un tipo di anemia che si accompagna a reticolocitopenia. Sono anemie per lo più normocromiche e
normocitiche cioè all’ematocrito riscontriamo un MCV normale ( chiede ad uno studente di fare un esempio: 85
μm³ o femtolitri) e un MCH normale ( cioè 29-30 picogrammi). Ovviamente se noi facciamo un esame del midollo
osseo ci rendiamo conto che gli eritroblasti sono molto ridotti. Questa è una caratteristica comune a tutte le anemie
di questo gruppo in cui includiamo le eritroblastopenie e l’anemia da insufficienza renale cronica. Le
eritroblastopenie vanno distinte in eritroblastopenia congenita ed eritroblastopenie acquiste, distinte in acute e
croniche. La eritroblastopenia congenita ( Anemia di Diamond- Blackfan) ha una trasmissione non sempre
standardizzata, alcune forme hanno una chiara trasmissione autosomica dominante, in altri casi la trasmissione è
recessiva. Il quadro clinico da cosa è rappresentato? [il prof dice che noi dobbiamo sempre tenere ben presenti i
segni e i sintomi di uno stato anemico. Il segni sono il pallore,la tachicardia, tachipnea. L’astenia è un sintomo in
quanto soggettivo]. Se il paziente ha una anemia severa e ponete il fonendoscopio sui focolai di auscultazione delle
valvole cardiache ( il prof ricorda che non corrispondono a quelli anatomici, ma sono le aree in cui apprezziamo
meglio il reperto), abbiamo un soffio sistolico generato dalla maggiore velocità del sangue dovuta ad una minore
viscosità. Il soffio è presente su tutti i 4 focolai; si tratta di un segno che è possibile riscontrare quando l’anemia è di
grado severo. Quindi nella eritroblastopenia congenita abbiamo un quadro clinico caratteristico dell’anemia, che può
essere severa anche a partire dai primi mesi di vita. Nel 30% di questi pazienti abbiamo una associazione con
malformazioni scheletriche, gonadiche, renali, cardiache, ecc. Laboratoristicamente nulla di più nulla di meno di
quello che abbiamo detto nelle caratteristiche generali delle anemie di questo gruppo, cioè una anemia di grado più
severo, normocromica, normocitica con reticolocitopenia. Se facciamo l’esame del midollo osseo c’è una ipoplasia
eritroblastica. Il trattamento si basa sui cortisonici, che possono dare una risposta parziale nel 60-70% dei casi, ma in
alcuni casi bisogna ricorrere alla ciclosporina che può dare dei risultati soddisfacenti. Se i risultati sono
insoddisfacenti il paziente può avere una Hb bassa tale da configurare uno stato di anemia severa e si deve ricorrere
ad un trattamento sostitutivo.
Accanto alla eritroblastopenia congenita ci sono le forme acquisite che possono essere acute e croniche. Le forme
acute hanno una insorgenza improvvisa e si verificano per lo più dopo infezioni virali. Un virus particolarmente
responsabile di queste forme è il parvovirus B19, perché ha un tropismo elettivo per gli eritroblasti. Il parvovirus B19
è l’agente eziologico della V malattia. Nei soggetti sani che hanno contratto questa infezione si ha una inibizione
transitoria della eritropoiesi che difficilmente dà luogo a quadri clinici eclatanti, tutto decorre in maniera subclinica.
Tuttavia, se questa infezione viene contratta da un paziente che già ha una emolisi cronica allora l’anemia può essere
anche acuta, si può addirittura avere una crisi aplastica. Oppure, nel paziente con immunodeficit acquisiti o congeniti
vi è una incapacità ad eliminare il virus e quindi l’infezione protratta darà luogo ad un quadro clinico ben evidente.
Questa condizione si può verificare anche in seguito alla somministrazione di farmaci come anti ipertensivi (alfametilDopa), FANS (indometacina), antibiotici (cefalotina che è una cefalosporina). Il quadro clinico, quindi, è evidente
WWW.SUNHOPE.IT
in coloro che hanno una emolisi cronica o un immunodeficit, negli altri la sintomatologia è sfumata e anche la
reticolocitopenia è transitoria. Agli esami di laboratorio abbiamo sempre anemia normocitica, normocromica con
reticolocitopenia e ipoplasia eritroblastica. In genere sono forme autolimitantesi e se si sospetta che alla genesi del
quadro clinico abbia contribuito uno dei farmaci sopra citati basta sospendere il farmaco. Si ricorre alla trasfusione
di emazie concentrate solo se insorge una anemia severa. Le eritroblastopenie croniche hanno un andamento
progressivo. Quello che dobbiamo sapere è che 1/3 di questi pazienti può nascondere un timoma. Possono avere un
ruolo importante anticorpi di classi IgG in grado di provocare una inibizione della crescita eritroide che, però,
potrebbe essere inibita anche da linfociti CD8, cioè T suppressor. Oppure può esserci una inibizione immunomediata della attività eritropoietinica. Segni e sintomi sono quelli tipici della anemia, il laboratorio non ci dice niente
di diverso da quello che ci saremmo aspettati, il decorso è cronico. È necessario che il paziente sia sottoposto alle
indagini strumentali necessarie per la diagnosi di un eventuale timoma, cui farà seguito la asportazione chirurgica.
Può essere necessario un supporto trasfusionale. Trattandosi di una forma cronica, nella maggior parte dei casi si ha
una buona risposta ai cortisonici, tuttavia, un cortisonico può essere utilizzato a lungo termine solo a basse dosi
altrimenti si hanno tutti gli effetti collaterali tipici.
L’altra anemia di questo gruppo è quella che compare nel paziente con insufficienza renale cronica. Ha una genesi
polifattoriale ma, sicuramente, il momento patogenetico più importante è il deficit della produzione di
eritropoietina. Questa viene prodotta dalle cellule iuxtaglomerulari del rene, dunque un rene che non funziona bene
ne produce una quantità ridotta soprattutto in risposta ad alcuni stimoli. Noi abbiamo dei sensori che entrano in
funzione durante uno stato di ipossia, dunque nel caso di anemia, questi sensori attivano il processo che porta alla
produzione di eritropoietina che stimola l’eritropoiesi e l’immissione in circolo di un numero maggiore di globuli rossi
( è un meccanismo di compenso). In questo caso la risposta eritropoietinica è insufficiente. La terapia nel paziente
con insufficienza renale cronica è l’eritropoietina ricombinante a basse dosi. Gli altri fattori che possono contribuire
alla genesi di questa anemia sono la ridotta sopravvivenza degli eritrociti e un cattivo funzionamento delle piastrine.
Il quadro clinico è dato dalla sommatoria dei segni e sintomi dello stato anemico e della IRC. L’anemia è sempre
normocromica e normocitica, anche se a volte i globuli rossi presentano una superficie molto irregolare con delle
spicole e sono definiti echinociti. Ci può essere anche iposideremia e ipoferritinemia perché il cattivo funzionamento
delle piastrine può determinare piccole emorragie croniche e occulte nel tubo digerente. Se facciamo il dosaggio
della eritropoietina sierica, il valore è piuttosto basso in rapporto al grado di anemia.
ANEMIE DEL II GRUPPO
Sono delle anemie in cui la eritroblastogenesi ( cioè il rifornimento degli eritroblasti a partire dai progenitori) è
conservata. Questi eritroblasti, però, non maturano in maniera corretta, perciò una parte muore all’interno del
midollo osseo, sicché noi osserviamo un accumulo delle forme di eritroblasti più immaturi rispetto alle forme più
mature. In ordine crescente di differenziamento abbiamo proeritroblasti, eritroblasti basofili, eritroblasti
policromatofili, eritroblasti ortocromatici che chiamiamo così perché il colore del loro citoplasma è molto simile a
quello degli eritrociti. A questo punto l’eritroblasto ortocromatico espelle il nucleo, passa nel sangue circolante
conservando ancora per qualche giorno dei residui di reticolo, dopodiché assume tutti le caratteristiche delle emazie
normali. In questo caso abbiamo un accumulo di pro eritroblasti ed eritroblasti basofili mentre sono scarsi quelli
policromatofili e ancor meno numerosi gli ortocromatici. È chiaro che nel sangue periferico saranno scarsi i
reticolociti, dunque si tratta di anemie caratterizzate da reticolocitopenia. In questo gruppo sono incluse due anemie
molto importanti: quella da carenza di vitamina B12 e quella da carenza di acido folico.
Il professore descrive uno schema. Al centro c’è il tetraidrofolato (THF) che è la forma attiva dell’acido folico, e tutte
le vie che partono da questo composto sono coinvolte nella sintesi del DNA. Ciò significa che se non si forma THF, è
compromessa tutta la sintesi di DNA. Il THF deriva dal metil-tetraidrofolato che può trasformarsi nella forma attiva
solo se c’è un composto che possa accettare il suo gruppo metilico (omocisteina). La reazione che comporta la
trasformazione del metil-THF in THF e l’omocisteina in metionina è catalizzata dalla metionina sintetasi che ha come
coenzima la cobalamina. Dunque il gruppo metilico del metil-THF viene trasferito sull’omocisteina che, in questo
modo, si trasforma in metionina. Il metil-THF, liberandosi del suo gruppo metile, si è trasformato nella sua forma
WWW.SUNHOPE.IT
attiva. Quindi, se c’è un deficit di cobalamina si ha una “trappola dei folati”(non viene liberata la forma attiva). La
vitamina B12 interviene anche in un’altra reazione importante: conversione del metilmalonilcoA in succinilcoA.
Questa reazione è importante perché porterà alla sintesi della mielina. Quindi, il deficit di vitamina B12 comporta
una alterata sintesi delle guaine mieliniche e questo ci spiega la sintomatologia neurologica presente nel quadro
clinico di una anemia da deficit di vit.B12. Inoltre, è opportuno considerare che in questa reazione non è
assolutamente coinvolto l’acido folico, quindi l’anemia da deficit dei folati non sarà complicata da sintomatologia
neurologica. La cobalamina partecipa alla prima reazione descritta come metil-cobalamina, alla seconda come
adenosil-cobalamina. Riassumendo, la carenza di vitamina B12 comporta una ridotta conversione della omocisteina
in metionina, ridotta formazione di tetraidrofolato, ridotta formazione di basi puriniche e compromissione della
sintesi di DNA, ridotta formazione di succinilcoA e compromissione della sintesi di guaine mieliniche.
Metabolismo della vitamina B12. La vitamina B12 non viene prodotta dall’organismo umano, quindi la ingeriamo
con gli alimenti in una forma legata alle proteine alimentari. Una volta nello stomaco, per effetto dei succhi gastrici,
viene staccata dalle proteine alimentari e si lega alla proteina R (prodotta dalle ghiandole salivari). Questo complesso
arriva nella seconda porzione del duodeno dove ci sono le proteasi pancreatiche che staccano la proteina R dalla vit.
B12 che può legarsi al fattore intrinseco; quest’ultimo era stato prodotto dalle cellule parietali gastriche del corpo e
del fondo. Questo complesso attraversa gran parte dell’intestino e giunge all’ileo distale. I villi dell’ileo distale hanno
recettori per il fattore intrinseco, quindi tutto questo complesso viene internalizzato all’interno della cellula ileale,
dove la vitamina B12 viene staccata e legata alla Transcobalamina II, quella proteina che veicola nel sangue la vit.
B12 consentendole di giungere nelle sedi in cui deve essere utilizzata o immagazzinata. Ragionando su queste
diverse fasi, si possono dedurre quali possono essere le condizioni in cui si instatura un deficit di vit. B12.
-
-
-
-
Dieta incongrua, ad esempio una dieta vegetariana molto stretta che esclude anche uova e latte (perché non
viene prodotta dall’organismo). Devono infatti ingerire la vit.B12 altrimenti col tempo andranno incontro ad
anemia
Mancanza del fattore intrinseco Possono essere presenti anticorpi inattivanti il fattore intrinseco o anticorpi
diretti contro le cellule parietali gastriche che determinano atrofia della mucosa gastrica, compromettendo
la sede di sintesi del fattore intrinseco. Ancora, si può trattare di un paziente gastrectomizzato o che abbia
subito una resezione gastrica, quindi gran parte della mucosa gastrica è stata asportata. Inoltre, si può
trattare di un paziente che ha tentato il suicidio ingerendo sostanze molto acide o molto basiche che hanno
determinato una distruzione della mucosa gastrica.
Alterazioni dell’assorbimento ileale Il paziente può presentare una patologia che interessa l’ileo terminale
come la malattia di Crohn, può aver subito un intervento chirurgico in cui è stato asportato il tratto ileociecale, oppure può essere presente una fistola che consente di bypassare la sede di assorbimento come una
fistola gastro-colica, digiuno-colico. Altre condizioni possono essere una sprue tropicale o non tropicale o,
ancora, la presenza di uno o più diverticoli che si complicano in una diverticolite e si ha una proliferazione
della flora batterica che utilizzerà per il proprio fabbisogno la vitamina B12.
Botriocefalosi. È una infestazione più diffusa nel nord Europa dove fanno maggiormente uso di pesce crudo
e uova di pesce. Dunque, se il nostro paziente riferisce di aver soggiornato nei paesi Baltici e dopo 5-6 mesi
presenta questa anemia macrocitica, è opportuno considerare anche questa possibilità.
Il prototipo della anemia megaloblastica è l’anemia perniciosa che è caratterizzata dalla presenza di anticorpi anti
parete gastrica, meno frequentemente anti fattore intrinseco. Alla fine il risultato è lo stesso perché quello che
manca è il fattore intrinseco. È possibile che il paziente abbia anche un’altra patologia autoimmune associata; ad
esempio, non è raro che un paziente con anemia perniciosa sia contemporaneamente affetto da tiroidite di
Hashimoto.
Quadro clinico Precedentemente è stato detto che il deficit di vit. B12 compromette la sintesi di DNA e delle guaine
mieliniche. Dunque, i tessuti maggiormente sensibili ad una ridotta di DNA sono quelli che presentano un ricambio
molto rapido: midollo osseo e il tratto gastrointestinale. In questi pazienti, oltre ai segni e sintomi caratteristici della
anemia può essere presente un subittero. Il subittero deve essere ricondotto ad un incremento della bilirubina non
WWW.SUNHOPE.IT
coniugata che proviene dal catabolismo dei precursori intramidollari. Questo paziente lamenterà una alterata
sensibilità gustativa e bruciori alla lingua configurando il quadro della glossite di Hunter: bruciore e parestesie
linguali, ipo/atrofia delle papille così che la lingua si presenta lucida con i bordi arrossati e infiammati. Possono
essere presenti disturbi digestivi, soprattutto la diarrea legata al malassorbimento riconducibile alla megaloblastosi,
cioè queste cellule voluminose che costituiscono l’epitelio intestinale. Può essere presente una sintomatologia
neurologica che comprende parestesie, cioè una alterazione della sensibilià (iperestesia, ipoestesia, disestesia),
alterazione della sensibilità profonda con perdita della capacità di localizzare gli arti nello spazio. Può essere
presente una andatura pareto spastica. All’esame obiettivo questi pazienti presentano una iperriflessia e se
avviciniamo un diapason alla loro superficie pretibiale, non saranno in grado di percepire le vibrazioni. Positività alla
prova di Romberg e del segno di Babinski. La prima si esegue facendo stare il paziente in piedi con i talloni uniti e le
braccia stese in avanti e invitandolo a chiudere gli occhi. Al soggetto normale non succede nulla, mentre il paziente
con deficit di vit. B12 può avere delle oscillazioni e addirittura cadere. Il secondo si esegue facendo strisciare una
superficie appuntita sotto la pianta del piede: nel soggetto normale c’è una flessione delle cinque dita, mentre in
questi pazienti avremo iperestensione dell’alluce e la flessione delle altre. RIASSUMENDO: c’è una triade composta
da segni e sintomi dell’anemia, sintomatologia digestiva e sintomatologia neurologica.
Il professore mostra una sezione di midollo spinale, dove si possono vedere aree chiare che interessano i cordoni
laterali e posteriori. Queste sono la conseguenza della demielinizzazione legata alla difettosa sintesi delle guaine
mieliniche.
L’alterata sintesi di DNA è responsabile di una eritropoiesi megalobastica. Il nucleo è grande e il citoplasma è esteso,
ma soprattutto c’è un asincronismo maturativo tra nucleo e citoplasma (il nucleo sta più indietro rispetto al
citoplasma). Il rallentamento maturativo comporta l’accumulo dei precursori eritroidi. All’esame
emocromocitometrico, oltre all’anemia (che può essere anche di grado severo), quello che spicca è l’incremento del
volume corpuscolare medio che può avere un valore superiore a 120 μm³ o femtolitri. Il valore di MCHC, cioè la
concentrazione di emoglobina rispetto a tutto il globulo rosso, è normale e può essere espresso in percentuale o in
g/dl. Se la concentrazione di Hb nel globulo rosso è normale, essendo il globulo rosso più grande, la quantità in peso
della Hb nel globulo rosso è aumentata. Ad esempio, se noi mettiamo 100 ml di un liquido colorato in una bottiglia
da 1 L diciamo che il liquido colorato ha una concentrazione del 10%, se noi prendiamo una bottiglia da 2L con 200
ml di liquido colorato, la concentrazione è sempre la stessa ma la quantità del liquido è maggiore (il doppio). In
questo caso la concentrazione di Hb in un globulo rosso è normale, ma se il globulo rosso è più grande, la quantità di
Hb (facendo riferimento al suo peso) è maggiore. Quindi il valore di MCH sarà nettamente aumentato. Tuttavia,
essendo poche le emazie, il valore complessivo di Hb sarà basso. Ci sarà reticolocitopenia poichè una buona parte
degli eritroblasti muore nel midollo senza mai diventare eritrociti maturi. Con il tempo anche la pastrinopoiesi e la
leucopoiesi saranno interessate dal deficit di vitamina B12 e con il tempo si instaura una piastrinopenia e una
leucopenia.
Guardando lo striscio di sangue è evidente una ANILOPOICHILOCITOSI, cioè una anisocitosi (emazie di dimensioni
differenti) con poichilocitosi (emazie di forma strana). All’interno di alcuni elementi cellulari si possono osservare
frammenti di cromosoma di colorito rossastro chiamati corpi di Jolly. Anche i neutrofili sono più voluminosi.
Normalmente il nucleo dei neutrofili presenta 2-4 restringimenti e maggiore è il numero dei restringimenti, più
vecchia è la cellula. In questo caso abbiamo neutrofili ipersegmentati (più vecchi) e questo significa che il ricambio
cellulare avviene in maniera insufficiente.
Il professore mostra una serie di immagini
1°striscio di sangue: queste emazie sono più grandi di quelle normali, ma soprattutto colpisce la diversità delle forme
e delle dimensioni. Alcune hanno aspetto ovalare e molto spesso questi elementi sono detti Megaloovalociti. Al
centro c’è un polimorfo nucleato neutrofilo il cui nucleo ha un numero elevato di restringimenti. Ci sono anche
piastrine molto più grandi rispetto ad uno striscio normale.
2° striscio: megalo ovalocita con corpo di Jolly
WWW.SUNHOPE.IT
3° striscio: ci sono due neutrofili, uno ipersegmentato e uno normalmente segmentato
Preparato di midollo osseo (ago aspirato): il colore preponderante è il viola-bluastro e questo spiega perché i primi
che osservarono il midollo di pazienti con anemia megaloblastica parlarono di midollo blu. Questo colore è dovuto
ad una concentrazione maggiore di pro eritroblasti ed eritroblasti basofili che hanno un citoplasma bluastro.
Quindi si deve notare la intensa basofilia citoplasmatica e l’intensa e omogenea colorazione del nucleo, espressione
del ritardo maturativo.
Iter diagnostico. Dosaggio della vitamina B12 che in questi pazienti è inferiore a 100 pg/dl. I parametri che ci
suggeriscono una eritropoiesi inefficace solo l’aumento delle LDH, la iperbilirubinemia indiretta e l’aumento della
sideremia. Dobbiamo sottoporre questi pazienti ad un esame endoscopico (EGDS che sta per
esofagogastroduodenoscopia) ed eseguire biopsie multiple perché, come detto precedentemente, una delle cause
della mancata attivazione del fattore intrinseco può essere una ipo/atrofia della parete gastrica, cioè sono presenti
degli anticorpi nei confronti della parete gastrica e contemporaneamente dei processi flogistici con infiltrazione
linfocitaria della mucosa e successivamente una sostituzione della normale mucosa gastrica con elementi che sono
più tipici dell’epitelio intestinale (metaplasia intestinale).
[Mostra un preparato in cui, grazie alla immunofluorescenza indiretta, sono stati messi in evidenza degli anticorpi
contro antigeni della parete gastrica. In questo caso la parete gastrica (di ratto) veniva essiccata e fissata su un
supporto solido. Si utilizzavano antianticorpi coniugati con fluorescina e, laddove questi si legavano agli anticorpi a
loro volta legati ad antigeni della parete gastrica, al microscopio era possibile notare il colore verde intenso.]
Inoltre, in questi pazienti c’è un accumulo dell’acido metilmalonico che sarà dosato nelle urine delle 24h. In questi
pazienti avremo dei valori anche 100 volte superiori (>300 mg/24 h) rispetto ai valori normali (3-4 mg/24h). A livello
del midollo osseo, grazie all’agoaspirato, troviamo un accumulo di pro eritroblasti ed eritroblasti basofili di
dimensioni aumentati, la povertà di elementi immaturi e maturi e l’asincronismo maturativo.
Terapia In passato venivano somministrate quantità esagerate di vitamina B12. È sufficiente somministrare 1mg/die
per via i.m. per 1 settimana, dove per 1mg si intende quello che commercialmente troviamo nella dobetin 1000.
Durante la seconda settimana sono sufficienti due iniezioni ( 1mg/die i.m. per due giorni), durante la III-IV settimana
basta una iniezione a settimane e se non si riesce a rimuovere la causa del deficit di vit. B12 sarà sufficiente una
inizione di 1mg al mese per evitare che il paziente diventi anemico. Possiamo correggere un deficit di vitamina B12
per via orale solo se la causa è una dieta incongrua.
Il professore mostra un grafico in cui sull’asse delle ordinate è presente il valore dell’ematocrito o il valore dei
reticolo citi e sull’asse delle ascisse il tempo. Nella prima settimana c’è un rapido incremento dei reticolociti
(l’eritropoiesi si sta riprendendo), ma è necessario più tempo affinchè ci sia un ripristino dell’ematocrito che avrà
valori normali dopo circa 2 mesi di terapia.
Durante la lezione(subito dopo le eritroblastopenie congenite) il professore ha parlato dell’esame del midollo e della
trasfusione che ho preferito riportare alla fine per dare una maggiore continuità al discorso.
ESAME DEL MIDOLLO OSSEO
Noi possiamo esaminare il midollo osseo mediante un agoaspirato e mediante una biopsia osteo-midollare. Questi
hanno delle finalità diverse. Il primo ci consente di fare un esame citologico ma non ci dice niente circa l’architettura
tissutale. Quindi solo con la biopsia osteo midollare, nella quale viene prelevato un cilindretto osseo, si può fare un
esame istologico ed eventualmente un esame immunoistochimico.
Sede L’ago aspirato può essere eseguito o dallo sterno o dalla cresta iliaca. Per quanto riguarda lo sterno si individua
la linea mediana e in corrispondenza della base del II-III spazio intercostale viene eseguito l’agoaspirato. Questa è
una procedura semplice, è infatti sufficiente una piccola quantità di anestetico locale. Oggi però gli ematologi non
eseguono più l’ago aspirato dallo sterno perché in alcuni pazienti in cui lo sterno è particolarmente fragile c’è stato
WWW.SUNHOPE.IT
qualche inconveniente relativo alla procedura. Ad esempio, nei pazienti con mieloma multiplo che hanno lo sterno
“a biscotto”, esercitando la pressione necessaria per entrare e superare il tavolato esterno si può verificare un
cedimento brusco della resistenza ossea e provocare in qualche caso la rottura dello sterno, con un possibile danno
ai grossi vasi situati posteriormente. Tutto ciò ha creato, sebbene raramente, qualche inconveniente. In questo caso i
risvolti sono stati soprattutto medico legali, perché la domanda che viene posta è “c’era la possibilità di eseguire lo
stesso esame in un’altra sede con meno rischi?” si, perché l’agoaspirato può essere fatto dalla cresta iliaca con rischi
notevolmente minori. Per la cresta iliaca viene scelto come punto di repere la spina iliaca posterior superiore, da qui
si sceglie un’area abbastanza pianeggiante che è il punto in cui noi dobbiamo affondare il nostro ago (dispositivo che
deriva da quello originario di Jamshidi). Mentre lo sterno è immediato, l’esecuzione dell’esame a livello della cresta
iliaca può risultare più difficoltosa, soprattutto se abbiamo un soggetto con un pannicolo adiposo molto
rappresentato. Bisogna fare l’anestesia locale con lidocaina all’1%. È, quindi, una procedura più lunga. Quando
dobbiamo eseguire la biopsia osteo midollare non abbiamo scelta, il prelievo viene eseguito dalla cresta iliaca. Noi da
dieci anni non eseguiamo più l’ago aspirato dallo sterno per un fatto anche didattico, perché gli specializzandi
devono familiarizzare con quella sede così da avere meno difficoltà per la biopsia osteo midollare. Poi c’è un altro
motivo: il paziente è più contento perché non vede. Quando l’esame veniva eseguito dallo sterno, il paziente
assisteva completamente alla procedura, invece quando gli viene fatto dalla cresta iliaca, è poco più di una iniezione
anche se più dolorosa. Insomma non c’è tutto quello che può essere legato alla suggestione individuale.
Cosa intendiamo quando parliamo di trasfusioni?
Nel caso delle anemie noi non trasfondiamo sangue intero ma le emazie concentrate. Innanzitutto, quando il sangue
viene raccolto, viene frazionato. È importante distinguere tra emocompenenti ed emoderivati. Gli emocomponenti
sono i globuli rossi concentrati, il plasma fresco congelato, il concentrato piastrinico, il crioprecipitato e riusciamo ad
ottenerli con tecniche semplici; basta, ad esempio, una centrifugazione a determinare un numero di giri e una forza
centrifuga, per cui dalla sacca principale collegata a sacche satelliti in cui sono fatti defluire gli emocomponenti.
Quindi è qualcosa che viene preparato direttamente dal centro trasfusionale. Invece gli emoderivati sono quelli che
si ottengono mediante frazionamento industriale, ad esempio sono utilizzati pool di plasmi da cui, con procedure
complesse, si ottengono immunoglobuline, albumina, fattori della coagulazione, ecc. Questi sono gli emoderivati.
Quando noi trasfondiamo i pazienti con emazie concentrate,la prima cosa noi gli iniettiamo delle emazie che
possono essere conservate fino a 40 giorni. Nelle sacche oltre all’anticoagulante ci sono dei substrati che consentono
al globulo rosso di mantere un suo metabolismo e una sacca standard è tale se al 40° giorno le emazie conservano
una vitalità di almeno il 70%. È chiaro che se voi ad un paziente trasfondete una unità di sangue raccolta 10 giorni
prima la percentuale di emazie vitali sarà superiore rispetto ad una sacca di sangue raccolto 35 giorni prima. Che
cosa dobbiamo aspettarci dalla trasfusione di una unità di sangue? Una unità di sangue standard si aggira intorno
450-500 ml di sangue intero, poi questa viene frazionata e si ottengono solo le emazie concentrate separate dagli
altri emocomponenti, si aggiunge l’anticoagulante. Si definisce unità standard di emazie concentrate quella in cui è
presente una quantità di emazie tale da consentire ad un individuo di 70 kg di ottenere un incremento della
emoglobina compreso tra 0,8-1 g. è chiaro che se noi abbiamo un paziente che pesa 100 kg, l’incremento di
emoglobina che avremo da una unità standard è inferiore, se è una donna di 50 kg avremo un incremento di Hb
superiore a quello atteso. È evidente che trasfondere una sola unità di sangue non ha molto senso, quindi bisogna
programmare che il paziente riceva almeno 2 unità di sangue. Se il paziente ha una condizione cardiovascolare in cui
si può temere un sovraccarico di volume, le due unità di sangue saranno trasfuse in momenti diversi. C’è indicazione
alla trasfusione quando l’anemia è severa, cioè quando la concentrazione di Hb è inferiore a 8 g/dl. Per esempio nei
pazienti che hanno un morbo di Cooley, cioè una β talassemia major, la trasfusione viene eseguita a concentrazioni
di Hb più elevate, oppure un paziente che ha 8g/dl e si è anemizzato molto lentamente (ad esempio una donna che
ha avuto flussi mestruali molto abbondanti) e si è adattato bene alla sua condizione non ha necessità di essere
trasfuso. Invece, se si tratta di un paziente anziano già vasculopatico e coronaropatico, un valore di 8 g/dl può già
essere considerato critico e il paziente ha necessità di trasfusione. Quindi qualunque terapia (anche quella
trasfusionale è una terapia) va sempre adattata al paziente.
WWW.SUNHOPE.IT
TESTI CONSIGLIATI:
-
Linfomi, mieloma e macroglobulinemia di Waldenstrom vanno bene dal testo di oncologia medica del
professore Ciardiello
Tura-Baccarani che è un testo molto semplice
Liso- Castolli della collana “core curriculum Ematologia”
Le anemie emolitiche emolitiche autoimmuni e la malattia emolitica del neonato non è fatta bene da nessuna
parte,quindi il professore dice che ci darà qualcosa a riguardo.
WWW.SUNHOPE.IT
Ematologia 21/03/2014
L’altra volta non ha parlato dell’anemia da deficit vitamina b12. Bisogna ricordare che la forma attiva dei
folati è il tetraidrofolato, perché è il composto da cui poi partono le vie che portano alla sintesi del dna, e
che se il metiltetraidrofolato non viene convertito in tetraidrofolato questo non è possibile. Abbiamo anche
visto che i folati non intervengono nelle reazioni biochimiche che portano alla sintesi della mielina, e quindi
nel caso di deficit di folati non vi è una sintomatologia neurologica. I folati sono presenti nelle verdure a
foglia verdi: spinaci, cavoli, lattuga, in alcuni frutti, banane, limoni, ma anche nel fegato e nel rene. Il
problema dei folati è che la cottura li degrada, per cui nella quota contenuta il 50%-60% viene demolita. Il
fabbisogno giornaliero (OMS) sono di 200 microgrammi nell’adulto e cinquanta nel bambino. Durante
l’allattamento 300 microgrammi al di, nella gravidanza il fabbisogno è addirittura doppio. I folati vengono
assorbiti nel duodeno e nel digiuno. all’interno delle cellule entrano come poliglutammati e vengono scissi
come monoglutammati , vengono trasportati fondamentalmente dall’alfa1macronodulina in minor misura
da albumina. Ricapitoliamo le condizioni che ci portano la carenza: la dieta (ad esempio gli anziani che
mangiano sempre le stesse cose), un aumentato fabbisogno per aumentato consumo (gravidanza e
accrescimento), in più in quei pazienti che hanno un’emolisi cronica: in questi casi abbiamo detto che vi è
una iperplasia eritroblastica, quindi c’è una maggiore quantità di folati. Alcuni farmaci possono agire
inibendo tetroidrofolato reduttasi, ad esempio metrotrexane (chemioterapico) , bilimetamina
(antimalarico), oppure dietro trattamento di alcune forme di toxoplasmosi, e poi il trimetropin (è un
composto largamente usato), specialmente
associato al sulfametrossazolo
(il nome commerciale è
bactrim. 160mg di trimetro pin +800 mg di sulfametrossazone). È chiaro che questo se assunto per lunghi
periodi di tempo può comportare un difettoso utilizzo dei folati. ci sono condizioni patologiche in cui vi è un
deficit delle scorte epatiche dei folati (alcolizzati e cirrotici). Il quadro clinico è del tutto analogo a quello
della vitamina b12 se si eccettua la sintomatologia neurologica, ma con i folati sierici ridotti (anche se
questi sono molto influenzati dalla dieta di tutti i giorni) per cui sarebbe meglio fare i dosaggi dei folati
intraeritrocitari (lo fanno in pochi laboratori, non è una procedura automatizzata). Come per la vitamina
b12 non vi è bisogno di alte dosi per correggere uno stato di carenza. La maggior parte delle specialità in
commercio sono dosate in 15 mg. Per correggere la carenza bastano da 5 a 15 mg al giorno, per il
mantenimento da 1 a 5 mg al giorno. Passiamo alle anemie del terzo gruppo. Sono caratterizzate dalla
compromissione della sintesi dell’emoglobina. L’emoglobina è formata da una parte proteica e da una
parte non proteica. Nella parte non proteica è costituita dall’eme (proto porfirina IX con al centro un
atomo di ferro). Capite bene come una carenza di ferro è responsabile di una ridotta sintesi dell’ HB, e
questo è alla base dell’ anemia sideropenia, ma il problema potrebbe anche riguardare la parte proteica, e
cioè le catene globiniche. Parleremo di emoglobinopatie facendo riferimento ad una alterazione qualitativa
WWW.SUNHOPE.IT
della catena globinica. Capite anche che se un amminoacido viene sostituito in un punto chiave le
caratteristiche chimico-fisiche dell’HB risultano modificate. Questo può portare a condizioni caratteristiche
che vedremo dopo. Parliamo di talassemie in quelle condizioni in cui è depressa la sintesi di una catena
globinica, o anche in più catene globiniche come nel caso delle doppie eterozigosi. Sono delle malattie
molto diffuse, descritte nel bacino del mediterraneo, anche se però questa distribuzione geografica perde il
suo significato visti i flussi migratori. (indica aree geografiche con beta e alfa talassemie). In italia abbiamo
aree che molte volte coincidono a zone che sono state bonificate e che prima erano zone malariche. A
parte sicilia e sardegna abbiamo percentuali elevate verso il delta del PO. In campania le percentuali
maggiori sono nella provincia di caserta. Indica schema: sull asse delle ordinate la concentrazione delle
varie emoglobine, sull’asse delle ascisse l’epoca prenatale, nascita, condizione a 3 e 6 mesi. Alla nascita il
bambino presenta quasi completamente hb fetale (tetrano alfa2 gamma2). Ma nei primi mesi di vita cessa
l’attività dei geni che codificano per le catene gamma e comincia l’attività dei geni che codificano per le
catene beta. Abbiamo uno switch tra catene gamma e beta. A 5-6 mesi la sostituzione è già considerevole
(osserva la curva dell’ emoglobina dallo schema. A 6 mesi la beta globina è già considerevole. Le catene
gamma invece scende fino a cadere a 6 mesi e raggiungere bassi livelli). In un soggetto normale l’hbF è
inferiore al 1%. Un ulteriore cosa da sottolineare è la curva che esprime la sintesi delle catene alfa. Queste
sono già molto rappresentate già prima della nascita. Questo discorso ci porta a capire che se un neonato
nasce da due genitori di cui non sappiamo nulla e questo bimbo ha una talassemia betaomozigota il quadro
clinico alla nascita sarà completamente muto, perché alla nascita c’è ancora l’hb fetale. Il problema si
verifica quando le catene delle hb fetale dovrebbero essere sostituite dalle catene beta, questo non si
verifica e si hanno tutte le conseguenze sul piano clinico. Mentre chi ha un alfa talassemia potrebbe avere
delle manifestazioni già alla nascita. Ovviamente tutto dipende da caso a caso. Ricapitolando i valori
normali delle varie emoglobine sono, l’HB A dell’adulto 96%, l’HB A2 ha una concentrazione dal 2 al 3,2 %.
L’ha F max fino all’1%. Nel caso della beta talassemia noi potremmo avere una condizione beta0 in cui non
vengono sintetizzate le catene globiniche beta e una condizione beta+ in cui vi è solo un deficit parziale
della sintesi delle catene beta. Quando diciamo deficit dell’HB beta è conseguente deficit dell’HB A
dell’adulto . nel caso delle beta talassemia è raro che si tratta di difetti “delezionale “ (genica) in realtà una
forma che è significamente si riscontra con maggiore frequenza è quella presenta in india. Nelle ns aree
sono solitamente mutazione puntiformi responsabili della trascrizione e della traslazione dell rna
messaggero. Tra due genitori portatori la probabilità che nasca un figlio affetto è del 25% (1 su 4) la
probabilità che sia portatore il neonato è 50%, che sia sano è 25%. Se invece un solo soggetto è portatore
le probabilità che nasca un figlio portatore è del 50% ma mai un soggetto malato. Cominciamo dalla beta
talassemia majior (m.di Coley). Riguardando la situazione italiana teniamo presente che abbiamo 3 milioni
di talassemici, la stragrande maggioranza sono portatori, poche migliaia hanno al forma grave (coley).
Comprendiamo nel morbo di coley condizioni in cui è in gioco una omozigosi del gene beta, ma può essere
WWW.SUNHOPE.IT
anche una eterozigosi complesse per diverse mutazioni ma che danno tutte un quadro clinico che è quella
del morbo di coley.in neonato alla nascita sta bene, ma poi nei primi mesi il bambino ha un accrescimento
stentato nei primi mesi, si nutre con difficoltà, diventa pallido e con colore cutanea con sfumature
giallognole a causa della Iperbilirubinemia indiretta. Se la sintesi delle catene beta è depressa noi abbiamo
un eccesso di catene alfa che non sono solubili e che precipitano all’interno dell’eritroblasto. dall’entità
delle precipitazioni questi eritroblasti potrebbero maturare, passare in circolo ed essere in tempi brevi
sequestrati dalla milza, ma potrebbero anche non giungere a maturazione ed essere distrutti a livello del
midollo osseo, realizzando una certa quota di eritropoiesi inefficace (anemia 2 gruppo). In questi soggetti in
realtà vie è anche questa condizione che giustifica l’Iperbilirubinemia non coniugata. Osservando un
bambino di 6-7 mesi quello che ci colpisce è anche la notevole epatosplenomegalia. Nei primi anni del
bambino poi notiamo alterazioni scheletriche, perché il midollo osseo sotto la spinta eritropoietina tende
ad espandersi in aree solitamente non adibite ad accogliere il midollo osseo. Abbiamo modificazioni
scheletriche caratteristiche: zigomi prominenti, radice del naso schiacciata, spazio tra le orbite allargate, poi
abbiamo alterazioni delle arcate dentarie. (descrive immagine di un bimbo in slide). Consideriamo una
bimba di 17 anni di circa 130 cm dove si evidenzia un ritardo dello sviluppo sessuale. Il danneggiamento è
legato all’eccesso di ferro che si deposita a livello delle gonadi e di altre ghiandole endocrine. L’eccesso di
ferro viene dalle trasfusioni che apporta un certo quantitativo di ferro che si deposita, inoltre in cuore,
fegato oltre che ghiandole endocrine. Quindi questi bambini nell’epoca in cui non si usavano farmaci che
chelavano il ferro in eccesso morivano nella seconda-terza decade di vita. Il deposito di ferro a livello del
miocardio da miocardiopatia dilatativa che non risponde al trattamento farmacologico. Il deposito in
eccesso a livello epatico dava una cirrosi epatica, poi c’era il danno endocrino con un quadro globale molto
complesso. Nella beta talassemia omozigote abbiamo un eccesso di catene alfa, perché non vi è sintesi di
catene beta (sbilanciamento a favore di alfa). Ma nel soggetto con beta talassemia omozigote vi è un
eccesso anche di HB F (che non è una HB patologica). In ciascuno di noi alcuni eritroblasti continuano a
sintetizzare HB F, tanto è vero che in ciascuno di noi la quota di HB F risulta a essere cmq presente. in quegli
eritroblasti in cui lo switch non si verifica, son eritroblasti che hanno una normale evoluzione (nda. non
capisco quello che vuole dire…sto trascrivendo letteralmente quello che ha detto il prof. A voi
l’interpretazione) . gli altri eritroblasti in cui c’è l’eccesso di catene alfa, sono eritroblasti che vanno avanti
con difficoltà, per cui si a livello midollare una lieve espansione di quegli eritroblasti che continuano a
sintetizzare l’HB F. quindi in questi soggetti troveremo queste di HB f anche del 15-20 & (nettamente più
alta). Però l’ HB F è l’HB del feto che ha una maggiore affinità per ossigeno, e quindi questo è ceduto con
maggiore difficoltà a livello dei tessuti periferici con ipossia (di questi tessuti) e sintesi di eritropoietina.
Però l’eccesso di catene alfa che abbiamo visto tende a precipitare; ora tenendo conto che due sono le
condizioni: o gli eritroblasti in cui si è verificata la precipitazione delle catene alfa vanno avanti riuscendo a
passare in circolo e venire sequestrati a livello splenico velocemente, oppure muoiono all’interno del
WWW.SUNHOPE.IT
midollo. In ciascuna delle due condizioni possiamo avere: da un lato una eritropoiesi inefficace se la
distruzione avviene all’interno del midollo. Se invece è la milza che sequestra questi eritroblasti abbiamo
una splenomegalia. In entrambe le condizioni cosa ne viene fuori: uno stato anemico. L’anemia stimola
ulteriormente l’eritropoietina, questa determina l’espansione del midollo, quindi ne vengono fuori le
deformazioni ossee, deficit di folati ecc. le anemie come abbiamo detto le trattiamo le trasfusioni e da qui
ne viene fuori un eccesso di ferro. Il tutto comporta un sovraccarico di ferro con danno a livello delle
ghiandole endocrine, cirrosi, insufficienza cardiaca (morte). Il midollo osseo si espande in aree dove non
dovrebbe essere presente in modo cosi rappresentato, (il prof osserva e commenta slide con la superficie
di taglio della volta cranica: la spongiosa ossea da l’impressione di essere come le setole di una spazzola
definita “cranio a spazzola”. Osserviamo inoltre una rx in proiezione postero-anteriore (e non anteroposteriore come normalmente si defisce….nda “mhà” ). La fonte di raggi x è alla spalle del paziente quindi
tali raggi lo attraversano da dietro (proiezione postero-anteriore). Osservando l’ombra mediana, il diametro
trasverso è notevolmente aumentato; questo è l’espressione della cardiopatia dilatativa di cui dicevamo
prima. È l’espressione del ferro in eccesso. Da un punto di vista ematologica abbiamo una anemia di grado
severo (HB tra 4 e 6 grammi/dl), ma quello che è importante è che guardando lo striscio vediamo una serie
di alterazioni caratteristiche: guardando i globuli rossi, notiamo che questi sono tutti “pallidi” , quindi
notiamo una ipoformia (??) che corrisponde al MCH (contenuto HB medio espresso in psicogrammi tra 27 e
31). Queste emazie inoltre sono prevalentemente piccole quindi facendo una media di tutti questi volumi
abbiamo un volume corpuscolare medio ridotto. Altra cosa, la notevole eterogeneità/variabilità del volume
di queste emazie e anche le strane forme ( a goccia, a bersaglio). Al centro addirittura un eritroblasto che
non dovrebbe essere presente nel sangue periferico ma dovrebbe stare nel midollo. Sempre al midollo
osseo possiamo vedere con un esame particolare l’accumulo di ferro. Spesso parliamo dell’elettroforesi dell
HB. L’elettroforesi dell’HB è importante per vedere se sono presenti bande di HB patologiche. Ma è un
esame poco attendibile per quantizzare queste bande. Per quantizzare noi facciamo riferimento ad una
tecnica cromatografica. In ogni caso lo studio dell’HB ci fa capire come detto prima, che vi è aumento dell
HB F, mentre invece vi è un assenza completa di HB A. come detto prima, quando non c’erano i farmaci
chelanti questi bambini morivano nella seconda-terza decade di vita . Negli ultimi decenni le cose sono
cambiante , in particolar modo quando l’eccesso di ferro è stato rimosso dai chelanti per il ferro. Il primo
farmaco utilizzato è deferoxamina B ”…questo farmaco viene somministrato per essere efficace per via
sottocutanea mediante infusore temporizzato. Si carica il microinfusore della dose stabilita, poi si stabilisce
il tempo di infusione. La cosa migliora è che l’infusione avvenga in 10-12 ore. Molti pazienti preferiscono
applicare questa infusione nelle ore notturne (per essere libere nelle altre ore). Gli aghi utilizzati sono adesi
ad una ventosa, i pazienti lo applicano sulla superficie anteriore dell’addome la sera e lo tolgono la mattina.
Questo consente di rimuovere gran parte del ferro in eccesso derivato dalle trasfusioni. Senza le trasfusioni
i pazienti non vanno avanti. Il paziente viene trasfuso quando la sua HB è inferire a 8 gr/dl, facendo
WWW.SUNHOPE.IT
ovviamente una valutazione paziente per paziente. (nda, Il proff dice qualcosa di incomprensibile)….. In
questi casi invece la trasfusione con emazie concentrate viene fatta con HB che scende al di sotto di 10,5-11
gr. Poi ci sono in commercio anche due preparati (zeferidone più altro che non si capisce) che possono
essere somministrati per via orale, con indicazione di seconda linee cioè in pazienti che non possono più
trarre giovamento dalla deferoxamina B, oppure sono diventati intolleranti alla deferoxamina. La soluzione
ideale per questi bambini, se hanno un fratello HLA identico è il trapianto con cellule staminali allogeniche.
La scuola italiana del professore Lucarelli di Pesaro è stata una delle prime ad avere una larga casistica con
risultati che facendo opportune selezioni e stabilendo precocemente di intervenire con questa procedura
ha ottenuto risultati lusinghieri. Tuttavia buoni risultati si ottengono anche con donatori HLA identici non
correlati dal punto di vista familiare. La prospettiva della risoluzione è legata alla terapia genica. Passiamo
adesso alla condizione opposta, cioè la stragrande maggioranza di quei tre milioni di italiani: il soggetto
portatore. Il soggetto portatore può anche non essere anemico ed avere un livello di HB di 14 gr/dl. Ma in
tal caso noi come c è ne accorgiamo? Il soggetto ha microcitosi e ipocloria, potrebbe anche non esservi
anemia. A questo punto il nostro problema è abbastanza ristretto perché quale anemie sono microcitiche e
ipocromiche a parte le sindromi talassemiche ?...è l’anemia sideropenica! . quindi valutiamo l’assetto
marziale di questi soggetti (sideremia, creatitinenemia e ??) come detto nella prima lezione e se li troviamo
normali passiamo alle indagini di secondo livello che ci portano poi a diagnosticare che si tratta di soggetti
con un tratto talassemico. Nel caso specifico del paziente con beta talassemia minor è di importanza
notevole dimostrare che l’HB A2 è superire dei valori normali, cioè praticamente >3.3%. Se guardiamo lo
striscio periferico ci rendiamo conto della macrocitosi , della ipocromia. Se facciamo la quantizzazione della
HB mediante cromatografia mostriamo che i valori sono superiori a 3,3% e spesso può essere aumentata
leggermente la concentrazione dell HB F: questa è la condizione del soggetto eterozigote. Poi abbiamo una
condizione clinica che definiamo talassemia intermedia. Abbiamo quindi il morbo di cooley, la beta
talassemia intermedia che è caratterizzata da livelli di HB compresa tra gli 8 e i 10gr, compatibile quindi con
una anemia di grado moderata. Questo nei soggetti che per lo più si sono adattati bene e che non hanno
bisogno di essere trasfusi. Però sviluppano negli anni un certo grado di emocromatosi per un aumentato
assorbimento del ferro. Come tutti i pazienti con anemia emolitica, ci può essere una tendenza alla
formazione di calcoli biliari, alcune volte possono presentare ulcere degli arti inferiori. La beta intermedia
può essere dovuta ad una mutazione che interessa il promoter del gene BETA e non il gene beta
direttamente. Questo riduce la trascrizione del gene beta e ne viene fuori la situazione clinica meno grave
del morbo di coley. oppure il paziente è portatore di una doppia eterozigosi, per esempio se è portatore di
alfa e beta talassemia, la depressione contemporanea di parte delle catene alfa, riduce lo sbilanciamento e
quindi parte di quelle conseguenze che abbiamo visto essere importanti nella beta talassemia mayor.
Un’altra doppia eterozigosi è quella dei pazienti che oltre al gene talassemico, hanno ereditato il gene per
la persistenza dell HB F; l HB F è pur sempre una HB fisiologica anche se non la migliore. Si vede sempre
WWW.SUNHOPE.IT
ridotto lo sbilanciamento delle catene globiniche e quindi vi è come conseguenze un attenuazione del
quadro clinico rispetto al paziente con beta talassemia mayor. Lo striscio periferico è simile a quello del
soggetto con morbo di cooley. Parliamo dei soggetti alfa talassemici. Il gene beta si trova sul cromosoma 11
(ciascuno dei due, sono due geni quindi). Nel caso dell’alfa talassemia i geni in questione sono 4: due per
ciascun cromosoma della coppia 16. Nella maggior parte degli alfa talassemici, è in gioco l’evento
“delezione”. Portatori silenti: vi è delezione o inattivazione di un solo gene alfa su 4. Questi soggetti
presentano una macrocitosi e una ipocromia analogamente a come visto nel soggetto beta minor. Però s
equi facciamo lo studio dell HB A2 non troviamo l’aumento di questa, anzi in alcuni casi può essere che
questa si è addirittura ridotta. In altri casi si ricorre alle indagini di terzo livello, ovvero alle indagini
molecolari. Nel caso in cui siano inattivati 2 geni alfa su 4 può esserci oltre a ipocromia e macrocitosi una
anemia di grado lieve. Sono soggetti che essenzialmente stanno bene. Anche qua l’elettroforesi delle HB
mostra HB F normali e la diagnosi di certezza di fonda su indagini molecolari. Terza condizione: delezioni o
inattivazione di 3 geni alfa su 4. Ne viene fuori la malattia da HB H. abbiamo una notevole delezione di
sintesi di catene alfa e un aumento di sintesi di catene beta. Questa non precipitano ma tendono ad
aggregarsi formando tetrameri beta4 (ovvero detta HB H). alla nascita quando le catene beta non sono
presenti, però già la situazione clinica è evidente, poi la sintesi delle catene alfa è già importante alla
nascita, e la depressa sintesi delle catene alfa porta un eccesso di catene gamma, con tetramero che forma
l HB di BART. Questi pazienti presentano una anemia emolitica cronica, moderata e sempre un connotato di
macrocitosi e ipocromia. Osservando bene lo striscio troviamo istociti (frammenti di emazie). Se sono
inattivati tutti e 4 geni alfa (o sono deleti) la condizione è incompatibile con la vita.
HB S è una
emoglobinopatia (è una alterazione qualitative; le talassemie sono alterazioni quantitative). L’HB S è
caratterizzata dalla sostituzione in posizione 6 amminoterminale della catena beta di un residuo di acido
glutammico con un residuo di guanina. Quando l HB passa dallo stato “ossi ” allo stato “desossi” queste
catene tendono a polimerizzare, formando strutture rigide che distorcono le membrane eritrocitarie. Le
conseguenze di questo quadro; i grossi rossi hanno grandi diametro tra 6 e 8 micron, mentre il diametro dei
vasi sanguigni può essere compreso anche tra i 2 e 3 micron e quindi il globulo rosso, passando se conserva
la sua elasticità non ha problemi, ma se ha una struttura rigida non può passare. Quindi vi sarà una
incapacità di ossigenazione nei tessuti periferici con possibilità di ischemia se non addirittura di necrosi con
quadri molto gravi nel soggetto omozigote. (nda..qui Il prof parla di genitori e switch…ma non capisco cosa
dice). oltre l’anemia possiamo trovare una iperbilirubinemia indiretta, questo può essere dovuto all’azione
dei sinusoidi splenici (nda. credo che abbia voluto dire questo..ma non sono sicuro), ma talora anche ad un
carenza di aptoglobina, perché queste strutture rigide possono anche rompersi in circolo. Se facciamo uno
studio dell HB con una semplice elettroforesi vedremo la comparsa della banda della HB S ma la andiamo a
quantizzare con una cromatografia. Se guardiamo lo strisci di questi soggetti possiamo notare la presenza di
emazie a falce. Il quadro clinico di questi soggetti omozigoti è un emolisi su cui si inseriscono poi delle crisi
WWW.SUNHOPE.IT
vaso occlusive, per cui potrebbero avere dei dolori ossei, quadri di addome acuto per occlusione dei vasi
addominali, dolori toracici. È una situazione che si vede poco, ma con i flussi migratori (africa
centrosettentrionale) è molto più facile vedere un bambino con crisi acuta di cui non sappiamo niente.
Bisogna sottolineare che avendo una emolisi cronica in un bambino se sopraggiunge una infezione da
parvovirus b19….ne può venire fuori una eritroblastonemia acquisita acuta. Durante la crisi di falcizzazione
queste emazie rigide possono ingorgare fegato e soprattutto milza la quale può andare incontro a infarti
ricorrenti. Si assiste ad una ipoplasia e quindi una atrofia della milza. Questi soggetti è come non avessero
la milza e quindi possono avere infezioni da parte di germi capsulati. Un'altra caratteristica è che questi
bambini possono avere un ictus celebrale (????). nel caso in cui si abbia una crisi occlusiva che interessi
determinate aree polmonari si può avere la sindrome toracica acuta con dolore violento. La cura sarebbe
trapianto con cellule staminali HLA da donatore identico (familiare, fratello, sorella). Si è visto che un certo
giovamento può essere ottenuto utilizzando l’idrossuria (credo), è un farmaco che inibisce ribonucleotide
(e un'altra cosa), che noi sfruttiamo nella malattie proliferative croniche. Si è vista che la idrossiuria è in
grado di aumentare la sintesi dell HB F, quindi in questi soggetti tramite un aumento dell HB F è possibili
mitigare la severità del quadro clinico. Il soggetto invece eterozigote è apparentemente sano in cui si può
mettere in evidenza la banda anomala della HB S mediante elettroforesi, ma con la quantizzazione fatta con
cromatografia vediamo circa un 40% di HB S. anche se è asintomatico è importante conoscere queste cose
per il soggetto doppio eterozigote. Fu descritto nel 1920 di soggetti anemici che avevano ereditato un gene
beta talassemico da uno dei due genitori ed un gene responsabile dell HB S (si chiama microdrepanocitosi,
è una doppia eterozigosi). In questo caso mancherà la sintesi dell HB A. il quadro clinico simula quello che
abbiamo visto nell anemia a cellule falciformi, e la gravità è tanto maggiore se l’eredità del gene falciforme
sia beta 0 o beta+. Lo studio elettroforesi ci permetterà di visualizzare le bande per diagnosi. L’HB E è una
HB in cui in nella catena beta posizione 26 amminoterminale un residuo di acido glutammico è sostituito da
lisina. Nei soggetti omozigoti determina una sintomatologia molto sfumata, e tanto più negli eterozigoti.
Quello che è importante sono le doppie eterozigosi, perché per esempio se un soggetto eredità da uno dei
due genitori il gene dell’HB E e dall’ altro il gene beta Talassemico ne può venire fuori una situazione che
clinicamente è qualcosa di intermedio tra il morbo di cooley e ma beta talassemia intermedia.
WWW.SUNHOPE.IT
Anemia Sideropenica (III gruppo)
La volta scorsa ci siamo fermati alle talassemie e alle emoglobinopatie.
Un'altra anemia del terzo gruppo molto importante, perché è quella più diffusa, è l’anemia sideropenica.
Diciamo subito che il fabbisogno del ferro nel sesso maschile è pressoché la metà di quello del sesso femminile,
considerando la donna nel periodo fertile, ovviamente per le perdite che si hanno nel flusso mestruale. Infatti, il
fabbisogno fisiologico reale giornaliero di ferro nei maschi è di 1 mg/die, mentre nella donna è di circa 1,8 mg /die.
Dove possiamo perdere il ferro, fisiologicamente, ogni giorno? Innanzitutto nelle feci, nella desquamazione
cellulare, una piccolissima parte anche con quelle cellule che troviamo nel sedimento urinario.
Col flusso mestruale, considerato normale, c’è una perdita dai 10 ai 20 mg/ die e nella gravidanza dai 700 ai 900
mg/die di ferro.
In quanto a contenuto, nelle carni e negli altri alimenti si ha un contenuto considerevole di ferro, però quello che è
importante notare è come la quota che viene assorbita, soprattutto dalle verdure, è una quantità nettamente più
bassa. Si tratta di un ferro difficilmente assorbibile. Quindi, in realtà, quello che è importate non è solo il contenuto
di ferro, ma anche in quale forma si trova questo ferro, se è facilmente assorbibile.
Dove si assorbe il ferro? La massima parte viene assorbito a livello del duodeno e della parte prossimale del
digiuno. La prima cosa che dobbiamo considerare è che, negli alimenti, il ferro si trova sotto forma di Ferro Ferrico
(Fe 3+) e per essere assorbito ha bisogno di essere ridotto a Ferro Ferroso (Fe 2+). E questo già ci dice una cosa
importante, perché tutte quelle condizioni che riducono l’acidità gastrica, con resezione gastrica o con trattamento
con anti-acidi…. (il prof non finisce la frase, ma credo volesse intendere che tutte queste condizioni possono
influenzare l’assorbimento del ferro.)
Ecco questo schema rappresenta la cellula duodenale… (la slide rappresenta una cellula enterica e il meccanismo di
assorbimento del ferro)
Ora nei villi vi è un enzima che è il citocromo p-prelipasi duodenale (non riesco a capire bene, però ricordavo si
chiamasse “ citocromo duodenale B”), che opera questa riduzione del ferro. A questo punto il ferro ferroso può
entrate nella cellula duodenale e viene veicolato ad opera di una proteina trasportatrice di metalli bivalenti. A
questo punto il ferro deve essere estromesso dalla cellula duodenale. L’unica proteina in grado di determinare
l’export verso il sangue è la ferroportina. Prima di passare nel sangue il ferro deve essere ri-ossidato. Così il ferro
trivalente viene legato dalla apoferritina, una proteina che può legare due ferro allo stato trivalente e può veicolare
il ferro o all’interno del midollo e degli eritroblasti, che hanno il recettore per la transferrina, o può essere
immagazzinato all’interno della cellula epatica.
Consideriamo che tutto questo meccanismo è regolato da un altro proteina sintetizzata dalla cellula epatica, che si
chiama Epcitina. Questa funziona legandosi alla ferroportina , degradandola. Quindi, se la Epcitina degrada la
ferroportina, è chiaro che sarà ridotta la quantità di ferro, la quale viene esportata dalla cellula duodenale verso il
sangue. Viceversa se l’Epcitina è ridotta, aumenta la quantità di ferro che passa il circolo e veicolata nei tessuti.
Fin’ora abbiamo parlato del ferro inorganico, quello che è maggiormente presente nella dieta. Però la quota di
questo ferro, che viene assorbita, non è preponderante. E’ molto meglio assorbito il ferro eme, cioè il ferro
dell’emoglobina e anche in questo caso, a livello della cellula duodenale, vi è un recettore specifico e un
trasportatore, che porta l’eme finché non viene degradato dall’eme- ossigenasi. Il ferro viene liberato e segue la
stessa via che abbiamo vista prima.
Ricapitolando, come vi dicevo il ferro nell’emoglobina, nella mioglobina, rappresenta, nella dieta, soltanto il 20%,
mentre l’80% è ferro inorganico.
Volevo dirvi qualcosa in più per quanto riguarda il meccanismo di regolazione dell’Epcitina. L’Epcitina viene
prodotta anche in maggiori quantità quando vie è un processo flogistico cronico. Perché è stimolata dall’IL-6.
Quando vi è una maggior produzione di Epcitina, da un lato è bloccato l’assorbimento a livello della cellula
duodenale, ma viene bloccata anche la liberazione a livello delle cellule del sistema macrofagico. Per cui, il ferro nei
depositi, della ferritina, che dovrebbe essere prontamente mobilizzato, non si mobilizza. Questo vi spiega il
paradosso di cui vi ho detto nella prima lezione. In realtà, nei pazienti che hanno una malattia cronica associata,
infiammatoria o neoplastica, c’è un’aumenta produzione di Epcitina e quindi abbiamo il paradosso di avere dei
depositi ben rappresentati, quindi una ferritina elevata e poi di una sideremia che risulta bassa e una transferrina
WWW.SUNHOPE.IT
che risulta ridotta o normale. Infatti, vi dissi guardatevi dal considerare l’assetto marziale fermandovi solo alla
sideremia. Perché altrimenti potreste fare l’errore di dare del ferro a questi pazienti, quando poi non serve.
Richiamiamo un poco le condizioni che possono aumentare o ridurre l’assorbimento del ferro.
Aumentato Assorbimento ferro:
Basso ph gastrico
Sostanze che favoriscono la riduzione del ferro
o Composti con SH ( cisteina)
o Acido ascorbico
o Rame
Ridotto Assorbimento ferro
Alterazioni anatomiche
o Assenza stomaco e intestino
Cause iatrogene
o Antiacidi
o Antibiotici
Alimenti
o Fosfati (uova)
o Acido fitico (cereali)
o Tannini (the, caffè)
o Amidi
Le cause della carenza.
Ridotto apporto alimentare
o Dieta Vegetariana stretta
o Dieta Lattea prolungata
o Dieta monotona nell’anziano
Ridotto assorbimento
o Achilia gastrica
o Gastroresezione
o Celiachia
Emorragie croniche
Vi voglio dire, che, in linea di massima, quando vi trovate davanti ad un soggetto si sesso maschile, con un anemia
iposideremica, pensate che la causa più probabile è uno stillicidio dal tubo digerente. Mentre, di fronte ad una
donna in periodo fertile, la causa più probabile è correlata al un flusso mestruale o a molteplici gravidanze.
Ricordatevi che i flussi mestruali vanno sempre analizzati per questa problematica, in base a com’è, il flusso
mestruale, abbondante, non lo è, quanto tempo dura, quanto tempo passa tra un flusso e quello successivo.
Per quanto riguarda il ridotto apporto alimentare, la dieta vegetariana espone ad una carenza di ferro, perché i
vegetali, anche se contengono una buona quantità di ferro, questa è poco assorbibile. Anche nel neonato che viene
allattato per periodi prolungati, c’è bisogno di un supplemento con il ferro. Oggi tutti i preparati sul commercio
presentano delle aggiunte. Nel soggetto anziano può essere trascurata una dieta equilibrata, soprattutto priva di
carne.
Le cause che portano ad un ridotto assorbimento di ferro già le abbiamo analizzate, soltanto tenete presente la
possibilità, che il paziente con l’anemia sideropenica possa avere una celiachia. Per cui, almeno in questo caso,
vanno richiesti gli anticorpi transglutaminasi per poi passare allo step successivo per la conferma.
WWW.SUNHOPE.IT
Per quando riguarda gli stillicidi che possono portare carenza di ferro, i più pericolosi sono quelli occulti, dal tubo
digerente, che possono essere causati da un’ernia iatale, un’ulcera peptica , un’erosione della mucosa gastrica o
duodenale o da una neoplasia.
-Sintomi
Voi sapete che il ferro non è presente soltanto nei globuli rossi e quindi non si trova solo nell’emoglobina, ma anche
nella mioglobina e in altre proteina. Questo significa che, nel paziente con anemia sideropenia, dobbiamo
distinguere un gruppo di sintomi che sono legati alla carenza di ferro nei tessuti e un gruppo di segni e sintomi
legati allo stato anemico. In realtà il gruppo di segni legati alla carenza tissutale precedono il quadro tipico
dell’anemia.
Abbiamo detto che il ferro è contenuto nella struttura della mioglobina. Questo spiega perché la facile esauribilità
muscolare di questi soggetti, la stanchezza precoce, può essere espressione della carenza del ferro nella
mioglobina e quindi precedere quell’astenia che dovrebbe essere invece legata all’anemia.
Inoltre sono caratteristiche le unghie, con la deformazione a “scodellina”, la presenza di striature, fragili. Anche i
capelli sono fragili. La glossite, lingua è arrossata, liscia. Possono esserci delle erosioni agli angoli della bocca dette
stomatite angolare.
I segni e sintomi dell’anemia li conosciamo benissimo e non li ripetiamo. Solo come vi ho detto nella prima lezione,
potrebbero arrivare nei vostri ambulatori donne con 7 g/dl di Hb e non lamentare la sintomatologia anemica. Sette
grammi di emoglobina rappresentano un’anemia di grado severo, ma questi pazienti vengono in ambulatorio con le
proprie gambe, senza lamentarsi. Questo è per dirvi quanto siano importanti i meccanismi di compenso
omeostatici, che si sono istaurati nel tempo, cosa che non avviene nell’anemia acuta anche di grado moderato.
Ormai dal 2000, si sono andate delineando delle relazioni tra l’anemia e le capacità cognitive.
-Clinica
In uno striscio di sangue con anemia sideropenica possiamo trovare:
Ipocromia (MCH ridotto)
Microcitosi (MCV ridotto)
Anisocitosi (RDW aumentato)
Poichilocitosi ( forme atipiche)
Per quanto concerne l’assetto marziale dobbiamo sempre chiedere la Sideremia, la Transferritinemia, la
Ferritinemia e il quadro tipico carenziale sarà:
Iposideremia
Ipertransferritinemia
Ipoferritinemia
Una volta che abbiamo diagnosticato con certezza che si tratti di anemia sideropenica, bisogna individuare la causa.
Dobbiamo cercarla tra le cause ovvie e quelle che potrebbero essere sconosciute.
-Terapia
La via di somministrazione principale per un trattamento completo è la via orale, sempre! Ricorreremo alla
somministrazione di ferro per via parenterale (solo per via endovenosa, quella intramuscolare non serve a niente)
solo in alcuni casi:
Deficit di assorbimento: resezione gastrica;
Terapia con EPO: maggiore fabbisogno di ferro, che crea una carenza funzionale;
Intolleranza alla terapia orale: i farmaci migliori spesso possono dare alcuni effetti collaterali (diarrea,
nausea, sapore metallico).
WWW.SUNHOPE.IT
Il migliore assorbimento del ferro avviene a digiuno, ad almeno 3 ore dai pasti, perché, altrimenti, il 40% si lega agli
alimenti e non viene assorbito. Anzi la cosa migliore è associare un po’ di acido ascorbico, Vit C, perché
ottimizziamo la conversione del ferro ferrico in ferroso.
La somministrazione per via endovenosa va prescritta solo in ambiente ospedaliero a causa di eventi avversi, tipo
shock anafilattico.
Se il ferro viene assorbito bene per via orale, passiamo al monitoraggio. Gli facciamo fare prima la conta dei
reticolociti, prima del trattamento e dopo una settimana. Se il ferro è stato assorbito in modo soddisfacente,
l’eritropoiesi si è rimessa in moto e avremo un aumento dei reticolociti dopo 7/8 giorni. Se invece non si verifica,
c’è qualche motivo di malassorbimento. Allora bisogna vedere cosa si può fare, cambiando preparato o ricorrendo
alla via parenterale.
L’incremento che ci dobbiamo anche aspettare è, per lo meno, di 1 g/dl di Hb dopo un mese. Se ciò non si verifica,
è probabile che ci sia più di una causa che giustifica la carenza di ferro. E’ possibile che una donna, che abbia dei
flussi mestruali abbondanti, abbia anche delle emorragie occulte della mucosa gastrica e quindi questo spiega il non
raggiungimento del livello di Hb, che ci saremmo aspettati.
Anemia Emolitica (IV gruppo)
Tutti i vari step precedenti sono normali: l’eritroblastogenesi è normale, la maturazione degli eritroblasti è
normale, la sintesi dell’emoglobina è normale, però queste emazie, quando passano in circolo, vivono poco.
Chiariamo prima un concetto. La differenza tra Emolisi e Anemia Emolitica:
Emolisi: riduzione della vita delle emazie < 120 gg
Anemia Emolitica: è quando la distruzione delle emazie supera la capacità di compenso del midollo
osseo, che è di circa 6 volte le sue funzioni basali. In questo caso la vita media eritrocitaria sarà < 20 gg.
Se invece la vita degli eritrociti è di 50/60 gg e c’è l’emolisi, l’anemia non compare e si parla di Emolisi compensata.
Abbiamo un insieme di caratteristiche che sono comuni a tutte le anemie emolitiche:
Espressione dell’emolisi:
o Vita media degli eritrociti <20 gg
o Aumento bilirubina non coniugata (sub itteo, ittero franco.)
o Aumento Stercobilinogeno e Urobilinogeno (Feci e urine ipercromiche.)
Espressione scompenso midollare:
o Iperplasia Eritroblastica midollare
o Reticolocitosi
o Aumento ricambio del ferro (da aumentato utilizzo del ferro da parte del midollo iperplastico e da
aumentata liberazione da emolisi.)
o Sideremia normale o aumentata
Queste caratteristiche sono essenziali delle anemie emolitiche extravascolari. Quando il globulo rosso invecchia,
l‘80% viene sequestrato dalla milza e abbiamo già detto l’emoglobina che fine fa: la parte proteica viene staccata
dall’eme, l’eme viene privato del ferro, la protoporfina 9 viene spaccata. Sappiamo anche che il 20% delle emazie si
rompe spontaneamente. Il livello massimo di questa emoglobina è di 0,3 g/dl. Questa emoglobina, che si libera dai
globuli rossi che si rompono in circolo, viene legata dall’aptoglobina. Essa può legare un’intera molecola di
emoglobina e tutto questo complesso viene rimosso dalla cellula epatica. Ma l’emoglobina libera in circolo si può
anche spezzare e l’eme derivante venire complessato o dall’emopessina o dall’albumina. Questi complessi vengono
rimossi dal circolo sempre dalla cellula epatica. Quell’emoglobina che si è liberata dalla rottura dei globuli rossi in
circolo, si presenta a livello del filtro glomerurale. Passa, arriva a livello tubulare, dove dei sistemi specifici
riassorbono questa emoglobina. Per questo motivo, nelle urine, non è presente emoglobina adultera.
WWW.SUNHOPE.IT
Voi capite, che, se la quota di globuli rossi che si rompesse in circolo fosse una quota enorme, tutti questi sistemi
verrebbero superati. Quindi una grande quantità di Hb arriverebbe a livello del rene, attraverserebbe il filtro
glomerurale e supererebbe le capacità di riassorbimento tubulare. Quindi troveremmo emoglobina nell’urina. Se
l’emolisi è di una certa entità, la evidenzieremo soltanto attraverso l’esame chimico fisico delle urine, ma se è
notevole la quantità di emazie distrutte, allora le urine diventeranno di un colorito rossiccio, rosso scuro,
emoglobinuria macroscopica. Quando abbiamo una crisi emolitica e la causa è una distruzione intravascolare, non
avremo più l’aptoglobina libera, perché sarà tutta impegnata a legare l’Hb liberata dai globuli rossi. Se prendiamo
un campione di sangue derivante da un’emolisi intravascolare, raccolto con un anticoagulante, e lo lasciamo
riposare, vedremo il plasma rossiccio, invece che giallo paglierino. Avremo anche un aumento della LDH.
Le anemie emolitiche possono essere dovuto a cause intracorpuscolari o extracorpuscolari. Noi vedremo soltanto
alcune delle prime.
Le anemie da causa intracorpuscolare o intraglobulare che tratteremo sono la Sferocitosi ereditaria e l’Anemia da
deficit della G6PD.
Sferocitosi Ereditaria.
La membrana plasmatica eritrocitaria è composta da un doppio strato lipidico, in cui il 70% è costituito da fosfolipidi.
Questo strato è attraversato, a tutto spessore, da proteine ed è ancorato a una rete proteica sottostante, che
costituisce il citoscheletro. Una delle proteine importanti che attraversa il doppio strato è la Proteina di banda 3.
Questa si lega con un’altra proteina di nome Anchirina, questo legame è ottimizzato dalla Proteina 4.2. L’Anchirina
ancora tutto quello che sta sopra al citoscheletro sottostante e, specificatamente, alla catena Beta della Spectrina.
Quindi, in questo caso, il doppio strato lipidico è ancorato al citoscheletro sottostante. Un’altra proteina integrale è
la Glicoforina C, che si lega a un filamento di actina, il quale, a sua volta, si lega alla Spectrina, in particolare,
all’estremità del tetramero della proteina. In ogni caso viene sempre realizzato un ancoraggio doppio strato lipidicocitoscheletro.
Nella Sferocitosi ereditaria vengono a mancare alcune di queste proteine. Ciò fa si che il doppio strato lipidico non
venga ancorato al citoscheletro sottostante. Quindi una quota del doppio strato viene perduta. Se ciò accade,
succede che la giunzione, che si verifica nello strato lipidico, fa in modo tale che il nostro globulo rosso si deforma,
non essendo più un disco biconcavo, ma una microsfera.
Con il termine Sferocitosi ereditaria indichiamo un insieme di difetti delle proteine appena citate. Queste sono
codificate da alcuni geni, cosa che ci spiega la non uniforme trasmissione ereditaria in alcuni soggetti rispetto ad altri.
Quando non avevamo queste conoscenze, dicevamo, orientativamente, che un 75% di questi pazienti aveva
trasmissione autosomica dominante e il 25% recessiva.
In realtà in questa patologia possiamo avere 4 condizioni:
Anchirina e Spectrina
Spectrina
Prot Banda 3
Prot 4.2
Ciascuna di queste condizioni può determinare perdita di parte della membrana e quindi Sferocitosi ereditaria.
-Fiosiopatologia
Le alterazioni proteiche determinano una disorganizzazione strutturale, con un perdita di una parte del doppio strato
lipidico. Di conseguenza, viene ridotta la superficie e mantenuto lo stesso volume. Questo spiega perché vengono
fuori le microsfere.
Inoltre queste emazie hanno un’aumentata fragilità osmotica e una minore deformabilità. Quando devono passare
attraverso le fenestrature dell’endotelio, a livello dei sinusoidi splenici, lo fanno con difficoltà, cosa che comporta un
WWW.SUNHOPE.IT
ulteriore danneggiamento della membrana eritrocitaria, finché alla fine la sfera viene sequestrata dai macrofagi
splenici e da’ un’ emolisi extravascolare.
-Clinica
Da tutto il ragionamento che abbiamo fatto, la triade caratteristica della Sferocitosi ereditaria sarà:
L’anemia: normocromica e normocitica;
Iperbilirubinemia indiretta (da emolisi);
Splenomegalia;
[qualcuno chiede perché prima il professore abbia parlato di microsferociti, se ora , invece, parla di anemia
normocitica. Il prof spiega che il volume medio degli eritrociti entra nel range fisiologico, ma tende verso il limite
inferiore. Quindi sono piccoli, ma non tanto da determinare microcitosi]
-Classificazione Clinica
Se noi abbiamo detto la condizione generale, la triade generale, ora dobbiamo considerare una cosa. Noi abbiamo
dei soggetti in cui l’anemia non c’è o potrebbe essere corpus colata (???) e questi soggetti vengono scoperti
nell’ambito di indagini familiari, perché è capitato un individuo in cui è stata fatta la diagnosi e quindi poi,
estendendo l’indagine in ambito familiare, sono stati trovati dei soggetti con delle caratteristiche molto sfumate o
non che non hanno affatto anemia. Questo vuol dire che, nell’ambito dei pazienti con Sferocitosi ereditaria, ci sono
pazienti asintomatici o oligosintomatici. La stragrande maggioranza di questi pazienti ha un’emoglobina compresa tra
8 e 12 g/dl. Questo significa che sono soggetti che possono avere un’anemia di grado moderato o lieve. C’è un certo
numero di pazienti, che, però, ha un livello di emoglobina al di sotto di 8 g/dl, cioè di grado severo, perché sono
trasfusione dipendente. Quindi, in realtà, questi sono i soggetti che rappresentano quella minoranza, la quale
necessita di un trattamento importante, quale la splenectomia. Questa non corregge il difetto biochimico, ma
elimina la serie di distruzione dei globuli rossi.
-Diagnosi
La diagnosi di Sferocitosi ereditaria si basa su:
Storia familiare positiva
Esame Clinico: Splenomegalia, Ittero
Esame di lab: iperbiliruminemia indiretta, aumento MCH, presenza di microsferociti
Test fragilità osmotica: emolisi in soluzione ipotonica.
-Complicanze
Litiasi biliare: è la più frequente, anche in altre anemie. Spesso possiamo avere una coeredità della sindrome
di Gilbert (il professore tiene a precisare che dire “ghilbert” sia sbagliato e che si pronuncia “ jilber”, perché
francese).
Crisi emolitiche: nel paziente, che ha già un’emolisi cronica, un’infezione può peggiorare l’entità dell’emolisi
e dell’anemia.
Crisi aplastiche: l’infezione con alcuni patogeni, come il parvovirus B19, agente eziologico della quinta
malattia, in un soggetto normale, può determinare un’inibizione transitoria dell’eritropoiesi con una
sintomatologia sfumata o del tutto inosservata. In un soggetto che ha già un’emolisi cronica, questo porta a
delle crisi aplastiche.
Crisi megaloblastiche: se il midollo osseo è cronicamente in attività, il fabbisogno di ac.folico risulta
aumentato. Quindi se non viene fornito, si può andare in contro a carenza e a macrocitosi.
-Terapia
Ac. Folico
Emazie concentrate ( solo in anemia di grado severo)
Splenectomia ( nei pazienti trasfusione dipendenti)
WWW.SUNHOPE.IT
Anemia da deficit della G6PD.
Richiamiamo qualche concetto di biochimica. Nei globuli rossi, gli enzimi, man mano che vanno in contro a perdita
di attività, non possono essere rimpiazzati, perché non è possibile la sintesi proteica, perché manca il nucleo. Il ciclo
di Krebs non è possibile, perché mancano i mitocondri. Sono possibili solo: la glicolisi anaerobia e il ciclo dei
pentosi.
Il primo serve per la produzione dell’ATP, importante per l’attività delle pompe di membrana, per mantenere
l’omeostasi; per la produzione del NADH, che serve per il mantenimento dell’enzima metaemoglobina reduttasi, il
quale fa si che l’emoglobina si mantenga allo stato ridotto; della 2-3 difosfoglicerato, la quale regola l’affinità
dell’emoglobina per l’ossigeno. Un suo deficit aumenta l’affinità dell’Hb per O2, determinando ipossia, produzione
di EPO, aumento dell’eritropoiesi e quindi poliglobulia.
Il ciclo dei pentosi, invece, è importante, perché, nella reazione che converte il glucosio-6-fosfato in glucosio-6fosfoglicerato, ad opera della G6PD, si produce il NAPDH, il quale mantiene allo stato ridotto il Glutatione. Questi è
importante, perché impedisce che avvenga l’ossidazione dell’emoglobina e, in particolare, dei gruppi sulfidrilici
liberi dell’emoglobina. Quindi capite che i livelli di glutatione ridotto sono scarsi, perché è carente il NAPDH, perché
manca l’enzima G6PD.
Questa è la causa di ciò che noi conosciamo come Favismo, che è un’anemia emolitica a trasmissione ereditaria Xlinked.
Conosciamo moltissime varianti della G6PD, ma quelle importanti sono divisibili in 3 gruppi:
Classe I: riguarda soprattutto la razza caucasica, dove il deficit enzimatico è importante (attività enzimatica
residua < 5%).
Classe II: sono ubiquitarie. Le crisi emolitiche si verificano sempre dopo ingestione di farmaci o di alimenti
che contengono sostanze ossidanti. L’attività enzimatica residua è un po’ più alta rispetto alla classe
precedente (attività enzimatica residua pari al 10%).
Classe III: l’attività enzimatica residua va dal 10 al 60 %. Le crisi emolitiche saranno clinicamente meno
importanti e sono autolimitanti.
La variante che conosciamo da noi è la variante mediterranea ed è quella che conosciamo come Favismo. Questi
soggetti, dopo aver ingerito fave e/o piselli, dopo un intervallo di tempo di 2-6 ore, vanno incontro ad una crisi
emolitica violenta intravascolare. (le emolisi intravascolari danno un quadro clinico sempre più grave di quelle
extravascolari). Nel caso d’ingestione di farmaci ad attività ossidante la crisi emolitica si verifica più tardivamente
rispetto alle fave, dopo circa 24 ore.
-Sintomi
Pallore
Ittero
Dolori addominali
Febbre
Urine scure
WWW.SUNHOPE.IT
Lezione di Ematologia
28-03-2014
Continuiamo con le anemie emolitiche , abbiamo visto quelle da causa intraglobulare ora vediamo quelle
extraglobulari di cui fanno parte le anemie immunoemolitiche . Quali sono le caratteristiche ? sono caratterizzate
dalla presenza di autoanticorpi adesi alla superficie eritrocitaria , se sono anticorpi adesi alla superficie
eritrocitaria è chiaro il perchè della positività al test di coombs diretto .Il quadro clinico e la sua gravità
dipendono dal tipo di anticorpo e dalle sue proprietà . Innanzitutto una prima differenziazione va fatta tra : le
anemie autoimmuni e quelle alloimmuni. Come suggerisce la definizione , le autoimmuni sono cosi chiamate
perché caratterizzate dalla presenza di un autoanticorpo e quindi non riconoscimento del self . Quello alloimmuni
invece sono delle anemie emolitiche in cui il sistema immunitario funziona correttamente è il caso della malattia
emolitica neonatale da incompatibilità Rh o la reazione emolitica trasfusionale ritardata . In questo gruppo molto
eterogeneo comprendiamo anche le anemie da isoagglutinine naturali come nel caso della MEN AB0 o come nel
caso di un errore trasfusionale quando a un soggetto viene trasfuso sangue non compatibile nell ambito del
sistema AB0.Sempre in questo gruppo poi abbiamo le anemie immunoemolitiche da farmaci .Innanzitutto
chiaramoci un po’ le idee riguardo il test di Coombs anzi ditemi voi cos è cosi ve lo ripetete ( nessuno risponde ).
Quando noi diciamo test di C. diretto che significa ? che cosa intendiamo? Vogliamo dire che..? che abbiamo
trovato ?.. chiariamole ora certe cose .. qual è la finalità del test di C. diretto ?valutare la presenza di anticorpi
adesi alla superficie del g. rosso, mentre invece la finalità del test di C. indiretto qual è? è quella di individuare
degli anticorpi che sono liberi nel sangue circolante .Allora quello che noi chiamiamo siero di Coombs , ma
impropriamente perché inizialmente era davvero un siero , ma oggi non è un siero , ma una miscela di due
anticorpi monoclonali diretti uno contro le IgG quindi anti-IgG e l'altro anticorpo monoclonale diretto contro la
frazione C3b del complemento. Quindi noi che abbiamo in questo caso ? una miscela di Coombs che continuiamo
a chiamare per consuetudine siero ma polispecifico proprio perché è una miscela di anticorpi .Esistono poi i
cosiddetti reattivi monospecifici costituito da anticorpi anti-IgG o anticorpo diretto contro la frazione C3b del
complemento .In prima battuta si utilizza la miscela polispecifica , in seconda battuta se positivo il test si utilizza il
reattivo monospecifico .test di Coombs indiretto , perché diciamo indiretto ? nello schemino – guardando la slide
– se sulla supeficie del g . rosso sono adesi gli anticorpi il nostro reattivo di Coombs avendo anticorpi anti-IgG Cosa
fa ? costituisce dei ponti tra le varie emazie che hanno adese sulla loro superficie molecole di IgG . Più ponti
costituiscono alla fine l’ agglutinato ed è quello che noi vediamo in basso a sn , e quello è un test di C. diretto
positivo . Se invece questa agglutinazione non si verifica , abbiamo una nubecola uniforme che si vede bene al
microscopio, certo oggi si ricorre alle tecniche su microcolonne .In realtà in queste colonne o c è del gel o delle
microbiglie che hanno delle proprietà standard : se è gel avrà una porosità standard . Allora il principio è sempre
lo stesso se si è formato l’agglutinato , l’ agglutinato è grosso e non passa e si accumula a monte test di C.diretto
(+) , se invece non si è avuta agglutinazione le emazie sono separate e passano e si accumulano sul fondo, quindi
test di C. diretto (-).
Nel caso in cui vogliamo identificare gli anticorpi liberi presenti nel siero che facciamo ? facciamo precedere tutta
questa fase da un’ altra fase ,fase in cui incubiamo il siero del pz in cui possiamo pensare ci siano degli anticorpi..
per esempio in una donna D(-), supponiamo abbia degli anticorpi anti-D supponiamo, facciamo incubare le
emazie con D(+). A questo punto gli anticorpi si andranno a legare alla superficie poi aggiungiamo il reattivo di C.
se si forma l’ agglutinato è positivo se no negativo. Quindi questo test perchè viene detto indiretto perché
facciamo precedere la prima fase suddetta alla fase in cui l anticorpo libero nel siero si possa legare al g. rosso
che possegga l’antigene verso cui l anticorpo si va a legare. Perciò test di C. indiretto .
Allora ragazzi io ci tengo molto a questo perché non è possibile che veda correntemente – io faccio lezione anche
agli specializzandi di medicina interna, geriatria , oncologia- e vedo comunque queste idee non sono chiare. Se voi
sapete come funziona la cosa vi rendete conto dell’uso razionale di questi test. Ora che abbiamo chiarito questo
cominciamo a parlare della malattia emolitica neonatale da incompatibilita Rh e da incompatibilità AB0.
WWW.SUNHOPE.IT
Questa è un’ altra storiella abbastanza semplice anche se poi alla fine non tutti hanno le idee chiare . Innanzitutto
quando noi diciamo incompatibilitò da Rh cosa vogliamo dire ?allora noi quando diciamo Rh non intendiamo un
antigene specifico ma intendiamo un sistema di antigeni . Nel sistema Rh infatti ce ne sono più di una 40ina e
noi non siamo qui a dirli tutti ma consideriamo il più importante. Quando diciamo che un soggetto è Rh (+) o Rh() che vogliamo dire ? che secondo la nomenclatura di Fisher questo soggetto possiede se è positivo o non
possiede se è negativo l’ antigene indicato con la D maiuscola . Lo stesso antigene Diner lo chiamò Rho ( Rh con
zero, con lo zero al pedice ). Quindi voi potete trovare nella nomenclatura americana l’ antigene D ,invece la
nomenclatura tedesca può anche indicare Rho, è la stessa cosa . Quindi Se un soggetto è A(+) vuol dire che
rispetto al sistema AB0 è di gruppo A rispetto al sistema Rh è positivo quindi ha l antigene . Nel caso specifico di
una donna D (-) supponiamo concepisca un feto D(+) . A questo punto cosa succede ? voi sapete bene che
durante la prima gravidanza non si ha nessun problema , alla nascita c’è maggiore probabilità di commistione. Ma
supponiamo che per un problema qualunque il contatto si sia verificato prima , nei primi mesi di gravidanza in
questo caso se i g. rossi del neonato sono passati nel circolo della mamma , il sistema immunitario di quest ultima
risponde producendo anticorpi che sono IgM , hanno struttura pentamerica tendono alla polimerizzazione per cui
non riescono ad attraversare la barriera feto- placentare e quindi non si verifica nulla .Se la donna poi ha altre
gravidanze incompatibili- non per forza la seconda - in linea teorica . Parliamo in linea teorica perché , perché
quando nasce da una donna D(-) un D(+) alla donna vengono somministate entro 72h max anticorpi contro l
antigene D di classe IgG. Queste IgG a che servono ? a legare le emazie del feto rimanenti circolanti nella
circolazione materna, ma essendo di classe IgG si fissano soltanto e non distruggono i g.rossi quindi cosa succede
? che i g. rossi ricoperti di anticorpi passano alla milza in quanto qui le cellule del sistema istocitario posseggono
recettori per la porzione Fc dell anticorpo ,vengono sequestrati , distrutti e spenta la risposta immunitaria .
Questo è un passo avanti che è stato fatto oggi la malattia emolitica del neonato è di rara osservazione . Questo
però è quanto riguarda la mamma . Però nel sangue del neonato quale test andiamo a fare per confermare la
possibilità di una malattia emolitica neonatale ? Test di C. diretto , perché gli anticorpi sono adesi alla superficie
del g. rosso, e questo lo possiamo fare direttamente sul sangue del cordone, sul sangue del funicolo. È chiaro che
il neonato verrà messo sotto osservazione, perché ? abbiamo una iperbilirubinemia non coniugata , normalmente
nel soggetto adulto il fegato può aumentare la captazione bilirubina fino a 4 volte ma nel neonato essendo il
fegato non al massimo della sua funzione , la bilirubina sale , sale il bambino viene messo sotto la lampada in
maniera tale che la bilirubina non coniugata venga trasformata in composti idrosolubili e eliminata. Cosa
importante è che se la bilirubina non coniugata raggiunge e supera il 20mg/dl essa attraversa a barriera ematoencefalica e causa ittero nucleare con lesione irreversibile dei gangli della base. Bisogna stare molto attenti e
prevenire questi problemi . oggi questi problemi pero si osservano molto raramente . ora vi faccio una domanda
in quale occasione il problema si potrebbe verificare già alla prima gravidanza ?risponde uno studente : “ dopo
una trasfusione “ , prof: esatto , dopo un intervento , o in seguito a evento traumatico ha avuto un emorragia è
stata portata in pronto soccorso sono state necessarie 4unità di sangue D(-) ma è disponibile solo D(+) quindi in
quel caso alla donna da un lato le si salva la vita dall altro la si condanna , perché se quella donna partorisce un
D(+) il problema insorge prima .quando una donna ha avuto una gravidanza e ha avuto d + ,nella seconda
gravidanza nella donna cosa andiamo a fare ? test di c. indiretto per vedere la concentrazione di questi anticorpi
che se tendono a aumentare vuol dire che c è una stimolazione in atto .
Men da incompatibilità AB0.
Voi conoscete sicuramente il vostro gruppo sanguigno… varie risposte .. Nell ambito del ambito del sistema AB0
non bisogna dire “0positivo “ oppure “ A negativo “ ma solo o A o B o 0, nell ambito del sistema Rh positivo o
negativo. La vostra collega è di gruppo 0 cosa significa ? che le emazie non hanno nessun antigene ma nel suo
sangue sono presenti anticorpi anti-A e anti-B, invece nel caso del gruppo A le emazie hanno antigeni anti-A e
anticorpi anti-B, lo stesso dicasi se qualcuno è di gruppo B , ha antigeni B e anticorpi anti-A . Questi anticorpi ,
queste agglutinine di che classe sono ? sono IgM.Ragion per cui teoricamente non ci sarebbe alcun problema visto
che non attraversano la barriera feto-placentare. In realtà si è visto che la donna di gruppo 0 che concepisca un
WWW.SUNHOPE.IT
feto di gruppo diverso, il neonato può avere problemi. Ma questo perché se gli anticorpi sono di classe IgM
?perchè ragazzi i soggetti di gruppo 0 oltre ad avere anticorpi anti-A e anti- B di classe IgM, possono avere una
piccola quota di anticorpi anti-A e anti-B di classe IgG che potrebbero creare qualche problema quindi già nella
prima gravidanza può verificarsi il problema, però la fortuna in un certo senso è che quasi mai quello che ci
saremmo aspettati si traduce in un evento clinico importante perché gli antigeni… supponiamo una donna di
gruppo 0 abbia concepito e poi partorito un neonato di gruppo A , gli antigeni A e B sulla superficie del neonato
durante la vita fetale e poi dopo la nascita sono antigeni che non sono completamente maturi, e quindi non
facilmente riconoscibili da parte degli anticorpi . D’altro canto voi sapete che gli antigeni A e B sono espressi non
soltanto sulla superficie dei g.rossi ma anche a livello di altri tessuti e sono presenti anche in determinate
secrezioni, per cui quando questi anticorpi della mamma attraversano la barriera feto placentare arrivano nell
organismo del neonato prima di raggiungere il bersaglio principale sulla superficie del g . rosso vengono in parte
neutralizzati da quegli antigeni tissutali o delle secrezioni del neonato . Quindi la quota di questi anticorpi che
arrivano al loro bersaglio sono pochi e per giunta non sono facilmente individualizzabili tutto questo porta a due
conseguenze . La prima sul piano clinico è che la bilirubina non coniugata aumenta ma non in maniera
drammatica e nella quasi totalità dei casi il neonato viene messo sotto la lampada e finisce lì. Ragazzi , se noi
andiamo a fare il test di C. diretto , in più della metà dei casi sarà negativo, ma sapete perché è negativo ?perchè
abbiamo detto che la quota di anticorpi che arriva al bersaglio è scarsa ed è al di sotto del limite di sensibilità del
test . Allora noi cosa dobbiamo fare quando c’è un’ incompatibilità AB0, dobbiamo rivedere anche il Test di
enuizione , che cerca di staccare questi anticorpi ,di concentrare questi anticorpi e utilizzarli. Ovviamente il fatto
che questi anticorpi si devono staccare in realtà che cosa significa ? come dobbiamo procedere ? ragazzi , la
reazione antigene <-/-> anticorpo è una reazione reversibile che ha un suo equilibrio e una volta raggiunto l’
equilibrio, all ‘ equilibrio chimico la quantità di antigene e anticorpo che si complessano a formare l’
immunocomplesso è uguale alla forma dissociata antigene – anticorpo. Significa quindi che questa è una reazione
reversibile e come tale c è un equilibrio tra la reazione diretta e quella inversa e ogni reazione ha una sua
costante, di associazione e dissociazione Ka e Kd .Se noi dobbiamo staccare questi anticorpi dalla superficie del g.
rosso , coinvolgiamo la costante di dissociazione quindi andiamo a spostare la reazione a sn e questo lo facciamo
o aumentando il calore o abbassando il Ph. Questo è il principio fondamentale su cui si basa il test di enuizione
.Dopo staccati andiamo a fare il test di C. indiretto.
Ragazzi ,ma chiedetevi una cosa voi sapete benissimo che la superficie eritrocitaria possiede tanti antigeni . Noi
quando diciamo gruppo sanguigno facciamo riferimento al sistema AB0 e Rh ma esistonosulla superficie del g.
rosso tanti altri gruppi , il problema- sebbene raro – si può verificare ogni volta per ogni gruppo sanguigno .
Reazione trasfusionale emolitica ritardata: supponiamo di avere un pz mielodisplastico( i pz mielodisplastici sono
pz che molto spesso vanno avanti a trasfusioni )quando facciamo la trasfusione la facciamo tenendo presenti i
sistemi AB0 e Rh ma non teniamo conto degli altri. Supponiamo che una donna o uomo politrasfuso sia negativo
per un antigene il DAF IA , dopo questa persona si sensibilizza e comincia a produrre anticorpi Inizialmente sono di
classe IgM. Poi supponiamo che in un futuro non lontano questo soggetto venga trasfuso nuovamente con un ‘
unità di sangue incompatibile nell ambito del sistema AB0 ma positivo per il DAF IA . Prima di una trasfusione
viene fatta una prova di compatibiltà pre trasfusionale , che deve essere fatta ogni volta prima di una trasfusione
. Si fa cimentando il siero del pz ricevente con il sangue del donatore . ela situazione del pz potrebbe cambiare
dopo ogni trasfusione perché ci potrebbe essere una stimolazione . Bene in questo caso al soggetto viene fatta la
prova di compatibilità e quest ultima è negativa , il pz viene trasfuso e dopo qualche giorno il pz continua a
sentirsi stanco gli facciamo l emocromo e non abbiamo quell incremento della bilirubina . vi dico che quando noi
trasfondiamo un ‘ unità standard, si tratta di emazie concentrate. Un’ unità standard in un individuo di 70Kg
determina un incremento della concentrazione dell Hb compreso tra 0,8-1 g di Hb, quindi è ovvio che in un pz che
pesa 100Kg l incremento è poco , viceversa in un soggetto dal peso minore .Questo soggetto viene trasfuso e
notiamo un subittero sclerale . Cos è successo ? in seguito alla nuova stimolazione immunitaria il sistema fa si che
vengano prodotte in breve tempo anticorpi di classe IgG che sono responsabili di un emolisi extravascolare .
Allora facciamo il nuovo prelievo al pz e lo inviamo al centro trasfusionale – che lo conserva per un max di 7ggWWW.SUNHOPE.IT
ritesta ciò che è stato fatto prima della trasfusione e conferma la negatività. Qualora però testa il sangue del pz
ottenuto dopo la trasfusione il test di compatibilità risulta positivo perché la concentrazione degli anticorpi , in
questo caso anti DAF IA , supera il valore soglia determinando la positività al testa.
Vediamo ora qualcosa sulle Malattie Emolitiche Alloimmuni. Come in tutte le anemie emolitiche sono
caratterizzate da :ridotta sopravvivenza degli eritrociti , al di sotto del limite che noi conosciamo che è intorno ai
20gg, presenza di anticorpi che mancano del riconoscimento del self e sono rilevabili mediante test di C. diretto.
Questi anticorpi possono avere un optimum termico a diverse temperature :nel caso della malattia emolitica da
alloanticorpo caldo vuol dire che esso ha un optimum di attività a 37°C, mentre quelli freddi a T più bassa .
esistono però anemie emolitiche in cui l alloanticorpo è bitermico cioè ha un optimum di attività a due T diverse.
Da cosa derivano ? ci potrebbe essere in seguito a un malattia infettiva , una modificazione della membrana
eritrocitaria che funge da autoantigene. È possibile quindi che durante un infezione virale vengano prodotti degli
anticorpi cross reattivi cioè diretti sia contro gli antigeni virali ma anche contro gli antigeni del g.rosso oppure
possono essere il frutto della comparsa di cloni proibiti , è quello che avviene nei linfomi , Leucemia Linfatica
Cronica.) . ricordate quando abbiamo parlato dell anemia perniciosa ? cosa abbiamo detto ? che l anemia
perniciosa è caratterizzata dalle presenza di anticorpi anti-fattore intrinseco che molto spesso questi pz
presentavano anche altri patologie importanti . Qualora il g rosso sia ricoperto da questi anticorpi esso viene
opsonizzato ,va a livello splenico, dove i macrofagi hanno il recettore per la porzione Fc delle Ig o del
complemento viene sequestrato e distrutto: distruzione extravascolare . ma è anche possibile che entrino in
gioco dei linfociti T , il g.rosso è opsonizzato il linfocita T che possiede o il recettore per Fc o per C3b in seguito al
legame , il linfocita T produce una certa quantità di citochine e il g rosso viene distrutto. Nel caso invece di
alloanticorpi di classe IgM freddi , le IgM sono in grado di provocare anche una possibile lisi intravascolare ,
perché possono attivare direttamente la cascata del complemento , quindi emolisi intravascolare . Le anemie
emolitiche da anticorpi caldi da un punto di vista clinico si distinguono in primitive e secondarie , le prime anche
dette idiopatiche , le secondarie possono essere secondarie a altre patologie ( LLC , Collagenopatie o infezioni ) in
genere queste a. e. hanno andamento più acuto nei bambini e nei soggetti giovani e invece in maniera meno
acuta negli anziani . la prognosi è nettamente migliore nei giovani. Come detto prima è un emolisi extravascolare
che avviene a livello splenico. Nel caso in cui a questa anemia si associa una piastrinopenia autoimmune (
Sindrome di Evans ) la diagnosi si basa sul test di C. diretto ovviamente quando questi alloanticorpi vanno a
saturare tutti i siti del g rosso non avendo più dove legarsi si troveranno liberi , per cui in un secondo momento si
potrà avere positività al test di C. indiretto.questi anticorpi sono diretti verso antigeni a larga diffusione . qual è il
trattamento di scelta ? è un trattamento steroideo con cortisonici e si ricorre alla splenectomia nei casi non
responsivi, un certo risultato in un 15-30% si ottiene somministrando endovena Ig – soprattutto nelle
piastrinopenie autoimmuni _ perché secondo voi ?perchè saturano i recettori per il frammento Fc a livell splenico
. risposta puo darla anche la ciclosporina oppure anticorpi monoclonali diretti verso CD20 espressa dai linfociti B .
che cosa accade invece nel caso delle anemie emolitiche da anticorpi freddi ?
Abbiamo visto si tratta di IgM, se noi prendiamo i sieri di quanti siamo qui incubiamo a 4°C, una certa quantità di
anticorpi IgM .. è importante la quantità che deve essere un titolo > o uguale a 1/64 a 4°C. queste IgM sono
dirette contro gli antigeni oligosaccaridici del g rosso. Questi anticorpi aderiscono a g rosso sempre a T < 37°C. in
realtà dove avviene l emolisi? Nelle a. e da anticorpi freddi questi hanno un andamento a “ pussè” cioè durante la
fase cronica l emolisa è extravascolare durante la riacutizzazione la emolisi è intravascolare . Nei bambini la a. e .
da anticorpi freddi si può verificare dopo infezioni in questo caso le IgM sono IgM policlonali, negli anziano sono
monoclonali e molto spesso è presente un apatologia linfoproliferativa di cui l a. e. fa parte del quadro clinico. La
prima cosa questi pz vanno tenuti al caldo , gli steroidi e la splenectomia sono poco efficaci. Terapeuticamente
una buona risposta si può avere con il clorambucil , ciclofosfamide e rituximab( risultati meno brillanti )
Emoglobinuria parossistica “ a frigore “ ( da freddo)
WWW.SUNHOPE.IT
È legata a una anticorpo bitermico che è in grado di fissarsi al g rosso a basse T ma è in grado di attivarsi solo a
alte T. Questo anticorpo si chiama anche emolisina bifasica di Donath-Landstainer. Un soggetto giovane ha
infezione virale guarisce , poi arriva il freddo e fin quando sta in ambiente freddo non succede niente pero nel
frattempo l anticorpo si lega al g rosso , a T più elevata attiva il complemento determina emolisi intravascolare e
quindi un emoglobinuria di un quadro acuto . in pratica la diagnosi si fa in laboratorio mettendo a contatto il siero
del pz con g rossi compatibili- emazie test- ( dello stesso gruppo sanguigno ) si incuba a 0°C per 30min dopo di che
la miscela viene a bagnomaria portata a 37°. Dopo di che la miscela assume un colorito rosso ciliegia perché si
sono rotte le emazie .
Anemia emolitica da protesi valvolare
Vi capiterà di osservare come in un soggetto a seguito di una sostituzione di unavalvola cardiaca presenterà
emolisi di lieve entità un abbassamento della concentrazione dell aptoglobina libera . cosa succede ?queste
emazie che vengono espulse con una certa v si rompono contro l anello valvolare . si ha emolisi intravascolare e
se questa è davvero importante si ha un a . e. intravascolare . possibilità questa da tener presente .
Malattie mieloproliferative croniche
Sono 4. E sono la Leucemia mieloide cronica , la policitemia vera , la trombocitemia essenziale e la mielofibrosi
idiopatica. La LCM sapete è caratterizzata da un ‘ alterazione citogenetica caratteristica che fu chiamato
cromosoma philadelphia , infatti le altre tre vengono indicate come filadelpia negative. Nella LMC è presente un
difetto molecolare definito ed è la mutazione di Jack2 ,codificato dall omonimo gene . in questo schema vedete
un recettore per l EPO . in condizione normali .. innazittutto jack2 si trova sul versante citoplasmatico . jack2 ha
diversi domini uno per il legame con il recettore uno catalitico e uno inibitore dell att catalitica . es EPO quando
sul recettore dell EPO arriva EPO si innescano una serie di modificazioni conformazionali che innescano una
cascata di trasduzione .jack2 innesca una serie di fosforilazione che signfica trasdurre il segnale . se per effeto di
una mutazione che consta in una sostituzione in posizione 617 di un residuo di valina con uno fenilalanina questo
determina la costante attivazione di jack2 e quindi un continuo stimolo proliferativo a livello nucleare . questo è
quanto accade nel 95% di policitemia vera , 50-60% dei casi di trombocitemia e anche nei casi di mielofibrosi
essenziale.una volta che abbiamo capito questo possiamo parlare della policitemia vera . per qeusta perenne
attivazione di jack2 . quando ci troviamo un npz che presenta ematocrito dell 55-60% che dobbiamo fare ? questa
è legata a una malattia linfoproliferativa o aumento dll EPO allora già la epo ci consente di sapere se è un aforma
primitiva o secondaria . quali sono le forme secondarie ? secondarie alla aumento di epo : ipossia , Bpco ,
dimunuzione della press parziale dell O2, cardiopatie congenite , vivere in alta montagna . L ipossia fa aumentare
la produzione di epo , quando la massa eritrocitaria aumenta , aumenta la viscosità e l ossigenazione diminuisce .
anche nei soggetti con diminuzione del fosfoglicerato l ossigeno viene ceduto con difficoltà, anche nei forti
fumatori , perche aumenta la carbossiemoglobina oppure inadeguata produzione di epo.
WWW.SUNHOPE.IT
LE MALATTIE MIELOPROLIFERATIVE
PROF. GUSTAFIERRO
01/04/2014
POLICITEMIA
La policitemia vera è caratterizzata da una proliferazione trilineare anche se la proliferazione eritroide è
preponderante, ciò porta un aumento della massa di eritrociti. Quindi abbiamo un paziente che anche se
fosse asintomatico, all’emocromo abbiamo un aumento dell’emoglobina e dell’ ematocrito. Dobbiamo
differenziare tra forme primitive e secondarie; l’aumento della massa eritrocitaria, può essere secondaria
ad un aumento dell’EPO, quindi il dosaggio di questa ci permette di capire se l’aumento è diretta
conseguenza di un aumento di EPO; ovviamente nelle forme primitive non c’è dipendenza con un aumento
di EPO. Le condizioni alle quali segue un aumento di Hb sono le Policitemie secondarie. Dobbiamo
considerare che le Hb patologiche con una elevata affinità per l’O₂ portano un aumento di produzione di
EPO, ma un aumento di EPO lo possiamo avere anche nelle neoplasie renali. Si parla di pseudo policitemia
quando c’è una riduzione del volume plasmatico insieme ad un valore percentuale dell’HCT aumentato.
Questa è una malattia della 5/6/7 decade di vita e nel rapporto tra i sessi i maschi sono leggermente più
colpiti. A livello del midollo osseo possiamo notare la presenza di due popolazioni di cellule staminali; il
clone neoplastico prolifera anche in assenza di EPO, inoltre questo clone patologico ha un alta affinità per
fattori di crescita IL-3, CSF, trombopoietina. In alcuni pazienti è iperespresso il gene PRV-1. Fondamentale è
la mutazione del gene JAK-2; questa mutazione è stata identificata nel 2005 e nel 95% dei casi è una
mutazione dell’esone 14, in una piccola percentuale di casi dell’esone 12. La mutazione consiste in una
sostituzione nell’esone 14 in posizione 917 della proteina JAK-2 con una valina che viene sostituita da una
fenilalanina. L’aumento di massa eritrocitaria è responsabile della sintomatologia dei pazienti a causa di
alterazioni emodinamiche (aumento della viscosità), ma anche proteine possono determinare alterazioni
della viscosità oltre alla massa eritrocitaria: fibrinogeno e una IgM (struttura pentamerica). Se consideriamo
un aumento di concentrazione di Hb, aumenta anche l’ossigenazione dei tessuti periferici, che è ottimale
tra i 12 e 16 g di Hb, ma all’aumentare dell’Hb l’ossigenazione peggiora e questo è legato proprio all’
aumento della viscosità. Il quadro clinico è caratterizzato da segni e sintomi minori e maggiori. Tra i segni e
sintomi minori: molti pazienti lamentano, dopo un bagno caldo, prurito (probabilmente dovuto a
liberazione di istamina), disturbi del microcircolo cerebrale (cefalea, vertigini, disturbi visivi, difficoltà di
concentrazione, ronzii auricolari), disturbi circolatori agli arti inferiori (tromboflebiti, claudicatio
intermittens, acrocianosi, fenomeno di Reynaud), parestesie. Segni e sintomi maggiori sono: ictus cerebrali,
infarto del miocardio, angina pectoris, embolia, attacco ischemico transitorio. Fattori predisponenti per
eventi trombotici sono età >60 anni, fumo, estro-progestinici, precedenti eventi trombotici, piastrinosi. [Il
prof fa vedere delle immagini] il viso è rosso vivo, acceso; le sclere sono rosee, le mani hanno aspetto lucido
(aspetto laccato). Un terzo dei pazienti presenta ipertensione arteriosa che si normalizza quando la massa
eritrocitaria è portata a livelli più bassi; si può presentare splenomegalia. La splenomegalia è rilevante nel
paziente con mielofibrosi idiopatica e nel paziente con leucemia mieloide cronica. Nella trombocitemia
essenziale e policitemia vera e secondaria la splenomegalia ci può essere ma non è rilevante. Sono presente
alterazioni citogenetiche, ma non abbiamo un marker specifico; si può presentare nel 30% dei casi c’è
delezione sul braccio lungo del cromosoma 20 oppure una monosomia del 9 o ancora una trisomia dell’8 o
una trisomia parziale del braccio lungo del cromosoma 1 (nessuna è caratteristica). Nel 2008 sono stati
definiti i criteri minori e maggiori. I criteri maggiori sono: 1) Hb >18,5 g/dl nel maschio e >16 g/dl nella
donna, 2) presenza di mutazione JAK-2. I criteri maggiori sono: 1) Biopsia osteomidollare che mostra
proliferazione trilineare eritroide, granulocitaria, megacariocitaria, 2) riduzione di EPO, 3) colonie eritroidi
spontanee in vitro. Per la diagnosi devono essere presenti i due criteri maggiori oppure un criterio maggiore
WWW.SUNHOPE.IT
e due minori. La sopravvivenza è di 10/15 anni in genere si hanno complicanze trombotiche; E’ possibile
una evoluzione o verso mielofibrosi o verso una leucemia mieloide acuta. L’evoluzione in mielofibrosi si
verifica in un 20% dei pazienti e avviene perché il midollo che inizialmente produce una grande quantità di
globuli rossi, dopo un po’ questa iperproduzione si spegne e si assiste ad una normalizzazione di Hb e HCT e
a volte si ha anche una riduzione di attività eritropoietica midollare facendo diventare i pazienti trasfusione
dipendenti a causa della fibrosi midollare. Nel 2-5% dei pazienti abbiamo una trasformazione in leucemia
mieloide acuta quasi sempre non linfoide dopo circa 8/10 anni. La terapia si avvale di salassi isovolemici con
i quali togliamo sangue intero e diamo una quantità equivalente di liquidi; possiamo utilizzare IFNα oppure
una terapia citoriduttiva con idrossiurea (HU) e pipobromano. L’algoritmo terapeutico della policitemia
vera se il paziente ha più di 60 anni si utilizza HU che è il farmaco di scelta; se inferiore a 50 anni IFNα; se
tra 50 e 60 anni si può pensare ad utilizzare HU. In tutti i pazienti si fa una profilassi per la trombosi con
antiaggreganti piastrinici (acido acetilsalicilico 100 mg/die). L’obbiettivo del salasso è ridurre la massa
eritrocitaria per diminuire la viscosità; nel maschio l’HCT deve essere del 45% nella donna 42%. Si deve
controllare anche l’assetto marziale perché l’iposideremia porta eventi trombotici. Si somministra HU da
500 a 1500 mg questa variabilità è dovuta alle diverse risposte interindividuali al farmaco e si può abbinare
ai salassi. Gli effetti collaterali sono: disturbi gastrointestinali, stomatite, pigmentazione, rischio di epatite.
Negli anziani che rispondono poco all’HU si utilizza il pipobromano. IFNα può essere somministrato
trisettimanalmente o con il Peg-IFN una monosomministrazione settimanale. Come antiaggreganti
piastrinici si usa Aspirina 100mg/die che riduce la mortalità del 50% se non ci sono emorragie
gastrointestinali.
TROMBOCITEMIA ESSENZIALE
Malattia clonale caratterizzata da iperplasia megacariocitaria e da trombocitosi periferica. Si deve avere
una piastrinosi stabile nel tempo (>450000/mm3). La patogenesi è dovuta ad una proliferazione
incontrollata della cellula totipotente a prevalente differenziazione megacariocitaria, c’è ipersensibilità dei
precursori megacariocitari a IL-3 e IL-6. La diagnosi spesso è occasionale e si riscontra un aumento delle
piastrine; nell’ 80% dei casi non è presente nessun sintomo. Ci può essere trombosi a vari distretti nel 15%
dei casi; nel 5% emorragie, quando aumentano le piastrine le emorragie sono più frequenti delle trombosi
perché le piastrine legano il fattore di Von Willebrand e quindi c’è una ridotta disponibilità nel sangue. Ci
possono essere disturbi del microcircolo come nella policitemia; splenomegalia può essere presente ma
non considerevole; alcuni possono lamentare dolore o bruciore alle dita dei piedi e si può avere
arrossamento, si parla di eritromelalgia ed è caratteristico. I criteri diagnostici sono: piastrine stabilmente
>450000/mm3 e quindi bisogna ripetere l’emocromo più volte e trovare il valore sempre superiore; biopsia
osteomidollare con iperplasia megacariocitaria, assente o lieve iperplasia delle altre linee; assenza di criteri
per diagnosi di altre malattie mieloproliferative. Presenza di mutazione JAK-2, in assenza dobbiamo
escludere condizioni di accumulo di piastrine. Tutti i criteri devono essere soddisfatti per poter porre la
diagnosi. La trombocitosi secondaria si distingue in: acuta (intervento chirurgico [700000-800000/mm3],
dopo una emorragia, crisi emolitica, infezioni, chemioterapia); cronica (splenectomia o agenesia splenica,
anemia sideropenia, neoplasia, processi infiammatori cronici, malattie renali croniche). In queste condizioni
si parla di trombocitosi. Si effettua una stratificazione dei pazienti in base al rischio di trombosi: basso (<60
anni, assenza di precedenti trombosi, piastrine <1500000/mm3, assenza di fattori di rischio
cardiovascolari); alto (> o uguale a 60 anni e pregressa trombosi); intermedio (<60 anni, nessun evento
trombotico, piastrine >1500000/mm3, presenza di FDR cardiovascolari). La sopravvivenza nei primi 10 anni
è normale alle persone non malate, poi peggiora; è possibile una evoluzione in leucemia mieloide acuta. La
terapia si avvale di HU, antiaggreganti (non indicato quando le PLT >1500000/mm3 a causa di eventi
WWW.SUNHOPE.IT
emorragici e trombotici), IFNα, anagrelide (inibitore della megacariocitopoiesi, inizialmente sviluppato
come antiaggregante).
LEUCEMIA MIELOIDE CRONICA
E’ una neoplasia ematologica caratterizzata dall’espansione maligna di cellule della linea granulocitaria.
Marker citogenetico è la traslocazione reciproca t(9;22)(q34;q11) quindi una traslocazione tra 9 e 22 che dà
origine ad un cromosoma 22 più piccolo detto cromosoma Philadelphia. Comprende il 15-20% di tutte le
leucemie. Il punto di rottura sui cromosomi non è sempre lo stesso, ma si verifica in determinate aree. Sul
cromosoma 9 la rottura può avvenire in una zona di 300 kilobasi (kb) che codifica per un protoncogene. Sul
cromosoma 22 possiamo avere 3 aree: 1) 5,8kb esone 12-13 (gene BCR); 2) esone e’2 ed e2 del gene BCR;
3)rottura a valle dell’esone 19. Tutte le nuove proteine hanno attività tirosinchinasica (TK) nettamente
maggiore rispetto a quella normale; 3 proteine sono più frequenti P190 (alta attività), P210(media attività),
P230(bassa attività). La leucemia linfoblastica acuta porta P190, la leucemia mieloide cronica P210, la
leucemia mieloide cronica con elementi maturi P230. Quindi maggiore è l’attività della TK minore è il grado
di maturazione degli elementi. La proteina presenta una tasca per l’ATP dove il gruppo P viene trasferito
alla Tyr del substrato e innesca le fosforilazioni a catena con trasduzione del segnale di proliferazione.
Quindi abbiamo uno stimolo proliferante associato alla inibizione dell’apoptosi da contatto. E’ una malattia
a 3 fasi: cronica (3/4 anni), accelerata (3/4 mesi), neoplastica (3/4 mesi), exitus. Domanda d’esame: Perché
riusciamo ad arrivare alla diagnosi? Abbiamo due possibilità perché il pz va dal medico: 1) completamente
asintomatico ma ha un aumento dei globuli rossi e elementi mieloidi circolanti a maturità intermedia nella
linea granulocitaria (mieloblasto -> promielocito -> mielocito -> metamielocito), il laboratorio generalmente
segnala mielociti e metamielociti. Compare sazietà precoce e peso epigastrico per splenomegalia. Poi
possiamo avere febbre, febbricola e calo ponderale. La malattia da cronica sfocia, se non trattata, in fase
terminale in cui in circolo abbiamo solo cellule blastiche. Effettuiamo quindi un agoaspirato midollare,
analisi citogenetica e andiamo a valutare le traslocazioni. Tramite gli alchilanti gli elementi immaturi
sparivano ma è definito effetto cosmetico, modificando l’apparenza i pazienti comunque evolvevano nella
fase blastica. L’IFNα a dosi alte funziona da antiproliferativo. Trapianto di midollo con età >55 anni ha una
mortalità elevata (in questo caso il midollo rigetta l’ospite e non, come negli altri casi, il contrario, perché il
midollo del donatore sviluppa nell’ospite anche il vecchio sistema immunitario. Poi sono subentrati gli
inibitori della TK: imatinib (Glivec) che va ad occupare la tasca dell’ATP sulla proteina e poi è stato
sviluppato ponatinib.
WWW.SUNHOPE.IT
Lezione di ematologia del 09/04/2014
Mielofibrosi idiopatica
Tra le malattie mieloproliferative la mielofibrosi idiopatica è la più rara,
ma è quella a prognosi peggiore perché non abbiamo ancora un farmaco
efficace.
Si distinguono forme primitive e secondarie; la forma primitiva è la vera e
propria malattia proliferativa idiopatica, la forma secondaria può essere
non neoplastica:
1. da infezioni: tubercolosi, istoplasmosi
2. ipo-iper paratiroidismo
3. malattia di Gaucher
4. morbo di Page
5. sclerodermia
neoplastica:
1. policitemia vera: dopo la prima fase con aumentata produzione dei
globuli rossi i pazienti hanno una fibrosi del midollo e devono
ricorrere alle trasfusioni
2. trombocitemia essenziale
3. malattie linfoproliferative
4. mielodisplasie
5. tumori solidi: carcinoma mammario, polmonare, dello stomaco,
prostata
Quindi per ogni malattia mieloproliferativa cronica Philadelphia
negative dobbiamo distinguere le forme primitive da quelle
secondarie
Quadro complessivo della mielofibrosi idiopatica:
spiccata fibrosi midollare reattiva progressiva fino ad un quadro di
osteosclerosi
iperplasia megacariocitaria e granulacitaria
spessissimo emopoiesi extramidollare
WWW.SUNHOPE.IT
il prof mostra delle immagini dove indica la fibrosi, l’osteosclerosi, la
collagenizzazione e l’angiogenesi. La collagenizzazione deriva dal
deposito di fibrille in conseguenza dell’iperattività dei fibroblasti i quali
non sono monoclonali, ma policlonali. Se andiamo a vagliare l’attività
di questi fibroblasti notiamo che questa è normale quindi l’iperattività
deriva da stimoli esterni quali citochine prodotte da megacariociti e
monociti attivati, per questo si dice che la fibrosi nella mielofibrosi
cronica è reattiva
Epidemiologia
La caratteristica in comune con le altre malattie mieloproliferative è
l’età di incidenza tra la sesta e la settima decade di età, è una patologia
rara con un’incidenza di 0.3-1.5 / 100.000 anno, le cause di morte
includono la trasformazione in leucemia mieloide acuta evento possibile
fino al 20-23 % dei pazienti, tale percentuale è molto più bassa per le
altre malattie mieloproliferative (policitemia vera, trombocitopenia
essenziale)
Sintomi e segni
asintomatico, scoperta occasionale
astenia e pallore (sintomi connessi all’anemia)
sazietà precoce, peso epigastrico a causa della splenomegalia
perdita di peso
febbricola
petecchie (emorragie molto piccole, macchioline rossastre di
diametro di pochi millimetri) o ecchimosi causate da
piastrinopenia
spleno ed epatomegalia
La splenomegalia è tipica della mielofibrosi idiopatica e della leucemia
mieloide cronica mentre manca o è molto lieve nelle trombocitopenia
essenziale e leucopenia. Mentre 40 anni fa la splenomegalia era
identificata nell’80% dei casi diagnosticati oggi è rilevata in percentuali
più basse non perché la malattia sia cambiata, ma perché si è anticipato
il tempo di diagnosi anche allo stato asintomatico o poco sintomatico
con lieve splenomegalia
WWW.SUNHOPE.IT
Reperti di laboratorio MI (mielofribrosi idiopatica)
anemia normocromica normocitica
leucocitosi > 20.000/mm³ o normale o bassa
piastrinopenia abbastanza frequente, piastrinosi rara. Nella MI si
distinguono 4 stadi, nello stadio 1 no piastrinopenia ma è possibile
piastrinosi quindi si deve fare diagnosi diff con trombocitopenia
tramite biopsia osteomidollare
precursori mieloidi e/o eritroidi nel sangue periferico
emazie a lacrima (dacriociti) nel sangue periferico
piastrine più grandi del normale nel sangue periferico
aumento della lattato deidrogenasi
aumento dell’acido urico per aumentato catabolismo delle purine
dalla degradazione cellulare
mutazione Jak 2 (V617F) che si ritrova in diversa percentuale in
tutte le malattie mieloproliferative Ph -. Nella policitemia vera nel
95% dei casi la mutazione riguarda V617F corrispondente
all’esone 14, mentre nel 3-4% c’è una mutazione dell’esone 12;
nella trombocitopenia essenziale e nella MI c’è la stessa
mutazione V617F nel 50-60% dei casi
Caratteristica essenziale della MI
PUNCTIO SICCA: aspirato midollare secco o inconsistente, si procede
quindi alla biopsia osteomidollare e si rileva un CILINDRO BIANCO
COMPATTO (nei soggetti sani il cilindro non è bianco compatto ma si
vede la quota spongiosa rossa) che evidenzia un’ipercellularità con
aumento di fibre reticolari, deposito di collagene e megacariociti.
Affinché si possa parlare di MI l’analisi citogenetica deve dimostrare
l’assenza del cromosoma Ph (traslocazione Bcr/Abl 9 22 e quindi la
presenza della proteina di fusione) per escludere che si tratti di leucemia
mieloide cronica. All’esame citogenetico possono essere presenti altre
diverse anomalie ma NESSUNA è indicativa di MI
13q- anche nelle leucemia linfatica cronica, più altre
20q- anche in policitemia vera
Trisomia 8 (+8)
WWW.SUNHOPE.IT
Monosomia 7 (-7)
Delezione braccio lungo 7 (7q-)
Per evidenziare le mutazioni genetiche possiamo utilizzare la Fish o il
metodo convenzionale; la Fish necessita di cellule in interfase, bisogna
usare sonde specifiche quindi dobbiamo sapere cosa ricercare, il metodo
convenzionale con la tecnica del bandeggio evidenzia tutti i cromosomi
colorati a bande, ma necessita di cellule in metafase cioè in ciclo
cellulare e pertanto in malattie in cui l’indice di moltiplicazione
cellulare è basso è difficile trovare cellule in metafase e tale tecnica non
ci da risultati. IN CONCLUSIONE non essendoci un marcatore
specifico di mutazione la Fish non da informazioni aggiuntive per la
diagnosi di MI e il metodo convenzionale non si usa perché non da
risultati.
La splenomegalia può essere tale da raggiungere la fossa iliaca sx anche
in seguito alle numerose trasfusioni e bisognerebbe ricorrere alla
splenectomia, intervento complesso perché la milza crea numerose
aderenze
La sopravvivenza è tra i 4 ed i 6 anni con notevoli oscillazioni
Cause di morte
insufficienza d’organo
ipertensine portale
trasformazione leucemica ( 8-23% di casi nella prima decade dalla
diagnosi)
Opzioni terapeutiche
Nessun trattamento è specifico
Pz asintomatico
Osservazione
Pz sintomatico anemico
Trasfusioni e terapia con
danazolo e cortisonici con
WWW.SUNHOPE.IT
Pz con splenomegalia
risposte parziali e non in più di
1/3 dei casi
Splenectomia o irradiazione
splenica
La splenectomia comporta tra i rischi la possibile emorragia, la trombosi e
l’infezione da parte di germi capsulati infatti il paziente deve essere
vaccinato con anti pneumococcico, meningococcico ed emofilo.
L’irradiazione splenica cerca di ridurre il volume della milza, i probabili
inconvenienti sono la citopenia, la sepsi e l’emorragia
Nel caso ci sia un aumento del numero dei globuli bianchi possiamo usare
l’hydroxy-urea che inibisce l’enzima ribonucleotide reduttasi e quindi
limita la sintesi di deossiribonucleotidi, oppure busulfano (agente
alchilante).
Nella terapia farmacologica della MI possiamo inserire anche gli agenti
citotossici tra cui la Talinomide nota per le proprietà teratogene, fu ritirato
dal commercio perché le donne che lo assumevano durante la gravidanza
avevano maggiore rischio di generare neonati affetti da focomelia; in
Germania fu usato per curare alcune forme di lebbra
Talinomide: il meccanismo d’azione principale è quello di ridurre
l’angiogenesi e la neoangiogenesi, entrambi necessari per la proliferazione
neoplastica perciò fu usato nella terapia del mieloma e della MI; inolte
inibisce il TNFα, TNFβ, IL-1, GMCSF e stimola la proliferazione dei
linfociti T con effetto immunomodulante
Il derivato con minori effetti indesiderati e molto più potente della
Talinomide è la Lenalidomide, si tratta di un farmaco che non ha
un’indicazione specifica. Oggi si devono usare farmaci indicati tra cui gli
inibitori di Jak2 (farmaci di uso non routinario, i pz vengono inseriti in uno
studio clinico) che consentono di limitare l’attivazione incontrollata dei
processi di fosforilazione a valle attivati da Jak2 mutato e quindi limitare
la trascrizione dei fattori di crescita Jak2 dipendenti.
Un’altra opzione terapeutica nel pz giovane con donatore HLA identico è
il trapianto con cellule staminali allogeniche
WWW.SUNHOPE.IT
LEUCEMIA MIELOIDE ACUTA
Le leucemie acute possono essere mieloidi o linfoidi.
A differenza della LMC caratterizzata dalla possibile assenza di sintomi e
quindi scoperta occasionale, cellule granulocitarie mediamente immature
nel sangue periferico ed aumento dei globuli bianchi la spia della LMA è
la presenza di cellule completamente immature ,i blasti, o pochi neutrofili
maturi; abbiamo lo IATUS LEUCEMICUS cioè assenza di tutte le forme a
maturità intermedia
DOMANDA D’ESAME: alla presenza di un emocromo e striscio di
sangue periferico come fai a distinguere la LMC dalla LMA? Le distinguo
perche nella LMC ci sono elementi granulocitari a maturità intermedia
mentre nella LMA ci sono elementi altamente immaturi, i blasti
Nella LMC ci possono essere dei blasti, ma gli elementi a maturità
intermedia sono i più rappresentati
BLASTI: sono cellule che hanno perso il controllo proliferativo e la
capacità di maturare e differenziare, tuttavia conservano caratteristiche
morfologiche, citochimiche, immunofenotipiche che ci consentono di dire
a quale linea mieloide appartengono. Si distinguono nella LMA in base a
questi caratteri forme da M0 a M7 che possono comprendere dei
sottogruppi
LMA può insorgere improvvisamente in soggetti che sono stati bene fino a
pochi mesi o poche settimane prima [forme primarie], oppure si può
verificare in pazienti che hanno avuto una sindrome mielodisplastica o in
pz trattati con chemio o chemio+radio terapia per una neoplasia [forme
secondarie]. Le forme secondarie hanno una prognosi peggiore rispetto
alle LMA a insorgenza de novo perché correlano con alterazioni
citogenetiche complex cioè complesse e multiple
Leucemia acuta significa proliferazione improvvisa di blasti che infiltrano
il midollo, il sangue periferico e altri organi e tessuti. A livello midollare
l’infiltrazione è responsabile della sindrome da spiazzamento e
compromissione delle linee maturative normali
WWW.SUNHOPE.IT
(eritropoiesianemia, megacariocitopoiesipiastrinopenia,
granulocitopoiesiimmunodeficienza. Questa triade è la caratteristica
clinica della LMA)
Epidemiologia
Incidenza in rapporto all’età: 60% > 60a 37.8% età giovanile
in età pediatrica
Per le linfoblastiche l’incidenza è esattamente speculare
6.3%
Sintomi e segni
infezioni ricorrenti che non rispondono ai comuni antibiotici e di
difficile diagnosi anche dopo ripetute colture
manifestazioni emorragiche (piastrinopenia) ecchimosi, petecchie,
gengivorragie, epistassi
febbre (da rilascio di citochine)….questa parte non è completa
perché il file in questo punto è danneggiato
I segni ed i sintomi possono anche non essere tutti presenti quindi
l’intervallo tra l’inizio dei sintomi e la diagnosi può andare da 2 o 3
settimane ad alcuni mesi
Anche se stiamo parlando di leucemia dobbiamo ricordare che i globuli
bianchi non sempre sono aumentati di numero, possono essere normali o
addirittura ridotti di numero quindi il criterio diagnostico principale è la
presenza di blasti nel sangue periferico
anemia presente nel 90% dei casi con gravità variabile
piastrinopenia molto frequente, ma non sempre presente
IMPORTANTE: la diagnosi di LMA non viene fatta sul sangue periferico,
ma sull’ago aspirato midollare che deve dimostrare la presenza di blasti
per più del 20%
Molto spesso all’analisi dell’ago aspirato troviamo un monomorfismo
cellulare cioè il midollo è infiltrato da tipi cellulari con le stesse
caratteristiche morfologiche cioè cellule blastiche (fino all’80%)
WWW.SUNHOPE.IT
Sull’ago aspirato effettuiamo un esame:
morfologico che mette in evidenza i blasti
citochimico
immunofenotipico con citometria a flusso sono utilizzati anticorpi
monoclonali per evidenziare le molecole adese a queste cellule e quindi
sapere se queste sono mieloidi o linfoidi
citogenetico che ha non tanto un’importanza diagnostica quanto
prognostica e quindi stabilire la terapia migliore per questi pazienti
di biologia molecolare per vedere se ci sono riarrangiamenti
RIASSUMENDO:
1. il quadro clinico è la conseguenza della sindrome da spiazzamento e
dell’aumento di citochine
2. accanto al quadro clinico ci deve mettere in allarme non il numero
dei globuli bianchi, ma la presenza bi blasti
3. la diagnosi si fa sul midollo osseo
All’osservazione di una provetta si sangue periferico con EDTA potassico
(tappo viola) dopo lieve centrifuga si osserva un buffy coat, cioè lo strato
presente tra il plasma e parte corpuscolata, che contiene globuli bianchi e
piastrine, particolarmente spesso; la parte corpuscolata è invece meno
rappresentata
Immagine: si notano lesioni da immunodeficit tra cui placca da candida
albicans sulla mucosa orale, lesione da herpes simplex sulla rima labiale,
placca sul palato molle da candida albicans
La terapia chemioterapica essendo non selettiva verso le cellule blastiche
rende nella prima fase i pz più anemici, piatrinopenici e leucopenici e
quindi più sensibili ad emorragie ed infezioni, può capitare un’infezione
profonda da Aspergillus che normalmente è rara. La piastrinopenia + CID
causano petecchie da ricercare sulle parti declivi quali caviglie, superficie
volare dell’avambraccio, sotto l’occhio.
WWW.SUNHOPE.IT
La CID può complicare alcune forme di LMA come la M3 cioè quella
dove sono presenti i blasti che somigliano un po’ ai pro mielociti perciò si
parla di LA promielocitica
Immagine del fondo oculare: evidenza di emorragie retiniche
Ripeti classificazione FAB
Fattori prognostici
Età
Conta leucocitaria
Anomalie citogenetiche: t(8,21) associata ad M2, t(15,17) M3*
Risposta iniziale alla terapia, è fortemente negativo la mancata
risposta alla terapia di induzione e quindi si ritenta con una
reinduzione
Sesso (m>f)
Coinvolgimento del SNC, raro nella LMA più frequente nella LA
linfoblastica
la traslocazione 15,17 causa mutazione del recettore dell’acido retinoico
Nei pazienti anziani c’è una prognosi peggiore perché la LMA è
conseguenza di un’evoluzione di una sindrome mielodisplastica, c’è
l’associazione con cariotipi sfavorevoli (monosomia 5, 7), la presenza
di comorbilità
Terapia
La prima fase del trattamento chemioterapico si chiama terapia di
induzione basata generalmente sull’impiego di Daunorubicina per 3 giorni
e Citosina Arabinoside per i 7 giorni successivi (protocollo 3 + 7).
Questa terapia da una risposta positiva nel 70% circa dei pazienti che
mostrano assenza di blasti nel sangue periferico, normalizzazione dei blasti
nel midollo osseo al di sotto del 5%, quadro clinico risolto e ricomparsa
delle varie linee midollari tali da dare un numero di piastrine > 75.000 e di
neutrofili > 1.500, anemia lieve fino alla risoluzione.
Nel 30% dei casi però non c’è la remissione o a causa di morte del pz
durante la terapia di induzione o perché resistente, nel secondo caso si
WWW.SUNHOPE.IT
procede con la re induzione della remissione con un nuovo protocollo
chemioterapico usando farmaci diversi da quelli dell’induzione.
Remissione non significa guarigione perché se blocco la terapia durante la
remissione la malattia evolverebbe verso la fase acuta e porterebbe a morte
il paziente, quindi si procede con un secondo step intermedio tra induzione
e consolidamento, tale step ci è indicato dalla citogenetica in quanto in
pazienti con mutazioni meno sfavorevoli si preferisce la terapia mente in
pz con mutazioni sfavorevoli e condizioni di compatibilità HLA si
preferisce un trapianto.
Nel caso in cui la citogenetica ci mostra la mutazione 15/17 (LMA M3) il
protocollo prevede Daunorubicina + ATRA (acido al trans retinoico) che
permette di superare l’alterazione causata dal blocco del recettore
dell’acido retinoico. Tale protocollo e la conoscenza della mutazione
recettoriale hanno migliorato la prognosi dei soggetti con LMA M3
Maria Ceparano
Montaggio: Emanuela Granata
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
Abbiamo parlato del grosso capitolo delle malattie emoproliferative e abbiamo fatto fino alle
mieleoproliferative croniche e poi per un ultimo abbiamo trattato le leucemie mieloidi acute. Adesso
seguiremo più o meno lo stesso iter e cioè facciamo per un attimo le sindromi linfoproliferative croniche e
poi faremo la leucemia linfoblastica acuta. Le sindromi linfoproliferative croniche possono essere ad
espressione linfomatosa o ad espressione leucemica. Quelle ad espressione linfomatosa, capirete bene,
sono il linfoma di Hodgkin e i linfomi non Hodgkin. Quelle ad espressione leucemica sono rappresentate da:
leucemia linfatica cronica (LLC), leucemia prolinfocitica, leucemia a cellule capellute. Non abbiamo il tempo
di trattarle tutte quindi di queste tre leucemie vi parlerò della più importante e della più diffusa che è la
leucemia linfatica cronica (LLC). Perché ci tengo che questa malattia venga fatta bene e venga conosciuta
da tutti i medici? Perché è sicuramente la leucemia più diffusa nel mondo occidentale con cui nella propria
attività tutti, e dico tutti, avranno a che fare prima o poi.
Leucemia linfatica cronica (LLC)
È una malattia ovviamente monoclonale; nel 95-97% dei casi è una leucemia a linfociti B, soltanto nella
piccola percentuale restante si tratta di linfociti T. La proliferazione di queste cellule leucemiche che
apparentemente sembrano normali, ma normali non sono, comporta un’infiltrazione del midollo osseo,
una colonizzazione del sangue periferico e successivamente un’infiltrazione degli organi linfatici nelle sedi
extralinfatiche.
Guardate i tassi di incidenza per 100000 abitanti sull’asse delle ordinate e sull’asse delle ascisse l’età. La
prima considerazione che fate è che si tratta di una malattia dell’età anziana, tant’è vero che oggi l’età
media è intorno ai 65-70 anni, più spostato verso i 70-79 anni di età, quindi ancora più in alto. Vedete
invece come è una malattia che non esiste nelle prime decadi di vita.
Vi voglio far vedere questo grafico (mostra il grafico) per farvi giungere a una conclusione; questi dati sono
stati riportati dal registro tumori e tengono conto dei casi segnalati in diverse zone, città, regioni italiane,
separatamente per il sesso maschile e per il sesso femminile. In entrambi i casi vedete che sul fondo c’è la
città di Napoli; ma non perché a Napoli non ci sono segnalazioni ma perché a Napoli c’è un ridotto numero
di LLC. Considerate che noi viviamo in una regione in cui l’inquinamento penso che non abbia eguali , o
quasi, in Italia. Questa leucemia, quindi, ha poco a che vedere con i fattori leucemogeni tradizionali.
Ricapitoliamo:
È la forma più comune nel mondo occidentale;
Diagnosi media 65-70 anni;
L’incidenza complessiva aumenta con l’età;
I maschi sono più colpiti (M/F=2:1);
La sopravvivenza mediana è di circa 9 anni ma non c’è nessun altra popolazione di pazienti che ha
un’eterogeneità in quanto a sopravvivenza maggiore della LLC. Diciamo che la sopravvivenza a 9
anni ma è un dato puramente teorico perché abbiamo pazienti che vivono solo qualche anno e
pazienti che vivono anche più a lungo, dopo cercheremo di capire perché.
Abbiamo appena detto che non è rilevante l’esposizione ai comuni inquinanti che hanno un ruolo
leucemogeno certo, per cui non è influenzata dall’esposizione a radiazioni ionizzanti o ad agenti chimici.
Quello che si sa è che vi è una certa familiarità: nei parenti di I grado dei pazienti con LLC l’incidenza è da 2
a 7 volte maggiore rispetto alla popolazione di controllo.
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
La LLC è una malattia che come abbiamo già visto per le malattie mieloproliferative croniche, può avere un
quadro clinico che sfugge completamente, che è completamente negativo e la scoperta viene fatta in modo
occasionale. Meno frequentemente possono essere presenti sintomi sistemici, come per esempio astenia,
febbre, dimagrimento, sudorazione notturna, ma le due principali modalità con le quali il paziente va dal
medico sono, o perché ha fatto esami per altri motivi ed è stata scoperta una leucocitosi assoluta o perché
il paziente ha notato l’ingrossamento di qualche stazione linfonodale. Queste sono le due modalità più
frequenti con le quali questi pazienti si rivolgono al medico. Alcuni pazienti hanno notato, oppure è il
medico di base che ha notato alcune infezioni ricorrenti o addirittura delle complicanze autoimmuni
ricorrenti. Ricordo un nostro paziente che con una notevole precisione fu in grado di dirci come le comuni
afte del cavo orale erano aumentate di frequenza man mano che il suo numero di linfociti era aumentato.
All’E.O. vi possono essere delle linfoadenomegalie ma possono anche mancare. Vi può essere
epatosplenomegalia ma non necessariamente. È possibile anche infiltrazione di organi non linfoidi. Le
complicanze autoimmuni sono la conseguenza dello squilibrio immunologico.
Quali sono i criteri che devono guidarci alla diagnosi? Certo se noi vediamo un paziente con linfocitosi,
come vi ho detto nella prima lezione, non dobbiamo guardare il valore percentuale ma dobbiamo sempre
guardare il numero assoluto. Certo dobbiamo tener presente che questo aumento al di là di un
determinato limite che è 5000/m3 deve essere stabile nel tempo; quindi se vediamo un paziente con
6000/m3 non è che pensiamo subito che abbia la LLC ma cerchiamo di confermare nel tempo questo dato,
vediamo se questo paziente dopo un mese, tre mesi, sei mesi presenta stabilmente più di 5000/ m3.
L’infiltrazione linfoide del midollo deve essere superiore al 30%.
Noi abbiamo detto che nel 95-97% dei casi queste cellule sono linfociti B quindi esprimono le molecole
CD19 e CD20. Poi c’è bisogno che questi linfociti B CD19+/CD20+ abbiano altre due molecole, CD5/CD23.
Quindi : CD19+/CD20+ e nello stesso tempo debbono coesprimere CD5 e CD23, è importantissimo perché ci
consente di differenziare la LLC dai linfomi leucemizzati cioè quei linfomi che in una fase avanzata infiltrano
il midollo e colonizzano il sangue periferico, ma quegli elementi circolanti linfoidi che vediamo non sono gli
elementi della LLC anche se morfologicamente sono molto somiglianti. Infatti fino a 15 anni fa
confondevamo spesso LLC con i linfomi leucemizzati.
Essendo linfociti B, esprimono sulla loro superficie delle Ig però a bassa densità con restrizione clonale. Se
prendiamo un soggetto sano, andiamo a vedere i suoi linfociti B, vediamo che ci sono tanti linfociti B che
esprimono Ig con catene leggere quanti linfociti B che esprimono Ig con catene leggere . In questo caso
i linfociti leucemici hanno come catene leggere o tutte o tutte . Questo è un criterio di restrizione
clonale. Alcune molecole invece non sono quasi mai espresse da questi linfociti leucemici, come KMC7,
mentre le molecole CD22 e CD79b sono intensamente positive in altre malattie linfoproliferative croniche
ma non nella LLC a cellule B. Se noi abbiamo cellule linfoidi che esprimono fortemente CD79b o CD22 già ci
fa porre tanti punti interrogativi che si possa trattare di LLC e ci spinge a riconsiderare la diagnosi.
Foto di un piccolo linfocita leucemico: dimostriamo che sono cellule CD19+ e CD20+ quindi sono linfociti B;
devono coesprimere contemporaneamente CD5 e CD23. Per quanto riguarda le altre molecole vale il
discorso appena fatto.
Conoscere queste molecole di consente di usare un sistema a punteggio lo “Scoring System” che ci
consente di dire che se abbiamo almeno 5 punti si tratta di LLC mentre con un punteggio più basso si tratta
di elementi linfoidi di altre malattie linfoproliferative croniche che hanno colonizzato il sangue periferico
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
con particolare riferimento ai linfomi leucemizzati.
Morfologicamente possiamo trovare tre varianti:
LLC tipica, variante in cui sono presenti piccoli linfociti con caratteristiche morfologiche
sovrapponibili ai piccoli linfociti normali del sangue periferico;
LLC atipica, possono essere presenti un certo numero di prolinfociti. Per poter parlare di variante
prolinfocitica della LLC dobbiamo dimostrare che il numero di prolinfociti sia tra il 10% e il 55 %,
non oltre il 55% altrimenti siamo nella leucemia a prolinfociti;
Mixed cell type, variante in cui i prolinfociti sono meno del 10% ma sono presenti cellule di forma
svariata, elementi linfoplasmocitoidi o elementi linfoidi di dimensione maggiori rispetto a quelle
che abbiamo visto.
Foto di prolinfocita: rispetto al normale è più grande, la rima citoplasmatica è molto ridotta.
Prima ho calcato la mano sulla sopravvivenza media di 9 anni dicendovi però come ci sia una variabilità
enorme tra paziente e paziente per cui si è sempre sentito il bisogno di cercare di individuare dei criteri
prognostici che ci potessero far capire già al momento della diagnosi quali pazienti avrebbero potuto avere
una prognosi migliore o quali una prognosi peggiore. I primi criteri furono criteri semplici e furono criteri
stadiativi. Uno fu la stadiazione proposta da un gruppo di Houston, stadiazione che poi è stata rivista
rispetto alla stesura originale del 1975; poi c’è stata la stadiazione di Binet, fatta da autori francesi. Oggi
teniamo conto di ambedue. Sono importanti perché l’inquadramento in uno di questi stadi ci consente di
ottenere informazioni prognostiche.
Vediamo innanzitutto gli stadi della scala americana.
Stadio 0: Stadio della diagnosi ; sono presenti quelle condizioni di cui abbiamo parlato cioè,
numero di linfociti superiore a 5000/m3, una infiltrazione linfoide superiore al 30%, linfociti B
CD19+ e CD20 +, devono coesprimere CD5 e CD23 , vi deve essere l’espressione di Ig di superficie a
bassa densità ma con restrizione clonale con catene leggere o tutte o tutte . Il paziente è
asintomatico.
Stadio 1: è presente linfoadenomegalia.
Stadio 2 : è presente epatosplenomegalia. Voglio dirvi che deve essere sempre confermato lo
stadio della diagnosi. L’epatosplenomegalia è indipendente se vi è o non vi è linfoadenomegalia.
Stadio 3: è presente anemia indipendentemente se vi è epatosplenomegalia e linfoadenomegalia.
Stadio 4: è presente piastrinopenia.
Il criterio stadiativo di Binet invece tiene conto del numero di stazioni linfonodali interessate. Binet ha
stabilito tre stadi:
stadio A: se le stazioni interessate sono meno di 3;
stadio B: se sono interessate 3 o più stazioni linfonodali;
stadio C: anemia di grado moderato severo con Hb<10g/dl e piastrinopenia <100000/m3 senza prendere più
in considerazione le stazioni linfonodali.
Si è visto che i pazienti con stadio 0-1 della stadiazione americana possono avere una sopravvivenza di 1415 anni mentre pazienti con alla diagnosi stadio 3-4 hanno una sopravvivenza media di 3 anni. Vi sono
pazienti che vanno peggio.
Vi devo dire che un criterio molto semplice per valutare la prognosi del paziente con LLC è valutare il tempo
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
necessario affinché i linfociti raddoppino. Si capì che due pazienti a cui veniva diagnosticato lo stesso stadio
della stadiazione americana, ad esempio stadio 2, entrambi alla diagnosi con 20000 linfociti. Se dopo un
anno,uno dei due pazienti mostrava un numero di linfociti più che doppio, maggiore di 40000, aveva una
prognosi peggiore rispetto al paziente sempre in stadio 2 che dopo un anno, partendo da 20000 linfociti ne
presentava 25000-26000. Questo è un criterio prognostico abbastanza semplice: la prognosi peggiora se i
linfociti raddoppiano in tempi brevi.
C’è stato sempre il bisogno di trovare altri marcatori prognostici per poter definire alla diagnosi quali
pazienti hanno necessità di essere trattati. La diagnosi di LLC non necessita per forza di trattamento, quindi
dobbiamo fare una stratificazione. Negli ultimi 15 anni sono comparsi in letteratura scientifica un migliaio di
lavori scientifici sulla valutazione di fattori prognostici che si potessero adoperare praticamente nei pazienti
con LLC ma ben pochi sono quei fattori prognostici che poi hanno dimostrato di avere un’utilità pratica.
Sicuramente una delle scoperte più importanti in questo ambito è stata quella di mettere a punto delle
metodiche che consentano di individuare alterazione citogenetiche in questi pazienti. Prima degli anni
2000, per effettuare un’analisi citogenetica dei pazienti con LLC facevamo ricorso alla tecnica tradizionale
del bandeggio, che è una tecnica che richiede che le cellule debbano trovarsi in metafase. Se queste cellule
sono a basso indice di crescita allora o vengono stimolate e mandate in metafase, cosa che non è facile,
oppure non succedeva nulla. Bene, fino al 2000 i pazienti con LLC con alterazioni citogenetiche non
superavano il 30% ma in realtà non era così. Nel 2000 un gruppo tedesco pubblicò sul New England Journal
of Medicine i risultati di uno studio. Questo gruppo tedesco non aveva adoperato la tecnica citogenetica
tradizionale, bensì la FISH cioè l’ibridazione in situ che può essere utilizzata con cellule che si trovano in
interfase utilizzando delle sonde ben definite, sapendo quindi quello che si va a cercare. Ebbene con un
pannello di sei sonde questo gruppo riuscì a dimostrare alterazioni citogenetiche in più dell’80% dei
pazienti con LLC, quindi il 30% era legato alla limitazione del metodo usato, alla limitazione tecnologica, ma
non alla realtà. Il merito più grande di questo gruppo tedesco fu quello di correlare le alterazioni
citogenetiche con la sopravvivenza.
Grafico: sulle ordinate la sopravvivenza, sulle ascisse i mesi. Le curve sono di colore diverso a seconda
dell’alterazione citogenetica riscontrata. I pazienti con delezione del braccio lungo del cromosoma 13 sono
la curva in verde. Vediamo la mediana tracciando la linea orizzontale che passa per il 50%. Questa va ad
intersecare la curva verde più o meno a livello di un valore che riportato sulle ascisse corrisponde
sicuramente a oltre i 130 mesi di sopravvivenza. Invece in pazienti che hanno a parità di stadio, una
delezione del braccio corto del cromosoma 17 la sopravvivenza è intorno a 30 mesi.
Quindi vi è una notevole variabilità a seconda dell’alterazione citogenetica. Questa è stata una scoperta di
notevole valore per la prognosi dei pazienti.
Un’altra scoperta è stata fatta nei primi mesi del 2000: i linfociti leucemici in pazienti con LLC non
originavano tutti da uno stesso stadio o momento maturativo. Voi sapete che i linfociti a un certo punto
devono passare attraverso il centro germinativo, incontrano l’antigene,subiscono delle mutazioni
somatiche. Hanno individuato che quei pazienti i cui linfociti leucemici originavano da linfociti che non
erano ancora passati per il centro germinativo e che avevano un assetto non mutato dei geni che codificano
per la parte variabile delle catene pesanti delle Ig, avevano una prognosi peggiore rispetto a quei pazienti i
cui linfociti leucemici erano originati da linfociti già passati per il centro germinativo e in cui si era già avuta
la mutazione somatica dei geni che codificano per le catene pesanti delle Ig. Questo significa originare da
due stadi, uno precoce e uno a maggiore maturazione per cui quelli che originavano da linfociti con assetto
più maturo avevano una prognosi migliore, quelli che originavano da linfociti naive avevano una prognosi
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
peggiore.
Altro criterio prognostico è dato dall’espressione della molecola CD38. Si è visto che pazienti con LLC i cui
linfociti esprimessero in più del 30% la molecola CD38,la prognosi era peggiore rispetto a pazienti i cui
linfociti CD38+ erano meno del 30%.
Vi ho detto solo una minima parte dei fattori prognostici, in realtà quello che vorrei che vi rimanesse è:i
criteri stadiativi; il tempo di raddoppiamento dei linfociti; le alterazioni citogenetiche; l’assetto mutato o
non mutato;l’espressione di CD48.
Come vedete da questo grafico il 70% dei pazienti al momento della diagnosi è costituito da pazienti
asintomatici, solo il 30% presenta già una sintomatologia e sono questi i pazienti che devono essere trattati
perché hanno una breve aspettativa di vita. Per tanti anni non avevamo mezzi efficaci per trattare pazienti
con LLC. Per decenni l’unico farmaco che avevamo era un alchilante, il Clorambucil, ma quando i pazienti
erano resistenti al Clorambucil oppure diventavano resistenti perché venivano trattati, rispondevano,
recidivavano a un certo punto rispondevano sempre di meno. Non avevamo alternative. L’unica possibilità
era associare al Clorambucil piccole dosi di cortisonici. Poi fu scoperta la Fludarabina, un analogo purinico.
Poi fu adoperata l’associazione fludarabina+ciclofosfamide, fin quando nel 2003 un gruppo statunitense di
Houston utilizzò in pazienti che non erano stati mai trattati precedentemente l’associazione ciclofosfamidefludarabina-anticorpo monoclonale che conoscevamo dal ’98,il rituximab,anticorpo monoclonale anti CD20,
che adoperavamo con successo nei linfomi non Hodgkin dolenti, soprattutto per il follicolare e
successivamente nel linfomi B aggressivi. L’associazione di questi tre farmaci rituximab-ciclofosfamidefludarabina consentì di ottenere un risultato che mai nessuno aveva riportato: 67% di risposta, che era un
risultato straordinario. Lo stesso gruppo che con enfasi aveva riportato questi risultati 3 anni dopo gettò
l’acqua sul fuoco. Arruolò in un nuovo studio solo pazienti con più di 70 anni proponendo la stessa
associazione. In molti casi la tossicità relativa al trattamento fu inaccettabile. Allora cosa si capì? Se
avevamo raggiunto un gold standard nella terapia di questi pazienti, non avevamo considerato che come
sempre la terapia non può prescindere dalle condizioni del paziente, e noi avevamo visto che sono solo
pazienti anziani. Allora dobbiamo fare prima una stratificazione dei pazienti in rapporto alle patologie
associate. D’altro canto sapete benissimo come l’età biologica può non corrispondere all’età anagrafica ,
posso avere una persona di 75 anni in ottime condizioni generali e uno di 60 anni che ha molte patologie.
Bisogna tener conto dell’età anagrafica e soprattutto biologica ovvero di quelle patologie associate, le
comorbidità. L’obiettivo allora deve essere differenziato.
Definiamo tre gruppi di pazienti in base al “fitness status”:
Paziente Fit: paziente completamente indipendente, non ha comorbidità associate. È un paziente
che ha una normale aspettativa di vita. L’obiettivo è di ottenere il massimo:prolungare la vita e
cercare di annullare la malattia minima residua, annullando la malattia minima residua migliora la
sopravvivenza;
Paziente unfit: paziente che ha qualche comorbidità associata, ha un “performance status” non
più ottimale però è un paziente che può ancora giovarsi di determinati trattamenti. Il nostro
obiettivo è di prolungare la sopravvivenza libera da malattia. La LLC è una malattia che pur
rispondendo ai trattamenti prima o poi ricade. Noi dobbiamo fare in maniera tale che le ricadute si
verifichino a intervalli sempre più lunghi. Questo è l’obiettivo in questi pazienti. Mentre nel
paziente fit noi possiamo adoperare il gold standard ovvero la triplice associazione, nel paziente
unfit possiamo aggiungere al rituximab un chemioterapico che ha una tossicità ridotta. Oggi quello
che gode della maggiore considerazione è la Bendamustina, in prima linea dai primi mesi di
WWW.SUNHOPE.IT
Ematologia, prof.Guastafierro.
11-04-2014
quest’anno. È un farmaco la cui formula di struttura per certi aspetti richiama gli alchilanti ma allo
stesso tempo ha un anello benzimidazolico che è tipico degli analoghi purinici. Capiamo, quindi,
dalla struttura che con questo farmaco possiamo unire gli effetti degli alchilanti e degli analoghi
purinici, a fronte di una tossicità accettabile. Un’altra possibilità è fare rituximab+clorambucil, il
vecchio farmaco che conosciamo e di cui abbiamo già parlato.
Paziente frail:gravi handicap, ridotta aspettativa di vita, molte comorbidità associate. L’obiettivo è
ottenere una palliazione dei sintomi, solo un miglioramento della qualità di vita. Parliamo di
trattamenti di supporto, se ha bisogno di essere trasfuso lo trasfondiamo, se ha l’anemia gli diamo
l’EPO, ma il nostro intento è esclusivamente quello di ottenere un miglioramento della qualità di
vita.
La terapia nei confronti di pazienti con LLC è così evoluta: prima non avevamo niente o quasi; poi abbiamo
identificato i farmaci veramente efficaci ma ci siamo accorti che non tutti i pazienti con LLC potevano
giovare di questi farmaci, allora la migliore stratificazione è in base al fitness status e in base alla
stratificazione abbiamo diversi obiettivi e diversi trattamenti.
WWW.SUNHOPE.IT
Lezione ematologia 29/04/2014
Prof.Guastafierro
LINFOMA DI HODGKIN
Le malattie linfoproliferative croniche possono essere ad espressione leucemica o linfomatosa.
In quelle ad espressione leucemica abbiamo incluso :leucemia linfatica cronica, leucemia a cellule
capellute, leucemia prolinfocitica. Le malattie linfoproliferative ad espressione linfomatosa comprendono i
linfomi: linfoma di Hodgkin e i linfomi non Hodgkin.
Thomas Hodgkin nel 1832 descrisse sette casi di pazienti che aveva seguito dall’inizio di questa malattia non
identificata e di cui aveva fatto anche l’autopsia, dopo la morte. Per più di 160anni non abbiamo saputo
quale fosse la cellula di origine di quella che allora si chiamava malattia di Hodgkin e che oggi, più
correttamente, chiamiamo linfoma di Hodgkin perché sappiamo che nel 98-99% dei casi deriva dai linfociti
B e nei casi restanti potrebbe originare dai linfociti T.
Hodgkin descrisse l’ ingrossamento dei linfonodi, la splenomegalia presente in questi pazienti ma non
spiegò la patogenesi della malattia. Verso la fine del XIX secolo ,nel 1898, Carl Sternberg descrisse una
cellula plurinucleata che frequentemente era riscontrabile nei preparati di questi pazienti.
Indipendentemente dalla sua descrizione Dorothy Reed descrisse la stessa cellula in preparati ottenuti da
linfonodi degli stessi pazienti. Questa cellula è stata etichettata, dal nome di coloro che l’hanno scoperta,
cellula di Reed-Sternberg. Entrambi i patologi diedero un’interpretazione diversa alla presenza di questa
cellula nei preparati: Sterberg pensò che la malattia descritta da Hodgkin avesse a che fare con il bacillo di
Koch; Reed,invece, si rese conto che questa cellula non aveva nulla a che fare con la tubercolosi perché
molti dei suoi preparati erano stati prelevati da pazienti che sicuramente non avevano avuto infezione
tubercolare.
Definizione:
Neoplasia di derivazione B-linfocitaria nel 98-99% dei casi caratterizzata dalla presenza di cellule
voluminose di Reed-Sternberg, ma anche dalle cellule di Hodgkin.
Quelle di Reed-Sterberg sono cellule giganti plurinucleate con citoplasma abbastanza basofilo, quelle di
Hodgkin sono mononucleate. Queste cellule sono circondate da una serie di cellule reattive, che non
hanno carattere patologico, ma consentono di giustificare varianti istologiche del linfoma di Hodgkin.
Eziopatogenesi:
Il virus di Epstein-Barr ha un suo ruolo,ma da solo non è in grado di determinare la malattia. C’è bisogno del
susseguirsi in step successivi di diversi fattori come: l’espressione di bcl-2,p53 ect.ect.
La variabilità di quadri istologici è spiegata dal fatto che l’ evento mutageno colpisce la cellula linfocitaria in
varie fasi di maturazione.
Modalità d’insorgenza:
Questa patologia ha un andamento bimodale. Vi sono due picchi: uno corrispondente alla terza decade di
vita, il più consistente, e un altro intorno alla sesta/settima decade di vita, di misura inferiore e più marcato
per il sesso maschile che per quello femminile.
WWW.SUNHOPE.IT
Quadro clinico:
Il paziente può essere completamente asintomatico ed essersi accorto di una linfoadenomegalia
superficiale. All’ ispezione la cute soprastante i linfonodi è normale, non presenta segni di flogosi; i
linfonodi sono indolenti spontaneamente e alla palpazione, hanno consistenza aumentata, sono
scarsamente spostabili sui piani superficiali e profondi e tendono a confluire in pacchetti. Caratteristiche
diverse dalla linfoadenomegalia della leucemia linfatica cronica in cui non si ha, almeno in fase iniziale, la
tendenza a riunirsi in pacchetti, i linfonodi sono discretamente mobili, sono aumentati di consistenza ma
non sono così duri come i linfonodi del linfoma di Hodgkin.
Il paziente può anche presentare un quadro clinico caratterizzato da:
linfoadenomegalia,
febbricola o febbre ondulante, raramente continua o remittente, il rialzo termico inizia nelle prime
ore del pomeriggio per poi abbassarsi nel corso della tarda sera accompagnandosi a profusa
sudorazione,
calo ponderale superiore al 10% del peso corporeo negli ultimi sei mesi,
prurito generalizzato e irrefrenabile, non associato ad alcuna manifestazione cutanea che possa
giustificarlo, è quindi un prurito “sine materia”,
tosse secca e stizzosa o dispnea, qualora vi sia interessamento delle stazioni linfonodali
mediastiniche.
Diagnosi:
L’ UNICO esame valido per la diagnosi è la biopsia escissionale di uno o più linfonodi, effettuando
successivamente l’esame istologico e l’esame immunoistochimico. La biopsia deve essere escissionale
perché deve essere rispettata la capsula. Facendo l’ago aspirato ,se siamo fortunati ,potremmo anche
evidenziare la presenza di cellule di Reed-Sternberg ma, è un esame citologico che non ci chiarisce quale
variante istologica abbia il nostro paziente. L ‘ esame istologico ci deve dare quindi le informazioni
strutturali per poter fare una buona diagnosi. Nell’ asportazione linfonodale, se il paziente presenta un’
interessamento di più stazioni linfonodali superficiali, è bene evitare di fare la biopsia ai linfonodi inguinali
e ai linfonodi laterocervicali perché sono quelli che più frequentemente sono sede di eventi flogistici
ripetuti e quindi possono dare quadri più difficili da interpretare.
L’ ago aspirato del midollo osseo ha finalità stadiative. Perchè se troviamo una infiltrazione linfoide del
midollo osseo possiamo già inquadrare il paziente al quarto stadio.
La TAC e la PET sono utili per comprendere quanto sia estesa la malattia.
La PET è una tomoscintigrafia globale corporea a emissione di positroni. Nell’esecuzione di questa tecnica
viene adoperato l’ isotopo18 del fluoro coniugato con desossiglucosio.I tessuti neoplastici , caratterizzati
dall’avere un metabolismo marcatamente più accentuato, captano il fluorodesossiglucosio . Esistono
condizioni in cui questo marcatore può essere captato da cellule non neoplastiche. Oggi la PET viene
effettuata in coregistrazione con una TAC a bassa risoluzione, per far in modo che si possa individuare con
una buona precisione l’ organo,il tessuto che è sede della captazione del fluorodesossiglucosio.
WWW.SUNHOPE.IT
La PET è molto importante nel linfoma di Hodgkin :
in fase di stadiazione
in fase di ristadiazione precoce dopo chemioterapia, dopo le prime 4 sedute.Se le condizioni
regrediscono allora la terapia sta facendo il suo effetto, ma se il quadro PET resta uguale o,
addirittura, peggiora allora questo è un segno che spinge il medico a cambiare protocollo
terapeutico.
Per distinguere il tessuto cicatriziale, esito della terapia antineoplastica, dal tessuto ancora sede di
malattia.
Ristadiazione al termine di malattia e follow-up
Stadiazione di Ann Arbor:
Stadio I: interessamento di un unico linfonodo o di un’unica stazione linfonodale o un unico sito
extralinfonodale
Stadio II: interessamento di due o più linfonodi o due o più stazioni linfonodale, ma tutte allo stesso lato
rispetto al diaframma, cioè o tutte stazioni sovradiaframmatiche o tutte stazioni sottodiaframmatiche
Stadio III: interessamento di stazioni linfonodali contemporaneamente sopra e sottodiaframmatiche
Stadio IV: malattia diffusa (interessa anche milza, fegato,polmone ectect.)
Ciascuno stadio è diviso in stadio A e B a seconda che siano assenti o presenti uno o più di questi segni e
sintomi :
presenza di febbre >38°
calo ponderale > 10% del peso corporeo totale degli ultimi sei mesi
profusa sudorazione notturna, se di lunga durata
Questi sintomi vengono definiti per questo “sintomi B”.
Revisione stadiazione di Cotswold:
Divisione delle stadio III in stadio
III 1 interessamento stazioni linfonodali profonde dell’addome superiore
III2 interessamento stazioni linfonodali profonde dell’addome inferiore
Concetto di massa Bulky:
si parla di massa Bulky addominale se il diametro della massa addominale supera i 10cm
si parla di mazza Bulky toracica quando il diametro della massa è 1/3 del diametro trasverso del torace.
N.B. Anche per i linfomi non Hodgkin usiamo lo stesso tipo di stadiazione.
WWW.SUNHOPE.IT
Varianti istologiche:
abbiamo un prima grossa divisione tra il tipo classico e la variante a predominanza linfocitaria.
La forma a predominanza linfocitaria è caratterizzata dalla presenza di cellule che in un primo tempo
furono chiamate cellule linfoistiocitiche, ma in realtà si capì che non si trattava di istiociti, queste cellula
sono circondate da un gran numero di linfociti,perciò è definita variante a predominanza linfocitaria, e nell’
80% dei casi questi linfociti, che circondano le cellule definite linfoistiocitiche,tendono a formare pseudo
noduli, ma la distribuzione dei linfociti può essere anche diffusa. La caratteristica delle cellule
linfoistiocitiche è data da un nucleo polilobato, tanto che vengono chiamate cellule “pop-corn”. Questa
forma è una variante che si discosta molto da quella classica come epoca d’insorgenza perché interessa la
quarta decade di vita ed ha un andamento clinico che ricorda molto quello dei linfomi non Hodgkin a bassa
aggressività, per molto tempo non richiede alcun trattamento, ma ha una peculiarità potrebbe evolvere nel
tempo in linfoma non Hodgkin.
Nel tipo classico di linfoma di Hodgkin vengono incluse quattro varianti istologiche:
la variante a sclerosi nodulare, la variante più descritta nel mondo occidentale ed a prognosi
migliore
variante a cellularità mista
variante a deplezione linfocitaria
variante ricca in linfociti caratterizzata dalla presenza di cellule di Reed-Sternberg o di Hodgkin,a
differenza della variante a predominanza linfocitaria
Terapia:
Una cosa da sapere prima dei diversi protocolli terapeutici è che non dobbiamo dare né più né meno della
terapia che necessita il singolo paziente.
In base ai fattori di rischio(rappresentati da presenza di massa bulky, l’ età superiore a 50anni, VES elevata,
coinvolgimento di 4 o più stazioni linfonodali) possiamo dividere i pazienti in:
stadio precoce favorevole
stadio precoce sfavorevole
stadio avanzato
Le possibilità terapeutiche sono :
chemioterapia
radioterapia
terapia ad alte dosi con trapianto di cellule staminali autologhe
terapia ad alte dosi con trapianto di cellule staminali allogeniche
Chemioterapia
Il primo protocollo efficace è stato il protocollo MOPP (Mecloretamina, Oncovin=Vincristina, Procarbazina,
Prednisone) ; la Mecloretamina ben presto non è stata più prodotta in Italia ed è stata sostituita dalla
ciclofosfamide per cui quel protocollo diventò protocollo COPP, ma attualmente in disuso.
Il protocollo ABVD( Adriamicina, Bleomicina, Vinblastina,Dacarbazina) è superiore e meno tossico del
protocollo COPP.
WWW.SUNHOPE.IT
Ogni ciclo di ABVD originariamente era suddivisa in due bracci (braccio A e braccio B),effettuate a due
settimane di distanza l’una dall’altra, tra le due l’unica differenza era nel dosaggio della dacarbazina.
Attualmente, pur parlando sempre di un ciclo a due bracci, non vi è più alcuna differenza tra il braccio A e il
braccio B.
Il protocollo BEACOPP(Bleomicina, Etoposide, Adriamicina, Ciclofosfamide,Oncovin= Vincristina,
Procarbazina e Prednisone) è il protocollo più articolato e tossico e viene adoperato nei pazienti in stadio
avanzato.
Quello che si adotta è il” BEACOPP-escalated” cioè una combinazione di questi sette chemioterapici la cui
posologia viene scalata nel corso del trattamento.
È importante stratificare i pazienti i maniera tale da dare la dose giusta per il singolo caso.
In stadio precoce favorevole (In stadio I o II di Ann Arbor senza nessun fattore di rischio)
È sufficiente fare una 2 cicli ABVD (cioè 4 sedute) con radioterapia esclusivamente sulle stazioni linfonodali
sedi di malattia( dose di radiazioni tra 20/30 Gy)
La sopravvivenza globale a 5 anni di questi pazienti è superiore al 95%
In stadio precoce sfavorevole (in stadio I o II ma con almeno un fattore di rischio)
È opportuno fare 6 cicli ABVD (cioè 12 sedute) con radioterapia sulle sedi linfonodali coinvolte da malattia
(dose di radiazioni tra 20/30 Gy che può essere aumentata a 30/36 se è presente massa Bulky)
La sopravvivenza globale a 5 anni è superiore al 90%
In stadio avanzato
Si possono fare 6-8 cicli del protocollo BEACOPP associato a radioterapia su sedi linfonodali coinvolte da
malattia perché PET positive post-chemioterapia.
Si può anche optare per 8 cicli ABVD( cioè 16 sedute).
Sicuramente il protocollo BEACOPP è più efficace,ma è anche più tossico.
Trapianto con cellule staminali autologhe
Si ricorre a questo tipo di terapia quando il paziente ricade dopo poco tempo dalla fine della terapia di
prima linea, oppure quando il paziente già durante il trattamento di prima linea dimostra refrattarietà.
Trapianto con cellule staminali allogeniche
Quando il paziente è recidivato dopo aver effettuato l’ allotrapianto, oppure quando, pur essendo un
paziente candidabile all’autotrapianto , non è stato possibile ottenere una quantità di cellule staminali
sufficienti dal suo midollo.
Problematiche successive alla terapia
L’80% dei pazienti con Linfoma di Hodgkin sopravvive bene, e gran parte di questi dopo il protocollo di
prima linea non ha più problemi.
WWW.SUNHOPE.IT
Confrontando nel tempo questi pazienti con soggetti di età comparabile è possibile notare un aumento del
rischio dello sviluppo di seconde neoplasie in questi pazienti. Questa maggiore suscettibilità a secondi
eventi neoplastici è legata proprio al trattamento effettuato per il Linfoma di Hodgkin.
Le seconde neoplasie sono costituite da:
1. tumori solidi
2. leucemia mieloide acuta secondaria o una malattia mielodisplastica
L’ insorgenza di una neoplasia solida si può verificare dopo una latenza dalla fine del trattamento per il
linfoma per lo più superiore ai 10 anni, valutando i casi che si verificano nel tempo la curva non raggiunge
mai un plateau, ma c’è sempre un incremento nel tempo, ciò significa che il rischio di avere una neoplasia
solida non diminuisce mai,anzi. Queste neoplasie solide si riscontrano più facilmente in pazienti trattati più
“pesantemente” con la radioterapia.
La leucemia mieloide acuta secondaria o una malattia mielodisplastica in genere insorge nella prima
decade dalla fine del trattamento, mediamente dopo 3-4 anni. Queste patologie invece sono da mettere in
rapporto alla chemioterapia.
Ecco perché è necessario stratificare i pazienti!!Per effettuare una terapia ad hoc per il paziente, e così
ridurre al minimo il rischio di seconde neoplasie.
Altre complicanze sono rappresentate da:
complicanze cardiache,
in particolare da associare alla tossicità miocardica dell’adriamicina a dosi superiori di 400-450 mg/m2.
Coronarosclerosi precoce in pazienti che hanno fatto radioterapia mediastinica
complicanze polmonari,
fibrosi polmonare per effetto della radioterapia , o fibrosi interstiziale per effetto della Bleomicina
endocrinopatie,
sterilità maschile, oggi con il protocollo ABVD si manifesta in minor percentuale rispetto al vecchio
protocollo MOPP/COPP, in ogni caso si può proporre al paziente di effettuare la criopreservazione del
liquido seminale
per quanto riguarda il sesso femminile le complicanze sono limitate e si manifestano spesso solo con
menopausa precoce
tireopatie
infezioni
nella fase che segue il trattamento, perché quest’ultimo li rende immunodepressi.
WWW.SUNHOPE.IT
Ematologia
29-04-2014
LINFOMI NON HODGKIN
Noi chiamiamo linfomi non Hodgkin un insieme di patologie diversissime, che si differenziano dal linfoma di
Hodgkin e complessivamente sono circa una trentina di forme diverse. In realtà sono delle neoplasie
maligne che derivano dalla replicazione clonale dei linfociti B,T e cellule NK. Possono avere una
localizzazione linfonodale,ma anche extranodale. Costituiscono il 4-5% di tutte le neoplasie:una stima per
l’Italia del 2013 era di circa 13000 casi. Ci sono alcuni linfomi che hanno una peculiare distribuzione
geografica, basta pensare per esempio al linfoma di Burkitt in Africa o al linfoma T di tipo nasale in Asia.
Certamente il sesso maschile è più colpito e il rapporto M:F è 1,4:1. L’incidenza aumenta con l’età anche se
non tutti i linfomi in rapporto con l’età hanno un andamento simile, ve ne sono alcuni più frequenti
nell’infanzia o nell’età giovanile,basti pensare al linfoma linfoblastico o lo stesso linfoma di Burkitt.
Vediamo adesso qualcosa che riguarda l’Italia sempre relativamente al periodo compreso tra il 2005 e il
2009 e vediamo come, globalmente considerati, senza scendere nei particolari di ciascuna variante,il picco
si trova dopo i 65 anni, quindi in realtà si tratta di malattie neoplastiche dell’età avanzata, fatta eccezione di
quelle forme che sono più facilmente osservabili nell’età giovanile e nell’infanzia. In realtà molteplici fattori
hanno un ruolo nel determinare l’insorgenza di un linfoma non Hodgkin, ma possiamo grosso modo dire
che tutti questi fattori possono agire in due modi diversi:o perché riducono la sorveglianza immunitaria o
perché vi è una stimolazione antigenica continua. Abbiamo detto riduzione della sorveglianza
immunitaria,bene… allora immunodeficienze congenite e immunodeficienze acquisite. Tra le congenite
ricordiamo la sindrome di Wiskott-Aldrich o la immunodeficienza con atassia-teleangesctasia;tra le
acquisite le PTLD post-transplant lynphoproliferative disease,che sono quelle patologie linfoproliferative
che insorgono in quei pazienti che sono stati sottoposti a trapianto di organo solido o cellule staminali
allogeniche oppure l’immunodeficienza presente nei pazienti affetti da virus dell’HIV1 e 2 ,che si
caratterizza per l’insorgenza di linfomi primitivi del sistema nervoso centrale. Un altro fattore è
l’autoimmunità ed è un’osservazione che doveva essere fatta alcuni decenni fa,cioè che pazienti con
malattie autoimmunitarie e in particolare con collagenopatie erano più spesso esposti o avevano un rischio
maggiore di andare incontro a un linfoma;questo può essere dovuto a un’attivazione cronica del sistema
immunitario oppure può anche essere legata alle terapie immunosoppressive di cui questi pazienti fanno
uso. Un rapporto molto stretto è documentato tra alcuni agenti infettivi e alcune forme di linfomi non
Hodgkin e in particolare il linfoma gastrico tipo MALT con l’Helicobacter pylori,il linfoma degli annessi
oculari con Clamydia psittaci oppure il virus dell’epatite C (HCV) associato con il linfoma marginale o
linfoplasmocitico con crioglobulinemia, HBV con linfoma di burkitt in Africa, il virus HTLV1 o HTLV2 correlati
con i linfomi T dell’adulto in Asia. Anche agenti fisici e chimici possono avere un loro ruolo nella genesi dei
linfomi non Hodgkin,in particolare le radiazioni ionizzanti,pesticidi,benzene, fumo di sigaretta, ma anche i
nitriti aggiunti negli alimenti(cosa che è sotto particolare osservazione). È stato più volte sottolineato negli
USA il rapporto dei coloranti per capelli con l’insorgenza dei linfomi. Quello dei linfomi non Hodgkin è
sempre stato un “mare magnum” in cui non ci si è capiti per decenni tra anatomopatologi,ematologi
oncologi di scuola europea o americana e ciò,unito alla difficoltà stessa della patologia,spiega il perché
dell’utilizzo di sempre nuove classificazioni. Il primo tentativo fu fatto da Rappaport nel 1966 ma lo stesso
si rese conto che la sua classificazione doveva essere rivista e la modificò dieci anni dopo. Nella mia
esperienza considerando da quando ero studente le ho viste, tutte tranne la prima versione di Rappaport
del 1966,quindi ci hanno fatto cambiare 50000 volte questo inquadramento,così abbiamo avuto la
classificazione di Lukes e Collins nel 1974, la Kiel ??? nel 1978, la Lendert,la Working formulation nel 1981,
la REAL classification nel 1994, poi abbiamo avuto quella del 2000 e poi finalmente quella del 2008. In
WWW.SUNHOPE.IT
quest’ultima (classificazione WHO) l’organizzazione mondiale della sanità ha grosso modo riunito tutti i
linfomi in 5 grossi gruppi:
1)
2)
3)
4)
5)
Derivati dai precursori dei linfociti,sia B che T
Derivati dai linfociti B periferici (quindi B maturi)
Derivati dai linfociti T periferici e dalle cellule NK
Linfomi di Hodgkin
Patologie linfoproliferative associate agli stati di immunodeficienza (es paziente trapiantato)
Questa classificazione comprende quindi tutti i linfomi, compresi quelli di Hodgkin. Sicuramente se noi
vogliamo distinguere grosso modo i linfomi B e T, vediamo subito che i linfomi B sono la stragrande
maggioranza e ,a seconda delle varie casistiche, i linfomi non Hodgkin che derivano dalle cellule T
rappresentano più o meno il 6-9%. Anche qua la diagnosi si basa esclusivamente sulla biopsia escissionale
con l’esame istologico e immunoistochimico, è inutile che stiamo a perdere tempo a fare l’agoaspirato:
molti possono essere anche bravissimi a eseguire l’agoasprirato però la stragrande maggioranza degli
ematologi è concorde nel ritenere che solo l’esame istologico e immunoistochimico possono portarci alla
diagnosi. La biopsia osteomidollare ha finalità stadiative, non diagnostiche, tuttavia però da essa si possono
ricavare alcune informazioni utili: per esempio possiamo fare delle indagini di immunocitochimica o di
immunoistochimica, che ci possono aiutare a capire quali molecole vengono espresse dalle cellule
linfomatose o studi molecolari, che ci permettono di identificare anche l’espressione di determinate
molecole, come pure gli studi citogenetici,per esempio noi sappiamo che la traslocazione 11-14 (t 11,14) è
tipica del linfoma mantellare con espressione della ciclina D1,la traslocazione 14-18 (t 14,18)è tipica del
linfoma follicolare con espressione di Bcl-2 , le alterazioni citogenetiche che coinvolgono il locus 3q27 si
ritrovano nel linfoma B diffuso a grandi cellule con espressione di Bcl-6 , la traslocazione 2-5 (t 2,5) con
espressione di ALK è tipica dei cosiddetti alkomi che sono linfomi non Hodgkin a grandi cellule
anaplastiche,la traslocazione 8-22 (t 8,22) con espressione di c-myc è la più importante tra quelle segnalate
(ci sono anche t 8,14 e t 2,8) per il linfoma di Burkitt. La rachicentesi con l’esame citologico del liquor è
importante quando abbiamo dei linfomi particolarmente aggressivi come il linfoma linfoblastico o anche
nei linfomi testicolari. Anche qui possiamo fare una stratificazione prognostica dei pazienti e uno degli
indicatori più diffuso è l’IPI( international prognostic index) che tiene conto di 5 fattori associati a un
maggior rischio: età>60 anni,stadio III-IV,valore della scala ECOG >2 (ricordiamo che questa scala valuta le
condizioni generali del paziente), LDH>norma,coinvolgimento di più di una stazione extranodale. Nei
pazienti con meno di 60 anni i fattori si riducono a 3,ovvero assumono significato prognostico negativo lo
stadio III-IV,ECOG>2, LDH>norma. Prima di passare agli aspetti della terapia, da un punto di vista clinico
dobbiamo fare alcune differenze; certamente ciascuna delle forme ha qualche peculiarità ma grosso modo
possiamo dire che tutti questi linfomi in rapporto all’andamento clinico possono essere raggruppati in:
1) Linfomi a bassa aggressività: sono detti anche indolenti,hanno un’evoluzione molto lenta,non
determinano problemi particolari per molti anni,non richiedono assolutamente di essere trattati
perché, pur rispondendo al trattamento, invariabilmente recidivano: è un po’ quello che abbiamo
visto per la leucemia linfoide cronica,un intervento terapeutico precoce non risolve la patologia che
tende a recidivare,quindi dovremmo fare ripetuti cicli di terapia e il paziente avrebbe più problemi
per le conseguenze della chemioterapia che per la malattia stessa, che ha una lenta evoluzione.
Quindi quand’è che interveniamo in un paziente con linfoma indolente? Quando compare la
sintomatologia generale, ovvero quando compaiono segni e sintomi linfoma-dipendenti quali
febbre,febbricola,calo ponderale,sudorazioni notturne(sintomi simili a quelli che abbiamo visto per
il linfoma di Hodgkin) oppure quando le linfoadenomegalie determinano problemi di tipo
WWW.SUNHOPE.IT
meccanico localmente,per esempio compressione delle vie biliari da parte di una
linfoadenomegalia a livello epatico. Quindi questi linfomi consentono una lunga sopravvivenza,per
molti anni possono non essere trattati ma non guariscono mai.
2) Linfomi ad alta aggressività: hanno caratteristiche diametralmente opposte,sono dei linfomi che
hanno rapida crescita,i pazienti sono spesso sintomatici alla diagnosi e vanno perciò trattati in
tempi brevi,altrimenti vanno incontro a morte; se il paziente non risponde al trattamento si passa a
una terapia di secondo livello ma già la mancata risposta al trattamento di prima linea è un fattore
prognostisco negativo, se invece il paziente risponde e soprattutto se è un linfoma a cellule B, può
anche guarire e questo si osserva nel 50% dei casi.
Quindi guardate un poco il paradosso: i linfomi indolenti consentono pure una lunga sopravvivenza ma non
guariscono mai, quelli aggressivi portano a morte il paziente se non sono trattati o se non si ha risposta al
trattamento ma possono anche guarire. Quindi è difficile dire quali sono benigni o maligni, per questo
adottiamo una terminologia relativa all’aggressività più che alla malignità. Quali sono i linfomi indolenti?
Sono il linfoma follicolare, quello linfoplasmocitico, quello marginale, quello a piccoli linfociti che ha
caratteristiche molto simili alla leucemia linfatica cronica. Tra i linfomi aggressivi ricordiamo il linfoma
mantellare, quello linfoblastico, quello a grandi cellule B, quello di Burkitt.
Quali sono i possibili approcci terapeutici? La chemioterapia, l’immunoterapia, la radioterapia,
l’autotrapianto e l’allotrapianto. Per quanto riguarda la chemioterapia il primo protocollo che è risultato
utile nel trattamento di questi linfomi è stato il protocollo CHOP con C che sta per ciclofosfamide, H per
adriamicina, O sta per oncovin ( nome commerciale per la vincristina) e poi P per prednisone; se da un lato
un certo numero di pazienti rispondeva alla terapia,dall’altro un numero ancora maggiore non rispondeva o
la risposta era insoddisfacente. Sono stati realizzati poi protocolli più aggressivi di seconda e terza
generazione con farmaci più aggressivi che consentivano nell’immediato di avere un miglior risultato però
erano gravati da alta tossicità. Alla fine degli anno ’90 si capì che la sopravvivenza globale,tenendo conto
della tossicità e degli effetti indesiderati della malattia, non era statisticamente diversa rispetto al
protocollo CHOP per cui questi protocolli aggressivi hanno perso la loro iniziale importanza. Certo i
protocolli aggressivi avevano dato ottimi risultati nei pazienti giovani, ad esempio il MACOP B, il VACOP B,
ma sappiamo che la maggioranza dei pazienti affetti da linfoma non Hodgkin è rappresentata da persone
anziane, che non giovavano molto di questi trattamenti. Nel 1998 entrò in terapia l’anti-CD20,rituximab,
cosa che consentì di dare una nuova vita al vecchio protocollo CHOP: associando nei linfomi a cellule B il
rituximab al CHOP (R-CHOP) si ottenevano risultati brillanti, superiori a quelli ottenuti dai protocolli
aggressivi di seconda e terza generazione e non gravati da quella tossicità,osservata invece in questi ultimi.
Per questi motivi il protocollo R-CHOP( oppure RCVP), utilizzato inizialmente nelle forme a bassa
aggressività e dopo anche per quelli ad alta aggressività, si è imposto come trattamento di prima scelta in
tutti i linfomi di tipo B ad eccezione del linfoma di Burkitt,del linfoma mantellare, del linfoma linfoblastico,
che sono altamente aggressivi e che non danno risultati soddisfacenti con il solo RCHOP ma necessitano di
protocolli diversi, e del linfoma indolente al primo stadio,che non viene proprio trattato (sarebbe come
sparare una mosca con un cannone!). Il linfoma linfoblastico richiama molto da vicino la leucemia acuta
linfoblastica ,di cui abbiamo parlato nelle scorse lezioni, e usufruisce degli stessi protocolli terapeutici; il
linfoma mantellare oggi è trattato con un protocollo chiamato R iperC vad, che associa il rituximab a
ciclofosfamide iperfrazionata, vincristina e desametasone, ai quali si alternano altre combinazioni come la
citosina arabinoside (araC);il linfoma di Burkitt è invece un linfoma altamente aggressivo,di cui conosciamo
almeno tre varianti,la variante endemica in Africa che colpisce soprattutto bambini e giovani adulti,
correlata a infezione da EBV,la variante sporadica del mondo occidentale, tipica di soggetti di età più
avanzata,matura o anziana,infine c’è la variante associata a virus dell’HIV. Per il linfoma di Burkitt il
WWW.SUNHOPE.IT
protocollo adoperato è l’R iperC vad (come per il linfoma mantellare) oppure il CODOX M IVAC, che è un
altro protocollo molto aggressivo in cui le sedute di chemioterapia vengono fatte a breve distanza e i
risultati oggi ottenuti sono migliori a quelli del recente passato, soprattutto quando vengono trattati
pazienti giovani, in buone condizioni; un protocollo modificato rispetto a CODOX M IVAC, con posologie più
leggere è stato disegnato per i pazienti anziani allo scopo di ridurre gli effetti tossici nel caso non solo dei
linfomi di Burkitt ma anche in quelli Burkitt-like, cioè quelli molto simili a quest’ultimo e che si osservano
nel mondo occidentale.
Mentre per i linfomi B abbiamo trattamenti efficaci, soprattutto da quando è stato aggiunto il
rituximab,purtroppo nei linfomi T la battaglia è ancora priva di risultati apprezzabili, infatti se si va a
valutare la sopravvivenza a tre anni di pazienti con linfomi T trattati con protocolli aggressivi o con
protocollo CHOP, che è il vecchio non ci sono differenze statisticamente evidenti, anzi talora i risultati sono
migliori con il vecchio CHOP e quindi sono da ricercare nuove strategie: non esiste un anticorpo
monoclonale che possiamo utilizzare e proprio trovare nuovi farmaci che possano modificare la prognosi di
questo tipo di linfomi è la sfida dell’oncologia e dell’ematologia moderne.
Carola Borrelli.
GAMMAPATIE MONOCLONALI
“Si definisce γ-patia monoclonale un disordine linfoproliferativo cronico, caratterizzato dalla produzione di
una Ig monoclonale, o componente M.”
La prima cosa che vi voglio dire è che quando si guarda un QPE non ci si deve attenere solo ai dati numerici
e ai range di riferimento, ma bisogna guardare sempre l’andamento della curva, derivata dalla separazione
elettroforetica delle varie bande proteiche. Grossomodo questo primo profilo (indica la slide) si può
definire normale, in questo secondo invece salta subito all’occhio un’anomalia: questo picco proteico nella
zona γ. Solitamente la zona γ ha l’aspetto di una cupola ancora più appiattita di quella dell’esempio
normale che qui abbiamo, nel quadro con anomalia invece notiamo un vistoso picco la cui altezza ci indica
l’elevata quantità di proteine migrate a questo livello del campo elettroforetico, mentre l’ampiezza ristretta
indica la stretta sovrapponibilità delle caratteristiche di queste proteine ( che le ha portate a migrare nella
stessa zona elettrica): ecco questa è l’espressione grafica di una γ-patia monoclonale. Allora, ripetiamo
insieme cos’è una componente o Ig monoclonale: si tratta di una serie di immunoglobuline prodotte da un
solo clone plasma cellulare, composte dallo stesso tipo di catena pesante- γ-, stesso tipo di catena leggera κ o λ- (IDENTITA’ ISOTIPICA), inoltre hanno anche la stessa regione ipervariabile (INDENTITA’ IDIOTIPICA),
che dovrebbe essere invece diversa per i vari prodotti dei diversi cloni sani, al fine di essere in grado di
riconoscere i molteplici Ag esistenti; ciò significa che questo anticorpo non può svolgere il suo ruolo
protettivo per l’organismo, essendo capace di riconoscere un’unica variabile antigenica. Questa completa
identità le fa migrare tutte nella stessa zona elettrica quando andiamo a fare il QPE.
Però una componente M può anche essere rappresentata solo da un frammento di Ig monoclonale o da
una singola catena leggera.
La catena leggera è definita tale in relazione al suo peso molecolare che si aggira intorno ai 23000- 25000
daltons, ciò significa che sono in grado di attraversare il filtro glomerulare, perciò noi le ritroviamo in grandi
quantità nelle urine poiché superano la capacità di riassorbimento, ricordiamo però che questo può
accadere anche in altre patologie, ematologiche e non.
WWW.SUNHOPE.IT
Una cosa che voglio ripetere insieme a voi è cos’è la proteinuria di Bence-Jones, in quanto ho notato che si
fa molta confusione su questo concetto. La proteinuria di Bence-Jones è il riscontro di una data
concentrazione urinaria (nelle 24h) di catene leggere MONOCLONALI! Non basta dire che si tratta di catene
leggere, perché altrimenti tutti noi avremo la proteinuria di Bence-Jones; perciò il test al calore, che di
solito viene usato per questa determinazione, è poco valido, in quanto esso non ci dà la certezza che si
tratti di catene monoclonali, tutt’ al più ci può far riscontrare un’elevata concentrazione di catene leggere,
magari tutte k o λ, facendoci solo supporre che si possa trattare di una catena monoclonale, quindi questo
test è gravato da un alto numero di falsi positivi e falsi negativi; la diagnosi di certezza di questa proteinuria
ce la dà soltanto l’IMMUNOFISSAZIONE.
Le γ-patie che si possono associare alla presenza di componente monoclonale sono: il mieloma multiplo, la
macroglobulinemia di Waldestrom, patologie linfoproliferative come la leucemia linfatica cronica, i linfomi
non hodgkin (patologie queste a carattere neoplastico, nelle quali è possibile, circa nel 5% dei pz, che ci sia
presenza di componente M), crioglobulinemie di tipo 1 e 2, le malattie delle catene pesanti, l’amiloidosi e la
MGUS (monoclonal gammopathy of undetermined significance).
La MGUS è una discrasia plasmacellulare con componente M, ma perché noi possiamo essere certi che si
tratti proprio di MGUS e non di un’altra delle γ-patie monoclonali suddette è necessario che siano
soddisfatti i seguenti criteri (che appunto ci fanno escludere le altre patologie con presenza di componente
M):
- il paziente deve essere ASINTOMATICO;
-se la componente M è una IgG il suo dosaggio nel siero deve essere ≤ 3,5g/dl;
- se la componente M è una IgA il suo dosaggio nel siero deve essere ≤ 2g/dl;
- la proteinuria di Bence-Jones deve essere assente o al massimo < 1g/24h;
-non devono esserci lesioni osteolitiche;
- anemia, ipercalcemia, insufficienza renale devo essere assenti;
- la % di plasmacellule in un ago aspirato midollare, o biopsia osteo-midollare, deve essere < 10%.
(condizione fondamentale!!!)
Solo se tutte queste condizione sono verificate noi possiamo affermare che il nostro pz ha una MGUS.
Una cosa che vorrei farvi notare è che se andiamo a guardare i dati numerici di un QPE questi sembrano
tutti normali, mentre andando a vedere il profilo noteremo nella zona γ una piccola accentuazione della
curva che appunto identifica la componente monoclonale; se poi andassimo a guardare proprio il
bandeggio su gel di agarosio noteremo che nella zona γ, invece della nubecola uniforme che ci
aspetteremmo di trovare in concordanza con i dati numerici, troviamo una traccia netta in corrispondenza
della zona più catodica del campo elettroforetico: questa è proprio la componente monoclonale; infatti la
caratterizzazione con immunofissazione ci dirà poi che questa componente M ha come catena pesante una
catena γ e come catena leggera una catena λ o k a seconda del caso. Tutto questo per sottolineare ancora
l’importanza di giudicare un QPE sia nel suo aspetto numerico,che in quello grafico e nel bandeggio.
Una volta questa patologia era chiamata γ-patia benigna, questo oggi non è più vero in ragione di uno
studio condotto al Mayo clinic, il quale ha dimostrato che il 25% dei pz affetti da MGUS dopo circa 10-30 aa,
WWW.SUNHOPE.IT
va incontro ad una progressione di questa Ig monoclonale, fino ad andare incontro ad un viraggio verso una
componente M a carattere francamente neoplastico, collocabile nell’ambito di una LLC, di un mieloma
multiplo, di una macroglobulinemia di Waldenstrom o di un linfoma. Ma noi possiamo, al momento della
diagnosi, predire quali pz andranno in contro a una trasformazione neoplastica, cioè se quel pz rientra in
quel 25%? Ebbene una certezza sicuramente non l’abbiamo, ma numerosi studi hanno evidenziato dei
fattori prognostici, che sono i seguenti:
- se al momento della diagnosi abbiamo una bassa concentrazione di componente M, c’è minore
probabilità che si possa avere una progressione verso una patologia maggiore (neoplastica), per esempio
questo grafico ci mostra l’andamento a 20 aa della componente M in vari pazienti e si è visto che se al
momento della diagnosi la concentrazione è 0,5g/dl il rischio è del 14%, ma se questa concentrazione
iniziale è di 2g/dl, il rischio sale al 41%, se poi la concentrazione iniziale è 3g/dl, il rischio sale addirittura al
64%, in base a questi dati quindi decidiamo quanto stretto deve essere il follow up di quel pz e quanto
dobbiamo temere un’evoluzione maligna;
- se nei primi 3aa di malattia non c’è aumento significativo della componente M, il pz ha una prognosi
buona;
-il rapporto tra le catene leggere libere nelle urine di norma è tra 0,26 e 1,65 (k:lambda), se questo valore al
momento della diagnosi e <0,26 o >1,65 la prognosi del pz è più sfavorevole.
Parliamo ora del MIELOMA MULTIPLO: è una neoplasia caratterizzata dalla proliferazione nel midollo osseo
di plasmacellule monoclonali, che producono e secernono nella maggioranza di casi una componente M,
esiste però una bassissima percentuale di pz in cui la componente M è prodotta ma non secreta dalle
plasmacellule.
Il mieloma multiplo rappresenta l’1%di tutte le neoplasie e il 10% di quelle ematologiche, i dati riportati dal
registro tumori del 2006 ci dicono che in Italia i nuovi casi di M.M. sono stati 4413, mentre i decessi 2525 e,
facendo una valutazione geografica, un rapporto fra le varie regioni, notiamo che esiste una sostanziale
omogeneità nella loro distribuzione.
È una malattia della sesta-settima decade di vita ed è rara nell’età giovanile,il rapporto tra uomini e donne
è maggiore di uno; esistono dei fattori di rischio, infatti è stata evidenziata una maggiore comparsa della
malattia in quei soggetti esposti per ragioni lavorative ad alcuni pesticidi, ai derivati del petrolio e a
radiazioni ionizzanti, andando però a non comprendere in questa categoria gli operatori sanitari, che al
giorno d’oggi sono ben protetti dall’esposizione.
Abbiamo qui un quadro latero-laterale Rx del cranio dove sono ben evidenti delle lesioni osteolitiche, qui
abbiamo un profilo QPE caratteristico e qui delle plasmacellule francamente atipiche nel midollo osseo,
piene di componente M evidenziata dall’immunofluorescenza con rodamina (n.d.s.: non sono sicura di aver
sentito bene questo nome).
Quali sono i quadri clinici che possiamo osservare?
-interessamento scheletrico
- insufficienza renale
-morbilità infettiva
WWW.SUNHOPE.IT
-ipercalcemia
-alterazioni neurologiche
-nel 2% dei casi sindrome da iperviscosità ematica.
Cominciamo dal quadro più frequente: le alterazioni scheletriche.
Si è visto che i linfociti B di questi pz esprimono una Ig uguale a quella monoclonale che troviamo in circolo
(a differenza di quanto accade in altre γ-patie), a indicare che tutto il comparto B è coinvolto. Cosa
troviamo nel microambiente midollare? Allora noi vediamo che le plasmacellule sono piene di molecole di
adesione e stringono un intimo rapporto con le cellule stromali. Vediamo come si esplica questo rapporto:
le cellule stromali sono coinvolte nella produzione di alcune citochine, tra cui IL-1β, IL-6, TNF-β, anche note
come fattori attivanti gli osteoclasti, esse infatti vanno a far convertire gli osteoblasti in osteoclasti e ad
attivare ancora le cellule stromali, le quali a questo punto, iniziano ad esprimere una molecola nota come
“ligando di RANK (RANK-L)” (RANK sta per “attivatore del recettore del fattore nucleare KB”), il cui
recettore si trova sugli osteoclasti. Di norma RANK-L è bloccato dall’osteoprotegerina, ma nei soggetti
affetti da M.M., l’osteoprotegerina è sequestrata ed internalizzata dalla proteina Syndecan1, espressa dalle
stesse cellule mielomatose, per cui RANK-L è libero di legare il suo recettore e attivare così gli osteoclasti,
dando inizio al processo di rimozione ossea. In definitiva viene squilibrato il rapporto RANKL/osteoprotegerina a favore di RANK-L portando alle lesioni scheletriche. Tali lesioni sono prevalentemente
a carico delle ossa corte e delle ossa piatte (vertebre, coste, bacino, ossa craniche), non mancano però le
lesioni anche a carico di omeri e femori. Vi faccio notare che quando una lesione dei corpi vertebrali
diventa importante, questi allora hanno difficoltà a sostenere il carico del corpo e possono pertanto
collassare provocando uno schiacciamento a carico delle radici nervose emergenti, ciò scatena una
sintomatologia dolorosa molto intensa che può essere improvvisa, ma talvolta può essere anche
progressiva, perché si assiste dapprima ad una cuneizzazione del corpo vertebrale con compressione lenta
e progressiva delle radici nervose, con la possibilità di neuropatie periferiche.
Questo è il motivo per cui ad un pz con mieloma dobbiamo far eseguire uno studio sistematico dello
scheletro. All’ Rx le lesione osteolitiche sono evidenti quando è stato rimosso almeno il 50% della massa
ossea e appaiono a stampo, con margini netti. Il quadro radiografico in alcuni casi può mostrare non lesioni
osteolitiche, bensì un profilo osteoporotico.
Per studiare bene la colonna vertebrale, l’indagine di scelta è l’RMN in proiezione laterale che rende ben
ragione delle lesioni osteolitiche vertebrali, della loro numerosità fino poi allo schiacciamento vertebrale
con relativa cuneizzazione ed eventualmente della manifestazione emorragica conseguente.
Filomena Boccia
WWW.SUNHOPE.IT
EMATOLOGIA
PROF. GUASTAFIERRO
05-05-14
Qual è il nostro atteggiamento di fronte ad una componente monoclonale? Su quale base
sospettiamo la presenza di una componente monoclonale? Guardando il profilo di un quadro
proteico elettroforetico. Per confermare il sospetto di una componente monoclonale ci serviamo
dell’ immunofissazione. Dobbiamo capire qual è la componente monoclonale , se è una IgA la
concentrazione deve essere superiore a 2g/dl , se una IgG la concentrazione deve essere superiore
a 3,5 g/dl. Dobbiamo considerare anche la protenuria di Bence-Jones . Che cos’è? Presenza di
catene leggere a carattere monoclonale . Un dosaggio immunochimico non ci dice se le catene
sono a carattere monoclonale , ci serviamo quindi dell ‘ immunofissazione , la quantità di questa
proteina nelle 24 h non deve superare 1 g . Cos’è che non ci deve essere ? non ci devono essere
lesioni osteolitiche , insufficienza renale ,ipercalcemia , anemia, l ’infliltrazione delle plasmacellule
al livello del midollo osseo deve essere inferiore al 10 %. Non parliamo di gammapatia benigna ma
gammatia monoclonale di significato indeterminato perché c’è la possibilità di degenerazione
maligna a 20 30 anni. Il mieloma multiplo è una patologia neoplastica caratterizzata dalla
presenza di una componente monoclonale prodotta da plasmacellule atipiche che la secernono
nel 90 % dei casi. Esiste una condizione rara in cui le plasmacellule atipiche sono in grado di
produrre ma non di secernere la componente monoclonale . Ci sono lesioni scheletriche. In
seguito all ‘ interazione delle plasmacellule atipiche e le cellule stromali del midollo , le cellule
stromali cominciano a produrre IL-6 , IL-1beta ,TNF , che sono fattori attivanti gli osteoclasti.
Questi fattori stimolano le cellule stromali a produrre il ligando per il recettore del fattore
nucleare Nkb (recettoreRANK) ,questo ligando è in equilibro con osteoprotegenina che cerca di
controbilanciare questo ligando impedendo che esso vada ad attivare il recettore RANK. Le lesioni
sono a margini netti, si trovano al livello delle ossa corte piatte (vertebre ,volta cranica ,bacino)
ossa lunghe (omero e femore). La risonanza magnetica nucleare è il migliore mezzo di diagnosi per
le lesioni ossee. Tutti i pazienti con mieloma hanno un certo coinvolgimento renale , in particolare
tre quadri clinici renali . Rene da mieloma, malattia da deposito delle catene leggere al livello delle
membrane basali e poi abbiamo l’amiloidosi . il coinvolgimento renale ha una patogenesi
multifattoriale. Il paziente con mieloma ha dolori ed assume farmaci FANS che precipitano la
situazione . La funzionalità renale può essere compromessa anche dalla disidratazione da episodio
febbrile.
RENE DA MIELOMA: le catene leggere attraverso il filtro renale raggiungono il tubulo contorto
distale, qui i sistemi di riassorbimento specifici non ce la fanno a riassorbire questo eccesso di
catene leggere che precipitano . Vengono richiamate in sede delle cellule infiammatorie con la
formazione di cilindri e dilatazione ed atrofia dei tubuli. Il rene perde la capacità di riassorbire le
proteine a basso peso molecolare e di concentrare le urine. DEPOSITO DELLE CATENE LEGGERE AL
LIVELLO DELLE MEMBRANE BASALI DI TUBULI E GLOMERULI(quadro clinico meno frequente) . Con
il tempo si ha una sclerosi delle membrane basali si instaura proteinuria ed insufficienza renale
cronica . AMILOIDOSI: produzione di amiloide al livello mesangiale e vascolare ,si può instaurare la
sindrome nefrosica. Le complicanze renali costituiscono la seconda causa di morte ,la prima è
dovuta alle infezioni .Nel mieloma abbiamo una iperproduzione di catene leggere ed una
WWW.SUNHOPE.IT
retroinibizione a carico delle catene normali. Le immunoglobuline normali quindi sono inefficienti
dal punto di vista difensivo. I pazienti inoltre sono immunodeficienti quando vengono trattati con
una polichemioterapia . La sintomatologia può essere di lieve entità :poliuria ,astenia , vomito
anoressia , disidratazione ,stato confusionale fino al coma . In questi pazienti molto spesso si
riscontra un anemia normocromica normocitica , a che cosa è dovuta? Deficit di EPO per la
compromissione della funzione renale e per infliltrazioni delle plasmacellule atipiche nel midollo
osseo . In questi pazienti si producono citochine infiammatorie che hanno un ‘ attività inibente l ‘
eritropoiesi . Nel mieloma micromolecolare abbiamo la mancanza della componente monoclonale
, anzi nel QPE la curva delle gamma globuline può risultare appiattita, tanto da farci porre la
diagnosi differenziale con un ‘ immunodeficienza . Nel micromolecolare la componente
monoclonale precipita al livello renale e viene eliminata con le urine compromettendo
gravemente la funzionalità renale , i pazienti spesso andavano direttamente in dialisi. Esami di
laboratorio importanti per la diagnosi sono: immunofissazione sierica ed urinaria , dosaggio delle
immunoglobuline sieriche , profilo chimico-clinico completo per appurare se il paziente ha
ipercalcemia e compromissione della funzione renale , emocromo completo con l ‘esame dello
striscio per verificare il quadro anemico .è raro ma è possibile che il mieloma di vecchia data possa
complicarsi con una colonizzazione periferica di plasmacellule ,dando luogo ad una leucemia
plasmacellulare . Proteinuria nelle 24 h e dosaggio delle catene leggere libere al livello sierico .è
fondamentale l ‘ ago aspirato midollare o la biopsia osso- midollare. Esami citogenetici . Immagini
di plasmacellule patologiche. Plasmacellula binucleata , a manico di specchio ,citoplasma infarcito
di vacuoli .I globuli rossi in paziente con mieloma , sono impilati c’è il cosiddetto rouleaux dovuto
alla massiva presenza della componente monoclonale che altera i globuli rossi .Il paziente con
mieloma presenta macroglossia ,deposito di amiloide che conferisce colorazione biancastra, ulcere
sui margini della lingua .La FISH evidenzia il 90% delle alterazione citogenetiche di questi pazienti .
Con la tecnica convenzionale è difficile trovare queste alterazioni perché con questa tecnica le
cellule devono essere in metafase .La FISH invece è una tecnica di ibridazione in situ che analizza le
cellule in interfase .Le alterazioni citogeniche coinvolgono il locus 14q32 , è il locus del gene che
codifica per la parte variabile delle catene delle immunoglobuline e ne comporta una
iperespressione . Queste trasformazioni non sono specifiche per il mieloma , la 11-14 la troviamo
nel caso del linfoma mantellare . Il gruppo internazionale per lo studio del mieloma ha stabilito tre
criteri che devono essere tutti verificati per fare diagnosi di mieloma multiplo . 1) plasmacellule al
livello del midollo osseo devono essere superiori del 10 % . 2) deve essere presente la componete
monoclonale sierica urinaria tenendo presente le due eccezioni , la componente monoclonale non
la troviamo nel mieloma micromolecolare e nel non secernente . 3) deve essere presente un
danno d’organo (acronimo CRAB). C sta per ipercalcemia ,calcemia maggiore di 11,5 mg/dl . R
per compromissione renale, creatinina superiore a 2mg /dl . A anemia normocromica normocitica
con Hg inferiore a 10 g/dl . B lesioni ossee nella maggiore parte dei casi lezioni osteolitiche ,in una
minore percentuale osteoporosi . i criteri CRAB sono importanti , una volta fatta la diagnosi se non
c’è un criterio CRAB è inutile cominciare il trattamento . Ci sono diversi criteri di stadiazione
quello più utilizzato è quello secondo Durie e Salmon . STADIO | : la calcemia è normale, al
massimo c’è una sola lesione osteolitica ,Hg superiore di 10mg /dl , la componente monoclonale
se è una IgG deve essere inferiore a 5g /dl , se è igA inferiore a 3g/dl , la protenuria di Bence –
WWW.SUNHOPE.IT
Jones deve essere inferiore a 4 g/dl .Il paziente si trova nel primo stadio se tutte queste condizioni
sono verificate . STADIO 3 ,quello più avanzato, si verifica quando anche solo una di queste
condizioni si verifica. Hg inferiore a 8,5 g/dl ,calcemia superiore a 12 mg/dl, più lesioni osteolitiche,
se la componente monoclonale è un IgG la concentrazione deve essere superiore a 7g/dl se è un
IgA deve essere superiore a 5g/dl , protenuria di Bence Jones superiore a 12 g/dl . STADIO 2 è uno
stadio intermedio tra il primo e il terzo .Questa classificazione permette di correlare lo stadio con
la massa plasmacellulare .
TERAPIA: se il paziente è asintomatico non lo trattiamo, i pazienti sintomatici sono valutati in base
ai criteri CRAB . Dobbiamo capire se il paziente sintomatico può essere avviato ad una procedura
trapiantologica o no. Se il paziente è eleggibile al trapianto cerchiamo di pulire il midollo dalle
plasmacellule atipiche, adoperiamo un protocollo VTD , V sta per verteib nome commerciale del
BORTEZOMIB inibitore del proteosoma 26s ,quindi non è un chemioterapico .T sta per talidomide
,D sta per desametasone . Dopo la pulizia del midollo tentiamo la raccolta delle staminali
autologhe e si fa il regime di condizionamento, cioè un regime che ci consente di distruggere il
midollo preesistente con alte dosi di melphalan. Si reinfondono al paziente le sue cellule staminali
, questa procedura può essere fatta una volta ma può essere ripetuta anche una seconda volta
attraverso un processo che si chiama tandem . Se il paziente invece non è eleggibile al trattamento
trapiantologico si segue un protocollo MP( mercolan più prednisone ),considerato protocollo di
base .In questo paziente si associa al MP il bortezomib oppure si associa la talidomide il protocollo
è MPT , se invece associamo la redamidomide il protocollo diventa MPR. Al momento il mieloma
multiplo non è una malattia guaribile .
MACROGLOBULINEMIA DI WALDENSTROM
È una malattia linfoproliferativa che è stata descritta per la prima volta nel 1944. È caratterizzata dalla presenza
di una componente monoclonale rappresentata sempre dalle IgM. Questa componente monoclonale è prodotta
essenzialmente da cellule linfoplasmacitoidi , cellule che hanno una maturità intermedia tra i linfociti e
plasmacellule . Il substrato anatomopatologico è la proliferazione di questi elementi linfoplasmacitoidi .
Istologicamente è un linfoma linfoplasmacitoide . Clinicamente abbiamo: splenomegalia , linfoadenopatia .
L’iperviscosità ematica da aumentata concentrazione di IgM è causa di TIA ,disturbi visivi, uditivi , vertigini ,
ronzii , cefalea , difficoltà alla concentrazione . Al livello renale si può verificare un quadro da sindrome nefrosica
, perdita massiva di proteine con le urine . La IgM si può comportare anche come una crioglobulina che precipita
a bassa temperatura, provocando dei problemi soprattutto al livello delle articolazioni . Le IgM possono
intervenire anche nei processi coagulativi dando luogo a delle manifestazioni emorragiche . Quando la viscosità è
tale da essere pericolosa ? quando la concentrazione supera i 4 g/dl . Molto spesso le IgM sono responsabili di
una neuropatia . Non vi sono mai lesioni scheletriche perché non c’è attivazione del network citochinico
responsabile delle lesioni osteolitiche del mieloma .
TERAPIA: se il paziente è asintomatico non si fa nulla. Se ha esclusivamente la neuropatia si tratta con un anti –
CD20 (rituximab) . Se c’è una sindrome di iperviscosità da un lato dobbiamo fare un plasma exchange che ci
riduca la presenza della componente monoclonale e subito dopo cominciare un trattamento citoriduttivo con
ciclofosfamide e desametasone . si eseguono sei cicli del protocollo RCD .
WWW.SUNHOPE.IT
MORBO DI WERLHOF
Al livello del midollo abbiamo i megacarioblasti e i megacariociti dalla cui frammentazione
citoplasmatica vengono fuori le piastrine . La trombopoietina stimola la profiliferazione dei
megacariociti. La sua importanza è stata dimostrata su studi condotti su topi. Mutazioni della
trombopoietica o del recettore che essa lega si manifesta con un’ importante piastrinopenia . I
valori normali di piastrine sono compresi da 150.00 e 350.00/microlitro e sono determinati con un
conta globuli automatico . Dobbiamo considerare se la piastrinopenia è isolata o associata ad
esempio ad un ‘anemia . Dobbiamo considerare se il paziente ha assunto farmaci e dobbiamo
effettuare un esame obiettivo . Cosa andiamo a cercare ? petecchie, micro emorragie che
appaiono come dei puntini rossastri di 1-2 mm. Ecchimosi ,stravasi di sangue quasi sempre post
traumatici. Le piastrinopenie vengono classificate in 5 gruppi . Il terzo gruppo è il più importante,
sono comprese piastrinopenie che riconoscono la base immunologica . Il quindi gruppo
comprende piastrinopenie da emarginazione ad esempio in paziente che hanno una grossa milza .
Dobbiamo distinguere una forma acuta e una cronica . L’acuta si verifica nei bambini o nei soggetti
giovani , tre settimane dopo un ‘ infezione grave . Vengono prodotti anticorpi con antigeni virali
che sono in grado di reagire anche con le glicoproteine delle piastrine. Sono forme che guariscono
senza l ‘intervento terapeutico . Clinicamente si possono avere emorragie gengivali , epistassi
,menorragia, emorragie congiuntivale e la formazione di bolle emorragiche al livello del cavo orale.
Per gli adulti la forma acuta è rara e le donne sono colpite più degli uomini . La forma acuta ha un
andamento stagionale inverno-primavera perché molto spesso sono conseguenti ad infezioni .Le
possibilità di remissione spontanea della forma adulta sono bassissime . Trattiamo il paziente se
per esempio deve subire un intervento chirurgico , lo trattiamo con cortisonici o meglio ancora
con immunoglobuline endovena in modo da saturare il recettore macrofagico con le
immunoglobuline ,così quando arrivano le piastrine opsonizzate non verranno più sequestrate. Le
immunoglobuline aumentano il numero delle piastrine almeno per 3 settimane , così da far
affrontare ad un paziente la fase post operatoria con tutta tranquillità . Posologia del prednisone si
parte da 1 mg per peso corporeo per 4settimane , questo dosaggio può essere aumentato se il
paziente non risponde . L ‘ altra possibilità è l uso di desametasone per via endovenosa . Per il
paziente refrattario facciamo la splenectomia . Abbiamo qualche possibilità anche con alcuni
agonisti che hanno un ‘azione simile a quella della trombopoietina .
P.s: Ultima lezione di ematologia .Non sono riuscita a riportare bene alcune cose , ovviamente fate
riferimento al libro di testo .Il professore a fine lezione ha raccomandato di studiare la parte dell '
emostasi che non è riuscito a spiegare . [cit.Maria Laura Conelli]
WWW.SUNHOPE.IT