Termodinamica
Uno degli elementi principali della termodinamica è l’esame
del bilancio energetico complessivo di un processo fisico (non
solo di natura meccanica).
Un sistema termodinamico è costituito microscopicamente
da un numero molto elevato di elementi (dell’ordine del numero di Avogadro NA = 6.022 · 1023 ), è quindi impossibile
descriverne il moto usando le equazioni usuali della dinamica
dei sistemi. Occorre trovare delle grandezze più adeguate
per la descrizione del sistema. Si tratta di grandezze di tipo
globale cioè riassuntive di ciò che avviene a livello microscopico. Esempi di queste grandezze macroscopiche sono la
temperatura, il volume, la pressione.
La parte di materia che intendiamo studiare è detta sistema termodinamico ed è separata mediante una superficie
chiusa, detta confine, dal resto dello spazio, detto ambiente
circostante. Il sistema interagisce con l’ambiente. Il confine
del sistema può essere fisso o mobile, può consentire il passaggio di massa. In questo caso il sistema termodinamico è
detto aperto, mentre se il confine non lascia passare massa
il sistema è detto chiuso. Considereremo sistemi chiusi. Un
sistema è detto isolato se tra il sistema e l’ambiente non
avvengono scambi né di energia né di materia.
Il numero minimo di variabili termodinamiche necessario per
descrivere il sistema non è fissato a priori. Lo stato termodinamico del sistema è dato dal valore di queste variabili.
P Equilibrio termodinamico: lo stato termodinamico di un
sistema è detto di equilibrio quando le variabili termodinamiche che lo descrivono sono costanti nel tempo.
In questo caso le variabili termodinamiche sono anche dette
variabili di stato.
Solo alcune variabili di stato sono indipendenti, le altre sono
determinate da relazioni, dette equazioni di stato. Ad esempio per un gas perfetto vale pV = nRT e delle tre variabili di
stato: pressione (p), volume (V ), temperatura (T ) solo due
sono indipendenti (n è il numero di moli e R = 8.31 J/mole·K
è la costante dei gas perfetti).
Gas perfetto: gas estremamente rarefatto in cui le interazioni tre le molecole avvengono solo tra urti elastici
pressione: modulo della componente normale della forza
agente su una superficie ∆S per unità di superficie
∆F⊥
∆S→0 ∆S
e si misura in N/m2 . Nel SI l’unità di misura della pressione
è il Pascal: 1 P a = 1 N/1m2 .
Un’altra unità usata è il bar: 1 bar = 105 P a.
mole: una mole di una sostanza contiene un numero di Avogadro (NA ) di unità elementari (atomi o molecole) di quella
data sostanza.
p = lim
L’equilibrio termodinamico si ottiene quando si ha contemporaneamente:
P equilibrio meccanico
P equilibrio chimico
P equilibrio termico (stessa temperatura in tutti i punti)
In particolare due corpi si trovano in equilibrio termico quando
le loro temperature sono uguali.
Principio zero della Termodinamica: due corpi messi a contatto tendono a raggiungere la stessa temperatura (equilibrio
termico).
Due sistemi in equilibrio termico con un terzo sono in equilibrio tra di loro.
Il principio zero della termodinamica permette di dare una
definizione oprerativa di uguaglianze tra due temperature e
viene utilizzato per misurare la temperatura degli oggetti
mediante un termometro.
Misura della temperatura
È noto che le proprietà di molti corpi variano al variare
dell’ambiente termico in cui sono posti (es. la lunghezza di
un’asta metallica o la resistenza elettrica di un filo). Possiamo utilizzare questa proprietà per costruire uno strumento
per confrontare le temperature dei corpi. Il fenomeno più
usato è quello della dilatazione termica (es. termometro
a mercurio, in cui la variazione della temperatura è proporzionale alla variazione dell’altezza del liquido nella colonna).
Per poter fare una misura della temperatura occorre scegliere
alcuni fenomeni riproducibili a cui attribuire una determinata
temperatura.
Esempio:
- ci si mette a pressione costante; si immerge un bulbo con
dentro un liquido in una bacinella con acqua e ghiaccio
- si segna una tacca dove arriva il liquido e lo si chiama zero
- si osserva che durante il cambiamento di stato (cioè quando
il ghiaccio si trasforma in acqua) il volume del liquido nel
bulbo non cambia
- si somministra calore all’acqua fino a quando l’acqua bolle
- si segna una tacca dove arriva il liquido nella colonna del
bulbo e la si chiama 100
- si divide la lunghezza tra le due tacche in cento parti
- il termometro cosı̀ fatto definisce la scala Celsius
La scala che abbiamo costruito dipende dal liquido scelto,
dalla scelta dei punti a cui fissare lo zero (fusione del ghiaccio) e il 100 (ebollizione): quindi la scala non è assoluta.
Nel costruire il termometro si assume che la variazione del
volume del liquido nel bulbo sia proporzionale alla temperatura t
dV = αdt
→
V = V0 (1 + αt)
α = coefficiente di dilatazione termica.
Questa assunzione è vera se la temperatura non varia troppo.
- α è molto più grande nei gas che nei solidi e nei liquidi
- α varia poco tra un gas e l’altro ma varia molto con la
pressione e se si prendono gas sempre più rarefatti si vede
che
1
o C −1
α → αGP =
273.15
lo stesso per tutti i gas perfetti
p
t
V
gas
perfetto
a
pressione costante
V0
V = V0 (1 + αGP t)
t
legge di Gay-Lussac
Legge di Boyle-Mariotte-Lussac: durante una trasformazione
isoterma (t = costante) si ha pV = costante
3000
p
2500
pV = cost
2000
1500
t1 < t2 < t3
1000
500
0
0
0.5
1
1.5
2
2.5
3
V
Seconda legge di Gay-Lussac: durante una trasformazione
isocora (V = costante) si ha p = p0 (1 + αt) con lo stesso α
della prima legge.
Supponiamo di partire dallo stato 0 in figura e di andare allo
stato 1 con una trasformazione isobara e poi allo stato 2 che
ha lo stesso volume dello stato 0. La trasformazione da 1 a
2 è isoterma (t1 = t2)
p
2
p2
pV = cost
isobara: 0 → 1:
V1 = V0 (1 + αt1 )
t1 = t2
p0
isoterma: 1 → 2:
p0 V1 = p2 V0
1
0
V0
V1
V
p0 V1
p0 V0 (1 + αt2 )
=
= p0 (1 + αt2 )
V0
V0
La seconda legge di Gay-Lussac è una conseguenza delle
altre due, quindi le tre variabili p, V , t non sono indipendenti.
p2 =
Relazione tra p, V , t
Sia 3 uno stato intermedio tra 1 e 2 sulla stessa isoterma
p
2
p2
pV = cost
isobara: 0 → 1:
V1 = V0(1 + αt1)
3
p3
t1 = t2 = t3
p0
isoterma: 1 → 3:
p0 V1 = p3 V3
1
0
V0
V3
V1
V
p3 V3 = p0 V0 (1 + αt1 ) = p0V0(1 + αt3 )
moltiplicando ambo i membri per 1/α
p3 V3
1
1
= p0 V0 ( + t3 )
α
α
→
p3 V3
p0 V0
=
1
( α1 + t3 )
α
Posto 1/α = T0
p3 V3
p0 V0
=
(∗)
T0 + t 3
T0
Fissiamo una nuova scala di temperatura, detta scala Kelvin
T = t + T0
Quando t = −T0 = −1/α = −273.15o C,
T = 0,
p = p0 (1 + αt) = 0 ,
V = V0(1 + αt) = 0
T = 0 è detto zero assoluto. Allo zero assoluto p e V si
annullano.
Usando la temperatura assoluta, l’equazione (*) diventa
p3 V 3
p0 V0
=
T3
T0
poichè 0 e 3 sono arbitrari, si ha
pV
= costante equazione di stato dei gas perfetti
T
La temperatura misurata con la scala Kelvin è detta assoluta.
La relazione che lega la temperatura della scala Celsius alla
temperatura assoluta T è:
t = T − 273.15
L’unità di misura della scala Kelvin (K) è uguale a quella
della scala Celsius.
Nel SI la temperatura è misurata con la scala Kelvin.
Il limite di temperatura più bassa è fissato pari allo zero della
scala Kelvin (zero assoluto). Sperimentalmente la temperatura minima raggiunta è dell’ordine di 10−9K.
Nella scala Kelvin al punto triplo dell’acqua (l’acqua liquida,
il ghiaccio solido e il vapor acqueo possono coesistere, in
equilibrio termico ad un solo valore di pressione e temperatura) si attribuisce una temperatura pari a 273.15K (= 0o C)
mentre l’unità Kelvin è pari a 1/273.15 della differenza di
temperatura tra il punto triplo dell’acqua e lo zero assoluto.
N.B.: nelle relazioni che scriveremo, come ad es. l’equazione
di stato dei gas perfetti, la temperatura è quella assoluta.
Solo quando si devono considerare differenze di temperatura
tra corpi è indifferente esprimere i valori delle due temperature in gradi centigradi o in kelvin.
pV
= costante ?
T
Sperimentalmente si osserva che fissati V e T , aumentando
la massa del gas aumenta la pressione, quindi la costante
deve aumentare con la massa del gas.
Quanto vale la costante in
Data una certa quantità di sostanza (ad es. 2 gr di idrogeno
molecolare H2 ) si trova un certo valore della costante. Per
ogni altra sostanza A si può determinare la quantità di sostanza
che dà lo stesso valore della costante: si trova che questa è
tale che
quantità di sostanza A
2 gr
=
peso molecolare sostanza A
peso molec.H2
questo rapporto è il numero N di molecole, quindi
pV = kN T
con k costante che ha lo stesso valore per tutti i gas.
D’altraparte il numero di molecole presenti in una data quantità di sostanza è legato al numero di moli n da N = nNA ,
dove NA è il numero di Avogadro (= numero di molecole
contenute in una mole di una data sostanza = 6.022 · 1023 ).
pV = knNA T = nRT
R = 8.31 J/(mole · K) ,
R ≡ kNA
k = 1.38 · 10−23 J/K
Temperatura e calore
Ogni cambiamento della temperatura avviene attraverso una
interazione tra sistemi che chiameremo scambio di calore.
Ad es. una tazza di caffé lasciata su un tavolo si raffredda
(diminuisce la sua temperatura), questo avviene perchè il
sistema caffé non è in equilibrio termico con l’ambiente esterno (aria) che è ad una temperatura inferiore. Il cambiamento di temperatura è dovuto al trasferimento di un tipo
di energia tra il sistema e il suo ambiente. Questa energia
è l’energia interna (o energia termica) dovuta al moto delle
molecole all’interno dell’oggetto. L’energia interna quando
viene trasferita è chiamata calore (Q). Il calore è considerato positivo quando l’energia interna è trasferita al sistema dall’ambiente (il sistema assorbe calore dall’ambiente),
negativo in caso contrario (si dice che il calore viene ceduto
dal sistema, anche se il calore non è una sostanza trasferita,
ma energia trasferita).
Il calore è l’energia che viene trasferita tra un sistema e
l’ambiente circostante a causa della differenza di temperatura esistente tra di essi.
Poichè il calore è energia trasferita, la sua unità di misura
nel sistema SI è il joule .
In passato questa identificazione non era nota e si era introdotta un’unità di misura per il calore, la caloria, come la
quantità di calore necessaria per innalzare la temperatura di
un grammo d’acqua da 14.5o C a 15.5oC.
La relazione tra queste due unità è 1 cal = 4.186J
Capacità termica (C)
J
K
è la costante di proporzionalità tra la quantità di calore e
la variazione di temperatura che essa produce. La capacità
termica è la quantità di calore che occorre fornire a un corpo
per aumentare la sua temperatura di un grado.
Calore specifico (c) è la capacità termica per unità di massa
Q = C∆T = C(Tf − Ti )
[C] =
J
kg · K
(spesso il calore specifico viene dato in cal/(gr · grado).)
Il calore specifico dipende dalla sostanza; assumeremo che
il calore specifico sia una costante, cioè che non dipenda
Q = c m(Tf − Ti )
[c] =
dalla temperatura, anche se questo è vero solo per intervalli
piccoli di temperatura.
Il calore specifico dell’acqua è definito pari a 1 ocal
tra 14.5 oC
C·g
e 15.5 o C.
Il calore specifico e la capacità termica dipendono da come è
avvenuto il trasferimento di calore al sistema. Infatti questo
trasferimento di calore può avvenire a pressione costante
oppure a volume costante. Per i solidi e i liquidi i calori
specifici corrispondenti (cp e cV ) differiscono poco, mentre
per i gas sono molto diversi.
Quando il calore viene assorbito da un campione non sempre
la sua temperatura aumenta. Il campione può cambiare di
fase (cioè passare da solido a liquido, o da liquido a gas): si
osserva che durante i cambiamenti di stato la temperatura
rimane costante.
Calore latente (L): è la quantità di calore per unità di massa
che occorre trasferire a un campione affinché subisca un
cambiamento di fase completo.
Se il campione ha una massa m la quantità di calore totale
trasferita per un cambiamento di fase è Q = mL.
Se il cambiamento avviene dalla fase liquida a quella aeriforme (il campione assorbe calore) o viceversa (il campione
cede calore) si parla di calore latente di evaporazione (LV );
se il cambiamento di fase avviene dalla fase solida a quella
liquida (il campione assorbe calore) o viceversa (il campione
cede calore) si parla di calore latente di fusione (LF ).
Nel seguito vedremo che si può trasferire energia al sistema
anche mediante un lavoro (L) associato ad una forza agente
sul sistema.
Trasformazioni termodinamiche
Se un sistema è in equilibrio termodinamico, il suo stato rimane immutato nel tempo. Quindi se lo stato varia deve
essere avvenuta un’interazione tra sistema e ambiente che
ha perturbato l’equilibrio e modificato il sistema. Si dice che
è avvenuta una trasformazione termodinamica. Lo stato iniziale e quello finale sono di equilibrio e quindi sono caratterizzati dai valori assunti dalle variabili di stato.
Consideriamo un sistema termodinamico in un dato stato iniziale,
caratterizzato da una pressione pi ,
da un volume Vi e da una temperatura Ti (ad esempio un gas confinato in un cilindro chiuso da un pistone mobile). Chiamiamo processo
termodinamico o trasformazione termodinamica il processo mediante il
quale il sistema passa da questo stato iniziale a uno finale
caratterizzato da una pressione pf , da un volume Vf e da una
temperatura Tf . Durante questo processo il calore può essere trasferito al sistema (calore positivo), ad esempio scaldando la base del pistone, o sottratto al sistema (calore
negativo). Inoltre il lavoro può essere compiuto dal sistema
per alzare (lavoro positivo) o per abbassare (lavoro negativo)
il pistone.
In generale durante la trasformazione il sistema passa attraverso infiniti stati che non sono di equilibrio. Tuttavia se
la trasformazione avviene mediante una successione di piccolissime perturbazioni ognuna delle quali porta il sistema da
uno stato di equilibrio ad un altro poco differente dal precedente, il sistema può essere considerato sempre all’equilibrio
termodinamico (trasformazione quasi-statica). La termodinamica classica studia queste trasformazioni che avvengono
molto lentamente in cui il sistema è sempre all’equilibrio.
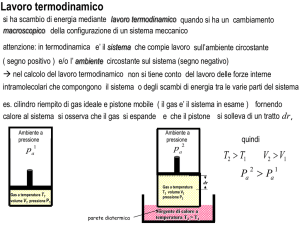
Ad es. lo stantuffo potrebbe contenere dei pallini di piombo.
~ il pisTogliendo un pallino il gas spinge con una forza F
tone verso l’alto che si sposta di un tratto d~s. L’intensità
di questa forza è pari a pA, dove p è la pressione del gas e
A è l’area della superficie del pistone. Il lavoro infinitesimo
compiuto dal gas è
~ · d~s = pAds = pdV
dL = F
dove dV è la variazione infinitesima del volume occupato dal
gas. Il lavoro totale compiuto dal gas per passare dal volume
Vi al volume Vf è
Z
Z Vf
L=
dL =
pdV
Vi
per calcolare questo integrale occorre sapere come varia la
pressione al variare del volume (e della temperatura del gas).
In generale ci sono tanti modi di andare da un dato stato
iniziale a uno finale, come mostrato in figura.
L’area tratteggiata rappresenta il lavoro compiuto dal sistema durante questo processo. Il
lavoro è positivo perchè la trasformazione procede verso destra. Nella figura (b) il passaggio
dal i a f avviene in due stadi. Nel primo tratto
la pressione è costante (non si tolgono pallini)
e l’aumento di volume è ottenuto innalzando
la temperatura del gas (scaldando il fondo del
cilindro). Quindi viene compiuto un lavoro
dal gas e viene fornito calore al sistema. Il
secondo tratto è a V costante (si blocca il
pistone) e la pressione scende riducendo la
temperatura del fondo. In questa fase il sistema trasferisce
calore al fondo. Il lavoro, positivo, viene
compiuto solo durante il primo stadio, mentre calore viene trasferito durante entrambi
gli stadi. Nella figura (c) i due stadi vengono
compiuti in ordine inverso. Il lavoro in questo
caso è minore. In generale un sistema può
essere portato da un dato stato iniziale a un dato stato
finale in diversi (infiniti) modi, ciascuno con lavoro e calore
trasferiti diversi: il lavoro e il calore sono quantità dipendenti
dal percorso seguito.
La figura (d) mostra un sistema che compie un lavoro negativo, quando una forza
esterna lo comprime riducendone il suo volume: il lavoro compiuto è ancora uguale,
in valore assoluto, all’area sottesa dalla
curva.
La figura (e) mostra un ciclo termodinamico nel quale un sistema passa da uno stato
iniziale i a uno stato f (espandendosi) e
quindi di nuovo allo stato i, riducendo il
suo volume. Il lavoro totale compiuto dal
sistema durante il ciclo è la somma del
lavoro positivo compiuto durante l’espansione e del lavoro
negativo compiuto durante la compressione ed è dato dall’area
della superficie tratteggiata.
Se il ciclo è percorso in senso orario L > 0, infatti Li,f > Lf,i ,
mentre se il ciclo è percoso in verso antiorario L < 0.
Esempi
P Lavoro svolto da un gas ideale a temperatura costante
Studiamo una trasformazione termodinamica reversibile in
cui la temperatura è mantenuta costante durante tutto il
processo (isoterma). Dalla legge dei gas ideali si ha
1
1
= ( costante )
V
V
a
temperatura
costante
la
pressione è inversamente proporzionale al volume (legge di
Boyle). Nel piano (p, V ) il luogo
dei punti che rappresentano gli
stati di equlibrio di un gas a una
p = (nRT )
data temperatura è costituito da un ramo di un’iperbole.
Il lavoro svolto dal gas durante un’espansione isoterma è
Z Vf
Z Vf
dV
Vf
L=
pdV = nRT
= nRT ln
V
Vi
Vi
Vi
quando il gas si espande Vf > Vi e L > 0, mentre L < 0
quando Vf < Vi .
P Lavoro svolto da un gas a volume costante: in una trasformazione isocora Vf = Vi quindi
Z Vf
L=
pdV = 0
Vi
P lavoro svolto da un gas a pressione costante: in una
trasformazione isobara
Z Vf
Z Vf
p
L=
pdV = p
dV
Vi
Vi
= p(Vf − Vi ) = p∆V
Vi
Vf
V
Prima legge della termodinamica
Sperimentalmente si vede che passando da uno stato iniziale
a uno finale il lavoro L compiuto dal sistema e il calore Q
trasferito dipendono dal tipo di trasformazione mentre la
quantità Q − L è sempre la stessa. A questa quantità si dà
il nome di variazione dell’energia interna
∆Eint = Eint,f − Eint,i = Q − L
Questo fatto è detto prima legge della termodinamica: per
andare da uno stato i a uno stato f il calore assorbito dal
sistema e il lavoro fatto dal sistema dipendono dal procedimento, ma lo loro differenza è sempre la stessa. Questo ci
dice che l’energia interna dipende solo dallo stato termodinamico in cui il sistema si trova. E’ una funzione di stato
(mentre il calore e il lavoro no).
Nel caso di una trasformazione infinitesima
dEint = δQ − δL
Per convenzione il calore è positivo o negativo a seconda
che sia assorbito o ceduto dal sistema, mentre il lavoro è
positivo o negativo a seconda che sia positivo o negativo il
lavoro fatto dal sistema. Perciò l’energia interna di un sistema cresce quando il sistema assorbe calore Q e diminuisce
quando il sistema compie un lavoro L > 0.
La prima legge della termodinamica è una generalizzazione
del principio di conservazione dell’energia studiato in meccanica.
Si possono distinguere diverse trasformazioni termodinamiche:
P trasformazioni adiabatiche: non si ha trasferimento di
calore tra il sistema e l’ambiente
∆Eint = −L
trasf. adiabatica
in questo caso se il sistema compie un lavoro positivo l’energia
interna del sistema diminuisce. L’unica interazione tra sistema e ambiente è attraverso il trasferimento di lavoro
(nell’esempio precedente anche la base del cilindro è isolante).
P trasformazioni a volume costante (isocòre): il sistema non
può compiere lavoro
∆Eint = Q
trasf. a volume costante
se viene fornito calore al sistema (quindi Q > 0) l’energia
interna del sistema aumenta.
P trasformazioni cicliche: sono trasformazioni in cui dopo
scambi di calore e lavoro il sistema torna allo stato iniziale:
sono quindi descritte da curve chiuse nel piano (p, V ). Le
proprietà intrinsiche del sistema devono essere le stesse e
quindi anche l’energia interna
∆Eint = 0 ⇒ Q = L
trasf. ciclica
P trasformazioni di espansione libera: sono trasformazioni
nelle quali non si ha né scambio di calore né di lavoro:
Q = L = 0. Non possono avvenire lentamente e quindi
durante il processo il sistema non è in equilibrio. Il gas
si trova in equilibrio solo negli stati iniziale e finale: nel
piano (p, V ) questi stati sono individuati da due punti ma
non possono essere disegnate le linee per andare da uno
all’altro.
Questo tipo di trasformazioni sono irreversibili. Le trasformazioni quasi-statiche (che avvengono attraverso stati di
equilibrio) sono invece trasformazioni reversibili: esse possono essere arrestate in un qualunque stato intermedio e
(variando di poco le condizioni esterne) si può invertire il
verso della trasformazione, ripercorrendo gli stessi stati già
attraversati.
Teoria cinetica dei gas
Si occupa di mettere in relazione le proprietà macroscopiche
dei gas (es. pressione e temperatura) con le proprietà microscopiche delle molecole del gas (come ad es. la loro velocità).
Poichè un sistema macroscopico è costituito da un numero
molto grande di molecole, si introducono le quantità medie.
Ad esempio la velocità media di un sistema di N molecole è
definita come
N
1 X
h~v i =
~vi
N i=1
Analogamente la velocità quadratica media è
N
N
1 X 2
1 X 2
2
2
hv i =
vi =
(vix + viy
+ viz
)
N i=1
N i=1
2
N
N
N
1 X 2
1 X 2
1 X 2
=
v +
v +
v
N i=1 ix
N i=1 iy
N i=1 iz
= hvx2i + hvy2 i + hvz2 i
Se una molecola urta contro una parete e se l’urto è elastico,
la variazione della quantità di moto è
y
∆px = −2mvx
z
∆py = ∆pz = 0
x
Nel caso di un gas perfetto il numero di molecole del sistema
è molto grande, ma il gas è cosı̀ rarefatto che si suppone che
le molecole non interagiscono fra di loro; si suppone inoltre
che le molecole urtano solo le pareti e che questi urti siano
elastici.
Se il gas è contenuto in una scatola cubica di lato L, il tempo
che intercorre tra un urto contro una parete e il successivo
è
2L
vx
La forza media che la
parete
esercita
sulla
molecola è
∆t =
z
L
2mvx
mvx2
hfxi = −
=−
∆t
L
quella sulla parete è
L
mvx2
hfx i =
L
Se ci sono N molecole (tutte con la stessa massa), con
velocità diverse, la forza media totale è
PN
N
2
1X
mN
mN 2
2
i=1 vix
hFxi =
mvix =
=
hvx i
L i=1
L
N
L
x
analogamente le forze medie che vengono esercitate sulle
altre pareti
mN 2
mN 2
hvy i
hFz i =
hvz i
L
L
Dato che non ci sono direzioni privilegiate hvx2 i = hvy2i =
hvz2 i = hv 2 i/3, quindi
hFy i =
mN 2
hv i
3L
La pressione esercitata sulle pareti si ottiene dividendo per
l’area della parete L2
hFx i = hFy i = hFz i =
mN 2
hv i
3
3L
Essendo il volume della scatola V = L3 , si ha
µ
¶
2
1
2
pV = N
mhv 2 i = N hKi
3
2
3
hpx i = hpy i = hpz i = p =
dove hKi è l’energia cinetica media. Confrontando questa
relazione con l’equazione di stato dei gas perfetti
pV = kN T
si trova
2
3
N hKi →
hKi = kT
3
2
cioè in un gas perfetto l’energia cinetica media è proporzionale
alla temperatura.
Ricordando il legame tra le costanti k e R (kNA = R) e che il
numero di molecole è N = nNA , dove n è il numero di moli,
si trova
3 nRT
3
hKi =
→
N < K >= nRT
2 N
2
kN T =
dove N hKi è l’energia cinetica totale del gas: in questo
modo abbiamo messo in relazione una grandezza macroscopica misurabile (la temperatura), con una grandezza non
misurabile (l’energia cinetica totale delle molecole del gas).
In questa trattazione si suppone che l’energia sia solo dovuta
al moto traslazionale delle molecole, si suppone cioè che le
molecole siano puntiformi.
L’ipotesi che gli urti contro le pareti siano elastici è ragionevole perchè l’atomo può assorbire energia solo se questa e pari
alla differenza di energia tra i livelli energetici dell’atomo:
siccome questa energia è molto grande l’urto a temperatura
ambiente è elastico.
E’ possibile considerare anche il caso in cui le molecole si
urtano tra di loro: il risultato non cambia
In conclusione se il gas perfetto è un gas monoatomico,
come l’elio, il neon , l’argo, l’energia interna del gas è data
dalla somma dell’energia cinetica traslazionale delle singole
molecole
3
Eint = nRT
gas ideale monoatomico
2
Se il gas perfetto è invece formato da molecole bi-atomiche,
oltre a traslare la molecola può anche ruotare: si può mostrare
in questo caso che i gradi di libertà su cui si equipartisce
l’energia sono 5 (3 traslazionali, più due rotazionali) e si
trova
5
Eint = nRT
gas ideale bi-atomico
2
Per molecole più complesse si hanno più gradi di libertà e
l’energia interna aumenta ma è sempre dipendente dalla temperatura del gas.
P Calore specifico (molare) a volume costante.
Nei gas ci si riferisce a moli di sostanze e si definisce il calore
specifico molare mediante la relazione
Q = nc∆T
dove n è il numero di moli del gas e Q è la quantità di
calore scambiato per far variare la temperatura di ∆T . In
altre parole il calore il calore specifico molare è la capacità
termica di una mole di sostanza: C = nc.
In un gas il calore specifico dipende dal modo con in avviene
lo scambio di calore. Consideriamo una trasformazione che
avviene a V costante: a partire dallo stato i si somministra
p
al gas del calore lentamente in
modo che la temperatura del
f
p + ∆p
gas aumenta da T a T + ∆T e
p
T + ∆T la pressione passa da p a p +
i
∆p, il gas si porta nello stato
T
finale f .
V
V
Il calore QV fornito durante la trasformazione è legato alla
variazione di temperatura da
QV = ncV ∆T
dove cV è il calore specifico molare a volume costante.
Per la prima legge della termodinamica
∆Eint = QV − L = ncV ∆T
(V = cost → L = 0)
Dalla teoria cinetica dei gas, se il gas ideale è monoatomico
Eint = 3nRT /2, quindi
∆Eint =
3
nR∆T
2
→
cV =
3
R = 12.5 J/(mole · K)
2
Analogamente nel caso di un gas ideale biatomico
5
R = 20.8 J/(mole · K)
2
Questi valori per cV sono in buon accordo con i dati sperimentali. Ad es. per He cV = 12.5, per Ar cV = 12.6,per O2
cV = 20.8.
cV =
Consideriamo ora una trasformazione in cui la temperatura
del gas aumenti della stessa piccola quantità ∆T considerata
prima ma che avviene a a pressione costante.
Il gas compie un lavoro
p
L = p∆V > 0
f
p + ∆p
e assorbe il calore
f0
p
T + ∆T
i
T
V
V + ∆V
Qp = ncp ∆T
V
dove cp è il calore specifico molare a pressione costante.
Per la prima legge della termodinamica
Qp = ∆Eint + p∆V
La variazione dell’energia interna è la stessa nelle due trasformazioni i → f e i → f 0 , in quanto Eint è proporzionale a T e
trasformazioni con ∆T uguali hanno ∆Eint uguali.
Nella trasformazione i → f , ∆Eint = QV , quindi Qp > QV : la
quantità di calore che bisogna fornire a una data quantità
di gas ideale per far aumentare la sua temperatura di ∆T è
maggiore se la trasformazione avviene a pressione costante
rispetto a quella a volume costante.
Possiamo legare cp a cV :
ncp ∆T = ncV ∆T + p∆V
Dall’equazione di stato dei gas ideali pV = nRT , in una
trasformazione isobara p∆V = nR∆T , quindi
ncp ∆T = ncV ∆T + nR∆T
cp = cV + R
→
relazione di Mayer
Sperimentalmente per i calori specifici dei gas ideali si trova:
a) per gas ideali monoatomici (gas rari come elio, neon, argon): cV = 32 R e cp = 52 R costanti per un ampio intervallo di
temperature;
b) per alcuni gas ideali biatomici (es. idrogeno, azoto):
cV = 52 R e cp = 72 R a temperatura ordinaria, mentre a temperature superiori, cp e cV crescono lentamente;
c) altri gas biatomici (ossigeno, fluoro, cloro, bromo) e i gas
ideali poliatomici hanno colori specifici variabili con la temperatura.
Questo diverso comportamento è spiegato dal fatto che,
a differenza dei gas monoatomici, all’energia interna contribuisce non solo il moto traslazionale delle molecole ma
anche altri moti (rotazionali, oscillatori).
Possiamo generalizzare l’equazione che dà l’energia interna
di un gas ideale qualunque introducendo cV
Eint = ncV T
dove cV è quello del gas considerato.
p
f 00
f
p + ∆p
f0
p
T + ∆T
i
T
V
V + ∆V
V
Per una trasformazione
qualunque in cui il gas
subisce una variazione di
temperatura ∆T si ha
∆Eint = ncV ∆T
Nelle tre trasformazioni in figura i valori di Q, L sono diversi, cosı̀ come i valori finali di p e V , mentre la variazione
dell’energia interna è sempre la stessa in quanto tutte e tre
le trasformazioni comportano la stessa variazione di temperatura e possiamo usare la trasformazione a V = cost per
calcolare ∆Eint .
P Trasformazione adiabatica di un gas perfetto
Si dice adiabatica una trasformazione che avviene senza
scambi di calore: Q = 0. Il sistema può solo scambiare
lavoro (cioè le pareti del contenitore sono isolanti e mobili).
Per la prima legge della termodinamica
L = −∆Eint
Se il gas si espande L > 0 quindi
∆Eint < 0 e ∆T < 0:
in una espansione adiabatica il gas
si raffredda.
T diminuisce perchè nell’espansione il gas compie un lavoro
a spese dell’energia interna.
Viceversa in una compressione adiabatica il gas si riscalda (il
lavoro è negativo, ∆Eint > 0 e T aumenta).
Ricaviamo le equazioni per una trasformazione adiabatica
reversibile di un gas perfetto. Per una trasformazione infinitesima con una variazione del volume dV , il lavoro è
nRT
dV
V
dove si è utilizzata l’equazione dei gas perfetti. Ma
L = pdV =
L = −dEint = −ncV dT
Uguagliando le due espressioni si trova
ncV dT = −
nRT
dV
V
→
dT
R dV
=−
T
cV V
Integrando questa relazione tra uno stato iniziale e uno finale
µ ¶R
Z Tf
Z Vf
dT
R
dV
Tf
R
Vf
V i cV
=−
→ ln
= − ln
= ln
T
c
V
T
c
V
Vf
i
i
V
V
Ti
Vi
Essendo
cp = cV + R
R
cp − cV
cp
=
=
−1=γ−1
cV
cV
cV
→
dove abbiamo definito
γ=
cp
cV
Quindi si ha
Tf
ln
= ln
Ti
µ
Vi
Vf
¶γ−1
Tf
→
=
Ti
µ
Vi
Vf
¶γ−1
cioè
Ti Viγ−1 = Tf Vfγ−1
Siccome gli stati iniziali e finale sono generici, durante una
trasformazione adiabatica si ha
T V γ−1 = costante
Utilizzando la legge dei gas perfetti (ricavando T e sostituendolo nell’equazione precedente), questa equazione è equivalente a
pV γ = costante
e anche a
Tp
1−γ
γ
= costante
Queste equazioni descrivono una trasformazione adiabatica
(reversibile) di un gas ideale.
N.B. anche l’espansione libera è una trasformazione adiabatica: in essa però il lavoro svolto è complessivamente nullo
e l’energia interna non varia. Le equazioni di prima non si
applicano all’espansione libera, anche se questa è adiabatica.
Nell’espansione libera il gas si trova in equilibrio solo negli
stati iniziali e finali, ma non si può disegnare un diagramma.
Dato che l’energia interna non cambia, anche la temperatura
non cambia:
Tf = Ti
espansione libera
per un gas ideale si ha quindi
pi Vi = pf Vf
espansione libera
P Trasformazioni isoterme di un gas ideale
Poichè T è costante, ∆Eint = 0 e Q = L. Abbiamo già calcolato il lavoro compiuto dal gas durante una trasformazione
isoterma
Z Vf
Z Vf
dV
Vf
pi
L=
pdV = nRT
= nRT ln
= nRT ln
V
Vi
pf
Vi
Vi
Si noti che una trasformazione isoterma reversibile comporta
sempre uno scambio di calore (Q 6= 0).
P Trasformazioni isocore di un gas ideale
V è costante, quindi L = 0. Il gas può scambiare solo calore
Q = ∆Eint = ncV ∆T = ncV
V ∆p
V ∆p
=
nR
γ−1
Se il gas assorbe calore dall’ambiente (Q > 0) la sua temperatura aumenta. D’altra parte poichè V è costante, dall’equazione dei gas si ha
pi
pf
=
Ti
Tf
⇐⇒
pi
Ti
=
pf
Tf
p e T sono direttamente proporzionali e anche la pressione del
gas aumenta. Viceversa quando il gas cede calore (Q < 0),
la sua temepratura e la sua pressione diminuiscono.
P Trasformazioni isobare di un gas ideale - Entalpia
Le pareti del contenitore in cui è contenuto il gas sono mobili e su di esse agisce una pressione esterna costante p.
Dall’equazione dei gas si ha
Vi
Vf
=
Ti
Tf
⇐⇒
Vi
Ti
=
Vf
Tf
T e V sono direttamente proporzionali. Il gas può scambiare
sia calore che lavoro:
Q = ncp (Tf − Ti )
nRTi ´
= nR(Tf − Ti )
L = p(Vf − Vi ) = p
−
p
p
∆Eint = Q − L = ncV (Tf − Ti )
³ nRT
f
se si cede calore al gas (Q > 0) la sua temperatura e il
suo volume aumentano e il gas compie un lavoro; se il gas
cede calore (Q < 0) la sua temperatura e il suo volume
diminuiscono e il gas subisce un lavoro.
Molti processi avvengono in natura a pressione costante. Per
le trasformazioni a pressione costante è utile introdurre una
nuova funzione, detta entalpia
H = Eint + pV
entalpia
l’entalpia è una funzione di stato, in quanto Eint , p e V sono
variabili di stato. In un gas ideale l’entalpia è funzione solo
della temperatura (come Eint e il prodotto pV ).
Per una trasformazione qualsiasi infinitesima
dH = dEint + d(pV ) = ncV dT + nR dT = ncp dT
(dove si è usata la relazione di Mayer cp − cV = R).
Questo permette di definire il calore specifico a pressione
costante in funzione dell’entalpia:
1 ∆H
n ∆T
Se la trasformazione avviene a p = cost, la variazione della
entalpia è pari al calore scambiato: ∆H = Qp
N.B.: È utile confrontare con trasformazione a V = cost
1 ∆Eint
∆Eint = QV = ncV ∆T =⇒ cV =
n ∆T
∆H = ncp ∆T
=⇒ cp =
P Entropia
Definiamo questa funzione di stato per il gas perfetto (la
definizione è però generale). In una trasformazione infinitesima reversibile di un gas ideale il calore scambiato
nRT
dV
V
Sappiamo che il calore dipende dalla trasformazione, infatti
Z f
Z Tf
Z Vf
dV
δQ = ncV
dT + nR
T
V
i
Ti
Vi
δQ = dEint + δL = ncV dT + pdV = ncV dT +
il primo termine dipende solo dagli stati i e f il secondo
dipende da come T varia nella trasformazione (cioè da come
T dipende da V ).
Se consideriamo la quantità
δQ
dT
dV
= ncV
+ nR
T
T
V
e integriamo tra gli stati i e f
Z f
Z Tf
Z Vf
δQ
Tf
Vf
dT
dV
= ncV
+ nR
= ncV ln
+ nR ln
T
T
V
Ti
Vi
i
Ti
Vi
il risultato dipende solo dagli stati i e f . Definiamo perciò
la funzione di stato S, detta entropia tale per cui la sua
variazione dS è
δQ
dS =
T
TB
VB
+ nR ln
(∗)
TA
VA
La variazione di entropia dipende dal calore scambiato nella
trasformazione e dalla temperatura a cui avviene lo scambio.
In particolare, in una trasformazione adiabatica ∆S = 0.
Usando R = (γ − 1)cV , la (*) diventa
∆S = SB − SA = ncV ln
SB − SA = ncV ln
TB VBγ−1
TA VAγ−1
Utilizzando l’equazione di stato si possono ottenere espressioni equivalenti (eliminando T o V )
pB VBγ
SB − SA = ncV ln
,
pA VAγ
(1−γ)/γ
SB − SA = ncp ln
TB p B
(1−γ)/γ
TA pA
trasf. isoterma (TA = TB ) :
trasf. isocora (VA = VB ) :
trasf. isobara (pA = pB ) :
SB − SA = ncV ln
VBγ−1
VAγ−1
TB
SB − SA = ncV ln
TA
TB
SB − SA = ncp ln
TA
N.B.: cp > cV → la variazione di entropia in una trasformazione isobara è maggiore di quella che si ha in una trasformazione isocora se la variazione di temperatura è la stessa
(esattamente come succede per il calore scambiato).
L’entropia è una variabile di stato (dipende solo dallo stato
del sistema) possiamo calcolare ∆S per un processo arbitrario da i a f , anche non reversibile.
Ad esempio, consideriamo l’espansione libera di un gas perfetto. Nel diagramma (p, V ) lo stato iniziale e finale del
sistema sono rappresentati da due punti. In un’espansione
libera la temperatura di un gas ideale non cambia, quindi i e
f devono stare sulla stessa isoterma. L’espansione isoterma
è un processo fisico diverso dall’espansione libera, ma se entrambi i processi hanno gli stessi stati
iniziale e finale, la variazione di entropia è la stessa
Z
1 f
Q
∆S = Sf − Si =
dQ =
trasf. isoterma
T i
T
dove Q è il calore ceduto dalla sorgente al sistema (cioè al
gas che si è espanso) durante il processo isotermo. Quindi
Q > 0 come la variazione di entropia.
L’entropia è una funzione di stato e può essere presa come
variabile indipendente (assieme ad un’altra) per descrivere il
sistema, es. S e T . Per trasformazioni reversibili possiamo
considerare i diagrammi nel piano (S, T ) (analogamente ai
diagrammi nel piano (p, V ) considerati finora). Integrando
dQ
⇒
dQ = T dS
T
tra lo stato iniziale e finale si trova il calore scambiato nella
trasformazione
Z f
Q=
T dS
dS =
i
il calore scambiato in una trasformazione
reversibile è dato dall’area superficie
sottesa dalla curva della temperatura nel
piano (S, T ).
Ovviamente il calore dipende dalla trasformazione.
Per una trasformazione ciclica reversibile, nel piano (S, T ) il
ciclo delimita un’area pari alla somma algebrica dei calori
scambiati in totale dal sistema: QA +QC ,
dove QA > 0 è il calore assorbito nella
trasformazione A → B (l’entropia aumenta) e QC < 0 è il calore ceduto
nella trasformazione B → A (l’entropia
diminuisce).
QA + QC è positivo se il ciclo è percorso in senso orario,
mentre è negativo se il ciclo è percorso in senso antiorario.
In tutti i casi, per il primo principio quest’area è uguale anche
al lavoro compiuto durante il ciclo.
Nel piano (S, T ) una trasformazione
isoterma reversibile è rappresentata
da una linea orizzontale (T = cost.);
una trasformazione adiabatica da una
linea verticale (S = cost. ⇔ dQ = 0);
T
T2
T1
S1
S2
S
Le variazioni di entropia nelle trasformazioni reversibili sono:
Q
T
∆S = 0
∆S =
∆S = 0
trasformazione isoterma
trasformazione adiabatica
trasformazione ciclica
P Energia libera
Si introduce un’altra funzione termodinamica che è utile soprattutto quando la trasformazione avviene a T = cost:
F = Eint − T S
energia libera
(o potenziale di Helmotz). La variazione dell’energia libera
per una trasformazione reversibile infinitesima a T = cost è
dF = dEint − T dS = δQ − δL − T dS = −δL
dove si è usato che δQ = T dS.
L’importanza dell’energia libera è legata al fatto che si può
calcolare dalla descrizione microscopica (in meccanica statistica), e da essa poi si derivano tutte le funzioni termodinamiche.
Riassumendo, delle varie funzioni termodinamiche introdotte,
l’energia interna rende conto degli scambi di calore e di lavoro, l’entropia di quelli di calore, l’entalpia dà lo scambio di
calore se la trasformazione avviene a p = cost, l’energia libera dà il lavoro fatto dal sistema se la trasformazione avviene
a T = cost.
P Trasformazioni cicliche
Sono trasformazioni in cui lo stato iniziale coincide con quello
finale: per il primo principio Q = L. Se durante il ciclo
viene prodotto lavoro (L > 0), assorbendo calore da sorgenti esterne, il ciclo è detto termico (macchina termica).
Se invece durante un ciclo viene richiesto un lavoro esterno
(L < 0) estraendo calore dal sistema, il ciclo è detto frigorifero (macchina frigorifera).
Il ciclo è costituito da varie trasformazioni e possiamo scrivere il calore complessivo scambiato come
Q = QA + QC
dove QA > 0 rappresenta la somma dei calori assorbiti e
QC < 0 la somma dei calori ceduti. Analogamente il lavoro
L = LF + LS
in cui LF > 0 è la somma dei lavori compiuti e LS < 0 è la
somma dei lavori subiti dal sistema.
Per un ciclo termico si definisce rendimento la quantità
L
QA + QC
QC
|QC |
=
=1+
=1−
QA
QA
QA
QA
il rendimento è quindi la percentuale di calore assorbito che
viene trasformata in lavoro.
η=
Dato che l’energia si conserva, se si somministra alla macchina
termica una certa quantità di energia termica, QA , in linea
di principio si potrebbe ottenere un lavoro che al massimo
è pari a QA . In realtà il rendimento è sempre minore di 1,
quindi non tutta l’energia termica data al sistema può essere
convertita in lavoro. Si ha cioè:
0≤η<1
cioè L < QA , |QC | ≤ QA , QC 6= 0
In un ciclo termico solo una frazione < 1 del calore assorbito
viene trasformata in lavoro, c’è sempre del calore ceduto.
Questo è ben evidente se si disegna il ciclo nel piano (S, T ).
Un ciclo termico (Q > 0) è percorso in verso orario.
Il lavoro totale è dato dall’area
racchiusa dal ciclo mentre il calore
assorbito è dato dall’area totale sottesa dalla curva superiore, quindi η che è il rapporto tra
queste due aree è sempre η < 1.
P Ciclo di Carnot
È costituito da quattro trasformazioni reversibili:
1)
2)
3)
4)
trasformazione
trasformazione
trasformazione
trasformazione
AB:
BC:
CD:
DA:
espansione isoterma reversibile
espansione adiabatica reversibile
compressione isoterma reversibile
compressione adiabatica reversibile
Nello stato A il gas è in equilibrio a contatto termico con
una sorgente di calore a temperatura T2 . Supponiamo che
l’espansione isoterma AB sia una serie di trasformazioni infinitesime in cui la pressione diminuisce di dp e il gas si espande di dV raffreddandosi di dT : si ha quindi una cessione
di calore dalla sorgente a temperatura T2 e il gas ritorna alla
temperatura T2 . Come risultato il gas passa dallo stato A
allo stato B assorbendo il calore QA pari al lavoro LAB fatto
dal gas nell’espansione isoterma
VB
LAB = nRT2 ln
= QA
VA
Nella trasformazione BC il gas è isolato da qualsiasi sorgente
di calore e il gas passa dallo stato B(pB , VB , T2 ) allo stato
C(pC , VC , T1 ) con T1 < T2 e
T2 VBγ−1 = T1 VCγ−1
(∗)
Il lavoro fatto dal gas è LBC = −∆Eint = ncV (T2 − T1).
Nella trasformazione CD il gas è a contatto termico con una
sorgente di calore alla temperatura T1 : se la pressione aumenta di dp, il gas si comprime di dV e aumenta la sua temperatura di dT ; il gas quindi cede dQ alla sorgente e ritorna
alla temperatura T1 . Il calore ceduto complessivamente è
VD
= LCD
VC
ed è negativo come il lavoro perchè VD < VC .
Nella trasformazione DA il gas è isolato termicamente e ritorna allo stato iniziale con
QC = nRT1 ln
T2 VAγ−1 = T1 VDγ−1
(∗∗)
Il lavoro del gas è LDA = −∆Eint = ncV (T1 − T2 ) = −LBC (si
assume γ costante). Sommando tutti i contributi
Q = QA + QC = L = LAB + LBC + LCD + LDA = LAB + LCD
Il rendimento del ciclo è
η =1+
QC
nRT1 ln(VD /VC )
T1 ln(VC /VD )
=1+
=1−
QA
nRT2 ln(VB /VA )
T2 ln(VB /VA )
Usando le relazioni (*) e (**) si ha VB /VA = VC /VD e quindi
T1
rendimento ciclo Carnot
T2
Il rendimento del ciclo dipende solo dalla temperatura delle
sorgenti con cui il gas scambia calore. Poichè T1 < T2 , si ha
0 < η < 1. Inoltre QA > |QC | e il gas assorbe complessivamente calore (Q > 0) e produce lavoro L = QA + QC < QA :
η =1−
il calore assorbito non si trasforma totalmente in lavoro, una
parte viene ceduta restando sotto forma di calore scambiato.
Anche se il primo principio della termodinamica non pone
limiti alle trasformazioni di energia da una forma all’altra,
la trasformazione del calore in lavoro appare limitata (mentre è sempre possibile trasformare integralmente il lavoro in
calore, per esempio sfruttando l’attrito). Questo fatto è una
espressione del secondo principio della termodinamica: non
esiste un ciclo di trasformazioni che dia come unico risultato l’acquisizione di calore da una sorgente termica e la sua
totale trasformazione in lavoro (enunciato di Kelvin).
Si può vedere che η < 1 anche dal calcolo della variazione
di entropia. Complessivamente nel ciclo ∆S = 0, perchè
l’entropia è una variabile di stato, quindi 0 = ∆SAB + ∆SCD ,
dato che nelle trasformazioni adiabatiche l’entropia non cambia. Dalla variazione di entropia lungo una isoterma si ha:
0=
QC
QA
+
T2
T1
→
QA
|QC |
=
→ QA > |QC | , (T1 < T2 )
T2
T1
Nel piano (S, T ) un ciclo di Carnot è rappresentato da un
rettangolo: l’espansione isoterma alla temperatura T2 è rappresentata dalla linea orizzontale T = T2 ; l’espansione adiabatica dalla linea verticale S = S2 ; la compressione isoterma
dalla linea orizzontale T = T1 ; la compressione adiabatica
dalla linea verticale S = S1 :
T
QA = T2 (S2 − S1)
T2
QC = T1 (S1 − S2 )
T1
S1
S2
S
L = QA + QC = (T2 − T1 )(S2 − S1 )
L
T1
η=
=1−
QA
T2
P Cicli firgoriferi
In un ciclo frigorifero il sistema complessivamente assorbe
lavoro e cede calore Q = L < 0. Nel caso più semplice il
sistema assorbe il calore Q0 dalla sorgente fredda e cede
calore QC a una sorgente calda: risulta sempre |QC | > Q0.
Quindi L = Q0 + QC = Q0 − |QC | < 0: occorre sempre compiere un lavoro sul sistema. Si definisce efficienza di un ciclo
frigorifero il rapporto
ξ=
Q0
Q0
=
|L|
|Q0 + QC |
Un ciclo di Carnot percorso in verso inverso costituisce un
esempio di ciclo frigorifero. Il gas
assorbe il calore
Q0 = nRT1 ln(VC /VD )
dalla sorgente alla temperatura
T1 (sorgente fredda) e cede il
calore
QC = nRT2 ln(VA /VB )
alla sorgente alla temperatura T2
(sorgente calda). L’efficienza è
Q0
nRT1 ln(VC /VD )
T1
=
=
|Q0 + QC |
nRT2 ln(VB /VA ) − nRT1 ln(VC /VD )
T2 − T 1
(si è usato VB /VA = VC /VD ).
In un ciclo frigorifero di Carnot tanto più le due temperature
a cui lavora il ciclo sono vicine tanto maggiore è l’efficienza
del ciclo. Inoltre
T2
|QC | =
Q0 > Q 0
T1
il calore ceduto dal sistema alla sorgente calda è sempre
maggiore (in modulo) di quello assorbito, cioè sottratto alla
ξ=
sorgente fredda e quindi il processo avviene sempre in presenza di lavoro fornito dall’ambiente al sistema:
Q0 − |QC | = |L| < 0.
Questo fatto è una espressione del secondo principio della
termodinamica: non esiste un ciclo di trasformazioni che dia
come unico risultato il trasferimento di calore da una data
sorgente termica a un’altra sorgente termica a temperatura
maggiore (enunciato di Clausius).
Nel piano (S, T ) un ciclo di Carnot frigorifero è rappresentato
dal rettangolo precedente percorso in verso opposto:
il calore assorbito dalla sorgente
T
fredda (T1 ) è Q0 = T1 (S2 − S1 ),
il calore ceduto alla sorgente calda è
T2
QC = T2 (S1 − S2 ),
T1
quindi
S1
S2
S
ξ=
Q0
T1
=
|L|
T2 − T1
L’esperienza ci fa notare che certi fenomeni avvengono spontaneamente solo in una data direzione: se consideriamo due
corpi a temperatura diversa posti in contatto, c’è sempre
una cessione di calore dal corpo più caldo a quello più freddo
fino a quando si raggiunge l’equilibrio termico: il calore non
passa mai spontaneamente dal corpo freddo a quello caldo.
Analogamente un gas che compie un’espansione libera riempiendo un recipiente vuoto a cui è connesso mediante un
rubinetto: le molecole del gas non si raccolgono spontaneamente in una sola metà del contenitore. Questi processi
si dicono irreversibili, cioè avvengono spontaneamente solo
in una data direzione (anche se il processo che va nella direzione contraria non sarebbe proibito dalla conservazione
dell’energia). Vedremo che per stabilire in quale direzione si
svolgono questi processi bisogna studiare l’entropia.
Confrontiamo una trasformazione reversibile con una irreversibile che avvengono tra gli stessi stati A e B con, ad es.,
TA = TB = T e il gas che viene compresso. Nella trasfor~est = −F
~int (la forza memazione reversibile in ogni istante F
dia che le particelle esercitano sul pistone è uguale alla forza
che bisogna applicare al pistone per farlo scendere), quindi
Z B
VB
int
Lest
=
−L
=
−
pdV
=
−nRT
ln
> 0,
(VB < VA )
rev
rev
V
A
A
Nella
trasformazione
irreFest
versibile all’inizio il sistema è
~est = −F
~int
in equilibrio quindi F
poi si abbassa velocemente il
R
pistone: non è possibile calcolare pdV perchè non si conosce
la pressione all’interno del gas, ma le particelle del gas hanno
rispetto al pistone una velocità maggiore che nel caso
reversibile (il pistone viene loro incontro più velocemente),
di conseguenza la forza media che le particelle esercitano sul
pistone sarà maggiore che nel caso reversibile. Quindi per
p
far scendere il pistone occorre ap~est maggiore rispetto a
plicare una F
B
quella che si applica nel caso reA
versibile, e per arrivare allo stesso
stato finale bisogna fare un lavoro
V
V
V
maggiore:
B
A
est
Lest
irr > Lrev
Se il gas si espande: nella trasformazione reversibile
VB
< 0,
(VB > VA )
VA
Nella trasformazione irreversibile, all’inizio il sistema è in
~est = −F
~int poi si allontana velocemente
equilibrio quindi F
il pistone: le particelle del gas hanno rispetto al pistone una
velocità minore che nel caso reversibile (il pistone si allontana più velocemente) di conseguenza la forza media che le
particelle esercitano sul pistone
Fest
sarà minore che nel caso reversibile. Quindi per far scorrere
il pistone occorre applicare una
~est minore rispetto a quella che si applica nel caso reversibile,
F
int
Lest
rev = −Lrev = −nRT ln
e per arrivare allo stesso stato finale bisogna fare un lavoro
minore:
p
est
|Lest
irr | < |Lrev |
A
però nell’espansione il lavoro esterno
è negativo quindi si ha ancora
B
VA
VB
V
est
Lest
irr > Lrev
Nelle due trasformazioni (quella reversibile e quella irreversibile)
la variazione dell’energia interna è la stessa (Eint è una variabile di stato), quindi
½
Qrev + Lest
rev
∆Eint AB =
Qirr < Qrev
Qirr + Lest
irr
Confrontiamo un ciclo di Carnot (termico) con un ciclo in
cui l’espansione isoterma A → B è sostituita da una
p
A
B
D
T2
C
T1
V
trasformazione irreversibile che
porta sempre il sistema da A → B.
Sia la trasformazione reversibile
che quella irreversibile avvengono
alla stessa temperatura T2 , quindi
Z B
Z B
δQirr
δQrev
<
≡ ∆SAB
T
T
A
A
la variazione dell’entropia è sempre maggiore della somma
(o integrale) delle quantità di calore scambiate irreversibilmente, ciascuna divisa per la temperatura a cui viene scambiata. Più in generale si ha
Z B
δQ
∆SAB ≥
disuguaglianza di Clausius
T
A
dove il segno uguale vale solo se la trasformazione è reversibile.
Un’importante conseguenza della disuguaglianza di Clausius
è che se un sistema è isolato, quindi non scambia né calore
né lavoro con l’esterno, si ha
Z B
δQ
∆SAB ≥
=0
T
A
dove il segno uguale vale solo se la trasformazione è reversibile: in una trasformazione di un sistema isolato l’entropia
aumenta o, se la trasformazione è reversibile, rimane costante.
L’entropia è perciò un indicatore dell’evoluzione temporale
dei sistemi isolati. In particolare nell’espansione libera di un
sistema isolato l’entropia aumenta.
La disuguaglianza di Clausius si generalizza al caso di un
ciclo:
I
δQirr
0 = ∆Sciclo >
T
dove l’integrale indica la somma algebrica di tutte le quantità
di calore scambiate nel ciclo, divise per la temperatura a cui
avviene lo scambio. Se tutte le trasformazioni del ciclo sono
reversibili l’integrale dà la variazione dell’entropia, quindi in
generale si ha
I
δQ
≤0
T
dove il segno di uguale vale solo se il ciclo è fatto di trasformazioni reversibili.
Confrontiamo il rendimento di una macchina di Carnot con
p
A
B
D
T2
C
T1
V
quello di una macchina in cui le
due isoterme sono sostituite da
due trasformazioni irreversibili (le
due macchine lavorano alle stesse
temperature).
η
rev
η irr
|Qrev
|
T1
=1− C
=
1
−
Qrev
T2
A
|Qirr
C |
= 1 − irr
QA
(T2 > T1 )
Per la disuguaglianza di Clausius
|
Qirr
Qirr
Qirr
|Qirr
|Qirr
T1
A
C
A
C |
+
<0 →
<
→
< C
T2
T1
T2
T1
T2
Qirr
A
Quindi
η irr < η rev
una macchina termica di Carnot reale ha un rendimento inferiore della corrispondente macchina di Carnot operante tra
le medesime temperature.
Terorema di Carnot:
¬ il rendimento di una macchina di Carnot reversibile è maggiore del rendimento di qualsiasi altra macchina, sia reversibile
che irreversibile
­ il rendimento di un ciclo di Carnot reversibile è sempre
η = 1 − T1/T2 , anche se la macchina lavora con un fluido che
non è un gas perfetto
® la definizione della variabile di stato entropia, che abbiamo
dato per il gas perfetto, è valida per un sistema qualsiasi
Z B
δQrev
∆SAB =
T
A
Non dimostriamo il teorema ma vediamo che ® è diretta
conseguenza di ­. Infatti se in una macchina di Carnot
reversibile il rendimento vale sempre
η =1−
T1
T2
indipendentemente dal gas utilizzato, allora dalla definizione
η =1−
|QC |
QA
→
|QC |
T1
=
QA
T2
|QC |
QA
QC
QA
=
→
+
=0
T1
T2
T1
T2
questo significa che in un ciclo reversibile, la somma dei calori
scambiati diviso la temperatura a cui avviene lo scambio è
zero
I
δQrev
=0
T
quindi si può introdurre una funzione di stato S la cui variazione infinitesima
δQrev
dS =
T
e tale che
I
Z B
δQ
dS = 0
e
= SB − SA
T
A
si ritrova quindi la definizione di entropia data per il gas
perfetto.