BROCHURE INFORMATIVA
AL FENILCHETONURIA
NE ,
CO N O S C E R L A B E
A LIM E NTA R S I
CO R R E T TA M E NTE
Copyright 2013 Dr. Schär AG/SPA,
Winkelau 9, 39014 Burgstall / Postal, Italia
Tutti i diritti riservati. La stampa o la riproduzione,
anche parziali, nonché la diffusione in Internet
sono consentite solo previa autorizzazione scritta
di Dr. Schär AG/SPA.
Immagini: pag. 18, 19, 20, 21, 22, 24 e 25 © Fotolia.com
INDICE
INTRODUZIONE
2
PKU (FENILCHETONURIA)
Che cosa è la PKU?
Che cosa è la fenilalanina (Phe)?
Quale è la frequenza di PKU?
Quali sono le conseguenze della PKU?
Come si diagnostica la PKU?
Quali sono le forme cliniche?
Come si cura la PKU?
3
4
4
5
5-6
6-7
7
LA NUTRIZIONE NELLA PKU
La dieta come terapia
Che cosa bisogna rispettare?
Quali sono i livelli di proteine raccomandati?
Quali sono i livelli possibili di fenilalanina?
Alimentazione equilibrata nella PKU
Il “bersaglio” alimentare per la PKU
Contenuto di fenilalanina in vari alimenti
Alimentazione dei neonati con PKU
Alimentazione dei bambini con PKU
PKU nell'adolescente e nell'adulto
PKU materna
8-9
10
11
12
13
14-15
16-17
18
19
20
21
LA VITA QUOTIDIANA NELLA PKU
Suggerimenti e trucchi
Attenzione all'aspartame!
Viaggiare e mangiare fuori casa
Alimenti dietetici: come possono aiutare?
ULTERIORI INFORMAZIONI
22-23
23
24-25
26
27
1
IL RUOLO
DELLA DIETOTERAPIA
GLI ERRORI CONGENITI DEL METABOLISMO
APPARTENGONO ALLA CATEGORIA DELLE MALATTIE
RARE (SE NE CONOSCONO CIRCA 600).
Sono malattie genetiche ed esordiscono
prevalentemente in età pediatrica.
La Fenilchetonuria (PKU) è la più frequente
delle malattie congenite del metabolismo
proteico.
L'intervento nutrizionale rappresenta la
soluzione terapeutica essenziale nell'approccio
clinico a numerose patologie congenite
dott.ssa Silvia Maria
del metabolismo. In alcune patologie
Bernabei - Dietista
Ospedale Pediatrico
(fenilchetonuria, leucinosi, acidemie
Bambino Gesù, Roma
organiche e altre) ha un ruolo chiave nella
gestione terapeutica, in altri casi è di ''supporto
alla terapia farmacologica''.
Le recenti ricerche in ambito chimico-industriale, e la conseguente
diffusione in commercio di prodotti dietetici a fini medico-speciali,
hanno permesso significative variazioni al trattamento dietetico ed
hanno migliorato notevolmente la compliance del bambino e della
famiglia. Il ruolo del dietista negli errori congeniti del metabolismo
è fondamentale per la corretta gestione della dieta domiciliare
per ottenere una buona compliance a lungo termine.
2
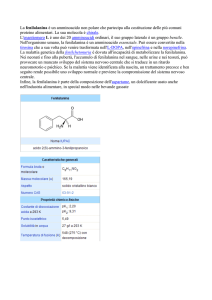
CHE COSA È LA PKU?
LA FENILCHETONURIA O PKU È LA PIÙ COMUNE
MALATTIA CONGENITA DEL METABOLISMO
DELLE PROTEINE.
La PKU è dovuta alla mutazione
di un gene deputato alla codifica
della fenilalanina idrossilasi (PAH),
enzima che converte l'aminoacido
fenilalanina in tirosina, un altro
aminoacido. L'assenza della fenilalanina
idrossilasi rende impossibile tale
reazione danneggiando il cervello.
Se identificata alla nascita, la
malattia beneficia di un trattamento
precoce che rende possibile uno
sviluppo normale, prevenendo
la compromissione del sistema
nervoso centrale.
MAMMA
PAPÀ
portatrice sana
portatore sano
PROBABILITÀ DI GENERARE UN FIGLIO/A:
sano/a
25%
portatore/trice sano/a
25%
portatore/trice sano/a
25%
malato/a
25%
3
CHE COSA È
LA FENILALANINA (PHE)?
GLI AMINOACIDI SONO I COMPONENTI DELLE PROTEINE.
LA FENILALANINA È UN AMINOACIDO ESSENZIALE.
Ciò significa che il corpo non è in
grado di sintetizzarlo e, pertanto,
deve essere introdotto attraverso
l'alimentazione. La fenilalanina è
presente in quasi tutti gli alimenti
e svolge compiti vitali nel corpo.
Normalmente metabolizzata a
tirosina dalla fenilalanina idrossilasi,
la fenilalanina può creare problemi
solo a chi è affetto da fenilchetonuria.
QUAL È LA FREQUENZA DI PKU?
LA PKU HA UNA FREQUENZA PARI A 1 INDIVIDUO
SU 10.000 NATI E FA PARTE DELLE MALATTIE “RARE”,
CHE GENERALMENTE SONO RICONDUCIBILI
AD UN’ORIGINE GENETICA.
4
QUALI SONO LE CONSEGUENZE
DELLA PKU?
LE ALTERAZIONI GENETICHE INCIDONO SULLA CARENZA
DELL’ENZIMA FENILALANINA IDROSSILASI, A SUA VOLTA
RESPONSABILE DEL METABOLISMO DELLA FENILALANINA.
La carenza di questo enzima comporta un accumulo di fenilalanina nel sangue,
nelle urine e nei tessuti, e può provocare un mancato sviluppo del sistema
nervoso centrale che si traduce in un ritardo neuromotorio e psichico.
Anche la cute risente dell'accumulo di fenilalanina per l'assenza di melanina.
Per questo motivo i soggetti fenilchetonurici non trattati presentano una pelle
chiarissima e gli occhi azzurri.
COME SI DIAGNOSTICA LA PKU?
Considerate le gravi conseguenze
cui i fenilchetonurici vanno incontro,
sia negli USA che in Europa, si sono
istituite indagini di massa su tutti
i neonati. Lo screening neonatale,
condotto tra le prime 72 ore di vita,
consente di diagnosticare
precocemente la PKU ed altre
patologie metaboliche
congenite.
DIAGNOSI PRENATALE
È anche possibile eseguire la diagnosi
prenatale per le gravidanze a rischio,
come ad esempio nelle famiglie
con uno o più figli fenilchetonurici.
In Italia, però, l'applicazione
della diagnosi prenatale è limitata,
in quanto si ricorre direttamente
allo screening neonatale.
5
Ciò permette di iniziare
tempestivamente il trattamento
idoneo e prevenire, così, qualsiasi
danno cerebrale.
L'analisi può essere fatta attraverso
il Test di Guthrie (una goccia di
sangue dal tallone del neonato viene
messo in contatto con un batterio)
o con tecniche più moderne come la
spettrometria di massa tandem.
QUALI SONO LE FORME CLINICHE?
IN BASE ALL'ATTIVITÀ RIMANENTE DELL'ENZIMA PAH
SI DISTINGUONO VARIE FORME CLINICHE.
Per chiarire quanta fenilalanina viene tollerata nella dieta di un paziente
la PKU viene distinta in 3 tipologie:
PKU LIEVE
i valori di fenilalanina nel sangue, a dieta libera,
risultano tra 2 mg/dl e 10 mg/dl
PKU MODERATA
i valori di fenilalanina nel sangue, a dieta libera,
risultano tra 10 mg/dl e 20 mg/dl. L'attività enzimatica è inferiore a 10%.
PKU CLASSICA O SEVERA
i valori di fenilalanina nel sangue, a dieta libera,
sono superiori a 20 mg/dl. L'attività enzimatica è inferiore a 5%.
6
In una persona non affetta da PKU i valori di fenilalanina non superano i 2
mg per decilitro di sangue. Anche il portatore sano del gene difettoso non
supera questo valore.
COME SI CURA LA PKU?
LA DIAGNOSI PRECOCE ED UNA DIETA INFLESSIBILE
POSSONO PREVENIRE L’INSORGENZA DEI SINTOMI
DA FENILCHETONURIA.
La dietoterapia, che viene monitorata dal medico la/il dietista dal centro
specializzato, consiste in un'alimentazione a basso contenuto di proteine
e di fenilalanina. I suoi benifici devono essere testati attraverso regolari analisi
dei livelli plasmatici di fenilalanina.
In alcuni casi è possibile
anche l'impiego di una terapia
farmacologica basata sul cofattore
dell'enzima PAH, il cosiddetto BH4.
Quest'ultimo supporta l'enzima nella
trasformazione della fenilalanina
in tirosina. Attraverso le adeguate
analisi, il medico è in grado di
stabilire se il paziente PKU risponde
o meno a questo trattamento
terapeutico. Il trattamento a base di
BH4 non sostituisce la dieta, tuttavia
permette di alleggerirla.
La costante assistenza del Centro
Malattie Metaboliche di riferimento
risulta di primaria importanza
per assicurare una terapia e un
trattamento efficacie
per il paziente.
7
LA DIETA COME TERAPIA
LA TERAPIA NUTRIZIONALE È, A TUTT’OGGI,
LA PRINCIPALE SOLUZIONE TERAPEUTICA NELLA
GESTIONE DI NUMEROSE PATOLOGIE METABOLICHE
CONGENITE, TRA CUI LA FENILCHETONURIA.
Il trattamento dietetico
della fenilchetonuria si basa su una
dieta a basso contenuto di fenilalanina e
limita, quindi, il suo consumo sia da fonti
naturali (alimenti proteici) che artificiali
(additivi alimentari), per prevenire il
danno funzionale e strutturale derivante
dall'accumulo di questo aminoacido.
Terapie alternative, come quella
genetica, sono tuttora in fase
sperimentale. La corretta gestione
della dietoterapia garantisce un
buono stato di salute e uno sviluppo
normale delle strutture tissutali
e nervose.
La dieta deve garantire, inoltre, un
consumo adeguato di aminoacidi
essenziali per la crescita. Diventa,
pertanto, necessaria l'assunzione
di integratori dietetici (miscele
aminoacidiche) con un contenuto equilibrato di aminoacidi (escluso la Phe),
sali minerali, vitamine ed oligoelementi.
La ricerca in campo nutrizionale
ha fatto passi da gigante e per
i pazienti affetti da PKU esistono
in commercio molti prodotti a
bassissimo contenuto proteico
(prodotti aproteici), come pasta,
pane, farina, biscotti, cracker e
sostituti del latte e delle uova.
8
Questi sono succedanei dei prodotti
normali e permettono di variare il
menù giornaliero senza incorrere
nella noiosità di pasti sempre uguali,
garantendo, inoltre, un idoneo
apporto calorico.
Intervenendo in questo modo,
si supera il problema metabolico
e si evitano gli accumuli intermedi
di sostanze tossiche.
Infine, il centro specializzato
monitora costantemente l'equilibrio
metabolico e i parametri di crescita
in base all'età e allo sviluppo
psicomotorio, per testare la validità
della terapia dietetica.
Vengono, inoltre, controllati
regolarmente gli indici nutrizionali in
modo da evitare squilibri o deficit di
alcuni nutrienti.
Se fino a ieri si riteneva che fosse sufficiente seguire la terapia nutrizionale
fino alla pubertà, oggi, gli esperti suggeriscono di osservare la dieta preposta
per tutta la vita. Il motivo della dieta inflessibile e permanente è valido:
un'interruzione della dieta in età adulta può portare tra l'altro ad una
riduzione del IQ (quoziente intellettivo) e ridotta capacità di concentrazione,
per cui è consigliato continuare la dieta per tutta la vita.
9
CHE COSA BISOGNA RISPETTARE?
NELLA TERAPIA DIETETICA BISOGNA CONSIDERARE
ALCUNE REGOLE DI BASE.
RIC
Innanzitutto, le concentrazioni
di fenilalanina nel sangue, devono
rientrare nei limiti previsti.
Tutte le proteine naturali contengono
il 4-6% di fenilalanina, pertanto,
seguendo una dieta normale,
è impossibile soddisfare le necessità
proteiche e non superare
il fabbisogno di fenilalanina.
• Alimenti con elevati contenuti
proteici, come carne, pesce, uova,
formaggio, soia e noci,
non sono raccomandati a pazienti
con fenilchetonuria classica o severa.
• Sono, invece, consigliati alimenti
naturali a basso contenuto
proteico come la frutta e le verdure.
I DI
CH
= Phe
10
L'apporto calorico ed energetico
viene, ulteriormente, definito e
bilanciato da alimenti aproteici come
farina, pane e pasta.
La terapia dietetica deve, inoltre,
limitare l'apporto alimentare di
fenilalanina, soddisfare il fabbisogno
di aminoacidi essenziali ed evitarne
gli eccessi. Infine, deve garantire
l'apporto di vitamine, minerali,
carboidrati, acidi grassi essenziali,
considerando l'età,
il sesso e il dispendio energetico
del paziente e prevenendo problemi
di malnutrizione. È, pertanto,
indispensabile l’assunzione di
integratori aminoacidici costituiti
principalmente da miscele di
L-aminoacidi, vitamine e sali minerali.
La dieta, quindi, deve consentire
un accrescimento e uno sviluppo
normali, evitando l'accumulo di
fenilalanina e dei suoi prodotti
terminali anomali.
QUALI SONO I LIVELLI DI
PROTEINE RACCOMANDATI?
LA DIETA A BASSO CONTENUTO PROTEICO DEVE COMUNQUE
COPRIRE IL FABBISOGNO PROTEICO DELL'INDIVIDUO
CHE VARIA CON L'ETÀ E OSCILLA TRA 1 E 3 g DI PROTEINE
PER kg DI PESO CORPOREO AL GIORNO.
Questo fabbisogno è leggermente
superiore a quello della popolazione
sana, a causa della minore qualità
nutritiva degli aminoacidi,
contenuti negli integratori, rispetto
a quella delle proteine alimentari.
La maggior parte del fabbisogno
proteico (circa l'80%) viene coperto
da miscele aminoacidiche, e solo il
20% da alimenti naturali e prodotti
aproteici.
20%
80%
• Frutta e verdura
• Prodotti dietetici aproteici
• Miscele
Aminoacidiche
PASTA
IPOPROTEICA
PKU
DRINK
11
QUALI SONO I LIVELLI POSSIBILI
DI FENILALANINA?
I LIVELLI DI ASSUNZIONE DI FENILALANINA VARIANO
SECONDO LA FORMA CLINICA DIAGNOSTICATA.
Maggiore è la gravità della PKU
e minore è l'attività enzimatica,
e di conseguenza varia anche la
tolleranza della fenilalanina. Per
calcolare la quantità di fenilalanina
negli alimenti si può considerare
che 1 g di proteine apporta circa
50 mg di fenilalanina. Questa regola
è utile per avere un orientamento
aprossimativo dei valori di
fenilalanina.
Idealmente l'assunzione di
fenilalanina viene sparsa nell'arco
della giornata nel modo di evitare
un'ingestione elevata in un'unica dosi.
12
Generalmente, due sistemi vengono
adottati per ripartire i fabbisogni
di fenilalanina e, quindi, monitorare i
suoi livelli ematici.
• Il primo sistema si incentra sulla
quantità giornaliera totale di
fenilalanina e prevede il calcolo
del contenuto di fenilalanina negli
alimenti consumati giornalmente.
• Il secondo sistema, invece, si
basa sul concetto degli equivalenti,
il cui criterio di calcolo varia a
livello mondiale. Un equivalente
corrisponde ad un quantitativo tra
10 e 50 mg di fenilalanina. Per es. 1
equivalente (20 mg fenilalanina)
= 50 g carote.
ALIMENTAZIONE EQUILIBRATA
NELLA PKU
ATTRAVERSO REGOLARI ESAMI VIENE INDIVIDUATA LA
QUANTITÀ DI FENILALANINA CHE OGNI SOGGETTO PKU
È IN GRADO DI TOLLERARE.
In base a queste informazioni
la dietista preparerà un piano
alimentare, che indicherà le quantità
degli alimenti speciali da assumere
durante la giornata. L'adesione alla
terapia nutrizionale e il rispetto delle
quantità giornaliere concesse di
fenilalanina viene monitorata dagli
specialisti del centro di malattie
metaboliche.
Per comprendere se l’alimentazione
è ben equilibrata, sarà necessario
monitorare in modo regolare i valori
ematici di fenilalanina. Questo sarà
reso possibile attraverso un piccolo
prelievo di sangue e nel porre
una piccola goccia su una carta
assorbente.
13
IL “BERSAGLIO”
ALIMENTARE
PER LA PKU
carne
e salumi
pizza
GLI ALIMENTI PIÙ VICINI
AL CENTRO CONTENGONO
MENO FENILALANINA.
QUELLI NEL CENTRO
NON CONTENGONO
FENILALANINA.
patate
pasta
succhi
frutta
secca
pane
formaggio
14
uova
Latte Latte
latte
e latticini
riso
e cuscus (cotto)
prodotti
aproteici
FARINA
APROTEICA
dolci
olio
mais
zucch
hero
acqu
ua
zzucchero
frutta
pesce e frutti
di mare
verdure e
vegetali
funghi
le
egumi
patatine
fritte
hamburg
ger
15
PANORAMICA DEL CONTENUTO DI FENILALANINA (PHE)
IN VARI ALIMENTI (per 100 g di alimento)
alto contenuto di PHE
> 200 mg
Cereali e prodotti
di derivazione
fiocchi d'avena, mais (chiccho
crudo), miglio, pane e pasta
tradizionale, riso, segale, trigo
riso cotto, mais
Contorni
amaranto, couscous
couscous cotto,
patate fritte (cotte)
Frutta
albicocche secche
avocado, banana chips,
fichi secchi
Verdura
aglio
broccoli, spinaci
Funghi
funghi secchi
Noci e semi
tutti i tipi di noci e semi
Legumi e soia
ceci, fagioli borlotti freschi,
lenticchie, piselli, soia
Latte e latticini,
uova
latte di pecora, formaggio,
mozzarella, parmigiano, yogurt
Carne e salumi
tutti i tipi di carne e
prodotti di salumeria
Pesce e
frutti di mare
tutti i tipi di pesce e
frutti di mare
Dolci
cacao, cioccolato, biscotti e
croissant tradizionali
Oli e grassi,
aceto
Bevande
16
100–200 mg
Additivi
aspartame
Altro
pocorn
latte di capra, latte di vacca
gelato
Per valori esatti si prega di consultare la/il dietista
50–100 mg
20–50 mg
1–20 mg
senza
PHE
prodotti aproteici
datteri secchi
arance, banane,
kiwi, mirtilli, cachi,
albicocche, fichi,
fragole, pesche,
mango
cocomero,
mele, papaya,
pere, prugne, uva,
cigliege, pompelmo
asparagi, cavolfiore,
fagiolini, lattuga, patate,
porri, zucchine, olive,
pomodori secchi,
melanzane, peperoni
gialli, radicchio, sedano
cipolle, pomodori
da insalata, zucca,
carote
cetrioli
funghi coltivati
latte umano
marmellate
miele
burro, margarina
zucchero
tutt i tipi di
oli, aceto
caffè, succo di
arancia
acqua, Tè
sale, pepe
tabella senza pretesa di completezza
17
ALIMENTAZIONE
DEI NEONATI CON PKU
QUALORA SI RISCONTRASSE FENILCHETONURIA IN
SEGUITO A SCREENING NEONATALE, È IMPORTANTE
INIZIARE DA SUBITO IL TRATTAMENTO DIETETICO.
Il latte materno, oltre ad apportare
aminoacidi ed acidi grassi essenziali
(principalmente omega 3 con effetto
neuroprotettivo), contiene piccole
quantità di fenilalanina.
Subito dopo la diagnosi si dovrà
inanzitutto cercare di abbassare
i livelli di PHE fino ad un range di
sicurezza.
18
L'allattamento al seno, dunque, è
consentito in neonati affetti da PKU
e contribuisce a creare un forte
legame biologico tra madre e figlio.
L'allattamento al seno dovrà essere
ulteriormente integrato con adeguate
miscele aminoacidiche.
Infine, sarà necessario monitorare
regolarmente i livelli ematici di
fenilalanina nel neonato.
ALIMENTAZIONE DEI
BAMBINI CON PKU
I BAMBINI SONO ORGANISMI IN CRESCITA, CARATTERIZZATI
DA UN ELEVATO FABBISOGNO CALORICO E PROTEICO.
Bisogna, pertanto, individuare l'equilibrio tra l'apporto minimo di fenilalanina
e la copertura del fabbisogno proteico ed energetico.
Il primo passo è prevenire, attraverso la dieta, l’accumulo di fenilalanina.
Questo non significa che la fenilalanina deve essere totalmente assente.
È sbagliato, dunque, escludere completamente i cibi proteici, mentre si
consiglia di introdurre quelli a moderato contenuto di proteine ma a basso
contenuto di fenilalanina. Questo approccio dietetico dipende dalla tolleranza
del bambino (ovvero a quale forma di fenilchetonuria ci si trova davanti).
Una dieta totalmente priva di
proteine provocherebbe una
crescita scarsa. Pertanto bisogna
garantire un apporto di aminoacidi
adeguato per mantenere in salute
il bambino e garantirne la crescita.
Tale apporto si ottiene attraverso la
supplementazione con miscele di
aminoacidi prive di fenilalanina ed
adatte all'età. È anche importante
porre attenzione all'equilibrata
introduzione, secondo il fabbisogno,
di vitamine (soprattutto Vit. D, Vit. A,
Vit. B12), sali minerali (calcio),
acidi grassi essenziali
(omega 3)
e micronutrienti (selenio, zinco, ferro).
In base al contenuto di fenilalanina
tollerato dal bambino, il fabbisogno
proteico sarà coperto da miscele
di aminoacidi e da proteine
naturali (cibi). L'effetto della dieta
verrà esaminato mediante prelievi
settimanali del sangue
per controllare i livelli
di fenilalanina.
AD ESEMPIO, NELLA
FENILCHETONURIA CLASSICA,
350 mg di fenilalanina al giorno,
corrispondono a circa 7 g
di proteine da alimenti.
19
PKU NELL
NELL̕ADOLESCENTE
ADOLESCENTE
E NELL̕ADULTO
NELL ADULTO
UNA DIETA CORRETTA CONSENTE UNO SVILUPPO
REGOLARE DELL'INDIVIDUO SIA A LIVELLO MENTALE
CHE FISICO, PERMETTENDOGLI DI AVERE UNO STILE DI
VITA ALQUANTO NORMALE SIA IN AMBITO LAVORATIVO
CHE NEL TEMPO LIBERO.
Nell'adulto il fabbisogno
degli aminoacidi non cambia
significativamente da quello del
bambino, ma ci sono alcuni nutrienti
altrettanto importanti.
Tra di essi ci sono:
• il calcio e la vitamina D
che prevengono l`osteoporosi
(perdita di massa ossea)
• l'acido folico che previene
l'arteriosclerosi (indurimento della
parete arteriosa)
• le fibre alimentari che influenzano
positivamente la digestione
e il senso di sazietà.
• i grassi essenziali omega 3
che hanno effetti immunitari
e neuroprottetivi.
20
Le miscele aminoacidiche per
gli adolescenti e gli adulti sono
disponibili in confezioni pratiche
per il consumo fuori casa,
permettendo di mantenere, così,
uno stile di vita attivo.
I controlli ematici di fenilalanina
possono essere meno frequenti
rispetto all'infanzia, ma comunque
regolari e con una frequenza
mensile.
PKU MATERNA
IL DESIDERIO DI GENERARE UN FIGLIO È LA
COSA PIÙ NATURALE CHE ESISTA. IN UNA DONNA
FENILCHETONURICA SI PRESENTA LA NECESSITÀ
DI ATTENZIONI COSTANTI E RIGOROSISSIME FIN DALLA
“DECISIONE” DI AVERE UN FIGLIO.
Ancora prima del concepimento si
rende necessaria una terapia dietetica
ristretta, idonea a mantenere, nei
limiti indicati, la concentrazione di
fenilalanina. Idealmente, nei sei mesi
che precedono il concepimento è
necessario raggiungere e mantenere
un livello di fenilalanina nel sangue
pari a 2 mg/dl (120 μmol/l).
I prodotti alimentari da escludere
completamente sono tutti i prodotti
d'origine animale (carne, pesce, uova,
latte e latticini in genere) e legumi. I
prodotti alimentari a base di cereali
(pane, pasta e prodotti da forno)
come anche l'uovo e il latte possono
essere sostituti con i relativi prodotti
dietetici aproteici.
La donna fenilchetonurica può
allattare, sia nel caso il figlio nasca
affetto da PKU sia quando non abbia
tale malattia, nonostante il latte
materno sia più ricco di fenilalanina
rispetto a quello di una
madre sana.
La dieta dovrà sempre essere
osservata e verrà valutato
regolarmente se l'alimentazione
è equilibrata attraverso prelievi
del sangue.
Se, sin dal concepimento, il tasso
di fenilalanina nel sangue della futura
mamma, è ben equilibrato, minore
di 5 mg/dl di sangue (ovvero 300
μmol/l), il bambino è nelle migliori
condizioni per il suo sviluppo
normale e nella maggior parte dei
casi (e comunque la probabilità è
minima) il nascituro non sarà affetto
da fenilchetonuria anche se sarà
portatore del gene alterato.
Quanto più si è motivate a seguire
un'alimentazione ristretta e rigorosa
tanto più alto sarà il successo della
dieta e garantirà un bambino che
non avrà alcuna conseguenza a causa
della fenilchetonuria materna (ritardo
mentale).
21
SUGGERIMENTI E TRUCCHI
LA DIETOTERAPIA E LA GESTIONE DELLA GIORNATA
ALIMENTARE PER I PAZIENTI FENILCHETONURICI
RAPPRESENTA UNA VERA E PROPRIA SFIDA.
Per la natura della malattia non esiste un trattamento universale poiché i
valori target di fenilalanina variano in rapporto alla tolleranza mostrata da
ogni singolo paziente. La dieta, che dovrà pertanto essere calibrata sul singolo
individuo, seguirà alcune regole:
• Gli alimenti sono in genere calcolati
a peso crudo e al netto degli scarti.
La parte edibile cui ci si riferisce è la
parte dell'alimento al netto di scarti e
pronto da cuocere (o da consumarsi
crudo).
• Il contenuto di fenilalanina e
proteine, per ciascun alimento, è
calcolato sulla base di diverse tabelle
di composizione degli alimenti.
• Dove possibile, è calcolato il
contenuto esatto di fenilalanina e
proteine anche per quegli alimenti
(frutta e verdura) che sono noti per il
loro basso contenuto in fenilalanina.
• Le dosi delle ricette, in particolare
la quantità degli alimenti aproteici
possono essere individualizzate a
seconda delle esigenze del soggetto.
• Le uova possono essere
sostitute con il prodotto dietetico
corrispondente (sostituto uova).
• Il latte è sostituito con un latte
a basso contenuto di proteine.
22
• Spesso è più semplice preparare
una quantità di alimento maggiore
di quella indicata; in questo caso
la dose degli ingredienti può essere
raddoppiata o triplicata, tenendo
però presente che per avere
la quantità di fenilalanina e proteine
indicata dalla ricetta si dovrà
somministrare la metà o un terzo
del piano preparato.
• La presentazione di un piatto è
importante; un piatto ben presentato
acquisisce un aspetto diverso e
invoglia al consumo.
• I cibi da consumarsi devono
avere una giusta temperatura:
da questa dipende parte del loro
sapore e della loro digeribilità.
Esempio: budini e frutta cotta si
serviranno freddi; paste asciutte,
verdure al burro, calde.
• La quantità di sale o zucchero, se
non specificata, può essere adeguata
a proprio gusto.
• Le erbe aromatiche così come
aceto e altri aromi sono facoltative
e seguono il gusto del paziente.
ATTENZIONE ALL'
ALL ASPARTAME!
ASPARTAME!
L'ASPARTAME È UN ADDITIVO ALIMENTARE AUTORIZZATO
A LIVELLO EUROPEO E CON IL NUMERO E951.
È un edulcorante, dolcificante ed esaltatore di sapidità artificiale che viene
usato in alimenti tipo bevande, prodotti di pasticceria e confetteria, prodotti
lattieri, gomme da masticare e prodotti per il controllo del peso.
È composto da due aminoacidi, la fenilalanina e l'acido aspartico
ed è per questo che in caso di PKU bisogna evitare
l'assunzione dell'aspartame controllando la lista
degli ingredienti. Nell'Unione Europea l'etichetta
degli alimenti deve dichiarare
la sua presenza.
23
VIAGGIARE E MANGIARE
FUORI CASA
LA PKU NON È UN OSTACOLO AL VIAGGIO.
Programmare il viaggio accuratamente significa anche avere tutte le
informazioni sulle località che si visiteranno conoscendo se ci sono centri
per la cura della PKU e portarsi tutto quello che può essere necessario per
la terapia. È buona norma preparare con anticipo una scorta dei prodotti
prescrivibili essenziali, necessari per il viaggio (integratori, alimenti aproteici,
latte speciale).
Nei viaggi all’estero può essere utile
portarsi una diagnosi del proprio
ospedale/medico curante/centro
per la PKU, su carta intestata
(possibilmente tradotta in inglese),
che riporti anche l’elenco di tutti i
prodotti speciali e il motivo per cui è
necessario portarli con sé: non solo si
velocizzeranno i controlli in dogana
ma, nel bisogno, sarete trattati in
modo idoneo. Una bilancia sarà utile
così come alcuni contenitori ermetici
in cui conservare gli alimenti o le
miscele di aminoacidi.
24
Sarà importante mantenere le
proprie abitudini di assunzione
di integratori e se non dovessero
esistere confezioni monodose,
prepararle personalmente in singole
porzioni chiudendole ermeticamente.
Gli integratori sono anche diluibili in
acqua o già liquidi, purché non se ne
perda una goccia.
Nulla vieta di accordarsi con gli
alberghi (chiedere di parlare con lo
staff di cucina) per concordare gli
alimenti che si possono consumare e
proporre di procurare gli alimenti di
cui si necessita.
Nei viaggi aerei è consigliato
scegliere il menù vegetariano previa
verifica di cosa viene proposto
e come è preparato.
Anche in questo caso é importante
portarsi una diagnosi.
È consentito portare il cibo e la
terapia necessaria nel bagaglio a
mano (meglio portare le quantità
sufficienti per qualche giorno, nel
caso le valige vadano smarrite).
È utile, inoltre, informarsi sui
parametri della fenilalanina nel
sangue negli altri Paesi e conoscere
frasi chiave sull’argomento nella
lingua locale.
È UTILE REDIGERE UN DIARIO
della propria giornata alimentare
così da capire quanta fenilalanina
si consuma.
25
ALIMENTI DIETETICI:
COME POSSONO AIUTARE?
L’ALIMENTAZIONE PER CHI È AFFETTO DI
FENILCHETONURIA DEVE ESSERE POVERA DI PROTEINE
E SOPRATTUTTO POVERA DI FENILALANINA.
Allo stesso tempo non devono però mancare gli altri aminoacidi, vitamine e
sali minerali e le calorie al fine di non danneggiare l’organismo.
I prodotti dietetici a basso contenuto di proteine rispondono a tale obiettivo e
sono studiati appositamente per chi deve seguire una dieta povera di proteine.
Esistono due tipi di prodotti:
• alimenti aproteici che contengono
proteine inferiore a 1%
• alimenti ipoproteici con un apporto
proteico tra 1 e 2%
La loro erogazione è regolata
dal Decreto del Ministero della
Sanità dell' 08.06.01. pubblicato
nella Gazzetta Ufficiale n° 154 del
05.07.2001. L'art.5, in particolare,
tratta le Malattie Metaboliche
Congenite.
Tra queste la Fenilchetonuria.
26
Per i costi relativi a questi
prodotti dietetici, i tetti di spesa,
adeguatamente alti e coperti
dall'Assistenza Sanitaria, variano
da regione a regione, cambiano a
seconda della patologia e vengono
aggiornati trimestralmente.
I PRODOTTI APROTEICI
facilitano l'alimentazione
povera di proteine, evitando
piatti monotoni.
ULTERIORI INFORMAZIONI
FONDAZIONI E ASSOCIAZIONI
Associazione Prevenzione Malattie Congenite www.apmmc.it
Associazione Malattie Metaboliche Congenite www.ammec.it
Associazione Italiana Sostegno Malattie Metaboliche Ereditarie www.aismme.org
Associazione Studio Malattie Metaboliche Ereditarie www.cometasmme.org
Associazione Rete Malattie Rare www.retemalattierare.it
Unione Malattie Rare www.umaronlus.org
CENTRI DI CURA
Centri Malattie Metaboliche Ereditari: www.cometaasmme.org
ULTERIORI SITI UTILI
Forum Fenilchetonuria: www.forum.fenilchetonuria.it/
Informazioni sulla Fenilchetonuria: www.pkualtervista.org; www.pkuinfo.it
www.mevalia.com
www.facebook.com/mevaliaIT
27
I CONSIGLI DEL NUTRIZIONISTA / NOTE
28
LA NUOVA
AL
LINEA
INEA AP
APROTEICA
PR
ROTEIC
PER LA TERAPIA NUTRIZIONALE DELLA
FENILCHETONURIA (PKU)
www.mevalia.com
Servizio Consumatori
800 847 081
LPIT06 14
Dr. Schär AG / SPA
Winkelau 9
39014 Burgstall / Postal (BZ) Italy
www.drschaer.com