La sindrome da microdelezione 22q11
Aspetti endocrini
R.Lala
Endocrinologo pediatra
Torino 19 marzo 2016
Incontro regionale per le famiglie
Prevalenza clinica delle
endocrinopatie J Guerrero Fernandez Ann Pediatr 2011
Disfunzione paratiroidea
(ipoparatiroidismo) 21-72%
Deficit di ormone della crescita 0-4,1%
(nel contesto di bassa statura che ha una
prevalenza del 16-41%)
Disfunzione tiroidea (ipotiroidismo
congenito, tireopatie autoimmuni : tiroidite
morbo di Graves Basedow) 0,7-7%
Quando porre il sospetto di
ipocalcemia
Sintomi aspecifici
Letargia
Eccitabilità
Ipertono
Crisi convulsive
QT lungo
Strabismo
Cianosi
Apnea
Laringospasmo
Sintomi specifici
Forma manifesta
Spasmo carpo-pedale
Opistotono
Trisma
Forma latente
Segno di Chwostek
Segno di Trusseau
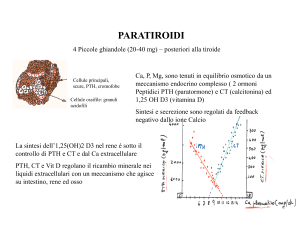
Prevalence of hypocalcaemia and its associated features in
22q11·2 deletion syndrome E Nin Man Cheung
Clin Endocrinol (Oxf). 2014 Jan; 81(2): 190–196.
Scatterplot of 397 simultaneously obtained intact PTH and ionized calcium levels available for 116 subjects with 22q11·2DS. Most
measurements (n = 364, 91·7%) were collected in adulthood. The dotted lines represent approximately normal ranges of intact PTH and
ionized calcium levels. The pink shaded area E represents 145 values in the normal range for both ionized calcium and PTH from 66
subjects, of whom 32 (48·5%) subjects had previously documented episodes of hypocalcaemia and 46 (69·7%) subjects had a lifetime
prevalence of hypocalcaemia. Area A (low calcium and high PTH levels) has 7 measurements from 7 subjects; area B (low calcium and
normal PTH levels) 153 measurements from 77 subjects; area C (low calcium and low PTH levels) 47 measurements from 21 subjects; area
D (normal calcium and high PTH levels) 4 measurements from 3 subjects; area F (normal calcium and low PTH levels) 35 measurements
from 23 subjects (of whom 19 (82·6%) had previously documented episodes of hypocalcaemia). Areas H (high calcium and normal PTH
levels) and I (high calcium and high PTH levels) together have 6 measurements from 6 subjects, all of whom had previously documented
episodes of hypocalcaemia. Area B contains 99 measurements representing relative hypoparathyroidism.
TRATTAMENTO
Ipocalcemia acuta :
CALCIO GLUCONATO 10% 2ml/kg in 10 minuti EV, eventualmente ripetuta
ogni 6-8 ore
Ipocalcemia cronica:
CALCITRIOLO (ROCALTROL 0.25-0.5 mg) ) 20-100 ng/kg/die in 2-3 dosi
+
CALCIO GLUCONATO 500-2000 mg/die in 3-4 dosi
• Il trattamento “convenzionale” ha bisogno di essere modulato in base alla
patologia, peso, altre situazioni con assorbimento alterato
• Il compenso metabolico è sempre una bilancia tra l’evitare le crisi acute e gli
effetti collaterali, in particolare l’ipercalciuria e la nefrocalcinosi
• Troppe compresse da assumere via orale per un bambino…e la compliance?
Servono nuove terapie!
J Pediatr Endocrinol Metab. 2014 Jan;27(1-2):53-9. doi: 10.1515/jpem2013-0159.
Teriparatide (rhPTH) treatment in children with syndromic
hypoparathyroidism.
Matarazzo P, Tuli G, Fiore L, Mussa A, Feyles F, Peiretti V, Lala R
IPOPARATIROIDISMO
TERIPARATIDE: hrPTH (Forsteo-Lilly)
• frammento N-terminale del PTH ricombinante con 34 aminoacidi.
• agisce legando il recettore sugli osteoclasti attraverso RANK-ligando
come il PTH umano.
• aumenta il calcio nel siero, parzialmente aumentando il riassorbimento
osseo, come fisiologicamente succede con il PTH umano in caso di
ipocalcemia.
Iniezioni sottocute con penne contenenti 600 mcg di teriparatide, ogni dose
eroga 20 mcg.
Indicazioni della FDA : donne con osteoporosi post-menopausale e in
uomini con alto rischio di fratture ossee
DATI ALLA DIAGNOSI
LA NOSTRA ESPERIENZA ( MAGGIO 2009 – MAGGIO 2010 )
SOGGETTO (SESSO)
DIAGNOSI
ETA’
GENETICA
P. R. (M)
SINDROME CHARGE
2 giorni
Non effettuata
B. A. (M)
SINDROME di Di George
2 giorni
Microdelezione cromosoma 22
B. A. (F)
SINDROME APECED
1 anno 7 mesi
Mutazione AIRE [260 T>C] e [967-979 del]
B. M. (M)
SINDROME APECED
6 anni 6 mesi
Mutazione AIRE [260 T>C] e [967-979 del]
C. F. (M)
SINDROME HDR
14 anni 2 mesi
Mutazione GATA3 gene
S. F. (F)
SINDROME APECED
5 anni 5 mesi
Mutazione AIRE [260 T>C] e [967-979 del]
Sindrome CHARGE
Sindrome di Di George
Sindrome APECED
• Coloboma
• Ipoparatiroidismo
• Ipoparatiroidismo
• Cardiopatia congenita
• Anomalie faciali
•Addison
• Atresia delle coane
• Deficit linfociti T
•T1DM
• Ritardo accrescitivo
• Cardiopatia congenita
• Candidosi mucocutanea cronica
• Anomalie uro-genitali
• Atresia esofagea
•Anemia perniciosa
• Sordità
• Ritardo mentale
• Epatite cronica attiva
• Ipoparatiroidismo
• Insufficienza gonadica
Sindrome HDR
• Ipoparatiroidismo
• Sordità neurosensoriale
bilaterale
• Displasie renale
TRATTAMENTO CON TERIPARATIDE
LA NOSTRA ESPERIENZA ( MAGGIO 2009 – MAGGIO 2010 )
Andamento della calcemia
Andamento della fosfatemia
10
7
8
Valori normali
5
3
Valori normali
4
1
-1
6
2
diagnosi
calcio serico
convenzionale (Ca + vit D)
diagnosi
0
fosfatemia
teriparatide
Andamento della calciuria
diagnosi
0,6
convenzionale (Ca + vit D)
0,5
teriparatide
0,4
0,3
0,2
Valore normale < 0.20
0,1
0
calciuria
convenzionale (Ca + vit D)
teriparatide
CONCLUSIONI
• Effetti a breve termine: miglior compenso metabolico, minor numero di
crisi tetaniche, riduzione della ipercalciuria, miglioramento della compliance.
• Effetti a lungo termine: prevenzione della nefrocalcinosi e della possibile
insufficienza renale
• Costo (570,71 euro per una penna che dura 28 giorni) , il farmaco è a carico
del SSN per i pazienti affetti da malattie rare.
• Somministrazione sottocutanea
• Off-label per l’età pediatrica
• Non ancora ben noti gli effetti induttori a lungo termine sull’osteosarcoma
(descritta in ratti trattati con alte dosi farmacologiche, 3-60 volte superiori a
quelle usate nell’uomo)
Limiti: nella osteoporosi post-menopausale il trattamento dura 2 anni ma nell’
ipoparatiroidismo il trattamento dovrebbe essere “a vita”.
Valutazione accrescitiva
American Journal of Medical Genetics Part A
Volume 158A, Issue 11, pages 2672-2681
Diagnosi di deficit di ormone della crescita
(nota AIFA 39)
Età evolutiva
Bassa statura da deficit di GH definito dai seguenti parametri clinico-auxologici e di laboratorio:
I. Parametri clinico – auxologici:
a) statura ≤ -3 DS
oppure
b) statura ≤2 DS e velocità di crescita/anno <-1,0 DS per età e sesso valutata a distanza di almeno
6 mesi o una riduzione della statura di 0,5 DS/anno nei bambini di età superiore a due anni.
oppure
c) Statura inferiore a -1,5 DS rispetto al target genetico e velocità di crescita/anno ≤-2 DS o ≤-1,5
DS dopo 2 anni consecutivi.
d) velocità di crescita/anno ≤-2 DS o ≤-1,5 DS dopo 2 anni consecutivi, anche in assenza di bassa
statura e dopo aver escluso altre forme morbose come causa del deficit di crescita; nei primi 2 anni
di vita, sarà sufficiente fare riferimento alla progressiva decelerazione della velocità di crescita (la
letteratura non fornisce a riguardo dati definitivi in termini di DS);
oppure
e) malformazioni/lesioni ipotalamo-ipofisario dimostrate a livello neuro-radiologico;associate a
II. Parametri di laboratorio:
a) risposta di GH < 8 µg/L a due test farmacologici eseguiti in giorni differenti
b) risposta di GH < 20 µg/L nel caso il test impiegato sia GHRH + arginina
Età evolutiva
In soggetti con statura < -3 DS oppure statura < -2 DS e velocità di
crescita/anno < -1 DS rispetto alla norma per età e sesso, misurata con
le stesse modalità a distanza di almeno 6 mesi e con normale
secrezione di GH, la terapia può essere effettuata solo se autorizzata
dalla Commissione Regionale preposta alla sorveglianza epidemiologica
ed al monitoraggio dell’appropriatezza del trattamento con GH in base
alle più recenti acquisizioni scientifiche in materia. Il dosaggio non dovrà
superare 50µg/kg/die (raccomandazione EMA). Nei casi autorizzati dalla
Commissione regionale, ma non compresi nelle indicazioni contenute
nella presente nota AIFA, l’uso è da ritenersi off-label ed è, pertanto,
soggetto alla normativa in materia.
Ipotiroidismo Congenito Primitivo :
una condizione eterogenea
Tiroide non in sede
Agenesia
Permanente
TITF1, PAX8, FOXE1
Ectopia
Permanente
TITF1, PAX8, FOXE1
Emiagenesia
Permanente
TITF1, PAX8, FOXE1
Ipoplasia
Permanente
TSHR
Disormonogenesi (volume
aumentato)
Permanente (?)
TPO, TG, NIS, PDS, DEHAL1, DUOX1, DUOX2
Anticorpi materni
Transitorio
-
Eccesso/difetto di iodio
Transitorio
-
Sostanze e farmaci
antitiroidei
Transitorio
-
Stato maturazione asse ITT
(IUGR/prematuranza)
Transitorio
-
Tiroide in sede
Altri
Sindrome di Down
Del 22q11
Conclusioni 1
La disfunzione endocrina prevalente è l’ipoparatiroidismo che si manifesta
con ipocalcemia cronica. L’ipocalcemia ed il suo trattamento possono
condizionare le performances cognitive e l’attività muscolare, inclusa quella
cardiaca. L’ipocalcemia determina contrazioni muscolari anche dolorose e
depressione del tono dell’umore ed è aggravata da un’eventuale carenza di
magnesio. L’ipocalcemia cronica inoltre può slatentizzare crisi epilettiche e
manifestarsi in occasione di stress, malattie intercorrenti, interventi chirurgici.
Il suo trattamento cronico può portare a deposizioni renali di calcio
(nefrocalcinosi) Sono in sperimentazione nuovi trattamenti che possono
ovviare a questo effetto indesiderato.
La bassa statura è condizione frequente, il deficit di ormone della crescita è
invece raro. Ove la crescita sia molto compromessa è possibile intervenire
con ormone della crescita off-label previa autorizzazione della specifica
Commisione Regionale.
L’elemento più rilevante del trattamento endocrino è il suo essere situato in
un progetto terapeutico globale, organizzato sui bisogni del singolo individuo
attraverso l’individuazione di un case manager.
Conclusioni 2
E’ probabile che gli individui con 22q11DS presentino
differenti priorità di tipo clinico e psico-sociale dalla nascita
alla maturità.
La care precoce è dominata dalla terapia chirurgica delle
malformazioni, dal supporto all’alimentazione, dal
trattamento delle infezioni; nella seconda infanzia,
l’attenzione prevalente va allo sviluppo neurologico, al
comportamento ed all’educazione; in adolescenza
predomina il monitoraggio della scoliosi con possibile
intervento chirurgico ed il supporto psico-sociale; nell’età
adulta, il supporto socioeconomico, medico generale e
psichiatrico
A.Habel Towards a safety net for management of 22q11.2 deletion syndrome: guidelines
for our times Eur J Pediatr 2014 173:757-765