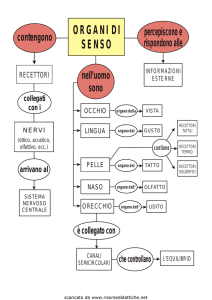
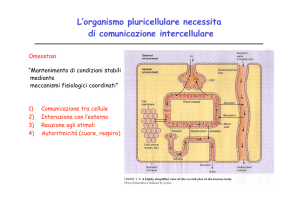
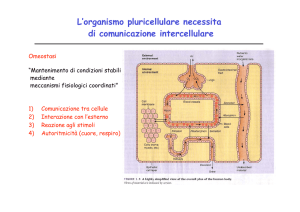
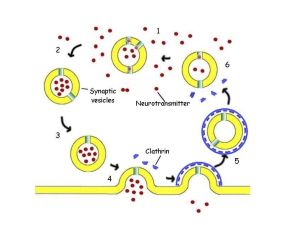
RECETTORI
Interazioni farmaco-recettore
La maggior parte dei farmaci interagisce con siti
specifici delle macromolecole (componenti della membrana
cellulare, enzimi, proteine) mediante interazioni
fisico-chimiche e steriche esatte tra specifici gruppi
chimici del farmaco e specifici siti della proteina.
Questi siti sono detti RECETTORI
Interazioni farmaco-recettore
Il binding di un ligando a un
recettore causa una risposta
positiva o negativa.
Una risposta positiva può
essere o un immediato effetto
fisiologico (apertura di un
canale ionico) oppure una serie
di eventi biochimici che portano
al rilascio di “secondi
messaggeri”.
Essi a loro volta promuovono
una nuova sequenza di eventi
biochimici che generano una
appropriata risposta biologica.
Ligando e recettore
N(CH3)2
N(CH3)2
Legame idrofobico
Legame dipolo-dipolo
O
O
O2N
O
N
Complesso con trasferimento
di carica
HO
Interazione ione-dipolo
H
Legame idrogeno
Cl
NH2
+
N
COO-
Legame ionico
HO
Esempi di alcune comuni forme di legame trovate nelle interazioni
farmaco-recettore
Ligando e recettore
Legame covalente:
è di gran lunga il più forte dei legami ligando-recettore.
Forma un legame irreversibile. Raramente viene cercato nell’azione di un
farmaco (eccezione: chemioterapici nel trattamento del cancro)
Legame ionico o elettrostatico:
è molto importante nelle interazioni ligando-recettore in quanto molti gruppi
funzionali dei recettori sono ionizzati a pH fisiologico.
Si formano interazioni reversibili.
Interazioni elettrostatiche:
in forma di forze attrattive ione-dipolo, interazioni dipolo-dipolo e legame H.
Formano legami più deboli del legame ionico.
Sono le interazioni ligando-recettore più diffuse.
Ligando e recettore
Complessi a trasferimento di carica:
si formano quando un gruppo elettron donatore è adiacente a un gruppo
elettron accettore. Il parziale trasferimento di carica crea un debole
legame elettrostatico.
I donatori sono specie ricche di e- p e gruppi chimici con doppietti
elettronici.
Legame idrofobico:
legame molto debole che si forma quando parti non-polari delle molecole
interagiscono in un ambiente privo di acqua.
Si crea un abbassamento dell’energia del sistema e, quindi, una struttura più
stabile.
Forze dispersive di London:
deboli interazioni dipolo-dipolo dovute alla formazione di dipoli transienti in
una struttura.
I dipoli transienti sono tempo-dipendenti e si formano a causa della
variazione nel tempo della distribuzione elettronica nelle molecole.
Ligando e recettore
Farmaco
Farmaco-Recettore
Il legame è dipendente dalla concentrazione [C] del farmaco:
man mano che [C] aumenta nei fluidi extracellulari, l’equilibrio si sposta a
destra e il farmaco si lega al recettore.
Quando [C] diminuisce, a causa del metabolismo e dell’escrezione, l’equilibrio
si sposta verso sinistra e il complesso si dissocia.
A basse concentrazioni un basso numero di recettori è impegnato dal
ligando.
I farmaci con lunga durata d’azione sono quelli che formano legami
stabili con il recettore.
Classificazione dei recettori
Tipi di recettori
Classificazione dei recettori
A) Recettori intracellulari
Ligandi endogeni: ormoni steroidei, ormoni della tiroide, vitamine (Vit. D e acido
retinoico)
Esempi: Ormone antidiuretico (ADH) o recettori della vasopressina
(sostanze lipofile in grado di attraversare la membrana plasmatica)
I recettori sono collocati nel nucleo cellulare.
Proteine (400-1000 aa) con una sequenza comune di circa 60 aa contenente
2 loops di 15 residui di aa noti come “zinc fingers”, in quanto ogni loop nasce
da 4 Cys coordinate a un atomo di Zn.
Classificazione dei recettori
Una volta formato il complesso
ormone-recettore avviene un
cambiamento conformazionale
che porta il complesso nel nucleo
dove si lega al DNA.
La risposta è un aumento
dell’attività RNA polimerasica e
dunque produzione di mRNA
specifico che, controllando la
sintesi di una specifica proteina,
dà origine alla risposta cellulare.
Classificazione dei recettori
Tipi di recettori
Classificazione dei recettori
B) Recettori legati a canali ionici
Ligandi endogeni: neurotrasmettitori
Esempi: nAChR (recettori colinergici nicotinici), recettori noradrenergici
Proteine di membrana con le zone C- e N-terminali extracellulari.
4 o 5 subunità intramembrana che circondano un poro centrale.
Ciascuna subunità è formata da 20-25 aa in forma di a-elica.
Residui di zuccheri sono attaccati alla catena N-terminale ma non sono
coinvolti nell’attività recettoriale.
I canali ionici “Voltage gated” sono attivati da alterazioni nel voltaggio
di membrana.
Ad es. i canali del sodio (Na+) si aprono quando la membrana viene depolarizzata
ad un potenziale basale ed essi stessi contribuiscono all’ ulteriore
depolarizzazione permettendo l’entrata di Na+ nella cellula.
Classificazione dei recettori
I canali ionici “Ligand gated” sono attivati dal binding di specifici ligandi o
farmaci al recettore.
Molti neurotrasmettitori e farmaci attivano i canali ionici Ligand gated,
tra cui molti tipi di recettori del glutammato.
Classificazione dei recettori
Classificazione dei recettori
Classificazione dei recettori
Tipi di recettori
Classificazione dei recettori
C) Recettori accoppiati alle Proteine G
Ligandi endogeni: ormoni e trasmettitori “lenti”.
Il recettore è accoppiato al sistema effettore mediante la proteina G.
Esempi: recettori colinergici muscarinici, noradrenergici e 7TM-GPCRs.
Singola catena polipeptidica formata da 400-500 aa.
Parte N-terminale extracellulare, parte C-terminale intracellulare.
La struttura del recettore include un dominio trans-membrana
a 7 eliche raggruppate attorno a un “pocket” centrale che
si pensa contenga il sito attivo del recettore.
Classificazione dei recettori
Elevata amplificazione del segnale biologico grazie all’attivazione delle
proteine G che, a loro volta, attivano canali ionici o, più comunemente,
altri enzimi (es. Adenilato ciclasi) causando l’attivazione di altri enzimi (es. PKA)
Questo sistema di amplificazione, che implica un’ estensione della durata
dell’attivazione della proteina G rispetto al legame del farmaco al recettore,
spiega perché gli effetti farmacologici massimali si osservano quando ormai
solo una piccola porzione di recettori è attivata.
Classificazione dei recettori
Classificazione dei recettori
Le proteine G, che trasducono il segnale
dai recettori, contengono 3 subunità,
a, b, e g. Le subunità b e g sono legate
covalentemente a lipidi di membrana.
Quando il recettore è libero,
la sottounità a della proteina Gs (Gsa)
lega il GDP ed è associata alle subunità
b e g.
Il GTP viene idrolizzato ed
il complesso Gsa-GDP si dissocia
dall'adenilato ciclasi e si riassocia
alle subunità Gsbg.
Il legame con l'ormone
induce un cambiamento
conformazionale nel
recettore.
Il recettore si lega alla
subunità Gsa.
Il legame del recettore
induce un cambiamento
conformazionale in Gsa.
Il GDP si stacca e viene
sostituito da una molecola di
GTP.
Il complesso attivo Gsa-GTP
dissocia dalle subunità Gsbg.
Il complesso Gsa-GTP
interagisce con l'adenilato
ciclasi che attivata catalizza
la sintesi del cAMP; l'ormone
si dissocia dal recettore.
Classificazione dei recettori
Tipi di recettori
Classificazione dei recettori
D) Recettori legati a enzimi
Ligandi endogeni: insulina e fattori di crescita.
Il recettore è accoppiato a una tirosin chinasi.
Esempi: recettori dell’insulina.
Caratterizzati da un solo dominio di transmembrana per subunità
di proteina. La subunità è legata ad ampi domini extra e intracellulari.
I domini extracellulari sono vari e dipendono dalla natura dei ligandi endogeni.
Tutti i domini intracellulari contengono un residuo tirosin chinasico
come parte integrante della struttura.
Per l’espressione dell’attività enzimatica è necessaria la dimerizzazione dei
recettori attivati causa del cambiamento conformazionale richiesto.
I residui di tirosina fosforilati agiscono come siti di legame per specifiche
proteine intracellulari che hanno in comune una sequenza conservata di circa
100 aa, chiamata “SH2 domain”, probabilmente la zona responsabile
delle successive risposte intracellulari.
Classificazione dei recettori
Teoria e assunzioni delle interazioni
farmaco-recettore
La combinazione o il legame a un recettore causa eventi che portano
a una risposta.
La risposta a un farmaco è “dose-dipendente”.
L’interazione farmaco-recettore segue semplici relazioni di
“massa-azione”, cioè una sola molecola di farmaco occupa un sito
recettoriale e l’interazione è reversibile.
Dato un farmaco, l’ampiezza della risposta è direttamente
proporzionale alla frazione di siti totali del recettore occupati
dalle molecole di farmaco (Teoria dell’occupazione – Clark, 1920 ca.).
Terminologia
Agonisti (o Agonisti Puri):
Farmaci che occupano i recettori e causano una
risposta piena o praticamente massimale.
La risposta massimale è di solito definita come
quella prodotta dagli agonisti più potenti, o
quella prodotta da un farmaco “classicamente”
associato alla risposta.
Terminologia
Agonisti Parziali:
Farmaci che occupano i recettori ma causano una
risposta inferiore a quella massima.
Ciò vuol dire che queste molecole sono meno
potenti e che l’occupazione del 100% dei siti
recettoriali provoca una risposta inferiore al
100% (in analogia con gli enzimi, corrispondono ai
substrati con una Vmax più bassa)
Terminologia
Antagonisti:
Farmaci che occupano o provocano cambiamenti
nel recettore, ma non causano alcuna risposta
recettoriale.
L’occupazione del recettore da parte di un
antagonista interferisce con l’occupazione da
parte dell’agonista.
Meccanismi delle interazioni
farmaco-recettore
Desensibilizzazione:
Ripetute esposizioni di un recettore alla stessa
dose di farmaco possono portare a una riduzione
della risposta.
E’ come se il farmaco, che inizialmente si comporta
da agonista puro, diventasse un agonista parziale
a causa dell’uso ripetuto.
Questo fenomeno è detto anche TACHIFILASSI