Lezione 21/10/2013
Prof. Teti
RISPOSTA AL DANNO CELLULARE
Ricordiamo che l'infiammazione è quel processo di difesa che nasce nei confronti di un'infezione
che però in alcune circostanze può diventare causa di danno al tessuto circostante e di necrosi
cellulare.
Noi invece stiamo seguendo un'altra linea direttiva che è quella delle risposte cellulari al danno,
infatti se ricordate vi ho detto che le cellule rispondono in maniera diversa a un danno causato da
agenti patogeni e la risposta è caratterizzata in primo luogo da risposte di tipo adattativo tra cui c'è
l'iperplasia, l'ipertrofia, la metaplasia e successivamente da un danno cellulare che prima può essere
reversibile e poi irreversibile.
Stavamo parlando, la volta scorsa, della patologia e delle alterazioni che riguardano l'equilibrio tra
proliferazione e morte cellulare, che interviene nei meccanismi dell'omeostasi nei tessuti, cioè
nell'equilibrio tra cellule che vengono prodotte e vengono perse dato dall'apoptosi e dal
differenziamento. Parlavamo anche dei meccanismi che attivano e che regalano la proliferazione,
perché indipendentemente dall'apoptosi ci sono meccanismi che attivano e inibiscono il processo
proliferativo. In particolare abbiamo accennato ai moduli -SH2 che sono specifici per ogni proteina
citoplasmatica e che sono specifici nel legame con una particolare proteina fosforilata per esempio
nella coda citoplasmatica.
Qui vedete messe in evidenza le varie fasi del ciclo proliferativo, le chinasi-ciclina-dipendenti
impegnate e viene messa in evidenza nella prima parte della fase G1 la ciclina g1, la ciclina d è
quella che da inizio alla progressione della fase g1 con le chinasi-ciclina-dipendenti 4 e 6 e poi c'è
la ciclina E ecc...
La fase G1 è una fase fondamentale del ciclo proliferativo, l'avevamo già sottolineato, perché è una
fase in cui intervengono molti fattori che regolano la progressione del ciclo mitotico o il blocco
della fase G1. E a questo riguardo la fase G1 è da considerare divisa in 2 metà: nella prima
intervengono i fattori di competenza, nella seconda i fattori di progressione. Però diciamo che anche
i geni attivati nella prima metà sono diversi da quelli che vengono attivati nella seconda metà. I
primi sono quelli rappresentati soprattutto da quei fattori di trascrizione che si chiamano c-myc, cfos, c-jun che sono fattori che a seguito di fosforilazione si legano ai loro siti di consenso dei geni
coinvolti nell'attivazione della proliferazione. Ora, questi geni sono molto precoci quindi sono
attivati nella prima metà della fase G1 e non richiedono una nuova sintesi cioè essendo geni precoci
sono immediatamente attivati mediante processo di fosforilazione senza che ci sia l'induzione della
sintesi di altre proteine, non attivazione dovuta ad altri geni. Mentre nella seconda parte della fase
G1 sono prodotti dei geni che sono attivati dai prodotti di questi geni precoci che si chiamano
IEGs, proprio per sottolineare il fatto che sono attivati in maniera istantanea. I prodotti codificati da
questi geni sono quelli che poi attivano gli altri geni che sono preponderanti nella seconda parte
della fase G1. Proprio questi geni precoci codificano per proteine fondamentali per attivare altri
geni coinvolti positivamente nella proliferazione. Quindi vedete è un processo complesso,
articolato, tra geni IEGs, della prima metà della fase G1, e geni attivati tardivamente, proprio i
prodotti di questi geni attivati direttamente dai fattori di crescita o dalle proteine chinasi.
I geni indotti dalle MAPK e da secondi messaggeri (CA2+, calmodulina) si distinguono in:
a) geni precoci, quali c-myc, c-fos, c-jun il cui RNA aumenta molto prima della metà della fase G1
e la cui induzione non necessità di sintesi proteica;
b) geni tardivi, il cui RNA aumenta dopo la metà della fase G1 e che dipendono dalla sintesi
proteica dei prodotti codificati dagli immediate early genes. Per questo motivo gli IEGs sono stati
chiamati “ingresso della risposta genomica” a stimoli quali virus o fattori di crescita.
Anche la differenziazione entra in gioco nell'equilibrio omeostatico che regola le dimensioni di un
tessuto e interviene in maniera diversa a seconda del tipo di tessuto. Per esempio la differenziazione
ha un certo ruolo per quanto riguarda le cellule del midollo osseo perché le cellule del midollo
osseo una volta che si differenziano non si dividono più. Voi lo sapete bene per quanto riguarda il
sangue che è l'unico tessuto umano in cui è nettamente distinguibile il compartimento di
proliferazione rispetto al compartimento di differenziazione. Perché il primo si trova a livello del
midollo osseo, il secondo nel sangue periferico degli altri tessuti di quei compartimenti che sono
fisicamente distinguibili. Quando le cellule del midollo osseo vanno incontro al un processo di
differenziazione ( il midollo osseo ha la peculiarità che le cellule che qui si dividono non solo si
differenziano ma anche maturano - compartimento di espansione-, le cellule che proliferano man
mano maturano e poi differenziano ) lasciano il midollo osseo, durante la cosiddetta fase di
emersione e vanno nel torrente circolatorio.
I compartimenti sono talmente distinti che le cellule una volta differenziate non è che maturano più
e quindi la differenziazione gioca un ruolo importante nel mantenimento dell'omeostasi proprio
perché impedisce che poi queste cellule proliferino ancora, interviene in senso negativo quindi nella
proliferazione. Mentre invece le cellule epatiche, essendo cellule stabili, anche se si differenziano
non perdono completamente la capacità di proliferare e se opportunamente stimolate comporta un
ritorno dalla fase G0 alla fase G1.
La differenziazione assume questo ruolo importante, nel tessuto ematico, nel regolare
negativamente la proliferazione: su questa base è comprensibile il meccanismo per cui nelle
patologie del midollo, le leucemie, l'aumento della massa dei blasti leucemici non è dovuto, come
tutti potreste credere, ad un aumento della loro attività proliferativa rispetto ai blasti normali (ci
sono vari parametri che valutano il grado di proliferazione: il tempo di permanenza nelle varie fasi
del ciclo mitotico, il numero di cellule che una volta lasciato il ciclo mitotico va verso la
differenziazione, presi in considerazione per valutare l'entità della proliferazione) ma è stato visto
che aumenta il pool delle cellule differenziate per una maggiore permanenza nel compartimento di
proliferazione. Nelle leucemie è alterato quindi il processo apoptotico, poiché come vedremo
meglio una delle caratteristiche dell'apoptosi è un processo fisiologico ma le sue alterazioni sia in
senso positivo che in senso negativo sono causa di patologia, nel caso dei tumori in genere ma
soprattutto delle leucemie dove c'è questo aumento del tempo di differenziazione: queste cellule
stanno a lungo nel compartimento di differenziazione perché c'è una ridotta apoptosi. Questo per
spiegare meglio nelle patologie neoplastiche il ruolo della differenziazione.
MECCANISMI CHE LIMITANO LA PROLIFERAZIONE
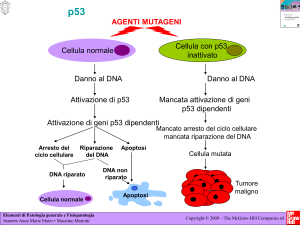
Una delle proteine normali che limitano il processo proliferativo è la proteina p53. Essa è codificata
da un gene onco-soppressore, è coinvolta nella regolazione del controllo del ciclo cellulare perché
va a determinare o un blocco di esso se c'è un danno al DNA in fase G1 oppure se il danno non è
riparabile manda in apoptosi la cellula (definizione correttamente esposta dalla collega A, nda).
E' un fattore di trascrizione perché si lega a siti di consenso di vari geni che sono diverso genere.
Dovete sapere che in più del 50% di tutti i tumori del mondo la p53 è mutata. Nel contempo è
rarissimo, forse non succede mai, che in un tumore sia mutato un solo gene infatti un tumore è la
risultante di una serie di mutazioni a livello di più geni che possono trovarsi anche su cromosomi
diversi; noi parliamo tra l'altro di cancerogenesi cioè esiste un processo dinamico in cui si passa
attraverso vari stadi in cui sono coinvolti vari geni; di solito la mutazione di p53 è un evento
abbastanza precoce che precede le mutazioni di altri geni.
Proprio a causa di questo importantissimo ruolo essa in condizioni normali è presente in
piccolissime quantità. D'altronde il punto è sempre là: ci tengo che voi abbiate chiaro il concetto che
tutte le risposte cellulari sono sotto il controllo di più fattori che tendono a tenere in equilibrio
omeostatico le varie funzioni della cellula. Per cui la p53 se fosse presente in grandi quantità
attiverebbe la morte cellulare, bloccherebbe la proliferazione in maniera molto significativa, per cui
in condizioni fisiologiche le quantità di p53 hanno livelli così bassi proprio perché servono
esclusivamente ad entrare in gioco con altri fattori che attivano la proliferazione. Però se c'è una
situazione in cui si ha un grave danno al DNA la p53 aumenta la sua concentrazione perché non
viene degradata. Essa infatti viene degradata quasi dopo la sua sintesi, ma in cellula sottoposta a
stress si accumula. Tutto questo perché viene fosforilata: viene attivata una protein-chinasi DNAdipendente dal danno del DNA (le cellule devono rispondere adeguatamente agli stress, per cui lo
stresso stress che altera il DNA è quello che attiva la p53) che fosforila p53 a livello di due serine
chiave per cui una volta fosforilata essa si accumula e può esercitare la sua funzione, maggiore in
una cellula non danneggiata. Parallelamente a questo meccanismo ne interviene un altro, in quanto
il livello di p53 è fondamentale per la cellula ne va della sua vita o morte: la p53 è regolata da
un'altra proteina che si chiama Mdm2 (Mouse double minute 2 homolog, conosciuta anche come E3
ubiquitin-protein ligase Mdm2), codificata dallo stesso gene, che inibisce la p53 con due
meccanismi (vedete quant'è importante la regolazione dei livelli di p53): da un lato agisce sul gene
di p53 bloccando la sua produzione (fattore di trascrizione negativo per p53), inoltre idrolizza la
p53 non fosforilata, per cui quando viene fosforilata la p53 da questa protein-chinasi DNAdipendente, la stessa p53 si copre di gruppi fosfato, si protegge dall'azione idrolitica di Mdm2. Per
cui mdm2 agisce a due livelli sul gene di p53 bloccandolo e sulla proteina degradandola sempre con
il sistema dell'ubiquitinazione. La regolazione dei livelli delle proteine intracellulari sia nell'atrofia,
sia in questo caso della p53 è mediato da un processo di distruzione non idrolitica, non dovuta
all'azione di enzimi idrolitici lisosomiali senno la cellula va incontro ad autofagia come succede in
alcune condizione patologiche. In questo caso vengono degradate singole proteine in maniera
silente, in maniera che non ci siano mutazioni cellulare, in base a un processo di ubiquitinazione,
cioè l'ubiquitina, una grossa molecola, che ricopre le proteine che devono essere degradate e le
inserisce in un complesso più ampio che si chiama proteasoma nell'ambito del quale le proteine
vengono degradate.
In cellule normali non sottoposte a stress p53 è una proteina altamente instabile che in genere viene
degradata subito per proteolisi dopo la sua stessa sintesi. Ha un'emivita di circa 20 minuti. Allo stato
stazionario la concentrazione di p53 in queste cellule è molto bassa. Essa si lega ai siti di consenso
presenti in vari geni; in seguito ad alcuni stress o danni al genoma, p53, può essere fosforilata nel
suo dominio N-terminale da alcune chinasi. La fosforilazione (serina 19 e 37) blocca il legame di
Mdm2 e salva p53 da ubiquitinazione e degradazione.
-Stress determina attivazione DNA-pk
-Mdm2 idrolizza la p53 non fosforilata
Il gene Mdm2 viene attivato da p53, la quale si autoregola, cioè attiva il gene di una proteina che la
distrugge; e questo è un altro esempio di meccanismo automatico di controllo delle risposte
cellulari. La proteina Mdm2 agisce sia come ubiquitina-ligasi (porta l'ubiquitina sulla p53) che
riconosce una lisina nel dominio trans-attivante (attiva i geni che sono sotto il suo controllo in
posizione trans) sulla p53. Quindi determina il legame dell'ubiquitina proprio al livello di questo
dominio trans-attivante e impedisce che la p53 svolga le sue funzioni perché ne attiva la
degradazione però d'altra parte, come ho già detto, inibisce la trascrizione di p53.
Vi avevo accennato l'altra volta che ci sono due tipi di geni coinvolti nella oncogenesi: i protooncogeni che attivano la proliferazione la cui mutazione comporta un guadagno di funzione e gli
onco-soppressori che invece hanno un ruolo di inibire la proliferazione per cui c'è una riduzione di
funzione.
PROTO-ONCOGENE-> più proliferazione (guadagno di funzione)
ONCO-SOPPRESSORI-> proliferazione inibita (riduzione di funzioni)
Le mutazioni che coinvolgono i proto-oncogeni sono mutazioni cosiddette dominanti perché basta
che cia sia una mutazione in uno dei due alleli per cui il prodotto codificato da quei geni sia mutato,
se c'è un guadagno di funzioni basta un allele per determinare un aumento della proteina. Per cui si
dice che è una mutazione dominante. Al contrario per i geni onco-soppressori si dice nella quasi
totalità dei quasi, tranne che per p53, che le mutazioni sono recessive. Le mutazioni che possono
riguardare gli onco-soppressori sono perlopiù delezioni o mutazioni puntiformi che determinano
un'alterazione della sequenza della proteina perché questa non è più funzionale; se questo accade a
livello di uno dei due alleli, l'altro allele funzionante può sopperire alle carenze dell'allele mutato, si
produce una quantità di proteina normale, non patologica, la quale può essere in grado di svolgere le
sue normali funzioni: perché ci sia una conseguenza a livello della proliferazione è necessario che la
mutazione riguardi tutti e due gli alleli. Il comportamento del gene tp53 è anomalo rispetto agli
altri onco-soppressori, perché si può comportare o come onco-soppressore recessivo o oncosoppressore dominante. Si comporta in maniera recessiva come tutti gli altri onco-soppressori: se
manca, è deleto o mutato, uno solo dei due alleli, l'altro allele è in grado di codificare per una
proteina normale, non mutata, fisiologica e quindi svolgere la sua funzione. Questo è il
comportamento recessivo che la tp53 condivide con tutti gli altri geni onco-soppressori.
La p53 si può comportare anche come gene dominante perchè appunto se si ha mutazione in un solo
allele e si produce una copia sola indenne per una proteina non mutata però si forma anche una
proteina mutata cioè questa non è il caso della delezione ma il caso di una mutazione. Il
comportamento recessivo è nel caso della delezione: è deleto un allele, l'altro funziona; ma se la
mutazione non è una mutazione per delezione ma una mutazione per sostituzione di basi avremmo
due copie del p53 una wild-type e un'altra mutata. Voi potreste pensare che quella wild-type
potrebbe funzionare e invece no perché la p53 per potere agire e funzionare deve essere sotto forma
di un tetramero omo tetramero, formato 4subunità di p53. Se in questo tetramero ci sono delle copie
mutate il tetramero non funziona, quindi in questo caso p53 si comporta da gene dominante. Quindi
se la mutazione del gene p53 è una delezione allora p53 si comporta da recessivo come tutti gli
onco soppressori di questo mondo, se invece la mutazione è a livello strutturale, la struttura del gene
è alterata, si producono delle copie wild-type e delle copie mutate che vanno a formare un tetramero
inefficiente, ecco quindi che la mutazione è mutante e il tetramero non essendo omotetramero non
funziona.
DELEZIONE p53-> si comporta da recessivo
ALTERAZIONE p53-> copie wild type e mutate, formazione di tetramero inefficiente (mutazione
dominante)
La p53 è una fosfoproteina molto versatile perché innesca reazioni che determinano la comparsa di
varie risposte cellulari. Essa è dotata di 4 domini:
a) il dominio trans-attivante, responsabile dell'attivazione dell'attivazione o della repressione del
gene, perchè questo dominio può essere sia in senso positivo che negativo, quindi agisce
sull'espressione del gene target
b) il dominio legante il DNA (Dna-binding)
c) dominio oligomerizzante, perchè la p53 agisce come tetramero, questo dominio facilita la
oligomerizzante
dominio responsabile della auto inibizione della sua attività trans-attivante (lap53 inibisce se stessa
grazie a questo dominio); questo ci fa vedere come le cellule mettano in atto molti meccanismi che
regolano le risposte cellulari, che intervengono in ogni fase dell'azione della proteina. Come
avviene per p53 che attiva il gene che codifica per una proteina che la inibisce e che si auto inibisce
da sola, inibendo la sua attività trans-attivante.
I segnali di sopravvivenza (soprattutto fattori di crescita che attivano Mdm2) e segnali mitogeni
(attivando altri fattori di crescita come Map chinasi che fosforilano soprattutto in tyr) causano la
rapida degradazione di p53 in quanto incrementano il livello della proteina Mdm2. Gli stimoli
mitogeni agiscono attraverso fattori di trascrizione che come p53 attivano la loro trascrizione del
gene Mdm2.
La p53 agisce come fattore di trascrizione su un gene chiave della proliferazione, ovvero, p21.
Questa proteina agisce a due livelli: da un lato blocca il PCNA (Antigene nucleare di cellule in
proliferazione che consentendo alla Dna-pol delta il contatto con il filamento danneggiato svolge un
ruolo importante sia per la sintesi di DNA che per la sua riparazione) e dall'altro agisce anche a
livello del complesso ciclina-chinasi-ciclina-dipendente, legandosi al complesso già formato,
bloccando la fosforilazione delle cicline causa un blocco del ciclo proliferativo. Quando non c'è p21
il complesso chinasi-ciclina-dipendente attiva la proliferazione. C'è un altro gene che codifica per
p15, un onco soppressore perchè blocca la proliferazione sempre a livello delle chinasi-ciclinadipendente, però a un diverso livello, in quanto in questo caso la p15 si lega alla chinasi-ciclinadipendente impedendo che sia attivata dal legame con la ciclina, si lega alla chinasi e impedisce che
la ciclina si leghi. Però la p53 agisce a più livelli, come fattore di trascrizione particolarmente
attivo. Poi c'è un altro gene presente in 3 isoforme, ovvero il GADD45 (Growth Arrest and DNA
Damage), il cui prodotto si lega anch'esso al Dna e attiva la riparazione di esso. Il suo legame col
Dna è mediato dalla p45 che viene ad essere espressa in condizioni di stress.
Inoltre p53 attiva l'apoptosi. Il gene BAX se prodotto attiva l'apoptosi non è l'unico ad essere
attivato da p53 ma ce ne sono altri. Per sommi capi possiamo dire che p53 attiva alcuni geni chiave
che facilitano il processo apoptotico ed ha un effetto negativo su un membro delle famiglia di BCL
che attiva la proliferazione. BCL la possiamo definire una sorta di famiglia allargata perché
contiene geni che attivano la proliferazione e geni che attivano l'apoptosi. In particolare BCL2 è un
membro di questa famiglia che attiva in maniera significativa la proliferazione, tanto è vero che il
suo gene è un proto-oncogene attivato in alcune leucemie. Altri membri di questa famiglia come
BAX invece attivano l'apoptosi.
La cellula risponde alle condizione di stress attivando le 3 isoforme del gene GADD45 che da un
lato può determinare la sopravvivenza cellulare dall'altro attivare le vie della morte e della
senescenza cellulare, perché nel caso in cui il GADD45 si lega a PCNA e questo Dna non è
eccessivamente danneggiato allora la cellula preferisce ripararlo, e le 3 isoforme codificate da esso
determineranno dunque la riparazione del Dna. Anche sotto questo aspetto la fase G1 è
fondamentale perché l'allungamento di essa da un po' di tempo alla cellula per riparare i danni al
Dna in modo che esso possa andare incontro alla sintesi e quindi di trasmettere alle cellule figlie il
Dna ricevuto. Quindi l'interazione con le chinasi-ciclina-dipendenti e la p21 determina un arresto
del ciclo cellulare alla fase G1 e permettere dunque la sopravvivenza della cellula e può influire
anche sul blocco del ciclo cellulare anche dopo la fase G0. D'altra parte, con l'attivazione di
determinate vie di trasduzione del segnale di MAP chinasi, questo gene GADD45 può attivare la
p28 e la jun chinasi che sono le vie dello stress cellulare che danno luogo all'apoptosi e alla
senescenza. Tra le altre proteine che inibiscono la proliferazione ce ne sono alcune fondamentali
come la p21 che è indotta dalla p53 e determina il legame con la chinasi-ciclina-dipendente e il
Dna. Ci sono poi p16, p15, p14 che sono proteine che bloccano il ciclo mitotico che vanno ad agire
sul complesso Cdk D 4-6, lo stesso attivo nella fase G1, agiscono quindi tutte allo stesso livello.
Essendo onco-soppressori spesso nelle neoplasie sono deleti.
Tutti questi fattori se son regolati da geni sono coinvolti nella trasformazione neoplastica, per cui io
faccio sempre il riferimento ai tumori per abituarvi a fare i collegamenti.
CARATTERISTICHE PRINCIPALI INIBITORI DELLE CHINASI CICLINE
DIPENDENTI:
p21-> La sua sintesi è indotta dall'anti-oncoproteina p53; agisce sui vari complessi Cdk tramite il
dominio N-terminale, sul PCNA tramite il dominio C-terminale.
P16-> modificato dal gene onco soppressore MTS1 che è deleto, ipermetilato o mutato in numerose
neoplasie umane
p15-> Codificato dal gene onco soppressore MTS2 che è deleto o ipermetilato in alcune neoplasie
umane; la sua sintesi è indotta da TGF-B.
P19-> La sua sequenza amminoacidica è parzialmente omologa (48%) a quella dell'inibitore p16.
P27-> E' degradato per proteolisi da ubiquitinazione quando la cellula deve entrare in fase S; il suo
rapporto stechiometrico con i complessi Cdk è modificato dal TGF-B.
P57-> Si presume che il gene codificatore sia un onco soppressore.
p18-> Debolmente attivo sulla proteina bersaglio Cdk6-D.
Finora abbiamo detto che gli onco-soppressori posso essere mutati o per delezione o per mutazione
puntiforme, però negli ultimi tempi si è messo in luce un meccanismo di alterazione dei geni
soprattutto geni onco-soppressori che non è dovuto ad alterazioni strutturali del gene, cioè delezioni
o sostituzioni di basi ma è dovuto a modificazioni chimiche delle basi dei geni; modificazioni che
hanno aperto un campo, per ora oggetto di molti studi ancora in progresso che però stanno dando
ottimi risultati, che si chiama epigenetica, che quindi si occupa non delle alterazioni strutturali ma
delle modificazioni chimiche delle basi azotate e delle proteine istoniche.
Si è visto che molto geni onco-soppressori non vengono espressi a causa proprio di modificazioni
epigenetiche. Il gene non è alterato quelle che spesso risultano alterate sono le citosine che sono
metilate, non citosine qualunque, ma citosine che fanno parte di alcune regioni del promoter che si
chiamano CPG Islands (P sta per fosforo) sono delle regioni con elevata frequenza seguite da
guanina. Di solito queste citosine metilate sono relative a citosine presenti in queste isole però
qualche volta a livello del gene dell'interleuchina 8, che come studierete nell'infiammazione è una
chemochina (una citochina che attira i neutrofili nella sede dell'infezione), in realtà sono solo 6
CPG nel promoter, la metilazione determina un silenziamento di IL-8. Essa è prodotta in grande
quantità non solo dai neutrofili ma anche in molti tumori perché in molte condizioni neoplastiche
c'è una iper attività a livello infiammatorio, c'è una stretta relazione tra infiammazione cronica e
neoplasie. Quindi la metilazione di queste citosine seguite da guanina può reprimere l'espressione
del gene. Molti geni onco soppressori hanno questa alterazione epigenetica e questo è un evento
molto importante perché è reversibile. Io vi ho detto l'altra volte che nei tumori ci sono delle
alterazioni strutturali dei geni coinvolti nella proliferazione che ovviamente se viene allontanata la
causa, l'agente cancerogeno, rimangono, la cellula rimane una cellula trasformata. Ma se
l'alterazione dei geni onco-soppressori è dovuta ad un'alterazione epigenetica allora si può
intervenire chimicamente e cercare di demetilare queste citosine e fare riesprimere i geni oncosoppressori. Un altra modificazione epigenetica molto importante nello sviluppo dei tumori è per
esempio il grado di acetilazione degli istoni, voi sapete perché ci sia una espressione genica è
necessario che i geni siano de acetilati. Vi son molti agenti utilizzati dal punto di vista terapeutico
che sono inibitori delle de acetilasi, mutati in diversi primers dei tumori, che agiscono a vari livelli
soprattutto a livello dell'istone H3H4. Questa nuova branca dello studio dei tumori, l'epigenetica, si
sta sviluppando perché in numerose neoplasie umane questi geni onco soppressori possono essere
mutati dal punti di vista epigenetico, per esempio ipermetilati. Poi ci sono altri geni coinvolti nella
regolazione degli inibitori delle chinasi-ciclina-dipendenti, tra cui il p19 omologo al p16, p27 che al
momento dell'ingresso della fase S viene degradato per ubiquitinazione ed è sotto il controllo del
TGF-B che è un inibitore della proliferazione, e poi altri geni un po' meno determinanti.
METAPLASIA
Un caso di risposta adattativa della cellula alle condizioni patologiche è costituito dalla metaplasia,
cioè da un cambiamento reversibile in cui un tipo cellulare adulto, già differenziato, è sostituito da
un altro tipo cellulare adulto. In tal modo, cellule più sensibili allo stress sono sostituite con cellule
più capaci di affrontare l'ambiente avverso. Esso è un evento reversibile, che può evolvere nelle
condizioni precedenti se lo stimolo che l'ha attivato cessa. Perché le cellule di un tessuto si
trasformano in cellule di un altro tessuto? Di solito il principio ritenuto comunemente valido è che
le cellule in seguito all'azione di stimoli nocivi cercano di opporre a questi delle cellule più
resistenti, meno sensibili al danno, che sicuramente sono ben differenziate ma che sono meno
specializzate, un po' più rozze, che sappiano affrontare più adeguatamente questi agenti nocivi.
Un esempio è dato dalla metaplasia squamosa del tratto respiratorio in seguito all'azione irritativa
cronica esercitata dal fumo di sigaretta. Vi è un tessuto epiteliale colonnare cilindrico sostituito con
l'epitelio piatto ben differenziato anche ma meno specializzato. Il primo svolge funzioni che il
secondo non svolge: intanto ha le ciglia e secerne muco, capacità fondamentali nel tratto bronchiale.
Però se la nostra mucosa bronchiale è sottoposta ad inquinamento ambientale o a fumo di sigaretta
(contiene idrocarburi policiclici) non è difficile trovare in soggetti esposti ad azione continua a
questi agenti si può determinare la metaplasia squamosa. Sostituzione di cellule funzionali e
protettive (produzione di muco, importante nella difesa contro le infezioni e contro altri agenti) con
cellule funzionalmente meno valide. Se lo stimolo persiste la metaplasia può evolvere in displasia,
proliferazione cellulare eccessiva e a lungo andare possono sfociare in tumori bronchiogeni (spesso
carcinomi in cellule squamose, segno che quella metaplasia è insorta sua una metaplasia squamosa).
Possono iniziare delle risposte cellulari ad altri stimoli a cui le cellule possono essere sottoposte che
possono determinare appunto la displasia.
Le cellule epiteliali colonnari della trachea e dei bronchi sono sostituite da cellule epiteliali
squamose stratificate. Condizioni in cui ci sono stimoli cronici come la presenza di calcoli, processi
infiammatori irritativi che inducono la sostituzione di cellule cilindriche con quelle epiteliali piatte.
Calcoli nei dotti escretori delle ghiandole salivari maggiori e del pancreas, nei dotti biliari possono
indurre la sostituzione dell'epitelio colonnare squamoso.
La deficienza di vitamina A ( la quale interviene nel processo di cheratinizzazione) induce
metaplasia squamosa delle cellule epiteliali, con comparsa iniziale di ipercheratosi generalizzata
non solo follicolare (corneificazione a livello dei follicoli piliferi) a cui segue frinoderma (pelli di
rospo) con perdita di cute sotto forma di grosse scaglie. C'è la cheratinizzazione a livello delle
mucose bronchiolari, quindi brochiolite, a livello delle bacinetto renale con pielonefriti e pieliti, a
livello della congiuntiva e della cornea con xeroftalmia (ispessimento e quindi secchezza a livello
dell'occhio, con conseguente perdita di lucentezza). L'eccesso di vitamina A sopprime la
cheratinizzazione.
Il problema della metaplasia squamosa è che si rimpiazzano le cellule più resistenti con cellule che
non svolgono nessuna funzione utile.
Si può comunque verificare una metaplasia da epitelio squamoso (piatto) a epitelio colonnare
(cilindrico) come nell'esofagite di Barret (non vi è infiammazione) in cui l'epitelio squamoso
esofageo è sostituito da cellule colonnari gastriche. È da tenere sotto controllo perché qualsiasi
forma di metaplasia, che comunque è reversibile, se persiste nel tempo può dare luogo a
trasformazioni tanto che per alcuni testi si tratta di lesione precancerosa che può regredire o
evolvere verso una neoplasia. Con questo termine si indica una lesione a livello della quale più
facilmente, da un punto di vista statistico, più frequentemente, c'è il passaggio a una condizione
neoplastica, cioè rispetto a un tessuto che non la presenta. Tutto questo però non ci dice che cos'è
una lesione precancerosa, per cui, secondo me una definizione più adeguata è quella di una lesione
in cui riscontriamo sia una metaplasia che un aumento dell'attività proliferativa. Si può passare
attraverso tanti passaggi da una condizione di difesa, di adattamento, a una condizione patologica,
neoplastica. E' una lesione che non necessariamente va incontro a neoplasia però a nessun sfugge il
fatto che cellule metaplastiche e cellule in attiva proliferazione possono essere più sensibili
all'azione degli agenti oncogeni, più facilmente di cellule non proliferanti, andranno incontro a
tumori. Cellule metaplastiche possono essere più sensibili ad agenti di natura oncogena.
Per esempio la leucoplachia a livello della mucosa orale è anch'essa una lesione precancerosa
perché abbiamo una prosoplasia, cioè metaplasia in cui il tessuto che sostituisce l'altro ha un più
altro grado di differenziazione (perché c'è cheratinizzazione) e c'è anche aumento della
proliferazione. Durante la prima lezione abbiamo parlato dello xeroderma pigmentosum che è anche
una lesione precancerosa perché abbiamo una metaplasia in quanto anche qui abbiamo una
cheratinizzazione eccessiva e aumenta anche la melanine perché aumenta il numero di melanociti
(per questo pigmentosum).
Per quanto riguarda il carcinoma cervicale in situ (il carcinoma non ha invaso le strutture
circostanti, la membrana basale) ci sono due diverse tendenze se considerarla tumore o lesione
precancerosa perché è una caratteristica dei tumori maligni è l'invasività ma deriva sempre dalla
vera differenza tra tumori maligni e tumori benigni che è quella dell'alterazione dei geni che
regolano la proliferazione e la differenziazione in quanto l'invasività è conseguenza dei geni che
codificano la differenziazione perché sono alterate le strutture di membrana. Il tumore è la
conseguenza di mutazioni strutturali o epigenetiche che coinvolgo geni per la differenziazione e la
proliferazione; nel carcinoma cervicale in situ noi abbiamo un aumento della proliferazione non per
alterazione genica per risposta a determinati stimoli è un processo di metaplasia. Esso può essere
un momento dello sviluppo del tumore, può essere il tumore che è radicato senza compromettere la
salute dell'individuo perché non ha invaso, non ha dato luogo a metastasi quindi l'eliminazione del
tumore può essere di valido aiuto, ma non perché non si tratti di tumore, ma perchè il tumore è in
uno stadio precedente; il tumore una volta instaurato passa per il processo di progressione
neoplastica. Per cui, anche se molti libri di testo ne parlano come lesione precancerosa, per me si
tratta di tumore; per me lesione precancerosa è laddove non c'è alterazione genetica o epigenetica
ma c'è metaplasia con aumento della proliferazione.
Anche le cellule del tessuto connettivo fibroso possono trasformarsi in osteoblasti (metaplasia
ossea) e condroblasti (metaplasia cartilaginea) soprattutto a livello dei foci alterati in seguit a
trauma.
La metaplasia potrebbe essere il risultato di una riprogrammazione genetica di:
a) cellule staminali, per esempio in un tessuto per cui esse sono programmate a evolversi verso un
tipo di cellula anziché verso un altro;
b) a livello di cellule mesenchimali già differenziate:
Ci sono altre sostanze che possono indurre l'insorgere di una metaplasia oltre al fumo di sigaretta
quali sostanze chimiche, vitamine, fattori di crescita; derivati dall'acido retinoico e citochine che
regolano crescita e differenziazione.
Fattori morfogenetici dell'osso appartenenti alla famiglia TGF-B determinano differenziazione
ossea. Alcuni farmaci citostatici, molto utilizzati nella terapia dei tumori, possono alterare la
differenziazione perché alterano i quadri di metilazione delle citosine. La differenziazione è dovuta
all'espressione di alcuni geni e al silenziamento di altri che è diversa nelle varie cellule, se sono
presenti fattori come questi farmaci citostatici che alterano i quadri di metilazione del DNA
ovviamente modifica il quadro di espressione genica, cioè alcuni geni vengono repressi e altri no.
Tutti fattori che modificano l'entità di espressione dei geni, modificano anche il grado di
differenziazione, il tipo di differenziazione. Per cui anche questi farmaci possono intervenire per
determinare la metaplasia, come altri agenti metilanti che sono in grado di modificare l'espressione
degli onco-soppressori e quindi indurre i tumori
Antonio Ferlito