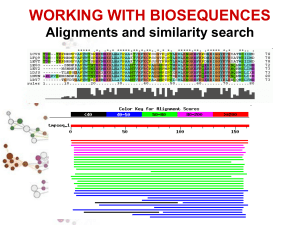
Ricerca di similarità di sequenza (FASTA e BLAST)
Allineamento di due sequenze
Allineamento multiplo di sequenze
RICERCA DI SIMILARITA’ E ALLINEAMENTO DI SEQUENZE
BLAST e PSI-BLAST
http://www.ncbi.nlm.nih.gov/blast/
FASTA
http://fasta.bioch.virginia.edu/ oppure http://www.ebi.ac.uk/fasta33/
Alcune caratteristiche dei tools più usati:
BLAST (Basic Local Alignment Search Tool), sviluppato dal National
Center for
Biotechnology Information, NCBI):
- allineamento locale
- estremamente veloce
- parte cercando brevi frammenti della sequenza, che poi prova ad
estendere
- usa una matrice di sostituzione in entrambe le fasi del processo di
allineamento (scansione del database e estensione della subsequenza):
più preciso ha quattro opzioni fondamentali:
BLASTP: confronta sequenze proteiche contro un database proteico
BLASTN: confronta sequenze nuclotidiche contro un database nucleotidico
TBLASTN: confronta una sequenza proteica contro un database
nucleotidico, traducendo ciascuna sequenza del database nucleotidico nei
suoi 6 frames di lettura
BLASTX: confronta una sequenza nucleotidica contro un database
proteico, dopo averla tradotta nei suoi 6 frames di lettura.
BLAST:
BLASTP
Seconda parte della pagina di BLAST:
Numero atteso di
HSP (Highscoring Segment
Pair) valutato su
base statistica
Dimensione delle
parole
Scelta della
matrice di
sostituzione
I valori di default usati da BLAST sono W=3, T=13, Matrice=BLOSUM 62
Penalità assegnata ai gap
Terza parte della pagina di BLAST:
FASTA: http://www.ebi.ac.uk/fasta33/
Ktup: lunghezza delle
parole
Align: numero di
allineamenti finali
Open e residue:
Penalità per i gap
Vari database
Sequenza in formato
FASTA
Allineamento di due sequenze:
BLAST: bl2seq
LALIGN: http://www.ch.embnet.org/software/LALIGN_form.html
EMBOSS: http://www.ebi.ac.uk/emboss/align/
LALIGN:
ALLINEAMENTO MULTIPLO DI SEQUENZE
Informazione biologica maggiore rispetto a quella riportata l’allineamento
di due sole sequenze: i residui più importanti dal punto di vista strutturale
o funzionale saranno estremamente conservati tra tutte le sequenze
dell’allineamento.
“Una sequenza amminoacidica fa la timida; un paio di sequenze omologhe
sussurrano; molte sequenze allineate gridano”.
Per essere informativo un allineamento multiplo dovrebbe contenere una
distribuzione di sequenze sia strettamente sia lontanamente correlate:
Svantaggi:
•tutte strettamente correlate => ridondanza
•tutte lontanamente correlate => allineamento inaccurato => inutilità
ALLINEAMENTO MULTIPLO DI SEQUENZE
Programmi per l’allineamento multiplo globale:
CLUSTALW: http://www.ebi.ac.uk/clustalw/ o scaricare il programma eseguibile
KALIGN
Multalin
TCOFFEE
http://msa.cgb.ki.se/cgi-bin/msa.cgi
http://bioinfo.genopole-toulouse.prd.fr/multalin/multalin.html
http://www.ch.embnet.org/software/TCoffee.html
CLUSTAL W:
-il tool più comune utilizzato per l’allineamento multiplo di sequenza:
- potenziato per allineamenti di sequenze proteiche divergenti favorisce
l’apertura di gaps in regioni in cui è potenzialmente presente un loop
piuttosto che una struttura secondaria ordinata (in base a una penalità
residuo-specifica e a una penalità ridotta in regioni idrofiliche) favorisce
l’apertura di gaps nelle stesse posizioni.