CUORE
Prof. Schiavone
17/04/2007
8.00- 10.00
SCOMPENSO CARDIOCIRCOLATORIO
È una delle patologie più frequenti nella popolazione e una di quelle in assoluto con la quale più
spesso vi troverete a dover fare i conti.
È una sindrome abbastanza complessa per poterne dare una definizione univoca e chiara; la più
generica e che più riproduce la sua realtà clinica è la seguente:
“lo scompenso cardiocircolatorio è una condizione fisiopatologica in cui un’anormalità della
funzione cardiaca è responsabile dell’incapacità del cuore a fornire una quantità di sangue adeguata
alle esigenze metaboliche dei tessuti periferici”.
Nelle condizioni di scompenso abitualmente, a meno che non ci sia un’estrinsecazione clinica
conclamata, la quantità di sangue che arriva ai vari organi e tessuti, in condizioni di riposo, è
sufficiente per un loro normale funzionamento. Mentre si manifesta in modo evidente quando venga
richiesto un aumento della portata cardiaca, per esempio in condizioni di sforzo, inteso in tutte le
sue accezioni, non solo fisico, quindi quando ci dovrebbe essere un implemento della portata
cardiaca, che non può essere assicurato dal cuore. Considerando quest’ultimo aspetto, forse la
definizione più giusta sarebbe quella di una condizione, in cui un’alterazione o funzionale o
strutturale cardiaca renda il cuore incapace di fornire ai tessuti periferici, in maniera istantanea, una
quantità di sangue necessaria per il corretto funzionamento degli stessi, nelle diverse esigenze
metaboliche.
FUNZIONE CARDIOCIRCOLATORIA
Substrati metabolici,ossigeno,
ormoni, catecolamine, etc…
contrattilità
distensibilità
frequenza
RAA
Portata cardiaca
Resistenze periferiche
(tono vasomotorio)
Una funzione circolatoria normale si esprime in ultima sostanza con una normale portata cardiaca,
risultato dell’interazione tra l’azione cardiaca e le resistenze periferiche, nel senso che il lavoro, che
il cuore svolge per pompare il sangue dall’interno della propria cavità all’esterno nei grandi vasi, è
modulato dalle seconde, espressione del tono vasomotorio delle arteriose, che creano una sorta di
barriera, più o meno variabile, al cuore stesso. Anche, però, caratteristiche proprie del cuore
contribuiscono a determinare l’entità della portata cardiaca: contrattilità, distensibilità e frequenza
cardiaca. Quando al cuore si richiede uno sforzo aggiuntivo, cioè un aumento della portata cardiaca,
la prima risposta da parte del cuore è un aumento della frequenza cardiaca e della forza di
contrazione. La variazione della distensibilità non può essere al servizio delle varie necessità perché
dipende da alterazioni organiche della parete miocardica, cioè dalla perdita di tessuto muscolare
attivo e sua sostituzione con materiale fibroso.
Su frequenza e contrattilità, invece, incidono tutta una serie di elementi regolatori di natura
neuroumorale, chimico, ormonale, substrati metabolici, la disponibilità di ossigeno; ma tutto ciò
agisce anche a livello delle resistenze periferiche. Tra portata cardiaca e resistenze periferiche esiste
un equilibrio estremamente complesso, su cui incidono numerosissimi parametri, anche con delle
variazioni che possono inficiare l’azione stessa del cuore; il cuore può aumentare le proprie
contrattilità e frequenza, ma, se contemporaneamente aumentano le resistenze periferiche in misura
notevole, il risultato finale è solo un aumento dello stress parietale del cuore, senza aumentare la
portata cardiaca. Quindi, perché questa modulazione avvenga realmente e proficuamente in
funzione delle esigenze dei tessuti periferici istante per istante, è necessario un sapiente rapporto tra
tutti i parametri che regolano il flusso circolatorio; non sempre tutti questi meccanismi sono diretti
verso lo stesso scopo.
Di fronte ad una condizione in cui il cuore viene sottoposto ad un eccesso di lavoro (per cause
intracardiache, come una stenosi valvolare aortica, od extracardiache, come la presenza di una
fistola artero-venosa), o pressorio o volumico, il cuore stesso risponde con un rimodellamento della
camera ventricolare.
Il sovraccarico presso rio e quello volumico costringono il cuore a lavorare in maniera antieconomica, con eccessiva richiesta e quindi dispendio di ossigeno ed energia. Un sovraccarico
pressorio si ha tutte le volte in cui si pone un ostacolo all’uscita del sangue dal ventricolo sx
nell’aorta e, appunto, le cause possono essere intrinseche al cuore stesso oppure estrinseche.
Poniamo l’esempio di un ostacolo intrinseco al cuore: nella stenosi aortica, per superare tale
ostacolo dato dalla riduzione dell’ostio valvolare, il cuore deve lavorare di più e le singole
fibrocellule muscolari cardiache sono sottoposte ad un maggiore stress. In tal caso, l’aumento dello
stress parietale si traduce in uno stimolo all’iperproduzione proteica , che a propria volta si traduce
in aumento del volume delle fibrocellule e, secondo le vedute più recenti, anche del proprio numero.
Il risultato ultimo è l’aumento dello spessore delle pareti, detto ipertrofia miocardica; normalmente
un cuore pesa tra i 250 e i 300 g, mentre cuori con spiccata ipertrofia possono raggiungere i 450500 g, e anche oltre. In questo modo si ottiene un’accresciuta forza lavoro, perciò l’ipertrofia
sarebbe un meccanismo finalisticamente ben orientato, perché avendo più unità lavorative, si
aumenta il lavoro producibile e si tende a pareggiare quel debito di fatica che ci sarebbe stato senza
questo tentativo di compenso, e che così viene invece ripartito tra più voluminose, e
presumibilmente numerose, fibrocellule. L’ipertrofia miocardica è, però, una condizione
antifisiologica e qui c’è da fare una differenziazione tra muscolo scheletrico e muscolo cardiaco, a
parità di richiesta funzionale: sottoponendo un muscolo scheletrico ad un allenamento continuo,
questo man mano diventa ipertrofico e non è malato, anzi probabilmente funziona molto meglio
della sua controparte normale, come resa energetica. Questo perché mantiene inalterate le proprie
caratteristiche strutturali e funzionali, nonostante l’aumento dello spessore e del numero delle fibre
muscolari. A livello cardiaco, invece, l’ipertrofia fibrocellulare non è accompagnata di solito da un
parallelo aumento della vascolarizzazione; la densità capillare del cuore normale, che è
approssimativamente di un capillare per 5 cardiomiociti, diventa la metà in un cuore ipertrofico,
cioè di un capillare per 10 cardiomiociti. Quindi le cellule sono potenzialmente più produttive, ma
in realtà sono mal nutrite, in quanto l’apporto di ossigeno che ricevono resta pari a quello
precedente l’ipertrofia e quindi causa di una ischemizzazione relativa.
In più, i sistemi neuroumorali od ormonali (angiotensina II, aldosterone, etc),che inducono
l’aumento della sintesi proteica, determinano anche un massivo incremento della quota collagene
del cuore, facendo assumere all’impalcatura fibrosa in percentuale un valore nettamente superiore
rispetto al normale. Anche qui, se nel cuore normale avevamo 5 fibre connettivali per ogni cellula
muscolare, nell’ipertrofia saranno 10 fibre per ogni cellula. Ricapitolando, abbiamo un deficit di
irrorazione ed un’alterazione nel rapporto tra fibrocellule muscolari e quota collagene.
L’alterazione di tale rapporto determina una ridotta distensibilità della massa muscolare; così, le
pressioni di riempimento ventricolare aumentano, perché il cuore non è più così elastico. Tanto per
far capire meglio il concetto di compliance e le sue implicazioni, considerate il cuore come fosse di
gomma; ne esistono diverse qualità, come quella dei palloncini del luna park che è particolarmente
elastica, potendoci insufflare dentro vari litri di gas o liquido distendendoli notevolmente, ma senza
comportare un eccessivo aumento della pressione al loro interno, perché il contenente si adatta
abbastanza bene al contenuto. Un altro tipo di gomma è quella dei copertoni delle automobili, in cui
anche modesti ingressi di aria determinano notevoli innalzamenti di pressione, proprio perché la
distensibilità del materiale è modesta. Quindi, aumentando nel cuore la quota fibrosa, esso
assomiglia sempre più alla gomma dei copertoni, piuttosto che a quella di un palloncino;a parità di
volume di riempimento diastolico, si genera una pressione telediastolica nettamente elevata.
Ne consegue che tale accresciuta pressione si esercita, in particolare, sugli strati subendocardici,
quelli più interni e più a stretto contatto col sangue, che è l’elemento che crea pressione. Per una
questione di fisiologia di circolazione coronaria, che vi verrà spiegata in un secondo momento, nella
parete endocardica la capacità di autoregolazione è zero, o comunque molto bassa, per cui, essa non
può rispondere a questa forza di compressione ab estrinseco aumentando il flusso, cioè
vasodilatando i propri capillari, perché lo sono già al massimo; quindi, si lascia progressivamente
schiacciare da questa pressione endocavitaria elevata e si creano fenomeni di necrosi
subendocardica. È una parete destinata, in un tempo più o meno lungo, a cedere per tutti questi
motivi; infatti, a lungo andare, l’ipertrofia si traduce progressivamente in scompenso
cardiocircolatorio. Quindi, quello che nasce come un meccanismo compensatorio, per
rinormalizzare la funzione cardiaca, ha in realtà in se stesso i germi della distruzione del cuore.
Nel sovraccarico volumico, poi, in fase diastolica arriva un volume di sangue al ventricolo
maggiore di quello che dovrebbe essere; una cavità ventricolare è costruita per contenere in diastole
circa 70, massimo 100 ml di sangue e, quando per diverse condizioni patologiche ne arriva una
quota maggiore, essa tende a dilatarsi. Comportano questa evenienza tutte le condizioni di rigurgito,
per esempio il rigurgito nell’insufficienza aortica, in cui una certa quota di sangue in diastole invece
di progredire in periferia, per la mancata giustapposizione dei lembi ricade all’interno del
ventricolo. Al ventricolo, in seguito al normale ritorno venoso e attraverso il circolo polmonare,
arriva anche la normale quota di sangue reflua dalla periferia, 50-60 ml; a questi si aggiunge quella
quota variabile che ricade nel ventricolo per l’insufficienza valvolare. Lo stesso succede
nell’insufficienza mitralica, in cui una quota di sangue, in aggiunta a quella della normale
circolazione, fa la spola tra l’atrio ed il ventricolo. In conseguenza di questo aumentato volume di
sangue, il ventricolo tende a dilatarsi e, volendo vedere in ciò un certo finalismo, potremmo dire che
lo fa per sfruttare al meglio la legge di Starling (la forza sviluppata in contrazione isometrica è
direttamente proporzionale alla lunghezza iniziale della fibra muscolare, ossia al pre-carico).
Quindi, con la dilatazione ventricolare si possono ottenere delle prestazioni meccaniche contrattili
superiori alla norma, tant’è vero che, nelle situazioni patologiche di rigurgito, inizialmente i pazienti
hanno delle frazioni di eiezione nettamente superiori alla norma: 70-75% contro il 55-60% del
normale.
A lungo andare, però, come avviene anche per gli elastici comuni, le fibre si sfibrano, per cui si
perde la capacità di ritorno alla posizione o dimensioni iniziali al termine della sollecitazione
meccanica, cioè si perde l’elasticità, la compliance del tessuto muscolare cardiaco. La curva che
esprime la legge di Starling ha infatti una iniziale fase ascendente, poi un plateau, dopo di che
decade pressoché verticalmente; quando si ha la rottura del sistema miofibrillare per eccessiva
distensione delle fibre, si ha un decadimento della pulsione contrattile. In più, il consumo di
ossigeno di tali fibrocellule è direttamente proporzionale alla loro lunghezza, e quindi alla misura
del raggio interno della cavità ventricolare; ma non aumenta adeguatamente l’apporto di ossigeno,
perciò si va in deprivazione di ossigeno, che conferisce alle cellule una sofferenza ischemica
cronica che le rende prima o poi prone ad una caduta di funzione e quindi allo scompenso.
Tutti questi fenomeni, che vanno sotto il nome di rimodellamento ventricolare, sono sostenuti da
una noxa consistente in alterazioni strutturali e/o funzionali cardiache, ed essi tendono a pareggiare
questi deficit, però in sostanza a lungo andare aggravano la situazione di partenza. Questo è comune
a tutti i meccanismi di compenso, i quali sono tutti antifisiologici: anche se nascono nell’intento di
parare le alterate funzioni cardiocircolatorie, tuttavia alla fine sono dannosi e tutta la fisiopatologia
dello scompenso cardiocircolatorio si basa sui meccanismi di compenso.
Infarto miocardico
ipertensione
valvulopatie
Sovraccarico funzionale
Stress parietale
Dilatazione
ipertrofia
Disfunzione
Aritmia
Attivazione
neuroumorale
Quando esiste un danno cardiaco qualsiasi (infarto, valvulopatia, ipertensione arteriosa, etc), che
produce una riduzione della portata cardiaca, l’organo che più di tutti risente nell’immediato di ciò è
il rene, che a livello dell’apparato iuxtaglomerulare produce renina, che converte
l’angiotensinogeno in angiotensina I, convertita dall’ACE in angiotensina II, uno dei più potenti
vasocostrittori che si conoscono. Essa produce un vasocostrizione a carico soprattutto degli organi
non vitali; volendo essere finalistici, ciò ha la finalità di ridurre l’apporto di sangue in tessuti che
non ne hanno un gran bisogno, come può essere la cute, il compartimento splancnico, e quindi
favorire un maggior afflusso in organi vitali, come cervello, cuore, rene.
In realtà, però, così facendo, non si fa che aumentare le resistenze vascolari periferiche; tant’è che
in queste fasi dello scompenso la pressione diastolica aumenta: abbiamo basse pressioni sistoliche,
che traducono la riduzione della portata cardiaca, con pressioni diastoliche relativamente più alte
(es. 120-125 mm Hg/ 90-95 mm Hg). L’aumento delle resistenze vascolari periferiche è una fase
del cosiddetto “compenso”; ciò mette il cuore nelle condizioni di un aggravamento del proprio
lavoro, che produce un aumento dello stress di parete, a sua volta determinante ulteriore danno
cardiaco. Si instaura un circolo vizioso che si ripercuote attraverso queste tappe in uno dei tanti
circoli viziosi della fisiopatologia dello scompenso cardiaco.
Danno cardiaco
Riduzione portata cardiaca
Danno
Ischemia renale
Consumo di ossigeno
Renina
Stress parietale
Angiotensina
Resistenze periferiche
Vasocostrizione organi
non vitali
Come abbiamo visto prima, l’aumento della renina determina un aumento dell’angiotensina I e II,
che da una parte potenzia l’ipertrofia e dall’altra induce un aumento della liberazione di
aldosterone, che concorre pure all’ipertrofia, ma soprattutto induce ritenzione di Na e una perdita di
K. La ritenzione di Na comporta ritenzione di liquidi, quindi ipervolemia; ciò potrebbe essere, tutto
sommato, desiderabile, vista la ridotta portata cardiaca, ma determina anche un sovraccarico
volumico, con conseguente aumento del lavoro cardiaco in un cuore che non ha più le capacità per
farlo. Anche l’ipersecrezione di aldosterone, perciò, si traduce in ultimo in un danno al cuore; ha
poi come effetto secondario l’aumentata secrezione di NE, che si aggiunge all’angiotensina
nell’indurre vasocostrizione. In conclusione, avremo un aumento della volemia associato ad un
aumento delle resistenze periferiche, quindi sovra-stress di parete.
L’ipervolemia, poi, quando esista un’ipertensione venosa, come accade in corso di scarso
funzionamento cardiaco, si traduce in una dispersione di liquidi negli interstizi: edemi periferici
nelle prime fasi, ascite più tardi.
Altra considerazione da fare riguarda il K: la sua perdita induce alterazione dell’equilibrio elettrico
delle cellule cardiache, rendendole più prone ad aritmie, danno funzionale che si somma a quello
strutturale. Esistono molti altri fenomeni legati alla perdita di K, come la riduzione delle capacità
del muscolo scheletrico; tutte conseguenze, comunque, innescate dal primum movens, che è la
riduzione della portata cardiaca.
Fisiopatologia dello scompenso cardiaco
Attivazione neurormonale
Renina
Lavoro cardiaco
Angiotensina I-II
Ipertrofia
Sovraccarico di volume
Aldosterone
Ritenzione Na
Perdita K
Ipervolemia
Edemi
NE
Vasocostrizione
La riduzione della portata cardiaca produce anche stimolazione dei barocettori aortici e carotidei,
che inducono in via riflessa aumentato rilascio di catecolamine, con stimolazione dei recettori e
2 cardiaci che comporta aumento della frequenza cardiaca, sia nel ritmo sinusale sia nella
fibrillazione striale o altre aritmie. Le catecolamine stimolano anche i recettori 1 e 2 periferici,
che si traduce in vasocostrizione e quindi aumento delle resistenze periferiche; ancora una volta i
due effetti combinati che sono deleteri per la condizione cardiocircolatoria. Inoltre la stimolazione
dei barocettori induce in risposta un aumento della contrattilità, cosa che comporta accresciute
richiesta e consumo di ossigeno, in una condizione già di ischemia relativa (per quanto detto prima),
che assommata alle conseguenze dell’ipocaliemia più facilmente porta ad aritmie e complicazioni
via via ingravescenti.
Riduzione portata
cardiaca
Ischemia, aritmie
danno cardiaco
Stimolazione barocettori
Catecolamine
Stimolazione recettori
1 e 2 cardiaci e
1 e 2 periferici
Consumo ossigeno
cardiaco
Contrattilità- tono vascolare
periferico
Ci sono altre attivazioni nefaste nell’ambito della fisiopatologia dello scompenso cardiaco, quali
l’iperproduzione di chinine, arginina e vasopressina, endotelina1, ottenendo: vasocostrizione
periferica, aumento dell’inotropismo cardiaco, vasocostrizione renale, ritenzione di Na ed acqua,
ipertensione polmonare, neoangiogenesi, aterosclerosi ed ipertrofia cardiaca; tutti effetti determinati
da queste sostanze e che concorrono al peggioramento dell’equilibrio cardiocircolatorio totale.
Quindi, ripetendo, i meccanismi attivati dalla riduzione della portata cardiaca si definiscono
compensatori, perché tendono al riequilibrio delle condizioni di circolo, ma in cronico esitano in un
progressivo danno del cuore: questo è il centro della fisiopatologia del cuore.
Non si può, quindi, definire un unico quadro di fisiopatologia dello scompenso cardiocircolatorio, e
vi è una serie di alterazioni più o meno concatenate tra di loro che determinano alla fine il
peggioramento continuo del quadro clinico.
L’eziologia prevalente di queste condizioni è quella ischemica, che da sola ricopre un 50% circa dei
casi, sottoforma di infarto miocardico in primis e di condizioni ischemiche croniche in seconda
istanza. Seguono le eziologie a carattere non ischemico: cardiomiopatia dilatativa idiopatica 18%;
cardiopatia vascolare 4%; ipertensione arteriosa 3,8%; cardiopatia alcoolica 1,8%; cardiomiopatia
post-partum 0,4%; cardiomiopatia infiltrativi 0,1%; altro (soprattutto uso di tossici, in particolare
cocaina) 21%.
Tra tutte queste, la cardiomiopatia dilatativa idiopatica è una forma di alterazione grave della
capacità contrattile cardiaca per cui esiste una familiarità: noxae patologiche varie che agiscono su
terreni metabolici predisposti possono in età più o meno giovanile produrre questo tipo di malattia.
Conosco una famiglia nella quale il nonno è morto all’improvviso a 40 aa probabilmente per
un’aritmia parossistica; suo figlio è morto a 60 aa per uno scompenso grave ed irreversibile legato
ad una cardiomiopatia dilatativi seria; la figlia di costui stava perfettamente bene fino a che, all’età
di 37-38 aa ha avuto un parto, prima del quale è stata adeguatamente controllata, proprio considerati
i precedenti familiari. L’ECG era assolutamente normale, non aveva alcun tipo di disturbo; a
distanza di un mese dal parto, questa paziente si presenta con una frazione di eiezione scesa dal
65% del pre-partum al 22%, con un quadro clinico di edema polmonare acuto ed un ECG che
evidenziava un blocco di branca sx completo e un QRS di durata particolarmente elevata. Il punto
di vista ecocardiografico evidenziava un volume ventricolare telediastolico sx che era passato da
82-83 del pre-partum a 180 del post-partum. Si conoscono le forme di miocardiopatia postpartum,
che non si sa bene come inquadrare dal punto di vista eziologico e fisiopatologico, ma in questa
donna probabilmente una noxa patogena, come probabilmente un fatto simil-influenzale, che
racconta nell’intervallo tra il parto ed il ricovero, noxa che in atre donne senza familiarità non
avrebbe sortito alcun effetto reale, è bastata per scatenare una reazione di questo genere. Vuol dire
che esiste una meiopragia d’organo, cioè una debolezza fondamentale, per cui l’organo risponde a
stimoli, che nel normale non dovrebbero esitare in alcuna forma particolare di cardiomiopatia, in
questa maniera perversa. In tal senso c’è una familiarità nelle cardiomiopatie dilatative idiopatiche
primitive.
Danno cardiaco
Ipossiemia
P telediastolica V sx
Ridotta portata cardiaca
RAA
RPV
P capillare polmonare
Meccanismi
neurormonali
Angiotensina II
Aldosterone
Dispnea- EPA
P arteriosa polmonare
Ischemia
Aritmie
Lavoro V dx
Dilatazione V dx
Insufficienza tricuspidalica
Ipervolemia
Epatomegalia
Insufficienza epatica
Ipoalbuminemia
P portale e P venosa
sistemica
Edemi periferici
Ascite
Questo schema tende a rapportare i momenti della fisiopatologia dello scompenso con alcune delle
sue manifestazioni cliniche più importanti. C’è un danno cardiaco, una ridotta portata cardiaca, un
primo circolo di cosiddetto compenso che passa per l’iperattivazione del sistema reninaangiotensina-aldosterone, con l’aumento delle resistenze periferiche e della volemia ed abbiamo
detto come questo si ritorca sul cuore stesso. Dall’ipervolemia derivano: primo, gli edemi periferici,
secondo l’ascite, che deriva anche da una condizione di epatomegalia e di insufficienza epatica
prodotta dall’aumento cronico della P nella vena cava inferiore, che si riverbera, attraverso le vv.
sovraepatiche, al fegato, il quale progressivamente si imbibisce di acqua, aumentando di volume.
L’epatomegalia, però, non è fine a se stessa, perché determina un aumento della pressione
intraepatica e quindi un’ipertensione portale, che a ritroso investe anche il microcircolo intestinale,
da cui inizia una trasudazione di liquido che forma l’ascite. L’eccesso cronico di liquido nel fegato
produce anche una sofferenza epatocitaria, che porta tra le altre cose ad ipoalbuminemia, ridotta
pressione oncotica e ulteriore trasudazione di liquido nel compartimento extravascolare,
peggiorando ulteriormente sia l’ascite sia gli edemi periferici.
Il danno cardiaco, abbiamo detto, produce un aumento della pressione telediastolica del ventricolo
sx e perciò della pressione capillare polmonare, responsabile della dispnea, una delle manifestazioni
cliniche più eclatanti di scompenso cardiaco. Essa è una sensazione soggettiva di respiro
difficoltoso, è un sintomo, non un segno; noi medici erroneamente diciamo dell’ammalato che è
dispnoico, quando rileviamo in lui un respiro difficoltoso, ma dovrebbe essere l’ammalato stesso a
riferircelo, mentre noi dovremmo distinguere tecnicamente, della dispnea che lui ci riferisce, se sia
una tachipnea, una iperpnea, un respiro rumoroso, etc.
La dispnea ha degli steps progressivi, relativamente all’entità dello scompenso che la genera; per
cui, nelle fasi iniziali, la dispnea è assente a riposo, mentre si manifesta dopo sforzo (dispnea da
sforzo). Progressivamente si riduce l’entità dello sforzo minimo capace di elicitarla, fino ad esserci
anche a riposo; segue l’ortopnea, cioè quella dispnea che obbliga all’ortostatismo o al platistatismo.
In clinostatismo, infatti, subentra un’alterata distribuzione di liquidi nel polmone, per cui il paziente
non riesce a respirare.
Dall’ortopnea si passa alla dispnea parossistica notturna, consistente in accessi improvvisi ed
autolimitantesi di dispnea, abitualmente notturni perché il sovraccarico di liquidi nel polmone, che
in ortostatismo si distribuisce prevalentemente alle basi polmonari, consentendo di respirare a
sufficienza, in clinostatismo lo fa a tutti i diversi livelli, compromettendo oggettivamente la capacità
respiratoria del paziente. In più, gli edemi e l’ascite nell’ortostatismo per gravità si trasferiscono
nelle parti declivi; mentre in clinostatismo, essi tendono a riassorbirsi, venendo meno l’effetto della
gravità, se non nelle parti inferiori degli arti, quindi c’è un ulteriore aumento della volemia, che il
cuore non può sopportare. Perciò, aumenta la pressione telediastolica, la pressione capillare
polmonare e quindi si hanno i sopraccitati accessi dispnoici notturni.
Ultimo step di questa condizione respiratoria è l’edema polmonare acuto, una condizione
premortale, se non adeguatamente trattato nel giro di pochi minuti; si instaura, di solito, su una
pregressa insufficienza cardiaca oppure in conseguenza di un fatto cardiaco acuto, come un infarto
miocardico acuto massivo. L’ammalato è pallido, con sfumatura cianotica, manifesta sudorazione
fredda, è orripilato, presenta un abbassamento dello stato di coscienza perché l’ossigenazione
diventa particolarmente bassa, è agitato, non riuscite a tenerlo fermo, né vuole stare fermo, è
obbligato a stare seduto, abitualmente sulla sponda del letto con le gambe appese, perché questo gli
consente un sequestro periferico di liquidi, con ridotto ritorno venoso e migliore respirazione. In
genere egli tende a piegare le gambe, in modo da comprimere al poplite le vene ed evitare che
molto sangue ritorni al cuore. Se auscultate, anche senza strumenti, sentite un gorgoglio, che viene
preceduto da una tossetta, dovuta all’edema della mucosa bronchiale, che pian piano si trasforma
nella sensazione acustica di gorgoglio, legato all’ingresso dell’aria in un volume contenente liquido.
A ciò, se l’ammalato non decide di lasciarci prima, segue l’emissione di liquido schiumoso rosato,
perché sempre nel liquido della trasudazione polmonare c’è qualche globulo rosso, e questa
schiuma esce dalla bocca e dalle narici perché, sempre per l’ipossia, c’è un’incoordinazione dei
movimenti di deglutizione ed antideglutizione, per cui si aprono tutte le vie di fuga per questo
liquido. Se ancora a questo livello non si interviene, l’ammalato è perduto, perché per avere la
fuoriuscita di tale schiuma dalla bocca vuol dire che i polmoni sono strapieni di acqua e quindi non
ossigena e l’ipossia gravissima produce danni cerebrali e cardiaci irreversibili, a cui segue la morte.
Andando a farne l’autopsia, a parte le condizioni cardiache, si ritrovano delle grosse masse
polmonari, mentre nella norma il polmone è molto piccolo, come delle spugne, molto pesanti e con
la sola pressione di un dito espellono una gran quantità di questa schiuma rosata.
Se però la condizione si instaura in cronico, l’aumento della pressione veno-capillare polmonare
produce un aumento della pressione arteriosa polmonare, in virtù della vasocostrizione delle
arteriose polmonari, con cui si riduce l’afflusso di sangue dal distretto arterioso a quello venocapillare. L’aumento della pressione arteriosa polmonare determina un sovraccarico di lavoro al
ventricolo dx, che non è per natura dotato di una massa muscolare tale da poter aumentare
esponenzialmente il proprio lavoro, come avviene a sx, quindi è in grado di sopportare solo modesti
aumenti del lavoro. Quando il sovraccarico è eccessivo, il ventricolo si dilata, stirando l’anello
valvolare della tricuspide e impedendone un’adeguata giustapposizione dei suoi lembi, con
insufficienza tricuspidalica. Quest’ultima impone un sovraccarico volumico al ventricolo dx, una
dilatazione ed un particolare aumento pressorio dell’atrio dx (6-10-15 mmHg contro i 2-3 mmHg
nella norma), il che si traduce immediatamente in aumento della pressione arteriosa centrale e in
particolare della vena cava inferiore; allora, per via retrograda, si producono gli stessi effetti
prodotti per via anterograda. L’ipertensione venosa, quindi, aggrava l’ipertensione portale,
l’epatomegalia, l’insufficienza epatica, l’ipoalbuminemia, alimentando e peggiorando il circolo.
Lo scompenso cardiaco è caratterizzato da alcuni sintomi soprattutto: dispnea da sforzo, ridotta
tolleranza allo sforzo, dispnea parossistica notturna, ortopnea, edema perimalleolare.
I segni invece sono, tra quelli più specifici: lo spostamento laterale dell’itto della punta,
l’incremento della pressione venosa giugulare, la presenza del III tono. Tra i segni meno specifici ci
sono: tachicardia, rantoli polmonari, ingrossamento epatico, edema periferico; tutti questi segni
possono originare anche da patologie completamente diverse, quindi hanno una specificità piuttosto
bassa. Il III tono è già più specifico, ma non totalmente, perché ad esempio è presente nel soggetto
giovane, in cui è assolutamente fisiologico; però, accoppiando il III tono allo spostamento laterale
dell’itto della punta, ha già un significato più specifico, perciò ogni considerazione deve essere
ritenuta come il tassello di un puzzle. Anche l’ipertensione venosa giugulare di per sé non è
patognomonica di scompenso cardiaco, perché può essere dovuta anche ad un tumore mediastinico,
o ad una trombosi della vena cava superiore; ma se avviene insieme al rilievo del III tono e
dell’aumento della volumetria ventricolare sx, assume caratteri di specificità molto più evidenti.
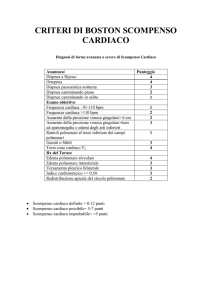
I criteri diagnostici di scompenso si dividono in maggiori e minori; i maggiori sono ortopnea,
dispnea parossistica notturna, turgore giugulare, rantoli polmonari, cardiomiopatia, edema
polmonare acuto, ritmo di galoppo (III tono oppure IV tono, nei casi più avanzati), pressione venosa
aumentata di più di 16 cm di acqua (circa 10 mmHg), tempo di circolazione superiore a 25 sec (il
normale è di 12 sec), reflusso epato-giugulare (comprimendo il fegato e aumentiamo ulteriormente
le pressioni venose, le giugulari diventano ancora più dilatate).
I criteri minori invece sono: edemi declivi, tosse notturna, dispnea da sforzo, epatomegalia,
versamento pleurico, ridotta capacità vitale, tachicardia. La dispnea da sforzo, ad esempio, può
essere presente nella banalissima anemia, nelle broncopneumopatie croniche, quindi non è
patognomonica dello scompenso e così tutti questi altri segni minori. La tachicardia è normalmente
presente in un paziente con lo scompenso, però c’è anche nell’anemia, nell’ipertiroidismo, nelle
disfunzioni neurovegetative, nei soggetti ansiosi, prima di un esame, quindi non si può definire
come segno maggiore dello scompenso.
A proposito dell’accuratezza diagnostica, la dispnea da sforzo ha una sensibilità del 75%, ma una
specificità del 50%; la dispnea parossistica notturna una sensibilità del 31% ed una specificità del
76%; l’ortopnea una sensibilità del 21%, ma una specificità del 81%. Cause non cardiache di
dispnea da sforzo sono malattie polmonari (BPCO, fibrosi), TEP; per la dispnea parossistica
notturna la sindrome nefritica, l’insufficienza renale, l’anemia, l’obesità; per l’ortopnea l’ansietà, il
decondizionamento fisico. Gli edemi declivi hanno bassa sensibilità e bassa specificità; in loro
presenza bisogna valutare la presenza di insufficienza venosa, trombosi venosa profonda,
ipoalbuminemia, farmaci (calcio-antagonisti) per un’accurata diagnosi differenziale. I calcioantagonisti sono dei vasodilatatori periferici ed aumentano la permeabilità dei vasi periferici, per cui
favoriscono la trasudazione di liquidi, che può essere tale da indurre rottura della cute, formazione
di ulcere e fuoriuscita di liquido cristallino (“acqua di rocca”).
A questo punto il prof. ci delizia con dei racconti in merito ai metodi non molto ortodossi di
risolvere gli edemi declivi conficcando degli aghi nell’arto del paziente in modo da renderlo un
colabrodo; l’epatomegalia concedendo a delle sanguisughe di fare bisboccia sull’ipocondrio dx del
paziente, magari lo stesso di prima. Dulcis in fundo, per fare i salassi non facevano mica dei
prelievi; piuttosto dei bei tagli a vivo nell’incavo del gomito, lasciando il paziente per un pò a
buttare il sangue, nel senso letterale del termine.
Diletta Contaldo





![Scompenso cardiaco- attività dell`Asl di Nuoro [file]](http://s1.studylibit.com/store/data/005106553_1-2acc9f03391e8aa6792037a95036da21-300x300.png)