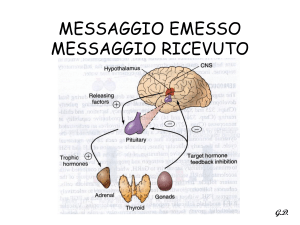
MESSAGGIO EMESSO
MESSAGGIO RICEVUTO
G.D.
Circuito controregolatore lungo
STRESS
Fisico
CRH
Emozione
Chimico (ipoglicemia)
Altri
ACTH
Feedback lungo
Corticotropi
Feedback lungo
Fisiologia della neuroregolazione dell’asse CRH-ACTHcorticosurrene
Esone 1
Esone 2
GENE
Introne 1
Introne 2
Trascrizione
mRNA
Regione Traslata
Traslazione e Rimozione del Segnale
POMC
Clivaggio Post-Traslazionale
N-POMC 1-76
J-PEPTIDE
B-LPH
ACTH 1-39
Processo Parziale
(30%)
Stati Patologici
N-POMC 1-48
g-MSH
g-LPH
b-END
• L’ACTH è secreto episodicamente dall’ipofisi e ha
un’emivita in circolo di circa 20 minuti.
• L’ampiezza, ma non la frequenza, dei picchi
secretori varia nell’arco delle 24 ore, con una
secrezione massima tra le 6 ed 8 del mattino e declina
dopo un nadir serale.
• Pattern di secrezione legato al ritmo sonno veglia.
• I fattori che stimolano la secrezione di ACTH sono il
CRF-41 e l’ormone Arginina Vasopressina.
• La 5 idrossitriptamina e l’acetilcolina stimolano la
secrezione di CRF, differentemente il GABA esercita
un’influenza inibitoria.
Le ghiandole surrenali
Situate in corrispondenza del polo renale superiore
ed adiacenti ad esso.
La vascolarizzazione deriva direttamente dall’aorta,
dalle arterie renali e dalle arterie freniche.
Le vene surrenaliche di destra confluiscono
direttamente nella cava inferiore, quelle di sinistra
sono tributarie della vena renale omolaterale.
Surreni
Costituiti da:
• ZONA CORTICALE
(derivazione mesodermica)
90%
• ZONA MIDOLLARE
10%
(derivazione neuroectodermica)
Compartimenti funzionali della
corteccia surrenale
glomerulare
fascicolare
reticolare
ORMONE SECRETO
aldosterone
cortisolo
DHEA
FATTORI DI
sistema reninaangiotensina
ACTH
ACTH
ZONA
PRINCIPALE
CONTROLLO
Corticosurrene: ormoni steroidei
Glucocorticoidi → promuovono la formazione del glucosio
dalle proteine, favoriscono la deposizione dei grassi,
incrementano il flusso ematico renale, riducono i processi
infiammatori, esercitano un’azione antiinsulinica diretta.
Mineralcorticoidi → favoriscono l’assorbimento del sodio e
l’eliminazione del potassio a livello dei tubuli renali, con
riassorbimento di acqua.
Androgeni → concorrono alla mascolinizzazione ed
all’anabolismo proteico, modificano le masse muscolari.
Cortisolo:
↑ Neoglucogenesi
↑ Lipolisi
↑ Glicogenosintesi
↓ Captaz. glucosio (muscolo)
↓ Sintesi proteica
↑ Catabolismo proteico
↑ Rilascio amino acidi
Fisiopatologia e clinica
corticosurrenalica
• Insufficienza corticosurrenalica
• Sindromi da iperfunzione
surrenalica
Insufficienza corticosurrenalica
Inadeguata secrezione di ormoni della corteccia
surrenale, in particolare di cortisolo, come
conseguenza della distruzione di più del 90% della
corticale del surrene (iposurrenalismo primario) o
di un deficit della secrezione ipofisaria di ACTH
(iposurrenalismo secondario*) o della secrezione
ipotalamica di CRH (iposurrenalismo terziario)
*spesso secondario a terapia prolungata con corticosteroidi in malattie
infiammatorie, allergiche, autoimmuni
Insufficienza corticosurreralica cronica
primitiva o Morbo di Addison: etiologia
• Adrenalite autoimmune
• Tubercolosi
• Altre
–
–
–
–
–
–
–
–
80%
19%
1%
Istoplasmosi, blastomicosi, sarcoidosi
Emocromatosi, amiloidosi
Malattie opportunistiche associate all’AIDS
Adrenomielodistrofia
Carcinomi metastatici
Deficit congenito familiare dei glucocorticoidi
Resistenza isolata ai glucocorticoidi
Iatrogene: ketoconazolo, mitotate, aminoglutemide, metirapone
Morbo di Addison
La forma più frequente è quella autoimmune caratterizzata
da un’infiltrazione di linfociti T citotossici, che porta
all’atrofia bilaterale della corticale dei surreni.
Sono stati individuati anticorpi circolanti contro la
21-idrossilasi ( raramente 17α-idrossilasi).
Talvolta si associa ad altre malattie autoimmuni:
• SPA di tipo 1 (m.di Addison, ipoparatiroidismo, candidosi
mucocutanea)
• SPA di tipo 2 (m. di Addison, tiroidite di Hashimoto,
DM1)
Morbo di Addison: patogenesi
Distruzione o atrofia primaria del corticosurrene
→ riduzione dei corticosteroidi (cortisolo) →
inefficace
controregolazione
ipotalamoipofisaria→ aumento della secrezione di CRH e
dei peptidi derivanti dalla pro-opiomelanocortina
ACTH e MSH (responsabile della melanodermia).
All’ipocortisolismo si associa ipoaldosteronismo.
Morbo di Addison: quadro clinico
•
•
•
•
•
•
•
•
•
•
•
•
Astenia e adinamia intensa
Stato depressivo e apatia
Anoressia
Disturbi gastrointestinali (vomito, diarrea, dolori
crampiformi)
Dimagramento
Disordini elettrolitici (iperpotassiemie, iposodiemia)
Disidratazione
Ipotensione arteriosa (vertigini in ortostatismo e lipotimie)
Desiderio di cibi salati
Ipoglicemia
Iperazotemia (da riduzione del filtrato glomerulare)
Melanodermia (iperpigmentazione)
Morbo di Addison: diagnosi
• Dosaggio cortisolo libero urinario 24 ore (ridotto)
• Dosaggio ACTH (utile per differenziare
l’ipocorticosurrenalismo primario da quello
secondario e terziario – aumentato nel primo caso)
• PRA (elevata, per diminuita produzione di
aldosterone)
• Elettroliti plasmatici (iperkaliemia, iposodiemia)
• Dosaggio Ab anti-surrene
Morbo di Addison: terapia
Terapia sostitutiva: somministrazione di
dosi adeguate di idrocortisone o cortisone
acetato; a volte utile l’associazione con
farmaci ad azione mineral attiva
(fluoroidrocortisone)
Sindromi da iperfunzione
surrenalica
La SINDROME di CUSHING è una
condizione di iperfunzione corticosurrenalica caratterizzata da una prevalente
iperincrezione di cortisolo.
Sindrome di Cushing
• ACTH-dipendente
– Morbo di Cushing
(ipersecrezione ipofisaria di ACTH)
– Sindrome da ACTH ectopico
( es. carcinoma a chicco d’avena del polmone)
68%
12%
• ACTH-indipendente
–
–
–
–
Adenoma surrenalico
Carcinoma surrenalico
Iperplasia surrenalica micronodulare
Iperplasia surrenalica macronodulare
10%
8%
1%
< 1%
Sindrome di Cushing: quadro clinico
•
•
•
•
•
•
•
•
•
•
Obesità centripeta
Pletora facciale (facies lunare)
Ridotta crescita lineare (nel bambino)
Oligo-menorrea
Irsutismo
Ipertensione
Ridotta tolleranza glicidica
Impotenza
Osteopenia con fratture
Debolezza muscolare miopatia
94%
84%
80%
76%
73%
72%
67%
67%
59%
58%
Sindrome di Cushing: quadro clinico
•
•
•
•
•
•
•
•
•
•
Strie rubre
Edema periferico
Acne, cute grassa
Facilità alle ecchimosi
Disturbi psichici
Polidipsia-poliuria
Insufficienza cardiaco-congestizia
Calcoli renali
Cefalea
Melanodermia (s.da ACTH ectopico)
57%
56%
53%
53%
48%
34%
22%
16%
14%
6%
Sindrome di Cushing: diagnosi
• Dosaggio del cortisolo libero urinario delle 24 ore
• Dosaggio cortisolo plasmatico (ritmo circadiano
8-16-23)
• Dosaggio ACTH plasmatico
• Test di inibizione con alte dosi di desametasone
(test di Liddle) per la diagnosi differenziale tra
Morbo di Cushing e S. dell’ACTH ectopico
Sindrome di Cushing: terapia
Chirurgica :
– asportazione adenoma ipofisario ACTHsecernente
– asportazione adenoma surrenalico monolaterale
cortisolo-secernente
– Surrenectomia bilaterale in caso di iperplasia
nodulare bilaterale (seguita da terapia
cortisonica sostitutiva)
Sintesi dei mineralcorticoidi
Zona glomerulosa
Stimoli: - ACTH
- Angio II
Azione dell’aldosterone sui
tubuli renali
Sistema renina-angiostensina aldosterone
(app. iuxtaglom.) Renina
ACE (tessuti:polmone)
↓
↓
Angiotensinogeno → Angiotensina I → Angiotensina II*
(fegato)
*↑P.A.
↑Vol. Plasm.
↑Vasocostriz.
+
Su.rrene (z. glomerulare) Secrez.
Aldosterone
↓
↑ Na+
↓ K+
↓−
Renina
Iperaldosteronismo
• Primitivo (Sindrome di Conn)
• Secondario
Fisiopatologia iperaldosteronismo
primitivo (S.di Conn)
↑ aldosterone → ↑ rit Na+ → ↑ perdita k+
↓
↓ renina
↓
↓ angio II
Iperaldosteronismo primitivo (S. di Conn)
• Aumentata ed inappropriata produzione di
aldosterone → ritenzione Na, ipertensione
arteriosa, bassa renina plasmatica, ipok
• Epidemiologia
- 2 % degli ipertesi
- F/M: 2 - 2,5/1 (adenoma; iperplasia M=F)
-Età: tutte (prev. 3a - 4a decade)
Iperaldosteronismo primitivo (S. di Conn)
• Anatomia patologica
– Adenoma (50%)
– Carcinoma (molto raro)
– Iperplasia bilaterale (nodulare o diffusa) (50%)
• Idiopatica
• Sensibile ai glucocorticoidi (ACTH“iperdipendente”)
• Iperplasia monolaterale (molto rara)
Iperaldosteronismo primitivo (S. di Conn):
Quadro Clinico
• Ipertensione arteriosa (da lieve a grave,
prevalentemente diastolica)
• Ipopotassiemia
• Alcalosi, tetania (rara)
• Poliuria, polidipsia (insensibilità ADH)
• Aritmie cardiache (ipertensione art., ipoK)
• Cefalea
• Edemi generalmente assenti
Iperaldosteronismo primitivo:
dati di laboratorio
•
•
•
•
Aldosterone plasmatico aumentato
Ipokaliemia
PRA soppressa
Evidenza per adenoma
– Livelli di aldosterone non modificabili con:
• Test posturali (3 ore stazione eretta)
• Carico di sale
• Somministrazione di captopril
– Studi morfofunzionali
• TAC/RMN
• Scintigrafia surrenalica con iodocolesterolo
• Cateterismo vene surrenaliche
Iperaldosteronismo primitivo: cenni
di terapia
• Adenoma
– Surrenectomia monolaterale
• Controllo ipertensione
– 90% a breve termine
– 70% a lungo termine
• Iperplasia bilaterale
– Surrenectomia bilaterale (rara)
– Terapia medica
• Spironolattone, canrenoato di K, amiloride
• Altri farmaci anti-ipertensivi: Ca antagonisti
Iperaldosteronismi secondari
• Con ipertensione arteriosa
(> produzione di renina)
– Ipertensione nefrovascolare
– Ipertensione maligna
– Reninoma
• Senza ipertensione arteriosa(> volume plasmatico (edemi)/< K+)
– Scompenso circolatorio
– S.nefrosica
– Cirrosi epatica
– Tubulopatie renali
– Diarrea, vomito
– Abuso di diuretici, lassativi
– S. di Bartter
Fisiopatologia e clinica della
midollare surrenalica
La midollare è la parte centrale delle ghiandole surrenali.
È una struttura altamente specializzata del sistema nervoso
simpatico.
Di origine neuroectodermica, fa parte del sistema
simpatocromaffine e sintetizza catecolamine: adrenalina
(80%), noradrenalina e, in piccola quantità, dopamina.
Cenni di Embriologia
Il tessuto cromaffine della midollare surrenalica
e dei gangli del sistema nervoso simpatico ha
un’origine comune, dalla cresta neurale.
I
precursori
del
sistema
simpatico,
i
simpatogoni, originano dalla cresta neurale
migrano ventralmente differenziandosi nei
neuroblasti paravertebrali e preaortici. Da
queste cellule originano quelle gangliari e i
feocromoblasti.
La
midollare
del
surrene
origina
dai
feocromoblasti, migrano dai gangli simpatici
nella corticale surrenale formando degli
agglomerati cellulari da cui origina la midollare
surrenale.
Il flusso ematico a livello della
midollare
surrenalica
deriva
dall’arteria
frenica
inferiore,
derivante dall’aorta, dall’arteria
renale e dal sistema portale
corticomidollare che origina dalla
zona reticolare della corteccia
surrenale.
NH2
Tirosina (alpha metilparatirosina)
OH
CO2H
Tirosina idrossilasi
(cAMP protein kinase, Glucocorticoid)
Sistema APUD
(amine precursor uptake
and decarboxylation)
Diidrossifenilalanina (DOPA)
Decarbossilasi aminoacida
L-aromatica
Dopamina
OH
Dopamina b-idrossilasi
OH
NH2
OH
Norepinefrina
Feniletanolamina
N-metiltransferasi (PNMT)
Epinefrina
OH
OH
H
N
OH
CH3
La
rimozione
delle
catecolamine
avviene
principalmente attraverso il meccanismo di captazione
delle stesse nel terminale presinaptico chiamato
uptake-I.
Le catecolamine possono anche essere rimosse dal
tessuto extraneuronale uptake-2, specialmente dal
fegato e dal rene, dove sono metabolizzate.
Le catecolamine vengono metabolizzate dalla CatecolO-metiltransferasi (COMT), che converte la NE in
normetanefrina e la epinefrina in metanefrina e dalla
monoammino ossidasi (MAO) che converte la
metanefrina e normetanefrina in acido vanilmandelico
e la NE e E in acido 3,4 diidrossi mandelico.
Le catecolamine sono immagazzinate in
granuli secretori attivamente da una pompa
H++ ATP e proteine trasportatrici quali la
vesicular monoamine transporters (VMATs)
Metaiodiobenzilguanidina (MIBG) I131 o I123 è
importato dalla VMATs nei granuli secretori
della midollare surrenalica
Ruolo fondamentale dei recettori
adrenergici a livello periferico
I recettori alpha hanno un effetto eccitatorio con
l’eccezione del tratto gastrointestinale, dove producono
effetti inibitori.
I recettori beta producono effetti inibitori eccezione fatta
del cuore, dove hanno uno stimolo eccitatorio.
Recettori a- Adrenergici
Sottotipo
Agonisti
a1
a2
epinefrina
norepinefrina
Fenilefrina
Metossamina
Clonidina
a-metil NE
Antagonisti
Fentolamina, Fenossibenzamina
Prazosina
Yohimbine
Terazosina
rauwolscine
Secondi
Messaggeri
Fosfatidilinositolo
↑ del Ca2+ intracellulare
↓ dell’AMP ciclico
↓ o ↑ del Ca2+ e dello scambio
Na + /H +
Effetti
sistemici
Vasocostrizione,
rilassamento intestinale,
contrazione uterina,
midriasi
↓ della NE presinaptica,
aggregazione delle
plasmacellule,
vasoconstizione, ↓ secrezione
insulinica
Recettori b- Adrenergici
Sottotipo
Agonisti
b1
b2
Isoprotenerolo, Epinefrina, NE
Denopamina
Terbutalina, Clenbuterolo
Albuterolo
Antagonisti
Propanololo, Nadololo, Timololo
Metoprololo
ICI 118551
Atenololo
Secondi
messaggeri
↑ dell’AMP
ciclico
↑ dell’AMP
ciclico
Effetti
Sistemici
↑ frequenza cardiaca
↑ contrattilità
↑ lipolisi
↑ secrezione di
renina
Rilassamento della
muscolatura liscia
↑ glicogenolisi nel
muscolo scheletrico
↑ NE presinaptica
Recettori b3 adrenergici aumentano
la termogenesi a livello del tessuto
adiposo bruno ed aumentano la
lipolisi
Qualsiasi
tumore
che
produce,
immagazzina e secerne catecolamine
ed innesca la patologia caratterizzata
da
un
eccessivo
rilascio
di
catecolamine, può essere considerato
un feocromocitoma ed essere trattato
come tale indipendentemente dalla
sua localizzazione
Feocromocitoma
•La più importante malattia della midollare surrenale
è il Feocromocitoma.
• Nella ghiandola surrenale si ha 85%- 90% dei tumori
• Le localizzazione extra surrenale può essere trovata in
organi che originano dalla cresta neurale –Corpo carotideo,
chemorecettori aortici, gangli simpatici, organo di
Zuckerkand- e sono detti paraganglioma, chemodectoma.
• Incidenza del 0.8 x 100000 per anno
•La frequenza è uguale in entrambi i sessi, sono più
frequenti tra i 30 e i 50 anni.
•Il 10% dei casi sono bilaterali nella forma sporadica .
•Il 50% dei casi sono bilaterali e generalmente
intrasurrenalici nella forma familiare .
Forma familiari di Feocromocitoma I
Neoplasia Endocrina Multipla tipo 2a (Sindrome di
Sipple):
• Mutazione del protoncogene RET Chr 10q11.2
•Carcinoma midollare della tiroide, Iperparatiroidsmo
Neoplasia Endocrina Multipla tipo 2b:
• Mutazione del protoncogene RET Chr 10
• Carcinoma midollare della tiroide, neuromi della
mucosa, ganglioneuroma intestinale, megacolon,
habitus Marfanoide
Feocromo e S. neuroectodermiche
Neurofibromatosi (malattia di von Recklinghasen)
tipo 1:
• Mutazione del gene tumore-soppressore NF1 Chr 17
• Neurofibromatosi periferica
Emangioblastoma Cerebelloretinale (sindrome di von
Hippel-Lindau) tipo 2:
• Mutazione erronea che interessa il gene tumore
soppressore VHL Chr 3p25-26
• Angioma retinico; emangioblastoma del cervelletto e
del midollo spinale; carcinoma renale a cellule chiare;
cisti pancreatiche, renali, epididimali e endolinfatiche
Manifestazioni Cliniche
• Ipertensione parossistica o sostenuta. Usualmente
l’ipertensione è labile. Sintomatologia associata a
cefalea, sudorazione profusa, palpitazioni con o senza
tachicardia, ansia, un senso di rovina, pallore cutaneo,
nausea e dolore addominale.
• L’associazione di cefalea, sudorazione e palpitazioni
(triade del feocromocitoma) permette la diagnosi con
una specificità del 93.8% ed una sensibilità del 90.9%
• L’ipertensione è sostenuta in circa metà dei paziente,
circa un terzo dei pazienti può avere crisi
parossistiche e meno di un quinto può avere pressione
normale
• Altri sintomi includono ipotensione ortostatica,
iperglicemia, ipermetabolismo, perdita di peso e
persino cambiamenti psichici
• L’iperglicemia è generalmente modesta
riscontra durante gli episodi ipertensivi.
è
si
• Il feocromocitma può precipitare un infarto del
miocardio; un prolungato eccesso di catecolamine
può portare ad una miocardiopatia (ipertrofia
cardiaca)
• Il feocromocitma può anche secernere la
calcitonina, il peptide vasoattivo intestinale e la
ACTH
Diagnosi differenziale
• Crisi ansiose, attacchi di panico, tireotossicosi,
sospensione repentina della terapia con clonidina, uso di
anfetamine, cocaina, ipoglicemia, angina pectoris o infarto
del miocardio, tumori del cervello, emorragia
subaracnoidea,
decondizionamento
cardiovascolare,
tossiemia della gravidanza, neuroblastoma (bambini).
• Tra i farmaci ricordiamo gli inibitori della
monoamminossidasi come la Selegilina e le benzodiazepine
tricicliche come la Clozapine
Diagnosi
• Test urinari: si misura il dosaggio della
metanefrina e dell’acido vanilmandelico.
Catecolamine
Metanefrine
Acido
vanilmandelico
Valori
normali
Catecolamine
totali
Epinefrina
<100 mg/
24 ore
< 20 mg/
24 ore
0.3-0.9 mg/24 ore
< 9 mg/24 ore
plasma: Normetanefrina 18-112 pg/mL
Metanefrina 12-61 pg/mL
< 300mg/24 ore
Norepinefrina
< 80mg/24 ore
< 500mg/24 ore
• Raccogliere le urine in un acido forte 20 mL 6N HCL o 25 mL di 50% acido acetico
Prelievo ematico
• Il paziente deve essere supino a digiuno e l’ago dovrebbe
essere inserito in vena per almeno 20’ prima del prelievo.
• I valori normali sono per la norepinefrina < 500 pg/mL e per
l’epinefrina < 100 pg/mL; valori superiori a 1500 pg/mL per la
norepinefrina e a 300 pg/ml per l’epinefrina sono diagnostici
per il Feocromocitoma
• La determinazione dei valori normetanefrina e metanefrina è
risultata utile, ma pochi laboratori fanno questo dosaggio
• La cromogranina A è un eccellente marker per il controllo nel
tempo di un paziente con un pregresso tumore neurendocrino.
Test farmacologici
(test inibitori)
• Test alla Fentolamina antagonista a-adrenergico:
Permette la diagnosi di feocromocitoma quando riduce la
pressione sistolica di 35 mm HG e quella diastolica di 25 mm Hg
dopo la somministrazione di 1 o 5 mg del farmaco. La pressione va
misurata ogni 30’’-60’’ per 10’.
•Test alla Clonidina agonista a2 adrenergico a livello centrale:
Si misurano le concentrazioni ematiche delle catecolamine prima e
tre ore dopo la somministrazione di clonidina (0.3 mg/70kg). Le
catecolamine di persone non affette dal feocromocitoma
diminuiscono; mentre quelle con il feocromocitoma o non hanno
variazioni o i valori delle catecolamine aumentano.
• Test di stimolo con Istamina, Glucagone, tiramina e
Metoclopramide (aumento di tre volte i valori di base nei soggetti
con feocromocitoma 12 mmol/L-2000 pg/mL)
Localizzazione
• Tomografia Computerizzata. Epinefrina urinaria maggiore al
15% delle catecolamine totali la localizzazione è surrenalica
• La risonanza magnetica permette di distinguere il
feocromocitoma da altre masse surrenaliche. MRI T2 pesata ha
un intensità tre volte superiore rispetto al fegato, MRI T1 pesata
ha un intensità uguale a quella epatica
• La scintigrafia con metaiodiobenzilguanidina I-131 permette
di
rilevare
l’ottanta-novantacinque
per
cento
dei
feocromocitomi. Questa metodica può anche visualizzare
neuroblastomi e tumori del sistema APUD. Octreotide-scan
(111In-DTPA) e tomografia ad emissione di positroni 6 (18F)
fluorodopamina
Trattamento
• Il trattamento di scelta è chirurgico
• Importante è la preparazione all’intervento per evitare
crisi ipertensive. Esistono diversi protocolli clinici
• La preparazione inizia sette giorni prima dell’intervento
con la somministrazione della fenossibenzamina abloccante non specifico . Il dosaggio iniziale è di 10 mg due
volte al giorno che può essere aumentato di 0.5-1.0 mg/kg
al giorno in due tre dosi giornaliere.
• Prazosina alla dose di 2-5 mg due volte al giorno;
Labetololo alla dose di 200-600 mg; Metirosina 250 mg
ogni sei ore con incrementi giornalieri di 250-500 mg
fino ad un massimo di 4 g al giorno; Nifedipina 10 mg è
usata per controllare le punte pressorie.
Trattamento
• Durante l’intervento chirurgico la fentolamina, la nicardipina o
la nifedipina dovrebbero essere somministrati per prevenire le
crisi ipertensive
• Se è presente tachicardia o aritmia indotta dalle catecolamine
Propanololo può essere usato per endovena a 0.1-1 mg
• Il paziente subito dopo l’intervento ha bisogno dell’infusione di
liquidi per recuperare il volume ematico
• Frequenti sono le crisi ipotensive che occorrono durante
l’intervento o subito dopo l’intervento chirurgico. In questa
circostanza si preferisce l’infusione di liquidi ma quando il
paziente è stato sottoposto ad una massiva terapia alfa bloccante
si può ipotizzare l’utilizzo di agenti pressori