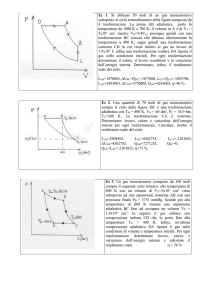
L’approssimazione adiabatica
Descriviamo un solido come insieme di nuclei (no struttura interna, solo massa e
carica totale) ed elettroni. Consideriamo i termini principali dell’operatore hamiltoniano:
Hˆ Tˆn Tˆel Vˆel el Vˆn el Vˆn n ;
ri xi , yi , zi ; pi p x ,i , p y ,i , p z ,i el
Ri X i , Yi , Z i ; Pi Px ,i , Py ,i , Pz ,i n
L’approssimazione adiabatica
Termini cinetici (per semplicità -> una sola specie atomica):
2
ˆ
Tn
Ri ;
i 1 2 M
N
2
2
ˆ
Tel
ri ;
i 1 2m
N el
2
L’approssimazione adiabatica
Termini di interazione:
Zi Z j e2
1
ˆ
Vn n
;
2 i j Ri R j
2
1
e
ˆ
Vel el
;
2 i j ri r j
Vˆel n
i, j
Zie2
;
Ri r j
L’approssimazione adiabatica
Zi Z j e2
1
ˆ
Vn n
;
2 i j Ri R j
Vˆel el
1
e2
;
2 i j ri r j
Vˆel n
i, j
Zie2
;
Ri r j
Questi termini sono “un disastro”!
Classicamente, l’energia non è somma delle energie
delle singole particelle. Il formalismo alla Boltzmann
non funziona; le equazioni del moto sono tutte
accoppiate.
Quantisticamente, l’operatore hamiltoniano non è
somma di operatori a particella singola; gli autovalori
non saranno somma di autovalori a particella singola;
la funzione d’onda del sistema non sarà il prodotto
di autofunzioni di particella singola.
In entrambe i casi, il problema è a molte (moltissime!)
particelle.
L’approssimazione adiabatica
Cosa vorrebbe dire, in linea di principio, studiare la dinamica di questo
sistema?
(r , R, t )
ˆ
H (r , R, t ) ih
;
t
(r , R, t ) drdR
2
Probabilità al tempo t di trovare gli
elettroni attorno ad r e i nuclei attorno a R
L’approssimazione adiabatica
Formalmente, la soluzione dipendente dal tempo è costruita a partire dalle
autofunzioni
(r , R, t )
ˆ
H (r , R, t ) i
;
t
( r , R , t ) cn u n ( r , R ) e
iWn t
;
n
Hˆ un (r , R ) Wnu n (r , R )
Per conoscere l’evoluzione temporale devo calcolare tutte le autofunzioni (non
basta lo stato fondamentale ..)
L’approssimazione adiabatica
E’ evidente che così non si va da nessuna parte. Per fortuna si può sfruttare la
grande differenza di massa tra nuclei ed elettroni
n
M
100 1000 (ps fs)
el
m
Posso quindi immaginare, istante per istante di trovare la distribuzione elettronica
corrispondente ad una data configurazione dei nuclei.
Inoltre (pensare alle tipiche temperature di Fermi), posso accontentarmi di calcolare la
distribuzione elettronica corrispondente allo stato elettronico fondamentale.
E’ importantissimo!!!
L’approssimazione adiabatica
L’idea è questa:
posizione ionica al tempo t
L’approssimazione adiabatica
L’idea è questa:
posizione ionica “modificata” al tempo t+dt con dt pari a una frazione piccola
del tipico tempo ionico. Per gli elettroni questo tempo è estremamente
lungo per cui posso pensare che essi “seguano adiabaticamente” il moto ionico,
ovvero che la distribuzione di carica elettronica cambi nel seguente modo
L’approssimazione adiabatica
L’idea è questa:
distribuzione di carica elettronica corrispondente allo stato fondamentale per la
configurazione ionica fissata al tempo t
L’approssimazione adiabatica
L’idea è questa:
distribuzione di carica elettronica corrispondente allo stato fondamentale per la
configurazione ionica fissata al tempo t+dt. Il moto ionico da t a t+dt sarà ovviamente
influenzato dalla distribuzione di carica elettronica al tempo t …
L’approssimazione adiabatica
Non ci interessa fare i conti; vogliamo capire il risultato. Per separare il moto
elettronico da quello ionico si impone una funzione d’onda fattorizzata in parte
ionica ed elettronica, la si inserisce nell’equazione agli autovalori, e si buttano parti
legate al rapporto tra le masse ..
u (r , R) el (r | R) ( R)
Hˆ el ( R) el , 0 (r | R) V ( R) el , 0 (r | R)
Hˆ el ( R) Tˆel Vˆel el Vˆel n ,ext ( R) C ( R);
Hˆ n Tˆn V ( R)
L’approssimazione adiabatica
Gli ioni sono descritti da un’hamiltoniana del tipo energia cinetica +
“potenziale inter-ionico” (o “interatomico”), in cui tale potenziale corrisponde
all’autovalore relativo allo stato fondamentale dell’hamiltoniana elettronica.
Hˆ n Tˆn V ( R);
Hˆ ( R) (r | R) V ( R)
el
el , 0
el , 0
(r | R)
L’approssimazione adiabatica
PASSAGGIO CHIAVE: IL MOTO NUCLEARE PUO’ ESSERE DESCRITTO
CON LA DINAMICA CLASSICA!!!!!
Hˆ n Tˆn V ( R);
Hˆ ( R) (r | R) V ( R)
el
el , 0
el , 0
(r | R)
Once the Schrodinger equation for the ground
state has been solved, then the “interatomic
potential” V(R) is known, and nuclei evolution
can in principle be investigated by considering
the nuclear hamiltonian:
H n Tn V (R);
• De Broglie wavelength:
Huge help:
h
2Mk BT
If << a (lattice parameter), nuclei follow Newton equation of motion
Usually true except for lightest elements and T->0
So, how do we study the dynamics?
1) Solve Schrödinger equation for the electrons
Density Functional Theory
Hohemberg and Kohn (1964)
Kohn and Sham (1965)
2) Solve Newton equations for the nuclei
We will learn how to do it
1+2 Ab initio Molecular Dynamics
Implemented in an easier way -> Car and Parrinello (1985)
Ionic (nuclear) configuration at time t
Ionic (nuclear) configuration at time t
Compute the electronic wavefunction for the ground
state: V(R) is known
Ionic (nuclear) configuration at time t
Solve the Newton equation of motion for the ions ….
Ionic (nuclear) configuration at time t +Dt
Solve the Newton equation of motion for the ions ….
Ionic (nuclear) configuration at time t +Dt
Re-compute the electronic wavefunction and start
again…..
This procedure is very demanding and allows to
simulate only short time scales and small systems, but it
is highly accurate.
Classical molecular dynamics (MD)
Classical Molecular Dynamics (this lecture)
1) Choose an “appropriate” form for V
2) Solve numerically Newton’s equations of
motion for the atoms
Classical MD is easier, faster, and it allows to
study larger systems, but
the reliability of the results totally depends on V