42527 MEDICINA GENETICA - C.I. (09/10)
Canale B
MALATTIA DI ALZHEIMER
Vincenzo Stanghellini (016074)
051.636-4101
[email protected]
MALATTIA DI ALZHEIMER (AD)
• Malattia neurologica del gruppo delle DEMENZE
• Caratterizzata da ETEROGENEITÀ GENETICA
mutazioni in geni diversi che portano allo stesso fenotipo
DEMENZA – DEFINIZIONE
Alterazione globale e spesso irreversibile e progressiva
delle funzioni corticali più alte includenti:
1.
apprendimento di abilità percettivo-motorie
2.
corretto uso di abilità sociali
3.
controllo
delle
reazioni
emotive
senza
evidente
ottundimento della coscienza
Royal College of Physicians 1981
AD - CLASSIFICAZIONE
FORME DI ALZHEIMER
Sporadico (75%) insorgenza tardiva (> 65 anni),
malattia multifattoriale
Familiare (25%) insorgenza precoce (< 65 anni)
malattia autosomica dominante
ALZHEIMER FAMILIARE
ad esordio precoce (<65 aa) APP, PS-1, PS-2
ad esordio tardivo
(>65 aa) APO E
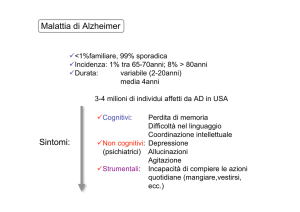
AD - EPIDEMIOLOGIA
Individui affetti
>4.000.000
>1.000.000
Costi/anno
50.000.000.000 USD
10.000.000.000 EURO
Prevalenza
>40anni
> 2/3 delle demenze >60 anni
10% >70 anni
AD - SEDI CEREBRALI COINVOLTE
• Gangli della base
Neuroni contenenti acetilcolina, importante per
l’acquisizione della memoria e l’apprendimento
• Ippocampo
Essenziale per la conservazione della memoria
• Corteccia cerebrale
Formazione del pensiero e del linguaggio
AD - CARATTERISTICHE CLINICHE
• Fasi precoci della malattia (ippocampo)
Progressiva perdita della memoria recente e della
capacità di svolgere compiti routinari
• Coinvolgimento della corteccia cerebrale:
Impoverimento del ragionamento, esplosioni
emotive, alterazione del linguaggio
• Morte di numerosi neuroni:
Alterazioni del comportamento (agitazione)
• Fasi finali:
Incapacità di riconoscere le facce e di comunicare,
perdita di controllo degli sfinteri anale e vescicale
• Tempo medio dalla diagnosi alla morte: 4-8 anni
sebbene la malattia talora possa durare > 20 anni
AD - DIAGNOSI
Diagnosi certa
• Caratteristiche anatomo-patologiche (biopsia / autopsia)
placche neuritiche “senili” e ammassi fibrillari in ippocampo,
corteccia temporale, nuclei della base
Diagnosi possibile o probabile
• Caratteristiche cliniche: deterioramento progressivo
• Laboratoristiche / strumentali: esami di laboratorio nella
norma; alterazioni precoci metaboliche nella corteccia parietale
alla PET; atrofia dell’ippocampo nelle fasi avanzate alla RMN
MALATTIA DI ALZHEIMER
• riduzione del flusso di sangue
• assottigliamento dei tessuti
• riduzione della massa
• anomalo utilizzo del glucosio
• Risonanza magnetica nucleare (RM)
• Tomografia computerizzata (TC)
• TC a emissione di fotone singolo (SPECT)
• Tomografia a emissione di positroni (PET)
AD - CARATTERISTICHE ANATOMO-PATOLOGICHE
Principali tipi di lesione istologica
• Placche amiloidi (extra-neuronali)
• Degenerazione neurofibrillare (intra-neuronale)
AD - CARATTERISTICHE ANATOMO-PATOLOGICHE 1
• Placche senili o placche neuritiche
Strutture di forma sferoidale, costituite da un
deposito
centrale
di
amiloide,
circondato
da
materiale granulare e filamentoso rappresentato da
neuriti rigonfi ed espansioni dendritiche, frammisti a
processi
gliali
e
cellule
microgliali.
La
composizione e la configurazione delle placche
senili variano a seconda del loro stadio di sviluppo.
La sostanza amiloide, componente essenziale delle placche senili, è principalmente
composta da una proteina ( A4) che deriva dalla proteolisi di un precursore di
maggiori dimensioni (Amyloid Protein Precursor - APP).
AD - CARATTERISTICHE ANATOMO-PATOLOGICHE 2
• Degenerazione neurofibrillare (grovigli neurofibrillari)
Masse di filamenti abnormi di forma globosa o a
fiamma, situate a livello del citoplasma perinucleare,
che spesso si estendono sino ai dendriti apicali; al
microscopio ottico presentano una struttura fibrillare
AD - FATTORI DI RISCHIO
• non genetici
potenzialmente modificabili
• genetici
non modificabili
AD - VALUTAZIONE DEL RISCHIO NON GENETICO
FATTORI DI RISCHIO VASCOLARI:
ipertensione sistolica >160 mmHg
colesterolo sierico > 6.5 mmol/L
FATTORI DI RISCHIO DOVUTI ALLO STILE DI VITA:
fumo di tabacco
scarsa attività fisica
nessuno / eccessivo consumo di alcool
traumatismi cranici
FATTORI DI RISCHIO SOCIO-DEMOGRAFICI:
età avanzata
sesso femminile
periodo di istruzione < 15 anni
lavoro che espone a tossine ambientali (es. metalli pesanti)
ALTRI FATTORI:
infezioni virali
depressione
PAN (anamnestica)
ipertiroidismo
familiarità per s. di Down
AD - CLASSIFICAZIONE EZIOPATOGENETICA
• 75% forme sporadiche
• 25 % forme familiari
• Forme precoci familiari (AD1 - APP, AD3 - PSEN1, AD4 - PSEN2): < 65 aa
• Forma tardiva familiare (AD2 - APOE): > 65 aa
AD FAMILIARE - GENI COINVOLTI
•
Gene della proteina amiloide (APP) - Cr21 - AD1
Mutazioni rare (20 famiglie nel mondo); esordio precoce (35-50 aa)
•
Gene della apolipoproteina E (APOE) Cr19 - AD2
Associato anche a forme sporadiche esordio tardivo (>65 aa)
•
Gene della presenilina 1 (PSEN1) Cr14 - AD3
> 50 mutazioni identificate; causa genetica più comune di MA familiare
ad esordio precoce (28-60 aa); forma grave
•
Gene della presenilina 2 (PSEN2) Cr 1 - AD4
Esordio meno precoce, andamento meno grave rispetto a PSEN1
AD - MUTAZIONE DEL GENE APP (amyloid precursor protein)
mutazione
APP
Cr21
Codifica per APP
(recettore tipo G)
adesione cellulare, crescita
sinaptica, riparazione neurale
Incapacità di formare l’intera proteina APP
Formazione di peptidi A più corti
Amiloide A,
Deposizione della proteina amiloide
nelle placche extratraneuronali
+ APOE, proteoglicani, anti-chimotripsina
Morte dei neuroni interessati
AD ad esordio precoce
Emorragie cerebrali secondarie ad amiloidosi vascolare
AD FAMILIARE AD ESORDIO PRECOCE
β-APP = amyloid beta precursor protein
recettore tipo G adesione cellulare, crescita sinaptica, riparazione neurale
AD1 mutazioni di β-APP (chr 21) catab. ad opera di -secretasi
AD3, AD4 mutazioni di PSEN-1 (chr 14) e PSEN-2 (chr 1) attività di γ-secretasi
produzione aggregati insolubili prot. β-amiloide (Aβ) (placche senili)
CMAJ, 2008;78:548–556
SECRETASI E AMYLOID PRECURSOR PROTEIN (APP)
Walter et al. Current Opinion in Neurobiology 2001, 11:585–590
EVIDENZE A FAVORE DEL RUOLO DEL GENE APP COME SEDE DELLE
MUTAZIONI RESPONSABILI DI AD
•
A4 (A peptide di 42 AA - prodotto di clivaggio proteolitico di APP):
principale componente della placca amiloidotica neuronale caratteristica
della AD
•
Sede del gene per AD: Cr 21 da studi di linkage genetico
•
I pazienti con trisomia 21 (doppia coppia del gene APP): AD entro i 40 aa
•
Mutazioni del gene APP identificate in pazienti con emorragie cerebrali
ereditarie ed amiloidosi vascolare (abbondante deposito perivasale di A;
sporadiche placche diffuse a tutto il parenchima cerebrale)
Preseniline
Famiglia di proteine di transmemembrana che
svolgono funzioni di proteasi (γ-secretasi)
PSEN 1 (cr. 14) molto simile a PSEN 2 (cr. 1)
Codificano per Pr simili probabilmente costituite da 7
domini di transmembrana (anche se studi recenti
avrebbero dimostrato la presenza di 8 domini)
Mutazioni missenso produzione di A4 con 42 AA
AD - GENI PRESENILINI
• Gene presenilino 1 (PS-1)
Localizzato sul cromosoma 14
Codifica per una proteina denominata S182
Mutazione: trasmissione autosomica dominante
Demenza a esordio precoce (45 anni < < 55 anni)
Andamento molto rapido con morte in pochi anni
Mutazioni identificate in numerose famiglie
AD - GENI PRESENILINI
• Gene presenilino 2 (PS-2)
Localizzato sul cromosoma 1
Codifica per una proteina chiamata STM2
Mutazioni ad esordio precoce, ma meno di PS-1 (media 53 anni)
Andamento un po’ meno grave (sopravvivenza media 11 anni)
Mutazioni identificate per la prima volta in una famiglia americana
di origini germaniche del Volga e successivamente in una
famiglia di Firenze.
Apolipoproteina E (APOE, cr 19)
Late - Onset
Proteina plasmatica di 299 aa coinvolta nel
trasporto del colesterolo
Lega la proteina β4-amiloide
Esistono 3 isoforme → ε2, ε3, ε4
ε2 158
Arg – Cys
ε3
wild-type
ε4 112
Cys – Arg
AD FAMILIARE AD ESORDIO TARDIVO
APOE (chr 19)
allele ε4 aumenta il rischio di Alzheimer
allele ε2 diminuisce il rischio di Alzheimer
Rischio di Alzheimer legato al genotipo di APO E
CMAJ, 2008;78:548–556
AD - GENE APOE
L’acquisizione con il patrimonio genetico dell’allele 4 del gene APOE
1. rischio di sviluppare AD nelle sue forme sia precoce che tardiva
2. dell’età di insorgenza della forma tardiva di MA
Non è ancora chiaro se l’allele 4 del gene APOE è sufficiente di per sé a
determinare la malattia e, a differenza delle mutazioni missenso nei geni APP,
PS1 e PS2, la sua scoperta in soggetti presintomatici è di significato incerto.
La scoperta dell’allele 4 del gene APOE in un soggetto con un quadro clinico
compatibile con AD può avere utilità clinica ancillare
AD - GENE APOE DEPOSITI AMILOIDOSICI EXTRA-CELLULARI
E NEUROFILAMENTI INTRANEURONALI (NFTi)
1. differenti alleli APOE potrebbero esercitare i loro diversi effetti
influenzando in modi diversi il metabolismo lipidico e/o la
riparazione neuronale
2. ε4 forte avidità di legame per A clearance
3. Proteina in presenza di ε4 agisce più liberamente alla formazione
dei neurofilamenti
Proteine Tau (cr 17q21)
•
MAPS (microtubule-associate proteins): proteine (6 isoforme 352-441 AA)
altamente solubili associate ai microtubuli neuronali (soprattutto SNC - in
porzioni distali di assoni x trasporto intracellulare di messaggeri molecolari)
•
Interagiscono con tubulina sintesi / stabilizzazione dei microtubuli
•
Fosforilazione di Ser262 o Ser214 precipitati insolubili separazione dai
microtubuli accumulo sotto forma di microfilamenti e funzione
•
Localizzazione coincide con le zone di danno neuronale ed è associata con
la presenza dell’allele 4 della apolipoproteina E (APOE).
MECCANISMI PATOGENETICI DI AD
FORMA SPORADICA
Fattori ambientali
Predisposizione genetica
produzione o
clearance di Aβ
FORMA FAMILIARE
Oligomerizzazione ed
iniziali depositi di Aβ 42
Alterate funzioni sinaptiche
Mutazione di β-APP
produzione di Aβ
rapporto Aβ 42/40
Risposta infiammatoria e formazione delle placche amilodosiche
Progressivo danno di sinapsi e assoni
Alterazione dell’omeostasi neuronale e stress ossidativo
Alterata oligomerizzazione ed iperfosforilazione di tau
Diffusione del danno neuronale, morte cellulare + deficit di neurotrasmettitori
Demenza e neurodegenerazione