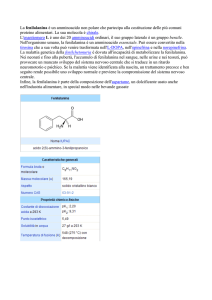
FENILCHETONURIA P.K.U.
La fenilchetonuria, detta anche
P.K.U., è una malattia che appartiene al ramo delle iperfenilalaninemie. Le iperfenilalaninemie derivano da un'alterata
trasformazione della fenilalanina in tirosina. Questa malattia che è caratterizzata da un'aumentata concentrazione
ematica di fenilalanina, da aumentate concentrazioni urinarie di fenilalanina e dei suoi derivati (particolarmente
fenilpiruvato, fenilacetato, fenillattato e fenilacetilglutamina) e da grave ritardo mentale.
Ognuna delle iperfenilalaninemie è causata dalla ridotta attività della
fenilalanina idrossilasi. Nell'uomo questo sistema enzimatico
completo è espresso solo nel fegato. La fenilalanina e l'ossigeno
molecolare sono i substrati dell'enzima che necessita, come cofattore,
di una pteridina ridotta, la tetraidrobiopterina. La tirosina e la
diidrobiopterina sono i prodotti di questo sistema catalitico;
quest'ultima viene poi riconvertita a tetraidrobiopterina da un secondo
enzima, la diidropteridina reduttasi. Nella fenilchetonuria classica
l'attività dell'apoenzima idrossilasi, il cui locus genico è stato mappato
sulla regione q22-q24.1 del cromosoma 12, è quasi totalmente assente.
Si riconoscono sei differenti mutazioni che portano a tale deficit
completo. Esse comprendono i cambiamenti "missense", i difetti di
splicing e le delezioni parziali. La PKU si presenta in circa 1 su 10000
nascite, e colpisce indistantemente sia biachi che neri. L'attività della
fenilalanina idrossilasi è bassa ma superiroe quella riscontrabile negli
omozigoti.
I portatori eterozigoti sono silenti dal punto di vista clinico ma
presentano concentrazioni plasmatiche di fenilalanina leggermente
aumentate. Dirette conseguenze dell'alterata idrossilazione sono
l'accumulo plasmatico e urinario di fenilalanina e la ridotta sintetasi di
tirosina. Nella PKU classica le concentrazioni dell'aminoacido si
elevano a sufficienza per attivare le vie metaboliche alternative portare
alla formazione di fenilpiruvato, fenilacetato, fenillattato e altri
derivati che sono rapidamente filtrati dal rene e escreti con le urine. Le
concentrazioni plasmatiche degli altri aminoacidi sono moderatamente
ridotte, probabilmente, come conseguenza dell'inibizione
dell'assorbimento intestinale o dell'alterazione del riassorbimento
tubulare renale. Il grave danno celebrale è dovuto alle molteplici
conseguenze dell'accumulo di fenilalanina: sottrazione di altri
aminoacidi necessari per la sintesi proteica, compromissione della
formazione o della stabilizzazione dei poliribosomi, ridotta sintesi di
mielina e insufficiente formazione di noradrenalina e serotonina.
La fenilalanina è un inibitore competitivo della tirosinasi, un enzima chiave nella via di sintesi della melanina. Questo
blocco, oltre alla ridotta disponibilità del precursore della melanina, la tirosina, spiega l'ipopigrnentazione dei capelli e
della pelle.
Alla nascita non è evidente alcuna anomalia. I bambini non trattati, affetti da fenilchetonuria, non riescono a
raggiungere le prime fasi dello sviluppo ed evidenziano progressive alterazioni della funzione cerebrale. Entro pochi
anni dalla nascita, la maggior parte necessita di un ricovero cronico in istituti a causa dell'ipereccitabilità e delle crisi
convulsive che accompagnano il grave ritardo mentale. Completano il grave quadro clinico anomalie
elettroeneefalografiche, un "odore di topo" della pelle, dei capelli e delle urine (dovuto all'accumulo di fenilacetato) e
una tendenza all'ipopigmentazione e all'eczema. Viceversa, i bambini cui la malattia viene diagnosticata alla nascita e
che sono immediatamente trattati non manifestano alcuno di questi sintomi. Più del 90% della prole è marcatamente
ritardata e in gran parte hanno altre anomalie congenite quali una microcefalia, ritardo di crescita e cardiopatie
congenite. Poiché questi bambini non sono omozigoti per la mutazione che pro voca fenilchetonuria, ma eterozigoti, le
manifestazioni cliniche vanno attribuite al danno prodotto dalle elevate concentrazioni materne di fenilalanina cui sono
stati esposti durante la vita intrauterina. Questa sindrome è denominata fenilchetonuria materna.
Alla nascita le concentrazioni plasmatiche di fenilalanina sono solitamente normali, ma aumentano rapidamente con
l'inizio dell'alimentazione contenente proteine e, di solito, sono anormali a partire dal quarto giorno. Poiché, se si vuole
prevenire il ritardo di sviluppo, la diagnosi e l'inizio del trattamento dietetico della fenilchetonuria classica devono
essere eseguiti prima che il bambino compia 30 giorni, nel Nord America e in Europa la maggior parte dei neonati viene
sottoposta alla determinazione delle concentrazioni ematiche di fenilalanina per mezzo del test di inibizione batterica di
Guthrie. I piccoli con valori anormali sono seguiti ulteriormente con dosaggi fluorometrici o cromatografici più precisi
dal punto di vista quantitativo. Nella fenilchetonuria classica. In qualsiasi bambino affetto da iperfenilalaninemia che,
malgrado una pronta diagnosi e un inizio immediato del trattamento dietetico, sviluppi un danno neurologico
progressivo, si deve prendere in considerazione un deficit di tetraidrobiopterina. Queste varianti che colpiscono dall'1 al
5% dei bambini fenilchetonurici, possono essere diagnosticate mediante un dosaggio enzimatico su colture di
fibroblasti. La somministrazione orale di carichi di tetraidrobiopterina può fare distinguere i bambini con
feniichetonuria classica (che non mostrano una risposta chimica) da quelli con deficit di tetraidrobiopterina (che
presentano una brusca caduta della fenilalanina piasmatica). La diagnosi prenatale della fenilchetonuria classica è
attualmente possibile impiegando le analisi, basate sul DNA, in grado di rilevare mutazioni specifiche oppure
polimorfismi di lunghezza dei frammenti di restrizione (linked restriction fragment length polymorphisms, RFLP). Il
deficit della diidropteridina reduttasi e i blocchi nella sintesi di tetraidrobiopterina possono essere rilevati in utero
impiegando analisi su ainniociti in coltura.
La fenilchetonuria classica è la prima malattia metabolica ereditaria in cui fu dimostrato che la prevenzione
dell'accumulo del metabolita nocivo previene le gravi conseguenze cliniche. Ciò è reso possibile da una dieta speciale
nella quale si sostituisce la maggior parte delle proteine con una mistura artificiale di aminoacidi a basso contenuto di
fenilalanina. Supplementando questa mistura con piccole quantità di cibo naturale, viene fornito con la dieta un apporto
di fenilalanina sufficiente per una crescita normale, ma che non origina livelli ematici di fenilalanina marcatamente
aumentati. Tale dieta deve essere istituita durante il primo mese di vita. Perfino in questo caso, sono spesso osservabili
modeste alterazioni del sistema nervoso. Poiché l'iperfenilalaninemia non controllata causa danni cerebrali durante
l'infanzia (e forse nell'età adulta), la restrizione dietetica dovrebbe essere mantenuta indefinitamente.
Tratto da ammec.it