Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 193-245
Audiologia (a cura di E. Marciano)
Lo screening neonatale dei disturbi permanenti dell’udito
L’impianto cocleare nel bambino
Forme ereditarie di ipoacusie neurosensoriali isolate e sindromiche nel bambino
Volume 39
156
Ottobre-Dicembre 2009
Neurologia (a cura di E. Mercuri)
Consenso sugli “Standard di cura”: un grosso passo avanti nel campo delle malattie
neuromuscolari
Nuovi standard di cura per le complicanze respiratorie e cardiologiche nel bambino
con distrofia muscolare di Duchenne
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
Frontiere (a cura di A. Cao, L.D. Notarangelo, A. Iolascon)
Le basi genetiche delle SCID
FOCUS SU: (a cura di G. Bona)
Obesità e sindrome metabolica in età pediatrica
Pacini
Editore
Medicina
Periodico trimestrale POSTE ITALIANE SPA - Spedizione in Abbonamento Postale - D.L. 353/2003 conv.in L.27/02/2004 n°46 art.1, comma 1, DCB PISA
Aut. Trib. di Milano n. 130 del 17/03/1971 - Stampa a tariffa ridotta - tassa pagata - Aut. Dirpostel Pisa n. 1/36131/4/1 del 10/09/1993 - Taxe perçue - Italia
Direzione
Generoso Andria, Napoli
Gianni Bona, Novara
Antonio Cao, Cagliari
Liviana Da Dalt, Padova
Alberto Martini, Genova
Pierpaolo Mastroiacovo, Roma
Luigi Daniele Notarangelo, Boston
Fabio Sereni, Milano
Luigi Titomanlio, Napoli
Alberto Villani, Roma
Redazione e Amministrazione
Pacini Editore S.p.A.
Via Gherardesca, 1
56121 Pisa
Tel. 050 313011 - Fax 050 3130300
[email protected]
Redattore Capo
Marina Macchiaiolo, Roma
Invio gratuito per i Soci SIP.
Comitato di Redazione
Salvatore Auricchio, Napoli
Stelvio Becchetti, Genova
Sergio Bernasconi, Parma
Andrea Biondi, Monza
Alessandro Calisti, Roma
Mauro Calvani, Roma
Antonio Correra, Napoli
Maurizio de Martino, Firenze
Pasquale Di Pietro, Genova
Alberto Edefonti, Milano
Renzo Galanello, Cagliari
Carlo Gelmetti, Milano
Achille Iolascon, Napoli
Riccardo Longhi, Como
Giuseppe Maggiore, Pisa
Paola Marchisio, Milano
Bruno Marino, Roma
Eugenio Mercuri, Roma
Paolo Paolucci, Modena
Luca Ramenghi, Milano
Franca Rusconi, Firenze
Volume 39
156
Ottobre-Dicembre 2009
Stampa
Industrie Grafiche Pacini, Pisa
Abbonamenti
Prospettive in Pediatria è una rivista trimestrale. I prezzi dell’abbonamento annuo sono i seguenti:
Italia € 54,00; estero € 68,00; istituzionale € 53,00; specializzandi € 30,00; fascicolo singolo € 28,00
Le richieste di abbonamento vanno indirizzate a: Prospettive
in Pediatria, Pacini Editore S.p.A., Via Gherardesca 1, 56121
Pisa – Tel. 050 313011 – Fax 050 3130300 – E-mail: [email protected]
I dati relativi agli abbonati sono trattati nel rispetto delle disposizioni contenute nel D.Lgs. del 30 giugno 2003 n. 196
a mezzo di elaboratori elettronici ad opera di soggetti appositamente incaricati. I dati sono utilizzati dall’editore per la
spedizione della presente pubblicazione. Ai sensi dell’articolo 7 del D.Lgs. 196/2003, in qualsiasi momento è possibile
consultare, modificare o cancellare i dati o opporsi al loro
utilizzo scrivendo al Titolare del Trattamento: Pacini Editore
S.p.A., Via Gherardesca 1, 56121 Pisa.
Le fotocopie per uso personale del lettore possono essere
effettuate nei limiti del 15% di ciascun fascicolo di periodico
dietro pagamento alla SIAE del compenso previsto dall’art.
68, commi 4 e 5, della legge 22 aprile 1941 n. 633.
Le riproduzioni effettuate per finalità di carattere professionale, economico o commerciale o comunque per uso diverso
da quello personale possono essere effettuate a seguito di
specifica autorizzazione rilasciata da AIDRO, Corso di Porta
Romana n. 108, Milano 20122, E-mail: [email protected]
e sito web: www.aidro.org.
© Copyright by Pacini Editore S.p.A.
Direttore Responsabile: Patrizia Alma Pacini
Finito di stampare nel mese di aprile 2010 presso le Industrie
Grafiche della Pacini Editore S.p.A., Pisa.
Pacini
Editore
Medicina
INDICE numero 156 Ottobre-Dicembre 2009
Audiologia (a cura di Elio Marciano)
Lo screening neonatale dei disturbi permanenti dell’udito
Alfredo Pisacane, Edoardo Arslan, Gennaro Auletta, Françoise Barrier, Luigi Barruffo, Grazia Isabella Continisio, Paola Continisio,
Monica Errichiello, Rita Malesci, Pasquale Riccardi, Fabiana Toscano, Elio Marciano...............................................................................193
L’impianto cocleare nel bambino
Gennaro Auletta, Ettore Cassandro, Elisabetta Genovese, Maria Consolazione Guarnaccia, Pasquale Iadicicco, Carla Laria,
Emma Landolfi, Giorgio Lilli, Pasquale Riccardi, Elio Marciano .................................................................................................................200
Forme ereditarie di ipoacusie neurosensoriali isolate e sindromiche nel bambino
Annamaria Franzè, Viviana Chinetti, Paolo Gasparini, Pasquale Giannini, Sandra Iossa, Carla Laria, Giorgio Lilli, Rita Malesci,
Alessandro Martini, Elio Marciano..............................................................................................................................................................205
Neurologia (a cura di Eugenio Mercuri)
Consenso sugli “Standard di cura”: un grosso passo avanti nel campo delle malattie neuromuscolari
Eugenio Mercuri, Flaviana Bianco, Gessica Vasco . ...................................................................................................................................210
Nuovi standard di cura per le complicanze respiratorie e cardiologiche nel bambino con distrofia muscolare di Duchenne
Angela Berardinelli, Marika Pane, Eugenio Mercuri....................................................................................................................................214
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
Adele D’Amico, Enrico Bertini ....................................................................................................................................................................219
Frontiere (a cura di Antonio Cao, Luigi D. Notarangelo, Achille Iolascon)
Le basi genetiche delle SCID
Fausto Cossu............................................................................................................................................................................................. 228
FOCUS SU: (a cura di Gianni Bona)
Obesità e sindrome metabolica in età pediatrica
Gianni Bona, Arianna Busti, Flavia Prodam, Simonetta Bellone.................................................................................................................. 239
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 193-199
Audiologia
Lo screening neonatale dei disturbi permanenti
dell’udito
Alfredo Pisacane, Edoardo Arslan*, Gennaro Auletta**, Françoise Barrier**, Luigi Barruffo***,
Grazia Isabella Continisio, Paola Continisio, Monica Errichiello**, Rita Malesci**,
Pasquale Riccardi**, Fabiana Toscano**, Elio Marciano**
Dipartimento di Pediatria, Università di Napoli Federico II, * Università di Padova; ** Unità di Audiologia, Dipartimento
di Neuroscienze, Università di Napoli Federico II; *** ASL Napoli 1 Centro
Riassunto
I disturbi permanenti bilaterali dell’udito in età pediatrica, definiti da una perdita uditiva ≥ 40 dB di soglia (HTL), sono relativamente frequenti e riguardano
circa 1,2 bambini ogni 1000 nati nei paesi industrializzati. Dato che essi possono avere effetti devastanti sulle competenze di comunicazione e sulla vita
scolastica e sociale, una diagnosi ed un intervento riabilitativo precoce, principalmente se entro il sesto mese di vita, sono misure rilevanti di salute pubblica, in quanto permettono competenze linguistiche e cognitive notevolmente migliori, in confronto a quelle ottenute da bambini nei quali la diagnosi è stata
fatta tardivamente. Per questo motivo i sistemi sanitari di molti paesi hanno avviato programmi di screening universale di tali disturbi.
Summary
Permanent childhood hearing impairment (PCHI) of ≥ 40 decibels relative to hearing threshold level (dB HTL) is relatively common, affecting 1.2 infants per
1000 live births in industrialised countries and can have a devastating impact on communication skills, education attainment, and quality of life. As early
identification and management of hearing impaired infants can improve language outcomes and no relevant harms are associated with programmes of
universal newborn hearing screening (UNHS), such programmes have been introduced in many countries.
Introduzione
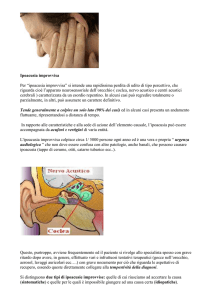
I disturbi permanenti dell’udito rappresentano una delle più frequenti disabilità in età evolutiva: la loro incidenza è infatti di circa 1,2-1,5
per mille nati in epoca neonatale, per diventare poi di circa il 2 per
mille in età scolare, in quanto una quota di ipoacusie si manifestano tardivamente o si acquisiscono nei primi 5 anni di vita (1, 2, 3).
Tra popolazioni maggiormente a rischio, come ad esempio i bambini
ricoverati nelle terapie intensive neonatali (TIN) o quelli con una familiarità positiva per ipoacusie infantili, la prevalenza di ipoacusia
può essere anche di 10 volte superiore.
Tenuto conto del numero di bambini che ogni anno nascono in Italia
(poco più di 500.000), il numero di soggetti tra 0 e 14 anni, affetti da
un’ipoacusia bilaterale clinicamente rilevante, dovrebbe aggirarsi tra
10.000 e 15.000. Fare in modo che la loro disabilità non si trasformi
in handicap è uno degli obiettivi delle iniziative di screening neonatale che, in anni recenti, sono divenute sempre più frequenti in tutti
i paesi industrializzati.
Eziologia delle ipoacusie
Le ipoacusie possono essere classificate, in base alla sede della lesione uditiva, in 3 tipi: ipoacusia trasmissiva, neurosensoriale, mista.
Accanto a queste vi sono anche cause eziologiche che possono provocare una lesione del sistema nervoso centrale, che può associarsi
o meno ad una perdita uditiva periferica (Tab. I).
Le ipoacusie trasmissive sono determinate da un problema dell’orecchio esterno o medio, mentre la perdita uditiva neurosensoriale è causata da patologie dell’orecchio interno o dell’ottavo nervo
cranico. Le ipoacusie centrali sono rare e spesso causate da problemi lungo la via uditiva centrale.
Le ipoacusie possono essere ereditarie o possono essere acquisite
in epoca prenatale, perinatale e dopo la nascita. Le lesioni specifiche
delle vie uditive centrali sono rare e di norma non si manifestano con
una perdita uditiva, ma come un disturbo di processing uditivo che si
evidenzia non prima dei 2-3 anni di vita.
Le ipoacusie ereditarie rappresentano circa il 60% del totale nei paesi sviluppati. In circa 1/3 dei casi, si tratta di condizioni sindromiche
(es. sindrome di Alport, di Usher, di Stickler ecc.), mentre gli altri 2/3
sono rappresentati da forme non sindromiche, più frequentemente
recessive, ma in alcuni casi, dominanti, X-linked o mitocondriali.
Tra le altre sordità, quelle associate al complesso TORCH (in particolare al CMV) sono le più frequenti; sono presenti in epoca
neonatale e in alcuni casi manifestano un andamento evolutivo,
con ipoacusia, mono o bilaterale, fluttuante e di gravità variabile,
fatto questo che consiglia un follow-up audiologico fino all’inizio
dell’età scolare nei pazienti dove è stata documentata l’infezione
(Grosse et al., 2008).
Negli ultimi 20 anni è andato sempre più aumentando il numero
dei bambini che sopravvivono, anche se gravemente prematuri o
con stati di sofferenza neonatale grave, grazie al sempre più efficace trattamento al quale vengono sottoposti nelle Terapia Neonatali
Intensive (TIN). Le sofferenze neonatali hanno perciò acquisito un
peso relativo sempre maggiore è sono oggi stimate attorno al 10%
di tutte le ipoacusie infantili. (Nafstad et al., 2002).
Infine tra le cause post-natali di sordità, la causa più frequente è
rappresentata dalla meningoencefalite.
Le ipoacusie ad insorgenza progressiva e tardiva
Le ipoacusie a sviluppo tardivo o con decorso progressivo possono
essere classificate in tre principali categorie:
• Acquisite: CMV sintomatico o non, sepsi neonatale, meningiti
batteriche o virali, agenti chemioterapici, trauma cranico.
193
A. Pisacane et al.
Tabella I.
Cause di ipoacusia congenita.
Ipoacusia trasmissiva
Ipoacusia neurosensoriale
Lesione del SNC
Malformazioni del condotto uditivo esterno, della membrana timpanica, della catena ossiculare, colesteatoma congenito
Genetiche, sindromi (es. di Waardensburg, di Usher, di Alport, di Turner), infezioni prenatali TORCH (CMV,
Sifilide, Herpes virus, rosolia, toxoplasmosi, sepsi streptococcica)
Iperbilirubinemia, ipossia, emorragia intraventricolare
• Strutturali: deformità strutturali della coclea congenite, ma non
inquadrate in una specifica sindrome; LVA (large vestibular
aqueduct), malformazione di Mondini.
• Genetiche: possono suddividersi in sindromiche e non sindromiche.
Le forme sindromiche comprendono la sindromi di Pendred, la brachio-oto-renale (BOR), la Alport, la Stickler, la Usher, la neurofibromatosi di tipo II, la Refsum.
Le forme non sindromiche comprendono, invece, le ipoacusie a carattere autosomico dominante a decorso progressivo ed alcune forme
autosomiche recessive da connessina 26 a decorso progressivo.
I seguenti fattori sono oggi considerati come possibili indicatori
di ipoacusie ad insorgenza tardiva: anamnesi di storia familiare di
ipoacusia infantile; ricovero in TIN per più di 5 giorni, infezioni uterine accertate, malformazioni cranio-facciali, malattie neurodegenerative, traumi cranici, peso alla nascita inferiore a 1500 g, con Apgar
minore di 3.
Il perché di uno screening neonatale universale
Scopo dello screening neonatale è l’identificazione precoce di una
patologia che nel neonato non dà segni o sintomi clinicamente rilevabili, dove per patologia si intende una perdita uditiva bilaterale,
presente alla nascita, con una soglia uditiva ≥ 40 dB HTL tra 0,5
e 4 kHz. I motivi che consigliano di eseguire un test di screening
a tutti i neonati, piuttosto che solo a quelli che hanno fattori di rischio per ipoacusia, sono rappresentati dal fatto che solo la metà
dei disturbi permanenti dell’udito si verifica in bambini con fattori
di rischio, mentre l’altra metà interessa bambini senza tali fattori.
Fino al momento in cui il test di screening neonatale si è diffuso ed è
stato adottato in praticamente tutti i paesi industrializzati, una prima
valutazione dell’udito veniva eseguita intorno agli 8 mesi con vari
test di distrazione (ad esempio il Boel test). Il Boel test è però poco
sensibile e poco specifico ed identifica solo poco più della metà dei
bambini con sordità. Questo spiega perché, fino a pochi anni fa, l’età
media alla quale un’ipoacusia neurosensoriale veniva diagnosticata
era intorno ai 2 anni (Kennedy, 2005). Tale ritardo nella diagnosi si
associava spesso con una scarsa efficacia della terapia riabilitativa.
I dati della letteratura mostrano che lo screening universale si mostra capace di abbassare significativamente l’epoca della diagnosi e
quindi l’inizio dell’intervento riabilitativo. L’età media della diagnosi,
nei contesti in cui viene eseguito lo screening neonatale, è di circa
6 mesi, mentre, laddove lo screening è condotto esclusivamente tra
i bambini a rischio, la diagnosi, tra i bambini senza fattori di rischio,
viene fatta tra i 15 ed i 25 mesi di età (Nelson, 2008).
I vantaggi di una diagnosi e di un intervento precoce
Anche se non sono disponibili al momento evidenze definitive e supportate da studi di grandi dimensioni sui vantaggi di un intervento
precoce, pur tuttavia esistono buoni dati, principalmente dagli Stati
Uniti e dal Regno Unito, che mostrano che benefici rilevanti sulle
194
performance comunicative, linguistiche, relazionali e cognitive derivano da una stimolazione acustica e da un intervento riabilitativo
instaurati entro il sesto mese di vita. Ciò è principalmente valido per
bambini ipoacusici senza altre patologie associate (Downs, 1999;
Moeller, 2000; Kennedy, 2006).
Sicurezza, fattibilità e rapporto benefici/costi dello
screening
I test attualmente impiegati nello screening uditivo neonatale sono
privi di qualsiasi rischio, rapidi, di facile esecuzione e molto sensibili e specifici. Con un opportuno addestramento degli operatori
dei punti nascita, sensibilità e specificità di un test di screening a 2
tappe (vedi i paragrafi seguenti) raggiunge il 98%. Ciò significa che,
su 100 bambini sani sottoposti al test di screening, ve ne saranno
circa 2 per i quali il test dà una risposta non esatta (falsi-positivi); i
falsi-negativi, nelle statistiche internazionali più accurate, sono invece circa 1 su 10.000 (Kennedy, 2004). Data la bassa incidenza
della malattia, è naturale attendersi che il valore predittivo del test
positivo sia basso (circa 5%); ciò significa che, tra i neonati che non
superano il test di screening e che vanno quindi sottoposti ad un
test di conferma della diagnosi, circa 1 su 20 sarà ipoacusico. Da
un punto di vista di efficienza, questo risultato è rilevante; passiamo
infatti da una probabilità pre-test di malattia di 1,2-1,5 per 1000 ad
una probabilità post-test del 5%, quindi circa 50 volte superiore.
Tenuto conto che i costi economici dello screening, peraltro non facilmente valutabili (Schroeder, 2006), sono comunque relativamente limitati ed in ogni caso inferiori a quelli per le cure di individui con grave
disabilità uditiva e che non vi sono effetti negativi associati al numero di
falsi positivi, la conclusione della maggioranza degli esperti è che i vantaggi di un test di screening universale siano rilevanti (Nelson, 2001). Il
punto critico invece è piuttosto l’organizzazione dello screening ed esso
verrà discusso nella sezione finale di questo articolo.
I pochi studi disponibili mostrano che gli effetti negativi sono nettamente inferiori in confronto ai potenziali vantaggi. Uno dei problemi
maggiori è lo stato di ansia dei genitori, in particolare nei casi falsi
positivi, dovuto alla incertezza e al timore, fino a quando la diagnosi
non viene confermata o esclusa (cosa che avviene tra il secondo ed
il terzo mese di vita). La situazione può essere contenuta da azioni di
counselling, da una comunicazione adeguata e da una presa in carico empatica fino al momento della diagnosi e nelle fasi successive,
nel caso il bambino risultasse realmente ipoacusico (Nelson, 2008).
ll problema delle forme ad insorgenza tardiva
o progressiva
Uno dei problemi più seri dello screening uditivo neonatale è costituito dai bambini che sviluppano forme di ipoacusia progressiva
e/o ad insorgenza tardiva. Non sono disponibili dati Italiani sull’incidenza di queste forme di ipoacusia, ma stime eseguite in altri paesi
suggeriscono che la percentuale di ipoacusie che si sviluppano dopo
Lo screening neonatale dei disturbi permanenti dell’udito
il periodo neonatale, e che non possono pertanto essere identificate
con il test di screening, possa rappresentare fino al 25% del totale
(Weichbold, 2006; Hutt, 2008). È quindi di fondamentale importanza il monitoraggio della funzione uditiva, per lo meno fino all’inizio
dell’età scolare e principalmente tra quei soggetti a rischio per l’insorgenza di forme tardive o progressive (familiarità per ipoacusia,
prematurità e ricovero in TIN, meningoencefaliti nei primi anni di
vita, ecc). In quest’ottica, i pediatri di famiglia costituiscono un anello
fondamentale di tale monitoraggio.
Test audiologici utilizzati per lo screening
Le procedure di screening sono rappresentate dalle emissioni otoacustiche (TEOAE e DPOAE) e dai potenziali evocati uditivi del tronco
(ABR), che vengono oggi effettuati con l’impiego di apparecchiature
completamente automatiche utilizzabili da personale non specializzato. Le otoemissioni si basano su un fenomeno fisiologico scoperto
da Kemp negli anni ’80. In un orecchio normale si verifica una eccitazione delle cellule neurosensoriali dell’organo del Corti: le cellule
ciliate esterne (OHC) e le cellule ciliate interne (IHC). Le OHC hanno
un ruolo di amplificatore meccanico, come dei piccoli muscoli che
si contraggono ed emettono un suono molto debole di ritorno che
può essere registrato con un microfono nel condotto uditivo esterno. Su tale base è stato elaborato l’impiego clinico delle OAE negli
screening uditivi. Nel condotto uditivo esterno si inserisce una sonda
attraverso la quale si invia un suono che giunge alla coclea; la stessa sonda è in grado di registrare il segnale di ritorno emesso dalla
contrazione delle cellule ciliate esterne cocleari. La mancanza di tale
segnale implica un’anomalia della funzione dell’organo del Corti e
quindi la possibile presenza di una ipoacusia neurosensoriale. La
presenza invece delle OAE dimostra una coclea funzionante e quindi
una normale capacità uditiva. La rapidità di esecuzione, l’assenza di
fastidio e l’affidabilità rendono questo test uno strumento valido per
lo screening delle ipoacusie in età neonatale. Nel caso in cui le emissioni otoacustiche siano presenti è possibile affermare che la coclea
funziona correttamente e, in genere, non è necessario eseguire altri esami, (escluse alcune situazioni particolari, ad es. la neuropatia
uditiva) o disfunzioni delle vie uditive dal tronco.
Viceversa se le OAE risultassero assenti (non dimenticando però che
vi è una significativa percentuale di neonati normali con OAE assenti,
i casi “falsi positivi” dovuti a difficoltà di registrazione, a caratteristiche anatomiche particolari del condotto uditivo esterno del neonato,
a presenza di meconio ecc.), si può ipotizzare una ipoacusia neurosensoriale e il neonato va avviato alla conferma della diagnosi, che
si esegue mediante la registrazione dei potenziali uditivi del tronco.
L’esecuzione delle TEAOE richiede pochi minuti e si esegue preferibilmente durante il sonno del neonato. L’ addestramento del personale medico o infermieristico che eseguirà il test non è particolarmente complesso. È da tener comunque presente che, anche se
con frequenza molto limitata, esistono anche casi “falsi negativi”,
in cui le otoemissioni sono presenti, ma è presente una ipoacusia
neurosensoriale; in questo caso ci troviamo quasi sempre di fronte
ad una neuropatia uditiva.
Gli ABR automatici (AABR) sono invece una indagine diagnostica di
recente introdotta nei programmi di screening audiologico neonatale. Si tratta della registrazione della risposta del tronco ad uno
stimolo uditivo con una apparecchiatura automatica che valuta la
presenza/assenza della V onda ABR per stimoli pari a 35-45 dB nHL.
L’identificazione dell’ onda V viene effettuata tramite una serie di
procedure statistiche e l’esito dell’ esame, così come per le otoemissioni, è di tipo pass/fail (cioè ha superato/fallito il test).
L’introduzione degli AABR, come test aggiuntivo alle OAE; ha permesso di ridurre il numero dei falsi positivi che dovranno esser sottoposti alle indagini audiologiche di approfondimento e soprattutto
di individuare parte dei rari casi di falsi negativi da neuropatia uditiva. (www.audiologia.unina.it/screening).
Lo status quo dei progetti di screening nei paesi
industrializzati
La storia dello screening neonatale delle ipoacusie è relativamente
giovane; partito come progetto-pilota nel Regno Unito alla fine degli
anni ’90, in meno di 10 anni lo screening si è esteso praticamente
a tutti i paesi industrializzati, con esperienze di grande interesse
condotte, oltre che nel Regno Unito, negli USA, in Australia e in Europa, inclusi alcuni paesi dell’Est Europeo (Kennedy, 2004; Grandori, 1999)). In Italia vi sono alcune Regioni, quali la Liguria (Calevo,
2007), la Campania (www.audiologia.unina.it), il Veneto (Suppiej,
2007) e l’Umbria, nelle quali lo screening è oramai una routine in
tutti i punti-nascita; in altre regioni è in corso di adozione, anche
perché è stato recentemente inserito tra le pratiche previste nei Livelli Essenziali di Assistenza.
Le criticità del processo dello screening
Teoricamente, lo screening delle ipoacusie ha un elevato rapporto
beneficio/rischi e costi/benefici. In realtà, le cose sono molto più
complesse ed il processo dello screening richiede un investimento
che non è solo economico, ma è di formazione interdisciplinare e di
cooperazione multisettoriale (McCracken, 2005).
Organizzazione e logistica complesse
Il modello organizzativo dello screening può prevedere una strutturazione in 3 livelli. Il primo livello è rappresentato dai punti nascita
e dai reparti di terapia intensiva neonatale; il secondo livello è costituito da strutture aziendali deputate alla conferma della diagnosi
(reparti di Audiologia e Foniatria, Otorinolaringoiatria), il terzo livello
è rappresentato dal Centro di Riferimento Regionale con compiti di
approfondimento ed indirizzo terapeutico-riabilitativo.
L’organizzazione dello screening è di una complessità notevole. La
nostra personale esperienza in Regione Campania è forse particolarmente complessa, per la esistenza di 78 punti nascita (di cui la metà
privati) nei quali si verificano circa 60.000 nascite ogni anno, 16 reparti di TIN ed una decina di unità operative audiologiche, dislocate a
livello provinciale e deputate ad una prima conferma della diagnosi.
Ma anche Regioni con un minor numero di nati e di strutture sanitarie possono incontrare difficoltà. Ne esaminiamo alcune.
Anche se di esecuzione rapida (2 o 3 minuti in un neonato che dorme o che succhia al seno), il test con le otoemissioni rappresenta
comunque un carico di lavoro ulteriore per il personale infermieristico. Per i bambini che non passano il test prima della dimissione
dal nido (che sono circa il 5% dei nati), va ripetuto un secondo test
al nido entro la terza settimana di vita, che rappresenta un ulteriore
carico di lavoro, ma che porta ad una riduzione della percentuale di
neonati che non passano il test da 5% a circa 2% (Kennedy, 2000).
I neonati che non passano il test vengono inviati presso una struttura audiologica di conferma della diagnosi con una prenotazione
telefonica immediata. Spiegare ai genitori il significato corretto del
risultato “fail” al test va fatto con calma ed in modo appropriato: va
fatta capire la necessità di eseguire il test di conferma, ma al tempo
195
A. Pisacane et al.
stesso va spiegato che solo 1 bambino su 20, tra quelli che non
passano il test, risulterà ipoacusico. Questo comporta una formazione ed una motivazione dei professionisti che lavorano nei puntinascita. La situazione nei reparti di TIN è più complessa, in quanto
il test con le otoemissioni non è sufficiente ed andrebbe integrato
con un test di ABR da screening (A-ABR). Poche TIN dispongono di
tale apparecchiatura al momento attuale: è quindi pratica comune
eseguire le sole otoemissioni ed inviare comunque ad un secondo
livello diagnostico non solo i neonati che non passano il test con le
otoemissioni, ma anche quelli che hanno i fattori di rischio associati
con un ricovero in TIN (peso molto basso, ipossia, ventilazione meccanica, sepsi, uso di farmaci ototossici, emorragie cerebrali, iperbilirubinemia significativa, ecc).
Un ulteriore punto di complessità gestionale è la manutenzione delle
apparecchiature e dei ricambi e l’invio periodico dei risultati dei test
effettuati presso un Centro di Riferimento regionale o provinciale.
Le strutture aziendali di conferma della diagnosi, dislocate a livello
provinciale, si sono trovate, in un arco di tempo relativamente breve, ad eseguire il test ABR ad un considerevole numero di bambini
piccoli. Ciò ha comportato (e comporterà, via via che lo screening
si diffonderà sul territorio nazionale) un riassetto organizzativo, la
necessità di addestramento del personale, un carico di lavoro per recuperare le famiglie che non si presentano all’appuntamento fissato
dai punti-nascita, uno scambio continuo di informazioni con il Centro
di Riferimento regionale o provinciale (Chia-ling, 2008).
Senza una committenza politica chiara e risorse dedicate, questo
processo potrebbe venire a cadere all’improvviso (dove esiste) o non
decollare mai (dove non esiste ancora).
Circa il 50% dei bambini ipoacusici sono bambini che, per le loro
caratteristiche (prematurità, al ricovero in TIN, presenza di sindromi complesse), richiedono approfondimenti diagnostici o interventi
terapeutici rappresentano una sfida organizzativa anche per i centri
più avanzati (per citare i più critici: strutture e personale per l’elettrococleografia, una batteria di test per valutare la funzionalità delle
protesi fino ai 18 mesi di vita, personale con competenze per il follow-up degli impianti cocleari).
Un sistema informativo da mettere a regime
Appare chiaro come una complessità di questo genere richieda un
sistema informativo dedicato, capace di archiviare i dati, di renderli
disponibili in rete alle varie strutture sanitarie coinvolte in modo da
far interagire tutte le persone coinvolte nello screening sia nelle attività più semplici, quali quelle di trasmissione delle informazioni e
di prenotazioni, quanto in quelle più complesse, quali quelle delle
discussione a distanza di indagini diagnostiche, di consulenze, ecc.
Anche per questo sono necessarie idee chiare, motivazione e risorse
finanziarie (Hinman, 2005).
gratuita solo le protesi analogiche e non quelle digitali, che sono
invece indispensabili per bambini piccoli, gli audioprotesisti non
appartengono al Servizio Sanitario Nazionale, ma sono dipendenti
di aziende private, i centri di riabilitazione deputati alle attività logopediche (in oltre la metà del nostro Paese) sono strutture private
accreditate, non esistono criteri nazionali per l’accreditamento delle
strutture deputate agli impianti cocleari, l’inserimento scolastico dei
bambini ipoacusici è spesso un problema, con notevole variabilità
nella reale presenza e soprattutto competenza degli insegnanti di
sostegno). Una famiglia, specialmente se il problema della sordità
non rappresenta l’unica patologia del bambino (Edwards, 2007), può
sperimentare notevoli difficoltà, se non esiste una centrale unica di
coordinamento delle attività. Gli inglesi definiscono questa rete dei
servizi a sostegno dei bambini ipoacusici friendly hearing services,
ma in Italia, purtroppo, non se ne parla ancora e ciascuno tenta di
creare percorsi adeguati alle realtà locali o alle singole famiglie. Se
lo screening, come si auspica, diventerà presto una delle prestazioni
prevista nei LEA, il Ministero della Salute e le Regioni dovranno affrontare rapidamente questo problema.
Il monitoraggio e la valutazione
La valutazione dell’efficacia dello screening va effettuata costantemente con un sistema informativo che permetta di ottenere indicatori specifici. Da quelli semplici (percentuali di copertura del test con
OAE, di refer, di neonati persi al follow-up, di bambini veri positivi,
età di prescrizione di protesi, ecc), a quelli più complessi, capaci di
fornire informazioni relative alle tipologie di approccio riabilitativo,
all’acquisizione delle competenze di linguaggio e di comunicazione, all’inserimento scolastico, alla partecipazione alla vita sociale
(Davis, 2001). Alleati importanti in questo percorso devono essere i
pediatri di famiglia, che vanno coinvolti fin dal momento del sospetto
diagnostico, per partecipare al momento della conferma della diagnosi e delle decisioni relative all’iter riabilitativo.
Anche qui la situazione è più complessa di quanto si possa trattare in un articolo sintetico. In grande sintesi, va valorizzato il ruolo
dei pediatri di famiglia nel monitoraggio dei bambini con fattori di
rischio di ipoacusia progressiva o tardiva, anche rivedendo l’utilizzazione di procedure attualmente utilizzate (ad esempio il Boel test
non è sufficientemente valido per ottenere informazioni oggettive
su una possibile perdita uditiva nel bambino nel secondo semestre
di vita). I pediatri di famiglia rappresentano inoltre una componente
essenziali della futura rete dei friendly hearing services e possono
giocare un ruolo fondamentale nel consigliare le famiglie verso i
percorsi diagnostici, protesici e riabilitativi più adeguati per il singolo bambino.
L’intervento precoce ed il percorso riabilitativo
Lo screening uditivo neonatale, secondo le procedure e i protocolli
La necessità dell’integrazione dei servizi sanitari, descritti precedentemente, costituisce il primo passo del processo
sociali e scolastici: quale possibilità in Italia di che deve portare alla diagnosi e alla successiva correzione protesica
del bambino, interventi che devono essere svolti in un arco di tempo
friendly hearing services?
L’esperienza del Regno Unito ha mostrato chiaramente che lo screening non ha funzionato bene fino a quando i servizi sociali, sanitari e
scolastici non hanno realizzato la necessità di una forte integrazione nella presa in carico dei bambini ipoacusici e delle loro famiglie
(Kennedy, 2004; McCracken, 2005). Il percorso riabilitativo è lungo e
spesso complesso ed i costi per le famiglie possono essere notevoli
(in Italia l’attuale Nomenclatore Tariffario concede in via totalmente
196
sufficientemente breve per garantire che il bambino sia in grado di
sviluppare in modo fisiologico la percezione uditiva (Holt, 2008).
Il sistema uditivo centrale ha un periodo limitato di plasticità, quindi
la potenzialità e l’efficienza nella organizzazione delle reti neurali,
che stanno alla base dei processi di percezione del linguaggio, vengono attivate e catalizzate dall’esperienza uditiva del bambino nei
primi anni di vita.
È ormai opinione comune che le vie uditive siano geneticamente
Lo screening neonatale dei disturbi permanenti dell’udito
predeterminate ed organizzate fin dalla nascita (Fallon et al., 2007),
almeno nel loro caratteristica principale e cioè nella identificazione
di popolazioni neurali univoche che collegano le diverse zone della
coclea fino alla corteccia uditiva (tonotopicità). La capacità invece di
creare i circuiti neurali dedicati alla identificazione dei tratti acustici
del linguaggio e successivamente alla percezione semantica della
lingua nativa si localizza nei primi due anni di vita nel periodo di
massima plasticità del sistema uditivo centrale e dipende criticamente da:
• da un normale funzionamento del recettore uditivo;
• da un ingresso acustico linguisticamente ricco che avvenga in
modo continuativo ed efficiente nel periodo da circa 4 mesi fino
a 3-5 anni.
Negli ultimi anni i meccanismi neurali alla base della plasticità uditiva sono stati oggetto di numerose ricerche, soprattutto dopo l’introduzione nella clinica degli impianti cocleari e la necessità di definire
procedure cliniche condivise nella loro applicazione ai bambini con
ipoacusie pre-verbali Per un approfondimento si rimanda a: Kral A
and Eggermont JJ, What’s to lose and what’s to learn: Development
under auditory deprivation, cochlear implants and limits of cortical
plasticity. Brain Research Reviews 2007; 56: 259-269 (28), eccellente review dove emerge il ruolo determinate che viene assunto
dalle cortecce associative e dalle connessioni top-down di controllo
sulla corteccia uditiva primaria.
Una condizione continuativa di assenza dell’ingresso uditivo in un
neonato per una lesione dell’orecchio interno provoca modifiche in
gran parte irreversibili nella struttura del sistema uditivo centrale
e di conseguenza nello sviluppo delle abilità linguistiche sia nella
percezione che nella produzione del linguaggio, che prende il nome
di deprivazione uditiva. Si tratta di quella gravissima situazione di
disabilità uditiva irreversibile e permanente che corrisponde alla si-
tuazione che una volta veniva chiamata “sordomutismo”, e che oggi
non ha più ragione di esistere data la disponibilità oggi di poter di
ripristinare la soglia uditiva in tutti i tipi e gradi di ipoacusie nei bambini. Dopo l’individuazione attraverso lo screening, l’età ottimale per
l’inizio dell’intervento protesico è attorno ai 3-4 mesi e, comunque,
non oltre i 10 mesi di vita, quando nel bambino il canale uditivo
diventa predominante nello sviluppo della percezione uditiva e deve
concludersi con il ripristino della soglia uditiva attraverso l’utilizzo di
protesi acustiche o con l’impiego di un impianto cocleare.
In termini schematici nella Figura 1 è riportato la sequenza temporale in funzione dell’età del bambino delle singole tappe diagnostiche
e terapeutiche per arrivare al ripristino della soglia uditiva nei bambini con ipoacusia identificati dallo screening.
È fondamentale sapere che oggi ai fini dello sviluppo del linguaggio
e per impedire l’instaurarsi di una condizione irreversibile di deprivazione centrale è possibile ripristinare una soglia uditiva normale,
con l’utilizzo di protesi acustiche o di impianti cocleari, in pressoché
in tutti i tipi e gradi di ipoacusie. Il rispetto quindi delle tappe di
acquisizione fisiologica delle competenze uditive di un bambino è
il punto cruciale sul quale si articola tutto il processo diagnosticoterapeutico che deve rispettare due esigenze che sono apparentemente in contrasto l’una con l’altra:
• la necessità di avere un tempo sufficiente lungo nel bambino per
ottenere misure cliniche affidabili sullo sviluppo della percezione
uditiva con l’utilizzo delle protesi acustiche, prima di sottoporre il
bambino ad un impianto cocleare (che è un intervento chirurgico
irreversibile);
• la necessità di non attendere troppo per non provocare nel bambino uno stato di deprivazione uditiva e quindi una condizione di
clinica nella quale l’utilizzo dell’impianto cocleare avrebbe minore efficienza.
Figura 1.
Percorso temporale delle procedure diagnostiche e terapeutiche nella sordità infantile.
197
A. Pisacane et al.
Come riportato nello schema le linee interrotte indicano due momenti significativi lungo l’asse dell’età del bambino, che ovviamente
non sono assoluti ma vanno interpretati all’interno della variabilità
che esiste nello sviluppo di un bambino soprattutto se ha avuto una
storia di sofferenza neonatale o altre comorbilità che ne hanno ritardato lo sviluppo.
Entro 6 mesi di vita il bambino con una perdita uditiva deve iniziare
a portare continuativamente protesi acustiche e quindi deve aver
completato l’iter di diagnosi audiologica.
Entro 12-18 mesi deve essere completato l’adattamento ottimale
delle protesi e si devono ottenere le misure audiologiche sulla percezione uditiva del bambino con le protesi sulle quali poter basare
l’indicazione ad un impianto cocleare.
Nei casi in cui la perdita uditiva del bambino è così grave da non
consentire, nemmeno con le protesi acustiche più potenti e regolate in modo ottimale, un ingresso uditivo sufficiente a sviluppare
il linguaggio, il piccolo viene avviato ad un impianto cocleare. La
procedura prevede una valutazione neuroradiologica delle strutture
dell’orecchio interno e delle vie uditive e se non vi sono controindicazioni anatomiche, l’inserimento chirurgico nella coclea di elettrodi
che stimolando direttamente le fibre del nervo VIII sono in grado di
ripristinare la percezione uditiva.
Il problema della decisione se avviare un bambino ad un impianto è
il contenuto deontologico ed etico della decisione stessa, in quanto
l’intervento ha una elevata probabilità di ledere le strutture neurosensoriali della coclea. Occorre quindi essere certi che la coclea non
funzioni, anche con la migliore correzione protesica disponibile, arrivando ad ottenere nel bambino una affidabile descrizione e valutazione del suo audiogramma con le protesi e delle sue prestazioni
nella percezione di stimoli linguistici.
Tutte le procedure diagnostiche riportate nel grafico successive alla
fase di screening, che comprende sia i test neonatali sia il II livello
nei casi fail entro i primi 2 mesi di vita; sono identificabili come
procedure di III livello e necessitano di strutture specialistiche con
competenze audiologiche pediatriche specifiche e con disponibilità di diverse professionalità sia mediche che tecniche. La diagnosi
in bambini così piccoli non si basa solo sull’esito di un unico test
audiologico strumentale, ma sulla concordanza di diversi esami e
sul convincimento clinico che deriva dall’esperienza e dal contributo
che diverse professionalità possono portare alla diagnosi.
Medici specialisti in audiologia e foniatria, audioprotesisti, audiometristi e logopedisti sono le figure professionali che ruotano attorno al
processo diagnostico e di abilitazione uditiva del bambino, ciascuna
con il suo specifico contributo. Gli audio protesisti sono responsabili della corretta applicazione e funzionamento delle protesi uditive, i logopedisti sono responsabili delle procedure di abilitazione
alla percezione uditiva e allo sviluppo del linguaggio nel bambino.
Il medico audiologo e foniatra è la figura di riferimento di tutto il
processo protesico e riabilitativo e si avvale nell’ambito ospedaliero
della attività degli audiometristi che sono responsabili delle misure
audiometriche nel bambino.
Il lavoro si svolge attraverso tutta la serie di controlli periodici, che
nei bambini al di sotto dell’anno di età sono programmati almeno
ogni 2 mesi, dove vengono valutate le competenze comunicative, la
percezione uditiva e lo sviluppo linguistico del bambino.
Il nucleo familiare è coinvolto in modo attivo nel percorso riabilitativo, riceve informazioni su come stimolare il bambino per
attivare, allenare e/o potenziare le sue competenze, in armonia
con le tappe fisiologiche dello sviluppo psico-motorio. Sono oggi
disponibili anche per la lingua italiana diversi test standardizzati
in funzione dell’età del bambino, per la misura della percezione
uditiva e fonetica, della percezione del linguaggio e per la misura
delle abilità del bambino sia nella percezione che nella produzione del linguaggio.
Conclusioni
Il percorso dallo screening alla terapia è complesso. Se eseguire
il test con le otoemissioni è semplice, ben più difficile è il coordinamento delle attività diagnostiche e riabilitative a livello regionale. Riteniamo però che sia una sfida da accettare e da vincere,
in quanto un intervento precoce può evitare che una disabilità,
spesso non ancora prevenibile, possa trasformarsi in un handicap
severo.
Box di orientamento
Quali sono le informazioni/novità che emergono da questo articolo:
• Lo screening universale dei disturbi permanenti dell’udito è un intervento che ha un rapporto favorevole costi/ benefici e benefici/rischi.
• Esistono buone evidenze scientifiche che un intervento precoce influenzi favorevolmente il linguaggio e le competenze cognitive, specialmente tra
i bambini ipoacusici senza altre patologie associate.
• La complessità del processo dello screening è notevole; è richiesta una notevole cooperazione tra varie figure professionali e la disponibilità di un
valido sistema informativo.
• Per la presa in carico delle famiglie è richiesta una stretta collaborazione tra servizi sociali, educativi e sanitari.
Bibliografia
American Academy of Pediatrics, Joint Committee on Infant Hearing. Year 2007
Position Statement: principles and guidelines for early hearing detection and
intervention programs. Pediatrics 2007;120:898-921.
** Una sintesi completa dello stato dell’arte.
Calevo MG, Mezzano P, Zullino E, et al. Ligurian experience on neonatal screening: clinical and epidemiological aspects. Acta Pediatrica 2007;96:1592-9.
Chia-ling L, Farrell J, MacNeil JR, et al. Evaluating loss to follow-up in newborn
hearing screening in Massachusetts. Pediatrics 2008;121:e335-e343.
198
Davis A, Yoshinaga-Itano C, Hind S. Universal newborn hearing screening: implications for coordinating and developing services for deaf and hearing impaired
children. BMJ 2001;323:539-40.
Downs MP, Yoshinaga-Itano C. The efficacy of early identification and intervention
for children with hearing impairment. Pediatr Clin North Am 1999;46:79-87.
Edwards LC. Children with cochlear implants and complex needs: a review of outcome research and psychological practice. J Deaf Stud Educ 2007;12:258-68.
Fallon JB, Irvine DRF, Shepherd RK. Cochlear implants and brain plasticity. Hearing Research 2007.08.004.
Fortnum HM, Summerfield AQ, Marshall DH, et al. Prevalence of permanent
Lo screening neonatale dei disturbi permanenti dell’udito
childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening. BMJ 2001;323:536-40.
Grandori F, Lutman M. The European Consensus Development Conference on
neonatal hearing screening. Am J Audiol 1999;8:19-20.
Grosse SD, Ross DS, Dollard SC. Congenital cytomegalovirus (CMV) infection as
a cause of permanent bilateral hearing loss: a quantitative assessment. J Clin
Virol 2008;41:57-62
Hinman AR, Eichwald J, Linzer D, et al. Integrating child health information systems. Am J Public Health 2005;95:1923-7.
* Per chi è interessato ai sistemi informative in età evolutiva.
Holt RF, Svirsky MA. An exploratory look at pediatric cochlear implantation: is
earliest always best? Ear and Hearing 2008;29:492-511.
Hutt N, Rhodes C. Post-natal hearing loss in universal neonatal hearing screening communities: current limitations and future directions. J Pediatr Child Health
2008;44:87-91.
* Ancora sull’importanza delle forme di sordità non presenti alla nascita.
Kennedy C, McCann D, Campbell MJ, et al. Universal newborn screening for
permanent childhood hearing impairment: an 8-year follow-up of a controlled
trial. Lancet 2005;366:660-62.
** Un contributo rilevante per comprendere come il Servizio Sanitario Inglese ha
organizzato e monitorato lo screening neonatale.
Kennedy C, McCann D. Universal neonatal hearing screening moving from evidence to practice. Arch Dis Child Neonatal Ed 2004;89:F378-F383.
** Una miniera di informazioni utili per quanti vogliano adottare lo screening a
livello regionale o nazionale.
Kennedy CR, Kimm L, Thornton ARD, et al. False positives in universal
neonatal screening for permanent childhood hearing impairment. Lancet
2000;356:1903-4.
Kennedy CR, McCann DC, Campbell MJ, et al. Language ability after early detection of permanent childhood hearing impairment. N Engl J Med
2006;354:2131-41.
** Le evidenze scientifiche a favore di un intervento precoce.
Kral A, Eggermont JJ. What’s to lose and what’s to learn: development under
auditory deprivation, cochlear implants and limits of cortical plasticity. Brain Research Reviews 2007;56:259-69.
McCracken W, Young A, Tattersall H, et al. The impact of the National newborn
screening programme on educational services in England. Deafness and Education International 2005;7:179-94.
* L’importanza dei friendly hearing services.
Mehl AL, Thomson, V. The Colorado Newborn Hearing Screening Project, 19921999: on the threshold of effective population-based universal newborn hearing
screening. Pediatrics 2002;109:1-8.
Moeller M. Early intervention and language development in children who are
deaf and herd of hearing. Pediatrics 2000;106/3/ e43.
Nafstad P, Samuelsen SO, Irgens LM, et al. Birth weight and hearing impairment
in Norvegians born from 1967 to 1993. Pediatrics 2002;110:e30.
* L’importanza della prematurità e del basso peso alla nascita come fattori di
rischio per l’ipoacusia neurosensoriale.
Nelson HD, Bougatsos C, Nygren P. Universal newborn hearing screening: systematic review to update the 2001 US Preventive Services Task Force recommendation. Pediatrics 2008;122:e266-e276.
** Una lettura di base per fare il punto sulla situazione.
Papsin BC, Gordon KA. Cochlear implants for children with severe-to-profound
hearing loss. N Engl J Med 2007;357:2380-7.
* Una buona introduzione agli impianti cocleari.
Schroeder L, Petrou S, Kennedy C, et al. The economic costs of congenital bilateral permanent childhood hearing impairment. Pediatrics 2006;117:1101-2.
Suppiej A, Rizzardi E, Zanardo V, et al. Reliability of hearing screening in high-risk
neonates: comparative study of otoacoustic emission, automated and conventional auditory brainstem response. Clin Neurophysiol 2007;118:869-76.
Universal screening for hearing loss in newborns: US Preventive Services Task
Force recommendation statement. Pediatrics 2008;122:143-8.
Weichbold V, Nekkahm-Heis D, Welzl-Mueller K. Universal newborn hearing
screening and postnatal hearing loss. Pediatrics 2006;117;e631-e636.
* Articolo importante per mettere a fuoco il problema delle sordità non identificate con il test neonatale di screening
www.audiologia.unina.it/screening.
* Tutti i documenti finora prodotti sull’esperienza dello screening nella Regione
Campania.
Corrispondenza
prof. Elio Marciano, Università di Napoli Federico II, Facoltà di Medicina e Chirurgia, via S. Pansini 5, 80131 Napoli. E-mail: [email protected].
199
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 200-204
audiologia
L’impianto cocleare nel bambino
Gennaro Auletta, Ettore Cassandro*, Elisabetta Genovese**, Maria Consolazione Guarnaccia**,
Pasquale Iadicicco, Carla Laria, Emma Landolfi, Giorgio Lilli, Pasquale Riccardi, Elio Marciano
Università di Napoli “Federico II”; * Università di Catanzaro; ** Università di Modena
Riassunto
Questo lavoro fornisce le linee generali sulle problematiche degli impianti cocleari nei bambini ipoacusici. Dagli anni ’90, si è avuta l’introduzione su larga
scala degli impianti cocleari che sono stati utilizzati nei soggetti ipoacusici che non ricevevano un adeguato beneficio dall’uso delle protesi acustiche convenzionali. Si riportano le informazioni sugli aspetti clinici e chirurgici degli impianti cocleari. Si affrontano, inoltre, le tematiche inerenti la selezione e la
riabilitazione dei soggetti impiantati. Si riporta, infine, un’ampia bibliografia ottenuta da letteratura internazionale inerente agli argomenti trattati.
Summary
This work aims to provide a general overview on the issue of the cochlear implantation in infancy. All the professionals involved in the health care of children
with permanent hearing disorders might hopefully find here useful and clear information about the general technical features of this devices and the clinical
significances it may concern. A general description of the clinical-surgical concerns about cochlear implantation as well as a the tracking period will be
issued as well. The paper is, moreover, provided of a wide tender from the most important literary production in the scientific field of the cochlear implant
so that one may study in deep any specific topic mentioned in the piece.
Obiettivo
Aspetti generali
Lo scopo di questa review è quello di fornire una panoramica dettagliata sulle attuali conoscenze sulle tematiche inerenti gli impianti
cocleari (IC) a tutti i professionisti socio-sanitari, soprattutto Pediatri,
coinvolti nell’assistenza del paziente e della sua famiglia.
La recente bibliografia fornisce un’evidente conferma della plasticità
del sistema uditivo, in quanto è in grado di riorganizzare la propria
struttura e le proprie funzioni.
La deprivazione acustica (ipoacusia) determina una mancata maturazione delle vie uditive ed una alterazione della organizzazione tonotopica dei nuclei tronco encefalici, delle stazioni intermedie (collicolo
inferiore, corpo genicolato mediale) e della corteccia uditiva primaria
e secondaria. (Eggermont et al., 2001; 2008). I cambiamenti strutturali nelle varie parti del sistema uditivo sono in relazione al periodo di
insorgenza della ipoacusia. (Sika et al., 2002; Kral et al., 2000; Leake
et al. 1988; 2001). L’esperienza uditiva gioca un ruolo fondamentale
nello stabilire la fine organizzazione del sistema uditivo. L’assenza di
una adeguata stimolazione sonora, in età preverbale, determina, infatti, una incompleta maturazione delle vie uditive che risulta, in ogni
modo, sufficiente per l’applicazione di impianto cocleare (Kral et al.,
2009). Particolarmente importanti sono le connessioni della corteccia
uditiva con le aree corticali frontale e prefrontale che hanno la funzione di stabilire un alto livello attentivo e cognitivo legato a processi
uditivi (Houston, 2003). Numerose ricerche hanno evidenziato che, nel
bambino, il maggiore guadagno nelle performance percettivo-verbali
si ottiene nei primi 9-12 mesi di utilizzo di protesi acustica (Sharma
et al., 2009, Arslan et al., 2003). Per quanto concerne l’attivazione
cerebrale dopo impianto cocleare molti studi attualmente si avvalgono
di metodiche obiettive (PET) che hanno evidenziato una ricomparsa
dell’attività metabolica dell’encefalo nell’area della corteccia uditiva
nei soggetti con IC (Fujiwara et al., 2008; Naito et al., 1995; Wong et
al., 1999; Nayto et al., 2000; Nishimura et al., 2000).
Introduzione
Negli anni ’70, grazie all’avvento della tecnologia digitale ed all’ampliamento delle conoscenze sulla fisiopatologia del sistema uditivo,
sono stati messi a punto dei dispositivi elettronici che, in diretto contatto con le strutture cocleari, sono in grado di evocare sensazioni
uditive valide in pazienti con gravi deficit uditivi neurosensoriali. La
sola stimolazione acustica fornita dagli apparecchi convenzionali, infatti, non arriva generalmente a sopperire ad una grave disfunzione
delle cellule ciliate, responsabili del fisiologico fenomeno di trasduzione del segnale acustico a livello dall’orecchio interno. È in questi
casi che trova indicazione l’impianto cocleare (IC), strumento in grado
di trasdurre i suoni in segnali elettrici che, opportunamente codificati,
possono stimolare le terminazioni del nervo acustico prossimali alla
coclea e dar via alla conduzione dello stimolo sonoro attraverso le
vie neurali afferenti al sistema nervoso centrale. Dagli anni ’90, si è
avuta l’introduzione su larga scala degli impianti cocleari che sono
stati utilizzati nei soggetti ipoacusici che non ricevevano un adeguato
beneficio dall’uso delle protesi acustiche convenzionali.
Metodologia della ricerca bibliografica
La bibliografia (review e articoli) riportata in questo articolo è stata reperita dalla banca bibliografica “Medline” mediante l’uso del
motore di ricerca “PubMed”, utilizzando parole chiavi quali: hearing
impairment and cochlear implant. Sono stati selezionati gli articoli
più rilevanti sugli impanti cocleari e le review in cui erano riportate
considerazioni significative sull’argomento.
200
Come è costituito un impianto cocleare
In generale l’impianto cocleare è costituito da (Fig. 1):
Parte esterna (processore esterno)
• microfono: raccoglie il segnale acustico e lo converte in segnale
elettrico;
L’impianto cocleare nel bambino
• processore del linguaggio: elabora il segnale acustico;
• antenna/magnete in collegamento con bobina trasmittente.
Parte interna (trasduttore)
• bobina ricevente: permette la comunicazione con il processore
esterno. Viene inserita chirurgicamente al di sotto della cute e
riceve l’informazione dal trasmettitore attraverso induzioni magnetiche (campi elettromagnetici);
• elettrodo/i intracocleare: produce stimolazione elettrica in prossimità delle vie periferiche del nervo acustico al fine di inviare
informazione ai centri nervosi.
Nella Figura 2 viene presentata la parte esterna dell’IC.
Nella Figura 3 si riporta lo schema completo della collocazione delle
diverse componenti (interne ed esterne) dell’IC.
I moderni IC sono costituiti da più elettrodi (array di elettrodi); gli elettrodi sono posizionati all’interno della coclea e la loro attivazione dipende
dalla frequenza del segnale d’ingresso. Un elettrodo posizionato nelle
aree apicali genera una sensazione sonora di tonalità grave; viceversa
avviene per gli elettrodi posizionati nelle aree basali, responsabili della
trasmissione dei segnali sonori in alta frequenza (Clark, 2003).
Da quanto detto si desume che il livello quantitativo di sensazione
uditiva descritto dal soggetto impiantato è determinato dalle caratteristiche della stimolazione elettrica, quali intensità e durata dell’impulso elettrico, nonché, dalla frequenza di stimolazione.
Generalmente gli impianti cocleari si differenziano:
• per la diversa strategia adottata nella trasduzione del segnale
sonoro;
• per tipologia di stimolazione.
Si utilizzano elettrodi di platino che evitano il rilascio di metalli o
materiali tossici per i tessuti neurali (Clark, 2003).
Il numero di elettrodi e la loro distanza incidono sulla risoluzione
della codifica tonotopica che dipende da due fattori fondamentali:
• dal numero di terminazioni nervose che possono essere stimolati in una particolare area della coclea;
• dall’area eccitata dalla stimolazione elettrica.
Nel caso in cui il numero di terminazioni nervose funzionanti è elevato, un aumento degli elettrodi attivi comporta un miglioramento
della codifica tonotopica; viceversa, se il numero è esiguo, ad un
aumento del numero di elettrodi potrebbe non corrispondere un miglioramento della percezione acustica (Saunders et al., 2002).
La comunicazione tra l’impianto ed il processore è per via transcutanea; in questo caso la connessione tra l’elemento esterno e quello
interno avviene utilizzando una bobina trasmittente esterna al cranio,
saldamente ancorata alla parete cranica grazie ad un magnete inserito sottocute. La bobina ricevente interna è posta in un alloggiamento
ricavato chirurgicamente nell’osso temporale. Per fare in modo che
le due bobine si trovino sullo stesso asse, in modo da ottenere la migliore trasmissione dei dati, si posiziona il magnete di sostegno della
bobina esterna in modo tale che le due bobine siano coassiali. Successivamente, la bobina ricevente invia (via cavo) il segnale elettrico
agli elettrodi.
Due sono le variabili grazie alle quali la stimolazione prodotta dallo
speech processor viene adattata alle necessità funzionali del nervo
acustico del singolo paziente: la strategia di codificazione e i livelli
elettrici che generano la sensazione sonora.
Aspetti chirurgici dell’impianto cocleare
L’intervento chirurgico di applicazione di impianto cocleare avviene secondo le moderne procedure intraoperatorie, ossia attraverso
Figura 1.
Elementi fondamentali dell’impianto cocleare.
Figura 2.
Elemento esterno (processore e bobina trasmittente).
Figura 3.
Collocazione dell’array di elettrodi all’interno della coclea.
un’anestesia generale, una durata di circa due ore ed un normale
periodo di ospedalizzazione.
L’accesso chirurgico avviene, generalmente, attraverso una incisione retroauricolare. Le modalità sono quelle di una timpanotomia
posteriore, procedura di largo uso in campo otochirurgico, che viene
effettuata dopo aver creato un letto osseo nella mastoide su cui appoggiare il processore interno. Attraverso la timpanotomia posteriore è possibili e identificare il promontorio, punto di repere essenziale
per l’esecuzione della cocleostomia, necessaria per l’inserimento
dell’array degli elettrodi all’interno della scala timpanica cocleare.
Al fine di garantire sia una corretta cicatrizzazione sia un buon fissaggio delle componenti interne ai tessuti (bobina trasmittente e array di
elettrodi), il collegamento tra processore del linguaggio (esterno) ed
201
G. Auletta et al.
antenna ricevente (interna) viene effettuato dopo circa trenta giorni.
Infine, nei pazienti portatori di IC, è necessario in caso di studio
neuroradiologico con risonanza magnetica, praticare particolari
avvertenze (ad esempio applicazione di bendaggio esterno all’area
di impianto) in modo da prevenire una eventuale dislocazione della
porzione interna del dispositivo (Clark, 2003).
Analisi costo-beneficio dell’utilizzo di un impianto cocleare
I benefici sociali indotti dall’impianto cocleare nel bambino sono una
maggiore integrazione nel sistema educativo e una successiva maggiore partecipazione al contesto sociale e professionale. Tali risultati
hanno indotto la Food and Drugs Administration (FDA) ad estendere
le indicazioni all’impianto cocleare dai quei pazienti che non avevano alcun beneficio dalla terapia protesica tradizionale a quelli che
avevano un beneficio insufficiente.
Negli ultimi anni, i principali centri europei ed internazionali hanno
iniziato ad applicare l’impianto bilateralmente. In particolare, per i
pazienti più piccoli (in età prelinguale) tale applicazione viene effettuata simultaneamente, ossia in un unico intervento chirurgico,
mentre per quelli più grandi la strategia applicativa degli impianti
bilaterali è più frequentemente di tipo sequenziale, due tempi chirurgici. Il passaggio da IC monolaterale a bilaterale è conseguente
a studi recenti dai quali emerge che l’udito binaurale può (in molti
casi) incrementare la capacità di comprensione verbale in ambiente
rumoroso nonché permettere la localizzazione della sorgente sonora
(van Hoese et al., 2002; 2003; Ching et al.; 2004, 2006; Valencia et
al., 2008; Offeciers et al., 2009; Beijen et al., 2007; Papsin et al.,
2008; Ching et al., 2006; Johnston et al., 2009).
La realizzazione di programmi screening uditivo universale neonatale e
di programmi di screening per i neonati a rischio ha facilitato la diagnosi
e l’intervento precoce per i bambini ipoacusici (JAMA, 2001; Pediatrics,
2002; Yoshinaga-Itano et al., 2001). Ciò ha determinato il recente cambiamento nei criteri di selezione per l’impianto cocleare da parte della FDA
che ha incluso i bambini al di sotto dei 12 mesi, intervento da praticare
in alcuni centri audiologici autorizzati. Prima di tale età l’IC potrà essere
eseguito in quei casi dove siano soddisfatti i criteri di certezza diagnostica
(definizione della soglia, della sede di lesione, dell’eziologia dell’ipoacusia)
e in casi particolari dove esiste il rischio di fibrosi/ossificazione precoce
della coclea, come ad esempio in caso di meningite batterica.
I piccoli pazienti con sordità neurosensoriale profonde (soglia uditiva
≥ 90 dB HL come media per le frequenze 500, 1000, 2000 Hz) rientrano generalmente nel gruppo di candidati all’intervento chirurgico.
Va valutato, in ogni modo, prima dell’indicazione all’IC, il beneficio
ottenuto dall’uso di protesi acustiche. Non è raro, infatti, che gruppi
di pazienti con analogo grado ipoacusia possano presentare evidenti
differenze nelle performance acustico-percettive. Rimane, inoltre, da
valutare, caso per caso, l’eventuale presenza di patologie associate:
un attento esame clinico unito ad uno studio neuro-radiologico dedicato risultano importanti per valutare la presenza di eventuali malformazioni (cocleari o del nel nervo acustico) che possano risultare
come controindicazioni. È da sottolineare, inoltre, che l’associazione
con altre disabilità (ipovisione, patologie della sfera neuro psicomotoria) non risultano essere motivo di esclusione dell’applicazione di
IC in quanto il dispositivo fornirebbe possibilità concrete di ulteriore
stimolazione neurosensoriale (Waltzman et al., 2000).
Iter riabilitativo del bambino con impianto cocleare
La selezione del bambino ipoacusico da sottoporre Il successo terapeutico del paziente con IC non può prescindere da un
adeguato programma riabilitativo ed ad una periodica verifica clinica
ad impianto cocleare
La precocità e la qualità di intervento in caso di ipoacusia grave o
profonda congenita giocano un ruolo importantissimo nell’abilitazione
uditiva del bambino (Svirsky et al., 2004, Holt et al., 2008; Tyler et al.,
1997) Una corretta protesizzazione acustica associata ad un corretto
iter riabilitativo avviati entro i primi sei mesi di vita consentono un
utilizzo massimale della plasticità delle vie uditive centrali ed offrono
un duplice vantaggio: il pieno utilizzo delle potenzialità percettivo-linguistiche del piccolo paziente e la garanzia di un intervento realmente
precoce in caso di selezione ad impianto cocleare (Yoshinaga-Itano et
al., 2001; 2003; Sharma et al., 2009; Nicholas et al., 2007).
L’età per la indicazione all’impianto cocleare varia a seconda dei vari
centri nazionali e internazionali, collocandosi mediamente intorno ai
12 mesi di vita. Ovviamente sono necessari tempi congrui di terapia
protesica (non meno di 6 mesi) e protocolli rigidi di valutazione del
beneficio protesico che comprendano sia la valutazione della percezione uditiva sia dello sviluppo linguistico-cognitivo.
delle abilità progressivamente conseguite. La presenza di centri clinici
di riferimento territoriale è essenziale al fine di garantire al paziente un
supporto specialistico multidisciplinare. La verifica dell’andamento delle
performance comunicativo-linguistiche di un bambino con impianto cocleare, difatti, dovranno essere valutate da un equipe clinico-riabilitativa
con diverse competenze (ad es. audiologo-foniatra, specialista ORL, tecnico audiometrista, logopedista, psicologo, neuropsichiatra infantile, pediatra, neuroradiologo, etc.) per elaborare comuni strategie riabilitative.
Particolare attenzione va, inoltre, posta al periodo immediatamente
successivo all’intervento chirurgico. È necessario, infatti, garantire una periodica programmazione personalizzata (mappaggio) del
dispositivo al fine di effettuare un corretto adattamento alle nuove
informazioni sonore percepite.
I controlli clinici nel periodo post-operatorio dovranno, pertanto,
essere a cadenza settimanale per i primi 30-40 giorni; diverranno,
successivamente, a cadenza mensile fino a quando, a circa 12-18
mesi dall’attivazione, si effettueranno 3/4 valutazioni in un anno.
Box di orientamento
• Una corretta protesizzazione acustica associata ad un idoneo iter riabilitativo avviati entro i primi sei mesi di vita consentono un utilizzo massimale
della plasticità delle vie uditive centrali e offrono un duplice vantaggio: sfruttano appieno le potenzialità uditive e linguistiche di un bambino e
garantiscono nel contempo una possibilità di intervento realmente precoce in caso di successiva scelta di impianto cocleare.
• Si ricorre all’impianto cocleare quando le protesi acustiche tradizionali non sono sufficienti al recupero dell’ipoacusia. Necessario, in ogni caso,
prima di passare all’impianto cocleare un trattamento con protesi di almeno 6 mesi.
• L’età per l’indicazione all’impianto cocleare varia, a seconda delle scelte dei vari centri nazionali ed internazionali, collocandosi mediamente intorno
ai 12 mesi di vita.
202
L’impianto cocleare nel bambino
Bibliografia
Arslan E, Genovese E, Santarelli R. Prevenzione e diagnosi precoce delle ipoacusie
infantili preverbale. Giornale di Neuropsichiatria dell’età evolutiva 2002;22:1-34.
Beijen JW, Snik AF, Mylanus EA. Sound localization ability of young children with
bilateral cochlear implants. Otol Neurotol 2007;28:479-85.
Papsin BC, Gordon KA. Bilateral cochlear implants should be the standard for
children with bilateral sensorineural deafness. Curr Opin Otolaryngol Head Neck
Surg 2008;16:69-74.
* L’applicazione di impianto bilaterale appare garantire migliori performance
comunicativo-linguistiche in quei pazienti impiantati precocemente, e simultaneamente, con dispositivo binaurale.
Fryauf-Bertschy H, Tyler RS, Kelsay DM, et al. Cochlear implant use by prelingually deafened children: the influences of age at implant and length of device
use. J Speech Lang Hear Res 1997;40:183-99.
Gantz BJ, Tyler RS, Woodworth GG, et al. Results of multichannel cochlear implants in congenital and acquired prelingual deafness in children: Five year follow up. Am J Otol 1994;15(Suppl. 2):1-7.
Ching TYC, van Wanrooy E, Hill M, et al. Performance in children with hearing
aids or cochlear implants: Bilateral stimulation and binaural hearing. Int J Audiol
2006;45(Suppl. 1):S4-15.
* La possibilità di essere forniti di udito binaurale permette ai pazienti con ipoacusia bilaterale profonda di sostenere in modo migliore richieste uditive in ambiente rumoroso od in condizioni ambientali difficoltose rispetto a quei pazienti a
parità di ipoacusia ma forniti di supporto protesico monoaurale.
Clark GM, Cochlear implants: fundamental & application. New York: AIP Press, 2003.
* Testo di fondamentale importanza per essere introdotti od approfondire, in
pratica, tutte le tematiche riguardanti l’impianto cocleare.
Eggermont JJ, Ponton CW, Don M, et al. Maturational delays in cortical evoked
potentials in cochlear implant users. Acta Otolaryngol 1997;117:161-3.
* Risposta dei potenziali evocati corticali in funzione del tempo di attivazione di
una stimolazione Sonora adeguata ed importanza sulla verifica degli outcome
percettivi del paziente con impianto cocleare.
Eggermont JJ. The role of soud in adult and developmental auditory cortical
plasticity. Ear Hear 2008;29:819-29.
Fryauf-Bertschy H, Tyler RS, Kelsay DMR, et al. Cochlear implant use by prelingually deafened children: the influence of age at implant and length of device
use. J Speech Language Hear Res 1997;40:183-99.
Fujiwara K, Naito Y, Senda M, et al. Brain. Children with profound deafness: a
visual language activation study by 18F-fluorodeoxyglucose positron emission
tomography. Acta Otolaryngol 2008;128:393-7.
Holt RF, Svirsky MA. An exploratory look at pediatric cochlear implantation: is
earliest always best? Ear Hear 2008;29:492-511.
Houston DM, Pisoni DB, Kirk KL, Ying EA, et al. Speech perception skills of deaf
infants following cochlear implantations: a first report. Intern Jour Ped Otorhinol
2003;67:479-95.
Kral A, Hartmann R, Tillein J, et al. Congenital auditory deprivation rediuces synaptyc activity within the auditory cortex in a layer-specific manner. Cereb Cortex
2000;10:714-26.
* Le proiezioni intercorticali appaiono danneggiate dalla deprivazione uditiva:
nella registrazione delle risposte da diversi strati della corteccia uditiva di gatti
ipoacusici hanno evidenziato una riduzione delle sinapsi dello strato infragranulare che invia impulsi in altre regioni corticali.
Kral A. Early hearing experience and sensitive developmental periods. HNO
2009;57:9-16.
Leake PA, Hradek GT. Cochlear pathology of long term neomycin induced deafness in cats. Hear Res 1988;33:31-4.
* A livello del sistema uditivo centrale, l’ipoacusia bilaterale determina una
riduzione delle sinapsi nel collicolo inferiore con conseguente compromissione
della selettività spaziale in funzione della durata della deprivazione sensoriale.
Si è dimostrato, in gatti sordi da diversi periodi di tempo e successivamente
impiantati, che la selettività spaziale degli impulsi elettrici a livello del collicolo
inferiore era danneggiata dal tempo di deprivazione e dal grado di degenerazione del ganglio spirale.
Leclerc C, Saint-Amour D, Lavoie ME, et al. Brain functional reorganization in
early blynd humans revealed by auditory event-related potentials. Neuroreport
2000;11: 545-50.
* I risultati delle registrazioni dei potenziali evocati uditivi rilevati nei soggetti
ciechi presentano delle onde N1 e P3 ben rappresentate di derivazione dalla
regione della corteccia occipitale.
Loizou P. Introduction to cochlear implants. IEEE Signal Processing Magazine
1998;101-30. *Analisi e descrizione della componentistica alla base del funzionamento elettroacustico dell’impianto cocleare.
Nayto Y, Okazawa H, Honjo I, et al. Cortical activation with sound stimulation in
cochlear implant users demonstrated by positron emission tomography. Cogn
Brain Res 1995;2:207-14.
Nayto Y, Tateya I, Fujiki N, et al. Increased cortical activation during hearing of
speech in cochlear implant users. Hear Res 2000;143:139-46.
* L’attivazione cerebrale alla PET con rumore non mostra significative differenze
tra soggetti impiantati e normoudenti; mentre con stimoli verbali (parole) si rileva una attivazione nei soggetti impiantati non solo nella corteccia temporale,
ma anche nell’area di Broca e nell’area omologa dell’emisfero destro, nell’area
motoria supplementare e nel giro cingolato anteriore. Quindi nei soggetti sordi
postlinguali l’ascolto di parole codificate dall’impianto può essere accompagnato da una attivazione di entrambe le cortecce temporale e frontale.
Nicholas JG, Geers AE. Will they catch up? The role of age at cochlear implantation in the spoken language development of children with severe to profound
hearing loss. J Speech Lang Hear Res 2007;50:1048-62.
* Sia le abilità percettive che quelle in output linguistico traggono giovamento da
una stimolazione precoce in tutte le aree del linguaggio espressivo.
Nishimura H, Doi K, Iwaki T, et al. Neural plasticity detected in short- and longterm cochlear implant users using PET. Neuroreport 2000;11:811-5.
* È stata studiata, utilizzando la PET, l’attivazione della corteccia uditiva con una
simultanea presentazione di segnali uditivi e visivi in due gruppi di soggetti:
un primo gruppo impiantato di recente e l’altro che utilizzava l’impianto già da
tempo. È stato riscontrato che la corteccia uditiva rimaneva inattiva dopo la
simultanea presentazione di stimoli uditivi e visivi nei soggetti da poco impiantati, mentre si attivava nel gruppo che utilizzava l’impianto da più tempo. Il
dato rilevato suggerisce la possibilità che l’interferenza di modalità diverse, quali
quella visiva ed uditiva, si possa ridurre nel tempo ed il ritardo nell’attivazione
della corteccia uditiva conferma la presenza di una certa plasticità neurale nella
corteccia uditiva dell’uomo adulto.
Papsin BC, Gordon KA. Bilateral cochlear implants should be the standard for
children with bilateral sensorineural deafness. Curr Opin Otolaryngol Head Neck
Surg 2008;16:69-74.
* L’applicazione di impianto bilaterale nel piccolo ipoacusico ha dimostrato
quanto le performance linguistico-comunicative così come quelle attentivo-percettive migliorino in caso di supporto elettroacustico binaurale.
Ponton CW, Eggermont JJ. Altered cortical maturation following profound deafness and cochlear implante use. Audiol Neurootol 2001;6:363-80.
* La differenzazione del tessuto corticale è marcatamente influenzata da un punto di vista morfofunzionale dall’entità e dalla durata della deprivazione uditiva.
Ponton CW, Don M, Eggermont JJ, et al. Auditory system plasticity in children
after long periods of complete deafness. Neuroreport 1996;8:61-5.
Ponton CW, Don M, Eggermont JJ, et al. Maturation of human cortical auditory
function: difference between normal hearing children and children with cochlear
implants. Ear Hear 1996;17:430-7.
Saunders E, Cohen L, Aschendorff A, et al. Threshold, comfortable level and
impedance changes as a function of electrode-modiolar distance. Ear Hear
2002;23(Suppl. 1):28S-40S.
* Una corretta applicazione intracocleare degli elettrodi garantisce migliori performance psicoacustiche ed un adeguata strategie di stimolazione elettroacustica.
Sharma A, Nash AA, Dorman M. Cortical development, plasticity and reorganization in children with cochlear implants. Jour Commun Disorders
2009;42:272-9.
* Una riorganizzazione di alcune aree corticali viene mantenuta se viene fornita
una adeguata stimolazione uditiva. esistono periodi-finestra entro i quali deve
essere garantita una utile stimolazione al fine di garantire delle sufficienti performance linguistiche al bambino con ipoacusia congenita.
Sika J. Plastic changes in the central auditory system after hearing loss, restoration of function and during learning. Physil Rew 2002;82:601-36.
* Negli animali in fase di sviluppo si determina, infatti, una riduzione del numero
di neuroni nei nuclei lungo le vie uditive e la formazione di nuove connessioni,
mentre negli animali adulti la perdita uditiva si accompagna a una riduzione sia
dell’attività dei circuiti inibitori corticali, e sia della attivazione della sintesi di
proteine normalmente presenti nel sistema uditivo durante lo sviluppo.
Svirsky MA, Teoh SW, Neuburger H. Development of language and speech perception in congenitally, profoundly deaf children as a function of age at cochlear
implantation. Audiol Neurootol 2004;9:224-33.
203
G. Auletta et al.
* Le competenze percettivo-linguistiche risultano evidentemente migliori in quei
pazienti con ipoacusia profonda congenita impiantati in età perlinguale.
Tyler RS, Fryauf-Bertschy H, Gantz BJ, et al. Speech perception in prelingually
implanted children after four years. In: Honjo I, Takahashi H (eds.). Cochlear Implant and related update: advanced in otorynolaringology. Basel: Karger 1997;
pp. 187-192.
* Viene dimostrato quanto le abilità linguistico-comunicative si rivelino vicine al
bambino normoudente per quei pazienti con sordità profonda a cui è stato applicato precocemente l’impianto cocleare.
Waltzman SB, Scalchunes V, Cohen NL. Performance of multiply handicapped
children using cochlear implants. Am J Otol 2000;21:329-35.
Wong D, Miyamoto RT, Pisoni DB, et al. PET imaging of cochlear implant and normal
hearing subjects listening to speech and nonspeech. Hear Res 1999;132:34-42.
* È stata confrontata l’attivazione cerebrale in soggetti normoudenti e sordi post-
verbali sottoposti ad IC, con stimoli verbali e non, evidenziando, in seguito a
stimolazione con frasi, più foci nella regione temporale destra, area in cui si
determinano i processi prosodici, nei soggetti impiantati rispetto ai normoudenti.
Questi, invece, mostravano un focus nel giro frontale inferiore, area coinvolta nei
processi semantici. Le medesime differenze nei due gruppi sono state riscontrate con la stimolazione tipo “cocktail party”.
Yoshinaga-Itano C, Coulter D, Thomson V. Developmental outcomes of childrenwith hearing loss born in Colorado hospitals with and without universal newborn
hearing screening programs. Semin Neonatol 2001;6:521-9.
* L’avvento degli screening uditivi universali ha apportato un evidente benificio
alla comunità attraverso una sempre più precoce diagnosi delle sordità congenite e quindi un intervento terapeutico efficace.
Yoshinaga-Itano C. From screening to early identification and intervention: discovering predictors to successful outcomes for children with significant hearing
loss. J Deaf Stud Deaf Educ 2003;8:11-30.
Corrispondenza
prof. Elio Marciano, Unità di Audiologia, Università di Napoli “Federico II”, via Pansini 5, 80131 Napoli. E-mail: [email protected]
204
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 205-209
Audiologia
Forme ereditarie di ipoacusie neurosensoriali
isolate e sindromiche nel bambino
Annamaria Franzè 1 2, Viviana Chinetti 1 3, Paolo Gasparini 4, Pasquale Giannini 3,
Sandra Iossa 1 3, Carla Laria 3, Giorgio Lilli 3, Rita Malesci 3, Alessandro Martini 5,
Elio Marciano 3
CEINGE-Biotecnologie Avanzate, Napoli; 2 Istituto di Genetica e Biofisica “A. Buzzati Traverso”, C.N.R., Napoli;
3
Unità di Audiologia, Dipartimento di Neuroscienze, Università di Napoli “Federico II”; 4 Genetica Medica, Università
di Trieste; 5 Dipartimento di Audiologia, Università di Ferrara
1
Riassunto
L’ipoacusia è una delle patologie più diffuse. Si stima infatti che circa 1 su 1000 nati presenta ipoacusia neurosensoriale congenita e che circa il 60% delle
ipoacusie neurosensoriali possono attualmente essere attribuite a cause genetiche. Esse sono distinte in forme sindromiche e forme isolate. La conoscenza
della genetica della sordità è aumentata notevolmente negli ultimi anni. Sono stati, infatti, già identificati numerosi loci e geni responsabili per sordità. A
dispetto di tale variabilità fenotipica si è però osservato che il gene GJB2, da solo o in associazione con il gene GJB6, è responsabile di un elevato numero
di casi, rendendo quindi lo screening molecolare per questi due geni uno strumento efficace per l’identificazione dell’etiologia in numerosi casi. Applicazioni di screening audiometrici precoci, in combinazione con test genetici possono così facilitare una diagnosi della sordità e migliorare la riabilitazione
audiologica.
Summary
Hearing impairment is the most prevalent sensory handicap (1/1000 newborns) and the genetic factors are of major importance in more than 60% of all
hearing loss. An important distinction is made between syndromic deafness and isolated deafness depending on the presence or not of associated manifestations from other organs. The knowledge about genetic deafness has increased dramatically in the last few years. In fact, several loci and genes for deafness of different types of monogenic inheritance have been identified. Because the GJB2 gene alone or together with the GJB6 gene is causative for many
cases, the molecular screening for these two genes is very efficient in determining etiology in several hearing loss cases. Application of early screening
tests in combination with genetic tests will facilitate early and specific diagnosis of hearing impairment and thereby improve audiological rehabilitation.
Obiettivo
Lo scopo di questa review è quello di fornire una panoramica dettagliata sulle attuali conoscenze delle forme ereditarie di ipoacusia
neurosensoriale a tutti i professionisti socio-sanitari, soprattutto Pediatri, coinvolti nell’assistenza del paziente e della sua famiglia.
Introduzione
L’ipoacusia è una delle patologie più diffuse. Si stima infatti che
circa 1 su 1000 nati presenta ipoacusia neurosensoriale congenita
(Morton, 1991); all’ipoacusia possono essere correlate molte complicazioni ed implicazioni cliniche e sociali con un forte impatto sulla
qualità di vita del paziente (Yoshinaga-Itano, 1999).
Nei bambini la determinazione del tipo di ipoacusia e la valutazione
etiologica sono quindi molto importanti. La valutazione etiologica è
importante non solo per determinare il rischio di sordità all’interno di una famiglia, ma può permettere l’anticipazione di potenziali
problemi di salute e offrire riferimenti per opzioni terapeutiche. Una
forma di ipoacusia grave, se non corretta, ha pesanti conseguenze
sullo sviluppo del linguaggio e può avere effetti negativi anche sullo
sviluppo cognitivo (Kochhar et al., 2007).
Negli ultimi anni, mediante lo screening neonatale, sono stati rea-
lizzati interventi precoci di riabilitazione (protesi acustiche ed impianti cocleari) che si sono dimostrati molto efficaci ed importanti
per assicurare uno sviluppo di linguaggio e cognitivo ottimale. In
assenza di screening neonatali, l’ipoacusia può infatti essere notata
dai genitori, insegnanti e pediatri solo quando il bambino comincia
ad avere problemi di linguaggio o di apprendimento, spesso intorno
ai 2-3 anni.
Negli ultimi tempi, grazie ai grandi progressi ottenuti nella biologia
molecolare, è risultato sempre più chiaro il contributo dei fattori genetici nelle ipoacusie. Attualmente si stima infatti che circa il 60%
delle ipoacusie neurosensoriali possono attualmente essere attribuite a cause genetiche (Rehm 2003); la rimanente parte è dovuta
a cause di natura ambientale. È importante però notare che non
sempre è facile escludere le cause genetiche, poichè esistono molti
casi di forme recessive sporadiche in cui non è presente una forma
familiare di ipoacusia e può quindi risultare difficile inquadrare queste forme di ipoacusia dal punto di vista eziopatogenetico. Inoltre
esistono casi in cui l’ipoacusia potrebbe essere attribuita a cause
ambientali (es. infezione da Citomegalovirus – CMV) ed invece è presente una causa genetica (Ross et al., 2007).
Le ipoacusie di tipo genetico possono essere congenite, ma svilupparsi a qualsiasi età, così come è possibile un aggravamento di un
deficit uditivo preesistente.
205
A. Franzè et al.
Attualmente si stima che il 70% delle forme genetiche di ipoacusia
neurosensoriale è non sindromico mentre il rimanente 30% ha una
delle circa 400 forme sindromiche finora identificate.
La conoscenza della genetica della sordità è aumentata notevolmente negli ultimi anni, ma, molto resta ancora da fare sia per quanto riguarda l’identificazione dei geni responsabili di patologie quali
le ipoacusie neurosensoriali, sia per quanto riguarda i meccanismi
molecolari alla base di queste patologie. I processi coinvolti nel meccanismo della funzione uditiva sono controllati da centinaia di geni
e l’ipoacusia su base genetica può essere causata da una grande
varietà di mutazioni in differenti geni. Le sordità non sindromiche
sono caratterizzate da un’elevata eterogeneità genetica, cioè dalla
presenza di geni diversi in grado, una volta mutati di dare origine a
quadri clinici del tutto sovrapponibili. Altra caratteristica comune ai
geni della sordità è la presenza di mutazioni in uno stesso gene in
grado di dare origine sia a forme trasmesse come carattere autosomico dominante sia a forme trasmesse come carattere autosomico
recessivo (Rehm, 2003). Inoltre, in alcuni casi mutazioni di uno stesso gene sono in grado di dare origine a forme di sordità sindromica
e non sindromica, come per esempio nel caso della connessina 26
o della miosina VIIA responsabili sia di forme di ipoacusia neurosensoriale non sindromiche recessive, sia dominanti ed anche di forme
sindromiche (Di Leva et al., 2006; Kelsell et al., 1997; Liu et al.,
1997; Richard et al., 2002; Weil et al., 1997).
Metodologia della ricerca bibliografica
La bibliografia (review e articoli) riportata in questo articolo è stata reperita dalla banca bibliografica “Medline” mediante l’uso del
motore di ricerca “PubMed”, utilizzando parole chiavi quali: hearing impairment and gene, hearing impairment and GJB2, connexin
26, connexin 30, syndromic hearing impairment, connexin 26 and
35delG; skin disease and connexin. Sono stati selezionati gli articoli
più rilevanti come quelli che citavano l’identificazione di nuovi geni o
nuove mutazioni implicate nell’ipoacusia e le review in cui erano riportate considerazioni significative sull’argomento. Inoltre sono stati
selezionati gli articoli in cui venivano dimostrate nuove correlazioni
tra mutazioni già note e nuovi fenotipi.
Ipoacusie neurosensoriali non sindromiche
Attualmente sono stati identificati un centinaio di loci, ma solo una
cinquantina dei corrispondenti geni malattia sono stati identificati.
Dati aggiornati in tempo reale sui loci e sui geni identificati finora
sono disponibili presso la “Hereditary Hearing loss Homepage” al
sito web http://webhost.ua.ac.be/hhh/.
L’evoluzione del deficit uditivo nelle ipoacusie genetiche isolate è molto
variabile sia per quanto riguarda l’entità del deficit che per quanto riguarda la modalità di progressione, potendo anche riscontrarsi differenze importanti non solo tra famiglia e famiglia, ma anche tra individuo ed
individuo con la stessa mutazione genetica (Martini et al., 1997).
Le forme di ipoacusia neurosensoriali non sindromiche vengono
classificate in base al tipo di trasmissione:
• autosomiche recessive (80% dei casi): sono in genere caratterizzate da una sordità neurosensoriale bilaterale profonda o totale già presente alla nascita o comunque ad insorgenza precoce
nel corso della prima infanzia. Per esse in genere non si verifica
un andamento progressivo;
• autosomiche dominanti (10-20% dei casi): sono generalmente
caratterizzate da un’ipoacusia neurosensoriale bilaterale mediograve che frequentemente si sviluppa nella prima adolescenza
206
o nell’età adulta. Sono caratterizzate da una estrema variabilità
di espressione clinica anche intrafamiliare con differenza di funzionalità anche tra un orecchio e l’altro. Hanno generalmente un
andamento progressivo;
• alterazioni del DNA mitocondriale (1-2% dei casi): l’ipoacusia
in questi casi è generalmente neurosensoriale, bilaterale e progressiva con insorgenza nella prima adolescenza;
• forme associate al cromosoma X (1-2% dei casi): l’ipoacusia in
questi casi è generalmente neurosensoriale o di tipo misto con
insorgenza e progressione variabile.
Recentemente è stata anche identificata una famiglia con trasmissione associata al cromosoma Y.
Mutazioni nel gene GJB2 (gap junction beta 2) sono responsabili di
circa il 50% delle sordità neurosensoriali recessive (Zelante et al.,
1997). Il gene GJB2 codifica per la connessina 26 (Cx26), che forma
canali nella membrana plasmatica tra le cellule cocleari (gap junctions). Nella popolazione Caucasica una singola mutazione, 35delG,
è responsabile per la maggior parte dei casi di sordità recessive
(Gasparini et al., 2000). Questa mutazione è quella prevalente negli
individui dell’Europa Mediterranea, con la frequenza dei portatori
stimata intorno a 1/30.
È stato osservato che anche i soggetti portatori di tale mutazione
possono rappresentare un gruppo a rischio, in cui la soglia uditiva può degenerare più rapidamente che nei soggetti non-portatori.
Studi effettuati hanno mostrato infatti differenze significative nelle
emissioni oto-acustiche dei soggetti portatori e in alcune frequenze
dell’audiogramma (Engel et al., 2002; Franzè et al., 2005).
Sono state inoltre identificate delezioni e mutazioni nel gene GJB6
(gap junction beta 6) nello stesso locus di Cx26 che codifica per
la connessina 30 (Cx30), espressa insieme alla connessina 26 nell’orecchio interno (Del Castillo et al., 2005). Quindi mutazioni nel
locus complesso DFNB1, che contiene due geni importanti per l’udito (GJB2 e GJB6), possono determinare un pattern di trasmissione
monogenico o digenico. In quest’ultimo caso a mutazioni nel gene
GJB2 identificate in eterozigosi sono state trovate associate sull’altro allele mutazioni nel gene GJB6. Certe altre volte mutazioni del
gene GJB2 possono costituire dei fattori aggravanti in portatori della
mutazione mitocondriale A1555G nel gene MTRNR1 codificante per
12s rRNA (Abe et al., 2001). La correlazione genotipo-fenotipo non
è semplice, in quanto, la perdita uditiva dovuta a mutazioni nel gene
GJB2 varia molto. Il primo report descriveva pazienti con una perdita bilaterale profonda e simmetrica (Kelsell et al., 1997; Zelante
et al., 1997). In seguito in molti pazienti è stata descritta una grande variabilità fenotipica inclusa l’asimmetria, variabilità di grado e
configurazione anche tra parenti portatori della stessa mutazione. Si
sono riscontrate forme progressive anche nel caso della mutazione
35delG riportata come causativa di forme stabili (Gopalarao et al.,
2008; Snoeckx et al., 2005). In diversi studi si riporta comunque
che generalmente persone omozigoti per la 35delG hanno perdite
uditive maggiori degli eterozigoti composti 35delG/non-35delG, in
cui ad una mutazione 35delG su un allele è associata sull’altro allele
una mutazione diversa dalla 35delG e persone con due mutazioni
diverse dalla 35delG hanno perdite uditive ancora meno gravi (Cryns
et al., 2004).
La grande variabilità fenotipica associata a mutazioni identiche potrebbe essere spiegata con l’intervento di geni modificatori ma, un
lavoro recente sembra smentire questa ipotesi almeno per quanto
riguarda la 35delG (Hilgert et al., 2009)
Inoltre, mutazioni nella connessina 26 sono state recentemente riscontrate anche come responsabili di forme di neuropatie uditive,
che sono caratterizzate da un normale funzionamento delle cellu-
Forme ereditarie di ipoacusie neurosensoriali isolate e sindromiche nel bambino
le ciliate estene come evidenziato dalla presenza delle emissioni
otoacustiche evocate (EOAE) e/o da microfonico coleare (CM) e da
potenziali evocati uditivi assenti o alterati (Santarelli et al., 2008).
Ipoacusie sindromiche
La maggior parte delle forme sindromiche, sono dovute a mutazioni mendeliane, altre sono associate a sbilanciamenti cromosomici
oppure a mutazione del genoma mitocondriale; non sempre comunque la causa è necessariamente genetica, è possibile distinguere anche eziopatogenesi ambientali (sindrome rubeolica); altre
volte ancora l’eziologia risulta sconosciuta. In base a ciò è possibile distinguere le sindromi in due grandi gruppi: quelle a causa nota
e quelle a genesi sconosciuta.
Nelle ipoacusie sindromiche rientrano numerosi quadri clinici in cui
Tabella I.
Forme sindromiche causate da mutazioni nel gene GJB2.
Sindrome
Cheratite-ittiosi sordità (KID)
Mutazione
G12R
Codice OMIM
148210
Ipercheratosi palmoplantare
sordità
Vohwinkel
G59A
148350
D66H
G130V
R75Q
124500
Cheratoderma palmoplantare
sordità (PPK)
121011
l’ipoacusia è associata ai più svariati tipi di disordini. Studi statistici hanno dimostrato che il 10% circa dei geni associati a malattie
umane, attualmente conosciute, quando mutati, provocano qualche
tipo di alterazione della funzionalità uditiva. Gorlin et al. (2004) hanno passato in rassegna 402 condizioni sindromiche, suddividendole
sulla base degli organi coinvolti. In questo articolo si riportano le
sindromi più diffuse suddividendole in due gruppi , quelle imputabili
a mutazioni nel gene GJB2 e quelle non imputabili a mutazioni in
tale gene.
A. Forme sindromiche imputabili a mutazioni nella
connessina 26
Mutazioni nel gene GJB2 sono state identificate anche a carico di
forme sindromiche. Diverse mutazioni coinvolte nella perdita di udito e problemi dermatologici sono stati infatti identificati nel gene
GJB2 (Cx26). Queste forme sindromiche generalmente hanno un
modello di ereditarietà dominante e clinicamente accompagnato a
manifestazioni ampiamente varianti (Kelsell et al., 2001; Iossa et al.,
2009; Richard, 2002; Heathcote et al., 2000; Maestrini et al., 1999;
Uyguner et al., 2002).
Nella Tabella I sono riportate le forme principali:
Recentemente è stato inoltre osservato che portatori obbligati
(eterozigoti) per mutazioni recessive nel gene GJB2 presentano un
ispessimento della cute (D’Adamo et al., 2009).
Inoltre, anche mutazioni a carico di altre connessine sono state
identificate come responsabili di forme sindromiche con ipoacusia e
problemi dermatologici (Remoto-Hasebe, 2009).
Tabella II.
Forme sindromiche causate da geni diversi da GJB2.
Sindrome
Crouzon
Codice
OMIM
123500
Gene
Principali caratteristiche audiologiche
FGFR2
Ipoacusia trasmissiva in più del 50% dei casi per atresia del condotto
uditivo esterno o malformazione della catena ossiculare
Apert
101200
FGFR2
Ipoacusia trasmissiva spesso da otite media nel 50% dei casi
Pfeiffer
101600
FGFR1
Ipoacusia trasmissiva o mista in più del 50% dei casi per stenosi/atresia del condotto uditivo esterno o malformazione della catena ossiculare
Saethtre-Chotzen
101400
TWIST 1
Ipoacusia trasmissiva o mista, da anchilosi stapediale e catena
fissa, ma anche neurosensoriale profonda
Treacher Collins (TCS)
154500
TCOF1
Ipoacusia di tipo trasmissivo o misto, raramente di tipo neurosensoriale, bilaterale nel 55% dei casi. Frequente compromissione
dell’apparato vestibolare
Nager
154400
Delezione1q12q21.3; traslocazione Malformazioni minori del padiglione e appendici preauricolari;
crom. (X;9) (p22.1;q32)
ipoacusia trasmissiva solitamente modesta
Stickler
108300;604841
COL2A1;COL11A2; COL11A1
Ipoacusia di tipo misto o neurosensoriale
Branchio-oto-renale (BOR) 113650;602588
EYA1; SIX1; SIX5
Deformità del padiglione auricolare ed alterazioni dell’orecchio mee branchio-otica (BO)
dio: Ipoacusia nel 75% dei casi, prevalentemente di tipo misto
Townes-Brocks
107480
SALL1
Ipoacusia neurosensoriale monolaterale o bilaterale di grado variabile
CHARGE
214800
Trisomia 22, delezione braccio lungo del Anomalie dell’orecchio esterno associate ad un’ipoacusia neurocrom. 9, 11 e 13, duplicazione parziale sensoriale monolaterale o bilaterale di grado variabile
del crom. 4 e 14; in molti casi è dimostrabile una aploinsufficienza del gene CHD7
Waarderburg
148820
PAX3; MITF; SLUG; EDNRB; EDN3;
Ipoacusia neurosensoriale
SOX10
Usher
276900
MYO7A; USH1C; CDH23; PCDH15;
Ipoacusia neurosensoriale in alcune forme associata a problemi
SANS;USH2A;VLGR1; WHRN; WHRN vestibolari.
Pendred
274600
SLC26A4; FOXI1
Ipoacusia neurosensoriale
207
A. Franzè et al.
B. Forme sindromiche non imputabili a mutazioni sorgenza dell’ipoacusia a cause ambientali, che invece possono non
essere la causa primaria e portare quindi ad una erronea stima del
nella connessina 26
In letteratura vengono descritte circa 30 differenti sindromi ereditarie. Molte sono sindromi caratterizzate da anomalie dell’orecchio
esterno (Gorlin et al., 2004) altre invece riguardano disfunzioni
dell’orecchio interno. Le anomalie dell’orecchio esterno variano
dall’anotia al semplice orecchio ad ansa e spesso fanno parte di
più complessi quadri di malformazione cranio-facciale che possono
essere distinte in craniosinostosi, disostosi mandibolari, condrodisplasie. Nella maggioranza dei casi l’ipoacusia in queste forme sindromiche è di tipo trasmissivo. Nella Tabella II sono riportate alcune
delle sindromi più comuni.
Conclusioni e prospettive per il futuro
La valutazione dell’etiologia dell’ipoacusia fornisce possibilità per la
scoperta della causa e la valutazione del rischio di ricorrenza nella
famiglia e dà anche un preavviso su potenziali problemi aggiuntivi
di salute.
A dispetto della grande variabilità fenotipica e dei molti geni coinvolti
nelle forme di ipoacusia sia sindromiche che isolate, la frequenza
delle mutazioni nel gene GJB2 è sufficientemente alta in diverse
popolazioni, tra cui quella mediterranea, da rendere l’analisi mutazionale in questo gene e nel gene GJB6 (Cx30), associata ad una
consulenza genetica appropriata, un test veramente molto utile. Il
test è molto utile anche per evitare di attribuire erroneamente l’in-
rischio di ricorrenza della malattia in altri figli. Dato comunque l’alto
numero di geni coinvolti nelle diverse forme di ipoacusia, è da tenere presente che un risultato negativo per i test molecolari nei geni
GJB2/GJB6 non indica necessariamente che la perdita uditiva non
sia di origine genetica. È importante perciò cercare di sviluppare ed
utilizzare metodiche sempre più avanzate, quali i “microarray” che
possono analizzare quanti più geni e più mutazioni possibili. Inoltre è
da tenere presente che poiché vi sono più di 400 sindromi associate
a perdite uditive è molto importante un approccio multidisciplinare
per una valutazione complessiva del bambino.
Un aumento delle informazioni sulla correlazione genotipo-fenotipo
potrà inoltre essere di grande aiuto ai clinici per la valutazione del
migliore approccio riabilitativo.
Dati salienti emersi dagli studi considerati
Dagli studi riportati in questo articolo si evidenziano diversi punti:
1. L’importanza del test genetico in diverse forme di ipoacusia
comprese quelle che sembrano imputabili a fattori ambientali
(es. infezione da CMV)
2. Il fenotipo è altamente variabile. Le forme di ipoacusia associate alla mutazione 35delG in omozigosi non sono sempre stabili
come si pensava in precedenza.
3. Mutazioni nel gene GJB2 (Cx26) sono associabili anche a neuropatie ed a forme sindromiche con fenotipo anch’esso molto
variabile.
Box di orientamento
Novità principali riportate in questo articolo:
• Informazioni aggiornate sulle attuali conoscenze delle forme di ipoacusia neurosensoriali sia isolate che sindromiche.
• Informazioni aggiornate sulla correlazione genotipo-fenotipo sia in soggetti portatori sani che in soggetti affetti.
• Indicazione delle principali indagini molecolari utili per una diagnosi etiologica corretta.
Bibliografia
Abe S, Kelley PM, Kimberling WJ, et al. Connexin 26 gene (GJB2) mutation modulates the severity of hearing loss associated with the 1555A-->G mitochondrial
mutation. Am J Med Genet 2001;103:334-8.
Cryns K, Orzan E, Murgia A, et al. A genotype-phenotype correlation for GJB2
(connexin 26) deafness. J Med Genet 2004;41:147-54.
del Castillo FJ, Rodriguez-Ballest eros M, Alvarez A, et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations
in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing
impairment. J Med Genet 2005;42:588-94.
D’Adamo P, Guerci VI, Fabretto A, et al. Does epidermal thickening explain GJB2 high
carrier frequency and heterozygote advantage? Eur J Hum Genet 2009;17:284-6.
Di Leva F, D’Adamo P, Cubellis MV, et al. Identification of a novel mutation in the
myosin VIIA motor domain in a family with autosomal dominant hearing loss
(DFNA11). Audiol Neurootol 2006;11:157-64.
Engel-Yeger B, Zaaroura S, Zlotogora J, et al. The effects of a connexin 26 mutation 35delG on oto-acoustic emissions and brainstem evoked potentials: homozygotes and carriers. Hear Res 2002;163:93-100.
Franzè A, Caravelli A, Di Leva F, et al. Audiometric evaluation of carriers of the
connexin 26 mutation 35delG. Eur Arch Otorhinolaryngol 2005;262:921-4.
Gasparini P, Rabionet R, Barbujani G, et al. High carrier frequency of the 35delG
deafness mutation in European population. Genetic Analysis Consortium of GJB2
35delG. Eur J Hum Genet 2000;8:19-23.
208
* Questo articolo fornisce utili informazioni riguardo alla percentuele di portatori
della mutazione 35delG nella popolazione Europea.
Gopalarao D, Kimberling WJ, Jesteadt W, et al. Is hearing loss due to mutations in
the Connexin 26 gene progressive? Int J Audiol 2008;47:11-20.
** Questo articolo può essere utile per approfondimenti su previsioni per la gravità e la progressione dell’ipoacusia associata ad alcune mutazioni.
Heathcote K, Syrris P, Carter ND, Patton MA. A connexin 26 mutation causes a
syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis. J Med
Genet 2000;37:50-1.
Hilgert N, Huentelman MJ, Thorburn AQ, et al. Phenotypic variability of patients
homozygous for the GJB2 mutation 35delG cannot be explained by the influence
of one major modifier gene. Eur J Hum Genet 2009;17:517-24.
Iossa S, Chinetti V, Auletta G, et al. New evidence for the correlation of the
p.G130V mutation in the GJB2 gene and syndromic hearing loss with palmoplantar keratoderma. Am J Med Genet A 2009;149:685-8.
* Questo articolo mette in risalto l’alta variabilità fenotipica associata alle mutazioni nel gene GJB2 anche in caso di forme sindromiche.
Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary nonsyndromic sensorineural deafness. Nature 1997;387:80-3.
Kelsell DP, Di WL, Houseman MJ. Connexin mutations in skin disease and hearing loss. Am J Hum Genet 2001;68:559-68. Review.
* Questa review fornisce indicazioni sulle forme sindromiche associate alla connessina 26.
Forme ereditarie di ipoacusie neurosensoriali isolate e sindromiche nel bambino
Kochhar A, Hildebrand MS, Smith RJ. Clinical aspects of hereditary hearing loss.
Genet Med 2007;9:393-408.
Liu XZ, Walsh J, Tamagawa Y, et al. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet
1997;17:268-9.
Maestrini E, Korge BP, Ocaña-Sierra J, et al. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum Mol Genet
1999;8:1237-43.
Martini A, Milani M, Rosignoli M, et al. Audiometric patterns of genetic non-syndromal sensorineural hearing loss. Audiology 1997;36:228-36.
Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci
1991;630:16-31.
Nemoto-Hasebe I, Akiyama M, Kudo S, et al. Novel mutation p.Gly59Arg in GJB6
encoding connexin 30 underlies palmoplantar keratoderma with pseudoainhum,
knuckle pads and hearing loss. Br J Dermatol 2009;161:452-5.
Rehm HL. Genetics and the Genome Project. Ear & Hearing 2003;24:270-274.
Richard G. Human connexin disorders of the skin. Cell Commun Adhes
2001;8:401-7.
Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness
syndrome. Am J Hum Genet 2002;70:1341-8.
Ross SA, Novak Z, Kumbla RA, et al. GJB2 and GJB6 mutations in children with
congenital cytomegalovirus infection. Pediatr Res 2007;61:687-91.
* Questo articolo mette in risalto l’utilità di effettuare il test genetico anche a
forme di ipoacusia apparentemente imputabili a fattori ambientali.
Santarelli R, Cama E, Scimemi P, et al. Audiological and electrocochleography
findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. Eur Arch Otorhinolaryngol 2008;265:43-51.
* Questo articolo mette in risalto l’utilità di effettuare il test genetico per il gene
GJB2 anche in caso di neuropatia.
Snoeckx RL, Huygen PL, Feldmann D, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 2005;77:945-57.
Uyguner O, Tukel T, Baykal C, et al. The novel R75Q mutation in the GJB2 gene
causes autosomal dominant hearing loss and palmoplantar keratoderma in a
Turkish family. Clin Genet 2002;62:306-9.
Weil D, Küssel P, Blanchard S, et al. The autosomal recessive isolated deafness,
DFNB2 and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene.
Nature Genet 1997;16:191-3.
Yoshinaga-Itano C. Benefits of early intervention for children with hearing loss.
Otolaryngol Clin North Am 1999;32:1089-102.
Zelante L, Gasparini P, Estivill X, et al. Connexin26 mutations associated with the
most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 1997;6:1605-9.
Corrispondenza
prof. Elio Marciano, Unità di Audiologia, Università di Napoli “Federico II”, via Pansini 5, 80131 Napoli. E-mail: [email protected]
209
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 210-213
neurologia
Consenso sugli “Standard di cura”:
un grosso passo avanti nel campo
delle malattie neuromuscolari
Eugenio Mercuri, Flaviana Bianco, Gessica Vasco
Servizio di Neuropsichiatria Infantile, Università Cattolica del Sacro Cuore, Roma
Riassunto
I notevoli progressi in campo genetico hanno migliorato notevolmente la conoscenza delle singole malattie neuromuscolari e delle complicanze a loro
legate.
Nell’ultimo decennio numerosi studi hanno dimostrato una modifica sostanziale della storia naturale di queste malattie con un notevole aumento della sopravvivenza ed una riduzione delle complicanze. Sebbene in questo campo gli studi basati sulla evidence based medicine siano, per vari motivi, scarsamente rappresentati, sono state fornite delle Linee Guida basate sul consenso di esperti nei diversi aspetti di cura legati alle varie complicanze. La traduzione e
diffusione di queste Linee Guida, sollecitate e promosse dalle associazioni di famiglie, hanno permesso un costante miglioramento della presa in carico di
questi bambini anche al di fuori dei centri altamente specializzati.
Un altro elemento importante proveniente dalle Linee Guida basate sul consenso è che, alla luce delle interessanti prospettive terapeutiche in questo campo, l’uniformazione degli standard di cura è fondamentale per disegnare ed attuare trial clinici multicentrici.
Summary
The dramatic advances in the understanding of the genetic background of neuromuscular disorders have greatly improved our knowledge about the individual diseases and their sequelae.
In the last decade many studies have shown a dramatic change of the natural history of these disorders with a substantial increase of survival rate and
a reduction of sequelae. Although there are only few studies based on evidence based medicine in this disease group, guidelines, based on a consensus
reached among the leading experts in specific fields, have become.
The translation and dissemination of these guidelines, promoted by parent groups, has brought a considerable improvement in the follow up of these
patients that has spread outside tertiary care centres.
Another important goal is that, in view of the forthcoming exciting new therapeutical prospective, an agreement on standards of care is essential when
planning and performing multicentric clinical trials.
Introduzione
Nuovi profili di storia naturale in distrofia muscolare
I notevoli progressi ottenuti nel campo della genetica delle malattie di Duchenne e atrofia muscolare spinale
neuromuscolari pediatriche hanno portato ad un miglioramento della loro classificazione e della conoscenza delle complicanze legate
alle singole forme, consentendo quindi anche una migliore pianificazione degli approcci di trattamento e di prevenzione.
Un chiaro esempio di questo processo viene dai pazienti affetti dalle
forme di distrofie muscolari congenite legate a mutazioni nel gene
SEPN1 (Ferreiro et al., 2002; Allamand et al., 2006) o ai geni del
collagene VI (Lampe, 2005). La possibilità di definire il gruppo di
pazienti sulla base delle mutazioni in questi gruppi ha permesso di
riconoscere l’alto rischio di sviluppare ipossie e ipercapnie notturne già alla fine della prima decade. Il migliore monitoraggio delle
attività respiratorie diurne e notturne in questi pazienti ha prodotto una drastica riduzione della loro mortalità in età pediatrica. Allo
stesso modo, il riconoscimento di mutazioni nel gene della lamina
AC (LMNA) nelle forme di distrofia muscolare Emery Dreifuss a trasmissione dominante ha consentito di identificare l’alto rischio di
aritmie e blocchi sopraventricolari in cui, a differenza di altri casi con
patologie neuromuscolari simili ma legate ad altri geni, l’inserimento
di un semplice pacemaker non è sufficiente e si richiede un defibrillatore per evitare morti improvvise (Bonne et al., 2000).
210
La possibilità di effettuare diagnosi genetiche precoci e la migliore conoscenza dell’evoluzione delle complicanze legate alle
singole forme, ha portato notevoli modifiche nel mangement delle forme di malattie neuromuscolari più conosciute e più comuni
in epoca pediatrica, ovvero la distrofia muscolare di Duchenne
(DMD) e l’atrofia muscolare spinale (SMA). Gli studi retrospettivi
e le raccolte prospettiche di dati longitudinali hanno chiaramente
dimostrato come nell’ultimo decennio ci sia stata una modifica
della storia naturale di queste malattie con un notevole aumento
della sopravvivenza ed una riduzione e minore progressione delle complicanze.
Una analisi retrospettiva della sopravvivenza dei pazienti affetti da
DMD nelle ultime decadi ha dimostrato chiaramente come vi sia stato un graduale e costante aumento dell’età di sopravvivenza. Mentre
negli anni ’60 la percentuale dei ragazzi affetti da DMD che arrivava
all’età di 25 anni era pari a poco più dello 0%, ai nostri giorni oltre il
53% di questi ragazzi supera i 30 anni con alcuni casi che arrivano
oltre i 40. L’aumento di sopravvivenza è stato parallelo all’introduzione ed alla diffusione di metodiche di ventilazione non invasiva
(Eagle et al., 2002 e 2007).
Negli ultimissimi anni si sono verificati anche notevoli miglioramenti,
Consenso sugli ‘Standard di cura’: un grosso passo avanti nel campo delle malattie neuromuscolari
sia nell’aspettativa di vita che nella gestione dei pazienti, anche per la
forma più grave di SMA, classificata come tipo I o malattia di Werdnig
Hoffmann. Fino a pochissimi anni fa una famiglia al momento della
diagnosi si trovava di fronte alla scelta di intervenire con modalità invasive, quali tracheotomia e ventilazione meccanica, o aspettare che
la malattia facesse il suo corso senza ricorrere a metodiche invasive,
con una sopravvivenza media di 8 mesi (Oskoui et al., 2007) e che
tranne rare eccezioni, non superava i due anni di vita. La graduale introduzione, in alcuni centri, di metodiche di ventilazione non invasiva
e di un atteggiamento più attento nei confronti dei problemi della deglutizione (con inserimento precoce di gastrostomie) ha fatto sì non
solo che la maggior parte dei bambini trattati con queste metodiche
superi i due anni di vita, ma ha anche migliorato notevolmente la
qualità di vita dei bambini e delle loro famiglie (Oskoui et al., 2007).
L’introduzione di queste metodiche, hanno avuto il vantaggio di aumentare il numero delle famiglie che sceglie un intervento attivo,
perché recepito come meno invasivo, più maneggevole nella routine
e meno traumatico rispetto alla tracheotomia.
Simili risultati si sono ottenuti anche nell’ambito delle forme intermedie di atrofia muscolare SMA, detta anche di tipo II, in cui il decorso della malattia, ritenuto chiaramente progressivo fino a pochi anni
fa, segue adesso un corso molto più lento con una riduzione drastica
dei decessi per malattie respiratorie e delle infezioni gravi. La migliore gestione delle complicanze respiratorie, con l’inserimento di
tecniche quali l’insufflator/exufflator (Bach, 1993) e di tecniche di
riabilitazione respiratoria, in associazione a tentativi terapeutici quali
quello con il Salbutamolo (Pane et al., 2008), hanno notevolmente
diminuito la frequenza e la gravità delle complicanze respiratorie
acute (Wang et al., 2007).
Nuovi standard di cura ed evidence based medicine
Se da un lato il miglioramento della sopravvivenza e della progressione della malattia appaiono indubbie, dall’altra appare difficile
stabilire in che misura le modifiche del management e della presa
in carico siano individualmente responsabili delle migliori condizioni
di questi pazienti.
A differenza di tante altre patologie, in cui sono presenti numerosi
studi basati sulla evidence based medicine, nel campo delle malattie
neuromuscolari tali studi sono scarsamente rappresentati.
A parte alcuni lavori su aspetti individuali di cura quali il trattamento
profilattico con ACE inibitori sul cuore in giovani pazienti affetti da
DMD (Duboc et al., 2007, Mercuri et al., 2008) o alcuni studi su
interventi ortopedici (Manzur et al., 1992), o sull’efficacia di alcune
metodiche ventilatorie ottenuti in piccole serie comprendenti pazienti affetti da patologie neuromuscolari diverse (Simonds, 2007),
non esistono grossi studi sistematici randomizzati sui diversi aspetti
di cura.
I motivi per tale differenza sono molteplici:
• queste malattie sono malattie rare in cui è difficile ottenere dei
numeri sufficientemente grandi per poter effettuare degli studi
randomizzati in doppio cieco in un unico centro o in un consorzio di pochi centri. Per questo i pochi trial clinici, in atto o in
programmazione, sono tutti studi multicentrici internazionali che
coinvolgono un alto numero di centri.
• In assenza di un accordo tra i vari centri sugli standard di cura,
appare difficile effettuare uno studio multicentrico per valutare
l’effetto di un intervento su uno degli aspetti cruciali della malattia in quanto entrano in gioco troppe variabili che rendono
difficile, se non impossibile, interpretare i risultati e stabilire con
esattezza se le eventuali modifiche dell’outcome siano da attribuirsi al fattore studiato piuttosto che ad altri fattori confondenti
quali diversi metodi di fisioterapia o di profilassi ecc.
• Un ulteriore problema è rappresentato dal fatto che alcune di
queste metodiche sono entrate nella pratica comune di alcuni centri anche se non precedute da studi di evidence based
medicine. Molti centri infatti avendo un’esperienza diretta di
quanto queste metodiche abbiano modificato lo stato di salute
e di qualità di vita, non trovano etico dover procedere a studi
randomizzati in cui è necessario sottoporre a placebo una parte
della popolazione studiata.
Nonostante queste limitazioni negli ultimi anni si è comunque avvertita l’esigenza di formalizzare queste esperienze acquisite sul
campo dai pochi esperti che avevano avuto l’ occasione di lavorare
in grossi centri di riferimento e di cercare un consenso, in modo da
fornire delle raccomandazioni sulle metodiche da usare e favorirne la diffusione anche in centri minori in cui il numero di pazienti
seguiti non era sufficientemente alto da poter acquisire lo stesso
livello di esperienza. Questo lavoro è stato condotto in Europa prima
dall’ENMC (European Neuro Muscular Consortium) e negli ultimi due
anni da TREAT NMD (Translational Research in Europe for the Assessment and Treatment of Neuromuscular Disease) due grosse istituzioni internazionali che hanno promosso una serie di workshops
coinvolgendo tutti i maggiori esperti in aspetti specifici delle malattie, producendo alla fine di ogni workshop, delle raccomnadazioni,
pubblicate su riviste inetrnazionali.
Questo sforzo europeo, è stato negli ultimissimi anni associato a
quello di altre istituzioni inetrnazionali, quali l’ICC (International
Coordinating Committee) o il Muscular Distrophy group che hanno
consentito di aumentare la partecipazione dei colleghi statunitensi
e di cercare un consenso ancora più largo. Seppure le modalità di
compilazione di queste raccomandazioni non seguano rigorosamente i principi dell’evidence based medicine o i criteri necessari per il
riconoscimento dell’efficacia di studi clinici dettati dalle review Cochrane, queste raccomandazioni rappresentano comunque il risultato di un consenso da parte degli esperti mondiali e la loro diffusione
sta portando a due risultati, ugualmente importanti per il futuro di
questi bambini.
Il primo è che la traduzione e diffusione di queste raccomandazioni, fortemente sollecitate e promosse dalle associazioni di famiglie,
stanno portando ad un lento ma costante miglioramento della presa
in carico di questi bambini anche al di fuori dei centri altamente specialistici con conseguente riduzione delle complicanze e della mortalità legate ad una scarsa conoscenza delle complicanze di queste
malattie. In molti centri vengono ormai forniti degli opuscoli con la
traduzione del consenso degli standard di cura o anche dei link sui
siti delle associazioni delle famiglie a cui le famiglie o medici con
poca esperienza su queste patologie, possano facilmente accedere. L’introduzione di tecniche di fisioterapia respiratoria, l’uso ormai
routinario di apparecchi in grado di ridurre il ristagno di secrezioni
quali l’insufflator-exufflator, o il trattamento precoce con antibiotici,
assieme ad un più attento monitoraggio di spirometria e dell’ossimetria notturna ha provocato una drastica riduzione della mortalità
in epoca pediatrica e del numero di ricoveri per infezioni respiratorie
gravi, non solo nelle atrofie muscolari spinali e nei ragazzi con DMD
ma anche nelle distrofie muscolari congenite e nelle miopatie congenite. In parte questo miglioramento è probabilmente anche dovuto
ad un più attento monitoraggio di altri fattori, quali deglutizione, alimentazione o possibili complicanze cardiache, che hanno comunque
contribuito al miglioramento dello stato di salute generale. Seppure
211
E. Mercuri et al.
sia difficile stabilire con esattezza quale sia il contributo di ciascun
elemento, appare indubbio che la combinazione di questi fattori e
un approccio multidisciplinare che coinvolge più specialisti hanno
portato ad un miglioramento globale di questi bambini.
L’altro elemento, ugualmente importante, è che il miglioramento dello stato di salute e l’uniformazione degli standard di cura è un passo
fondamentale nel quadro più generale degli studi clinici terapeutici.
Nonostante non vi sia ancora una cura per nessuna malattia neuromuscolare su base ereditaria, negli ultimi anni vi sono stati numerosi trial clinici, molti dei quali ancora in corso, e vi sono numerose
prospettive terapeutiche che, per la prima volta, fanno intravedere la
possibilità di curare alcune di queste malattie. I progressi più grossi
si sono avuti nel campo della DMD dove, dopo anni di lavoro in laboratorio per stabilire efficacia e safety nei modelli animali, si stanno
finalmente effettuando i primi studi clinici fase 2/3 utilizzando diverse metodiche, per valutare l’efficacia e la safety di queste metodiche
su bambini e ragazzi. Oltre al lavoro sulle cellule staminali, che ha
prodotto risultati sorprendenti nel cane distrofico (Tonlorenzi et al.,
2007) che è in fase di preparazione nell’uomo, vi sono studi avviati
in cui l’impiego di nuove tecniche, come l’exon skipping, è stato
recentemente usato in bambini DMD in due studi parallei in Olanda
e nel Regno Unito. Entrambi gli studi, effettuati adoperando lo steso
principio ma molecole diverse, hanno chiaramente dimostrato come
iniezioni locali di oligonuceleotidi antisenso possano provocare una
ricomparsa della distrofina in pazienti distrofici che, a causa della
malattia hanno un assenza totale della proteina (Aartsma-Rus et al.,
2007 e 2009; Kinali et al., 2009). In seguito a questa fase iniziale
che ha dimostrato sia l efficacia locale che la mancanza di reazioni
di rigetto di entrambe le molecole, sono in corso degli studi pilota
per valutare la safety di entrambe le molecole per uso sistemico
(sottocute o i.v.) e, nei prossimi mesi, verranno lanciati due grossi
studi multicentrici ed internazionali, randomizzati e a doppio cieco
per valutare l’efficacia delle molecole.
Sono in corso altri approcci che sfruttano meccansimi simili, ma
specializzati per alcuni tipi specifici di mutazioni come le mutazioni
nonsense. La PTC Therapeutics sta portando avanti uno studio clinico di fase 2/3 dove si cerca di confermare l’efficacia del nuovo
composto di origine post-trascrizionale, progettato per ignorare il
segnale che genera l’interruzione prematura dei meccanismi di lettura del DNA creando in questo modo un percorso alternativo che
conduce ad una nuova produzione di distrofina e, quindi, ad un probabile beneficio terapeutico (Wilton, 2007).
Alla luce della velocità con cui queste proposte terapeutiche stanno
diventando disponibili, e in previsione di studi in fase 3 su base multicentrica, appare quindi ancora più importante eliminare tutti i fattori
confondenti, che potrebbero derivare da diversi standard di cura utilizzati nei diversi centri. Nello stesso tempo, proprio in previsione di
terapie che potrebbero diventare disponibili nei prossimi anni, le famiglie diventano ancora più esigenti e attente ai livelli di cura quotidiana
in modo da contrastare quanto possibile l’evoluzione della malattia ed
essere nelle condizioni migliori al momento di una possibile cura.
In Appendice viene riportata una lista di link e voci bibliografiche
utili per accedere ai siti di queste organizzazioni ed alle Linee Guida
disponibili.
Box di orientamento
• Nell’ultimo decennio numerosi studi hanno dimostrato una modifica sostanziale della storia naturale delle malattie neuromuscolari pediatriche con
un notevole aumento della sopravvivenza ed una riduzione delle complicanze.
• Sebbene gli studi basati sulla evidence based medicine siano scarsamente rappresentati, sono state fornite delle Linee Guida basate sul consenso
di esperti nei diversi aspetti di cura legati alle varie complicanze.
• La traduzione e diffusione di queste Linee Guida appare essenziale per promuovere una migliore presa in carico di questi bambini anche al di fuori
dei centri altamente specializzati.
• Alla luce delle interessanti prospettive terapeutiche in questo campo, l’uniformazione degli standard di cura appare ancora più importante per
disegnare ed attuare trial clinici multicentrici.
Bibliografia
Aartsma-Rus A, Janson AA, van Ommen GJ, et al. Antisense-induced exon
skipping for duplications in Duchenne muscular dystrophy. BMC Med Genet
2007;8:43.
Aartsma-Rus A, van Ommen GJ. Less is more: therapeutic exon skipping for
Duchenne muscular dystrophy. Lancet Neurol 2009;8:873-5.
* Gruppo olandese che illustra i dati dello studio sperimentale in corso.
Allamand V, Richard P, Lescure A, et al. A single homozygous point mutation in
a 3’untranslated region motif of selenoprotein N mRNA causes SEPN1-related
myopathy. EMBO Rep 2006;7:450-4.
Bach JR. Mechanical insufflation-exsufflation: comparison of peak expiratory flows with manually assisted and unassisted coughing techniques. Chest
1993;104:1553-62.
* Questo è il primo autore/articolo che ha sperimentato l’effetto benefico della
ventilazione non invasiva in queste patologie.
Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of
autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of
the lamin A/C gene. Ann Neurol 2000;48:170-80.
212
Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in
Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J 2007;154:596-602.
* Follow-up a 10 anni sull’uso dell’ace-inibitore come prevenzione cardiologica.
Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy-the additive effect of spinal surgery and home nocturnal ventilation in improving
survival. Neuromuscul Disord 2007;17:470-5.
Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home
nocturnal ventilation. Neuromuscul Disord 2002;12:926-9.
Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N
gene, which is implicated in rigid spine muscular dystrophy, cause the classical
phenotype of multiminicore disease: reassessing the nosology of early-onset
myopathies. Am J Hum Genet 2002;71:739-49.
Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study.
Lancet Neurol 2009;8:918-28.
* Studio che ha valutato la sicurezza e la tollerabilità dell’iniezione intramuscolo
di AVI-4658.
Consenso sugli ‘Standard di cura’: un grosso passo avanti nel campo delle malattie neuromuscolari
Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet
2005;42:673-85.
Manzur AY, Hyde SA, Rodillo E, et al. A randomized controlled trial of early surgery
in Duchenne muscular dystrophy. Neuromuscul Disord 1992;2:379-87.
Mercuri E, Mayhew A, Muntoni F, et al. Towards harmonisation of outcome measures for DMD and SMA within TREAT-NMD; report of three expert workshops:
TREAT-NMD/ENMC workshop on outcome measures, 12th--13th May 2007,
Naarden, The Netherlands; TREAT-NMD workshop on outcome measures in experimental trials for DMD, 30th June--1st July 2007, Naarden, The Netherlands;
conjoint Institute of Myology TREAT-NMD meeting on physical activity monitoring
in neuromuscular disorders, 11th July 2007, Paris, France. TREAT-NMD Neuromuscular Network. Neuromuscul Disord 2008;18:894-903.
* Lavoro di armonizzazione delle outcomes majores nelle malattie neuromuscolari.
Oskoui M, Levy G, Garland CJ, et al. The changing natural history of spinal muscular atrophy type 1. Neurology 2007;69:1931-6.
Pane M, Staccioli S, Messina S, et al. Daily salbutamol in young patients with
SMA type II. Neuromuscul Disord 2008;18:536-40.
* Trial pilota che mostra effetti positivi sulla forza nei pazienti SMA II.
Peltz SW, Welch EM, Jacobson A, et al. Nonsense suppression activity of PTC124
(ataluren). Proc Natl Acad Sci U S A 2009;106:E64.
* Studio che mostra i primi dati del farmaco sperimentale PTC 124.
Simonds AK. Respiratory support for the severely handicapped child with
neuromuscular disease: ethics and practicality. Semin Respir Crit Care Med
2007;28:342-54.
* Importanza della ventilazione non invasiva sulla sopravvivenza dei pazienti affetti da distrofia muscolare di Duchenne.
Tonlorenzi R, Dellavalle A, Schnapp E, et al. Isolation and characterization of
mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell
Biol 2007;Chapter 2:Unit 2B.1
* Descrizione dell’isolamento dei mesangiobalsti.
Wang CH, Finkel RS, Bertini ES, et al.; Participants of the International Conference on SMA Standard of Care Consensus statement for standard of care in
spinal muscular atrophy. J Child Neurol 2007;22:1027-49.
Wilton S. PTC124, nonsense mutations and Duchenne muscular dystrophy. Neuromuscul Disord 2007;17:719-20.
* Primi approcci dell’utilizzo della nuova molecola nella distrofia di Duchenne.
Appendice
Link
Sito Parent project
http://www.parentproject.org/italia/index.php?option=com_content
&task=view&id=311&Itemid=84
Sito Famiglie SMA
http://www.famigliesma.org/index.php?lang=1&view=000000000
008&colore=blu&callermenu=2#
Sito TREAT NMD
http://www.treat-nmd.eu/patients/patient-care/care-standards/
http://www.treat-nmd.eu/userfiles/file/general/TREAT-NMD_DMD_
interim_recommendations.pdf
http://www.treat-nmd.eu/patients/patient-care/sma/
Corrispondenza
prof. Eugenio Mercuri, Neuropsichiatria Infantile, Policlinico Gemelli, largo Gemelli, 00168 Roma. E-mail: [email protected]
213
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 214-218
neurologia
Nuovi standard di cura per le complicanze
respiratorie e cardiologiche nel bambino
con distrofia muscolare di Duchenne
Angela Berardinelli*, Marika Pane**, Eugenio Mercuri**
Dipartimento di Clinica Neurologica e Psichiatria dell’età evolutiva, Fondazione “C. Mondino” I.R.C.C.S., Pavia;
Servizio di Neuropsichiatria Infantile Università Cattolica del Sacro Cuore, Roma
*
**
Riassunto
La distrofia muscolare tipo Duchenne con una incidenza di circa 1: 3500 maschi nati vivi è la più comune malattia neuromuscolare infantile. È dovuta all’assenza completa della proteina distrofina. L’evoluzione clinica della malattia prevede il progressivo aggravarsi del deficit di forza muscolare, con crescenti
difficoltà nella deambulazione, nei passaggi posturali, fino alla perdita della deambulazione autonoma in genere prima dei 13 anni seguito dallo sviluppo
della scoliosi. Il coinvolgimento della muscolatura respiratoria e cardiaca determina l’insorgenza del deficit respiratorio e il coinvolgimento cardiaco. Nonostante non esista ancora una terapia risolutiva per la distrofia muscolare di Duchenne, si può comunque affermare che negli ultimi 20 anni le condizioni
cliniche, la sopravvivenza e la qualità della vita dei ragazzi affetti da distrofia muscolare di Duchenne sono notevolmente cambiate, soprattutto in relazione
al miglioramento della gestione delle complicanze cardiorespiratorie ed ortopediche. In questo articolo riportiamo alcune linee guida sugli aspetti respiratori
e cardiologici di questi pazienti.
Summary
Duchenne muscular dystrophy has an incidence of 1 in 3500 male live births and is the most common childhood muscular dystrophy. The course of Duchenne
muscular dystrophy is characterized by the progression of muscle weakness and contractures leading to loss of ambulation before 13 years, generally followed
by onset of progressive scoliosis. The involvement of heart and respiratory muscles are responsible for progressive cardiac and respiratory impairment. Although
there is no effective cure for patients with Duchenne muscular dystrophy, there has been a progressive increase in survival over the last few decades that
has been correlated to a better management of cardiac and respiratory complications. In this article we report respiratory and cardiologic guidelines on
some aspects of the management of these patients
Definizione, epidemiologia e inquadramento diagnostico
Gestione clinica del paziente in cui è stata posta
La distrofia muscolare tipo Duchenne (DMD) è una malattia X-linked, diagnosi di distrofia muscolare di Duchenne
con una incidenza di circa 1:3500 maschi nati vivi. È dovuta all’assenza completa della proteina Distrofina, una proteina che stabilizza
la membrana sarcolemmatica.
Il motivo che più frequentemente conduce alla consultazione del
neuropsichiatra infantile o del pediatra è il riscontro da parte dei
genitori di difficoltà motorie nel piccolo riconducibili ad un deficit
di forza prevalente a carico della muscolatura del cingolo pelvico:
andatura “anserina”, evidente soprattutto quando il bambino tenta
di correre, difficoltà nel rialzarsi da terra, il piccolo, da supino, passa
prono, poi gattoni, poi solleva il bacino e poi si spinge con una mano
sul ginocchio (manovra di arrampicamento o Gowers) a quell’età ancora incompleta, difficoltà nel salire le scale, impossibilità a saltare.
Una parte delle consultazioni viene richiesta a causa del riscontro
occasionale di marcata iperckemia.
La diagnosi si basa essenzialmente sulla biopsia muscolare, che
mostra un quadro istopatologico di tipo distrofico e, alla determinazione immunoistochimica che mostra l’assenza completa della
proteina distrofina. L’iter diagnostico verrà competato dall’analisi
molecolare del gene.
L’evoluzione clinica della malattia prevede l’aggravarsi continuo
del deficit di forza muscolare, fino alla perdita dell’autonomia nel
cammino, prima dei 13 anni e un progressivo coinvolgimento della
muscolatura respiratoria e del miocardio.
214
Al momento, nonostante gli straordinari progressi nella conoscenza della malattia e i molteplici tentativi sperimentali non esiste una
terapia risolutiva per la DMD. L’introduzione della terapia steroidea
(unico presidio che abbia dimostrato fino ad ora una certa efficacia) ha prolungato in media la durata della deambulazione di circa
1 anno se somministrata con regimi non continui (giorni alternati,
10 giorni sì/10 no) o anche di diversi anni se somministrata, laddove non insorgano effetti collaterali importanti, quotidianamente.
Si può comunque affermare che negli ultimi 20 anni le condizioni
cliniche, la sopravvivenza e la qualità della vita dei ragazzi affetti
da DMD si sono notevolmente modificati, soprattutto in relazione al
miglioramento della gestione delle complicanze cardiorespiratorie.
L’aspettativa di vita è, attualmente, in media superiore ai 30 anni
(Eagle et al., 2006).
Come nel caso delle SMA (atrofia muscolare spinale) è sempre necessario prospettare ai genitori l’importanza di un approccio multidisciplinare inetgrato e fornire le seguenti informazioni: le problematiche ortopediche e riabilitative sono abbastanza simili a quelle
discusse nell’articolo precedente sulle atrofie muscolari spinali, seppure la storia naturale della malattia sia chiaramente diversa per
la natura più progressiva della malattia di Duchenne. Maggiore
importanza rivestono invece le complicanze cardiorespiratorie che
meritano un approfondimento specifico.
Nuovi standard di cura per le complicanze respiratorie e cardiologiche nel bambino con distrofia muscolare di Duchenne
Follow-up e trattamento delle complicanze
cardiologiche e polmonari
Complicanze cardiache
I primi segni di coinvolgimento cardiaco nella DMD sono generalmente costituiti dall’insorgenza di una cardiomiopatia dilatativa. In
uno studio condotto su 328 pazienti affetti da DMD il coinvolgimento
cardiaco è divenuto sintomatico mediamente intorno o dopo i 10
anni di età, mentre in precedenza sia i rilievi ecocardiografici sia la
clinica erano pressoché normali (Nigro et al., 1990). I primi segni di
interessamento miocardico sono rappresentati da anomalie elettrocardiografiche minori che possono essere individuate all’esordio del
quadro clinico, ma restano asintomatiche fino ai 10 anni di età; la
più frequente è la tachicardia sinusale (Bhattacharyya et al., 1997).
Altre anomalie elettrocardiografiche riscontrate sono battiti atriali
premature, che diventano più frequenti con l’età, oltre a tachicardia
atriale ectopica e fibrillazione/flutter atriale. Nel 40% dei soggetti vi
è inoltre un intervallo PQ più corto della norma (Nigro et al., 1995),
onda P allungata e PQ aumentato. Le aritmie ventricolari sono infrequenti negli stadi precoci ma la loro incidenza aumenta con la
progressione della malattia (Chenard et al., 1993). L’evoluzione è
verso una cardiomiopatia con dilatazione delle camere cardiache e
depressione della frazione di eiezione del ventricolo sinistro dovuta
ad una diffusa fibrosi tissutale che interessa anche il tessuto di conduzione cardiaco (Duboc et al., 2007).
Dal punto di vista ecocardiografico, ispessimenti parietali sono presenti in un terzo dei pazienti entro i 14 anni, nella metà entro i 18 anni
e sono costantemente presenti dopo i 18 anni (Manzur et al., 2008).
La cardiopatia dilatativa coinvolge primitivamente il ventricolo sinistro. Il ventricolo destro può essere coinvolto secondariamente ai
problemi respiratori e alla conseguente ipertensione polmonare.
Una situazione di scompenso cardiaco è presente in circa il 35% dei
pazienti DMD in fase terminale (Corrado et al., 2002).
Monitoraggio clinico-strumentale
La scarsa o nulla sintomatologia clinica riferibile alle problematiche
cardiache da un lato e la rilevanza di queste dall’altro impongono
che i soggetti affetti da DMD siano sottoposti a sorveglianza clinicostrumentale costante.
La letteratura scientifica (Bushby et al., 2003) suggerisce di effettuare una valutazione completa alla diagnosi e poi una ogni due
anni fino ai 10 anni e poi una all’anno o più spesso se sono state
individuate anomalie.
I pazienti vanno inoltre valutati in modo completo prima di qualsiasi
anestesia generale, a qualsiasi età.
Gli strumenti abituali del monitoraggio cardiaco comprendono, oltre naturalmente all’esame obiettivo, l’elettrocardiogramma (ECG),
l’ECG Holter e l’ecocardiogramma.
Nei pazienti in fase più avanzata o comunque con gravi malformazioni della gabbia toracica l’esecuzione dell’ecocardio può risultare
particolarmente difficile. Recentemente sono state proposte altre
metodiche di valutazione, quali studi elettrofisiologici endocavitari
(invasivi) per evidenziare più precocemente il rischio di disturbi del
ritmo e l’ecocardiografia Doppler tissutale (non invasiva) ma si tratta
di metodiche attualmente ancora solo usate a scopo di ricerca. La
valutazione cardiologia deve sempre associarsi a quella respiratoria
per le possibili reciproche influenze. Le anomalie del ritmo cardiaco
dovrebbero essere ricercate e trattate rapidamente. Un periodico
monitoraggio con ECG Holter va inserito nel follow-up dei pazienti
con disfunzione cardiaca.
Negli ultimi anni sta acquisendo rilievo l’utilizzo della risonanza
magnetica cardiaca, che sembrerebbe più efficace nell’individuare
eventuali anomalie e che è in ogni caso indicata nei soggetti con
cattiva finestra ecocardiografica.
Terapia
Seppure non vi sia univocità sulle modalità di trattamento farmacologico della cardiomiopatia dilatativa nella DMD, né su quando sia
opportuno iniziarlo, negli ultimi anni alcuni studi hanno fortemente
suggerito l’importanza di un trattamento precoce.
Gia da diversi anni, le conclusioni di alcuni workshop di esperti nel
campo suggerivano, sulla base della loro esperienza personale l’impiego di ACE inibitori e beta-bloccanti, associate a diuretici all’esordio dello scompenso cardiaco (Bushby et al., 2003).
Queste indicazioni sono state fortemente rafforzate da studi più recenti che indicano chiaramente come un precoce trattamento con
l’ACE inibitore Perindopril avrebbe una influenza positiva rallentando
l’insorgenza della cardiomiopatia dilatativa e aumentando la durata
della vita. Secondo Duboc, l’autore del lavoro, per ottenere questi
risultati è tuttavia fondamentale che il Perindopril sia introdotto tra i
9,5 e i 13 ani di età, con una frazione di eiezione superiore al 45%.
Dopo il primo lavoro con un follow up relativamente breve, pubblicato nel 2005 (Duboc, 2005) l’autore ha recentemente pubblicato la
prosecuzione del follow-up a 10 anni, confermando i primi risultati
(Duboc, 2007) in cui il 92,9% dei pazienti trattati fin dall’inizio con
Perindopril erano ancora vivi dopo 10 anni di trattamento, rispetto al
65,5% del gruppo di soggetti inzialmente trattati con placebo.
Sulla base di questi risultati, replicati anche in altri centri, è quindi
ormai accettato che, in assenza di una terapia risolutiva, sembra
opportuno intervenire con il Perindopril, che peraltro ha scarsi effetti
collaterali, prima che il danno miocardico diventi importante.
Se dai monitoraggi con ECG Holter emergono aritmie ventricolari maggiori, andrebbe introdotto un trattamento antiaritmico, considerando
tuttavia il possibile effetto inotropo negativo del farmaco scelto.
Follow-up e trattamento delle complicanze polmonari
Le problematiche respiratorie fondamentali nella DMD sono rappresentate dall’insorgenza di una progressiva sindrome disventilatoria
restrittiva, frutto dell’ingravescente perdita di forza delle muscolatura respiratoria, associato alle deformità toraciche che insorgono per
il deficit di forza muscolare generalizzato e dell’immobilità
I segni più importanti correlati a questo difetto di forza ingravescente sono rappresentati dalla progressiva comparsa di disturbi respiratori sonno-relati con ritenzione di CO2, atelettasie e la difficoltà ad
espettorare.
Da uno studio condotto recentemente su 58 pazienti affetti da DMD
(Phillips et al., 2001) è emerso che anomali risultati alle prove di
funzionalità respiratoria nei bambini affetti da DMD sono individuabili a partire dagli 8 anni di età, ma la perdita dei volume respiratori
è particolarmente evidente solo dal momento in cui i bambini perdono la deambulazione autonoma (Gozal, 2006); successivamente,
in adolescenza, si osserva l’insorgenza di episodi di ipossiemia durante il sonno (Khan et al., 1994) l’insufficienza respiratoria diviene
conclamata entro i 18-20 anni di età.
I primi segni di disturbi respiratori sonno-relati sono l’ipopnea e le
desaturazioni in sonno REM, che nelle successive fasi di evoluzione
sono presenti anche in sonno non REM.
La capacità vitale (CV) e la forza della muscolatura respiratoria declinano nei soggetti affetti da DMD in modo prevedibile e abbastanza omogeneo, tuttavia l’età alla quale i singoli pazienti raggiungono
215
A. Berardinelli et al.
una certa CV, per es. < 1 L ( valore considerato critico) può essere
diversa da soggetto a soggetto (Hukins et al., 2000). Eventi acuti
quali infezioni respiratorie anche banali possono diventare particolarmente problematiche, soprattutto per la difficoltà ad espettorare
(Manzur et al., 2002).
Sintomi e monitoraggio dei pazienti
La maggior parte dei pazienti e dei loro familiari non sono consapevoli della loro ridotta funzionalità respiratoria e non segnalano
spontaneamente sintomi suggestivi di tale problema fino a quando una banale infezione respiratoria delle vie aeree non determina una evidente difficoltà nella espettorazione, con conseguente
evoluzione in un focolaio polmonare di lunga e difficile risoluzione. In merito alla frequenza delle valutazioni nelle varie fasi della
malattia, le Linee Guida dell’American Thoracic Society pubblicate
nel 2004 raccomandano: almeno una visita/anno dallo pneumologo pediatra nelle prime fasi della malattia, in bambini tra i 4 e i 6
anni e prima della perdita della deambulazione autonoma, al fine
di ottenere una valutazione-base della funzionalità polmonare per
meglio indirizzare il successivo management del paziente. Dopo
la perdita della deambulazione, di fronte al rilevamento di una
CV inferiore all’80% del predetto e/o al raggiungimento dei 12
ani di età i pazienti dovrebbero essere sottoposti ad almeno due
valutazioni specialistiche pneumologiche/anno. Ogni valutazione
dovrebbe comprendere: esame obiettivo, determinazione dalla
saturazione ossiemoglobinica con elettrodo a dito, esame della
funzionalità respiratoria con metodo spirometrico, (Phillips et al.,
2001) e, quando possibile, la determinazione del picco di tosse
(Bach et al., 1997). Ad ogni visita è importante associare una attenta valutazione anamnestica dei disturbi respiratori sonno-relati (Finder et al., 2004).
È fondamentale, ad ogni incontro con i pazienti, ricercare una storia di infezioni respiratorie frequenti, inappetenza, sonno interrotto
da risvegli frequenti, incubi, difficoltà al risveglio mattutino, cefalea, inappetenza e nausea mattutine. Anche in presenza di valori
FVC (capacità vitale forzata accettabili, la presenza di uno o più
di questi segni rappresenta una indicazione all esecuzione di una
ossimetria notturna.
trattenendo il fiato (Kang et al., 2000), o l’applicazione di pressione
positiva con un Ambu e maschera a pressione positiva intermittente
o un ventilatore meccanico.
Le tecniche meccaniche di tosse assistita: l’apparecchio in ed exsufflator (Fig. 1) simula un colpo di tosse attraverso un respiro con
pressione positiva seguito da una pressione negativa (Segal et al.,
1994). Questa tecnica ha dimostrato di generare un flusso espiratorio del picco di tosse più elevato sia di quello prodotto dalle tecniche
manuali sia con le ispirazioni forzate (Bach, 1993).
L’in ed exsufflator è risultato particolarmente importante nel prevenire le ospedalizzazioni o la necessità di tracheostomia nei soggetti
DMD con picco di flusso espiratorio della tosse intorno a 160 l/min,
specialmente quando la gravità della curvatura scoliotica rende difficile e non ottimale l’uso delle tecniche manuali (Bach et al., 1997).
Nei pazienti tracheostomizzati l’in ed exsufflator offre diversi vantaggi rispetto alla aspirazione tradizionale, inclusa la clearance delle
secrezioni dalle vie aeree periferiche, evitando così il trauma della
suzione diretta alla mucosa tracheale ed è meglio tollerato dal paziente (Garstang et al., 2000).
Strumenti per la mobilizzazione del muco: la ventilazione percussiva intrapolmonare dà scariche di oscillazioni di alta frequenza,
bassa ampiezza che si sovrappongono ad una continuo flusso di
pressione positiva nelle vie aeree. Un recente lavoro condotto su
un gruppo di pazienti, uno solo dei quali affetto da DMD ha riportato l’efficacia di questa tecnica nella risoluzione di persistenti
secrezioni resistenti alle tecniche convenzionali (Birnkrant et al.,
1996).
L’uso degli steroidi ormai comune nella DMD, oltre ai risultati positivi
sulla funzionalità motoria, sembra determinare anche una miglioramento della FVC e della capacità di tossire misurata mediante il
PCF stando ai risultati di uno studio condotto su 35 pazienti, 10 dei
quali in steroide e 25 non trattati di età compresa tra i 7 e i 21 anni
(Daftary et al., 2007) ed a un altro studio condotto su 79 pazienti di
età compresa tra i 10 e i 18 anni di età, 40 dei quali trattati 34 no
(Biggar et al., 2006).
Terapie
Il trattamento delle problematiche respiratorie nei DMD prevede la
prevenzione degli episodi acuti, l’addestramento del paziente e della
famiglia a riconoscere i primi segni di difficoltà respiratoria, e – nella
fasi più avanzate della malattia – il supporto ventilatorio meccanico,
dapprima notturno e poi diurno. Alcune semplici accortezze possono
aiutare a diminuire i rischio di episodi acuti:
• per prevenire le complicanze legate ai possibili episodi infettivi a
carico delle vie aeree tutti i soggetti affetti da DMD dovrebbero
essere sottoposti annualmente a vaccino antiinfluenzale e antipneumococcico ( Finder et al., 2004);
• pur non essendoci dati certi relativi sulla possibile efficacia della
fisioterapia e del training respiratorio nella DMD nella prognosi
a lungo termine, queste metodiche aiutano nella vita quotidiana
i pazienti a liberarsi delle secrezioni ed a diminuire il rischio di
infezioni legate al loro ristagno (Eagle et al., 2006).
Le tecniche manuali di tosse assistita comprendono le inspirazioni
assistite seguite dall’incremento della espirazione forzata. Un aumento della capacità inspiratoria può essere ottenuto attraverso
l’uso del respiro glossofaringeo (forzare l’aria nei polmoni usando la
bocca), o facendo una serie di respiri quanto più profondi possibile
216
Figura 1.
Macchina della tosse.
Nuovi standard di cura per le complicanze respiratorie e cardiologiche nel bambino con distrofia muscolare di Duchenne
Insufficienza respiratoria clinica e ventilazione
Nei soggetti affetti da DMD l’insufficienza respiratoria cronica (IRC)
evolve attraverso 4 fasi: stadio 1, caratterizzato da disturbi respiratori sonno-relati senza ipercapnia; stadio 2, caratterizzato da disturbi respiratori sonno-relati durante le fasi di sonno REM; stadio 3:
disturbi respiratori sonno-relati con ipercapnia durante il sonno REM
e non-REM; stadio 4: ipercapnia diurna (Ragette et al., 2002). Negli
stadi 2 e 3 la ventilazione meccanica corregge l’ipercapnia, migliora
la sopravvivenza e la qualità della vita (Toussaint et al., 2007).
Quando si parla di ventilazione si intende ventilazione non invasiva a
pressione positiva (NIPPV), erogata per lo più attraverso apparecchi
a pressione positiva, con maschera nasale.
Complessivamente, è stato dimostrato che, nei pazienti DMD, l’uso
della ventilazione meccanica allunga l’aspettativa di vita di almeno
5-10 anni (Simonds et al., 1998).
Introdotta negli anni ’80 da Rideau et al. la maschera nasale resta
tuttora la prima scelta come interfaccia per la ventilazione notturna
(Ward et al., 2005). Sebbene negli anni recenti stia diventando via
via meno utilizzata, per i pazienti che richiedano la ventilazione 24
ore/die, la Tracheostomia rappresenta un mezzo più efficace e più
sicuro della NIPPV (Baydur et al., 2000).
Problematiche anestesiologiche nella distrofia
muscolare di Duchenne
L’aumentata durata della vita dei pazienti DMD fa sì che sia più probabile che necessitino di interventi chirurgici, sia per la correzione della
curvature scoliotica, sia per altre possibili cause. L’insieme del deficit
respiratorio connesso all’evoluzione della malattia, della suscettibilità a reazioni avverse ai farmaci anestetici (che possono ricordare
l’ipertermia maligna, pur costituendo due condizioni differenti), delle
problematiche di dismotilità gastro-intestinale conseguenti alla malattia di base, della macroglossia, del possibile blocco dell’articolazione
temporo-mandibolare che spesso si instaura nelle fasi più avanzate della malattia, delle problematiche cardiache, fanno sì che questi
ragazzi presentino un rischio connesso alle procedure chirurgiche e
anestesiologiche superiore alla media. Per questa ragione recentemente l’ACCP (American College of Chest Physicians) ha prodotto un
documento di Consensus sulle procedure anestesiologiche e chirurgiche al quale si rimanda per i dettagli (Birnkrant et al., 2007).
Problemi psico-sociali
Un altro aspetto importante legato all’assistenza ma più generalmente a vissuto di malattia è rappresentato dall’impatto che la malattia, l’assistenza e le varie terapie ha sulla integrazione psicosociale di questi ragazzi. I ragazzi affetti da DMD spesso hanno difficoltà
nell’elaborazione della loro immagine, momenti di ansia, di fragilità
emotiva nell’accentuarsi delle loro difficoltà e di angoscia e difficoltà
ad accettare nuove modalità terapeutiche e ad utilizzare nuovi ausili
per la loro autonomia. Il malato neuromuscolare potrà avere bisogno
di un aiuto nell’elaborazione del lutto per un corpo che non corrisponde a quanto immaginato e la cui funzionalità viene continuamente messa in discussione. Una presa in carico globale e multidisciplinare dal punto di vista psichico non può non riferirsi e prestare
adeguata attenzione anche ai familiari o alle persone coinvolte sia
da dinamiche affettive che sociali al malato neuromuscolare.
Una più ampia revisione può essere trovata in una recente pubblicazione basata su un consenso internazionale che tratta in maniera dettagliata molti aspetti di standard di cura nei pazienti affetti da DMD.
Box di orientamento
• Nell’ultimo decennio numerosi studi hanno dimostrato una modifica nella storia naturale delle malattie neuromuscolari pediatriche con un notevole
aumento della sopravvivenza e della qualità della vita dei ragazzi affetti da DMD, soprattutto in relazione al miglioramento della gestione delle
complicanze cardiorespiratorie ed ortopediche.
• La rilevanza delle problematiche cardiache impone che i soggetti affetti da DMD siano sottoposti a sorveglianza clinico-strumentale costante con
possibilità di intervento precoce prima della comparsa di una sintomatologia grave.
• Il trattamento delle problematiche respiratorie nei DMD prevede la prevenzione degli episodi acuti, l’addestramento del paziente e della famiglia a
riconoscere i primi segni di difficoltà respiratoria, e – nella fasi più avanzate della malattia – il supporto ventilatorio meccanico, dapprima notturno
e poi diurno.
Bibliografia
Bach JR, O’Brien J, Krotenberg R, et al. Management of end stage respiratory
failure in Duchenne muscular dystrophy. Muscle Nerve 1987;10:177-82.
** Questo è il primo autore/articolo che ha sperimentato l’effetto benefico della
ventilazione non invasiva in queste patologie.
Bach JR. Mechanical insufflation-exsufflation: comparison of peak expiratory flows with manually assisted and unassisted coughing techniques. Chest
1993;104:1553-62.
Bach JR, Ishikawa Y, Kim H. Prevention of pulmonary morbidity for patients with
Duchenne muscular dystrophy. Chest 1997;112:1024-8.
* Esprime l’importanza della prevenzione dei problemi respiratori nei pazienti
affetti da DMD.
Baydur A, Layne E, Aral H, et al. Long term non-invasive ventilation in the community for patients with musculoskeletal disorders: 46 year experience and review. Thorax 2000;55:4-11.
Bhattacharyya KB, Basu N, Ray TN, et al. Profile of electrocardiographic changes
in Duchenne muscular dystrophy. J Indian Med Assoc 1997;95:40-2.
Birnkrant DJ, Pope JF, Lewarski J, et al. Persistent pulmonary consolidation
treated with intrapulmonary percussive ventilation: a preliminary report. Pediatr
Pulmonol 1996;21:246-9.
Birnkrant D, Panitch H, Benditt J, et al. American College of Chest Physicians
Consensus Statement on the Respiratory and Related Management of Patients
With Duchenne Muscular Dystrophy Undergoing Anesthesia or Sedation. Chest
2007;132:1977-86.
Biggar WD, Harris VA, Eliasoph L, et al. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscular Disorders 2006;16:249-55.
** Questo articolo ha modificato l’approccio dei clinici alla malattia, aumentato
il tempo di deambulazione dei pazienti mediante l’effetto benefico dei cortisone
somministrato tutti i giorni.
Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th–9th June 2002, Naarden, the Netherlands. Neuromuscul Disord
2003;13:166-72.
** Linee guida sui problemi cardiologici delle malattia neuromuscolari.
217
A. Berardinelli et al.
Bushby K, Finkel R, Birnkrant DJ, et al.; for the DMD Care Considerations
Working Group. Diagnosis and management of Duchenne muscular dystrophy : implementation of multidisciplinary care. Lancet Neurol 2010;9:17789.
** Articolo generale basato su un consenso di un alrgo gruppo di esperti.
Chenard AA, Becane HM, Tertrain F, et al. Ventricular arrhythmia in Duchenne
muscular dystrophy: prevalence, significance and prognosis. Neuromuscul Disord 1993;3:201-6.
Corrado G, Lissoni A, Beretta S, et al. Prognostic value of electrocardiograms,
ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. Am J Cardiol
2002;89:838-41.
Daftary AS, Crisanti M, Kalra M, et al. Effect of long-term steroids on cough efficiency and respiratory muscle strength in patients with Duchenne muscular
dystrophy. Pediatrics 2000;119:e320-4.
Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and
progression of left ventricular dysfunction in Duchenne muscular dystrophy. J
Am Coll Cardiol 2005;45:855-7.
** Esprime l’importanza della prevenzione farmacologica nella progressione del
coinvolgimento cardiaco nella DMD.
Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in
Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J 2007;154:596-602.
** Follow-up a 10 anni sull’uso dell’ace-inibitore come prevenzione.
Eagle M, Chatwin M. Respiratory muscle training in children and adults with
neuromuscular disease (Protocol). Cochrane Database of Systematic Reviews
2006, Issue 3. Art. No.: CD006155.
** Linee guida sugli aspetti respiratori nelle malattie neuromuscolari.
Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home
nocturnal ventilation. Neuromuscul Disord 2002;12:926-9.
** Primi dati di storia naturale di ventilazione nella distrofia di Duchenne.
Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy-the additive effect of spinal surgery and home nocturnal ventilation in improving
survival. Neuromuscul Disord 2007 ; 17:470-5.
** Importanza della ventilazione notturna e della chirurgia della scoliosi nel management dei pazienti affetti da DMD.
Finder JD, Birnkrant D, Carl J, et al. American Thoracic Society Respiratory care
of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am
J Respir Crit Care Med 2004;170:456-65.
Garstang SV, Kirshblum SC, Wood KE. Patient preference for in-exsufflation for secretion management with spinal cord injury. J Spinal Cord Med
2000;23:80-5.
Gozal D. Pulmonary manifestations of neuromuscular disease with special ref-
erence to Duchenne muscular dystrophy and spinal muscular atrophy. Pediatr
Pulmonol 2000;29:141-50.
Hukins CA, Hillman DR. Daytime predictors of sleep hypoventilation in Duchenne
muscular Dystrophy. Am J Respir Crit Care Med 2000;161:166-70.
Kang SW, Bach JR. Maximum insufflation capacity. Chest 2000;118:61-5.
Khan Y, Heckmatt JZ. Obstructive apnoeas in Duchenne muscular dystrophy.
Thorax 1994;49:157-61.
Manzur AY, Muntoni F, Simonds A. Muscular dystrophy campaign sponsored
workshop: recommendation for respiratory care of children with spinal muscular atrophy type II and III. 13th February 2002, London, UK. Neuromusc Disord
2003;13:184-9.
** Linee guida sulla gestione delle problematiche respiratorie nei pazienti con
atrofia muscolare spinale II e III.
Manzur AY, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child 2008;93:986-90.
* Linee guida sul management sulla Distrofia muscolare di Duchenne.
Nigro G, Comi LI, Politano L, et al. The incidence and evolution of cardiomyopathy
in Duchenne muscular dystrophy. Int J Cardiol 1990;26:271-7.
* Studio molto ampio che documenta la presenza della cardiomiopatia dilatativa
già intorno a 10 anni.
Nigro G, Comi LI, Politano L, et al. Evaluation of the cardiomyopathy in Becker
muscular dystrophy. Muscle Nerve 1995;18:283-91.
Philipps M, Quinlivan R , Edwards R et al. Changes in spirometry over time as a
prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir
Crit Care Med 2001;164:2191-4.
Ragette R, Mellies U, Schwake C, et al. Patterns and predictors of sleep disordered breathing in primary myopathies. Thorax 2002;57:724-8.
Segal M, Salomon A, Herschfus J. Alternating positive-negative pressures in
mechanical respiration (the cycling valve device employing air pressures). Dis
Chest 1954;25:640-8.
Simonds AK, Muntoni F, Heather S, et al. Impact of nasal ventilation on survival in
hypercapnic Duchenne muscular dystrophy. Thorax 1998;53:949-52.
* Importanza della ventilazione non invasiva sulla sopravvivenza dei pazienti
affetti da Distrofia muscolare di Duchenne.
Toussaint M, Chatwin M, Soudon P. Mechanical ventilation in Duchenne patients
with chronic respiratory insufficiency: clinical implications of 20 years published
experience. Chron Resp Dis 2007;4:167-77.
Sottolinea l’importanza della ventilazione meccanica nei DMD.
Ward S, Chatwin M, Heather S, et al. Randomised controlled trial of non-invasive
ventilation (NIV) for nocturnal hypoventilation in neuromuscular and chest wall
disease patients with daytime normocapnia. Thorax 2005;60:1019-24.
** Importanza della ventilazione non invasiva sulla sopravvivenza dei pazienti
con malattie muscolari: studio randomizzato.
Link
Sito Parent Project:
http://www.parentproject.org/italia/index.php
Sito American Thoracic Society
http://www.thoracic.org/
Corrispondenza
prof. Eugenio Mercuri, Neuropsichiatria Infantile, Policlinico Gemelli, largo A. Gemelli 8, 00168 Roma. E-mail: [email protected]
218
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 219-227
neurologia
Nuovi standard di cura per la presa in carico
del bambino con atrofia muscolare spinale
Adele D’Amico, Enrico Bertini
Unità di Medicina Molecolare per Malattie Neuromuscolari e Neurodegenerative, Ospedale Bambino Gesù, Roma
Riassunto
L’atrofia muscolare spinale è una malattia neurodegenerativa geneticamente determinata che necessita di un approccio multidisciplinare per la molteplicità
dei problemi medici che si devono affrontare. Con la necessità di iniziare i trial clinici è sorta l’esigenza di generare linee standardizzate di presa in carico dei
pazienti affetti da atrofia muscolare spinale non solo per curarli nel modo migliore, ma anche per ridurre i fattori di variabilità confondenti che interferiscono
con la valutazione dei trial clinici.
In questa revisione andremo a descrivere le aree di presa in carico dei pazienti affetti da atrofia muscolare spinale che hanno riscosso il massimo consenso
tra gli esperti internazionali coinvolti in una Consensus Conference pubblicata nel 2007: l’iter diagnostico, la gestione delle complicanze acute e croniche della
insufficienza respiratoria e le problematiche nutrizionali. Riassumeremo anche le aree di minor consenso riguardanti l’intervento riabilitativo ed ortopedico.
Summary
Spinal muscular atrophy is a neurodegenerative and genetic determined disease that requires multidisciplinary medical care. Efforts for building up clinical
trials in spinal muscular atrophy are recent, however standardization of care for patients is a preliminary important issue not only to deliver the best practice
for patients but also to reduce confounding factors when conducting clinical trials. A first consensus guideline for the care of patients with spinal muscular
atrophy has been generated with international effort in 2007.
In this review we will cover the topics that received the best consensus for standard of care in patients with spinal muscular atrophy among the participating
international experts to a Consensus conference published in 2007: the diagnosis, the respiratory and nutritional management and we will also summarize
the issues that received a lesser amount of agreement between experts, rehabilitation and bone surgery.
Introduzione
L’atrofia muscolare spinale (SMA) è una malattia neurodegenerativa
geneticamente determinata che necessita di un approccio multidisciplinare per la molteplicità dei problemi medici che si devono
affrontare. Con la necessità di iniziare i trials clinici è stata consequenziale l’idea di condividere linee standardizzate di presa in carico
dei pazienti affetti da SMA non solo per seguire i pazienti nel modo
migliore ma anche per ridurre i fattori confondenti di variabilità che
si devono considerare nei trials clinici (Crawford, 2004; Hirtz et al.,
2005; Bertini et al., 2005). Un primo documento è stato generato nel
2007 frutto di un impegno internazionale di esperti clinici (Wang et
al., 2007). Questo studio preliminare ha permesso di evidenziare le
aree di scarso consenso come quella della gestione ortopedico chirurgica e fisiatrica della SMA fornendo indicazioni per ricerche future. In questa revisione andremo a riassumere le indicazioni emerse
dalla Consensus Conference publicate nel 2007 (Wang et al., 2007)
e descriveremo con maggiore dettaglio le aree di presa in carico che
hanno riscosso il massimo consenso tra gli esperti internazionali
interpellati: l’iter diagnostico, la gestione delle complicanze acute e
croniche della insufficienza respiratoria e le problematiche nutrizionali dei pazienti SMA. Si rimanda al testo originario della Consensus
Conference per maggiori dettagli (Wang et al., 2007).
La diagnosi
Quando un pediatra si confronta con un bambino che presenta una
ipotonia o debolezza muscolare dovrebbe sempre sospettare una
SMA, una delle malattie genetiche autosomico recessive più frequenti con una prevalenza stimata intorno a 1 su 10.000 nati vivi
ed una frequenza dei portatori di 1/40-1/60 (Prior, 2008). Alcune caratteristiche cliniche posso aiutare nella formulazione della diagnosi. La debolezza muscolare è solitamente simmetrica e coinvolge
prevalentemente i distretti prossimali piuttosto che la muscolatura
distale. Gli arti inferiori sono in genere maggiormente interessati rispetto agli arti superiori. I riflessi osteotendinei (ROT) sono assenti o
diffusamente deboli e le sensibilità sono conservate. La partecipazione ambientale è sempre buona e lo sguardo di questi bambini è
sempre vigile e attento.
L’entità e la gravità della debolezza muscolare in genere correlano
con l’età di esordio.
La Tabella I mostra la classificazione delle diverse forme di SMA
e le loro principali caratteristiche cliniche come concordata dalla Consensus Statement for Standard of Care in Spinal Muscular
Atrophy pubblicata nel 2007 (Wang et al., 2007) che in gran parte
è ripresa da precedenti classificazioni (Munsat et al., 1992; Dubowitz et al., 1999).
Alcuni lavori riportano anche una forma di SMA tipo IV, che si distingue per un esordio in età adulta, dopo i 18 anni di età, e per
un’evoluzione più lieve.
Tale classificazione, tuttavia, è solo indicativa e nella pratica clinica
alcuni pazienti possono nel corso della evoluzione presentare livelli
di severità funzionale crescente, arrivando a perdere abilità funzionali precedentemente acquisite (la posizione seduta o la capacità di
camminare).
Il trattamento ed il follow-up dei pazienti affetti da SMA dovrebbe
essere pianificato tenendo conto dello stato funzionale dei pazienti
al momento della visita, piuttosto che sulla base della severità clinica all’esordio.
219
A. D’Amico, E. Bertini
Tabella I.
Classificazione clinica delle atrofie muscolari spinali.
Tipo di SMA
Età di esordio
Miglior prestazione motoria
acquisibile
Non raggiunge la posizione
seduta autonoma
Età media al
decesso
< 2 anni
Tipo I (severo)
sindrome di
Werdnig-Hoffman
0-6 mesi
Tipo II (intermedio)
7-18 mesi
Non raggiunge la posizione eretta e la deambulazione autonoma
> 2 anni
Tipo III (lieve)
Malattia di
Kugelberg-Welander
> 18 mesi
Acquisisce la deambulazione
autonoma
Età adulta
Iter diagnostico
La Figura 1 mostra l’algoritmo diagnostico in caso di sospetta SMA.
Il primo test diagnostico che dovrebbe essere eseguito è l’analisi
genetica per la ricerca di delezioni nel gene SMN. L’identificazione di
una delezione in omozigosi nell’esone 7 del gene SMN1 (associato
a meno delezioni nell’esone 8) conferma la diagnosi di SMA (SMA
associata a SMN, 5q-SMA) (Zerres et al., 1997).
Gli altri test diagnostici mostrati nella Figura 1 dovrebbero essere eseguiti solo nei pazienti in cui la ricerca di delezioni nel gene
SMN 1 sia risultata negativa.
SMA con quadro
clinico tipico
Non delezione
omozigote SMN 1
-
Neuropatia
demielinizzante o
assonale
-
Sindrome
miastenica
-
Miopatia
- Distrofia muscolare
Test genetico
SMN 1
Marcata ipostenia e ipotonia muscolare; incompleto
controllo del capo; pianto e colpo di tosse deboli; difficoltà alla suzione e deglutizione; morbidità precoce a
causa di infezioni respiratorie ricorrenti, polmoniti ab
ingestis e insufficienza respiratoria
Ritardo nell’acquisizione delle tappe motorie; scarsa
crescita ponderale; colpo di tosse debole; fine tremore distale agli arti superiori; sviluppo di retrazioni
tendinee e scoliosi
Ipostenia muscolare di grado variabile; crampi
muscolari; perdita della deambulazione autonoma in
età adulta
Gestione clinica generale del paziente in cui è stata
posta diagnosi di atrofia muscolare spinale
Data la complessità delle problematiche mediche associate alla
SMA la presa in carico di un paziente deve essere ad opera di un
medico, pediatra o neurologo esperto in problemi neuromuscolari in
grado di affrontare con il paziente e i familiari le diverse implicazioni
conseguenti alla diagnosi.
Durante il primo incontro è importante offrire ai familiari informazioni su:
Delezione omozigote
SMN 1
Conferma diagnosi SMA 5q
95% sensibilità
100% specificità
Debolezza prossimale > distale,
CK normale, EMG neurogeno
Ripetere esame clinico,
EMG, VCN, CK
Segni inusuali di
SMA con EMG
neurogena,
normale CK
Debolezza diffusa,
normale EMG eVCN,
CK, RMN midollo ed
enceali
Altre malattie del motoneurone:
SMARD, X-SMA, ALS giovanile
Biopsia muscolare o
nervo, test specifici
per miopatie o
distrofie o neuropatie
ereditarie
Figura 1.
Iter diagnostico per le atrofie muscolari spinali.
220
Caratteristiche cliniche
Caratterizzare copie di SMN 1
2 copie di
SMN1
1 copia di SMN1
Sequenziare SMN 1 per mutazioni
puntiformi
Non mutazione SMN 1
Diagnosi di SMA con difetto SMN
1 non confermato
Mutazione identificata
Diagnosi di SMA 5q
confermata
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
• aspetti generali della malattia e del suo decorso (classificazione
clinica e prognosi delle diverse forme di SMA);
• possibili approcci terapeutici (Informazioni riguardo trial clinici in
corso o in programmazione);
• contatti con le associazioni delle famiglie e dei pazienti affetti da
SMA (siti internet e/o recapiti telefonici per poter trovare informazioni relative alla malattia).
Il medico curante dovrebbe inoltre pianificare, insieme ai familiari,
un follow-up multidisciplinare che coinvolga i seguenti specialisti:
• Neurologo;
• Pneumologo;
• Gastroenterologo e Nutrizionista;
• Ortopedico e Fisiatra;
• Genetista.
Il ruolo del genetista medico è importante per garantire ai familiari
una corretta informazione sulle basi genetiche della malattia. In particolare i familiari dovrebbero ricevere le seguenti informazioni:
• tipo di ereditarietà (autosomico recessiva) e rischio di ricorrenza
nella famiglia;
• alcune nozioni sulla struttura del gene SMN, differenza tra SMN1
e SMN2 e significato del numero di copie SMN2;
• limiti del valore prognostico del numero di copie del gene SMN2:
sebbene un elevato numero di copie del gene SMN2 correlari
con un fenotipo clinico più lieve c’è comunque un’ampia variabilità clinica e non è raccomandato utilizzare tale dato come indice
prognostico;
• importanza e limiti del test per lo stato di portatore sano;
• informazioni riguardo alla diagnosi prenatale e alla pianificazione di future gravidanze (diagnosi prenatale e pre-impianto).
Follow-up e trattamento delle complicanze polmonari
Complicanze respiratorie nei pazienti affetti da SMA
Le complicanze respiratorie sono la principale causa di morbilità e
mortalità nelle SMA di tipo I e II, e possono presentarsi in una minoranza dei casi di SMA III. I principali problemi respiratori nei pazienti
affetti da SMA si possono così riassumere:
1. colpo di tosse debole con conseguente difficoltà alla espettorazione ed eliminazione delle secrezioni delle basse vie respiratorie;
2. immaturità polmonare;
3. ipoventilazione notturna;
4. infezioni ricorrenti.
Una disfunzione dell’apparato orofaringeo, con perdita del riflesso
orofaringeo e conseguente difficoltà di deglutizione, gioca un ruolo
importante nella determinazione delle complicanze respiratorie. Un
supporto per la tosse dovrebbe essere fornito se il colpo di tosse
risulta ipovalido ed insufficiente nell’eliminazione delle secrezioni.
L’interessamento respiratorio ed i sintomi ad esso correlati tendono
a progredire nel tempo. Inizialmente si possono manifestare infezioni
ricorrenti ed episodi di desaturazione notturna, fino a determinare un
quadro di persistente ipoventilazione notturna con ipercapnia diurna.
Quando compaiono i primi disturbi di ipoventilazione durante il sonno
dovrebbe essere garantito un supporto ventilatorio notturno.
La pervietà delle vie aeree è fondamentale nella gestione della insufficienza respiratoria acuta e cronica nei pazienti affetti da SMA.
Monitoraggio e follow-up della funzione respiratoria
I pazienti affetti da SMA dovrebbero effettuare una valutazione clinica ogni 3-6 mesi, a discrezione del medico referente sulla base della
severità clinica delle problematiche da affrontare.
A. Pazienti che non sono in grado di mantenere la posizione seduta
autonoma (SMA I)
• Raccolta anamnestica riguardo la frequenza di infezioni
polmonari e delle vie respiratorie e i trattamenti antibiotici
ricevuti nei 6 mesi precedenti.
• Esame clinico per valutare il colore della cute, possibili deformazioni/malformazioni della gabbia toracica, l’efficacia
del colpo di tosse e le caratteristiche del respiro, (frequenza
respiratoria presenza o meno di respiro paradosso).
• Saturimetria notturna per monitorare il livello di saturazione
di ossigeno nel sangue mediante sensore transcutaneo.
• Polisonnografia con misura della CO2 al fine di identificare
segni di ipoventilazione durante il sonno.
• Rx torace di base e durante gli episodi di insufficienza respiratoria acuta.
• Studio della deglutizione: nei casi di deterioramento acuto
della funzione respiratoria senza causa apparente, o in caso
di infezioni polmonari ricorrenti.
B. Pazienti che sono in grado di mantenere la posizione seduta in
autonomia, ma non hanno acquisito la stazione eretta e la deambulazione autonoma (SMA II)
• Valutazione clinica e strumentale come riportato sopra per i
pazienti affetti da SMA tipo I.
• Valutazione clinica e radiologica delle deformità della colonna
vertebrale per monitorare un’eventuale comparsa di scoliosi.
C. Pazienti deambulanti (SMA III):
Generalmente questi pazienti sviluppano problemi respiratori
solo nelle fasi più avanzate della malattia per cui i controlli clinici e strumentali raccomandati sopra per le forme di SMA più
severe potranno essere effettuati con una frequenza annuale.
In questi pazienti inoltre è possibile eseguire anche prove di funzionalità respiratoria per la valutazione dei volumi polmonari e la
forza dei muscoli respiratori.
Prevenzione delle complicanze respiratorie
Per una corretta gestione delle problematiche respiratorie nei pazienti
affetti da SMA è fondamentale istruire le famiglie sulla gestione delle complicanze respiratorie acute e croniche. I familiari dovrebbero
avere tutte le informazioni necessarie riguardanti le diverse possibilità
di trattamento a lungo termine (ventilazione non invasiva, macchina
per l’assistenza della tosse e delle secrezioni) oltre che conoscere le
complicanze associate ad eventuali interventi chirurgici.
Per i bambini affetti da SMA tipo I, considerata la rapida progressione della malattia, è importante affrontare precocemente con i familiari le possibilità di intervento, in particolare la possibilità di iniziare
una ventilazione non invasiva e l’assistenza per la gestione delle
secrezioni. È opportuno discutere con i familiari le loro aspettative.
La gestione del paziente dovrebbe infatti consistere in un compromesso tra le aspettative della famiglia e gli obiettivi raggiungibili.
Gestione delle problematiche quotidiane dei pazienti affetti da
SMA tipo I
La gestione quotidiana dei pazienti affetti da SMA I dovrebbe prevedere:
• una particolare attenzione ad ogni cambiamento clinico del
bambino rispetto alla sua condizione di partenza;
• la conoscenza dei sintomi o segni che possano suggerire uno stato
di ipoventilazione, al fine di permetterne il precoce riconoscimento;
• la gestione delle complicanze acute con particolare attenzione
alla possibilità di avere un rapido accesso a strutture mediche
specializzate;
221
A. D’Amico, E. Bertini
• la conoscenza delle tecniche per la gestione delle secrezioni e il
mantenimento della pervietà delle vie aeree;
• il supporto respiratorio (compresa la ventilazione non-invasiva).
• un’adeguata nutrizione e idratazione;
• un precoce trattamento antibiotico in caso di infezione delle vie
aeree;
• le vaccinazioni correntemente in uso, inclusi i vaccini per l’influenza stagionale e lo pneumococco ed un trattamento immunostimolante come profilassi (palivizumab).
Gestione a lungo termine dei problemi respiratori
È fondamentale discutere con i familiari quali sono i loro obiettivi
principali e valutarli di volta in volta in relazione alle condizioni del
bambino e al progredire della malattia.
Gli obiettivi a lungo termine sono: garantire una buona ossigenazione e normalizzare gli scambi gassosi, migliorare la qualità del sonno,
agevolare la gestione domiciliare del paziente, ridurre gli accessi
alle unità di terapia intensiva e le ospedalizzazioni e limitare il più
possibile l’impatto della malattia sulla vita quotidiana.
Interventi precoci possono prolungare l’aspettativa di vita senza
compromettere la qualità della vita stessa.
Vie aeree
• L’assistenza alla tosse, manuale o utilizzando una macchina tipo
in-exsufflator, è raccomandata quotidianamente nella maggior
parte dei pazienti che presentano un quadro clinico severo. I familiari e coloro che si prendono cura del paziente dovrebbero
essere istruiti su come eseguire tali tecniche.
• La fisioterapia respiratoria e il drenaggio posturale per favorire
l’espettorazione e l’eliminazione delle secrezioni.
• L’aspirazione delle secrezioni nelle alte vie respiratorie.
• La saturimetria notturna per monitorare la ventilazione diurna e
notturna.
Supporto ventilatorio
• Un supporto ventilatorio è indicato in tutti i casi in cui viene evidenziata una ipercapnia. La ventilazione notturna non invasiva
riduce i sintomi da ipoventilazione, garantisce un buon riposo
notturno e migliora la qualità della vita.
• L’utilizzo di un ventilatore a due livelli a pressione positiva (BiPAP), rappresenta il miglior supporto ventilatorio non invasivo
per questi bambini e il suo utilizzo, anche per brevi periodi durante la giornata, può stimolare lo sviluppo polmonare e ridurre
le deformità sternali e della gabbia toracica.
• La ventilazione meccanica a pressione positiva continua (CPAP)
può essere utilizzata in una fase precoce e di transizione, ma
l’obiettivo deve essere la sua sostituzione con un ventilatore tipo
BiPAP.
• Tracheostomia: nei pazienti che non acquisiscono la posizione
seduta in autonomia, l’esecuzione della tracheostomia è un
argomento tutt’ora controverso oltre che un dilemma etico. In
questi bambini, che presentano una rapida evoluzione della malattia, bisogna considerare la possibilità di attuare diversi livelli
di assistenza che vanno dalle cure palliative senza un supporto
ventilatorio fino alla tracheotomia, con respirazione meccanica
assistita. La ventilazione non invasiva può essere considerata
come intervento terapeutico o come cura palliativa. Se la famiglia decide per un supporto ventilatorio questo tipo di assistenza
può rappresentare un compromesso ottimale avendo l’obiettivo
principale di limitare la permanenza nelle unità di terapia intensiva pediatrica ed evitare la tracheotomia, ove possibile.
222
Problematiche da considerare in caso di interventi
chirurgici
Gli interventi chirurgici sui pazienti affetti da SMA presentano un alto
rischio di complicazioni postoperatorie e post-anestesia generale,
quali infezioni nosocomiali, intubazione prolungata, necessità di tracheotomia e decesso.
Prima di ogni intervento chirurgico è quindi fondamentale ottimizzare e stabilizzare le funzioni respiratorie.
Un’accurata valutazione pre-operatoria deve quindi prevedere:
• esame clinico generale;
• misurazioni ripetute della funzione respiratoria e valutazione
dell’efficacia del colpo di tosse;
• radiografia del torace;
• valutazione dell’eventuale presenza di disturbi ventilatori durante il sonno;
• considerare la presenza di altri fattori di rischio quali anchilosi
mandibolare, disfunzione orofaringea, reflusso gastro-esofageo,
stato nutrizionale inadeguato, asma, ecc.
Se le prove di funzionalità respiratoria e/o lo studio del sonno risultano alterati, è importante valutare la necessità di iniziare una
ventilazione non invasiva notturna e tecniche di assistenza della
tosse prima di procedere ad un intervento chirurgico programmato.
Il paziente dovrebbe essere istruito su tali tecniche e l’intervento
dovrebbe essere eseguito solo dopo che il paziente abbia preso
confidenza e si sia adattato alle nuove procedure. Se il paziente presenta anchilosi mandibolare, l’intubazione dovrebbe essere eseguita
mediante broncoscopia a fibre ottiche.
Gestione post-operatoria
• Se la funzione respiratoria e l’efficacia del colpo di tosse sono nei
limiti di norma e le funzioni muscolari sono relativamente conservate non vi sono significativi rischi di complicanze postoperatorie.
• Se la funzione respiratoria è compromessa alla valutazione preoperatoria, è necessario effettuare uno stretto monitoraggio del
paziente durante il post-operatorio e considerare l’eventuale necessità di misure più invasive atte a garantire la ventilazione.
• Se il paziente è in supporto ventilatorio durante il sonno prima
dell’intervento, il medesimo supporto deve essere garantito anche nell’immediato post-operatorio.
• L’estubazione deve essere attentamente pianificata e la ventilazione non invasiva può essere considerata come passaggio
intermedio nello svezzamento del paziente, con lo scopo di ristabilire la condizione respiratoria pre-esistente. Se il paziente
richiede un supporto ventilatorio continuo prima dell’intervento
o se durante l’intervento sono stati utilizzati agenti paralizzanti,
il paziente dovrebbe essere trasferito direttamente in una Unità
di terapia intensiva.
• Consigliare ai pazienti di portare con sé i propri apparecchi
(ventilatore non invasivo e in-exsufflator) da utilizzare nel post
operatorio, in considerazione della limitata disponibilità di tali
dispositivi nelle strutture ospedaliere.
• Il monitoraggio della CO2 e l’emogas analisi possono aiutare a
garantire un uso appropriato dell’ossigeno. L’ossigeno deve essere somministrato con precauzione nei pazienti affetti da SMA
poiché una ipossiemia secondaria a ipoventilazione può essere
erroneamente valutata come ipossiemia dovuta ad altre cause
quali secrezioni mucose o ateletassia.
• Attuare una terapia del dolore per prevenire l’ipoventilazione secondaria ad una scarsa escursione toracica, causata dalla sintomatologia dolorosa. In alcuni casi può rendersi utile un transito-
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
rio incremento dell’assistenza ventilatoria meglio per controllare
il dolore nel post-operatorio.
Gestione dei problemi acuti
Nella gestione delle complicanze acute nei pazienti affetti da SMA
deve essere garantita una buona funzionalità respiratoria, prevenendo la formazione di ateletassie ed incrementando gli scambi gassosi, ove possibile senza supporto ventilatorio non invasivo.
Favorire la pervietà delle vie aeree
• Tecniche di assistenza della tosse, manuali o meccaniche (tramite apparecchio tipo in-exsufflator); aspirazione delle secrezioni nelle prime vie respiratorie e bronco aspirazione. Le tecniche
volte a favorire l’espettorazione e il colpo di tosse attraverso la
fisioterapia respiratoria e il drenoaggio posturale sono preferibili
all’aspirazione profonda e alla broncoscopia in quanto meglio
tollerate dal paziente.
• Monitoraggio degli scambi gassosi e del livello di ossigenazione
mediante saturimetria e eroga analisi.
Supporto respiratorio
(i) Per i pazienti non deambulanti:
L’uso della ventilazione non invasiva in acuto è sconsigliato in quanto può portare a scompenso respiratorio dovuto all’instaurarsi di un
circolo vizioso tra sovraccarico ventilatorio, indebolimento della muscolatura respiratoria e inefficace espettorazione delle secrezioni. Tuttavia se il paziente è già in ventilazione assistita notturna, può essere
utile utilizzare la ventilazione non invasiva anche durante le ore diurne
sempre associata a tecniche volte a favorire l’espettorazione.
• La somministrazione di ossigeno attraverso il circuito della ventilazione non invasiva dovrebbe essere iniziata al fine di correggere gli episodi di desaturazione, solo dopo aver opportunamente regolato l’apparecchio a pressione positiva inspiratoria
ed espiratoria e in associazione con le tecniche atte a garantire
la pervietà delle vie respiratorie.
• Se l’approccio non invasivo risulta insufficiente, è necessario
procedere con intubazione e ventilazione meccanica. Tuttavia,
l’intubazione rappresenta una parte importante della gestione
del paziente affetto da SMA e tale decisione dovrebbe sempre
discussa con i familiari e dove possibile dovrebbe essere presa
preventivamente.
• Dopo la completa risoluzione della complicanza acuta e il paziente
può essere estubato e riadattato alla ventilazione non invasiva.
• Se la ventilazione non invasiva risultasse insufficiente si dovrà
procedere ad una tracheotomia.
(ii) Per i pazienti deambulanti
• L’ossigenoterapia. l’assistenza ventilatoria non invasiva e l’intubazione, anche se per un breve periodo di tempo, dovrebbero
essere eseguite con le stesse precauzioni e raccomandazioni
descritte per i pazienti non deambulanti.
• La possibilità di avere a domicilio un apparecchio per la ventilazione non invasiva dovrebbe essere presa in considerazione in
quei pazienti che hanno avuto bisogno di un supporto ventilatorio durante un episodio acuto.
Ulteriori interventi consigliati
In corso di malattie acute, in tutti i pazienti affetti da SMA, indipendentemente dal loro stato funzionale, è raccomandato impostare
un’adeguata terapia antibiotica, fornire un adeguato supporto nu-
trizionale, garantire una buona idratazione, valutare e trattare un
eventuale reflusso gastroesofageo.
Problemi nutrizionali e dell’apparato gastrointestinale
Le principali complicanze nutrizionali e gastrointestinali nei pazienti
affetti da SMA comprendono:
1. difficoltà di nutrizione e di deglutizione. La disfunzione bulbare e
dell’apparato orofaringeo è comune nei pazienti affetti da SMA.
Tale disfunzione può causare polmoniti ab ingestis, che rappresentano la principale causa di morte in questi pazienti.
2. Disfunzione dell’apparato gatrointestinale. I pazienti affetti da
SMA spesso presentano un’alterata motilità gastro-intestinale,
che può causare costipazione, stitichezza, ritardo nello svuotamento gastrico e reflusso gastroesofageo.
3. Crescita e problemi nutrizionali. In assenza di opportune misure
di supporto, i pazienti affetti da SMA di tipo I presentano un ritardo di crescita con scarso incremento ponderale conseguente
a iponutrizione. Viceversa i pazienti affetti dalle forme più lievi
tendono a sviluppare sovrappeso o obesità come conseguenza
della scarsa mobilità.
4. Problemi respiratori. Lo sviluppo di complicanze respiratorie
(colpo di tosse ipovalido, dispnea, infezioni polmonari, cianosi
o episodi di desaturazione durante i pasti) possono favorire o
incrementare i problemi di alimentazione ed aumentare il rischio
di polmoniti ab ingestis, mettendo potenzialmente a rischio la
vita del paziente. Le difficoltà respiratorie inoltre, richiedendo un
maggiore sforzo alla muscolatura toracica, aumentano il dispendio energetico e il fabbisogno calorico.
Problemi nutrizionali e di deglutizione
Le problematiche nutrizionali di questi pazienti sono differenti in
base alla severità della malattia. Nei bambini che non arrivano a
mantenere la posizione seduta autonoma è costante la comparsa di
un disturbo della deglutizione che peggiora nel tempo e determina
un rallentamento della crescita, episodi di polmonite ab ingestis e
come vedremo in seguito pone quasi sempre l’indicazione ad una
gastrostomia percutanea. Problemi di alimentazione e nutrizione
sono tuttavia molto comuni anche in pazienti affetti da SMA tipo II.
Un recente lavoro di Messina et al. (2008) condotto su una popolazione di circa 120 pazienti affetti da SMA tipo II di età compresa tra 1 e
47 anni dimostra infatti come circa il 30% di questi pazienti presenti
una difficoltà di alimentazione conseguente ad una limitata apertura
della mandibola e a una difficoltà e affaticamento nella masticazione.
In circa il 25 % di questa popolazione viene inoltre rilevato un disturbo
della deglutizione tale da determinare nel tempo una modifica della
dieta verso la scelta di cibi semisolidi. Più rari sono invece in questa
popolazione episodi di polmonite ab ingestis. Nella maggior parte di
questi pazienti si osserva inoltre una tendenza al basso peso.
Le problematiche nutrizionali dei pazienti affetti da SMA tipo III sono
al contrario più spesso quelli di sovrappeso o obesità come conseguenza della ridotta attività fisica e conseguente ridotta domanda
metabolica (Sproule et al., 2009).
1. Sintomi associati alle problematiche nutrizionali e alle difficoltà di deglutizione
• Necessità di tempi prolungati per terminare il pasto.
• Affaticamento durante l’alimentazione.
• Episodi acuti di soffocamento o colpi di tosse durante o immediatamente dopo la deglutizione.
• Infezioni respiratorie (polmoniti) ricorrenti rappresentano un
223
A. D’Amico, E. Bertini
campanello d’allarme per possibili episodi di ab ingestis, che
possono essere altrimenti silenti e verificarsi in assenza di
sintomi più tipici quali tosse o sensazione di soffocamento.
• Paralisi delle corde vocali: può essere un segno diagnostico
di aspirazione laringea.
2. Principali cause delle problematiche nutrizionali
Fase pre-orale
• Limitata apertura mandibolare dovuta a ridotta motilità dell’articolazione mandibolo-mascellare.
• Difficoltà nel portare il cibo alla bocca conseguente all’ipostenia muscolare degli arti.
Fase orale
• Morso debole.
• Facile affaticabilità della muscolatura masticatoria.
Fase di deglutizione
• Scarso controllo del capo.
• Disfunzione della muscolatura orofaringea con insufficiente
motilità faringea durante ladeglutizione.
• Scarsa coordinazione tra deglutizione e respirazione.
3. Valutazione dello stato nutritivo e delle difficoltà di deglutizione
• Valutazione dello stato nutritivo da parte di uno specialista
nutrizionista/dietologo.
• Osservazione del paziente durante il pasto (valutazione del
controllo e della posizione del capo durante l’alimentazione
in relazione al loro effetto sull’efficienza dell’alimentazione e
della deglutizione).
• Esame clinico dell’apparato masticatorio e buccale.
• Lo studio video-fluorangiografico dell’apparato oro-faringeo
è indicato nei casi in cui si sospettino difficoltà di deglutizione (disfagia) al fine di identificare appropriate strategie di
trattamento.
4. Gestione delle difficoltà di alimentazione e deglutizione
Trattamenti specifici dovrebbero essere finalizzati a ridurre il rischio di
ab ingestis, ad ottimizzare l’efficienza della nutrizione e a rendere più
piacevole il momento del pasto. Tali interventi terapeutici dovrebbero
essere valutati obiettivamente tramite esame video-fluorangiografico.
• Modificare la consistenza del cibo. Una dieta semi-solida
può compensare le difficoltà di masticazione e ridurre il tempo necessario per terminare il pasto. L’utilizzo di addensanti
per i liquidi può ridurre gli episodi di ab ingestis.
• Correggere la postura in posizione seduta e utilizzare se necessari appropriati ausili, (e.g. Neater Eater®, supporti per
gli arti superiori, cannucce valvolate) al fine di facilitare l’alimentazione in autonomia e aumentare l’efficienza della deglutizione e dell’alimentazione. In alcuni casi una valutazione
o un trattamento da parte di un terapista occupazionale o
un fisioterapista possono essere utili al fine di identificare
l’intervento più adatto alle specifiche esigenze del paziente.
• Associare integratori alimentari e valutare la necessità di tecniche alternative per l’alimentazione appena si riconoscono
segni di insufficiente apporto per via orale. La decisione se
posizionare una sondino gastrico in un bambino affetto da
SMA deve essere presa sulla base di un accordo tra coloro, medici e familiari, che si prendono cura del bambino. La
nutrizione mediante sondino naso-gastrico o naso-digiunale
dovrebbe essere considerata in attesa della gastrostomia. Il
sondino naso-digiunale è preferibile nei casi in cui si sospetti
o sia stato accertato un reflusso gastro-esofageo con ab in-
224
gestis, specialmente se il paziente è in ventilazione assistita.
Tuttavia le difficoltà tecniche legate al suo posizionamento
posso limitarne l’applicazione.
• La gastrostomia è la soluzione ottimale nei casi in cui non sia
possibile garantire un’adeguata alimentazione per via orale
o quando sia elevato il rischio di ab ingestis. Essa riduce la
morbilità e le difficoltà di utilizzo della ventilazione non invasiva assistita associate all’utilizzo del sondino naso-gastrico
o naso-digiunale (per scarsa aderenza della maschera). Le
tecniche di chirurgia laparoscopica per il posizionamento del
tipo gastrostomico rappresentano la migliore soluzione per
l’estubazione immediata o precoce nel post-operatorio. Devono tuttavia essere prese cautele al fine di ridurre al minimo il
tempo di digiuno pre-operatorio e impostare un’adeguata alimentazione il più precocemente possibile dopo l’intervento.
Disfunzione gastro-intestinale
I problemi gastro-intestinali più frequenti nei pazienti affetti da SMA
sono: reflusso gastro-esofageo, costipazione, gonfiore e distensione
addominali. Il reflusso gastro-esofageo gioca un ruolo importante
nel determinare la morbilità e la mortalità nei pazienti con SMA. Cibi
ricchi di grassi dovrebbe essere evitati in quanto ritardano lo svuotamento gastrico e aumentano il rischio di reflusso gastro-esofageo.
1. Principali sintomi di reflusso gastro-esofageo:
• emesi;
• dolore, senso di pesantezza o di bruciore toracici o addominali;
• alitosi;
• frequenti episodi di rigurgito o vomito post-prandiali;
• rifiuto del cibo dovuto a sensazione di fastidio o dolore durante la deglutizione.
2. Valutazione dei problemi gastro-intestinali:
• ricerca dei segni precoci di reflusso gastro-esofageo;
• uno studio radiografico funzionale, al fine di identificare anomalie anatomiche e documentare la presenza di reflusso
gastro-esofageo;
• studio della motilità gastrointestinale mediante scintigrafia
al fine di documentare un ritardo nello svuotamento gastrico
che può contribuire al precoce senso di sazietà e allo sviluppo di reflusso gastro-esofageo.
3. Gestione del reflusso gastro-esofageo:
• uso di neutralizzanti dell’acidità (es. magnesio o calcio carbonato) e/o di inibitori delle secrezioni acide (es. bloccanti
istaminici e inibitori della pompa protonica quali famotidina, ranitidina, omeoprazolo) come sintomatici. Tali farmaci
dovrebbero essere somministrati solo per brevi periodi di
tempo in quanto il loro uso prolungato può incrementare il
rischio di gastroenteriti e polmoniti;
• se il paziente presenta un ritardo dello svuotamento gastrico o
una riduzione della motilità gastroesofagea, può essere indicato
l’uso di agenti procinetici (es. metoclopramide, eritromicina);
• l’uso di probiotici quali acidophilus o lactobacillus al fine di
mantenere una buona flora intestinale, in particolare durante
e dopo trattamenti antibiotici o in associazione a trattamento
prolungato con anti-acidi, è tutt’ora oggetto di studio;
• l’intervento di funduplicatio secondo Nissen per reflusso
gastro-esofageo per via laparoscopica durante il posizionamento del tubo per la gastrostomia può rappresentare una
valida alternativa nei pazienti che presentano reflusso ga-
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
stro-esofageo refrattario alle terapie mediche o per i quali
si ritenga che il beneficio derivante da tale procedura sia
maggiore rispetto ai rischi chirurgici e anestesiologici.
Gestione dei problemi di crescita e di sotto-/sovra-peso
• L’obiettivo deve essere quello di mantenere il bambino nella sua
normale curva di crescita.
• Monitorare le curve di velocità di crescita nel tempo (peso, altezza, peso/altezza).
• Ad ogni visita è raccomandata la valutazione dello stato nutrizionale da parte di un dietologo/dietista o di uno specialista dell’alimentazione. Un diario alimentare dei 3 giorni precedenti alla
visita è un metodo semplice ed accurato per valutare il tipo di
alimentazione e valutare la necessità di integrazioni alimentari.
• I pazienti a rischio di obesità dovrebbero mantenere i parametri
di crescita di peso, altezza e BMI (body mass index) ai percentili
inferiori per sesso ed età.
• È importante assicurare un adeguato apporto di calcio e vitamina D.
• Il dosaggio dei livelli sierici di pre-albumina può essere utile per
valutare l’adeguatezza dell’apporto proteico.
Gestione della nutrizione in corso di patologie acute nei pazienti
affetti da SMA
• I pazienti affetti da SMA, in particolare i pazienti non deambulanti, sono particolarmente suscettibili a condizioni di digiuno e tendono a sviluppare più frequentemente uno stato di ipoglicemia in
seguito a scarso apporto alimentare. In questi pazienti, è quindi
importante evitare periodi prolungati di digiuno, specialmente in
corso di malattie acute.
• In caso di ospedalizzazione l’intero apporto calorico dovrebbe essere
somministrato nelle prime ore successive al ricovero, per via enterale
e/o parenterale, ove necessario, al fine di ripristinare un apporto alimentare adeguato alle richieste.
• In caso di interventi chirurgici, è raccomandato un precoce supplemento calorico nel postoperatorio al fine di evitare il catabolismo
muscolare, soprattutto nei bambini con scarsa riserva di grassi.
Interventi ortopedici e trattamenti riabilitativi
Interventi ortopedici e trattamenti riabilitativi nei pazienti affetti da SMA:
A. Problemi principali: l’ipostenia muscolare può portare allo sviluppo di retrazioni tendinee e deformità della colonna vertebrale,
che aumentano il rischio di sintomatologia dolorosa, articolare e
muscolare, osteopenia e fratture.
B. Principali procedure valutative:
• range di movimento (ROM);
• misurazione della forza;
• test per valutare la postura e le abilità funzionali;
• valutazione di eventuali ortesi;
• esame radiografico della colonna vertebrale o delle articolazioni;
• DEXA scan;
• interventi ortopedici chirurgici.
Valutazione attraverso livelli funzionali e trattamento
A. Pazienti che non mantengono la posizione seduta autonoma:
Valutazione:
• valutazione funzionale attraverso terapia fisica e occupazionale;
• valutazione logopedica se l’apparato deglutitorio, il linguaggio o il tono della voce sono compromessi a causa di retrazioni o malposizionamento mandibolari.
Principali interventi:
• supporto e correzione della postura: la postura del paziente
dovrebbe essere supportata e corretta al fine di migliorare lo
stato funzionale del paziente;
• gestione delle contratture muscolari e delle retrazioni tendinee: l’utilizzo di tutori o doccette può essere indicato al fine
di mantenere una buona mobilità articolare e prevenire la
sintomatologia dolorosa;
• gestione del dolore;
• strumenti adattati alle necessità del paziente: adeguati
supporti per il gioco e le attività occupazionali dovrebbero
includere giochi leggeri per peso, sistemi di attivazione e
tecnologie di assistenza a controllo variabile;
• carrozzina: deve garantire una corretta postura e una maggior indipendenza del paziente;
• ortesi: adeguate ortesi per gli arti superiori, quali supporti
mobili o bende elastiche, possono agevolare il paziente nelle
attività quotidiane, aumentando il range dei movimenti ed
incrementando la funzionalità degli arti superiori;
• modificazioni adattative del domicilio al fine di garantirne una
sicura accessibilità e aumentare l’indipendenza del paziente.
B. Pazienti che mantengono la posizione seduta autonoma:
Valutazione:
• valutazione funzionale (Hammersmith Functional Motor
Scale for SMA, Modified-Hammersmith functional motor
scale for SMA, Gross Motor Function Measure (GMFM) e Motor Function Measure (MFM);
• misura quantitativa delle retrazioni tendinee con goniometro;
• misura della forza muscolare mediante valutazione manuale
(scala MRC) o miometro;
• studio radiologico (Rx) della colonna vertebrale e delle anche;
• valutazione della postura, della mobilità e dell’autonomia.
Appropriati sistemi per la postura e la mobilità, manuali od
elettrici, dovrebbe essere considerati per i bambini di età
superiore ai 18-24 mesi.
Principali interventi (fisioterapia, terapia occupazionale e interventi
ortopedici):
• carrozzina. Deve garantire una corretta postura e una maggior indipendenza del paziente;
• modificazioni adattative del domicilio al fine di garantirne
una sicura accessibilità e aumentare l’indipendenza del paziente;
• gestione delle retrazioni tendinee. La prevenzione e la gestione delle retrazioni tendinee costituiscono il principale obiettivo del trattamento riabilitativo e devono includere esercizi
di stretching e programmi di supporto al fine di preservare
una buona mobilità articolare. Un adeguato trattamento delle
retrazioni tendinee può migliorare la tollerabilità delle ortesi
e permettere l’utilizzo di supporti (standing) per mantenere
la stazione eretta. Tutori tipo AFO possono ritardare lo sviluppo di retrazioni a livello dell’articolazione tibio-tarsica. Ortesi
per gli arti superiori con supporti mobili o bende elastiche
aumentano il range dei movimenti e le abilità funzionali;
• una regolare attività motoria, quale il nuoto o attività sportive
adattate alle esigenze del paziente, dovrebbe essere incoraggiata al fine di mantenere una buona mobilità e un certo
grado di resistenza nelle prestazioni motorie;
• il mantenimento della posizione eretta deve essere incoraggiato. Tutori lunghi leggeri per gli arti inferiori (KAFOs) o ortesi di
supporto per il mantenimento della posizione eretta o la deam-
225
A. D’Amico, E. Bertini
bulazione assistita dovrebbero essere forniti ai pazienti con adeguata forza muscolare. Ove questo non fosse possibile a causa
della severa ipostenia, dovrebbe essere considerato l’utilizzo di
ausili per mantenere la posizione eretta (standing frame);
• ortesi per la colonna vertebrale;
• chirurgia vertebrale (vedi paragrafo successivo).
C. Pazienti deambulanti:
Valutazione:
• esame della deambulazione e dell’equilibrio per valutare la
presenza di atteggiamenti compensatori e la necessità di dispositivi adattativi;
• valutazione della mobilità articolare e dell’allineamento della
colonna vertebrale;
• valutazione da parte di un fisioterapista e di un terapista occupazionale al fine di individuare la necessità di opportuni
supporti, che facilitino la mobilità e la funzionalità, e di adattamenti nell’ambiente domiciliare, lavorativo o di studio, che
ne garantiscano l’accessibilità;
• valutazione degli ausili necessari per lo svolgimento delle
attività quotidiane;
• Rx di specifici distretti e DEXA scan possono essere considerati
in caso di eventi traumatici acuti a carico dell’apparato muscolo
scheletrico, conseguenti a cadute, incidenti o sovra-uso.
Principali interventi:
• carrozzina per lunghi i spostamenti al fine di garantire al paziente una maggior indipendenza;
• gestione delle retrazioni tendinee ed educazione alla loro
prevenzione e al mantenimento di una buona mobilità articolare;
• fisioterapia e terapia occupazionale per garantire la sicurezza e l’indipendenza del paziente o per prolungare la deambulazione autonoma;
• la deambulazione dovrebbe essere incoraggiata, fornendo
l’assistenza necessaria o specifiche ortesi ove richiesto;
• un’attività motoria regolare deve essere incoraggiata al fine
di mantenere le performance motorie, la resistenza e la forza
muscolare. Questa può includere il nuoto, l’idroterapia, l’ippoterapia o sport adattati alle necessità e alle possibilità del
paziente;
• educazione alla guida di un’autovettura al fine di aumentare
l’indipendenza del paziente. Controlli e adattamenti personalizzati possono essere considerati in relazione alle capacità motorie del paziente;
• modificazioni adattative del domicilio al fine di garantirne una
sicura accessibilità e aumentare l’indipendenza del paziente;
• ortesi e ausili per la colonna vertebrale e gli arti se il paziente
sviluppa scoliosi e retrazioni tendinee;
• chirurgia vertebrale (vedi paragrafo successivo).
Ortesi
• È importante che il tecnico ortopedico, il fisioterapista e i familiari collaborino nell’identificare l’ausilio più idoneo per raggiungere
l’obiettivo funzionale desiderato e garantire la comodità al paziente.
• Il tecnico ortopedico dovrebbe avere una buona conoscenza ed
esperienza al fine di poter consapevolmente scegliere i materiali
più adatti e di poter adattare l’ausilio alle specifiche esigenze del
paziente.
• Supporti per la colonna vertebrale possono essere utilizzati per
sostenere la postura ma sembrano essere inefficaci nel prevenire
o nel ritardare la progressione delle deformità della colonna verte-
226
brale. Quando utilizzati, dovrebbero essere realizzati con un taglio
addominale al fine di non ostacolare l’escursione diaframmatica e
garantire l’accesso al tubo della gastrostomia se presente.
Interventi chirurgici ortopedici
1. Sublussazione dell’anca e retrazioni tendinee:
• La sublussazione dell’anca è raramente dolorosa nei pazienti affetti da SMA. La riduzione chirurgica e l’osteotomia sono
frequentemente seguite da una ri-dislocazione. Nella maggior parte dei casi, quindi, tale intervento non è indicato.
• Deformità del piede e della caviglia, conseguenti a severe
retrazioni tendinee, possono rendere difficoltoso l’utilizzo
di normali scarpe e possono rappresentare un’indicazione
all’intervento di allungamento tendineo. Nei pazienti deambulanti, un precoce ed intenso trattamento fisioterapico iniziato subito dopo l’intervento di allungamento tendineo ne
garantisce un miglior risultato.
2. Intervento correttivo per la scoliosi:
• L’intervento chirurgico correttivo della scoliosi ha benefici
sulla postura e sull’equilibrio in posizione seduta, sulle performance motorie oltre che dal punto di vista estetico. Migliori risultati sembrano ottenersi quando l’intervento viene
eseguito precocemente.
• L’intervento correttivo della scoliosi (atrodesi vertebrale)
sembra avere effetti positivi nei pazienti che sopravvivono
oltre i 2 anni di età e presentano una scoliosi severa e progressiva. Deve essere eseguito quando la funzionalità respiratoria è adeguata a sopportare l’intervento e l’anestesia.
• Argomento controverso è l’effetto dell’intervento correttivo
della scoliosi sulla funzionalità respiratoria. La rapida progressione del deterioramento della funzionalità respiratoria,
tuttavia, sembrerebbe essere rallentata in seguito a correzione chirurgiche delle deformità vertebrali.
• Durante la procedura chirurgica possono verificarsi complicanze conseguenti ad un’ eccessiva perdita di sangue. Le
più frequenti complicanze post-operatarie includono il fallimento della correzione della curva scoliotica, pseudo-artrosi, necessità di supporto ventilatorio prolungato o a lungo
termine, infezioni respiratorie e delle ferite chirurgiche.
• Particolare attenzione deve essere posta nei pazienti deambulanti, in quanto l’alterazione delle abilità funzionali e dell’equilibrio o un deterioramento della funzionalità respiratoria conseguenti all’intervento, potrebbero portare alla perdita
della deambulazione autonoma.
Gestione del paziente affetto da SMA nel pre-operatorio.
1. Gestione del paziente nel pre-operatorio:
• valutare le modificazioni delle ortesi che saranno necessarie
dopo l’intervento e pianificare i tempi per la loro realizzazione. Una nuova carrozzina o modifiche alla carrozzina in uso
(seduta, schienale, sostegni per la testa e gli arti inferiori)
saranno probabilmente necessarie dopo l’intervento;
• dare istruzioni per gli spostamenti e i trasferimenti del paziente nel post-operatorio, provvedendo alla disponibilità di un
sollevatore meccanico, qualora dovesse essere necessario.
2. Gestione del paziente nel post-operatorio:
• assicurare adeguate e tempestive modifiche delle ortesi in
uso e gli adattamenti necessari, eseguire trattamenti di fisioterapia al fine di mantenere una buona mobilità articolare;
• garantire un appropriato supporto ventilatorio (ventilazione
non invasiva);
Nuovi standard di cura per la presa in carico del bambino con atrofia muscolare spinale
• istruire il personale infermieristico ed i familiari in merito alle
modalità di mobilizzazione, trasferimento, vestizione, pulizia
e uso della toilette del paziente;
• mobilizzare il paziente il più precocemente possibile in rispetto dell’intervento subito e delle indicazioni date dal chirurgo.
Cure palliative
• L’assistenza clinica dei pazienti affetti da SMA dovrebbe sempre
prendere in considerazione il potenziale conflitto degli obiettivi
terapeutici. Questo conflitto è reso più difficile dal naturale coinvolgimento di più persone (genitori, fratelli, altri familiari, tutori,
datore di lavoro e la società in genere) nelle scelte che condizionano la vita di un bambino.
• Vi è una profonda responsabilità da parte del medico curante nel
presentare in modo corretto, obiettivo ed equilibrato le diverse
opzioni di trattamento/intervento, al fine di poterli iniziare il più
precocemente possibile dopo la diagnosi.
• La scelta a favore o a sfavore di un determinato intervento non è
una scelta a binario unico, nè deve essere definitiva e irrevocabile. Concedere tempo per pensare, parlare onestamente delle
diverse opzioni di trattamento e dei loro effetti, riconoscere la
possibilità di rivedere le scelte fatte ed avere un rapporto umano
e personale con il paziente ed i familiari sono aspetti fondamentali nella presa in carico di un paziente affetto da SMA.
• La gastrostomia dovrebbe essere eseguita precocemente, quando i
•
•
•
•
rischi chirurgici e anestesiologici associati sono ancora bassi al fine
di garantire un adeguato e stabile stato nutrizionale per il momento
in cui il bambino presenterà maggiori difficoltà di alimentazione.
La scelta riguardo gli interventi da eseguire in caso di insufficienza
respiratoria acuta,come la rianimazione in condizioni di emergenza e
l’intubazione endotracheale, andrebbe discussa e presa in anticipo.
La decisione su tempi e modalità di interruzione delle cure deve essere discussa e definita insieme al paziente e ai familiari, in accordo
con le normative/leggi vigenti in materia. I genitori o parenti devono
essere consapevoli, preparati ed informati in merito a queste possibilità al fine di non ritardare la scelta ed evitare di imporla aggressivamente e inaspettatamente in un momento particolarmente
difficile e traumatico, quale può essere l’emergenza acuta.
La presa in carico di un paziente affetto da SMA dovrebbe avvenire da parte di un team multi specialistico che prenda in
considerazione gli aspetti medici, sociali e psicologici a seconda
delle specifiche necessità. Strutture per la cura a lungo termine
o altre soluzioni per la gestione del paziente nelle fasi terminali,
un supporto psicologico per i familiari sono aspetti che svolgono
un ruolo importante.
Nella situazione in cui si decida di non intervenire con supporto ventilatorio meccanico, un’adeguata preparazione alla gestione della
dispnea nella fase terminale può essere di conforto per il paziente
e i familiari. L’uso di narcotici può ridurre la sofferenza del paziente,
evitando che un sovradosaggio del farmaco possa contribuire a determinarne la morte.
Box di orientamento
• L’atrofia muscolare spinale (SMA) è una malattia neurodegenerativa che necessita di un approccio multidisciplinare per la presa in carico dei pazienti.
• Con la necessità di iniziare i trials clinici è sorta l’esigenza di generare linee standardizzate di presa in carico dei pazienti SMA non solo per curarli
nel modo migliore, ma anche per ridurre i fattori di variabilità confondenti che interferiscono con la valutazione dei trials clinici.
• Le aree di presa in carico dei pazienti SMA che hanno riscosso il massimo consenso tra gli esperti internazionali coinvolti in una Consensus conference
pubblicata nel 2007 sono l’iter diagnostico, la gestione delle complicanze acute e croniche della insufficienza respiratoria e le problematiche nutrizionali.
• Le aree di presa in carico dei pazienti SMA che hanno riscosso il minor consenso sono quelle riguardanti l’intervento riabilitativo ed ortopedico e
per queste si apre uno spazio per ricerche future.
Bibliografia
Bertini E, Burghes A, Bushby K, et al. 134th ENMC International Workshop: Outcome Measures and Treatment of Spinal Muscular Atrophy, 11-13 February
2005, Naarden, the Netherlands. Neuromuscul Disord. 2005;15:802-16.
* Conclusioni del primo workshop internazionale dedicato alla pianificazione dei
trials clinici sulla SMA.
Crawford TO. Concerns about the design of clinical trials for spinal muscular
atrophy. Neuromuscul Disord 2004;14:456-60.
* Prima review sulle problematiche legate ai trials clinici sulla SMA.
Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding
clinical phenotype. Eur J Paediatr Neurol 1999;3:49-51.
Hirtz D, Iannaccone S, Heemskerk J, et al. Challenges and opportunities in clinical trials for spinal muscular atrophy. Neurology 2005;65:1352-7.
Messina S, Pane M, De Rose P, Vasta, et al. Feeding problems and malnutrition in
spinal muscular atrophy type II. Neuromuscul Disord 2008;18:389-93.
* Interessante studio sui problemi nutrizionali dei pazienti con SMA II.
Munsat TL, Davies KE. International SMA consortium meeting (26-28 June 1992,
Bonn, Germany). Neuromuscul Disord 1992;2:423-8.
* Prima proposta concordata sulla classificazione di severità funzionale della
SMA.
Prior TW; Professional Practice and Guidelines Committee. Carrier screening for
spinal muscular atrophy. Genet Med 2008;10:840-2.
Sproule DM, Montes J, Montgomery M, et al. Increased fat mass and high incidence of overweight despite low body mass index in patients with spinal muscular atrophy. Neuromuscul Disord 2009;19:391-6.
Wang CH, Finkel RS, Bertini E, et al. Participants of the International Conference
on SMA Standard of Care Consensus statement for standard of care in spinal
muscular atrophy. J Child Neurol 2007;22:1027-49.
* Documento di riferimento che risssume le conclusioni del Consensus statement interanazionale sullo Standard of Care nella SMA.
Zerres K, Wirth B, Rudnik-Schoneborn S. Spinal muscular atrophy - clinical and
genetic correlations. Neuromuscul Disord 1997;7:202-7.
Corrispondenza
dott. Enrico Bertini, Unità di Medicina Molecolare per Malattie Neuromuscolari e Neurodegenerative, Ospedale Bambino Gesù, p.zza S. Onofrio 4, 00165
Roma. E-mail: [email protected]
227
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 228-238
frontiere
Le basi genetiche delle SCID
Fausto Cossu
Centro Trapianti Midollo Osseo, II Clinica Pediatrica dell’Università, Ospedale Microcitemico, Cagliari
Riassunto
Le SCID (malattie con immunodeficienza combinata severa) sono primariamente malattie prenatali della differenziazione dei linfociti T, che richiede nei
suoi vari stadi l’espressione di numerosi geni. Negli ultimi anni si sono identificati nuovi geni-malattia causa di SCID e si sono definiti meglio i meccanismi
molecolari e le corrispondenti caratteristiche immunologiche delle varie forme.
Attualmente si classificano le SCID in 6 grandi gruppi in base ai meccanismi patogenetici prevalenti: aumentata apoptosi linfocitaria; difetti di segnali mediati da citochine; difetti del pre-T cell receptor; difetti dei canali del calcio; difetti di embriogenesi del timo; altri meccanismi.
Le SCID presentano sia aspetti generali comuni che aspetti peculiari delle diverse forme (es. presenza o meno di manifestazioni extra-immunologiche), e
le nuove conoscenze sulle basi genetiche consentono una migliore comprensione anche riguardo a diagnosi (es. fenotipo linfocitario) e terapia (es. utilizzo
e tipo di condizionamento pre-trapianto di cellule staminali ematopoietiche).
Summary
Severe combined immunodeficiency diseases are primarily prenatal illnesses of the development of T lymphocytes, that requires the expression of numerous genes. In the last years novel genetic defects causing severe combined immunodeficiency diseases have been discovered, and the molecular mechanisms and immunological characteristics of the various forms of severe combined immunodeficiency diseases have been defined better. Currently severe
combined immunodeficiency diseases are classified into six groups with different prevalent pathogenic mechanisms: increased lymphocyte apoptosis;
impaired cytokine-mediated signaling; impaired signaling through the pre-T cell receptor; impaired calcium flux; defects of thymus embryogenesis; other
mechanisms. The various forms of severe combined immunodeficiency diseases show both common general and peculiar characters (e.g., absence or
presence of extra-immunological manifestations). New knowledge of the genetic bases allow a best understanding also respect to diagnosis (e.g., lymphocyte phenotype) and therapy (e.g., use and type of pre-hematopoietic stem cell transplant conditioning).
Introduzione
Le SCID (severe combined immunodeficiency diseases – malattie
con immunodeficienza combinata severa; gruppo eterogeneo con
incidenza di circa 1:50.000 neonati) sono la forma più grave di
immunodeficienza primitiva, presente già alla nascita anche se le
prime manifestazioni cliniche si osservano più spesso nel piccolo
lattante (Ochs et al., 2007; Rezaei et al., 2008).
Argomento di questo articolo, basato sulla revisione della letteratura
più recente ed esperienza personale dell’Autore, sono i principali
aggiornamenti riguardo a: scoperta di nuovi geni-malattia la cui mutazione è causa di SCID, meccanismi molecolari ed immunologici
delle forme già note, classificazione dei genotipi associati a SCID,
correlazioni tra basi genetiche ed aspetti clinici.
Aspetti generali (aggiornati) delle SCID
Per la prima volta al mondo, dal Gennaio 2008 nello stato del
Wisconsin (USA) si esegue lo screening del neonato per le SCID (su
Guthrie card, dosaggio dei T Cell Receptor Excision Circles – TRECs,
Fig. 1) (Baker et al., 2009); il poster dello screening (Fig. 2) descrive
gli aspetti fondamentali delle SCID: i bambini non producono linfociti T (o, comunque, T funzionali) e acquisiscono infezioni multiple
batteriche, virali e fungine (anche da eventuali vaccini con microrganismi attenuati, es. bacillus Calmette-Guerin – BCG); le SCID sono
un’emergenza pediatrica: con diagnosi e HSCT (hematopoietic stem
cell transplantation, trapianto di cellule staminali ematopoietiche;
228
Box 1) o terapia genica (Box 2) tempestivi questi bambini possono
guarire, altrimenti il decorso è rapidamente fatale. È utile ricordare
altri aspetti generali delle SCID (per i riferimenti alle singole forme
di SCID, si veda oltre):
• Alla nascita il neonato affetto da SCID appare normale nella
maggioranza dei casi; talvolta ci sono segni cutanei, ancora
lievi, simil-GvHD (Graft versus Host Disease) da engraftment di
linfociti T materni; invece, basso peso e lunghezza ridotta per
l’età gestazionale, microcefalia, facies dismorfica, condrodisplasia metafisaria, ecc. sono manifestazioni extra-immunologiche
delle forme in cui l’alterazione genetica colpisce oltre ai linfociti
anche altri tipi cellulari ed apparati (Tab. I).
• Ogni SCID è comunque una malattia prenatale dello sviluppo timico dei linfociti T, già presente alla nascita anche se asintomatica
(lo screening TRECs non dosa un enzima né cerca mutazioni, ma
conta i linfociti T naive normali, già assenti o assai ridotti).
Da notare che, a differenza del topo, nell’uomo in assenza di SCID
lo sviluppo del sistema immunitario è precoce già dall’embrione: intensa “T lymphopoiesis” prenatale del timo; l’esposizione in utero
ad antigeni estranei genera risposta immunologica e non tolleranza,
a parte quella verso gli alloantigeni materni non condivisi dal feto
(tolleranza mediata da linfociti T regolatori CD4+CD25highFoxP3+TReg
specifici: 15-20% dei linfociti T CD4+ negli organi linfoidi periferici
del feto umano; Mold et al., 2008). E infatti, con l’HSCT in utero:
successo (parziale, in genere ricostituzione limitata alla linea T) nei
feti affetti da SCID, ma risultato del tutto negativo – 0% di successo
(!), con oltretutto il 24% di procedure-related death – in 17/17 feti
Le basi genetiche delle SCID
Figura 1.
T-cell Receptor Excision Circles (TRECs) prodotti nei timociti (linfociti T in differenziazione nel timo) dai riarrangiamenti nel locus TCRδ/α, posto in
14q11 (da Spits, 2002, mod.).
I TRECs sono circoli di DNA episomico (non integrati nel genoma), stabili ma non duplicati dalla mitosi e quindi diluiti ad ogni divisione cellulare; la
PCR (Polymerase Chain Reaction) quantitativa del TREC δRec-ψJα misura nel sangue periferico i linfociti T naive αβ recentemente dismessi dal
timo: nel neonato un valore < 25 TRECs/mL indica SCID.
Figura 2.
Il poster del Wisconsin State Laboratory of Hygiene che illustra lo screening neonatale per SCID (http://www.slh.wisc.edu/posters/080918_Updated_Scid_Poster.pdf).
Il bambino della fotografia è David Phillip Vetter (21 Settembre 1971 – 22 Febbraio 1984), il “bubble boy”.
229
F. Cossu
Tabella I.
Classificazione delle SCID.
T / B / NK
Gene
Locus
Trasmissione
Proteina**
Manifestazioni
extraimmunologiche
Aumentata apoptosi linfocitaria
Disgenesia reticolare
T-B-NK(“aleukocytosis”)
ADA SCID
T-B-NK-
AK2
1p34
AR
Adenilato kinasi 2
Sordità neurosensoriale
ADA
20q13.11
AR
Adenosina deaminasi
PNP SCID
PNP
14q13.1
AR
Purina nucleoside
fosforilasi
Anomalie condrocostali, displasie
scheletro, epatite neonatale, sordità
neurosensoriale, disturbi neurologici
Disturbi neurologici
IL2RG
JAK3
IL7RA
Xq13.1
19p13.1
5p13
XL
AR
AR
Catena γ common
Janus chinasi 3
Catena α del recettore
dell’IL-7
11p13
11p13
10p13
8q11.21
13q33.3
AR
AR
AR
AR
AR
RAG1
RAG2
Artemis
DNA-PKcs
DNA ligasi IV
T-B-NK-
Difetti di segnali mediati da citochine
Difetto di catena γc
T-B+NKDifetto di JAK3
T-B+NKDifetto di IL-7Rα
T-B+NK+
Difetti del pre-T cell receptor (pre-TCR)
Difetti di ricombinazione
V(D)J
Difetto di RAG1
T-B-NK+
RAG-1
Difetto di RAG2
T-B-NK+
RAG-2
Difetto di Artemis
T-B-NK+
DCLRE1C
Difetto di DNA-PKcs
T-B-NK+
PRKD
Difetto di DNA ligasi IV
T-B-NK+
LIG4
Difetto di Cernunnos/XLF
Difetti di trasmissione
del segnale del pre-TCR
Difetto completo di CD3δ
Difetto completo di CD3ε
Difetto completo di CD3ζ
Difetto completo di CD3γ
Difetto di CD45
Difetto di ZAP-70
T-B-NK+
NHEJ1
2q35
AR
Cernunnos/XLF
T-B+NK+
T-B+NK+
T-B+NK+
T-B+NK+
T-B+NK+/T+B+NK+
CD4+CD8T-B+NK+
CD3D
CD3E
CD3Z
CD3G
PTPRC
11q23
11q23
1q24.2
11q23
1q31.3
AR
AR
AR
AR
AR
CD3δ
CD3ε
CD3ζ
CD3γ
CD45 (LCA)
ZAP70
LCK
2q11.2
1p35.1
AR
AR
ZAP-70
p56LCK
ORAI1
STIM1
12q24
11p15.5
AR
AR
ORAI1
STIM1
Miopatia, note di displasia ectodermica
Miopatia, note di displasia ectodermica
WHN
17q11.2
AR
FOXN1
Alopecia
> 35 geni
22q11.2
AD
TBX1, e altre
Dismorfie facciali, malformazioni
cardiache, malformazioni di altri orga
ni, assenza paratiroidi con ipocalcemia
neonatale
AD
CHD-7
Associazione CHARGE
Difetto di p56LCK
Difetti dei canali del calcio
Difetto di ORAI1
T+B+NK+
Difetto di STIM1
T+B+NK+
Difetti di embriogenesi del timo
Sindrome Nude/SCID
T-B+NK+
Anomalia di DiGeorge
T-B+NK+
completa:
del22q11.2
CHARGE
Embriopatia da madre diabetica
Radiosensibilità
Radiosensibilità
Radiosensibilità, facies dismorfica,
microcefalia, ritardo di accrescimento
e psicomotorio
Radiosensibilità, facies dismorfica,
microcefalia, ritardo di accrescimento
e psicomotorio
CHD7
Malformazioni cardiache, renali,
intestinali; difetti del tubo neurale, agenesia lombosacrale, oloprosencefalia;
ipoglicemia neonatale
segue Tabella I.
230
Le basi genetiche delle SCID
continua Tabella I.
T / B / NK
Altri meccanismi
Difetto di coronina 1A
T-B+NK+
Difetto delle molecole MHC T+B+NK+
di classe II
CD4-CD8+
Sindrome CHH (cartilage hair
hypoplasia)
Sindrome di Hoyeraal-Hreidarsson (HHS)
HFM (hereditary folate malabsorption)
T-B+NK+
T+B-NKT+B+NK+
Gene
Locus
Trasmissione
Proteina**
CORO1A
CIITA
RFXANK
RFX5
RFXAP
RMRP
16p11.2
16p13.13
19p13.11
1q21.2
13q13.3
9p13.3
AR
AR
AR
AR
AR
AR
DKC1
?
SLC46A1
Xq28
?
17q11.2
XL
AR
AR
Coronina 1A
CIITA
RFXANK
RFX5
RFXAP
**RNA dell’RNase MRP
complex
Discherina
?
PCFT
affetti da emoglobinopatie (talassemia, drepanocitosi) e quindi con
sistema immunitario normale (Muench, 2005).
• Per definizione, in tutte le SCID sono compromessi sviluppo e
funzioni dei linfociti T (congenital severe T cell immunodeficiencies). Però, linfociti T, linfociti B e linfociti NK (a differenza di T e
B, i NK non riarrangiano il loro DNA “germline” per creare i geni
di recettori antigene-specifici) hanno in comune linee cellulari,
segnali di sviluppo e di funzione, vie metaboliche, ecc. Perciò
sono spesso compromessi di per sé anche linfociti B e/o linfociti
NK, e si distinguono le SCID in base alle diverse “combinazioni”
delle conte T/B/NK (– indica assenza o conte assai ridotte): T-BNK-, T-B+NK+, T-B+NK-, T-B-NK+.
Senza le varie sottopopolazioni di linfociti T CD4+ helper (TH1, TH2,
TReg, TFH, TH17 e, di recente identificazione, TH22 e TH9) non possono comunque funzionare, anche se presenti e “normali”: linfociti B
(nelle SCID di regola: agammaglobulinemia), macrofagi, ed anche
eventuali linfociti T “residui”.
• L’assenza dei linfociti T nelle SCID causa linfopenia (da ricordare
sempre che nell’adulto linfopenia = linfociti totali <1000/µL; invece, nel primo anno di vita il limite inferiore normale è 2000/µL
nel neonato e 4000/µL dai 6 ai 9 mesi).
• Esistono però molti casi di SCID con conte linfocitarie T presenti (ridotte – normali – alte): es., SCID T+B+NK+ “funzionale” nei
difetti dei canali del calcio (Feske, 2009); SCID T+(CD4+ CD8-)
B+NK+ nel deficit di ZAP70 (Turul et al., 2009); SCID T+B-NK- nella
Sindrome di Hoyeraal-Hreidarsson (Cossu et al., 2002), ecc.
Le SCID T+ o T++ però si hanno soprattutto per linfociti T anomali
ed oligoclonali che modificano le conte da T-B-NK- a T+B-NK-, da
T-B+NK- a T+B+NK-, da T-B+NK+ a T+B+NK+, o da T-B-NK+ a T+B-NK+.
Tali linfociti anomali (Vβ families oligoclonali; linfociti T memory
CD4+CD45RO+ e linfociti T attivati CD3DR+ alti; linfociti T naive
CD4+CD45RA+, proliferazione linfocitaria in vitro e TRECs molto
bassi – importante per lo screening neonatale) sono presenti in
due situazioni fondamentali:
1. SCID con “engraftment” massivo di linfociti T materni: utili per la
diagnosi di SCID (HLA, DNA e/o cromosomi materni nel sangue
del neonato affetto), i T materni possono causare ad es.: GvHD
cutanea ed epatica; piastrinopenia o pancitopenia autoimmuni
(pre- o post- trapianto); rigetto dell’HSCT dal padre o altro donatore diverso dalla madre (Palmer et al., 2007); gammopatia
monoclonale da espansione clonale di linfociti B (del bambino o
anch’essi di origine materna) in assenza di TReg (Kobrynski et al,,
2006); quadro clinico attenuato in caso di compatibilità HLA tra
feto e madre (Tezcan et al., 2005).
Manifestazioni
extraimmunologiche
Nanismo con arti corti, ipotricosi
Ipoplasia cervelletto, microcefalia,
ritardo accrescimento
Anemia megaloblastica, convulsioni,
rischio di danni neurologici
2. Sindrome di Omenn: causata da mutazioni ipomorfiche (“leaky
SCID”) non solo di RAG1-RAG2 (recombination activating genes)
ma anche di numerosi altri geni le cui mutazioni null causano invece SCID tipica (Villa et al., 2008) (Fig. 3). La Omenn è una SCID
T+ o T++ estremamente grave, con linfociti CD4+TH2 oligoclonali
del bambino (non materni) iper- auto- reattivi, prodotti grazie a
carattere “leaky” della mutazione e carenza di tolleranza immunologica centrale (timo: difetto di cellule epiteliali e dendritiche e
di espressione di AIRE – Autoimmune Regulator Element) e periferica (difetto di TReg) (Cavadini et al., 2005; Liston et al., 2008).
Oltre alle infezioni tipiche delle SCID si hanno infiammazione aggressiva, IgE elevate, eosinofilia, “dermatite” (d.d. con “eczema
atopico”!) ed eritrodermia severa, alopecia, perdita di proteine da
cute e intestino, diarrea intrattabile, edema, disturbi metabolici,
linfoadenomegalia ed epatosplenomegalia (segni e sintomi spesso evolvono nel tempo e non compaiono simultaneamente).
• Dunque, per mutazioni di uno stesso gene sono possibili sia
SCID “tipica” che sindrome di Omenn, ma anche altri quadri atipici e, all’estremo, difetti immunologici modesti con
manifestazioni più tardive. La variabilità dei fenotipi clinici
è un aspetto comune (interferenza di altri fattori genetici,
fattori ambientali, ecc.), talvolta osservato anche in fratelli
con stessa mutazione. In certi casi il fenotipo variante è attenuato per un mosaicismo da reversione genetica spontanea
(correzione della mutazione) in una linea cellulare somatica
che poi si espande (Wada et al., 2008).
Genotipi associati a SCID e aspetti clinici
A parte il caso della anomalia di DiGeorge completa, le SCID sono disordini monogenici a trasmissione autosomica recessiva (AR-SCID)
o X-linked recessiva (XL-SCID). Una classificazione aggiornata si
basa sulla genetica e sui derivanti meccanismi patogenetici prevalenti (Tab. I).
SCID da aumentata apoptosi linfocitaria
La Disgenesia reticolare associa una SCID T-B-NK- ad una neutropenia severa congenita (blocco allo stadio promielocitico): leucociti
totali < 400/µL (“aleukocytosis”), sepsi fatali già nei primi giorni di
vita, assente risposta al G-CSF, sordità neurosensoriale nei lattanti
che sopravvivono grazie all’HSCT. Due gruppi (Lagresle-Peyrou et
al., 2009; Pannicke et al., 2009) ne hanno di recente identificato la
causa: mutazioni (missense; delezioni) del gene AK2 codificante la
adenilato kinasi 2 (AK2) mitocondriale. Le adenilato kinasi catalizza-
231
F. Cossu
Figura 3.
Sindrome di Omenn (iniziale misdiagnosis di “eczema atopico”; paziente negativa per mutazioni di RAG1-RAG2).
La SCID Omenn è stata osservata in associazione con mutazioni ipomorfiche (“leaky SCID”) di numerosi geni: RAG1-RAG2, DCLRE1C-Artemis,
ADA, LIG4-DNA Ligasi IV, RMRP-CHH, IL2RG, IL7RA, anomalia di DiGeorge, WHN-FOXN1, ZAP-70. Si noti l’assenza nella lista, per il momento, di
JAK3. In circa il 50% dei pazienti Omenn la mutazione resta sconosciuta, probabilmente anche per il gran numero di geni da considerare e per il
ruolo di geni ancora non identificati.
no la trasformazione reversibile 2 ADP D ATP + AMP (già dai batteri,
una cellula sopravvive solo se concentrazioni di ATP/ADP/AMP entro
range strettamente controllati). Esistono nelle cellule umane 7 diversi isozimi di adenilato kinasi, con diverse localizzazioni tessutali;
le principali sono AK1 (cytosol) e AK2 – AK3 – AK4 (mitocondriali),
espresse in tutte le cellule nucleate a parte le linee mieloidi e linfocitarie e le cellule della stria vascolare dell’orecchio interno, che
invece esprimono soltanto la AK2. La AK2 è localizzata nello spazio
intermembranoso mitocondriale, come la HAX1 (HCLS1 associated
protein X) il cui difetto causa la neutropenia severa congenita delle
famiglie descritte in origine da Kostmann.
ADA-SCID (Deficit di Adenosina deaminasi): la adenosina deaminasi
(ADA) trasforma adenosina (Ado) in inosina e deossiadenosina (dAdo)
in deossinosina, e il suo deficit causa intossicazione metabolica da
accumulo di Ado, dAdo e dATP (deossiadenosina trifosfato). Oltre ai
noti effetti intracellulari di dAdo e dATP (apoptosi linfocitaria generalizzata "SCID T-B-NK-), l’Ado extracellulare in eccesso (normalmente
è prodotta dai linfociti CD4+TReg) agisce su recettori specifici con ulteriore inibizione linfocitaria (Cassani et al., 2008) e probabile ruolo
nelle manifestazioni autoimmunitarie (anemia emolitica, piastrinopenia, diabete, ipotiroidismo, ecc.) frequenti nella ADA-SCID anche
dopo trattamento (Sauer et al., 2009).
La ADA ha espressione massima nel timo ma comunque ubiquitaria e
nella ADA-SCID sono frequenti importanti manifestazioni extra-immunologiche: anomalie delle giunzioni costocondrali, displasie scheletriche
(vertebre, ossa iliache, ossa lunghe; specifico difetto di rimodellamento
osseo, Sauer et al., 2009); epatite neonatale; sordità neurosensoriale;
anomalie neurologiche (deficit motorio, cognitivo e del comportamento)
con prognosi poco buona anche dopo la correzione del difetto immunitario (Honig et al., 2007) (QI < 2 SDs correlato ai livelli di dATP alla
diagnosi; tra l’altro il deficit isolato di S-adenosil-omocisteina idrolasi,
inibita dalla dAdo, causa di per sé grave ritardo psicomotorio).
232
Per la ADA-SCID esistono realmente diverse opzioni terapeutiche
(Gaspar et al., 2009; Fig. 4):
• l’HSCT senza condizionamento da donatore familiare HLAmatched resta il trattamento di scelta, con successo > 90%;
invece, l’HSCT (con condizionamento mieloablativo o a ridotta
intensità) da donatore HLA-mismatched, specie genitore aploidentico, ha successo solo nel circa 40% e dovrebbe essere
evitato;
• terapia sostitutiva enzimatica: iniezione i.m. 1-2 volte/settimana di PEG-ADA (polyethylene-glycol-modified calf intestinal
ADA; ADAGEN®), protegge dall’intossicazione metabolica sia i
linfociti immaturi (buona ricostituzione immunologica entro 24 mesi) che gli altri tessuti ed è spesso terapia salva-vita alla
diagnosi. Se prolungata > 6 mesi, dà sopravvivenza a 20 anni
di circa 80%; nel 20% restante: mortalità precoce per condizioni
già gravi alla diagnosi, tardiva specie per anemia emolitica refrattaria (1-3 anni), e insufficienza respiratoria cronica, malattie
linfoproliferative, tumori epatici (5-15 anni). A lungo termine (810 anni) si è inoltre osservato un graduale declino di conte e
funzioni linfocitarie.
• terapia genica: come descritto di recente (Aiuti et al., 2009) ha
raggiunto per la ADA-SCID i risultati più promettenti (Box 2).
Deficit di PNP (Purina nucleoside fosforilasi): anche il difetto dell’enzima successivo all’ADA nella “purine salvage pathway” può presentarsi
con un quadro di SCID (la dGTP, deossiguanosina trifosfato, accumulata causa apoptosi dei linfociti, specie T). Da ricordare che le cellule non
producono acido urico: uricemia tipicamente < 2 mg/dL.
È una forma con prognosi grave (Aytekin et al., 2009): la PEG-PNP
non è commercializzata, la terapia genica è ancora in fase di ricerca;
l’unica possibilità terapeutica è l’HSCT, che peraltro non corregge i
gravi disturbi neurologici in genere presenti (iper- o ipotonia, atassia,
ritardo psicomotorio); frequenti anche le manifestazioni autoimmuni
Le basi genetiche delle SCID
Figura 4.
Flow chart per il trattamento della ADA-SCID (PEG-ADA – polyethylene-glycol-modified calf intestinal ADA; HSCT – hematopoietic stem cell
transplantation; da Gaspar et al., 2009, mod.).
(anemia emolitica, piastrinopenia, neutropenia, artriti, ecc.) ed inoltre neoplasie.
STAT5; la maggior parte dei pazienti sono composti eterozigoti di
due mutazioni diverse (Pesu et al., 2005).
SCID da difetti di segnali mediati da citochine
La SCID da difetto di catena gamma common (γc) colpisce solo i maschi in quanto XL-SCID T-B+NK- da mutazioni del gene IL2RG posto in
Xq13.1 e codificante la catena γ del recettore per l’IL-2. David Phillip Vetter (Fig. 2) era affetto da questa forma (con mutazione IL2RG
nonsense exon 7 C937A S308X -62 aa. of 369 aa.); il cosiddetto
“bubble boy paradox” (ad es., sia il topo knockout per IL-2 che i rari
pazienti con difetto di IL-2 non presentano SCID ma invece grave
difetto dei linfociti TReg con autoimmunità severa, ecc.) si è risolto
con la scoperta che la catena γ è condivisa (γ common chain – γc)
anche dai recettori di IL-4, IL-7, IL-9, IL-15, e IL-21; IL-7 e IL-15
sono essenziali per lo sviluppo, rispettivamente, di linfociti T e NK
(Berg, 2008; Rochman et al., 2009).
La XL-SCID è la SCID più frequente (almeno negli USA) ed è una
forma esclusivamente immunologica. Anche i cheratinociti cutanei
però esprimono i recettori citochinici con γc (e JAK3), importanti per
l’immunità innata locale: a distanza di molti anni dall’HSCT (e anche
dopo terapia genica) è frequente una papillomatosi cutanea severa
da infezione cronica da HPV dei cheratinociti nei pazienti con XLSCID o AR-SCID da difetto di JAK3.
L’HSCT senza condizionamento da donatore familiare HLA-matched
ha nella XL-SCID successo > 95%, però con frequente mancata ricostituzione immunologica della linea B (Box 1). La storia della terapia genica per la XL-SCID è invece controversa (Box 2).
Invece, la SCID da Difetto di IL-7Rα (catena α del recettore dell’IL-7) è
una SCID T-B+NK+ in quanto la funzione dell’IL-15 ("NK) è normale ed
è compromessa solo la funzione dell’ IL-7 e della TSLP (thymic stromal lymphopoietin) che condivide la stessa catena α nel suo recettore
(Giliani et al., 2005; Rochman et al., 2009). Nell’uomo l’IL-7, prodotta
dalle cellule stromali dei tessuti linfoidi e dagli epatociti, appare essere
il vero “T cell growth development factor”, e infatti il suo dosaggio
nell’eluato da Guthrie card è un metodo alternativo o complementare
ai TRECs per lo screening di SCID (aumenta per “feedback” in caso di
assente risposta dei linfociti T; IL-7 > 15 pg/mL indica SCID).
La SCID da difetto di JAK3 è l’equivalente AR del Difetto di γc, dato
che la tirosin-chinasi JAK3 (Janus chinasi 3) si associa appunto alla
catena γc nella “signaling pathway” IL-2, 4, 7, 9, 15, 21 - JAK3 -
SCID da difetti del pre-T cell receptor (pre-TCR)
Durante la “T lymphopoiesis” nel microambiente del timo (stadio
di “large pre-T cell”) ha un ruolo essenziale il pre-TCR (Nemazee,
2006), formato da una catena TCRβ (gene TCRβ riarrangiato) e dalla
catena invariante pTα (pre-TCR α chain) codificata dal gene PTCRA
posto in 6p21.2 (quest’ultimo davvero un molto sospetto “gene
candidato” per SCID). Il pre-TCR trasmette il suo segnale mediante
numerose altre molecole comuni anche al TCR (T cell receptor dei
linfociti T maturi): molecole del complesso recettoriale (CD3γ, CD3δ,
CD3ε, CD3ζ), protein-tirosina chinasi (Fyn, Lck, ZAP-70), proteinfosfotirosina fosfatasi (CD45).
C’è evidente analogia con il pre-B cell receptor (pre-BCR) della “B
lymphopoiesis”: i difetti del pre-BCR (Igµ, λ5, VpreB, CD19, BTK)
bloccano lo sviluppo dei linfociti B e causano agammaglobulinemia;
quelli del pre-TCR, distinti in difetti di ricombinazione V(D)J e difetti
di trasmissione del segnale del pre-TCR, bloccano lo sviluppo dei
linfociti T e causano perciò SCID (Fig. 5).
233
F. Cossu
Difetti di ricombinazione V(D)J
La parte variabile, antigene-specifica, della catena TCRβ del preTCR, catena Igµ del pre-BCR e poi catene di TCR, BCR e immunoglobuline è codificata dai relativi segmenti genici riarrangiati mediante
la ricombinazione V(D)J del DNA. Questa avviene in due fasi: una
specifica dei linfociti T e B (“trasposasi” RAG1 e RAG2, codificate
dai RAG-1 e RAG-2); una successiva svolta dalle proteine del Non
Homologous End-Joining (NHEJ: Ku70/80, DNA-PKcs, Artemis, Cernunnos/XLF, DNA ligase IV, XRCC4), complesso che ripara in tutte le
cellule le “double-strand breaks” del DNA (Xu, 2006). Dal difetto di
queste molecole derivano SCID T-B-NK+, con nel primo caso (RAG1,
RAG2) patologia limitata ai linfociti T e B e nel secondo interessante invece tutte le cellule con problemi simili ad altre sindromi con
difetto di riparazione del DNA: radiosensibilità, particolare suscettibilità ai chemioterapici (agenti alchilanti) anche dei condizionamenti
pre-HSCT, predisposizione a neoplasie, e (ma solo in alcune forme:
difetto di DNA ligase IV, difetto di Cernunnos/XLF) facies dismorfica,
microcefalia, malformazioni dell’encefalo, ritardo psicomotorio.
Il difetto di RAG1 o RAG2 causa SCID tipica, Sindrome di Omenn, ed
inoltre rare forme peculiari es. con infezione precoce e generalizzata
da CMV, citopenia autoimmune ed espansione di linfociti T γ/δ, o con
granulomi di cute, mucose, organi interni ed EBV-linfoma (Sobacchi
et al., 2006; Schuetz et al., 2008).
Sono finora noti nell’uomo i difetti di quattro molecole del NHEJ.
La SCID da difetto di Artemis, in cui è mutato il gene DCLRE1C (DNA
cross-link repair protein 1C), è presente con varie mutazioni in tutte
le popolazioni; un tempo era però conosciuta come “SCIDA” cioè
SCID delle popolazioni di lingua Athabaskan (Apache, Nazione Navajo) nelle quali è assai frequente (1/2000 nati) per l’esistenza di un
effetto fondatore (eterozigote per la particolare mutazione exon 8
C576A - Artemis Y192X ben 1 persona ogni 10) (Li et al., 2002).
Il difetto di DNA ligasi IV causa la “Ligase IV syndrome” (simile alla
Nijmegen breakage syndrome, NBS) con, oltre a possibile SCID tipica
o Omenn, facies dismorfica, microcefalia, ritardo di accrescimento
e psicomotorio, anomalie della cute, pancitopenia, predisposizione
(anche negli eterozigoti) a leucemie e altre neoplasie (Chistiakov et
al., 2009).
Il difetto di Cernunnos/XLF causa immunodeficienza combinata con
compromissione immunologica di solito più tardiva rispetto ad una
SCID tipica, ed inoltre dismorfie, microcefalia, ritardo psico-motorio,
insufficienza midollare e mielodisplasia (Buck et al., 2006).
Recentemente in una bambina di origine turca, di genitori consanguinei, con diagnosi di SCID T-B-NK+ all’età di 5 mesi, è stato
poi identificato il primo caso di difetto di DNA-PKcs per mutazione
(missense ipomorfica T9185C L3062R) del gene PRKDC codificante
la DNA-dependent protein kinase catalitic subunit (DNA-PKcs). La
mutazione di tale gene è la nota causa della SCID “naturale” del topo
oltre che di altri animali (cavalli arabi, cani Jack Russell terriers) ed
era perciò stata ricercata a lungo nell’uomo, con notevoli difficoltà
dato che gene e proteina sono davvero grandissimi (86 esoni; 4096
aminoacidi) ed inoltre ad es. l’espressione della proteina può essere
normale con soltanto difetto di funzione (van der Burg et al., 2009).
Tutte le SCID da difetto di ricombinazione V(D)J hanno particolari
problemi con il HSCT (Box 1).
Figura 5.
Rappresentazione schematica del pre-T cell receptor (pre-TCR; timo:
large pre-T cell, con gene TCRβ riarrangiato) e del pre-B cell receptor
(pre-BCR; midollo osseo: large pre-B cell, con gene IgHμ riarrangiato).
I difetti del pre-TCR, distinti in difetti di ricombinazione V(D)J e difetti di
trasmissione del segnale del pre-TCR, bloccano lo sviluppo dei linfociti
T e causano perciò SCID; i difetti del pre-BCR bloccano lo sviluppo dei
linfociti B e causano agammaglobulinemia.
Fyn, Lck, Blk, Lyn, Syk, e Btk (Bruton tyrosine kinase): protein-tirosina chinasi; CD45: protein-fosfotirosina fosfatasi (da Nemazee, 2006, mod.).
234
Difetti di trasmissione del segnale del pre-TCR
AR- SCID T-B+NK+ può aversi per un difetto completo di CD3δ, CD3ε,
CD3ζ o CD3γ (circa 1% di tutte le SCID). Le diverse subunità CD3,
in forma di dimeri γε, δε, e ζζ , si associano al pre-TCR e poi al TCR
e sono indispensabili per assemblaggio nella membrana cellulare,
trasmissione del segnale, e quindi T lymphopoiesis e attivazione dei
linfociti T maturi; sono possibili, per vari meccanismi, fenotipi di gravità assai diversa (Recio et al., 2007; Rieux-Laucat et al., 2006).
Assai rara è anche la AR-SCID T-B+NK+ da difetto di CD45 (Tchilian et
al., 2001). CD45 (LCA –Leucocyte Common Antigen) è una proteintirosina fosfatasi essenziale per la trasmissione del segnale di preTCR e TCR (e anche BCR), costituisce circa il 10% delle proteine di
membrana dei linfociti T e B, ed esiste in multiple isoforme prodotte
da complessi splicing alternativi degli esoni codificanti la parte extracellulare. L’espressione delle diverse isoforme dipende dal tipo di
cellula ed inoltre dallo stato di differenziazione e attivazione cellulare
(es., come noto, linfociti CD4 naive o memory); CD45 è una molecola
di straordinario interesse anche per l’associazione tra polimorfismi e
suscettibilita o resistenza ad es. verso malattie autoimmuni (Tchilian
et al., 2006).
Il difetto di ZAP-70 causa un caratteristico deficit di linfociti CD8+
e può manifestarsi con una AR-SCID T+(CD4+ CD8-)B+NK+ o invece
quadri più attenuati (Turul et al., 2009).
È stato finora descritto un singolo caso di AR SCID T-B+NK+ da difetto
di p56LCK (non identificate mutazioni del gene, ma splicing aberrante con assenza nell’RNA trascritto dell’esone 7 ed espressione della
Le basi genetiche delle SCID
proteina molto ridotta); il difetto di p56LCK si associa anche a CD4
linfopenia idiopatica.
SCID da difetti dei canali del calcio
Le variazioni dei livelli di Ca2+ intracellulare sono un meccanismo
fondamentale per la trasduzione del segnale in tutte le cellule (Feske, 2007). Nei linfociti T l’attivazione del complesso TCR/CD3 causa
un rilascio di Ca2+ dalle scorte del reticolo endoplasmatico (ER-stores), cui segue una store-operated Ca2+ entry (SOCE) cioè un cospicuo ingresso nella cellula dallo spazio extracellulare di altri ioni
Ca2+ per l’apertura di canali della membrana plasmatica (CRAC: Ca2+
release-activated calcium channels); tra i principali effetti finali dell’aumento del Ca2+ intracellulare è la traslocazione del NFAT (nuclear
factor of activated T cells) nel nucleo, con attivazione di uno specifico gruppo di geni . In rari pazienti con AR-SCID si è individuato un
difetto di SOCE-CRAC causato da mutazioni dei geni codificanti due
proteine altamente conservate: ORAI1 (nome ORAI: Ore – Stagioni
della mitologia; è la subunità formante i pori dei CRAC) e STIM1
(stromal interaction molecule-1; è il sensore della concentrazione di
Ca2+ nell’ER ed attiva ORAI1-CRAC).
I fenotipi clinici del difetto di ORAI1 e del difetto di STIM1 sono
simili (Feske, 2009): SCID severa, però T+B+NK+ (differenziazione
e numero normale dei linfociti T, B e NK) con nei linfociti T grave
difetto di influsso di Ca2+ e di proliferazione dopo stimolazione con
mitogeni; ipergammaglobulinemia, ma con deficit di produzione
anticorpale specifica; miopatia congenita non progressiva (lattante
ipotonico); aspetti di displasia ectodermica (anidrosi, difetto dello smalto). Nel difetto di STIM1 si hanno inoltre deficit di linfociti
CD4+TReg e gravi manifestazioni autoimmuni (epatosplenomegalia,
linfoadenomegalia, anemia emolitica autoimmune, piastrinopenia)
(Picard et al., 2009).
SCID da difetti di embriogenesi del timo
Le interazioni tra le cellule timiche (cellule stromali; cellule epiteliali
– TECs; cellule dendritiche midollari) ed i timociti appaiono sempre
più importanti nella patogenesi delle SCID (Poliani et al., 2009; Rodewald, 2008). Due forme di SCID riconoscono un primitivo difetto
embriologico del timo:
Sindrome Nude/SCID
Il gene WHN (wingled-helix-nude) codifica il fattore di trascrizione FOXN1 (forkhead box N 1) espresso selettivamente nell’epitelio di cute e timo, e le sue mutazioni causano il noto fenotipo
“Nude/SCID” del topo e anche l’equivalente umano (alopecia totale
congenita; distrofia ungueale; assenza del timo, con tipica SCID TB+NK+ - Frank et al., 1999). La forma umana è stata identificata in
due sorelle di un piccolo paese della Campania (Acerno), dove per
un effetto fondatore il 6.52% della popolazione è portatrice della
mutazione C792T R255X; FOXN1 appare essenziale anche per lo
sviluppo embrionale del tubo neurale (anencefalia e spina bifida
in un feto affetto; elevata incidenza di aborti precoci nelle coppi di
eterozigoti) (Amorosi et al., 2008).
Anomalia di DiGeorge
La anomalia di DiGeorge completa (circa 1% di tutti i casi di DiGeorge), caratterizzata dalla assenza del timo con conseguente SCID TB+NK+, ed inoltre dismorfie facciali, malformazioni cardiache, assenza delle paratiroidi con tetania ipocalcemica neonatale, ha diverse
eziologie (Markert et al., 2009): circa 50%, “classica” del 22q11.2
di circa 3 Mb interessante > 35 geni, tra cui TBX-1 coinvolto nello
sviluppo di cuore, timo, paratiroidi, palato, viso; circa 25%, asso-
ciazione CHARGE (coloboma, heart defects, atresia choanae, retardated growth and development, genital hypolasia, ear anomalies/
deafness; nella maggioranza dei pazienti mutazione del gene CHD7,
codificante la chromodomain helicase DNA-binding protein-7); circa
15%, embriopatia da madre diabetica. Del 22q11.2 e CHD7 hanno
trasmissione autosomica dominante (mutazione de novo in > 80%);
in circa 1/3 dei casi si ha un quadro tipo Omenn.
La assai particolare terapia per correggere il difetto immunologico
determinato dalla atimia è il trapianto di timo (timo neonatale prelevato durante interventi di cardiochirurgia e posto in coltura; poi
inserimento di “slide” multiple di tessuto in incisioni profonde nei
muscoli quadricipiti del bambino).
SCID da altri meccanismi
Difetto di Coronina 1A: i topi con mutazione del gene CORO1A presentano un severo deficit di linfociti T periferici; questo ha suggerito
la recente scoperta di un difetto di Coronina 1A in una paziente di 13
mesi con quadro di AR-SCID T-B+NK+ e con in un gene CORO1A una
delezione di 2 bp, l’altro gene interamente assente per una delezione di 600 kb in 16p11.2 (Shiow et al., 2009).
Le Coronine (1A, 1B, 1C, 2A, 2B, 7) sono una famiglia di proteine
altamente conservate e antagonizzano (all’opposto della WASP –
Wiskott-Aldrich Syndrome Protein) la polimerizzazione dell’actina,
partecipando alla regolazione delle complesse variazioni dinamiche del citoscheletro. La Coronina 1A è soprattutto espressa nella
linea linfocitaria T: il suo difetto causa in particolare un eccesso di
F-actina nei timociti con drastica riduzione della motilità cellulare,
blocco della dismissione dal timo e ritenzione intratimica dei linfociti T maturi “single positive” CD4+CD8− o CD4−CD8+ (Burkhardt
et al., 2008).
Mutazioni dei geni (CIITA, RFXANK, RFX5, RFXAP) codificanti quattro
fattori che regolano promoter e trascrizione del cluster HLA DR, DP,
DQ (posto in 6p21.3) causano il difetto delle molecole MHC di Classe
II, normalmente espresse da cellule epiteliali timiche, linfociti T attivati e cellule (linfociti B, cellule dendritiche, monociti/macrofagi) che
presentano l’antigene ai linfociti T CD4+. Ne deriva un grave deficit
immunitario con, a parte una minoranza di casi “attenuati”, quadro
di SCID T+(CD4- CD8+)B+NK+ a prognosi particolarmente severa: nei
pazienti finora descritti, successo <50% con l’HSCT (anche da donatore HLA-matched) (Renella et al., 2006).
Mutazioni del gene RMRP (ribonuclease mitochondrial RNA processing), altamente polimorfico, codificante non una proteina ma
l’RNA dell’ “RNase MRP ribonucleoprotein complex” (composto
inoltre da almeno 10 diverse proteine) causano difetto del processing dei ribosomi e di replicazione mitocondriale e cellulare ed un
ampio spettro fenotipico (Notarangelo et al., 2008): la sindrome
CHH (Cartilage hair hypoplasia), AR, frequente negli Amish e nei
Finlandesi (rispettivamente 1:19 e 1:76 individui portatori della
mutazione 70 A>G da effetto fondatore) è una condrodisplasia
metafisaria con nanismo, capelli chiari ipotrofici, e immunodeficienza variante da SCID T-B+NK+ (anche Omenn) a deficit selettivo
di linfociti T CD8+, immunodeficienza combinata senza alterazioni
scheletriche, e all’altro estremo deficit immunitario non significativo. Oltre a CHH, mutazioni di RMRP (in genere in altri punti della
molecola di RNA, lunga 267 nt) causano altre tre displasie ossee:
displasia metafisaria senza ipotricosi (MDWH), displasia cifomelica, e displasia anauxetica.
Nella sindrome di Hoyeraal-Hreidarsson (HHS) è patognomonica l’associazione tra immunodeficienza (SCID T+B-NK-) e ipoplasia del cervel-
235
F. Cossu
letto; la forma XL, da mutazione del gene DKC1 codificante la discherina (componente della telomerasi) è la variante severa infantile della
discheratosi congenita (Cossu et al., 2002); sono stati descritti anche
casi di HHS, ancora più rari, a trasmissione autosomica recessiva.
Infine, si è di recente definito che il difetto di PCFT (proton-coupled folate transporter) con malassorbimento intestinale dei folati
(HFM – hereditary folate malabsorption) può presentarsi con un
fenotipo SCID T+B+NK+ ai 3-4 mesi di età (deplezione delle riserve
acquisite per via transplacentare; sono particolarmente sensibili
i linfociti ed il sistema nervoso in formazione). Si hanno arresto
dell’accrescimento, diarrea, gravi infezioni, agammaglobulinemia,
assente proliferazione linfocitaria ed inoltre anemia (megaloblastica; però mascherata a normocitica se concorrente carenza di
ferro), convulsioni, gravi rischi per il sistema nervoso. L’acido folico plasmatico è indosabile. È una SCID speciale, “reversibile”:
la terapia con acido folinico per via parenterale consente una
drammatica correzione del difetto immunologico ed impedisce,
se tempestiva, l’instaurarsi di danni neurologici irreversibili (Borzutzky et al., 2009).
Box 1 di orientamento
Trapianto di cellule staminali ematopoietiche (hematopoietic stem cell transplantation – HSCT)
È l’unica possibilità di cura per la quasi totalità dei bambini affetti da SCID; recenti lavori (Mazzolari et al., 2009; Railey et al., 2009; Neven et al., 2009)
ne descrivono i risultati a lungo termine in oltre 300 pazienti. Le percentuali di successo, assai migliorate negli ultimi decenni, sono del 70-95%, con
però variabile incidenza anche di importanti complicanze. Diversi principali fattori influiscono sul risultato del trapianto:
• Condizioni cliniche al momento della diagnosi: si conferma sempre più l’esito migliore in caso di diagnosi e HSCT entro i primi 3-4 mesi di vita,
con quindi bambino in condizioni migliori, assenza di infezioni virali, ecc.; è evidente a questo riguardo l’importanza dello screening neonatale.
• Tipo di donatore: un familiare HLA-matched è ovviamente il donatore ideale, però disponibile solo in una minoranza di casi; l’alternativa è un donatore HLA-matched non familiare o, se anche questo non disponibile in tempi brevi, un genitore HLA-aploidentico (con midollo rigorosamente depleto
di linfociti T).
• Non condizionamento versus condizionamento e genotipo/fenotipo della SCID: l’attecchimento senza condizionamento è in teoria possibile
nella maggioranza dei pazienti, in particolare se SCID NK- quali difetto di γc e di JAK3, con però alta incidenza di mancata ricostituzione funzionale
della linea B e quindi necessità di trattamento con Ig a tempo indefinito. Gli attuali protocolli di condizionamento non mieloablativi (es., con fludarabina)
comportano anche in questi bambini di pochi mesi di età una minore tossicità, e presumibilmente meno effetti a lungo termine es. sull’accrescimento
e sulle ghiandole endocrine. D’altra parte, nelle SCID con difetto di ricombinazione V(D)J (soprattutto forme da difetto di NHEJ) il difetto molecolare
causa problemi di riparazione del DNA e quindi una particolare sensibilità al condizionamento (soprattutto agenti alchilanti). Non solo per la diversa
sensibilità al condizionamento, il genotipo/fenotipo delle SCID influenza il risultato dell’HSCT: ad es., risultati migliori nelle SCID da difetto di γc e JAK3
(a parte frequente papillomatosi da HPV) e IL-7Rα, peggiori nei difetti di ricombinazione V(D)J (elevato rischio di tossicità acuta da condizionamento; e,
pur in presenza di ricostituzione immunitaria soddisfacente, frequenti manifestazioni croniche autoimmuni, problemi intestinali, ecc.) e nella ADA-SCID
(frequenti complicanze neurologiche permanenti: ritardo mentale, disfunzioni motorie, iperattività, sordità).
Box 2 di orientamento
Terapia genica (TG).
L’inserzione mediante un vettore virale (al momento, Moloney murine leukemia virus – MLV –, defettivi) del gene normale nel DNA delle cellule staminali
ematopoietiche CD34+ (midollo autologo) da cui poi si differenziano anche i linfociti, ha dato risultati differenti nelle due forme di SCID in cui ha avuto
finora applicazione pratica (Quasim et al., 2009):
• XL-SCID da difetto di γc: a partire dal 1999, due gruppi europei (Parigi; Londra) hanno eseguito TG su rispettivamente 11 e 10 pazienti (di età da
1 mese a 33 mesi); si è ottenuta ricostituzione immunologica in 17/21 pazienti, con indipendenza dalle Ig in 12/21 pazienti; però, 5/21 pazienti (4
nello studio francese, 1 in quello inglese), hanno presentato a distanza di 24 - 68 mesi dal trattamento una leucemia acuta linfoblastica T (T-ALL) per
principalmente inserzione (mutagenesi inserzionale) di una singola copia intatta del retrovirus defettivo contenente il gene IL2RG ma anche il proprio
enhancer in prossimità del promoter del LMO2 (noto oncogene dei linfociti T) con sua aberrante trascrizione ed espressione. Un paziente è deceduto
per la leucemia, mentre gli altri sono stati curati con chemioterapia; l’occorrenza di questa grave complicanza ha portato alla sospensione della TG per
XL-SCID in Europa (Howe et al., 2008).
Negli USA è attualmente in corso un trial di TG (al momento chiuso; NCT00028236) che ha reclutato 8 pazienti già sottoposti senza successo a trapianto
allogenico di cellule staminali: nei primi tre pazienti, di età 10-14 anni, la ricostituzione immunologica è stata scarsa, probabilmente in rapporto con
l’età più avanzata ed involuzione timica, infezioni croniche virali, ecc.
• Nella ADA-SCID la TG ha invece dato risultati molto promettenti ed è realmente indicata nei pazienti privi di donatore HLA-matched, come mostrato
in particolare dalla casistica del gruppo di Alessandro Aiuti (Gaspar et al., 2009): TG con un migliorato protocollo di transduzione delle cellule CD34+ da
parte di un vettore MLV defettivo, un condizionamento non-mieloalblativo (busulfano 2 mg/kg ev a -3 e -2) per “fare spazio nel midollo”, e la sospensione della PEG-ADA per dare un vantaggio selettivo ai linfociti con il gene corretto, in 15 pazienti di età da 6 mesi a 5.6 anni. Tutti i 15 pazienti sono
vivi, con nei primi 10 (Aiuti et al., 2009): in 9/10 buona ricostituzione T, in 8/10 espressione di ADA persistente con “detossificazione sistemica” senza
più necessità di PEG-ADA fino a 8 anni dopo la TG, in 5/10 non più necessità di trattamento con Ig. Risultati simili si sono osservati nei pazienti di altri
gruppi; in particolare, in nessun paziente con ADA-SCID trattato con TG si è finora osservata la complicanza della proliferazione clonale leucemica da
mutagenesi inserzionale, e questo nonostante il vettore si integri comunque “pericolosamente” in prossimità di LMO2 o altri noti oncogeni (Sauer et
al., 2009).
Attualmente c’è un’intensa ricerca per altri vettori virali che non presentino problemi di mutagenesi inserzionale: i SIN (self-inactivating) Lentivirus si
integrano anche nelle cellule non in mitosi e (al contrario dei gammaretrovirus quali il MLV) non hanno come target preferenziale di inserzione le regioni
promoter dei geni attivi. Una recente osservazione (espansione clonale da integrazione del vettore Lentivirus nel gene della proteina DNA-binding
HMGA2 in un paziente del trial di TG per talassemia in corso in Francia; Williams, 2009) impone però ulteriore cautela.
236
Le basi genetiche delle SCID
Bibliografia
Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to
adenosine deaminase deficiency. N Engl J Med 2009;360:447-58.
** Descrive i risultati assai promettenti della terapia genica dell’ADA-SCID.
Amorosi S, D’Armiento M, Calcagno G, et al. FOXN1 homozygous mutation associated with anencephaly and severe neural tube defect in human athymic
Nude/SCID fetus. Clin Genet 2008;73:380-4.
Aytekin C, Dogu F, Tanir G, et al. Purine nucleoside phosphorylase deficiency with
fatal course in two sisters. Eur J Pediatr 2010;169:311-4.
Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn
screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol 2009;124:522-7.
** Lo screening del neonato per SCID nello stato del Wisconsin.
Berg LJ. The “bubble boy” paradox: an answer that led to a question. J Immunol
2008;181:5815-6.
Borzutzky A, Crompton B, Bergmann AK, et al. Reversible severe combined immunodeficiency phenotype secondary to a mutation of the proton-coupled folate
transporter. Clin Immunol 2009;133:287-94.
Buck D, Malivert L, de Chasseval R, et al. Cernunnos, a novel nonhomologous
end-joining factor, is mutated in human immunodeficiency with microcephaly.
Cell 2006;124:287-99.
Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation.
Annu Rev Immunol 2008;26:233-59.
Cassani B, Mirolo M, Cattaneo F, et al. Altered intracellular and extracellular signaling leads to impaired T-cell functions in ADA-SCID patients. Blood
2008;111:4209-19.
Cavadini P, Vermi W, Facchetti F, et al. AIRE deficiency in thymus of 2 patients
with Omenn syndrome. J Clin Invest 2005;115:728-32.
Chistiakov DA, Voronova NV, Chistiakov AP. Ligase IV syndrome. Eur J Med Genet
2009;32:573-8.
Cossu F, Vulliamy TJ, Marrone A, et al. A novel DKC1 mutation, severe combined
immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant
with Hoyeraal-Hreidarsson syndrome. Br J Haematol 2002;119:765-8.
Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 2007;7:690-702.
Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated
Ca2+ entry in the immune system and beyond. Immunol Rev 2009;231:189-209.
** Le SCID da difetti dei canali del calcio.
Frank J, Pignata C, Panteleyev AA, et al. Exposing the human nude phenotype.
Nature 1999;398:473-4.
Gaspar HB, Aiuti A, Porta F, et al. How I treat ADA deficiency. Blood
2009;114:3524-32.
** Le linee guida per il trattamento dell’ADA-SCID.
Giliani S, Mori L, de Saint Basile G, et al. Interleukin-7 receptor alpha (IL7Ralpha) deficiency: cellular and molecular bases. Analysis of clinical, immunological, and molecular features in 16 novel patients. Immunol Rev
2005;203:110-26.
Honig M, Albert MH, Schulz A, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of
CNS complications. Blood 2007;109:3595-602.
** I problemi neurologici permanenti anche dopo trattamento nell’ADA-SCID.
Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene
therapy of SCID-X1 patients. J Clin Invest 2008;118:3143-50.
** La mutagenesi inserzionale osservata dopo terapia genica in pazienti con
SCID da difetto di γc.
Kobrynski LJ, Abramowsky C. Monoclonal IgA gammopathy due to maternal B cells in an infant with severe combined immunodeficiency (SCID) prior
to hematopoietic stem cell transplantation. J Pediatr Hematol Oncol 2006;
28:53-6.
Lagresle-Peyrou C, Six EM, Picard C, et al. Human adenylate kinase 2 deficiency
causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet 2009;41:106-11.
** La scoperta del gene che causa la disgenesia reticolare.
Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like
protein, causes SCID in Athabascan-speaking Native Americans. J Immunol
2002;168:6323-9.
Liston A, Enders A, Siggs OM. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat Rev Immunol 2008;8:545-58.
Markert ML, Devlin BH, Chinn IK, et al. Thymus transplantation in complete DiGeorge anomaly. Immunol Res 2009;44:61-70.
Mazzolari E, de Martiis D, Forino C, et al. Single-center analysis of long-term outcome after hematopoietic cell transplantation in children with congenital severe
T cell immunodeficiency. Immunol Res 2009;44:4-17.
Mold JE, Michaëlsson J, Burt TD, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 2008;322:1562-5.
Muench MO. In utero transplantation: baby steps towards an effective therapy.
Bone Marrow Transplant 2005;35:537-47.
** I risultati dettagliati del trapianto in utero nelle varie patologie.
Nemazee D. Receptor editing in lymphocyte development and central tolerance.
Nat Rev Immunol 2006;6:728-40.
Neven B, Leroy S, Decaluwe H, et al. Long-term outcome after hematopoietic
stem cell transplantation of a single-center cohort of 90 patients with severe
combined immunodeficiency. Blood 2009;113:4114-24.
Notarangelo LD, Roifman CM, Giliani S. Cartilage-hair hypoplasia: molecular basis and heterogeneity of the immunological phenotype. Curr Opin Allergy Clin
Immunol 2008;8:534-9.
Ochs H, Smith CIE, Puck J eds. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. Second Edition. New York: Oxford University
Press 2007.
Palmer K, Green TD, Roberts JL, et al. Unusual clinical and immunologic manifestations of transplacentally acquired maternal T cells in severe combined immunodeficiency. J Allergy Clin Immunol 2007;120:423-8.
Pannicke U, Hönig M, Hess I, et al. Reticular dysgenesis (aleukocytosis) is caused
by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet
2009;41:101-5.
** La scoperta del gene che causa la disgenesia reticolare.
Pesu M, Candotti F, Husa M, et al. Jak3, severe combined immunodeficiency, and
a new class of immunosuppressive drugs. Immunol Rev 2005;203:127-42.
Picard C, McCarl CA, Papolos A, et al. STIM1 mutation associated with a syndrome
of immunodeficiency and autoimmunity. N Engl J Med 2009;360:1971-80.
Poliani PL, Vermi W, Facchetti F. Thymus microenvironment in human primary
immunodeficiency diseases. Curr Opin Allergy Clin Immunol 2009;9:489-95.
** Le alterazioni del timo nelle diverse forme di SCID e di immunodeficienze
primitive.
Qasim W, Gaspar HB, Thrasher AJ. Progress and prospects: gene therapy for
inherited immunodeficiencies. Gene Ther 2009;16:1285-91.
** Il punto attuale sulla terapia genica delle SCID (e malattia granulomatosa
cronica).
Railey MD, Lokhnygina Y, Buckley RH. Long-term Clinical Outcome of Patients
with Severe Combined Immunodeficiency Who Received Related Donor Bone
Marrow Transplants without Pretransplant Chemotherapy or Post-transplant
GVHD Prophylaxis. J Pediatr 2009;155:834-40 [Epub ahead of print].
Recio MJ, Moreno-Pelayo MA, Kiliç SS, et al. Differential biological role of CD3
chains revealed by human immunodeficiencies. J Immunol 2007;178:255664.
Renella R, Picard C, Neven B, et al. Human leucocyte antigen-identical haematopoietic stem cell transplantation in major histocompatiblity complex class II
immunodeficiency: reduced survival correlates with an increased incidence of
acute graft-versus-host disease and pre-existing viral infections. Br J Haematol
2006;134:510-6.
Rezaei N, Aghamohammadi A, Notarangelo LD (eds.). Primary immunodeficiency
diseases: definition, diagnosis, and management. Berlin Heidelberg: SpringerVerlag, 2008.
Rieux-Laucat F, Hivroz C, Lim A, et al. Inherited and somatic CD3zeta mutations
in a patient with T-cell deficiency. N Engl J Med 2006;354:1913-21.
Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by
γc family cytokines. Nat Rev Immunol 2009;9:480-90.
Rodewald HR. Thymus organogenesis. Annu Rev Immunol 2008;26:355-88.
Sauer AV, Aiuti A. New insights into the pathogenesis of adenosine deaminasesevere combined immunodeficiency and progress in gene therapy. Curr Opin
Allergy Clin Immunol 2009;9:496-502.
Sauer AV, Mrak E, Jofra Hernandez R, et al. ADA-deficient SCID is associated with
a specific microenvironment and bone phenotype characterized by RANKL/OPG
imbalance and osteoblast insufficiency. Blood 2009;114:3216-26.
237
F. Cossu
Schuetz C, Huck K, Gudowius S, et al. An immunodeficiency disease with RAG
mutations and granulomas. N Engl J Med 2008;358:2030-8.
Shiow LR, Paris K, Akana MC, et al. Severe combined immunodeficiency (SCID)
and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A
mutation and a chromosome 16p11.2 deletion. Clin Immunol 2009;131:24-30.
** Il primo caso di SCID umana da difetto di Coronina 1A.
Sobacchi C, Marrella V, Rucci F, et al. RAG-dependent primary immunodeficiencies. Hum Mutat 2006;27:1174-84.
Spits H. Development of α/β T cells in the human thymus. Nat Rev Immunol
2002;2:760-72.
Tchilian EZ, Wallace DL, Wells RS, et al. A deletion in the gene encoding the CD45
antigen in a patient with SCID. J Immunol 2001;166:1308-13.
Tchilian EZ, Beverley PC. Altered CD45 expression and disease. Trends Immunol
2006;27:146-53.
Tezcan I, Ersoy F, Sanal O, et al. Long-term survival in severe combined immune deficiency: the role of persistent maternal engraftment. J Pediatr 2005;146:137-40.
Turul T, Tezcan I, Artac H, et al. Clinical heterogeneity can hamper the diagnosis
of patients with ZAP70 deficiency. Eur J Pediatr 2009;168:87-93.
van der Burg M, van Dongen JJ, van Gent DC. DNA-PKcs deficiency in human:
long predicted, finally found. Curr Opin Allergy Clin Immunol 2009;9:503-9.
** Il primo caso umano di SCID da mutazione del gene PRKDC.
Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky
severe combined immunodeficiency. J Allergy Clin Immunol 2008;122:10826.
** La sindrome di Omenn è causata da mutazioni “leaky” non solo di RAG1-RAG2
ma di molti altri geni delle SCID.
Wada T, Candotti F. Somatic mosaicism in primary immune deficiencies. Curr
Opin Allergy Clin Immunol 2008;8:510-4.
Williams DA. Gene therapy continues to mature and to face challenges. Mol Ther
2009;17:1305-6.
Xu Y. DNA damage: a trigger of innate immunity but a requirement for adaptive
immune homeostasis. Nat Rev Immunol 2006;6:261-70.
Corrispondenza
dott. Fausto Cossu, Centro Trapianti Midollo Osseo, Clinica Pediatrica II, Ospedale Microcitemico, via Jenner, 09121 Cagliari. E-mail: [email protected]
238
Ottobre-Dicembre 2009 • Vol. 39 • N. 156 • Pp. 239-245
FOCUS SU:
Obesità e sindrome metabolica in età pediatrica
Gianni Bona, Arianna Busti, Flavia Prodam, Simonetta Bellone
Clinica Pediatrica, Dipartimento di Scienze Mediche, Università del Piemonte Orientale Novara
Riassunto
In Italia la prevalenza del sovrappeso e dell’obesità sono tra le maggiori in Europa, con un tasso di crescita annuo pari agli Stati Uniti. L’interessamento
precoce dell’infanzia ha reso necessario parlare di Sindrome Metabolica anche in età pediatrica, presente nel 30% dei bambini obesi. Alla base di tale
patologia, meccanismi ambientali, metabolici, ormonali e, di recente scoperta, genetici sono responsabili di un circolo vizioso che porta alla comparsa di
pubertà precoce, insulinoresistenza e patologie cardiovascolari precoci. Per far fronte a tale problema sono necessari strumenti diagnostici e terapeutici
precisi e validati, che possano essere monitorati nel tempo. In campo terapeutico il ruolo fondamentale è svolto dalla dietoterapia e dall’attività fisica, ma
la definizione di obesità come malattia multifattoriale, sta aprendo il campo a nuovi farmaci, tra cui la metformina, già approvata per diabete mellito 2. La
terapia dell’obesità e complicanze implica una diagnosi ed una definizione precisa di obesità e Sindrome Metabolica, spesso difficile in età pediatrica in
cui permangono difficoltà legate alle modificazioni somatiche e metaboliche durante l’accrescimento. Negli anni le società scientifiche hanno sviluppato
classificazioni e parametri metabolici e fisici che si adattassero a tali esigenze, spesso modificando classificazioni redatte per l’età adulta. In particolare, la
scoperta del ruolo chiave dell’obesità centrale e delle modificazioni epatiche nell’insulinoresistenza, ha ridotto l’importanza di parametri quali il body mass
index a favore della circonferenza vita e del nuovo rapporto vita/altezza e dello studio ecografico del fegato. Molto resta ancora da fare, soprattutto per la
definizione di sindrome metabolica.
Summary
In Italy the prevalence of overweight and obesity are among the largest in Europe, with an annual growth rate equal to the United States. Involvement early
childhood has made it necessary to speak of metabolic syndrome in childhood also, present in 30% of obese children. Environmental factors, metabolic
mechanisms, hormonal, and newly discovered, genetic mechanisms, are responsible for a cycle that leads to the onset of early puberty, insulin resistance and cardiovascular disease. To resolve this problem are necessary accurate and validated diagnostic tools and appropriate therapies, which can be
monitored over time. The key role is played by diet therapy and physical activity, but the definition of obesity as a multifactorial disease, is opening the field
to new drugs, including metformin, which is already approved for diabetes mellitus 2. Treatment and complications of obesity involves a diagnosis and
a precise definition of obesity and metabolic syndrome, often difficult in children, where there are still problems associated with somatic and metabolic
changes during growth. Over the years, scientific societies have developed new classifications and metabolic and physical parameters, often by changing
classifications prepared for adulthood. In particular, the discovery of the role of central obesity for the insulin insensitivity and liver changes, reduced the
importance of parameters such as body mass index and give much importance to waist circumference, to the new waist/height ratio and to ultrasound for
the study of liver. Much remains to be done, especially for the definition of metabolic syndrome in childhood.
Introduzione
Modalità di ricerca bibliografica
L’obesità venne inserita per la prima volta nella Classificazione Internazionale delle Malattie, a partire dal 1948. Spesso risulta ancora
attuale la convinzione che il grasso corporeo sia solo un deposito di
energia, senza funzioni ormonali e metaboliche, e che il suo aumento possa costituire solo un problema estetico o al più un impaccio
per le prestazioni motorie, piuttosto che una malattia con ripercussioni sulla salute. La maggior parte dei genitori non conosce le
conseguenze metaboliche dell’obesità ed è convinta che una buona
dieta possa risolvere da sola il problema. I dati epidemiologici citati
evidenziano il carattere epidemico dell’obesità in età pediatrica e
come la sua insorgenza nei primi anni dell’infanzia, abbia reso necessario considerare anche l’età pediatrica a rischio di sviluppare la
sindrome metabolica (SM).
Scopo di tale revisione, è stimare le reali dimensioni del problema
“obesità/SM” in età pediatrica e fare chiarezza sui criteri diagnostici
a disposizione del medico per una corretta diagnosi ed intervento.
Altro scopo è presentare i numerosi progressi della conoscenza in
campo metabolico, guardando al bambino obeso non solo come un
soggetto “inattivo e mangione”, ma come un soggetto in cui sia fattori genetici/ambientali, sia modificazioni metaboliche, possano determinare l’insorgenza di un circolo vizioso che porterà allo sviluppo
di SM e patologie cardiovascolari.
La ricerca degli articoli rilevanti su “Obesità e Sindrome Metabolica”
è stata effettuata sulla banca bibliografica Medline utilizzando come
motore di ricerca PubMed e la funzione MeSH database. Come parole chiave: Metabolic Syndrome, Body Mass Index, Classification,
NCEP, school program, Oveweight, Obesity, Polycystic Ovary Syndrome, Insuline Resistence. Sono stati utilizzati i seguenti filtri: bambini
da 0-18 anni.
Obesità in età pediatrica
Le dimensioni del problema
L’Organizzazione Mondiale della Sanità (OMS) afferma che circa un
miliardo di persone nel modo è in sovrappeso e 300 milioni sono
invece obesi. L’epidemiologia dell’obesità infantile, pur presentando aspetti di difficile interpretazione, mancando sicuri standard di
riferimento, sottolinea il trend in crescita negli ultimi 20 anni, raggiungendo i caratteri di un’epidemia nei Paesi industrializzati (Stati
Uniti, Europa), ma coinvolgendo progressivamente anche Paesi in
via di sviluppo (Asia, India, Sud America). In Europa la prevalenza del
sovrappeso in età pediatrica è del 15-17% e del 3% per l’obesità.
Negli Stati Uniti, la prevalenza dell’obesità in età pediatrica (18%) è
in rapido incremento, ipotizzando che nel 2050, i soggetti obesi di
239
G. Bona et al.
età inferiore ai 20 anni saranno circa il 25% (Kipping et al., 2008;
Termizy et al., 2009). In Italia il quadro epidemiologico è in parte
sovrapponibile agli altri Paesi industrializzati, ma negli ultimi anni
il problema sta assumendo dimensioni sempre maggiori, tra le più
preoccupanti in Europa. Gli ultimi dati a disposizione (1999-2002)
mostrano come la prevalenza del sovrappeso sia del 20%, mentre la
quota degli obesi sia pari al 4%. Il problema interessa soprattutto gli
adolescenti di sesso maschile, età in cui si raggiunge un picco per
il sovrappeso (36,8%), ed il Sud Italia (17% al Nord, 21% al Centro
e 25% al Sud, composta per il 66% da maschi) (fonte ISTAT 2006).
In Italia la prevalenza di sovrappeso in età pediatrica è superiore di
circa 3 punti percentuali rispetto alla media Europea, con un tasso
di crescita/annua di circa 0,5-1%/anno, raggiungendo un incremento annuo pari a quello degli Stati Uniti. Anche in età pediatrica
le complicanze ad essa correlate (diabete mellito tipo 2 – DM2 –,
ipertensione arteriosa) sono in netto aumento, presentandosi più
precocemente ed aggravando lo stato di salute della popolazione.
Si stima infatti che il 2-8% dei costi globali per la Sanità sia legato
proprio all’obesità.
Definizione e diagnosi
L’obesità è definita come un eccessivo accumulo di grasso corporeo.
Tale definizione presuppone un’accurata misurazione della massa
adiposa e valori di riferimento validati nei due sessi e nelle varie età,
per poter definire una condizione di normalità o di patologia. Entrambe questi presupposti non sono disponibili per la comune pratica clinica. Infatti la misurazione del grasso corporeo è complessa
e le metodiche più accurate (densitometria DEXA) non sono adatte
ad un uso routinario. Alternative sono la bioimpedenziometria e la
misurazione delle pliche di grasso sottocutaneo, entrambe limitate
dall’assenza di cut off di riferimento per età e sesso. Durante l’adolescenza lo spessore delle pliche sottocutanee è il miglior predittore
del grasso corporeo in età adulta, ma è spesso impreciso per la
variabilità intraoperatore. A sua volta l’accuratezza e riproducibilità
dell’impedenziometria è limitata dalle variazioni dello stato di idratazione del soggetto (assunzione di cibo, età, stadio dello sviluppo).
Nella pratica clinica quotidiana si preferisce utilizzare degli indici
surrogati ed indiretti di stima del tessuto adiposo, tra cui il body
mass index (BMI), la circonferenza vita (CV), il rapporto vita/fianchi
ed il più recente rapporto vita/altezza. In età pediatrica non esistono
cutoff validati per tali indici, in quanto non terrebbero conto dei cambiamenti durante lo sviluppo e la crescita. Il valore calcolato viene
confrontato così con curve di riferimento.
Il più utilizzato è il BMI (peso in kg/quadrato altezza in metri) che,
rispetto al solo peso, meglio descrive la distribuzione della massa
corporea. Le curve Italiane per i soggetti dai 2 ai 20 anni sono state
recentemente pubblicate nel 2006 (Cacciari et al., 2006) e definiscono
sovrappeso un BMI superiore al 75° centile ed obeso un soggetto con
BMI > 95°. Non tutte le società scientifiche concordano sui centili di
riferimento: per esempio, l’American Academy of Pediatrics e l’Endocrine Society (Kavey et al., 2006) definiscono un soggetto sovrappeso
se > 84° centile ed obeso se > 94°. Inoltre, tutte le curve di BMI
sono basate sull’età e sesso ma non sugli stadi di Tanner e possono sovrastimare, o sottostimare il sovrappeso in soggetti con pubertà
anticipata, o ritardata. Considerate tali limitazioni, alcuni autori hanno
proposto l’utilizzo di un indice standardizzato, il BMI z score. Questo
rappresenta il numero di deviazioni standard sopra o sotto la media
del BMI calcolata per età e sesso. Le difficoltà all’uso del BMI z score
dipendono dallo scarso utilizzo nella pratica clinica e dalla non immediata comprensione da parte dei genitori, rendendolo meno utile
nell’educazione del nucleo famigliare (Daniels et al., 2009).
240
La misurazione della CV nella pratica clinica è andata aumentando
negli ultimi anni (misura della circonferenza orizzontale passante
per le creste iliache con un metro flessibile, nel soggetto in posizione eretta a piedi uniti). In Italia vengono utilizzate le curve redatte
nel 1996, precedenti però alla pubblicazione delle nuove carte di
crescita italiane, non rispecchiando la reale distribuzione della popolazione pediatrica e denunciando la necessità di costruirne delle
nuove. Numerosi studi hanno invece utilizzato le più recenti curve
europee (Zanolli et al., 1996, Savva et al., 2000, Fernández et al.,
2004) (Tab. I).
La CV si è dimostrata utile nel predire le complicanze dell’obesità,
fornendo indicazioni sufficienti, seppur surrogate, sul tessuto adiposo viscerale, maggiormente correlato al rischio cardiovascolare.
Studi su adulti hanno dimostrato infatti che una CV > 95° centile
correla meglio con alterazioni (99%) della risposta insulinica e del
quadro lipidico rispetto al gruppo con CV nei limiti (34%) (Savva et
al., 2000).
Per meglio correlare la CV con la distribuzione della massa grassa,
questo parametro viene spesso rapportato ad altre misure antropometriche (fianchi, altezza). Il rapporto vita/fianchi identifica una distribuzione androide o ginoide della massa grassa, ma è fortemente
influenzato dalla struttura ossea, dalla costituzione corporea e dalla
postura, non utile nel predire il rischio cardiovascolare.
Attualmente si sta affermando l’utilizzo del rapporto vita/altezza (vita
in cm/altezza in cm). I valori ottenuti vengono confrontati con curve e considerati a rischio quando superiori al 75° centile (rapporto
pari a 0,5). Il rapporto vita/altezza correla meglio del BMI e della
sola CV con le alterazioni del quadro lipidico, in particolare con la
riduzione del colesterolo HDL (high density lipoprotein) ed il livello di
aterosclerosi, oltre che con il grado di resistenza insulinica sia in età
adulta sia pediatrica (Savva et al., 2000; Maffeis et al., 2009).
Nessuno di questi parametri da solo sembrerebbe essere migliore
degli altri nella definizione di obesità, ma l’utilizzo in associazione è
fortemente raccomandato per poter identificare soggetti con grasso
Tabella I.
Centili della circonferenza vita per sesso ed età europei (misure in cm)
(da Fernandez et al., 2004, mod.).
Percentile maschi
Percentile femmine
Età
75°
90°
75°
90°
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
48,6
51,2
53,8
56,5
59,1
61,7
64,3
67,0
69,6
72,2
74,9
77,5
80,1
82,8
85,4
88,0
90,6
50,6
54,0
57,4
60,8
64,2
67,6
71,0
74,3
77,7
81,1
84,5
87,9
91,3
94,7
98,1
101,5
104,9
49,6
51,9
54,2
56,5
58,8
61,1
63,4
65,7
68,0
70,3
72,6
74,9
77,2
79,5
81,8
84,1
86,4
52,5
55,4
58,2
61,1
64,0
66,8
69,7
72,6
75,5
78,3
81,2
84,1
86,9
89,8
92,7
95,5
98,4
Obesità e sindrome metabolica in età pediatrica
prevalentemente viscerale ed indirizzare ad ulteriori esami di approfondimento (glicemia a digiuno, quadro lipidico, carico orale di glucosio) i pazienti che risultassero predisposti ad un maggiore rischio
cardiovascolare, nell’ottica di ridurre la medicalizzazione ed avere a
disposizione parametri facilmente monitorabili nel tempo.
Tali indicazioni sono valide, seppur discusse, sia in età adulta sia
pediatrica (de Kroon et al., 2008).
La sindrome metabolica in età pediatrica
Definizione ed epidemiologia
La SM è stata per la prima volta definita nel 1988 da Reaven et al.,
come un’insieme di alterazioni metaboliche o un insieme di fattori di
rischio cardiovascolare (resistenza insulinica, ipertensione arteriosa,
alterazioni del quadro lipidico ed incremento ponderale a distribuzione centrale), in grado di determinare una patologia conclamata
come il DM2 o patologie cardiovascolari.
Studi osservazionali retrospettivi Europei in età pediatrica (19982004) hanno dimostrato che la prevalenza della SM in età pediatrica
cresce parallelamente all’aumentare del grado di obesità, risultando
circa del 4-6% nei soggetti sovrappeso e del 28,7-30% nei soggetti obesi. Il sesso femminile è maggiormente colpito, soprattutto
durante la pubertà. Tali dati sono però imprecisi, non considerando l’incrementata prevalenza dell’obesità infantile negli ultimi 5-10
anni e basandosi su criteri non univoci, differenti nei diversi gruppi
di studio, non sempre validati per l’età pediatrica (de Kroon et al.,
2008; Cook et al., 2003).
Classificazione e diagnosi
La diagnosi di SM viene posta sulla base della combinazione di dati
clinici, fisici (BMI, pressione arteriosa, CV) ed esami laboratoristici
(colesterolo totale, colesterolo low density lipoprotein (LDL) e HDL,
trigliceridi, glicemia, carico orale di glucosio).
L’assenza di criteri e parametri validati in età pediatrica è un problema attuale, che rende spesso difficile classificare il “quadro meta-
bolico” di un paziente in una categoria ad alto, medio o basso rischio
cardiovascolare futuro. A tale problema differenti gruppi di studio
e società scientifiche hanno cercato di dare una risposta, creando
classificazioni, la maggior parte redatte per l’età adulta (OMS, 1992;
European group for the study of insuline resistence, 2005).
Tali criteri non possono essere utilizzati in età pediatrica senza le opportune modifiche, in quanto non tengono conto del processo di accrescimento e delle relative modificazioni somatiche e metaboliche, in
particolare nel periodo dell’adolescenza, durante il quale si presenta
fisiologicamente un’aumentata resistenza insulinica senza necessariamente sviluppare SM (Cook et al., 2003; Alberti et al.; 2005).
La prima classificazione pediatrica risale al 1992, a cura del National Cholesterol Education Program (NCEP), modificando i cut-off di
riferimento per il colesterolo e per i trigliceridi dell’adulto dell’ATPIII
(Adult Tratment Panel III) (Tab. II), senza suddivisione per età e sesso.
Nel 2007 la International Diabetes Federation (IDF) ha pubblicato
criteri pediatrici (Tab. III) (Zimmet et al., 2007), ispirandosi in parte ai criteri IDF per l’adulto e ponendo come elemento centrale la
circonferenza vita (CV), condizione senza la quale non può essere
formulata la diagnosi di SM. I differenti criteri sono poi stati suddivisi in base al sesso ed all’età, considerando adulti soggetti con età
maggiore di 16 anni.
Tali classificazioni sono attualmente quelle più utilizzate, ma differenti studi, tra cui il National Health and Nutrition Examination Study – NHANES 1999-2000 (Duncan et al., 2004), non le hanno mai
completamente validate per l’età pediatrica, mancando dati a lungo
termine e confronti tra le differenti etnie. Gli studiosi denunciano il
rischio di sovrastimare la SM con entrambe le classificazioni, ponendo l’una l’attenzione soprattutto sulla famigliarità (NCEP) e l’altra
sulla CV (IDF), entrambe non in grado di giustificare da sole la SM.
L’IDF inoltre, considerando la CV un dato sufficiente per la diagnosi,
definisce affetti da SM un maggior numero di soggetti rispetto alla
classificazione NCEP (differenza 5-7%) includendo quasi tutti i sovrappeso ed obesi, avendo quest’ultimi una CV > 95° centile. Tale
presupposto rischia di sovrastimare l’effettiva prevalenza di SM nei
soggetti con obesità centrale, ma contemporaneamente rischia di
Tabella II.
Criteri NCEP ATPIII modificati per l’età pediatrica. Definizione di sindrome metabolica quando presenti almeno tre criteri (NCEP, 1992-2005).
Criteri National Colesterol Educational Pannel
≥ 95° centile per età e sesso (curve Cacciari 2006)
BMI
Pressione arteriosa
Pressione sistolica o diastolica ≥ 90° centile per età e sesso (Fernandez et al., 2004)
Trigliceridi
≥ 90° centile per età e sesso
Colesterolo HDL
≤ 10° centile per età e sesso
Alterata glicemia a digiuno (IFG)
o dopo due ore dal carico di glucosio (IGT)
IFG glicemia a digiuno ≥ 100 mg/dL
IGT glicemia dopo due ore dal carico orale di glucosio ≥ 140 e < 200 mg/dl
Tabella III.
Criteri IDF 2007 per la diagnosi di sindrome metabolica in età pediatrica. La definizione di Sindrome Metabolica viene formulata sulla presenza
di circonferenza vita > 90° centile, più almeno due degli altri criteri (Zimmet et al., 2007).
Anni
Circonferenza vita
Trigliceridi
Colesterolo HDL
6-9
≥ 90° centile
10-15
≥ 90° centile
≥ 150 mg/dL
< 40 mg/dL
≥ 16
Maschi ≥ 94 cm,
femmine ≥ 80 cm
≥ 150 mg/dL o un
trattamento specifico
Maschi < 40 mg/dL,
femmine < 50 mg/dL o un
trattamento specifico
Pressione arteriosa
Glucosio
Sistolica ≥ 130 o
diastolica ≥ 85 mmHg
Sistolica ≥ 130 o
diastolica ≥ 85 mmHg o
in trattamento specifico
Glicemia a digiuno ≥
100 mg/dL o DMT2
Glicemia a digiuno ≥
100 mg/dL o DMT2
241
G. Bona et al.
sottostimare notevolmente il rischio di SM in soggetti ipertesi ed
ipercolesterolemici, ma con CV nella norma.
Pur presentando numerosi limiti, queste classificazioni devono essere introdotte nella pratica clinica, rappresentando un guida valida
per identificare soggetti con elevato rischio cardiovascolare e quindi
indirizzarli ad ulteriori esami o terapie più aggressive. Non solo, ma
scorporando tali classificazioni, possiamo ottenere importati strumenti per la pratica clinica, tra cui i valori di riferimento sesso ed
età dipendenti per il colesterolo ed i trigliceridi, in parte basati sui
criteri NCEP modificati per l’età pediatrica e pubblicati recentemente sia dall’American Academy of Pediatrics, sia dall’American Heart
Association (Nader et al., 2006) ed i centili per la pressione arteriosa
determinati dalla seconda Task Force del National Heart, Lung and
Blood Institute (NHLBI) di Bethesda (MD, USA), sesso, età ed altezza
dipendenti (Task Force Report, 1996).
Conseguenze non endocrine dell’obesità e della sindrome metabolica
Particolare attenzione deve essere posta alle conseguenze durante
l’accrescimento dovute al peso ed alla SM (Tab. IV). Numerosi studi
hanno recentemente puntato l’attenzione sulle complicanze epatiche;
il 40% dei bambini obesi presenta alterazioni ecografiche ed elevazione degli enzimi di necrosi e di stasi epatica. Tali condizioni sono
descritte come “steatopatie non alcoliche (non-alcoholic fatty liver
disease – NAFLD” secondarie ad un eccessivo rilascio di acidi grassi
non esterificati in circolo ed iperafflusso di substrati lipidici al fegato.
Le NAFLD sono associate soprattutto a SM ed insulinoresistenza
piuttosto che al solo sovrappeso, ma non è ancora chiaro se siano
una causa od un effetto della SM, e si ipotizza un loro futuro utilizzo
come ulteriore criterio diagnostico per la SM sia in età pediatrica sia
adulta. In età pediatrica, in particolare, la NAFLD sembra possedere
caratteristiche istologiche differenti con più precoce fibrosi e più veloce esaurimento funzionale epatico (Freemark et al., 2007).
L’insulino resistenza
Definizione
L’obesità è frequentemente caratterizzata da una condizione di insulino resistenza (IR), ovvero da una riduzione della risposta dei tessuti
bersaglio all’azione dell’insulina, con conseguenze metaboliche sull’intero organismo essendo un ormone ad azione pleiotropica.
Insulinoresistenza ed obesità
La relazione causale tra obesità ed IR è ormai dimostrata. Entrambe i fattori danno origine ad un ciclo che si auto-mantiene. L’IR
dipende infatti in gran parte dall’azione del grasso viscerale, che
presenta una maggiore sensibilità agli stimoli lipolitici, soprattutto
indotti dalle catecolamine, con aumentata dismissione in circolo di
acidi grassi liberi (FFA) e conseguente aumento dell’ossidazione
lipidica nell’organismo. L’aumento degli FFA circolanti e lo stress
ossidativo da essi indotto determina nel tessuto adiposo un aumento della secrezione di resistina (aumento resistenza insulinica)
e riduzione dell’adiponectina (ormone insulino-sensibilizzante).
Aumenta, inoltre, la secrezione di citochine infiammatorie (TNF-α,
IL-6, PCR), inducendo uno stato infiammatorio cronico responsabile
esso stesso di IR (Weiss et al., 2004). A livello muscolare si riduce
l’espressione in membrana dei trasportatori per il glucosio (GLUT4)
e compaiono alterazioni del segnale intracellulare del recettore per
l’insulina. Il fegato, in risposta allo stress ossidativo ed alla cronica
iperinsulinemia, riduce l’espressione e la funzionalità dei recettori
dell’insulina, non produce glicogeno e favorisce l’incremento dei
livelli FFA per mancato utilizzo degli stessi. Per ottenere gli stessi
effetti biologici è necessario mantenere un compenso metabolico, inducendo un’incremento dell’insulinemia, configurando così
un quadro di iperinsulinismo associato a normoglicemia. L’iniziale compenso instaurato dalle β-cellule pancreatiche nel tempo
tende ad esaurirsi, con comparsa, dapprima di alterata glicemia
postprandiale, e successivamente iperglicemia a digiuno, instaurandosi precocemente un quadro di diabete mellito di tipo 2. L’IR
stessa contribuisce ad un danno diretto delle β-cellule attraverso meccanismi di glucotossicità e lipotossicità con conseguenze
anche per il metabolismo lipidico e proteico. Conseguenze dell’IR
sono infatti anche le dislipidemie (ipetrigliceridemia, aumento del
colesterolo LDL, riduzione del colesterolo HDL), l’infiammazione
sistemica con danno endoteliale precoce e l’aterosclerosi (Vigneri
et al., 2007; Maffei et al., 2009).
Insulinoresistenza ed apparato endocrino
L’IR è in grado di spiegare anche alcune alterazione endocrine che caratterizzano i soggetti sovrappeso ed obesi. In particolare, le bambine
e le adolescenti obese, sono predisposte ad anticipo della pubertà,
manifestando telarca e/o pubarca precoce e/o menarca anticipato, in
parte spiegabile per effetto dell’iperinsulinismo sulla leptina. La leptina, ormone prodotto dagli adipociti, informa l’ipotalamo sullo stato
energetico, impedendo l’incremento ponderale, riducendo l’appetito
e promuovendo il dispendio energetico. Svolge inoltre un ruolo chiave
nell’inizio della pubertà e nella regolazione del ciclo mestruale, promuovendo la pulsatilità del GnRH. L’iperinsulinismo riduce l’effetto
della leptina sull’ipotalamo, facilitando un ulteriore incremento ponderale ed innescando un’aumentata secrezione di leptina a scopo compensatorio. I conseguenti elevati livelli di leptina favoriscono quindi
l’inizio della pulsatilità del GnRH, l’aumento degli ormoni sessuali e
l’inizio della pubertà. Gli estrogeni e gli androgeni circolanti promuo-
Tabella IV.
Principali conseguenze non endocrine dell’obesità.
Disturbi dell’accrescimento
Disturbi respiratori
Problemi psicologici
Disturbi cardiovascolari
Ghiandole non endocrine
242
Difetti dello scheletro, scoliosi, piede piatto, coxa vara, ginocchia in atteggiamento di valgismo, malattia di
Osgood-Shlatter, artrosi precoce
Deposizione di grasso sottodiaframmatico ed addominale, infiltrazione grassa della muscolatura del torace,
ipoventilazione con riduzione volume di riserva espiratoria, sindrome delle apnee ostruttive notturne
Ridotta autostima e socializzazione
Ipertensione, inspessimento intima arterie, dislipidemie, cardiopatia ischemica in giovane età, scarsa tolleranza agli sforzi fisici
Steatopatia non alcolica, fibrosi epatica, caclolosi biliare
Obesità e sindrome metabolica in età pediatrica
vono così la secrezione e l’azione dell’ormone della crescita (GH) e
del suo effettore IGF-I (insulin-like growth factor I). L’azione sinergica
di questi tre ormoni è alla base della fisiologica resistenza insulinica
durante la pubertà, che nel soggetto obeso va ad aggravare l’IR già
presente. Nei maschi sovrappeso il quadro non è del tutto sovrapponibile: l’anticipazione della pubertà non è così marcata, anche se presentano spesso saldatura precoce delle cartilagini di accrescimento,
con riduzione della statura definitiva (Agirbasli et al., 2009).
Obesità ad insorgenza precoce, alterazioni endocrine e scarsa risposta alla terapia dietetica o farmacologica, devono però farci sospettare anche forme di obesità secondaria a patologie endocrine (sindrome di Cusching), a mutazioni genetiche (Tab. V). Recentemente
sono stati infatti scoperte mutazioni in alcuni loci genetici in grado
di determinare IR ed obesità, di cui le principali sono mutazioni del
gene codificante per: leptina, recettore della leptina, proopiomelanocortina e recettore delle melanocortine.
Diagnosi
Alla luce di quanto detto, l’obesità non può però essere banalizzata al semplice controllo del peso, ma fondamentale è il controllo
dell’IR e quindi in generale del metabolismo, in particolare durante
l’adolescenza, in quanto più facilmente si possono instaurare le basi
fisiopatologiche di malattie come il DM2, la sindrome dell’ovaio policistico (PCOS) e patologie cardiovascolari precoci.
Il gold standard per la misurazione della IR è il clamp euglicemico,
metodica costosa, indaginosa ed invasiva. Dagli studi di clamp si
sono ottenuti degli indici surrogati di IR, facilmente calcolabili, che si
ottengono da formule sui livelli di glicemia ed insulina a digiuno (indici HOMA – Homeostasis Model Assessment – e QUICKI) o durante
carico orale di glucosio (indice ISI) (Tab. VI). Non vi sono ancora dati
chiari in termini di prognostici in età pediatrica nell’uso di tali indici
(Maffeis et al., 2009).
L’attenzione, anche se con alcune delusioni, si sta spostando verso
gli indici e le citochine infiammatorie, elevate nell’IR. Un esempio è
il dosaggio della PCR, spesso elevata in soggetti obesi. Recenti studi
ne hanno però messo in dubbio l’utilità nella diagnosi di IR, limitandone l’uso a predittore di rischio cardiovascolare globale.
Terapia dell’obesità e della sindrome metabolica
Essendo l’obesità una patologia multifattoriale le scelte terapeutiche
sono diverse e si basano su differenti combinazioni di dieta, attività
fisica, terapia comportamentale, terapia farmacologica e chirurgica.
Terapia dietetica ed attività fisica
Non esistono studi sistematici che quantifichino la riduzione del peso ottenuta con la sola dieta. È invece dimostrata l’efficacia sul lungo termine
di diete impostate rispetto ai soli consigli alimentari. In età pediatrica
devono essere somministrate diete normocaloriche per l’età, basandosi
sulla correzione delle errate abitudini alimentari senza eliminare nessun
elemento nutritivo, che rallenterebbe la crescita e ridurrebbe la massa
magra. Importante, per la buona riuscita della dieta è il follow-up del
paziente, ottenendo migliori risultati con un programma strutturato di visite ad intervalli di 2 settimane-2 mesi. A tale scopo il ruolo del pediatra
di famiglia è fondamentale nel monitorare e modificare la dietoterapia
a seconda delle esigenze. L’incremento dell’attività fisica è però indispensabile per indurre un permanente cambiamento nel metabolismo
del soggetto, infatti non solo facilita il calo ponderale, ma promuove l’insulinosensibilità e migliora il fitness cardio-respiratorio. Per ottenere tali
effetti l’attività fisica deve essere quotidiana, e mantenuta per almeno
40 minuti consecutivi (Brufani et al., 2008). In età pediatrica, per indurre
miglioramenti significativi di complicanze come l’ipertensione e le dislipidemie, è sufficiente una riduzione del 10% del peso corporeo, spesso
ottenibile mantenendo stabile il peso e sfruttando la crescita in statura,
migliorando quindi il rapporto peso/altezza. La riduzione del peso corporeo svolge di per sé un’azione diretta sulle complicanze dell’obesità,
evitando l’assunzione di farmaci specifici per le singole complicanze. Il
rischio di sviluppare SM e NALFD decresce in modo significativo quanto più precocemente venga instaurata una terapia per il sovrappeso e
l’obesità (August et al., 2008; Love-Osborne et al., 2008).
Terapia farmacologica
La terapia farmacologica rappresenta il passo successivo, quando dieta ed attività fisica non siano sufficienti. Sconsigliate durante l’infanzia,
tali terapie sono concesse in casi eccezionali durante l’adolescenza,
quali obesità complicate su base genetica o in soggetti con un’obesità
Tabella V.
Caratteristiche cliniche dei pazienti affetti da obesità monogenica.
Gene mutato
LEP (leptina)
Meccanismo di
trasmissione ereditaria
Recessivo
Epoca di comparsa
dell’obesità
Primo anno di vita
Recessivo
Primo anno di vita
Ridotta
Recessivo/dominante
Dominante
Infanzia
Infanzia/età adulta
Normale/aumentata
Aumentata
LEPR (recettore della leptina)
POMC
MC4R
Velocità di crescita
Normale
Endocrinopatie
associate
Ipogonadismoipogonadotropo,
Ipotiroidismo
Ipogonadismoipogonadotropo,
Ipotiroidismo
Deficit di ACTH
Nessuno
Tabella VI.
Calcolo degli indici di insulina resistenza.
Indice
HOMA
QUIKI
ISI
Glicemia digiuno (mg/dL) x Insulinemia digiuno (mcU/mL)/22,5
1/Log (insulinemia digiuno) + Log (glicemia digiuno)
10000/ RadQ (glicemia digiuno x insulinemia digiuno) x (media glicemie x media insulinemie durante curva da carico)
243
G. Bona et al.
grave ed importanti complicanze ad essa associate. La terapia farmacologica deve essere sempre preceduta da un attenta valutazione del
rapporto rischio/beneficio ed attento follow-up del paziente, visti gli
effetti collaterali e la possibile interferenza con la crescita.
I farmaci autorizzati sono attualmente: orlistat e metformina nel diabete mellito tipo 2.
Non esistono studi di posologia ed efficacia di tali farmaci in età pediatrica, gli unici studi confermano l’utilità della metformina nel diabete
mellito di tipo 2, nella clinica e nella IR della PCOS (off-label). Oltre
ad un miglioramento del peso e del quadro metabolico, la metformina riduce la steatosi epatica e migliora la funzionalità epatica stessa
(Freemark et al., 2007).
L’orlistat impedisce l’assorbimento del 30% dei grassi assunti con
la dieta, inibendo l’azione della lipasi pancreatica. In commercio dal
1999, è da pochi anni approvato in età pediatrica Il periodo di trattamento non deve superare i 6 mesi e la terapia deve essere interrotta in assenza di calo ponderale nei primi 3 mesi. L’EMEA (European
Medicines Agency) e la FDA (Food and drug administration) nel 2008
ne sconsigliano l’utilizzo in casi di obesità complicata da epatopatia,
visto il rischio di pancreatite ed epatite. L’utilizzo a lungo termine può
determinare alterazioni gastro-intestinali e ridotto assorbimento delle
vitamine liposolubili.
Terapia chirurgica
Terapia di terzo livello, alcuni autori concordano nel riservarla a casi gravi di obesità complicata (ipoventilazione, BMI > 40, grave ipertensione
arteriosa con danno cardiaco). Nonostante ciò il numero annuale di interventi di chirurgia bariatrica in adolescenti è incrementato di circa 5
volte negli Stati Uniti ed in Australia dal 1997 al 2003. Studi prospettici
e caso-controllo condotti su adolescenti e giovani adulti hanno dimo-
strato come la chirurgia permetta di ottenere una maggiore riduzione
del BMI e, rispetto ai soggetti non operati, un maggiore e più rapido
miglioramento di marker metabolici. Le procedure utilizzate sono il Bypass gastrico (Stati Uniti) ed il bendaggio gastrico per via laparoscopica
(Australia ed Europa). La procedura chirurgica è comunque da evitare
prima del completo sviluppo, viste le conseguenze sull’assorbimento di
nutrienti e sullo sviluppo e stabilizzazione delle funzioni metaboliche/endocrine dell’apparato digerente durante la crescita.
Conclusioni
La prevalenza dell’obesità nei bambini e negli adolescenti continua a
crescere. Gli epidemiologi hanno allertato sulla possibilità che i nati in
questo secolo possano avere una durata di vita inferiore a quella dei
genitori proprio a causa dell’obesità e delle patologie cardiovascolari conseguenti (in media 10 anni nei prossimi 50 anni) (Jasik et al.,
2008). L’obesità infantile quindi non può essere considerata un problema del singolo individuo, ma più in generale una sfida per la Sanità
mondiale. Le strategie sinora messe in atto non sono risultate efficaci.
Tra le principali motivazioni vi è un’importante sottostima da parte dei
genitori del sovrappeso, del danno fisico (spesso silente nelle prime
fasi) e la sottostima di fattori obesogeni (pubblicità, sedentarietà) a cui
tutti siamo sottoposti. Per poter affrontare questo problema è importante considerare l’obesità come un profondo cambiamento metabolico, che necessita di disporre di punti di riferimento chiari e condivisi
(classificazioni, nuovi indici metabolici e terapie, ecc.). Mentre questi
sono già presenti in età adula, nonostante gli ultimi progressi, molto
resta ancora da fare in età pediatrica prima di raggiungere un approccio sistematico e validato, unico in grado di ottenere risultati nella
pratica clinica. A tale scopo è fondamentale coinvolgere la società, i
genitori e stimolare gli specialisti che si occupano di tale complicata
patologia a formulare delle linee di azione clinica comuni e condivise.
Box di orientamento
Obesità
• In Italia la prevalenza di sovrappeso in età pediatrica è superiore di circa 3 punti percentuali rispetto alla media Europea, con
un tasso di crescita/annua pari a quello degli Stati Uniti.
• Il problema interessa soprattutto il Sud Italia e gli adolescenti di sesso maschile.
• Circonferenza vita e rapporto vita/altezza correlano con il rischio cardiovascolare.
• L’uso combinato di BMI, circonferenza vita, rapporto vita/altezza, BMI z score sono fortemente raccomandati, rispetto al
monitoraggio di un solo parametro.
Sindrome
• Il 30% dei bambini obesi è affetto da sindrome metabolica, soprattutto femmine.
metabolica
• Non esistono criteri diagnostici validati per l’età pediatrica, le due classificazioni disponibili sono: Adult Tratment Panel III
(1992) e la IDF (2007), opportunamente modificate per l’età pediatrica.
• Il loro utilizzo è raccomandato per identificare soggetti con elevato rischio cardio-vascolare ed avere un approccio sistematico a tale patologia.
• Alterazioni ecografiche ed enzimatiche della funzionalità epatica sono frequenti in caso di sindrome metabolica e vanno
indagate.
Insulino resistenza • L’insulino-resistenza, soprattutto durante l’adolescenza, deve essere valutata mediante l’uso degli indici HOMA, QUIKI ed ISI,
anche in soggetti in leggero sovrappeso.
• La IR è alla base di pubertà precoce e riduzione della statura definitiva.
• Endocrinopatie, obesità insorta durante l’infanzia e resistenza alla terapia dietetica e/o farmacologica devono far pensare ad
obesità su base genetica.
Terapia
• La terapia per l’obesità e la sindrome metabolica coincidono.
• I maggiori benefici si ottengono con diete strutturate, normocaloriche, incentrate sulla correzione delle abitudini alimentari,
seguite da follow-up ogni 2 settimane - 2 mesi e quando servisse, esami ematochimici semestrali/annuali.
• L’attività fisica è indispensabile, deve essere quotidiana, per almeno 40 minuti.
• I farmaci approvati sono metformina e orlistat. Non sono ancora disponibili dati sugli effetti a lungo termine nel bambino, ma
possono essere utilizzati in caso di obesità grave, complicata nell’adolescente. L’epatotossicità ne limita l’utilizzo in pazienti
affetti da steatopatia.
• La chirurgia è riservata a casi gravi di obesità complicata (ipoventilazione, BMI > 40, grave ipertensione arteriosa con danno
cardiaco). Attualmente non è raccomandato il suo utilizzo prima del completamento dello sviluppo.
244
Obesità e sindrome metabolica in età pediatrica
Bibliografia
Agirbasli M, Agaoglu NB, Orak N, et al. Sex hormones and metabolic syndrome
in children and adolesents. Metabolism 2009;58:1256-62.
* Lavoro approfondito sulla fisiopatologia della correlazione tra pubertà anticipate ed isulino-resistenza.
Alberti KG, Zimmet P, Shaw J. De metabolic syndrome – A new worldwide definition. Lancet 2005;366:1059-62.
August GP, Caprio S, Fennoy I, et al. Prevention and treatment of pediatric obesity: an Endocrine Society Clinical Practice Guideline based on Expert Opinion. J
Clin Endocrinol Metab 2008;93:4576-99.
** Revisione accurate delle single complicanze dell’obesità e delle possibili
strategie terapeutiche. Poco approfondita la terapia farmacologica dell’obesità
stessa.
Boney CM, Verma A, Tucker R, et al. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus.
Pediatrics 2005;115:e290-e296.
Brufani C, Grossi A, Fintini D, et al. Cardiovascular fitness, insulin resistance and
metabolic syndrome in severely obese prepubertal italian children. Horm Res
2008;70:349-56.
Cacciari E, Milani S, Balsamo A, et al. Directive Councils of SIEDP/ISPED for
1996-97 and 2002-03. J Endocrinol Invest 2006;29:581-93.
Cook S, Weitzman M, Auinger P, et al. Prevalence of a metabolic syndrome phenotype in adolescents. Arch Pediatr Adolesc Med 2003;157:821-7.
Daniels RS. The use of BMI in the Clinical Setting. Pediatrics 2009;124:S35-S41.
* L’articolo offre numerosi spunti di riflessione sui pregi e difetti degli indici di
misurazione del grasso corporeo a disposizione (BMI, BMI z score, ecc.).
Duncan GE, Li SM, Zhou XH. Prevalence and trends of metabolic syndrome phenotype among U.S. Adolescents, 1999-2000. Diabetes Care 2004;27:2438-43.
De Kroon MLA, Renders CM, Kuipers ECC, et al. Identifying metabolic syndrome
without blood test in yang adults – The Terneuzen birth cohort. Eur J Public
Health 2008;18:656-60.
Fernández JR, Redden DT, Pietrobelli A, et al. Waist circumference percentiles in
nationally representative samples of African-American, European-American, and
Mexican-American children and adolescents. J Pediatr 2004;145:439-44.
Freemark M. Liver dysfunction in paediatric obesity: a randomized, controlled
trial of metformin. Acta Paediat 2007;96:1326-32.
Jasik BC, Lustig RH. Adolescent obesity and puberty: The “perfect storm”. Ann
NY Acad Sci 2008;1135:265-79.
Kipping RR, Jago R, Lawlor DA. Obesity in children. Part 1: epidemiology, measurement, risk factors, and screening. BMJ 2008;337:a1824.
** Accurata revisione dell’epidemiologia Europea dell’obesità e sovrappeso.
Kipping RR, Jago R, Lawlor DA. Obesity in children. Part 2: Prevention and management. BMJ 2008;22:337:1848.
Love-Osborne K, Sheeder J, Zeitler P. Addition of metformin to a lifestyle modification program in adolescents with insulin resistance. J Pediatr 2008;152:817-22.
Maffeis C, Manfredi R, Trombetta M, et al. Insulin sensitivity is correlated with
subcutaneous but not visceral body fat in overweight and obese prepubertal
children. J Clin Endocrinol Metab 2008;93:2122-28.
Maffeis C. L’obesità del bambino: aspetti clinici e fisiopatologici. Torino:Centro
Scientifico Editore 2009.
** Libro curato nella stesura, approfondisce tutti gli aspetti legati all’obesità e alla
sindrome metabolica nel bambino. Numerosi paragrafi dedicati alla fisiologia lo
rendono utile non solo come testo di consultazione, ma come vero libro di testo.
Nader PR, O’Brien M, Houts R, et al. Identifying risk for obesity in early childhood.
Pediatrics 2006;118:e594-e601.
National cholesterol education program: report of the Expert Panel on population
strategies for blood cholesterol reduction: executive summary. Arch Intern Med
1991;151:1071-84.
Savva SC, Tornaritis M, Savva ME, et al. Waist circumference and waist-to-height
ratio are better predictors of cardiovascular disease risk factors in children than
body mass index. Int J Obes 2000;24:1453-58.
Termizy HM, Mafauzy M. Metabolic syndrome and its characteristics among
obese patients attending an obesity clinic. Singapore Med J 2009;50:390-4.
Task Force Report on High Blood Pressure in Children and Adolescents, Update: a
working group report from the National High Blood Pressure Education Program.
National High Blood Pressure Education Program Working Group on Hypertension Control in Children and Adolescents. Pediatrics 1996;98:649-58.
Vigneri R, Ghigo E. Obesità addominale. Il Diabete 2007; Suppl. 1.
Weiss R, Dziura J, Burget TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 2004;350:2362-74.
Zanolli R, Morgese G. Waist percentiles: a simple test for atherogenic disease?
Acta Paediatr 1996;85:1368-9.
Zimmet P, Alberti K, Kaufman F, et al. The metabolic syndrome in children and
adolescents – an IDF consensus report. Pediatr Diabetes 2007;8:299-306.
* In questo lavoro vengono riportati i criteri pediatrici per la sindrome metabolica
redatti dall’IDF. Il testo approfondisce anche gli studi che hanno portato a compiere tali modifiche rispetto ai criteri per l’età adulta.
Corrispondenza
prof. Gianni Bona, Dipartimento Scienze Mediche, via Solaroli 17, 28100 Novara. E-mail: [email protected]
245