ISSN 2038-2839
Editor in chief
Giorgio Lambertenghi Deliliers
Anno 8
Numero 1
2011
Seminari
di Ematologia
Oncologica
NEL PROSSIMO NUMERO
CELLULE STAMINALI
Biologia e medicina rigenerativa •
Plasticità delle staminali neurali •
Staminali e rigenerazione cardiaca •
Staminali e malattie autoimmuni •
Terapie cellulari nei tumori solidi •
Linfomi
aggressivi
EDIZIONI
INTERNAZIONALI srl
Edizioni Medico Scientifiche - Pavia
Linfomi
aggressivi
Meccanismi patogenetici
5
MARCO FANGAZIO, SILVIA RASI,
ALESSIO BRUSCAGGIN, DAVIDE ROSSI,
GIANLUCA GAIDANO
Vol. 8 - n. 1 - 2011
Editor in Chief
Giorgio Lambertenghi Deliliers
Fondazione IRCCS Ca’ Granda
Ospedale Maggiore Policlinico di Milano
Editorial Board
Sergio Amadori
Università degli Studi Tor Vergata, Roma
Linfomi non Hodgkin
a grandi cellule
Mario Boccadoro
Università degli Studi, Torino
17
Alberto Bosi
Università degli Studi, Firenze
Federico Caligaris Cappio
Università Vita e Salute, Istituto San Raffaele, Milano
ANNALISA CHIAPPELLA, DAVIDE ROSSI,
UMBERTO VITOLO
Antonio Cuneo
Università degli Studi, Ferrara
Marco Gobbi
Università degli Studi, Genova
Fabrizio Pane
Linfoma mantellare
29
Università degli Studi, Napoli
Mario Petrini
Università degli Studi, Pisa
MARCO LADETTO, SIMONE FERRERO,
SARA BARBIERO
Giovanni Pizzolo
Università degli Studi, Verona
Giorgina Specchia
Università degli Studi, Bari
Linfoma linfoblastico dell’adulto
47
Direttore Responsabile
Paolo E. Zoncada
Registrazione Trib. di Milano n. 532
del 6 settembre 2007
STEFANO LUMINARI, ALESSANDRA DONDI,
GINO SANTINI
Linfomi non Hodgkin T/NK
ANNALISA PELI, GIUSEPPE ROSSI
61
Edizioni Internazionali srl
Divisione EDIMES
Edizioni Medico-Scientifiche - Pavia
Via Riviera, 39 - 27100 Pavia
Tel. +39 0382 526253 r.a. - Fax +39 0382 423120
E-mail: [email protected]
Seminari
2
Periodicità
Quadrimestrale
Scopi
Seminari di Ematologia Oncologica è un periodico di aggiornamento che nasce come servizio per i medici con l’intenzione di
rendere più facilmente e rapidamente disponibili informazioni su
argomenti pertinenti l’ematologia oncologica.
Lo scopo della rivista è quello di assistere il lettore fornendogli in maniera esaustiva:
a) opinioni di esperti qualificati sui più recenti progressi in forma
chiara, aggiornata e concisa;
b) revisioni critiche di argomenti di grande rilevanza pertinenti gli
interessi culturali degli specialisti interessati;
NORME REDAZIONALI
1) Il testo dell’articolo deve essere editato utilizzando il programma
Microsoft Word per Windows o Macintosh.
Agli AA. è riservata la correzione ed il rinvio (entro e non oltre 5
gg. dal ricevimento) delle sole prime bozze del lavoro.
2) L’Autore è tenuto ad ottenere l’autorizzazione di «Copyright»
qualora riproduca nel testo tabelle, figure, microfotografie od
altro materiale iconografico già pubblicato altrove. Tale materiale illustrativo dovrà essere riprodotto con la dicitura «per
concessione di …» seguito dalla citazione della fonte di provenienza.
3) Il manoscritto dovrebbe seguire nelle linee generali la seguente
traccia:
Titolo
Conciso, ma informativo ed esauriente.
Nome, Cognome degli AA., Istituzione di appartenenza senza
abbreviazioni.
Nome, Cognome, Foto a colori, Indirizzo, Telefono, Fax, E-mail del
1° Autore cui andrà indirizzata la corrispondenza.
Introduzione
Concisa ed essenziale, comunque tale da rendere in maniera chiara ed esaustiva lo scopo dell’articolo.
Parole chiave
Si richiedono 3/5 parole.
Corpo dell’articolo
Il contenuto non deve essere inferiore alle 30 cartelle dattiloscritte
(2.000 battute cad.) compresa la bibliografia e dovrà rendere lo stato
dell’arte aggiornato dell’argomento trattato. L’articolo deve essere
corredato di illustrazioni/fotografie, possibilmente a colori, in file ad
alta risoluzione (salvati in formato .tif, .eps, .jpg).
Le citazioni bibliografiche nel testo devono essere essenziali, ma
aggiornate (non con i nomi degli AA. ma con la numerazione corrispondente alle voci della bibliografia), dovranno essere numerate con il numero arabo (1) secondo l’ordine di comparsa nel testo
e comunque in numero non superiore a 100÷120.
di Ematologia
Oncologica
Periodico di aggiornamento
sulla clinica e terapia
delle emopatie neoplastiche
Bibliografia
Per lo stile nella stesura seguire le seguenti indicazioni o consultare
il sito “International Committee of Medical Journal Editors Uniform
Requirements for Manuscripts Submitted to Biomedical Journals:
Sample References”.
Es. 1 - Articolo standard
1. Bianchi AG, Rossi EV. Immunologic effect of donor lymphocytes in bone marrow transplantation. N Engl J Med. 2004; 232:
284-7.
Es. 2 - Articolo con più di 6 autori (dopo il 6° autore et al.)
1. Bianchi AG, Rossi EV, Rose ME, Huerbin MB, Melick J, Marion
DW, et al. Immunologic effect of donor lymphocytes in bone marrow transplantation. N Engl J Med. 2004; 232: 284-7.
Es. 3 - Letter
1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocytes
[Letter]. N Engl J Med. 2004; 232: 284-7.
Es. 4 - Capitoli di libri
1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocytes. In: Caplan RS, Vigna AB, editors. Immunology. Milano:
MacGraw-Hill; 2002; p. 93-113.
Es. 5 - Abstract congressi (non più di 6 autori)
1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocytes
in bone marrow transplantation [Abstract]. Haematologica.
2002; 19: (Suppl. 1): S178.
Ringraziamenti
Riguarda persone e/o gruppi che, pur non avendo dignità di AA.,
meritano comunque di essere citati per il loro apporto alla realizzazione dell’articolo.
Edizioni Internazionali Srl
Divisione EDIMES
EDIZIONI MEDICO SCIENTIFICHE - PAVIA
Via Riviera, 39 • 27100 Pavia
Tel. 0382526253 r.a. • Fax 0382423120
E-mail: [email protected]
3
Editoriale
GIORGIO LAMBERTENGHI DELILIERS
Fondazione IRCCS Ca’ Granda,
Ospedale Maggiore Policlinico di Milano
Le neoplasie del tessuto linfatico comprendono
un largo spettro di forme che, anche se simili sul
piano morfologico, presentano aspetti clinici differenti. Pertanto, sul piano pratico, è utile distinguere linfomi maligni indolenti e aggressivi sulla
base della sintomatologia alla diagnosi e dell’aspettativa di vita, se il paziente non viene trattato. Seminari di Ematologia Oncologica dedica
questo primo numero del 2011 ai linfomi aggressivi la cui classificazione si basa attualmente su
criteri multidisciplinari, integrati dalla identificazione di specifiche lesioni molecolari che hanno
consentito di chiarire il processo di linfomagenesi. Quest’ultimo è riconducibile ad un processo
multifasico che nasce da lesioni ai proto-oncogeni ed ai geni onco-soppressori, e viene, poi,
ulteriormente sostenuto dalla formazione di proteine di fusione. Un ruolo fondamentale sembra
avere anche il microambiente che, attraverso
meccanismi ancora in gran parte sconosciuti,
favorisce lo sviluppo del processo neoplastico.
Queste ricerche stanno avendo un significativo
impatto clinico per la valutazione della malattia
minima residua e la formulazione di nuovi modelli prognostici, rivolti alla ottimizzazione delle strategie terapeutiche.
Un esempio significativo di integrazione tra biologia e clinica sono i linfomi a grandi cellule dove
gli studi di espressione genica hanno permesso di riconoscere due sottogruppi di pazienti a
prognosi differente. Grazie a questa ricerca traslazionale sono stati chiariti anche alcuni aspetti della patogenesi del linfoma mantellare, dove
è stata identificata una signature propria, indipendentemente dalla iperespressione di ciclina
D1. Viceversa nei linfomi T sistemici e nel linfoma linfoblastico, sia per la loro eterogeneità biologica, sia per la relativa rarità delle casistiche,
gli studi sono attualmente in corso e i risultati
preliminari.
5
Meccanismi patogenetici
MARCO FANGAZIO, SILVIA RASI, ALESSIO BRUSCAGGIN, DAVIDE ROSSI,
GIANLUCA GAIDANO
Divisione di Ematologia, Dipartimento di Medicina Clinica e Sperimentale,
Università degli Studi del Piemonte Orientale “Amedeo Avogadro”
e Azienda Ospedaliero-Universitaria Maggiore della Carità, Novara
Gianluca Gaidano
n INTRODUZIONE
La classificazione dei linfomi aggressivi si è evoluta nel corso degli anni e da un approccio esclusivamente morfologico si è passati alla odierna
classificazione WHO (World Health Organization),
che identifica entità cliniche definite in base a criteri multidisciplinari, in grado di combinare dati clinici, morfologici, istologici, immunofenotipici e
genetici. Tuttavia, anche nell’ambito delle categorie nosologiche dei linfomi non-Hodgkin aggressivi identificate dalla classificazione WHO, vi è la
presenza di un’estrema eterogeneità sia per quanto riguarda la risposta al trattamento somministrato sia per quanto riguarda la sopravvivenza dei
pazienti. Deriva quindi la necessità di perseguire
la ricerca di nuovi marcatori molecolari che consentano l’identificazione di sottogruppi di pazienti che possano beneficiare di approcci terapeutici differenziati. Ad oggi sono state identificate
numerose alterazioni genetiche, che hanno permesso di chiarire la patogenesi della malattia.
Parole chiave: linfomi aggressivi, patogenesi molecolare, marcatori biologici.
Indirizzo per la corrispondenza
Prof. Gianluca Gaidano
Divisione di Ematologia
Dipartimento di Medicina Clinica e Sperimentale
Università degli Studi del Piemonte Orientale
“Amedeo Avogadro”
Via Solaroli, 17 - 28100 Novara
E-mail: [email protected]
Classicamente, la patogenesi dei linfomi nonHodgkin aggressivi è riconducibile ad un processo multifasico in cui l’insorgenza in una cellula di
una particolare alterazione genetica predispone
la cellula stessa all’insorgenza di altre alterazioni
genetiche. Queste alterazioni sono per lo più rappresentate da lesioni molecolari che apportano
danni a proto-oncogeni e geni onco-soppressori. Nell’ambito delle lesioni molecolari dei protooncogeni, il principale meccanismo di deregolazione è rappresentato dalla traslocazione cromosomica. Mediante questo meccanismo, il protooncogene può essere allontanato dalle proprie
strutture regolatorie ed essere posto sotto nuovi
elementi di controllo, che ne determinano la deregolazione della espressione. Alternativamente, la
traslocazione cromosomica può determinare la
formazione di un trascritto di fusione, derivante
dalla fusione dei due geni coinvolti nella rottura
cromosomica. I proto-oncogeni attivati dalla formazione di proteine di fusione generano proteine chimeriche che mostrano nuove proprietà biologiche in grado di sostenere il processo di linfomagenesi.
Solitamente, l’inattivazione bi-allelica di geni
onco-soppressori avviene per mutazione inattivante di un allele e delezione dell’altro allele, secondo un processo multifasico. In una minoranza di
casi, invece, l’inattivazione bi-allelica è causata da
una doppia mutazione su entrambi gli alleli o è
sostenuta da una delezione in omozigosi del gene.
Un meccanismo di acquisizione di mutazioni tipico dei linfomi è caratterizzato dalla ipermutazione somatica, che fisiologicamente riguarda i geni
6
Seminari di Ematologia Oncologica
delle immunoglobuline, ma può estendersi in
maniera aberrante (ipermutazione somatica aberrante) anche ad altri geni. Inoltre, l’inattivazione
dei geni onco-soppressori può avvenire anche
mediante il meccanismo di metilazione delle regioni regolatorie del gene, che ne determinano una
ridotta espressione.
In questa rassegna, saranno considerati dal punto di vista patogenetico i seguenti linfomi aggressivi:
1. linfoma diffuso a grandi cellule B (Diffuse Large
B Cell Lymphoma, DLBCL);
2. linfoma mantellare (Mantle Cell Lymphoma,
MCL);
3. linfoma a cellule T periferiche (Peripheral T-cell
Lymphoma, PTCL);
4. linfomi aggressivi dell’ospite immunodeficiente.
Ampio spazio nella trattazione sarà dedicato al
DLBCL, data la rilevanza epidemiologica di questo tipo di linfoma.
n PATOGENESI MOLECOLARE
DEL LINFOMA DIFFUSO
A GRANDI CELLULE B
A livello mondiale, i linfomi rappresentano la quinta neoplasia più diffusa: in particolare, il DLBCL
è la variante più frequente e rappresenta il più
comune linfoma aggressivo (1). I dati del Registro
della Fondazione Italiana Linfomi (FIL) dimostrano che il DLBCL rappresenta in Italia circa il 40%
delle nuove diagnosi di linfoma.
La prevalenza del DLBCL è maggiore nel sesso
maschile, e l’età mediana alla diagnosi è intorno
alla sesta decade di vita (1, 2).
Il DLBCL si presenta morfologicamente in modo
eterogeneo, e la classificazione nel corso degli
anni è stata progressivamente affinata. Secondo
la classificazione della WHO, il DLBCL è definito
come una neoplasia delle cellule B mature, caratterizzate da un profilo di proliferazione diffusa e
da una dimensione nucleare maggiore o uguale
a quella dei normali macrofagi o più di due volte
quella di un normale linfocita (1). Ad oggi, si riconoscono tre varianti morfologiche più comuni:
centroblastico, immunoblastico e anaplastico.
Il DLBCL può insorgere de novo o rappresenta-
re la progressione/trasformazione di un precedente linfoma indolente. Nella metà dei casi di evoluzione da linfoma indolente, i DLBCL trasformati evolvono da un precedente linfoma follicolare;
in altri casi, originano da una precedente leucemia linfatica cronica/linfoma a piccoli linfociti, prendendo così l’eponimo di sindrome di Richter.
Nonostante questa evidenza, ad oggi non è possibile definire con certezza se esista un comune
percorso molecolare alla base della trasformazione in DLBCL a partire da tutte queste differenti
condizioni cliniche, oppure se la trasformazione
da un disordine linfoproliferativo B indolente ad
uno aggressivo segua strade diverse a seconda
del tipo iniziale di malattia.
Tra i fattori di rischio noti per lo sviluppo di un
DLBCL, vi sono le condizioni di immunodeficienza, tra cui l’infezione da virus dell’immunodeficienza umana (HIV), il trapianto d’organo solido e le
terapie immunosoppressive prolungate.
La caratterizzazione immunofenotipica del DLBCL
mostra l'espressione di marcatori della linea B,
quali CD19, CD20, CD22 e CD79a, mentre
l’espressione del CD30 è tipica delle varianti anaplastiche (1, 3). Nel 50-75% dei casi, si può riscontrare l'espressione delle immunoglobuline di
superficie e/o citoplasmatiche, e nel 10% dei casi
dell’antigene CD5 (1). L’espressione di bcl-2 è eterogenea nelle diverse casistiche (25-80%).
L’espressione di bcl-6 si riscontra nel 70% dei casi
circa, ed è consistente con l’origine dal centro germinativo del linfonodo (1, 3, 4).
Dal punto di vista clinico, il DLBCL è una malattia a decorso aggressivo, con possibilità di interessamento di sedi linfonodali e/o extranodali. Le
sedi nodali interessate con maggiore frequenza
sono le sedi laterocervicali e sovraclaveari. I linfonodi sedi di malattia hanno diametro variabile
e possono raggiungere dimensioni superiori ai dieci centimetri; qualora questa soglia venga superata, l’adenopatia viene definita bulky (5).
Eterogeneità molecolare
Dal punto di vista puramente istologico, non è
possibile rendere conto della eterogeneità del
DLBCL. Il processo di trasformazione maligna è
differente a seconda del sottotipo molecolare considerato, e ad anomalie genetiche differenti corrispondono differenze nella presentazione clinica,
Meccanismi patogenetici
nei tassi di guarigione dopo chemioterapia, e nella potenziale responsività a target therapies.
Per definire l’eterogeneità istogenetica del DLBCL,
è stato utilizzato lo studio del profilo di espressione genica (GEP). Tale approccio ha permesso
di suddividere i DLBCL in due sottogruppi principali: germinal center B-like (GCB-like), a indicare un’origine dal centro germinativo, e activated
B cell-like (ABC-like) a indicare un’origine da linfociti post-centro germinativo (7, 8). Il sottogruppo GCB-like si caratterizza per elevati livelli di
espressione di LMO2, BCL6, CD10, CD38 e Amyb, tutti marcatori tipici delle cellule del centro
germinativo (4, 7). Nel sottogruppo ABC-like, si
ritrova invece principalmente l’espressione di
XBP1 (regolatore della secrezione immunoglobulinica) (9, 10) FLIP, e BCL2. L’attivazione costitutiva della via di NF-κB induce i linfomi ABC-like
ad esprimere il fattore di trascrizione IRF4
(MUM1/LSIRF), e questo potrebbe indurne, almeno parzialmente, la differenziazione immunoblastica (11, 12).
È importante osservare, comunque, che i linfomi
ABC-like frequentemente acquisiscono lesioni
genetiche che inattivano BLIMP-1, bloccando così
la differenziazione del clone neoplastico a plasmacellula (7, 14-18).
Altri studi di gene expression profiling hanno suddiviso i DLBCL secondo altri profili di espressione genica delineando tre gruppi distinti:
1. Oxidative phosphorylation (Ox Phos);
2. B-cell receptor/proliferation (BCR);
3. Host Response (HR) (19).
Il primo gruppo presenta aumentati livelli di
espressione di geni associati alla fosforilazione
ossidativa, alla funzione mitocondriale, ed al trasporto degli elettroni (19). Si tratta principalmente di DLBCL caratterizzati da lesioni genetiche
coinvolgenti i membri della famiglia di BCL2. Il
secondo gruppo, presenta invece un’aumentata
espressione di geni coinvolti nel signaling del
recettore delle cellule B, nella proliferazione e replicazione cellulare, nel riparo del DNA e coinvolge
anche fattori di trascrizione specifici della cellula
B, tra cui BCL-6 (19).
In ultimo, il sottogruppo HR presenta un’elevata
espressione di geni associati ai pathways delle cellule T e geni correlati alla risposta immune/infiammatoria (19).
Meccanismi di lesione molecolare
Durante la normale maturazione dei linfociti B, due
distinte modificazioni del DNA alterano il recettore delle cellule B: l’ipermutazione somatica e la
class switch recombination. Entrambi questi
meccanismi richiedono l’intervento dell’enzima
activation-induced cytidine deaminase (AID) (20).
La class switch recombination cambia la classe
della catena pesante delle immunoglobuline da
IgM a IgG, IgA o IgE, mentre l’ipermutazione
somatica agisce modificando la regione variabile delle immunoglobuline, creando una popolazione di cellule B con affinità aumentata (o ridotta) per un particolare antigene. Queste modificazioni genetiche sono essenziali per una risposta
immune normale, ma sono anche una fonte di
danno al DNA che può diventare patologico, o
meglio patogenetico, nei linfomi.
L’enzima AID gioca numerosi ruoli nella linfomagenesi. È stato ben dimostrato in modelli murini
che lo sviluppo del DLBCL richiede AID (21), e
d’altra parte la sovraespressione di AID è all’origine dello sviluppo di linfomi a cellule B in modelli transgenici (22-25). I DLBCL accumulano mutazioni AID-dipendenti in molti geni, inclusi gli oncogeni c-MYC, RhoH/TTF, PAX5, e PIM1 (26).
Queste mutazioni possono accumularsi per un
difetto nel meccanismo di riparazione del danno
al DNA e/o per selezione di cellule che portano
mutazioni oncogenetiche (24).
La class switch recombination, che è mediata da
AID, introduce rotture della doppia elica del DNA
nelle regioni di ricombinazione dei geni che codificano per le catene pesanti delle immunoglobuline.
Queste, quindi, possono determinare traslocazioni con il gene c-MYC e rotture all’interno del locus
c-MYC (27-32). Il sottotipo ABC-like di DLBCL non
solo ha livelli estremamente elevati di AID, ma
subisce anche una class switch recombination
aberrante in cui le regioni di ricombinazione dei
geni codificanti per le catene pesanti delle immunoglobuline sostengono delezioni, inserzioni, e
mutazioni senza partecipare ad un evento di class
switch fisiologico (31).
I normali meccanismi della ricombinazione VDJ
delle catene immunoglobuliniche, della ipermutazione somatica, e della class switch recombination possono alterare il genoma dei linfomi, cre-
7
8
Seminari di Ematologia Oncologica
ando in questo modo il potenziale per traslocazioni in cui, conseguentemente alle rotture del
DNA, i loci genici delle immunoglobuline, o di altri
geni costitutivamente espressi nelle cellule B del
centro germinativo, forniscono sequenze regolatorie che causano la deregolazione trascrizionale dei proto-oncogeni ad esse giustapposti a
seguito della traslocazione cromosomica (33). Nel
contesto del DLBCL, le traslocazioni di BCL6 sono
un valido esempio di quanto appena affermato.
Queste traslocazioni avvengono in una significativa proporzione di DLBCL (prevalentemente nel
sottotipo ABC-like, e in minor proporzione nel sottotipo GCB-like) e pongono il gene BCL6 sotto il
controllo del promotore dei geni immunoglobulinici o di altri geni normalmente espressi nelle cellule B del centro germinativo.
Altri meccanismi di lesione molecolare nel DLBCL
sono rappresentati da mutazioni puntiformi che
attivano proto-oncogeni o inattivano geni oncosoppressori. Molteplici geni sono colpiti da questo meccanismo mutazionale.
Del tutto recentemente, una nuova classe di geni,
rappresentata da acetiltransferasi, si è rivelata
essere frequentemente inattivata tramite mutazioni puntiformi (34).
DLBCL GCB-like e ABC-like:
aspetti fenotipici e vie oncogenetiche
Oltre che per eterogeneità morfologica, il DLBCL
si caratterizza anche per eterogeneità fenotipica
e, come già riportato per gli studi di espressione
genica, l’espressione di specifici marcatori rivela
una diversa istogenesi delle cellule linfomatose.
È stato delineato un modello fenotipico basato sull’analisi di tre marcatori immunoistochimici: CD10
e BCL6, che fisiologicamente identificano le cellule B appartenenti al centro germinativo, e IRF4
che invece è comunemente espresso nelle cellule maturate oltre il centro germinativo (35, 36).
Utilizzando un algoritmo basato sulla diversa
espressione di tali marcatori, è possibile identificare i due sottogruppi di DLBCL riconosciuti dal
GEP:
1. fenotipo tipico delle cellule del centro germinativo (CD10 +/BCL-6+/-/IRF4+/- o CD10-/BCL6+/IRF4-), corrispondenti alla categoria GCBlike identificata dagli studi di gene expression
profiling;
2. fenotipo non-centro germinativo (CD10 /BCL-6-/IRF4+/- o CD10-/Bcl-6+/IRF4+), corrispondenti alla categoria ABC-like identificata dagli studi di gene expression profiling (35,
36) (Figura 1).
Dal punto di vista patogenetico, i due sottogruppi istogenetici di DLBCL identificati in base a GEP
e immunofenotipo sono caratterizzati da lesioni
molecolari differenti. Nel sottogruppo GCB-like,
si riscontrano traslocazioni coinvolgenti BCL2,
delezioni a carico del gene oncosoppressore
PTEN, amplificazioni di microRNA (miR-17-92 che
reprimono l’espressione di PTEN), e mutazioni
puntiformi del gene EZH2, che codifica per un
enzima coinvolto nella metilazione istonica (18, 37,
38). Del tutto recentemente, è stato dimostrato
come l’inattivazione di CREBBP/EP300 sia associata allo sviluppo di DLBCL GCB-like e, inoltre,
di una frazione di linfomi follicolari (34).
Dall’osservazione che le lesioni riscontrate a livello di CREBBP/EP300 avvengono nella maggior
parte dei casi in condizione di eterozigosi, si deduce l’aploinsufficienza nell’attività oncosoppressiva di queste proteine (34).
Le mutazioni inattivanti di CREBBP/EP300 producono un deficit dell’attività acetilante su BCL6
e TP53, che si traduce in un’attivazione costitutiva dell’oncoproteina BCL6 e in una riduzione
dell’attività oncosoppressiva di p53, determinando un incremento della tolleranza cellulare al danno del DNA contestualmente ad una diminuzione della via apoptotica e dell’arresto del ciclo cellulare (34).
Le lesioni molecolari di acetiltransferasi, quali
CREBBP e EP300, sono di particolare rilievo come
bersagli terapeutici per farmaci con attività di inibitori delle istondeacetilasi (HIDAC inhibitors).
Nel sottogruppo di DLBCL ABC-like, le alterazioni molecolari più significative dal punto di vista
patogenetico sono le traslocazioni del protoncogene BCL6, le mutazioni e/o delezioni del gene
oncosoppressore BLIMP1, l’amplificazione del
locus di BCL2, che porta ad una iperespressione del gene, e le delezioni del locus IRF4A-ARF,
che codifica per gli oncosopressori p16 e p14ARF
(18, 39).
Caratteristica del sottogruppo ABC-like è l’attivazione costituzionale della via di segnalazione di
NF-κB, un evento patogenetico in grado di pro-
Meccanismi patogenetici
FIGURA 1 - Algoritmo di definizione
istogenetica e prognostica mediante
immunoistochimica sul tessuto bioptico, secondo Hans (35), che permette di suddividere i DLBCL in centro
germinativo-like (GC) e non-centro
germinativo-like (non-GC).
muovere la proliferazione cellulare e inibire l’apoptosi. L’interferenza con il segnale di NF-κB uccide le cellule ABC-like ma non quelle GCB-like, e
ciò dimostra che il sottotipo ABC-like dipende dall’attività costitutiva di questa via di trasduzione del
segnale (11, 12).
L’iperattivazione della via di segnalazione di NFκB può essere secondaria a diverse lesioni genetiche, più frequentemente a carico del gene oncosoppressore A20, ma anche degli attivatori della
via di signaling, come CARD11 e TRAF2, e alla
attivazione cronica del B-cell receptor secondaria a mutazioni dei domini ITAM di CD79A e
CD79B (40, 41). Le aberrazioni di A20 non avvengono comunemente nel DLBCL GCB-like, ma
sono presenti in altri linfomi con attività NF-κB (41,
42, 44-46).
Marcatori biologici come fattori prognostici
La prognosi del DLBCL è estremamente eterogenea e, nonostante sia sensibilmente migliorata negli ultimi anni, i fattori predittivi della risposta alla terapia non sono ancora del tutto noti. Il
principale modello prognostico applicato al
DLBCL è l’International Prognostic Index (IPI) che,
in base alla valutazione di cinque variabili cliniche
(LDH elevata, età maggiore di 60 anni, stadio
secondo Ann Arbor maggiore o uguale a III, coinvolgimento di due o più sedi extranodali, e performance status secondo ECOG maggiore o
uguale a 2) consente di assegnare i pazienti a
quattro categorie di rischio di recidiva (basso
rischio, intermedio-basso, intermedio-alto, alto)
(47). Tali categorie di rischio correlano con una
diversa probabilità di sopravvivenza globale a
quattro anni, variabile oltre l’80% per il DLBCL a
basso rischio a meno del 60% per il DLBCL ad
alto rischio, e con una diversa sopravvivenza libera da progressione a quattro anni, variabile tra
l’85% e il 50% (48).
Un ulteriore indice prognostico è l’International
Prognostic Index assessed at time of Relapse (IPIR), utile nell’indicare la sopravvivenza globale e
la sopravvivenza libera da progressione in pazienti in prima recidiva. IPI-R identifica due categorie di rischio di fallimento della terapia di seconda linea contenente derivati del platino, seguita
da trapianto di cellule staminali emopoietiche
autologhe (49).
Lo stato attuale degli indicatori clinici di prognosi nel DLBCL induce la necessità di generare nuovi marcatori prognostici, in particolare volti a identificare i pazienti ad alto rischio di fallimento della terapia di prima linea. I nuovi fattori prognostici proposti negli ultimi anni derivano dall’analisi
delle caratteristiche immunoistochimiche e molecolari della malattia. Inoltre, è stato suggerito che
anche il profilo genetico dell’ospite possa rivestire rilevanza prognostica per il DLBCL.
Uno dei nuovi e più semplici modelli prognostici
da applicare nella pratica clinica è l’algoritmo di
Hans, che dimostra come la suddivisione dei
DLBCL in centro germinativo-like e non-centro
germinativo-like si traduca in una sensibile diffe-
9
10
Seminari di Ematologia Oncologica
renza di sopravvivenza globale a cinque anni,
variabile tra il 76% per i casi centro germinativo
e il 34% per i casi non-centro germinativo-like (36).
In maniera analoga, la caratterizzazione del profilo di espressione genica distingue due sottogruppi di DLBCL, GCB-like e ABC-like, con una
sopravvivenza globale a cinque anni del 76% per
il primo e del 16% per il secondo sottogruppo (7).
Tra i marcatori molecolari, la presenza di riarrangiamento di BCL2 e BCL6 non ha rilevanza prognostica (50, 51).
Al contrario, alcuni studi suggeriscono che le
mutazioni di BCL6 e la metilazione del promotore di MGMT siano correlate con un decorso clinico favorevole (52, 53). In particolare, la metilazione del promotore di MGMT sembrerebbe esse-
re un indicatore di potenziale risposta alla terapia (52).
Altri marcatori prognostici favorevoli sono rappresentati dall’espressione di LMO2 e di HIF1 (54,
55), mentre le mutazioni di TP53 (56) e i riarrangiamenti del protoncogene c-MYC (57) correlano con una riduzione della sopravvivenza globale. I riarrangiamenti di c-MYC hanno rilevanza nell’identificare pazienti con una prognosi particolarmente severa, e spesso sono presenti nei casi di
DLBCL cosiddetti double hit, che portano la traslocazione di BCL6 o BCL2 contemporaneamente alla traslocazione di c-MYC. Le mutazioni di
TP53, come anche in altre neoplasie linfoidi, sono
un classico marcatore di refrattarietà ai farmaci
contenuti nel programma terapeutico RituximabFIGURA 2 - MLH1 codifica per una
proteina coinvolta nei meccanismi di
riparazione del DNA. Il genotipo
MLH1 rs 1799977 AA si associa a normali livelli cellulari della proteina
MLH1. Questa, in caso di danno al
DNA, è in grado di promuovere
l’apoptosi mediata da p53 (pannello
A). I genotipi di MLH1 rs1799977
AG/GG si associano a riduzione dell’espressione di MLH1 con conseguente riduzione della capacità di attivare la via apoptotica e determinando così farmacoresistenza (pannello
B). Sulla base di questo modello biologico, nel DLBCL trattato con RCHOP21 i genotipi di MLH1 rs
1799977 AG/GG hanno una probabilità cumulativa di sopravvivenza globale a quattro anni significativamente inferiore rispetto al genotipo AA.
Meccanismi patogenetici
CHOP, e impongono la necessità per il futuro di
disegnare schemi terapeutici in grado di vincere
la chemiorefrattarietà (R-CHOP) indotta dalle
mutazioni di TP53.
Marcatori molecolari dell’ospite come
predittori di farmacoresistenza
Per quanto riguarda l’impatto del profilo genetico dell’ospite sulla sopravvivenza e sulla risposta al trattamento chemioterapico, osservazioni
interessanti stanno emergendo dall’analisi dei polimorfismi che coinvolgono singoli nucleotidi (single nucleotide polymorphism, SNP). In particolare, alcuni studi hanno evidenziato come SNP dei
geni GSTA1 e CYBA, coinvolti nella farmacocinetica e nella farmacodinamica dei chemioterapici
utilizzati nello schema R-CHOP comunemente
impiegato nella terapia dei DLBCL, siano fattori
prognostici indipendenti di sopravvivenza libera
da eventi (58). Altri studi hanno evidenziato che
SNP del gene dell’interleuchina 10 sono correlati alla prognosi dei DLBCL (59, 60).
Un recente studio condotto su due coorti di
pazienti (una di training e una di validazione) ha
analizzato 35 polimorfismi a singolo nucleotide
(SNP) di geni coinvolti nella riparazione del danno del DNA, in base alla possibile influenza di
tale meccanismo sull’attività citotossica dei farmaci utilizzati nella terapia del DLBCL (61). Sia
nella coorte di training, sia nella coorte di validazione, il genotipo MLH1 rs1799977 AG/GG è
stato selezionato come predittore indipendente
di sopravvivenza globale (61).
La ridotta sopravvivenza globale associata al
genotipo MLH1 rs1799977 AG/GG è espressione di un aumentato rischio di fallimento del trattamento di prima linea (R-CHOP) e di seconda
linea (schemi contenenti composti del platino).
È stato dimostrato che il polimorfismo di MLH1
rs1799977 mantiene un valore prognostico indipendente anche rispetto ai fattori prognostici
standard presenti alla diagnosi, ed è in grado di
definire due sottogruppi di rischio, sia tra i
pazienti con IPI score basso o intermedio-basso, sia tra quelli con IPI score intermedio-alto o
alto (61). MLH1 rs1799977 codifica per una proteina coinvolta nei meccanismi di riparazione del
DNA (Figura 2), e, se validato da studi prospettici, potrebbe rappresentare un fattore progno-
stico indipendente di sopravvivenza globale e di
rischio di fallimento della terapia con schemi contenenti antracicline e derivati del platino, entrambi ampiamente utilizzati nella terapia dei DLBCL.
n PATOGENESI MOLECOLARE
DEL LINFOMA MANTELLARE
Il linfoma mantellare (MCL) rappresenta approssimativamente il 3-10% dei linfomi non-Hodgkin.
È caratterizzato dalla traslocazione t(11;14) coinvolgente il locus BCL-1 (ciclina D1), un fattore di
controllo del ciclo cellulare, ed il locus delle immunoglobuline (1). La traslocazione provoca una
sovraespressione del proto-oncogene BCL-1
che determina un’alterazione del ciclo cellulare e
il conseguente sviluppo tumorale.
Mediante analisi GEP è stato possibile identificare una signature propria dei pazienti con MCL,
indipendentemente dalla iperespressione di ciclina D1.
Tra questi vi sono geni che hanno un ruolo nella
regolazione dell’apoptosi, nel controllo del ciclo
cellulare e nella trasduzione del segnale.
In particolare, sono stati riscontrati deregolati geni
coinvolti nei pathways di TNF, NF-κB, TGFβ, WNT
e PI3K/AKT, mentre è stata osservata una correlazione tra la presenza del recettore di IL10
(IL10R) e una più lunga sopravvivenza dei
pazienti (62). Infine, l’analisi GEP ha permesso il
riconoscimento molecolare della variante blastoide di MCL, caratterizzata da iperespressione di
cyclin-dependent kinase (CDK) 4 e di CDC28 protein kinase 1.
CDK4 si associa con ciclina D1 e favorisce la progressione del ciclo cellulare attraverso il checkpoint G1/S.
L’iperespressione di CDC28 protein kinase 1 blocca l’inibizione del complesso ciclina D1/CDK4 da
parte dell’inibitore CDK p27/Kip1 (63).
n PATOGENESI MOLECOLARE
DEI LINFOMI A CELLULE T
PERIFERICHE
I linfomi a cellule T periferiche (PTCL) costituiscono
circa il 10-15% di tutti i linfomi non-Hodgkin. In cir-
11
12
Seminari di Ematologia Oncologica
ca il 50% dei casi si parla di PTCL non specificato
(unspecified PTCL, PTCL-U), mentre gli altri casi sono
suddivisi in linfoma T a grandi cellule anaplastiche
(Anaplastic Large Cell Lymphoma, ALCL), linfoma T
angio-immunoblastico (Angioimmunoblastic T-cell
Lymphoma, AILT), linfoma e leucemia T dell’adulto
(Adult T-Cell Leukemia and Lymphoma, ATLL) (1).
Nei pazienti ALCL è tipica la traslocazione t(2;5) che
determina la formazione di una proteina di fusione
codificata dai geni NPM e ALK (64).
NPM codifica per una proteina nucleolare, mentre
ALK codifica per una tirosino kinasi normalmente
espressa nelle cellule T.
Vi sono poi altre anomalie citogenetiche ricorrenti,
quali la trisomia del cromosoma 3, 5, 8 e X, le delezioni del 6q, i riarrangiamenti del 7q, la monosomia
13 o la delezione di 13q14.
Mediant e analisi GEP sono stati identificati due
sottogruppi di PTCL: un gruppo a prognosi favorevole, associato alla espressione dei geni della
via di NF-κB, e un secondo gruppo a prognosi
sfavorevole, associato ad una alta espressione di
geni coinvolti in pathways correlati alla proliferazione cellulare (65).
Da analisi di GEP è stato anche identificato
PDGFRA come gene potenzialmente coinvolto
nella patogenesi dei PTCL (66). Inoltre sono stati caratterizzati tre sottogruppi prognostici di PTCL
in base al profilo di espressione citochinica: prognosi sfavorevole in associazione all’espressione
di CCR4, intermedia per l’espressione di CXCR3,
e favorevole in associazione ad espressione di
CCR3 (67).
Mediante analisi GEP è stato osservato che AILT
si associa tipicamente ad un fenotipo Th1 caratterizzato dalla espressione di citochine quali
CXCR3, TNF receptor OX40, e CXCL13. In particolare, quest’ultimo marcatore è uno tra i geni
maggiormente espressi da parte delle cellule T
regolatorie del centro germinativo; da qui l’ipotesi che l’istogenesi del AILT sia riconducibile a questo tipo cellulare. Gli ALCL sono invece associati ad un fenotipo Th2 caratterizzato dall’espressione delle citochine CCR3 e CCR4, e dei geni
IL13R, FOS e JUNB (65).
Ad oggi però, l’analisi GEP nei linfomi T ha fornito solo risultati preliminari poiché effettuata su
casistiche ridotte, e si tratta quindi di modelli che
necessitano di ulteriore validazione.
n PATOGENESI DEI LINFOMI
AGGRESSIVI DELL’OSPITE
IMMUNODEFICIENTE
In base alla classificazione WHO, i linfomi associati a infezione da HIV sono entità clinico-patologiche distinte rispetto alle malattie linfoproliferative dell’ospite immunocompetente (1).
I linfomi HIV-correlati sono generalmente linfomi
non-Hodgkin (HIV-NHL) di origine B e presentano istologia ad alto grado di malignità, disseminazione extranodale e comportamento clinico
aggressivo (1). In termini patologici, i linfomi HIVcorrelati sono distinti in: DLBCL, linfoma di
Burkitt/Burkitt-like (BL/BLL), linfoma primitivo del
sistema nervoso centrale (PCNSL), linfoma plasmablastico del cavo orale (PBL), linfoma primitivo delle cavità sierose (PEL) e linfoma di
Hodgkin (HL) (1).
Nell’ambito dei HIV-NHL a cellule B, le informazioni riguardanti l’istogenesi derivano dall’applicazione di un modello basato su marcatori genetici e immunofenotipici in grado di distinguere i
linfociti B maturi in:
1. cellule B vergini,
2. cellule B del centro germinativo (CG),
3. cellule B post-CG.
Le mutazioni dei geni variabili delle immunoglobuline (IGV) si accumulano fisiologicamente
durante il transito dei linfociti B attraverso il CG
(mutazioni ongoing), per quindi rimanere stabili nelle fasi di differenziazione post-CG (68). Pertanto,
le mutazioni dei geni IGV rappresentano il più affidabile marcatore genotipico di istogenesi: la positività per mutazioni dei geni IGV ongoing identifica l’origine del clone neoplastico dai linfociti B
del CG, mentre la positività per mutazioni stabili
identifica l’origine del clone neoplastico dai linfociti B post-CG (68).
L’applicazione di tale modello istogenetico ai HIVNHL ha rivelato che, a differenza di quanto avviene nei soggetti immunocompetenti, solo una frazione di HIV-BL e HIV-DLBCL riflettono i linfociti B
del CG in base alla presenza di mutazioni ongoing
dei geni IGV e al fenotipo BCL6+/MUM1-/CD138.
La maggior parte di HIV-NHL originano invece dai
linfociti B post-CG, portano mutazioni stabili dei
geni IGV ed esprimono il fenotipo BCL6/MUM1+/CD38+ (69). Infine, una parte di HIV-PBL,
Meccanismi patogenetici
pur in assenza di mutazioni dei geni IGV, esprime i marcatori fenotipici delle cellule B post-CG
e, dunque, verosimilmente origina da cellule B differenziatesi senza transitare attraverso il CG.
Le differenze istogenetiche dei HIV-NHL possono avere rilevanza clinica. L’espressione di CD138
e di altri marcatori del fenotipo post-GC è risultata associata a sopravvivenza libera da malattia
e sopravvivenza globale, mentre l’espressione di
marcatori del CG (ad esempio, BCL6) è risultata
associata a sopravvivenza libera da malattia e
sopravvivenza globale inferiori più favorevoli. Il
valore prognostico sfavorevole del profilo postcentro germinativo è stato confermato come marcatore indipendente da fattori prognostici convenzionali. La prognosi sfavorevole associata al profilo post-CG osservata nei HIV-NHL è per altro
coerente con quanto osservato nei linfomi diffusi a grandi cellule B della popolazione immunocompetente (7).
Una peculiarità dei linfomi aggressivi associati a
immunodeficienza è rappresentato dal caso dell’infezione virale. I virus oncogeni possono agire
tramite meccanismi diretti, come EBV e HHV8, e
indiretti, come HIV. I virus oncogeni che agiscono con meccanismo diretto sono in grado di infettare i linfociti B e indurne la trasformazione tramite la produzione di proteine virali. Ne sono
esempio le proteine virali di EBV:
1. EBNA2, un co-fattore trascrizionale che interagisce nelle cellule umane con la via di
NOTCH1, regolando la trascrizione di numerosi geni umani fra cui c-MYC;
2. LMP1, una proteina di membrana in grado di
mimare l’azione del CD40 umano, garantendo un segnale di sopravvivenza e proliferazione tramite la via di NF-κB;
3. LMP2A, una proteina di membrana in grado
di attivare la trasduzione del segnale delle tirosin-kinasi associate al recettore per l’antigene delle cellule B e fornire un importante
segnale di sopravvivenza;
4. EBERs, RNA non tradotti in grado di indurre
stimolazione autocrina da IL10.
Esempi di proteine virali di HHV8 coinvolte nella
trasformazione includono:
1. LANA1, in grado di inibire la via di p53 e interferire con la via di Rb, favorendo la progressione del ciclo cellulare;
2. ciclina virale, in grado di mimare l’azione della ciclina D2 umana e tuttavia insensibile ai
meccanismi regolatori della ciclina D2 umana;
3. IL6 virale, in grado di mimare l’azione antiapoptotica e proliferativa della IL6 umana.
n CONCLUSIONI E PROSPETTIVE
FUTURE
Numerosi esempi dimostrano come le alterazioni genetiche individuate nei linfomi maligni rappresentino importanti marcatori molecolari sia di
diagnosi che di prognosi e siano strumenti validati e indispensabili nella pratica diagnostica. I
marcatori molecolari hanno anche un ruolo fondamentale nella costruzione di modelli prognostici che possano consentire di adattare la terapia
a ciascun paziente.
Per una più completa caratterizzazione delle diverse classi di linfomi, risulta quindi indispensabile
ampliare le conoscenze riguardo le lesioni genetiche, anche mediante l’utilizzo di nuove tecnologie in particolare la metodica di sequenziamento dell’intero genoma.
Sebbene il meccanismo mediante il quale il microambiente possa favorire la crescita dei linfomi non
sia stato ancora del tutto chiarito, è certo che
anche questo meccanismo, oltre alla presenza di
lesioni genetiche, riveste un ruolo fondamentale
nella linfomagenesi.
È necessario quindi comprendere meglio l’interazione tra linfoma e microambiente per una
migliore comprensione dello sviluppo del linfoma
stesso.
Identificare il ruolo e le interazioni fra le diverse
componenti cellulari presenti nei linfomi potrebbe permettere l’individuazione di nuovi target terapeutici per questo tipo di malattia.
n BIBLIOGRAFIA
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA,
Stein H, et al. World Health Organization Classification
of Tumours, Pathology and Genetics of Tumours of
Haematopoietic and Lymphoid Tissues. IARC Press:
Lyon; 2008.
2. Evans LS, Hancock BW. Non-Hodgkin Lymphoma.
Lancet. 2003; 362: 139-146.
13
14
Seminari di Ematologia Oncologica
3. Hoffman R, Benz EJ, Shattil SJ, Furie B, Silberstein LE,
McGlave P, et al. Hematology basic principles and practice. 5 ed. Philadelphia: Churchill Livingstone; 2009.
4. Küppers R, Klein U, Hansmann ML, Rajewsky K.
Cellular origin of Human B-Cell Lymphomas. N Eng J
Med. 1999; 341: 1520-1529.
5. Lister TA, Crowther D, Sutcliffe SB, Glastein E,
Canellos GP, Young RC, et al. Report of a committee
convened to discuss the evaluation and staging of
patients with Hodgkin’s disease: Cotswolds meeting.
J Clin Oncol. 1989; 7: 1630-1636.
6. Carbone PP, Kaplan HS, Musshoff K, Smithers DW,
Tubiana M. Report of the Committee on Hodgkin’s
Disease Staging Classification. Cancer Res. 1971; 31:
1860-1861.
7. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS,
Rosenwald A, et al. Distinct types of diffuse large Bcell lymphoma identified by gene espression profiling.
Nature. 2000; 403: 503-511.
8. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao
H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008; 359: 2313-2323.
9. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A,
Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell
lymphoma. Proc Natl Acad Sci USA. 2003; 100: 99919996.
10. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH,
Qian SB, Zhao H, et al. XBP1, downstream of Blimp1, expands the secretory apparatus and other
organelles, and increases protein synthesis in plasma
cell differentiation. Immunity. 2004; 21: 81-93.
11. Davis RE, Brown KD, Siebenlist U, Staudt LM.
Constitutive nuclear factor kappaB activity is
required for survival of activated B cell-like diffuse
large B cell lymphoma cells. J Exp Med. 2001; 194:
1861-1874.
12. Lam LT, Davis RE, Pierce J, Hepperle M, Xu Y, Hottelet
M, et al. Small molecule inhibitors of IkappaB kinase
are selectively toxic for subgroups of diffuse large Bcell lymphoma defined by gene expression profiling.
Clin Cancer Res. 2005; 11: 28-40.
13. Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt
LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000; 13: 199-212.
14. Tam W, Gomez M, Chadburn A, Lee JW, Chan WC,
Knowles DM. Mutational analysis of PRDM1 indicates
a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006; 107: 4090-4100.
15. Pasqualucci L, Compagno M, Houldsworth J, Monti
S, Grunn A, Nandula SV, et al. Inactivation of the
PRDM1/BLIMP1 gene in diffuse large B cell lymphoma.
J Exp Med. 2006; 203: 311-317.
16. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM,
Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked
by BCL6 gene alterations in B cell lymphoma. Cancer
Cell. 2007; 12: 280-292.
17. Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J,
et al. Distinctive patterns of BCL6 molecular alterations
and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia.
2007; 21: 2332-2343.
18. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave
SS, Davis RE, et al. Molecular subtypes of diffuse large
B-cell Lymphoma arise by distinct genetic pathways.
Proc Natl Acad Sci USA. 2008; 105: 13520-13525.
19. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P,
Mihm M, et al. Molecular profiling of diffuse large Bcell lymphoma identifies robust subtypes including one
characterized by host inflammatory response. Blood.
2005; 105: 1851-1861.
20. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S,
Shinkai Y, Honjo T. Class switch recombination and
hypermutation require activation-induced cytidine
deaminase (AID), a potential RNA editing enzyme. Cell.
2000; 102: 553-563.
21. Pasqualucci L, Bhagat G, Jankovic M, Compagno M,
Smith P, Muramatsu M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet.
2008; 40: 108-112.
22. Pasqualucci L, Migliazza A, Fracchiolla N, William C,
Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci USA.
1998; 95: 11816-11821.
23. Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation
of BCL-6 gene in normal B cells by the process of
somatic hypermutation of Ig genes. Science. 1998; 280:
1750-1752.
24. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC,
Kleinstein SH, et al. Two levels of protection for the B
cell genome during somatic hypermutation. Nature.
2008; 451: 841-846.
25. Robbiani DF, Bunting S, Feldhahn N, Bothmer A,
Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell
lymphomas with reciprocal chromosome translocations. Mol Cell. 2009; 36: 631-641.
26. Pasqualucci L, Neumeister P, Goossens T, Nanjangud
G, Chaganti RS, Küppers R, et al. Hypermutation of
multiple proto-oncogenes in B-cell diffuse large-cell
lymphomas. Nature. 2001; 412: 341-346.
27. Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL,
Kuehl WM. Promiscuous translocations into
immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci USA. 1996; 93:
13931-13936.
28. Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio
S, Chen-Kiang S, Muramatsu M, et al. AID is required
for c-myc/IgH chromosome translocations in vivo. Cell.
2004; 118: 431-438.
29. Ramiro AR, Jankovic M, Callen E, Difilippantonio S,
Meccanismi patogenetici
Chen HT, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations.
Nature. 2006; 440: 105-109.
30. Franco S, Gostissa M, Zha S, Lombard DB, Murphy
MM, Zarrin AA, et al. H2AX prevents DNA breaks from
progressing to chromosome breaks and translocations.
Mol Cell. 2006; 21: 201-214.
31. Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W,
Wright GW, et al. Aberrant immunoglobulin class switch
recombination and switch translocations in activated
B cell-like diffuse large B cell lymphoma. J Exp Med.
2007; 204: 633-643.
32. Robbiani DF, Bothmer A, Callen E, Reina-San-Martin
B, Dorsett Y, Difilippantonio S, et al. AID is required for
the chromosomal breaks in c-myc that lead to cmyc/IgH translocations. Cell. 2008; 135: 1028-1038.
33. Callén E, Jankovic M, Difilippantonio S, Daniel JA, Chen
HT, Celeste A, et al. ATM prevents the persistence and
propagation of chromosome breaks in lymphocytes.
Cell. 2007; 130: 63-75.
34. Pasqualucci L, Dominguez-Sola D, Chiarenza A,
Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma.
Nature. 2011; 471: 189-95.
35. Colomo L, Lopez-Guillermo A, Perales M, Rives S,
Martınez A, Bosch F, et al. Clinical impact of the differentiation profile assessed by immunophenotyping
in patients with diffuse large B-cell lymphoma. Blood.
2003; 101: 78-84.
36. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD,
Delabie J, Ott G, et al. Confirmarion of the molecular
classification of diffuse large B-cell lymphoma by
immunohistochemistry using a tissue microarray.
Blood. 2004; 103: 275-282.
37. Xiao C, Srinivasan L, Calado DP, Patterson HC,
Zhang B, Wang J, et al. Lymphoproliferative disease
and autoimmunity in mice with increate miR-17-92
expression in lymphocytes. Nat Immunol. 2008; 9:
405-414.
38. Morin RD, Johnson NA, Severson TM, Mungall AJ,
An J, Goya R, et al. Somatic mutations altering EZH2
(Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;
42: 181-185.
39. Tagawa H, Suguro M, Tsuzuki S, Matsuo K, Karnan S,
Ohshima K, et al. Comparison of genome profiles for
identification of distinct subgroups of diffuse large Bcell lymphoma. Blood. 2005; 106: 1770-1777.
40. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright
GW et al. Oncogenic CARD11 mutations in human diffuse large B-cell lymphoma. Science. 2008; 319: 16761679.
41. Compagno M, Lim WK, Grunn A, Nandula SV,
Brahmachary M, Shen Q, et al. Mutations of multiple
genes cause deregulation of NF-kappaB in large B-celllymphoma. Nature. 2009; 459: 717-721.
42. Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi
K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009; 430: 694-649.
43. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM,
Romesser PB, et al. Chronic active B-cell-receptor signaling in diffuse large B-cell lymphoma. Nature. 2010;
463: 88-92.
44. Honma K, Tsuzuki S, Nakagawa M, Tagawa H,
Nakamura S, Morishima Y, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009; 114:
2467-2475.
45. Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI,
Hartmann S, Mechtersheimer G, et al. TNFAIP3 (A20)
is a tumor suppressor gene in Hodgkin lymphoma and
primary mediastinal B cell lymphoma. J Exp Med. 2009;
206: 981-989.
46. Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM,
Compagno M, et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood. 2009; 113: 4918-4921.
47. The International Non-Hodgkin’s Lymphoma Prognostic
Factors Project. A predictive model for aggressive nonHodgkin’s lymphoma. N Engl J Med. 1993; 329: 987994.
48. Sehn LH, Berry B, Chhanabhai M, Fitzgerald C, Gill K,
Hoskins P, et al. The revised International Prognostic
Index (R-IPI) is a better predictor of outcome than the
standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007; 109:
1857-1861.
49. Lerner RE, Thomas W, Defor TE, Weisdorf DJ, Burns
LJ. The International Prognostic Index assessed at
relapse predicts outcomes of autologous transplantation for diffuse large-cell non-Hodgkin’s lymphoma in
second complete or partial remission. Biol Blood
Marrow Transplant. 2007; 13: 486-492.
50. Gascoyne RD, Adomat SA, Krajewski S, Krajewska M,
Horsman DE, Tolcher AW, et al. Prognostic Significance
of Bcl-2 Protein Expression and Bcl-2 Gene
Rearrangement in Diffuse Aggressive Non-Hodgkin’s
Lymphoma. Blood. 1997; 90: 244-251.
51. Vitolo U, Gaidano G, Botto B, Volpe G, Audisio E, Bertini
M, et al. Rearrangements of bcl-6, bcl-2, c-myc and
6q deletion in B-diffuse large-cell lymphoma: Clinical
relevance in 71 patients. Ann Oncol. 1998; 9: 55-61.
52. Esteller M, Gaidano G, Goodman SN, Zagonel V,
Capello D, Botto B, et al. Hypermethylation of the DNA
Repair Gene O6-Methylguanine DNA Methyltransferase
and Survival of Patients With Diffuse Large B-Cell
Lymphoma. J Nat Cancer Ist. 2002; 94: 26-32.
53. Vitolo U, Botto B, Capello D, Vivenza D, Zagonel V,
Gloghini A, et al. Point mutations of the BCL-6 gene:
clinical and prognostic correlation in B-diffuse large cell
lymphoma. Leukemia. 2002; 16: 668-675.
54. Natkunam Y, Farinha P, Hsi ED, Hans CP, Tibshirani R,
Sehn LH, et al. LMO2 protein expression predicts sur-
15
16
Seminari di Ematologia Oncologica
vival in patients with diffuse large B-cell lymphoma
treated with anthracycline-based chemotherapy with
and without rituximab. J Clin Oncol. 2008; 26: 447-454.
55. Evens Am, Sehn LH, Farinha P, Nelson BP, Raji A, Lu
Y et al. Hypoxia-inducible factor-1 {alpha} expression
predicts superior survival in patients with diffuse large
B-cell lymphoma treated with R-CHOP. J Clin Oncol.
2010; 28: 1017-1024.
56. Young KH, Leroy K, Møller MB, Colleoni GW, SánchezBeato M, Kerbauy FR, et al. Structural profiles of TP53
gene mutations predict clinical outcome in diffuse large
B-cell lymphoma: an international collaborative study.
Blood. 2008; 112: 3088-3098.
57. Savage KJ, Johnson NA, Ben-Neriah S, Connors JM,
Sehn LH, Farinha P, et al. MYC gene rearrangements
are associated with a poor prognosis in diffuse large
B-cell lymphoma patients treated with R-CHOP
chemotherapy. Blood. 2009; 114: 3533-3537.
58. Rossi D, Rasi S, Franceschetti S, Capello D, Castelli
A, De Paoli L, et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with RCHOP21. Leukemia. 2009; 23: 1118-1126.
59. Lech-Maranda E, Baseggio L, Bienvenu J, Charlot C,
Berger F, Rigal D, et al. Interleukin-10 gene promoter
polymorphisms influence the clinical outcome of diffuse large B-cell lymphoma. Blood. 2004; 103: 35293534.
60. Kube D, Hua TD, Von Bonin F, Schoof N, Zeynalova
S, Klöss M, et al. Effect of interleukin-10 gene polymorphisms on clinical outcome of patients with
aggressive non-Hodgkin’s lymphoma: an exploratory
study. Clin Cancer Res. 2008; 14: 3777-3784.
61. Rossi D, Rasi S, Di Rocco A, Fabbri A, Forconi F,
Gloghini A, et al. The host genetic background of DNA
repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood. 2010; 117:
2405-13.
62. Hofmann WK, de Vos S, Tsukasaki K, Wachsman W,
Pinkus GS, Said JW, et al. Altered apoptosis pathways
in mantle cell lymphoma detected by oligonucleotide
microarray. Blood. 2001; 98: 787-794.
63. De Vos S, Krug U, Hofmann WK, Pinkus GS, Swerdlow
SH, Wachsman W, et al. Cell cycle alterations in the
blastoid variant of mantle cell lymphoma (MCL-BV) as
detected by gene expression profiling of mantle cell
lymphoma (MCL) and MCL-BV. Diagn Mol Pathol. 2003;
12: 35-43.
64. Morris SW, Kirstein MN, Valentine MB, Dittmer KG,
Shapiro DN, Saltman DL, et al. Fusion of a kinase gene,
ALK, to a nucleolar protein gene, NPM, in nonHodgkin’s lymphoma. Science. 1994; 263: 1281-1284.
65. Martinez-Delgado B. Peripheral T-cell lymphoma gene
expression profiles. Hematol Oncol. 2006; 24: 113-119.
66. Piccaluga PP, Agostinelli C, Zinzani PL, Baccarani M,
Dalla-Favera R, Pileri SA. Expression of platelet-derived
growth factor receptor alpha in peripheral T-cell lymphoma not otherwise specified. Lancet Oncol. 2005;
6: 440.
67. Asano N, Suzuki R, Ohshima K, Kagami Y, Ishida F,
Yoshino T, et al. Linkage of expression of chemokine
receptors (CXCR3 and CCR4) and cytotoxic molecules
in peripheral T cell lymphoma, not otherwise specified
and ALK-negative anaplastic large cell lymphoma. Int
J Hematol. 2010; 91: 426-435.
68. Capello D, Martini M, Gloghini A, Cerri M, Rasi S,
Deambrogi C, et al. Molecular analysis of immunoglobulin variable genes in human immunodeficiency virusrelated non-Hodgkin’s lymphoma reveals implications
for disease pathogenesis and histogenesis.
Haematologica. 2008; 93: 1178-1185.
69. Carbone A, Gloghini A, Larocca LM, Capello D,
Pierconti F, Canzonieri V, et al. Expression profile of
MUM1/IRF4, BCL-6, and CD138/syndecan-1 defines
novel histogenetic subsets of human immunodeficiency virus-related lymphomas. Blood. 2001; 97: 744-751.
17
Linfomi non Hodgkin
a grandi cellule
ANNALISA CHIAPPELLA1, DAVIDE ROSSI2, UMBERTO VITOLO1
1
S.C. Ematologia 2, Dipartimento di Oncologia, Azienda Ospedaliera ed Universitaria
San Giovanni Battista, Torino, Italia;
2
Divisione di Ematologia, Dipartimento di Medicina Clinica e Sperimentale,
Università degli Studi del Piemonte Orientale Amedeo Avogadro, Novara, Italia
n INTRODUZIONE
I linfomi diffusi a grandi cellule B (DLBCL) rappresentano il 30% di tutti i linfomi non-Hodgkin nell’adulto e il tasso di incidenza è in costante incremento (Figura 1); l’età mediana di insorgenza è
55-60 anni (1, 2). Lo schema CHOP (ciclofosfamide, doxorubicina, vincristina, prednisone) ha
rappresentato per molti decenni il cardine della
terapia dei linfomi. L’introduzione dell’anticorpo
monoclonale anti-CD20 rituximab in associazione alla chemioterapia standard ha permesso di
migliorare l’outcome dei pazienti affetti da DLBCL.
Lo studio randomizzato condotto dal Groupe
d’Etude des Lymphomes de l’Adulte (GELA) ha
dimostrato un vantaggio significativo per i pazienti anziani affetti da DLBCL trattati alla diagnosi con
R-CHOP21 rispetto a CHOP21, con un tasso di
remissione completa (RC) del 75% vs 63% (3). Ad
un follow-up di dieci anni, l’overall survival (OS)
è del 43.5% vs 27.6% e la progression-free survival (PFS) è del 36.5% vs 20.1% per R-CHOP21
vs CHOP21 rispettivamente (4).
Parole chiave: linfoma diffuso a grandi cellule B, RCHOP, fattori prognostici, trattamento di prima linea,
trattamento recidivati/refrattari
Indirizzo per la corrispondenza
Umberto Vitolo, MD
S.C. Ematologia 2
Azienda Ospedaliera e Universitaria S. Giovanni Battista
Corso Bramante - 10126 Torino, Italy
E-mail: [email protected]
Umberto Vitolo
16%
28%
6%
30%
20%
n Linfomi follicolari
n Linfomi indolenti non follicolari
n Linfomi diffusi a grandi cellule B
n Linfomi a cellule T
n Altri tipi di linfoma
FIGURA 1 - Incidenza dei vari sottotipi istologici di linfoma non
Hodgkin.
Il tentativo di migliorare l’outcome dei pazienti
affetti da DLBCL e l'impiego dei fattori di crescita granulocitari, ha favorito l’introduzione dei regimi di chemioterapia dose-dense, quali R-CHOP14
(ogni due settimane), con risultati superiori al solo
CHOP14 nei pazienti anziani (5).
Nel tentativo di migliorare ulteriormente la prognosi, sono stati utilizzati regimi di chemioterapia ad
alte dosi con reinfusione di cellule staminali autologhe periferiche, ma i risultati sono stati contrastanti nell’era pre-rituximab (6).
Nonostante i vari schemi utilizzati il 40% circa dei
pazienti tende a recidivare o è refrattario alla terapia di prima linea. È quindi indispensabile una
caratterizzazione accurata del rischio prognosti-
18
Seminari di Ematologia Oncologica
co alla diagnosi, al fine di identificare i pazienti a
prognosi veramente sfavorevole, per poter attuare strategie terapeutiche mirate.
n FATTORI PROGNOSTICI
Alla diagnosi, l’identificazione di fattori clinici,
radiologici e molecolari è necessaria per discriminare pazienti a diversa prognosi.
L’International Prognostic Index (IPI), basato su
cinque fattori prognostici negativi (età>60, stadio
III-IV, LDH elevata, PS >1 e interessamento di più
di una sede extralinfonodale) permette di identificare quattro diversi gruppi di rischio, con una OS
a 5 anni compresa tra 26% e 73% (7). L’IPI, disegnato per pazienti trattati secondo schemi
CHOP/CHOP-like, risulta valido anche nel contesto dei moderni regimi di immunochemioterapia che includono rituximab (7).
La tomografia ad emissione di positroni (18F-FDG
PET) si è dimostrata un ottimo strumento nel valutare la risposta al trattamento dei DLBCL, in considerazione dell’avidità di tale linfoma. La valutazione della risposta finale con PET è altamente
predittiva della PFS e OS nei linfomi aggressivi con
o senza masse residue alla TAC. Sulla base
dell’International Workshop Criteria (IWC) e
dell’International Harmonization Project per la PET,
sono state formulate le raccomandazioni riguardo i criteri di risposta per i linfomi aggressivi. La
PET negatività diventa quindi indispensabile per
definire la risposta completa alla terapia (9). Il valore della valutazione intermedia precoce con PET
come predittore della risposta finale è invece controverso e argomento di dibattito (10-12).
Un limite dei fattori clinici prognostici è però determinato dal non prendere in considerazione l’eterogeneità biologica dei DLBCL e i meccanismi
patogenetici che ne regolano la proliferazione.
La classificazione WHO del 2008 riconosce tale
eterogeneità e in primo luogo sottolinea la necessità di determinare l’indice di proliferazione MIB1
(13). I DLBCL con MIB1 >80-90% pongono un
problema di diagnosi differenziale con il linfoma
di Burkitt e le nuove entità clinico-patologiche
individuate nella classificazione WHO come
unclassified aggressive lymphomas, double hit
lymphomas, con caratteristiche intermedie tra lin-
foma di Burkitt classico e DLBCL. In questi casi,
sono indispensabili una revisione istopatologica accurata e uno studio mediante FISH al fine
di individuare la presenza della traslocazione di
c-MYC. Tali pazienti, infatti, hanno una prognosi infausta se trattati con la chemioimmunoterapia standard R-CHOP. Tuttavia la miglior
opzione terapeutica per questo sottotipo di linfomi aggressivi non è ancora stata identificata
e al momento non esiste una linea guida riconosciuta di trattamento (Tabella 1).
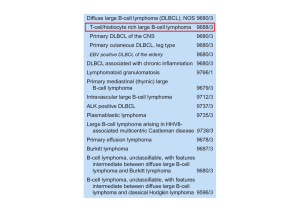
DLBCL, not otherwise specified (NOS)
• Common morphologic variants
- Centroblastic
- Immunoblastic
- Anaplastic
• Rare morphologic variants
• Molecular subgroups
- Germinal center B cell-like (GCB)
- Activated B cell-like (ABC)
• Immunohistochemical subgroups
- CD5-positive DLBCL
- Germinal center B cell-like (GCB)
- Nongerminal center B cell-like (non-GCB)
Diffuse large B-cell lymphoma subtypes
• T-cell/histiocyte-rich large B-cell lymphoma
• Primary DLBCL of the CNS
• Primry cutaneous DLBCL, leg type
• EBV-positive DLBCL of the elderly
Other lymphomas of large B cells
• Primary mediastinal (thymic) large B-cell lymphoma
• Intravascular large B-cell lymphoma
• DLBCL associated with chronic inflammation
• Lymphomatoid granulomatosis
• ALK-positive LBCL
• Plasmablastic lymphoma
• Large B-cell lymphoma arising in HHV8-associate
multicentric Castelman disease
• Primary effusion lymphoma
Borderline cases
• B-cell lymphoma, unclassifiable, with features
intermediate between diffuse large B-cell lymphoma
and Burkitt lymphoma
• B-cell lymphoma, unclassifiable, with features
intermediate between diffuse large B-cell lymphoma
and classical Hodgkin lymphoma
ALK indicates anaplastic lymphoma receptor tyrosine kinase; and HHV8,
human herpesvirus 8.
TABELLA 1 - Classificazione WHO 2008 dei DLBCL.
Linfomi non Hodgkin a grandi cellule
L’analisi tramite gene expression profiling (GEP)
ha permesso di risolvere a livello massimo di sensibilità la eterogeneità biologica del DLBCL,
identificando due categorie maggiori sulla base
di patterns di espressione genica:
- una categoria di DLBCL caratterizzata da profilo di espressione genica delle cellule B del
centro germinativo (Germinal Center B Cell);
- una categoria di DLBCL con profilo di
espressione genica simile a quello delle cellule B periferiche attivate (Activated B Cell)
(14) (Figura 2).
Al fine di trasferire i risultati degli studi di espressione genica nella pratica clinica, il gruppo di Hans
(15) ha studiato mediante immunoistochimica su
tissue microarray il pattern di espressione delle
proteine CD10, Bcl-6, IRF4/MUM1, Bcl-2, ciclina D2, e FOXP1, la cui espressione a livello di
mRNA era fortemente associata con i gruppi GCB
o ABC. I risultati sono stati usati per sottoclassificare i casi di DLBCL in due sottogruppi, GCB e
non-GCB (reminiscente della categoria ABC), in
GCB (42 cases)
+
+
CD10
+
-
Non-GC (27 cases)
MUM1
-
BCL-6
-
GCB (22 cases)
Non-GCB (61 cases)
FIGURA 3 - Albero decisionale di Hans per la classificazione dei
DLBCL sulla base dell’immunoperossidasi/tissue microarray (15).
base alla espressione dei tre marcatori CD10,
BCL6 e IRF/MUM1 (Figura 3).
La rilevanza clinica della distinzione tra GCB e
ABC deriva dalla osservazione che, se identificato mediante GEP, il gruppo di linfomi ABC ha
una prognosi più sfavorevole. Il gruppo ABC presenta un’attivazione costitutiva del pathway di
NF-kB sostenuta da lesioni genetiche che colpiscono diversi geni apparteneti a questo
pathway tra cui TNFAIP3/A20, CARD11, CD79A,
CD79B, MYD88. Su tale base, Dunleavy (16) ha
testato l’associazione di bortezomib, inibitore di
NF-kB, alla chemioterapia di prima linea (DAEPOCH) e ha dimostrato un possibile vantaggio
dell’associazione nel gruppo ABC rispetto al
gruppo GCB.
Oltre alla biologia della cellula tumorale, rivestono un ruolo determinante anche le caratteristiche
genetiche dell’ospite che sono alla base dello studio della farmacogenetica. Studi di farmacogenetica hanno documentato che i polimorfismi dell’ospite sono coinvolti nel metabolismo, nella
detossificazione dei farmaci e sono responsabili, almeno in parte, della variabilità in termini di efficacia e tossicità dello stesso trattamento in soggetti diversi (17, 18).
n TERAPIA DI PRIMA LINEA
FIGURA 2 - Tecnologia del gene array. Due patterns caratteristici dei DLBCL: Germinal Center B cell e Activated B Cell.
L’aggiunta del rituximab alla chemioterapia standard CHOP21 o alla chemioterapia dose-dense
CHOP14 ha migliorato significativamente la prognosi dei DLBCL rispetto all’era pre-rituximab.
19
20
Seminari di Ematologia Oncologica
Tuttavia, i pazienti a prognosi sfavorevole hanno
una probabilità di cura solo nel 45-55% dei casi;
in tali gruppi di pazienti devono essere presi in
considerazione approcci terapeutici sperimentali, nell’ambito di studi clinici, al fine di incrementare le loro chances terapeutiche.
Pazienti giovani
Nei pazienti affetti da DLBCL a basso rischio (IPI
0-1), in accordo con i risultati dello studio MInT, lo
standard di terapia è rappresentato da 6 cicli RCHOP21 con consolidamento radioterapico sulle
masse bulky o sulle localizzazioni extranodali (19).
Nei pazienti ad alto rischio, numerosi studi di fase
II hanno dimostrato che l’associazione di rituximab alla chemioterapia dose-dense CHOP14like è fattibile ed efficace in pazienti giovani affetti da DLBCL. Brusamolino et al. (20) hanno dimostrato la fattibilità di R-CHOP14 con supporto di
Pegfilgrastin in 50 pazienti affetti da linfoma
aggressivo di nuova diagnosi; la dose-intensity
del trattamento è stata del 95% con una bassa
incidenza di neutropenie febbrili. Tuttavia, da
questo studio è emerso il rilevante rischio di polmoniti da pneumocystis in corso di chemioimmunoterapia; per tale motivo, la profilassi con
cotrimoxazolo è obbligatoria in questo gruppo
di pazienti.
Al fine di migliorare la prognosi nei pazienti affetti da DLBCL di nuova diagnosi ad alto rischio,
sono stati sperimentati schemi di chemioterapia
ad alte dosi con reinfusione di cellule staminali
autologhe periferiche (HDC+ASCT).
Ad oggi, l'approccio HDC+ASCT è raccomandato nei pazienti giovani eligibili a trapianto che non
hanno ottenuto una remissione completa al trattamento di prima linea o in pazienti che hanno una
recidiva chemiosensibile della malattia. Secondo
le linee guida della Società Italiana di Ematologia
(SIE), HDC+ASCT in prima linea è indicato solo
all’interno di protocolli clinici sperimentali (21). Il
rischio di recidiva a livello del sistema nervoso
Author
Treatment
Inclusion
FFS/OS
CR% TD%
Brusamolino (20)
RCHOP14
<71 yr, stage II-IV
2 yr FFS 72W% OS 68%
74
2
Coso (47)
RISC
<61 yr, aa-IPI 2-3,
stage II-IV
5 yr FFS 63% OS 65%
72
3
Glass (48)
MegaCHOEP
<61 yr, aa-IPI 1-2-3,
stage III-IV
5 y FFS 62%, OS 67%
70
4.5
Intragumtornchai
(49)
CHOP-ESHAP-HDT
RCHOP-ESHAP
<66 yr, aaIPI2-3,
stage III-IV
5 yr FFS 16% OS 24%
5 yr FFS 61% OS 61%
36
67
8
11
Rueda (50)
RCHOP14
<71 yr, stage II-IV
30 m PFS 72% OS 86%
73
1
Stewart (51)
CHOP+DICEP+BEAM
<65 yr, aa-IPI 2-3
4 yr EFS 72% OS 79%
n.a.
1.8
Tarella (52)
RHDS-maps
<66 yr, aa-IPI 2,3,
stage II-IV
4 yr FFS 73% OS 76%
80
5
Arranz (53)
MegaCHOP ± IFE
+ BEAM
18-65 yr, low IPI with
beta2microglobulin or
intermediate/high risk
5 yr PFS 56% OS 64%
n.a.
3.5
Haioun (54)
ACE + HDT+ASCT ± R
ADVBP + HDT+ASCT ± R
18-60 yr, aa-IPI 2-3
4 yr EFS 71-80% OS 48-53
72
4
Vitolo (22)
RMegaCEOPRMAD-BEAM
<61 yr, aa-IPI 2-3,
stage III-IV
4 yr FFS 73% OS 80%
82
5
FFS: Failure-Free Survival; OS: Overall Survival; EFS: Event-Free Survival; CR: complete remission; TD: toxic death; aa-IPI: age-adjusted International
Prognostic Index; n.a. not applicable.
TABELLA 2 - Studi clinici; DLBCL a cattiva prognosi trattati con (rituximab) dose-dense chemioterapia +/- HDC+ASCT.
Linfomi non Hodgkin a grandi cellule
centrale (SNC) è circa il 5%. Per prevenire tale
rischio le linee guida SIE sottolineano la necessità di eseguire una profilassi con punture lombari medicate nei pazienti affetti da DLBCL a
rischio di tale recidiva. I pazienti a rischio sono
considerati quelli con coinvolgimento midollare,
con LDH elevata e due sedi extranodali coinvolte e quelli con interessamento al di sopra della
linea pterigopalatina, orbita, seni paranasali,
palato duro, in presenza di masse endocanalari
o paravertebrali o in caso di coinvolgimento testicolare del linfoma.
Nell’era pre-rituximab i risultati di studi randomizzati tra chemioterapia vs HDC+ASCT erano contrastanti, con tassi di sopravvivenza sovrapponibili nei due bracci di trattamento (6).
Dal 2002 al 2005 il Gruppo Italiano MultiRegionale
Linfomi e Leucemie GIMURELL, ha condotto uno
studio di fase II (clinicaltrials.gov: NCT00556127)
su 94 pazienti affetti da DLBCL alla diagnosi e IPI
sfavorevole per valutare la fattibilità e l’efficacia di
una chemioimmunoterapia dose-dense RMegaCEOP seguita da intensificazione con chemioterapia ad alte dosi con citarabina ad alte dosi
e mitoxantrone e da consolidamento con trapianto autologo condizionato con BEAM. I risultati sono
stati incoraggianti: RC 82%, 4-year PFS 73% e
4-year OS 80% (22). Diversi studi clinici sono stati condotti in pazienti affetti da DLBCL a prognosi sfavorevole per testare l’efficacia di schemi di
chemioterapia dose-dense con o senza rituximab
e con o senza intensificazione con HDC+ASCT
(Tabella 2).
Questi studi suggeriscono che uno schema di
chemioimmunoterapia ad alte dosi con trapianto autologo è efficace in pazienti giovani affetti da
DLBCL a prognosi sfavorevole. Tuttavia solo studi clinici randomizzati di fase III potranno rispondere all’interrogativo se una terapia ad alte dosi
è superiore alla chemio immunoterapia standard
R-CHOP21/R-CHOP14. I risultati dello studio randomizzato di fase III condotto dal gruppo tedesco di confronto tra dose-dense R-CHOEP14 vs
dose-escalated R-CHOEP+ASCT e quelli dello
studio condotto dal gruppo francese tra RCHOP14 e R-CEEP+HDC+ASCT hanno evidenziato o una superiorità del braccio standard, o una
sua sostanziale equivalenza rispetto al braccio
intensificato; ciò potrebbe essere dovuto all’eccessiva tossicità e alla non fattibilità dello schema intensificato (23, 24).
La Fondazione Italiana Linfomi (FIL) ha condotto
uno studio randomizzato di fase III
(clinicaltrials.gov NCT00499018) dal 2005 al
2009; 400 pazienti affetti da DLBCL a prognosi
sfavorevole sono stati randomizzati alla diagnosi a ricevere rituximab-chemioterapia dose-dense R-CHOP14 o R-MegaCHOP14 con o senza
intensificazione con HDC+ASCT. I risultati dello
studio sono in fase di elaborazione (Figura 4).
FIGURA 4 - Studio randomizzato di fase III (Fondazione Italiana
Linfomi).
21
22
Seminari di Ematologia Oncologica
Pazienti anziani
Il 50% dei pazienti affetti da DLBCL ha un’età
maggiore di 60 anni; un trattamento appropriato
è potenzialmente in grado di indurre le stesse
risposte nei pazienti giovani e nei pazienti anziani; è quindi essenziale trattare il maggior numero di pazienti anziani con una terapia convenzionale adeguata. A tal fine è d’obbligo differenziare tra età anagrafica e biologica i pazienti anziani, distinguendo tra fragili e non fragili utilizzando i parametri del Comprehensive Geriatric
Assessment (CGA): età ≥80 anni, capacità a svolgere le attività quotidiane (ADL), indice di comorbilità geriatriche cumulative (CIRS-G), sindrome
geriatrica (25, 26) (Tabella 3).
In base ai risultati dello studio randomizzato
CHOP21 vs R-CHOP21 (3), il trattamento standard per i pazienti anziani affetti da DLBCL è RCHOP21.
Lo studio RICOVER60 coordinato dal gruppo
German High Grade Non Hodgkin’s Lymphoma
Study Group (DSHNHL), è stato condotto su 1.222
pazienti randomizzati alla diagnosi a ricevere 6 o
8 cicli di R-CHOP14 con o senza rituximab e successiva radioterapia di consolidamento sulle
localizzazioni bulky (5). I risultati dello studio hanno dimostrato che 6 R-CHOP14 con 8 infusioni
di rituximab determinano un significativo miglio-
ramento della prognosi in termini di event-free survival (EFS), PFS e OS rispetto a 6 cicli CHOP14
senza rituximab (3-year OS 78.1% vs 67.7%). Non
è invece emersa alcuna differenza con l’aggiunta di ulteriori 2 cicli di chemioterapia CHOP14.
Studi randomizzati di confronto tra R-CHOP21 e
R-CHOP14 sono stati condotti dal gruppo francese GELA (27) e dal gruppo inglese British
National Cancer Research Institute (NCRI) (28);
l’obiettivo era determinare se R-CHOP14 per sei
cicli poteva diventare lo standard terapeutico in
questo gruppo di pazienti. Nello studio francese
randomizzato tra 8 cicli R-CHOP21 e 6 cicli RCHOP14, i risultati di R-CHOP14 sono stati inferiori all’atteso per scarsa aderenza alla terapia di
supporto con fattori di crescita granulocitari.
Tuttavia, il dato di confronto derivava dalla popolazione tedesca del RICOVER60, con caratteristiche non sovrapponibili completamente al gruppo di pazienti inseriti nel trial francese. Lo studio
inglese randomizzato tra 6 cicli R-CHOP14 e 8
R-CHOP21, pur non avendo la potenza statistica per determinare un vantaggio tra uno dei due
schemi, ha permesso di dimostrare che non ci
sono differenze statisticamente significative in termini di tossicità tra i due schemi.
In conclusione, R-CHOP21 e R-CHOP14 possono essere entrambi utilizzati nella pratica clinica
Diagnostic work-up
Exclusion/confirmation of EBV-positive DLBCL
CNS diagnostics only for patients at high risk for CNS disease or with testicular DLBCL
Echocardiogram and lung function test mandatory
Exclusion of other relevant organ dysfunctions
Determination of performance status
Prognostic assignation according to classical IPI
Patients >80 y (both chronologically and biologically)
Geriatric self-assessment (25)
Treatment and supportive measures
Prephase treatment mandatory
CNS prophylaxis with systemic high-dose MTX for patients at high risk for CNS disease only
Not less than 6 x R-CHOP.14 + 2R or 8 x R-CHOP-21 outside clinical trials
G-CSF or pegfilgrastim mandatory after R-CHOP
Infection prophylaxis with cotrimoxazole mandatory, with levofloxacin and acyclovir if severe neutropenia
Visit approximately day 8 after first R-CHOP
Hydrocortisone substitution in patients with fatigue after prednisone tapering
No radiotherapy to patients in complete remission after R-CHOP
TABELLA 3 - Strategie terapeutiche specifiche per età nel trattamento dei DLBCL (26).
Linfomi non Hodgkin a grandi cellule
per il trattamento in prima linea di pazienti anziani affetti da DLBCL; la scelta tra cicli somministrati a due diversi intervalli di tempo può essere basata sulla scelta del centro e in base alle comorbilità e al performance status del paziente.
Il gruppo tedesco DSHNHL ha testato uno schema dense-R-CHOP14, che prevede una dose
supplementare di rituximab a dosi intensificate
durante i primi due cicli R-CHOP14, per un totale di dodici infusioni di rituximab in associazione
a sei cicli di chemioterapia, con l’obiettivo di
migliorare l’efficacia di R-CHOP14 nei pazienti
anziani (29). Il confronto storico tra il gruppo trattato con il dense-R-CHOP14 e il gruppo dello
schema RICOVER60 ha evidenziato un netto
incremento dei livelli sierici di rituximab, una maggior efficacia per i pazienti ad alto rischio (1-year
EFS 74% vs 65%), ma anche un significativo
incremento dell’incidenza di infezioni, in particolare polmoniti da pneumocystis jiroveci nel gruppo dense-R-CHOP14 che non si è più osservato dopo l’introduzione di adeguata profilassi.
Un diverso approccio per migliorare l’outcome dei
pazienti anziani non fragili affetti da DLBCL è introdurre nel trattamento di prima linea farmaci biologici, precedentemente testati in un setting di
pazienti recidivati, in associazione alla chemioimmunoterapia standard. Tra questi farmaci rivestono particolare interesse gli agenti immunomodulatori (IMiDs)®, quali la lenalidomide (30).
Sulla base di questa ipotesi, la Fondazione Italiana
Linfomi sta conducendo uno studio pilota di fase
I-II che prevede il trattamento con lenalidomide
in associazione a R-CHOP21, per valutare la fattibilità e l’efficacia di tale associazione come trattamento di prima linea nei pazienti anziani affetti da DLBCL (clinicaltrials.gov NCT00907348).
L’analisi della fase I, disegnata con il Continual
Reassessment Method, ha permesso di concludere che la massima dose tollerata di lenalidomide assunta dal giorno 1 al giorno 14 in associazione con R-CHOP21 è di 15 mg (31).
n TERAPIA DEI PAZIENTI
IN RECIDIVA O REFRATTARI
La prognosi dei pazienti affetti da DLBCL in recidiva o refrattari alla terapia di prima linea è infau-
sta, con una prospettiva di sopravvivenza libera
da malattia inferiore al 10% nei pazienti recidivati o refrattari alla terapia di prima linea trattati con
sola chemioterapia di salvataggio; nei giovani in
recidiva, come dimostrato dai risultati del PARMA trial, la terapia standard è rappresentata da
una chemioterapia di induzione seguita da
HDC+ASCT; con questa strategia, circa il 50% dei
pazienti può ottenere un outcome favorevole (32)
(Figura 5).
Fattori prognostici alla recidiva, da valutare come
predittori di risposta ad una seconda linea terapeutica, sono la durata della risposta alla prima
linea, l’IPI alla recidiva e la chemiosensibilità alla
terapia di induzione di seconda linea pre-ASCT.
L’IPI, infatti, si è dimostrato efficace nel predire
OS e PFS anche nei pazienti refrattari e recidivati (33). Un’altra analisi ha inoltre dimostrato che
la recidiva della malattia a meno di dodici mesi
dalla diagnosi, correla significativamente con un
outcome più infausto (34). Tale dato è stato dimostrato anche in una recente analisi condotta nello studio CORAL (35), in cui l’EFS nelle recidive
precoci (meno di dodici mesi) è 20% vs 45% nelle recidive tardive (p<0.0001) e EFS per IPI ad alto
rischio alla recidiva è 18% vs 40% nel basso
rischio (p<0.0001). Dallo studio emerge inoltre che
il precedente trattamento con rituximab influisce
negativamente sulla EFS a 3 anni (21% vs 47%
p<0.0001).
Un altro indice prognostico affidabile nel predire
l’outcome del paziente in recidiva, è la negatività della valutazione PET pretrapianto-autologo. In
uno studio di fase II, la PET positività pre-ASCT
era associata ad una scarsa durata della risposta e ad un alto rischio di recidiva; se associata
ad una PET-positività persistente post autologo,
acquistava un ulteriore significato sfavorevole (36,
37). Tale affermazione è supportata da Cheson (9),
che sostiene che l’ottenimento della remissione
completa pre-autologo, definita come PET negatività, è un forte fattore discriminante per l’efficacia della HDC+ASCT.
Obiettivo della terapia di salvataggio è indurre una
remissione completa prima del consolidamento
con HDC+ASCT, superando eventuali resistenze
ai chemioterapici e favorendo la mobilizzazione
di cellule staminali autologhe periferiche. Molti
schemi sono stati utilizzati come trattamento di
23
100
90
80
70
60
50
40
30
20
10
0
P=0.001
Transplantation
Conventional treatment
0
15
30
45
60
75
90
Months after randomization
Overall survival (%)
Seminari di Ematologia Oncologica
Event-free survival (%)
24
100
90
80
70
60
50
40
30
20
10
0
P=0.001
Transplantation
Conventional treatment
0
15
30
45
60
75
90
Months after randomization
FIGURA 5 - PARMA Trial (31).
seconda linea nei pazienti affetti da DLBCL in recidiva. L’aggiunta del rituximab al trattamento chemioterapico ha permesso di migliorare la prognosi dei pazienti in recidiva/refrattari. In uno studio
condotto dal gruppo HOVON in 239 pazienti con
DLBCL in recidiva/refrattari, i pazienti erano randomizzati a ricevere uno schema di chemioterapia di salvataggio DHAP-VIM-DHAP con o senza rituximab seguito da autologo.
I risultati dello studio hanno evidenziato un netto vantaggio a favore del braccio con rituximab
rispetto al braccio senza rituximab, in termini di
RC (75% vs 54%) di Failure Free Survival (FFS)
a 2 anni (50% vs 24%) (38).
Resta tuttavia l’interrogativo su quale sia il miglior
schema di chemioterapia di salvataggio. Nello studio CORAL (35), studio clinico randomizzato condotto su 396 pazienti, i pazienti venivano randomizzati alla diagnosi a ricevere R-ICE o R-DHAP
pre-ASCT. I risultati hanno evidenziato che non vi
è alcuna differenza tra i due schemi di trattamento. Tuttavia, in un’analisi combinata di fattori prognostici clinici e biologici, si è evidenziato come
nel pattern GCB si evidenzia un vantaggio nel trattamento con R-DHAP rispetto a R-ICE, mentre nei
DLBCL con profilo ABC la prognosi è infausta sia
con R-DHAP che con R-ICE (39). È quindi necessario studiare delle strategie che possano permettere di migliorare la prognosi anche in questi sottogruppi più sfavorevoli e meno responsivi.
Per migliorare l’outcome dei pazienti affetti da
DLBCL in recidiva e refrattari, sono stati sperimentati ulteriori approcci terapeutici. Uno di questi è
rappresentato dall’integrazione della radioimmunoterapia con Zevalin nel consolidamento BEAM
per il trapianto autologo. Da questi studi è emerso che l’associazione Z-BEAM è fattibile, con una
tossicità sovrapponibile a quella del solo BEAM
e una possibile maggior efficacia (40, 41).
Nei pazienti con malattia refrattaria e/o recidivati dopo HDC+ASCT, è da valutare l’indicazione al
trapianto allogenico; i risultati di studi condotti in
questo ambito sono incoraggianti, con ottenimento di minori recidive e di un periodo libero da
malattia più lungo per l’allogenico rispetto al trapianto autologo. Tuttavia, l’alta incidenza di mortalità correlata al trapianto contrasta con i dati
positivi ottenuti sull’outcome (42, 43). Dati incoraggianti sono emersi anche da studi con l’utilizzo di trapianto allogenico a intensità ridotta (44).
I pazienti giovani non eligibili a terapie ad alte dosi
con procedura trapiantologica o i pazienti anziani, in presenza di DLBCL in recidiva/refrattario,
sono i candidati d’elezione per terapie sperimentali e farmaci biologici.
Tra questi farmaci rivestono un particolare interesse gli agenti immunomodulatori (IMiDs)®,
quali la lenalidomide, gli inibitori di m-TOR, quali temsirolimus ed everolimus, gli inibitori del proteasoma, come il bortezomib, e gli inibitori della istone-deacetilasi, come il vorinostat e il panobinostat (30). In pazienti a prognosi sfavorevole,
refrattari e non eligibili ad HDC+ASCT, sono stati utilizzati nuovi farmaci biologici, con risultati
incoraggianti (Figura 6).
La lenalidomide è stata testata come agente singolo in monoterapia in una serie di 73 pazienti con
DLBCL recidivato/refrattario; la risposta globale
ottenuta è stata del 29%, con una tossicità moderata (45).
Linfomi non Hodgkin a grandi cellule
vata nel 35% dei casi, con un tasso di RC del 12%
e tossicità prevalentemente ematologica (46).
Studi clinici che prevedono l’utilizzo di farmaci biologici quali lenalidomide, enzastaurin, nuovi anticorpi monoclonali anti-CD20 e anti-CD22 in
monoterapia o in associazione a chemioterapia
sono tuttora in corso.
DLBCL single-agent efficaci
Lenalidomide
SGN-40
mTOR inhibitors
Anti-Survivin
n CONCLUSIONI
Bortezomib
Vorinostat
Bevacizumab
Syk inhibitor
Enzastaurin
“Strength” of mechanism
FIGURA 6 - Nuovi agenti biologici per la terapia di DLBCL (30).
In un’altra serie di pazienti affetti da DLBCL in recidiva/refrattari, 49 pazienti fortemente pretrattati
hanno ricevuto lenalidomide 25 mg/die per 21 gg
a cicli mensili; una risposta globale è stata osser-
R-CHOP21 e R-CHOP14 rappresentano la terapia standard per il trattamento dei DLBCL alla diagnosi. L’identificazione di fattori prognostici permette di identificare classi di rischio pazienti a prognosi sfavorevole. In tale gruppo di pazienti è consigliato l’arruolamento in studi clinici (Figura 7). I
pazienti affetti da DLBCL in recidiva o refrattario
dopo R-CHOP hanno una prognosi più sfavorevole rispetto all’esperienza passata. È necessario migliorare l’efficacia della terapia di salvataggio con l’ausilio di nuovi farmaci sia nei giovani
DLBCL
≤60 years
>60 years
aa-IPI 0-1
aa-IPI 2-3
R-CHOP 21x6
Encourage enrollement
in clinical trials
If clinical trials not available
standard therapy
R-CHOP 21x8 or R-CHOP 14x6 (Rx8)
all IPI
If available
clinicals trials:
R-CHOP ±
novel drugs
Standard therapy
R-CHOP21 x 8 or R-CHOP14 x 6 (Rx8)
Rituximab - dose dense chemotherapy (R-CHOP like) ± R-HDC and ASCT
Rituximab - dose dense chemotherapy (R-CHOP like) ± novel drugs
FIGURA 7 - Algoritmo di terapia di prima linea nei DLBCL.
25
26
Seminari di Ematologia Oncologica
pre-ASCT e sia nei pazienti non candidabili a
HDC+ASCT.
n BIBLIOGRAFIA
1. Greiner TC, Medeiros LJ, Jaffe ES. Non-Hodgkin’s lymphoma. Cancer. 1995; 75: 370-80.
2. The non-Hodgkin's Lymphoma Classification Project.
A clinical evaluation of the International Lymphoma
Study Group classification of non-Hodgkin's
Lymphoma. Blood. 1997; 89: 3909-18.
3. Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H,
Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients
with diffuse large-B-cell lymphoma. N Engl J Med.
2002; 346: 235-42.
4. Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G,
Plantier I, Castaigne S, et al. Blood. 2010; 116: 204045.
5. Pfreundschuh M, Schubert J, Ziepert M, Schmits R,
Mohren M, Lengfelder E, et al. Six versus eight cycles
of bi-weekly CHOP-14 with or without Rituximab in elderly patients with aggressive CD20+ B-Cell
Lymphomas: a randomised controlled trial (RICOVER60). Lancet Oncol. 2008; 9: 105-16.
6. Greb A, Bohlius J, Trelle S, Schiefer D, De Souza CA,
Gisselbrecht C, et al. High-dose chemotherapy with
autologous stem cell support in first-line treatment of
aggressive non-Hodgkin lymphoma - Results of a comprehensive meta-analysis. Cancer Treat Rev. 2007; 33:
338-46.
7. Shipp MA, Harrington DP: A predictive model for
aggressive NHL: the International non-Hodgkin’s lymphoma prognostic factors project. N Engl J Med. 1993;
329: 987-94.
8. Ziepert M, Hasenclever D, Kuhnt E, Glass B,
Schmitz N, Pfrendschuh M, et al. Standard international prognostic index remains a valid predictor of
outcome for patients with aggressive CD20+ B-cell
lymphoma in the rituximab era. J Clin Oncol. 2010;
28: 2373-80.
9. Cheson BD, Pfistner B, Juweid ME, Gascoyne RD,
Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007; 25:
579-86.
10. Haioun C, Itti E, Rahmouni A, Brice P, Rain JD, Belhadj
K, et al. [18-F]fluoro-2-deoxy-D-glucose positron
emission tomography in aggressive lymphoma: an early prognostic tool for predicting patients outcome.
Blood. 2005; 106: 1376-81.
11. Cashen A, Dehdashti F, Luo J, Bartlett NL. Poor predictive value of FDG-PET/CT performed after 2 cycles
of R-CHOP in patients with Diffuse Large B-Cell
Lymphoma (DLCL). Blood. 2008; 112: 371.
12. Vitolo U, Chiappella A, Bellò M, Passera R, Botto B,
Castellano G, et al. The outcome of patients with
Diffuse Large B-Cell Lymphoma (DLBCL) treated with
Rituximab-CHOP (R-CHOP) is not predicted by 18FDG-Positron Emission Tomography/Computerized
Tomography (PET) performed at intermediate incourse evaluation, but only by PET assessed at the end
of therapy. Blood. 2010; 116: 2819.
13. World Health Organization. WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues.
Geneva, Switzerland: WHO Press. 2008.
14. Alizadeh AA, Elsen MB, Davis RE, et al. Distinct types
of diffuse large B-cell lymphoma identified by gene
expression profiling. Nature. 2000; 403: 504-11.
15. Hans CP, Weisenburger DD, Greiner TC, et al.
Confirmation of the molecular classification of diffuse
large B-cell lymphoma by immunohistochemistry
using a tissue microarray. Blood. 2004; 103: 275-82.
16. Dunleavy K, Pittaluga S, Czuczman MS, et al.
Differential efficacy of bortezomib plus chemotherapy
within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009; 113: 6069-76.
17. Rossi D, Rasi S, Franceschetti S, Capello D, Castelli
A, De Paoli L, et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with RCHOP21. Leukemia. 2009; 23: 1118-26.
18. Rossi D, Rasi S, Di Rocco A, Fabbri A, Forconi F,
Gloghini A, et al. The host genetic background of DNA
repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood 2011; 117
(8): 2405-13.
19. Pfreundschuh M, Trumper L, Osterborg A, Pettengell
R, Trneny M, Imrie K, et al. CHOP-like chemotherapy
plus Rituximab versus CHOP-like chemotherapy alone
in young patients with good-prognosis Diffuse LargeB-Cell Lymphoma: a randomised controlled trial by the
MabThera International Trial (MInT) Group. Lancet
Oncol. 2006; 7: 379-91.
20. Brusamolino E, Rusconi C, Montalbetti L, Gargantini
L, Uziel L, Pinotti G, et al. Dose-dense R-CHOP-14 supported by pegfilgrastim in patients with diffuse large
B-cell lymphoma: a phase II study of feasibility and toxicity. Haematologica. 2006; 91: 496-502.
21. Barosi G, Carella A, Lazzarino M, Marchetti M, Martelli
M, Rambaldi A, et al. Management of nodal diffuse
large B-cell lymphomas: practice guidelines from the
Italian Society of Hematology, the Italian Society of
Experimental Hematology and the Italian Group for
Bone Marrow Transplantation. Haematologica. 2006;
91: 96-103.
22. Vitolo U, Chiappella A, Angelucci E, Rossi G, Liberati
AM, Cabras MG, et al. Dose-dense and high-dose
chemotherapy plus Rituximab with autologous stem
cell transplantation for primary treatment of diffuse
large-B-cell lymphoma at poor prognosis: a phase II
multicenter study. Haematologica. 2009; 94: 1250-8.
23. Schmitz N, Nickelsen M, Ziepert M, Haenel M,
Linfomi non Hodgkin a grandi cellule
Borchmann P, Viardot A, et al. Aggressive chemotherapy (CHOEP-14) and Rituximab or High-Dose Therapy
(MegaCHOEP) and Rituximab for young, high-risk
patients with aggressive B-cell lymphoma: results of
the MegaCHOEP trial of the German High-Grade NonHodgkin Lymphoma Study Group (DSHNHL). Blood.
2009; 114: 404.
24. Milpied NJ, Legouill S, Lamy T, Delwail V, Gressin R,
Guyotat D, et al. No benefit of first-line Rituximab (R)
- High-Dose Therapy (R-HDT) over R-CHOP14 for
young adults with Diffuse Large B-Cell Lymphoma.
Preliminary results of the GOELAMS 075 prospective
multicentre randomized trial. Blood. 2010; 116: 685.
25. Tucci A, Ferrari S, Bottelli C, Borlenghi E, Drera M, Rossi
G. A comprehensive geriatric assessment is more effective than clinical judgment to identify elderly diffuse large
cell lymphoma patients who benefit from aggressive
therapy. Cancer. 2009; 115: 4547-53.
26. Pfreundschuh M. How I treat elderly patients with diffuse large B-cell lymphoma. Blood. 2010; 116: 5103-10.
27. Delarue R, Tilly H, Salles G, Gisselbrecht C, Mounier
N, Fournier M, et al. R-CHOP14 Compared to RCHOP21 in Elderly Patients with Diffuse Large B-Cell
Lymphoma: Results of the Interim Analysis of the
LNH03-6B GELA Study. Blood. 2009; 114: 406.
28. Cunningham D, Smith P, Mouncey P, et al. A phase III
trial comparing R-CHOP 14 and R-CHOP 21 for the
treatment of patients with newly diagnosed diffuse large
B-cell non-Hodgkin’s lymphoma [Abstract]. J Clin Oncol.
2009; 27: 435s.
29. Pfreundschuh M, Zeynalova S, Poeschel V, et al.
Improved outcome of elderly patients with poor prognosis diffuse large B-cell lymphoma (DLBCL) after
dose-dense Rituximab: results of the DENSE-RCHOP-14 trial of the German High-Grade NonHodgkin Lymphoma Study Group (DSHNHL) [Abstract].
Ann Oncol 2008; 19 (Suppl 4): iv99.
30. Leonard JP, Martin P, Barrientos J, Elstrom R. Targeted
treatment and new agents in diffuse large B-cell lymphoma. [Abstract]. Semin Hematol. 2008; 45 (S2): 1116.
31. Vitolo U, Chiappella A, Carella AM, Baldi I, Ciccone G,
Congiu A, et al. Prospective, multicenter phase I-II pilot
trial to evaluate efficacy and safety of Lenalidomide plus
Rituximab-CHOP21 (LR-CHOP21) for elderly patients
with untreated Diffuse Large B-Cell Lymphoma (DLBCL): interim analysis of the Intergruppo Italiano Linfomi
(IIL) REAL07 study. Blood. 2010; 116: 2871.
32. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous
bone marrow transplantation as compared with salvage
chemotherapy in relapses of chemotherapy-sensitive
non-Hodgkin's lymphoma. N Engl J Med. 1995; 333:
1540-45.
33. Blay JY, Gomez F, Sebban C, et al. The international
prognostic index correlates to survival in patients with
aggressive lymphoma in relapse: analysis of the PARMA trial. Blood. 1998; 92: 3562-68.
34. Guglielmi C, Gomez F, Philip T, Hagenbeek A, Martelli
M, Sebban C, et al. Time to relapse has prognostic value in patients with aggressive lymphoma enrolled into
the Parma trial. J Clin Oncol. 1998; 16: 3264-19.
35. Gisselbrecht C, Glass B, Mounier N, Gill D, Linch
DC, Trneny M, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the Rituximab era. J Clin Oncol. 2010; 28:
4184-90.
36. Spaepen K, Stroobants S, Dupont P, et al. Prognostic
value of positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose ([18F]FDG) after first-line
chemotherapy in non-Hodgkin's lymphoma: is
[18F]FDG-PET a valid alternative to conventional diagnostic methods? J Clin Oncol. 2001; 19: 414-19.
37. Filmont JE, Gisselbrecht C, Cuenca X, Deville L, Ertault
M, Brice P, et al. The impact of pre- and post-transplantation positron emission tomography using 18-fluorodeoxyglucose on poor-prognosis lymphoma
patients undergoing autologous stem cell transplantation. Cancer. 2007; 110: 1361-69.
38. Vellenga E, Notenboom A, van’t Veer M, et al.
Rituximab (Mabthera) improves the treatment results
of DHAP-VIM-DHAP and ASCT in relapsed/progressive aggressive CD20+ NHL. A prospective randomized HOVON trial. Blood. 2008; 111: 537-43.
39. Thieblemont C, Briere J, Mounier N, et al. Prognostic
Impact of Germinal Center (GC)/ Activated B-Cell
(ABC) Classification Analysed by Immunochemistry,
FISH Analysis and GEP, In Relapsed/Refractory
Diffuse Large B-Cell Lymphoma (DLBCL): The BioCORAL Study. [Abstract]. Proc Am Soc Hematol.
2010; abstr 993.
40. Shimoni A, Zwas ST, Oksman Y, et al. Yttrium-90ibritumomab tiuxetan (Zevalin) combined with highdose BEAM chemotherapy and autologous stem cell
transplantation for chemo-refractory aggressive
non-Hodgkin's lymphoma. Exp Hematol. 2007; 35:
534-40.
41. Krishnan A, Nademanee A, Fung HC, Raubitschek AA,
Molina A, Yamauchi D, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan
and high-dose chemotherapy in patients with nonHodgkin’s lymphoma. J Clin Oncol. 2008; 26: 90-5.
42. Chopra R, Goldstone AH, Pearce R, Philip T, Petersen
F, Appelbaum F, De Vol E, Ernst P. Autologous versus
allogeneic bone marrow transplantation for non
Hodgkin’s lymphoma: a case-controlled analysis of the
European Bone Marrow Transplant Group Registry
data. J Clin Oncol 1992; 10: 1690-5.
43. Peniket A J, Ruiz de Elvira Peniket AJ, Ruiz de Elvira
MC, Taghipour G, Cordonnier C, Gluckman E, de Witte
T, Santini G, Blaise D, Greinix H, Ferrant A, Cornelissen
J, Schmitz N, Goldstone AH. An EBMT registry
matched study of allogeneic stem cell transplants for
lymphoma: allogeneic transplantation is associated with
a lower relapse rate but a higher procedure-related
27
28
Seminari di Ematologia Oncologica
mortality rate than autologous transplantation. Bone
Marrow Transplant. 2003; 31: 667-78.
44. Kirsty Thomson KJ, Morris EC, Bloor A, Cook G,
Milligan D, Parker A, Clark F, Yung L, Linch DC,
Chakraverty R, Peggs KS, Mackinnon S. Favorable
long-term survival after reduced-intensity allogeneic
transplantation for multiple-relapse aggressive nonHodgkin's lymphoma. J Clin Oncol. 2009; 27: 426-32.
45. Czuczman MS, Vose JM, Zinzani PL, et al. Confirmation
of the efficacy and safety of lenalidomide oral
monotherapy in patients with relapsed or refractory diffuse large-B cell lymphoma: results of an international study (NHL-003). [Abstract] Blood. 112: 268.
46. Wiernik PH, Lossos IS, Tuscano JM, Justice G, Vose
JM, Craig E, et al. Lenalidomide monotherapy in
relapsed or refractory aggressive non Hodgkin’s lymphoma. J Clin Oncol 2008; 26: 4952-57.
47. Coso D, Sebban C, Boulat O, Biron P, Rey J, Aurran
T, et al. A phase II trial of rituximab as adjuvant to intensive sequential chemotherapy in patients under 60
years with untreated poor-prognosis diffuse large Bcell lymphoma. Bone Marrow Transplant 2006; 38 (3):
217-22.
48. Glass B, Kloess M, Bentz M, Schlimok G, Berdel WE,
Feller A, et al. Dose-escalated CHOP plus etoposide
(MegaCHOEP) followed by repeated stem cell transplantation for primary treatment of aggressive highrisk non-Hodgkin lymphoma. Blood 2006; 107 (8):
3058-64.
49. Intragumtornchai T, Bunworasate U, Nakorn TN,
Rojnuckarin P. Rituximab-CHOP-ESHAP vs CHOPESHAP-high-dose therapy vs conventional CHOP
chemotherapy in high-intermediate and high-risk
aggressive non-Hodgkin's lymphoma. Leuk Lymphoma
2006; 47 (7): 1306-14.
50. Rueda A, Sabin P, Rifá J, Llanos M, Gómez-Codina J,
Lobo F, et al. R-CHOP-14 in patients with diffuse large
B-cell lymphoma younger than 70 years: a multicentre,
prospective study. Hematol Oncol 2008; 26 (1): 27-32.
51. Stewart DA, Bahlis N, Valentine K, Balogh A, Savoie
L, Morris DG, et al. Upfront double high-dose
chemotherapy with DICEP followed by BEAM and
autologous stem cell transplantation for poor-prognosis aggressive non-Hodgkin lymphoma. Blood 2006;
107 (12): 4623-27.
52. Tarella C, Zanni M, Di Nicola M, Patti C, Calvi R,
Pescarollo A, et al. Prolonged survival in poor-risk diffuse large B-cell lymphoma following front-line treatment with rituximab-supplemented, early-intensified
chemotherapy with multiple autologous hematopoietic stem cell support: a multicenter study by GITIL
(Gruppo Italiano Terapie Innovative nei Linfomi).
Leukemia 2007; 21 (8): 1802-11.
53. Arranz R, Conde E, Grande C, Mateos MV, Gandarillas
M, Albo C, et al. Dose-escalated CHOP and tailored
intensification with IFE according to early response and
followed by BEAM/autologous stem-cell transplantation in poor-risk aggressive B-cell lymphoma: a
prospective study from the GEL-TAMO Study Group.
Eur J Haematol 2008; 80 (3): 227-35.
54. Haioun C, Mounier N, Emile JF, Ranta D, Coiffier B, Tilly
H, et al. Rituximab versus observation after high-dose
consolidative first-line chemotherapy with autologous
stem-cell transplantation in patients with poor-risk diffuse large B-cell lymphoma. Ann Oncol 2009; 20 (12):
1985-92.
29
Linfoma mantellare
MARCO LADETTO, SIMONE FERRERO, SARA BARBIERO
Divisione di Ematologia, Dipartimenti di Medicina e Oncologia Sperimentale,
Università di Torino
Marco Ladetto
n INTRODUZIONE
Il linfoma mantellare (MCL) rappresenta un’entità relativamente rara tra le neoplasie ematologiche, con un’incidenza che si aggira intorno al
6% di tutti i linfomi non Hodgkin (1) e circa 300
casi diagnosticati in Italia ogni anno (2).
Nonostante ciò questo tipo di linfoma è stato
estremamente ben caratterizzato dal punto di
vista biologico, genetico, istologico, immunofenotipico e clinico. Infatti la sua identificazione
come entità clinica distinta risale all’inizio degli
anni 90 (fu inserito nella classificazione REAL nel
1994 (3)).
Contestualmente alla sua definizione istopatologica sono immediatamente emerse le specifiche caratteristiche cliniche di questa patologia,
in particolar modo la marcata chemioresistenza che giustificava la pessima prognosi dei
pazienti affetti da questa patologia (4).
Nel corso degli anni 90 e nella prima decade del
millennio i progressi nel campo del MCL sono
stati però molto rapidi e hanno investito il campo biologico, biotraslazionale e clinico. A tutt’oggi è forse il linfoma in cui si è raggiunta la masParole chiave: linfoma mantellare, malattia minima residua, chemioterapia intensificata, nuove molecole
Indirizzo per la corrispondenza
Divisione di Ematologia 1 U.
A.O.U. San Giovanni Battista di Torino
Via Genova 3 - 10126 Torino
E-mail: [email protected]
sima integrazione tra questi aspetti e proprio
questa integrazione rappresenta la base dei
significativi successi ottenuti in termini di ris posta clinica e sopravvivenza (5).
Per questi motivi il MCL è una neoplasia il cui
trattamento adeguato richiede elevata accuratezza diagnostica, attento monitoraggio clinico
e scelte terapeutiche mirate e spesso intensive
ed è pertanto meritevole di trattamento in
ambito specialistico assai più di altri istotipi di
linfoma di più comune riscontro.
L’obiettivo principale di questa pubblicazione
è dunque quello di illustrare gli importanti progressi sinora ottenuti grazie ad una efficace
integrazione tra ricerca biologica, traslazionale e clinica.
n CENNI DI PATOGENESI
MOLECOLARE
Fin verso la fine degli anni ’90 si pensava che il
MCL originasse da linfociti B naïve non transitati attraverso il centro germinativo (6).
Questa ipotesi era suffragata dalla caratteristica coespressione dell’antigene T-cellulare CD5
insieme al tipico immunofenotipo B-cellulare:
CD10–, CD19+, CD20+, CD22+, CD43+, CD79a+
(7), ma (a differenza della leucemia linfatica cronica) usualmente CD23– e CD200– (8). Diversi
studi sul recettore immunoglobulinico hanno in
parte smentito questa ipotesi, evidenziando
come almeno un’ampia porzione dei casi derivi da cellule B che hanno incontrato l’antigene.
30
Seminari di Ematologia Oncologica
In effetti circa un quarto dei pazienti presenta un
recettore immunoglobulinico con più del 2% di
mutazioni somatiche e anche nei casi non ipermutati è verosimile che, per la maggior parte di
essi, l’origine istopatogenetica risieda in cellule
B memoria che hanno comunque avuto l’incontro con l’antigene, seppur non nel contesto del
centro germinativo (Figura 1) (9,10). Nonostante
ciò, il ruolo ricoper to dalla stimolazione antigenica nel MCL è ancora ben lungi dall’essere definito (11, 12).
La patogenesi del MCL è caratterizzata dalla
contemporanea deregolazione del ciclo cellulare e dei meccanismi di risposta al danno del DNA
(13). Infatti la caratteristica genetica fondamentale di questo linfoma è la traslocazione cromosomica t(11;14)(q13;q32), responsabile dell’espressione aberrante di ciclina D1, importante regolatore del ciclo cellulare (14). I pochi casi
di MCL negativi per la traslocazione t(11;14) condividono comunque simili caratteristiche cliniche,
morfologiche e di espressione genica e spesso
mostrano un’iperespressione delle cicline D2 e
D3 (15).
Ulteriori eventi genetici incrementano il potenziale oncogenetico delle cicline, spesso attraverso l’inattivazione dei meccanismi di risposta al
danno del DNA: l’inattivazione delle chinasi cicli-
MIDOLLO OSSEO E
SANGUE PERIFERICO
Precursore B
cellulare
LINFONODI E ORGANI
LINFOIDI SECONDARI
Linfociti B naive
MCL???
Linfociti B naive
MATURAZIONE
EXTRA CG
MATURAZIONE
INTRA CG
MCL non
ipermutato
(75% dei casi)
Linfociti B memoria non
transitati attraverso il CG
Centro
germinativo
Zona
mantellare
MCL
ipermutato
(25% dei casi)
Linfociti B memoria
transitati attraverso il CG
Zona
marginale
FIGURA 1 - Ontogenesi cellulare del MCL. La maggior parte dei casi origina da linfociti B memoria che sono andati incontro a maturazione
al di fuori del centro germinativo (CG) del follicolo linfoide e pertanto non hanno accumulato ipermutazioni somatiche nel gene delle
immunoglobuline (MCL non ipermutato). Circa un quarto dei casi di linfoma mantellare origina invece da linfociti B memoria che
sono andati incontro a maturazione all’interno del centro germinativo, accumulando così ipermutazioni somatiche (MCL ipermutato).
Meno verosimile l’ipotesi secondo la quale il linfoma mantellare deriverebbe da linfociti B naive.
Linfoma mantellare
nadipendenti (p16INK4a/p16ARF) incrementa infatti la degradazione della proteina pro-apoptotica p53 (16, 17), così come la mutazione del gene
ATM (presente fino al 75% dei casi) dà come esito un blocco dei normali processi di arresto del
ciclo cellulare, riparazione del DNA e apoptosi,
fisiologicamente mediati da p53 (18).
L’insieme di tutti questi eventi contribuisce alla
progressione del ciclo cellulare, favorendo inoltre una marcata instabilità genetica. Inoltre la
sopravvivenza e la crescita tumorale sono promosse dall’alterazione di diverse altre vie di
segnalazione intracellullare: fra queste l’attivazione costitutiva del pathway PI3K/AKT/mTOR
(19) e delle vie proliferative di WNT (20),
Hedgehog (21) e NF-κB (22).
Per contro, gli studi dei profili di espressione
genica hanno recentemente identificato un pannello di geni proliferativi (tra i quali SOX11) in grado di discernere i classici casi di MCL da quel
sottogruppo di pazienti caratterizzati da una prognosi favorevole e definiti pertanto linfomi mantellari indolenti (23).
In conclusione, il MCL rappresenta il paradigma
di una neoplasia caratterizzata da un alto grado di instabilità genomica e da un numero elevato di alterazioni cromosomiche secondarie che
interferiscono con la regolazione del ciclo cellulare, i meccanismi di risposta al danno del DNA,
l’apoptosi e le vie di trasduzione del segnale. Il
crescente interesse per le basi molecolari di questa patologia si sta traducendo in questi anni nel-
LinfocitaT
BCR
Ciclina D1
CDk4/6
Proteasoma
26S
Microambiente
Midollare
FIGURA 2 - Bersagli molecolari del MCL e relativi approcci farmacologici. Nella figura sono illustrate schematicamente le principali
vie proliferative che caratterizzano la cellula di MCL e alcuni bersagli molecolari colpiti da alcuni dei più importanti tra i nuovi farmaci
attualmente in sperimentazione clinica (per i dettagli si faccia riferimento alla trattazione). BCR = recettore delle cellule B.
31
32
Seminari di Ematologia Oncologica
la messa a punto di nuove strategie farmacologiche, che porteranno in un futuro prossimo a
un miglioramento della gestione clinica dei
pazienti. In effetti già oggi alcuni dei più efficaci agenti terapeutici impiegati nella terapia di salvataggio del MCL agiscono sui bersagli molecolari sopracitati, in particolare il bortezomib, che
interferisce sul pathway di NF-κB, e il temsirolimus, che agisce
sul pathway di
PI3K/AKT/mTOR (Figura 2).
n FATTORI PROGNOSTICI
CLINICI E BIOLOGICI
A partire dall’inserimento del MCL all’interno della REAL classification nel 1994 (3) molti lavori
hanno descritto su serie limitate di pazienti le
caratteristiche clinico-patologiche, l’andamento
clinico e i fattori prognostici di questo tipo di linfoma (24-29). Se in generale in quegli anni i risultati terapeutici non erano soddisfacenti, alcuni
gruppi di pazienti presentavano però un andamento clinico migliore di altri, e vi era un piccolo sottogruppo di pazienti caratterizzati da
remissioni cliniche di lunga durata. Nonostante
ciò, per molto tempo non è stato possibile definire per questa patologia un indice prognostico
uniformemente accettato, a differenza di quanto successo per il linfoma diffuso a grandi cellule (IPI) (30) e per il linfoma follicolare (FLIPI (31)
e FLIPI2 (32)). Poiché l’applicazione di tali strumenti al MCL presentava serie limitazioni (24, 25,
29, 33), è stato costruito recentemente, grazie
al supporto dello European Mantle Cell
Lymphoma Network, un indice prognostico
specifico per il MCL, denominato MIPI (Mantle-
Cell-Lymphoma International Prognostic Index)
(34). Il MIPI si basa su quattro fattori prognostici indipendenti (età, ECOG performance status,
LDH e numero di leucociti), facilmente disponibili nella quotidiana pratica clinica. Dal momento che il MIPI si calcola attraverso una complessa formula matematica, è stata sviluppata per
l’uso clinico una versione semplificata del MIPI
che ne riproduce molto bene il valore prognostico (Tabella 1). Grazie all’utilizzo di questo nuovo strumento è possibile suddividere i pazienti
in tre gruppi ben bilanciati caratterizzati da prognosi decisamente differenti in termini di sopravvivenza: (alto rischio con overall survival (OS)
mediana di 29 mesi; rischio intermedio con OS
mediana di 51 mesi; basso rischio con OS
mediana non raggiunta) (34).
Pertanto il MIPI potrà rivelarsi utile nel prendere decisioni terapeutiche individualizzate sul livello di rischio di ciascun paziente e permetterà una
migliore stratificazione dei pazienti negli studi clinici, fornendo così una base comune per valutare il ruolo dei nuovi marcatori prognostici biologici.
Diversi studi istopatologici hanno riconosciuto
nell’ultimo decennio come il grado di proliferazione tumorale, valutato attraverso l’espressione dell’antigene correlato alla proliferazione Ki67, sia il miglior fattore biologico predittivo di
sopravvivenza nei pazienti con MCL (35, 36, 26,
29) in maniera indipendente dal MIPI (34). Il tentativo di integrazione di questo parametro all’interno del MIPI, nell’intento di costruire un indice combinato (MIPI-b, ossia MIPI biologico), non
ha però sostanzialmente migliorato il potere prognostico del MIPI classico (34).
Pertanto, considerando anche che il valore del
Età (anni)
ECOG
LDH/massimo valore limite
Leucociti (x109/L)
0
<50
0-1
<0,670
<6,700
1
50-59
-
0,670-0,999
6,700-9,999
2
60-69
2-4
1,000-1,499
10,000-14,999
3
>69
-
>1,499
>14,999
Punteggio
Si applica un punteggio da 0 a 3 per ogni fattore prognostico, fino a un massimo di 11 punti. I pazienti la cui somma sia <4 sono classificati come basso
rischio; i pazienti la cui somma sia 4-5 sono classificati come rischio intermedio; i pazienti la cui somma sia >5 sono classificati come alto rischio.
TABELLA 1 - Calcolo del MIPI semplificato.
Linfoma mantellare
Ki-67 non è facilmente ottenibile per motivi tecnici in tutti i pazienti e che la standardizzazione
e riproducibilità di questo marcatore molecolare necessitano ancora di essere affinate, al
momento attuale il MIPI-b non è particolarmente utile al di fuori del contesto di studi sperimentali. Fra gli altri fattori prognostici biologici vale
la pena di ricordare la mancata espressione di
SOX11, recentemente identificata come diagnostica di quel sottogruppo di MCL indolenti, caratterizzati da una prognosi decisamente più favorevole (23).
La malattia minima residua
A differenza dei fattori prognostici descritti in precedenza, indagati alla diagnosi e direttamente
correlati all’aggressività del linfoma, la malattia
minima residua valuta in maniera molto precisa
la risposta alla terapia di ogni singolo paziente,
identificando la presenza anche di una minima
quantità di malattia residuata dopo un trattamento efficace e in grado di dare origine a una recidiva. Il marcatore molecolare che indaga la
malattia minima residua è costituito da un tratto di DNA caratteristico della cellula tumorale e
non presente nelle cellule sane; per quanto
riguarda il MCL, anche se i primi studi di malattia minima residua sono stati condotti sul trascritto Bcl-1 (prodotto specifico della traslocazione
t11;14), ultimamente il marcatore maggiormente utilizzato è il riarrangiamento delle catene
pesanti delle immunoglobuline e le metodologie
di analisi si basano a livello tecnico su approcci di PCR (reazione polimerasica a catena) qualitativi e quantitativi (37-40).
Durante gli ultimi anni l’impatto clinico della
malattia minima residua si è reso particolarmente importante nel campo del MCL, riflettendo i
considerevoli progressi ottenuti nella gestione di
questa patologia. Tali successi erano già stati
annunciati agli inizi del millennio da diversi studi di malattia minima residua, che documentavano come queste nuove terapie abbattessero
in maniera significativa la carica tumorale e come
tale risultato si traducesse in un netto miglioramento prognostico (41, 42, 38, 40). Sulla base
della dimostrazione dell’elevato valore prognostico della malattia minima residua in questo tipo
di linfoma, sono stati recentemente pubblicati i
promettenti risultati di studi sperimentali incentrati sul trattamento della persistenza o della recidiva molecolare di questa patologia (39, 43).
Le attuali conoscenze derivate dagli studi clinici di malattia minima residua sul MCL ci consentono di trarre le seguenti conclusioni:
- prima dell’avvento del rituximab non si riuscivano a ottenere remissioni molecolari né con la
chemioterapia convenzionale (44, 38) né con
programmi basati sul trapianto autologo di cellule staminali (37). Quest’ultimo concetto si contrappone alle remissioni molecolari ottenibili
invece nel linfoma follicolare e sottolinea la caratteristica chemioresistenza dell’istotipo mantellare (37);
- l’integrazione del rituximab e della citarabina
ad alte dosi in programmi autotrapiantologici ha
consentito il raggiungimento di livelli di citoriduzione mai osservati in precedenza e ha dimostrato come la risposta molecolare possa effettivamente essere considerata un obiettivo raggiungibile anche nei pazienti affetti da MCL (41,
45, 42);
- il trapianto autologo di cellule staminali in associazione al rituximab può ridurre ulteriormente la
carica tumorale residua dopo chemio-immunoterapia, innalzando il tasso di remissioni molecolari fino al 70% dei casi e confermando dunque la nota attività anti-linfomatosa della chemioterapia ad alte dosi in questo tipo di linfoma. Inoltre una remissione molecolare può
essere raggiunta anche in circa il 60% dei
pazienti non candidabili ad un programma trapiantologico, se trattati con chemioterapia convenzionale supplementata da rituximab (40);
- la remissione molecolare è un valido predittore prognostico indipendente in diversi contesti
terapeutici, dal trapianto autologo alla chemioimmunoterapia convenzionale ai regimi di mantenimento. Inoltre il concetto di remissione molecolare supera il valore dello stato di remissione
clinica: infatti i pazienti in remissione parziale, ma
negativi per la malattia minima residua, mostrano una durata di risposta superiore rispetto ai
pazienti in remissione completa, ma persistentemente positivi a livello molecolare (38, 40);
- nei pazienti trattati con rituximab l’aspirato
midollare è risultato essere il campione più adeguato su cui valutare la malattia minima residua.
33
34
Seminari di Ematologia Oncologica
Anche il sangue periferico può essere utilizzato
per simili analisi, ma il suo valore predittivo è inferiore: pertanto il suo utilizzo deve essere limitato a contesti terapeutici molto particolari, come
ad esempio per i pazienti anziani compromessi
da un punto di vista generale (40);
- è possibile impiegare l’analisi della malattia
minima residua per offrire terapie personalizzate ad ogni singolo paziente: infatti è stato dimostrato come il trattamento delle recidive molecolari con rituximab sia in grado di reindurre
remissioni molecolari (46) e dai primi risultati
sembra, di conseguenza, incidere in maniera
benefica sull’andamento clinico (43, 39).
n ATTUALI STRATEGIE
TERAPEUTICHE
Contrariamente a quanto accade per altri tipi di
malattie linfoproliferative, una strategia di watchful waiting non è raccomandata per il MCL,
considerato il decorso clinico aggressivo e la prognosi infausta nella maggior parte dei casi. Fa
eccezione un limitato gruppo di pazienti la cui
malattia, presentandosi con un andamento
meno aggressivo e una prognosi decisamente
più favorevole (in qualche maniera più simile a
quella di una leucemia linfatica cronica), è stata definita come MCL indolente (47, 23). Questi
pazienti si caratterizzano in genere per la frequente leucemizzazione accompagnata da splenomegalia in assenza di localizzazioni adenopatiche di rilievo. Proprio per evitare un trattamento troppo precoce a questi pazienti è raccomandato di monitorare attentamente nel tempo tutti i pazienti asintomatici con massa tumorale limitata e iniziare un trattamento solo al momento
di una rapida progressione o in caso di comparsa di sintomatologia correlata alla malattia (48).
D’altra parte la maggioranza dei pazienti con
diagnosi di MCL giunge all’attenzione dell’ematologo con la classica presentazione di malattia adenopatica avanzata e sintomatica che
necessita rapidamente di un trattamento citoriduttivo. A questo punto le decisioni terapeutiche devono essere prese essenzialmente sulla base dell’età, delle condizioni generali e delle comorbilità del paziente: i soggetti di età infe-
riore ai 60 anni con un ECOG performance status (non causato dalla malattia) (49) <4 oppure
soggetti fino a 65 anni con buon performance
status (ECOG <3) sono da considerare giovani
e da avviare a una terapia aggressiva, che nella maggior parte dei Centri è oggi basata sul trapianto autologo.
Gli altri pazienti rappresentano un gruppo decisamente più eterogeneo. Sicuramente nei
pazienti anziani in buone condizioni generali
l’obiettivo del trattamento è quello di ottenere il
massimo livello di citoriduzione possibile, al fine
di migliorare l’aspettativa di vita. Per contro, nei
pazienti con comorbilità importanti l’obiettivo del
trattamento è di natura palliativa. Al fine di ottimizzare il trattamento di questi pazienti è pertanto considerata di sempre maggior utilità la
valutazione geriatrica multidimensionale (CGA:
Comprehensive Geriatric Assessment) (50), che
ha l’obiettivo di identificare i soggetti in grado
di affrontare una terapia volta al controllo a medio
e lungo termine della patologia (pazienti fit) e
distinguerli invece dai pazienti in grado di tollerare solo una terapia palliativa, basata sul contenimento dei sintomi del linfoma (pazienti unfit
o fragili).
Terapia di prima linea nei pazienti giovani
Rispetto al pessimismo che contraddistingueva
l’approccio clinico al linfoma mantellare negli anni
90, la prima decade del millennio ha offerto significativi progressi.
L’uso più razionale degli schemi chemioterapici e l’introduzione del rituximab hanno infatti
aperto nuovi e interessanti scenari di trattamento che hanno portato a significativi progressi nelle possibilità di cura di questo tipo di neoplasia
(5). Al momento diversi nuovi regimi terapeutici
hanno dimostrato la loro efficacia nei confronti
del MCL e la maggior parte dei pazienti ottiene
risposte considerevoli e spesso durature, in taluni casi associate a persistenti remissioni molecolari. Nonostante ciò, occorre sottolineare
come la maggior parte dei pazienti vada tuttora incontro a recidiva e che pertanto il MCL rappresenta ancora la principale causa di morte per
i soggetti che ne sono affetti. Infatti, contrariamente ad altri tipi di linfomi, nel MCL i regimi polichemioterapici convenzionali non garantiscono
Linfoma mantellare
il controllo a lungo termine della patologia (4).
Di seguito cercheremo di sintetizzare i principali schemi terapeutici per il paziente giovane utilizzati nella quotidiana pratica clinica:
- tenendo conto dei limitati successi ottenibili con
regimi polichemioterapici contenenti antracicline (15-20% di risposte complete), classici
schemi terapeutici come CHOP o MCP (mitoxantrone, clorambucile e prednisone) non sono
considerati adeguati di per sé per il trattamento del MCL in un paziente giovane (4). L’aggiunta
del rituximab allo schema CHOP ha permesso
di ottenere risposte cliniche nella maggior parte dei pazienti, anche con un discreto numero
di risposte complete (35%) (51). Infine dati
recentemente presentati suggeriscono come l’inserimento della citarabina nei regimi di induzione (cicli alternati R-CHOP/R-DHAP) possa ulteriormente incrementare il tasso di risposte complete fino anche al 50-60% (52);
- nonostante i tassi di risposta anche elevati ottenibili in induzione con questi schemi, la durata
dei periodi di remissione sarebbe modesta
senza un’adeguata terapia di consolidamento.
Partendo dai successi ottenuti con l’impiego di
alte dosi di citarabina e dal trapianto autologo
(45, 53, 54), lo standard terapeutico attuale per
il paziente giovane è rappresentato da un regime immunochemioterapico di induzione seguito da consolidamento con chemioterapia ad alte
dosi contenente citarabina e supporto di cellule staminali emopoietiche. I dati positivi di simili regimi sono stati confermati da differenti studi multicentrici che testimoniano tassi di eventfree survival (EFS) e overall survival (OS) rispettivamente fino a 65% e 80% a quattro anni, a
fronte però di una non trascurabile mortalità legata al trattamento (fino al 5%) (43, 55, 56, 57, 52);
- in alternativa ai regimi trapiantologici sono stati messi a punto negli USA schemi di induzione
con intensificazione di dose; tali schemi, basati sull’impiego di alte dosi di citarabina, sempre
supplementati da rituximab (R-Hyper-CVAD/MA),
si sono rivelati altamente efficaci (con tassi di
risposta completa fino all’87% e tempo mediano al fallimento della terapia (TTF) di circa sei
anni), pur non essendo scevri da importanti tossicità, con un tasso di mortalità legata al trattamento intorno all’8% (58, 59). Il principale van-
taggio del regime R-Hyper-CVAD/MA è quello di
offrire un’elevata intensità di trattamento senza
imporre l’uso del trapianto autologo; tuttavia
occorre considerare che tale regime non ne risolve le problematiche principali. In effetti la mortalità connessa al trattamento è analoga se non
superiore ai programmi autotrapiantologici e il
rischio di complicanze clonali tardive rimane
comunque elevato (Tabella 2). Inoltre al momento l’uso del regime R-Hyper-CVAD/MA non è
s upportato da studi clinici di fase III e da un’ampia validazione su base multicentrica. In effetti
gli eccellenti risultati ottenuti presso l’MD
Anderson Cancer Center non sono stati pienamente confermati da uno studio multicentrico
successivo, in cui il progression-free survival
(PFS) a due anni si attestava soltanto al 60% (60);
- purtroppo, nonostante le risposte cliniche
(anche di media-lunga durata) ottenibili con le
terapie ad alte dosi descritte, praticamente tutti i pazienti con MCL alla fine recidivano e muoiono a causa della loro malattia. Per questo motivo vi è ancora la necessità di migliorare la durata della remissione e prolungare il PFS attraverso una terapia continuativa di mantenimento della risposta ottenuta grazie agli schemi ad alte
dosi. Al momento attuale non ci sono dati convincenti che dimostrino l’effettivo beneficio di una
terapia di mantenimento post-trapianto, anche
se interessanti risultati potranno pervenire dagli
studi in corso sull’impiego in questo ambito di
farmaci non chemioterapici: in particolare all’interno dello European Mantle Cell Lymphoma
Network si sta indagando in due studi di fase III
l’efficacia del mantenimento post-trapianto con
rituximab (GOELAMS LyMa - Clin Trials Gov
Number NCT00921414) e lenalidomide (studio
della Fondazione Italiana Linfomi attualmente in
corso IIL-MCL0208: EudraCT Number 2009012807-25).
Pertanto, allo stato attuale, al di fuori di un trial
clinico lo standard terapeutico per i pazienti giovani privi di comorbilità significative è un trattamento aggressivo, basato su regimi mieloablativi con supporto di cellule staminali emopoietiche (trapianto autologo) oppure, sia pur con una
evidenza meno ampia, su regimi di induzione
altamente intensificati (come lo schema HyperCVAD/MA), entrambi supplementati con anticor-
35
36
Seminari di Ematologia Oncologica
po monoclonale anti-CD20 (rituximab). Infatti,
come si evince dalla Tabella 2, i trattamenti sinora dimostratisi più efficaci sono caratterizzati da
un’elevata intensità di dose di citarabina.
L’importanza di questo farmaco nel trattamento del MCL è stata ulteriormente dimostrata dai
risultati dell’attuale trial dello European Mantle
Cell Lymphoma Network (Clin Trials Gov Number
NCT00209222) presentati al recente congresso
dell’American Society of Hematology (52).
Terapia di prima linea nei pazienti anziani
I pazienti che non sono candidabili per età o
comorbilità a regimi terapeutici aggressivi possono comunque essere trattati con successo con
diversi tipi di chemioterapia convenzionale supplementata con rituximab, anche se la durata
delle remissioni così ottenute è purtroppo piuttosto modesta (51). Quale che sia lo schema
terapeutico utilizzato, per migliorare la prognosi di questi pazienti rimane cruciale il supporto
di un regime di consolidamento che faccia seguito alla prima linea di terapia. I migliori strumenti terapeutici al momento disponibili per l’ematologo sono i seguenti:
- regimi di induzione: al momento non esiste uno
standard terapeutico per il paziente anziano,
soprattutto perché i dati di efficacia disponibili
provengono per lo più da studi effettuati in
pazienti recidivati o refrattari oppure sono estrapolati dai risultati dei regimi di induzione pre-alte
dosi (61, 62, 51, 63). In ogni caso gli schemi più
ampiamente utilizzati nella pratica clinica sono
a base di fludarabina (FC/FCM), antracicline
(CHOP) o citarabina (DHAP), sempre supplementati dal rituximab. Nell’ottica di limitare al massimo le tossicità ricercando comunque una risposta clinica il più possibile duratura, sono stati utilizzati anche schemi più blandi come CVP o rituximab in monoterapia (64-66). Interessanti risultati in questo senso arriveranno dallo studio prospettico randomizzato dello European Mantle
Cell Lymphoma Network che per la prima volta
paragona l’efficacia di R-CHOP vs R-FC nel
paziente anziano (Clin Trials Gov Number
NCT00209209). D’altra parte, considerando i dati
di efficacia della citarabina provenienti dagli studi sui pazienti giovani, sarebbero da considerare anche per pazienti anziani schemi terapeuti-
ci basati su questo farmaco, magari con riduzioni di dosaggio;
- recentemente la sorprendente attività e la limitata tossicità di uno schema di induzione contenente bendamustina e rituximab sono state
messe in luce dallo studio prospettico randomizzato del gruppo tedesco StiL (67), pertanto
anche questo regime è oggi da considerare fra
le terapie di prima linea del paziente anziano. È
prevedibile nei prossimi anni un incremento nell’utilizzo di questo farmaco, anche in schemi integrati con nuove molecole (come descritto in una
sezione successiva);
- la discussione su quale sia il migliore regime
di consolidamento da offrire ai pazienti anziani
con MCL è tuttora aperta: le evidenze di un possibile ruolo dell’IFN-α nell’allungare la PFS sono
poco convincenti (24, 68) e in particolare in Italia
si registra meno entusiasmo per questo farmaco rispetto ad altri Paesi, anche a causa dei suoi
effetti collaterali e della sua scarsa tollerabilità.
Diverso è il discorso per il mantenimento con
rituximab: se i primi dati in pazienti recidivati
sono piuttosto favorevoli (69), i risultati del primo studio prospettico randomizzato dello
European Mantle Cell Lymphoma Network (Clin
Trials Gov Number NCT00209209) che paragona il mantenimento con rituximab a quello con
IFN-α in prima linea nell’anziano saranno presto resi disponibili;
- la radioimmunoterapia (RIT) con Y90 Ibritumomab
tiuxetan (Zevalin®) potrebbe rappresentare un valido strumento terapeutico alternativo: infatti i risultati preliminari di uno studio sul consolidamento
con RIT dopo R-CHOP mostrano un netto
miglioramento dei tassi di risposte complete e un
prolungamento della PFS (70), ma al momento
dati più solidi a sostegno di questo tipo di trattamento non sono ancora disponibili.
Pertanto allo stato attuale al di fuori di un trial
clinico lo standard terapeutico per i pazienti non
candidabili per età o comorbilità a regimi terapeutici aggressivi dovrebbe essere basato o su
un regime R-CHOP-like oppure su un regime
contenente fludarabina e rituximab (R-FM o RFC).
Un discorso diverso deve essere fatto invece per
quanto riguarda i soggetti anziani con molteplici comorbilità, giudicati unfit o fragili da una valu-
Linfoma mantellare
tazione geriatrica multidimensionale. Per questo
sottogruppo di pazienti le armi a disposizione
sono al momento davvero poche e il trattamento deve essere inteso in senso meramente palliativo e di controllo a breve-medio termine dei
sintomi del linfoma. In questo senso sono state proposte terapie per via orale a limitata tossicità con clorambucile, monoterapie con rituximab oppure, tenendo conto dei recenti dati
tedeschi, schemi a base di bendamustina a dosi
ridotte (71).
Infine alcuni di questi pazienti potrebbero giovarsi in prima linea delle nuove molecole non
chemioterapiche, valutate al momento attuale da
diversi protocolli clinici in fasi più avanzate di
malattia (e descritte più dettagliatamente in una
sezione successiva).
Terapia di salvataggio
Il MCL è una malattia non eradicabile, pertanto
il verificarsi della recidiva è purtroppo la regola.
La criticità della recidiva è evidenziata dal difficile approccio terapeutico a questo momento
inevitabile della storia naturale della patologia,
caratterizzato da un’aumentata chemioresistenza e dall’assenza di validi strumenti farmacologici in grado di controllare a lungo termine la proliferazione neoplastica. Inoltre un’ulteriore problematica può essere rappresentata dalle condizioni generali di questi pazienti al momento della recidiva, tenendo presente che l’età, le
comorbilità e le sequele delle tossicità determinate dalla prima linea di trattamento (generalmente aggressiva) possono limitare di molto le
scelte terapeutiche successive.
Non esiste al momento attuale una terapia standard di salvataggio per il MCL in recidiva o refrattario: molti regimi terapeutici inducono ottime risposte nei confronti del tumore ma quasi nessuno è
in grado di eliminare le successive ricadute di
malattia, responsabili nella maggioranza dei casi
della morte del paziente. Appare importante sottolineare come per questi pazienti l’arruolamento
in studi clinici di ricerca possa rappresentare una
scelta estremamente appropriata, poiché consente di metter loro a disposizione gratuitamente e
precocemente farmaci efficaci, costosi e in molti
casi non ancora prescrivibili.
Cercheremo di seguito di sintetizzare il difficile
approccio terapeutico a questa fase critica della patologia, mettendo in evidenza i punti sui
quali vi è maggiore accordo tra i clinici:
- il trapianto allogenico di cellule staminali, grazie all’effetto graft-versus-lymphoma rimane
l’unica opzione in grado di controllare a lungo
termine il MCL recidivato. Pertanto tale approccio va considerato per ogni paziente giovane che
recidivi in seguito a una terapia di prima linea
appropriata (71).
Grazie all’impiego dei regimi di condizionamento a intensità ridotta e alla selezione di pazienti chemioresponsivi, sono stati ottenuti, nel contesto di esperienze unicentriche, tassi di risposta completa molto elevati (superiori al 90%) e
una PFS che sfiora il 50% a 6 anni, a prezzo di
una mortalità legata alla procedura di circa il 10%
e di un’incidenza di graft-versus-host-disease
cronica in circa il 60% dei pazienti (72, 73).
Rimane però da considerare come il trapianto
allogenico di cellule staminali possa essere applicato solo ad una minoranza di soggetti affetti da
MCL, in dipendenza dall’età del paziente, dalla
disponibilità di un donatore e dalla presenza di
una malattia non chemiorefrattaria;
- i regimi di reinduzione sono vari e molto efficaci, con tassi di risposta che raggiungono il 6080% (con un 30-50% di risposte complete): gli
schemi più utilizzati comprendono il rituximab e
si basano su fludarabina (R-FC/R-FCM), bendamustina (R-B), citarabina-platino (R-DHAP) e
gemcitabina-platino (R-GEMOX) (62, 74, 75);
- nei pazienti non allotrapiantati, risulta essenziale offrire un consolidamento o un mantenimento,
a causa dell’elevato rischio di successiva recidiva a breve termine. A questo scopo il mantenimento con rituximab ha dimostrato la propria efficacia (69), mentre sono meno convincenti i dati
sulla radioimmunoterapia (RIT) (76). È ormai evidente invece come un regime autotrapiantologico in seconda remissione non offra garanzie adeguate che ne controbilancino il costo in termini
di tossicità (73). Numerosi nuovi farmaci, poco tossici e di promettente attività, potranno in futuro
entrare a far parte di efficaci programmi di mantenimento della risposta, ma al momento sono
ancora in fase di sperimentazione (e saranno
descritti in dettaglio più avanti);
- per le recidive successive alla prima si può
37
38
Seminari di Ematologia Oncologica
Regime di
trattamento
Numero di
pazienti
CR
Follow-up
mediano
(anni)
PFS
OS
Quantità di
citarabina
somministrata
(mg/mq)
TRM
Incidenza
di secondi
tumori
R-Hyper
CVAD/R-MAa
65
89%
8
52%
a 10 anni
64%
a 10 anni
36000-48000
8%
5%
R-HDSb
27
100%
10
46%
a 10 anni
72%
a 10 anni
24000
4%
3,7%
NLGc
160
90%
3,9
66%
a 6 anni
70%
a 6 anni
8000-12000
5%
1,3%
EuMCLNet
(exp.arm B)d
195
65%
2,3
65%
a 3 anni
80%
a 3 anni
18000
4%
nd
CR = tasso di risposte complete; PFS = progression-free survival; OS = overall survival; TRM = tasso di mortalità legata al trattamento; R-HDS = terapia
sequenziale ad alte dosi supplementata da rituximab; NLG = Nordic Lymphoma Group; EuMCLNet = European Mantle Cell Lymphoma Network; nd = non
disponibile. aRomaguera et al., BJH 2010; bMagni et al., BMT 2009; cGeisler et al., Blood 2008; dHermine et al., Blood abstr 2010.
TABELLA 2 - Confronto efficacia/tossicità fra i più comuni regimi ad alte dosi per pazienti con MCL <65 anni.
semplicemente considerare la ripetizione di una
terapia precedente, qualora questa abbia garantito un periodo di remissione prolungato (71). In
alternativa dovrebbe essere considerato un
approccio molecolare all’interno di uno studio clinico controllato, qualora disponibile.
Al momento attuale i dati più maturi che sono
stati pubblicati sui nuovi farmaci riguardano in
particolare il bortezomib, la lenalidomide e il temsirolimus e verranno descritti più dettagliatamente nella sezione successiva.
n NUOVI APPROCCI TERAPEUTICI E
FUTURE POSSIBILITÀ DI SVILUPPO
Nonostante i successi terapeutici che negli ultimi anni hanno migliorato di molto la prognosi dei
pazienti affetti da MCL, questa malattia è considerata ancora oggi non eradicabile e il problema di come evitare/posporre la recidiva e di
come trattarla una volta che si sia verificata rimane ancora del tutto aperto.
Le conoscenze che si vanno accumulando sulla
biologia e sulla patogenesi hanno consentito
l’ideazione e lo sviluppo di nuovi approcci terapeutici mirati a differenti bersagli molecolari, i più
importanti dei quali sono semplicisticamente rappresentati nella figura 2. Questi nuovi farmaci non
chemioterapici, grazie alla loro efficacia sulla
malattia e ai profili di tossicità relativamente favorevoli, sono stati sperimentati sia nell’intento di
incrementare il tasso di risposta alle terapie di
induzione, sia all’interno di regimi di mantenimento della risposta, sia per trattare recidive di malattia non più responsive alle terapie convenzionali. Abbiamo cercato di schematizzare nella parte
seguente e nella Tabella 3 le principali nuove
opzioni terapeutiche già sperimentate in trial clinici controllati e attualmente in corso di studio su
serie numerose di pazienti; infine offriremo un rapido excursus sulle più promettenti molecole sintetizzate di recente in grado di rivestire in futuro
un ruolo clinico significativo (77).
1) Bortezomib (Velcade ®). È un potente inibitore selettivo e reversibile del proteasoma 26S che
ha mostrato fin dalle prime applicazioni in
monoterapia risultati incoraggianti nei confronti del MCL recidivato o refrattario (78); tali risposte obiettive sono verosimilmente legate all’inibizione della via di segnalazione del fattore
nucleare NFκB, costitutivamente attivata in
questa neoplasia (79). La limitata tossicità di questa molecola (essenzialmente moderata trombocitopenia, neuropatia e diarrea) e le evidenze in
vitro di sinergia farmacodinamica con agenti chemioterapici (80) ne hanno favorito la sperimentazione in associazione alla citarabina e al rituximab (R-DHAB) in seconda linea di terapia, con
tassi di risposta obiettiva nella metà dei pazien-
Linfoma mantellare
ti (81). Il bortezomib dunque sembra pronto per
essere lanciato in prima linea nel MCL in associazione con la chemioterapia, come suggerito
dai brillanti risultati di uno studio appena pubblicato (tassi di risposta completa fino al 70%
indotti dall’associazione di bortezomib con chemioterapia R-CHOP) (82).
2) Talidomide e Lenalidomide (Revlimid ®). La talidomide, farmaco dalle note attività sull’angiogenesi e sul microambiente midollare, è attiva contro il MCL se utilizzata in combinazione col rituximab (83). In maniera ancora più marcata la
lenalidomide, molecola immunomodulatoria di
seconda generazione, determina in monoterapia tassi di risposta fino al 50% in casi di MCL
plurirecidivato (84, 85). Basandosi su questi dati,
sulla limitata tossicità (neutropenia e trombocitopenia moderate, lieve anemia) e sulla maneggevolezza della formulazione per os, la lenalidomide potrebbe rivelarsi un ottimo farmaco per
il mantenimento post-trapianto autologo, come
indagato dallo studio in corso della Fondazione
Italiana Linfomi IIL-MCL0208 (EudraCT Number
2009-012807-25).
3) Temsirolimus (Torisel ®). Il suo meccanismo di
azione è piuttosto complesso: sinteticamente
questa molecola blocca la traduzione dell’mRNA
della ciclina D1 attraverso l’inibizione del media-
Farmaco
tore mTOR (bersaglio della rapamicina nei
mammiferi). Gli studi su pazienti plurirecidivati
riportano risultati interessanti, paragonabili in termini di efficacia agli inibitori del proteasoma, con
tossicità prevalentemente di tipo ematologico
(86, 87). Per questo motivo in Germania è stato attivato uno studio di fase II sull’utilizzo di temsirolimus in combinazione con bendamustina e
rituximab (BeRT - Clin Trials Gov Number
NCT01078142) per MCL o linfoma follicolare in
recidiva. Infine un composto simile ma in formulazione orale (everolimus/ rad001) ha dato risultati incoraggianti in diversi tipi di linfomi ed è in
corso di sperimentazione anche nel linfoma mantellare (88, 89).
4) Nuovi farmaci in corso di sviluppo (Figura 2 e
Tabella 3):
a) il flavopiridolo inibisce in maniera diretta le
CDK (chinasi ciclina-dipendente) 4 e 6,
determinando una deregolazione della ciclina D1: in uno studio di associazione con fludarabina e rituximab ha fornito interessanti
risultati su un ristretto gruppo di pazienti (90);
b) il CAL-101, un inibitore del PI3Kd (fosfatidilinositolo-3-chinasi d) che blocca il signaling
B-cellulare, ha dimostrato un’impressionante attività soprattutto nel linfoma mantellare
(91, 92);
Meccanismo di azione
Referenza
Fase di studio
Numero di pazienti
ORR
CR
Bortezomib
Inibitore proteasoma
(78)
II
141
33%
8%
Talidomide*
IMiD
(83)
II
16
81%
31%
Lenalidomide
IMiD
(85)
II
39
41%
13%
Temsirolimus
Inibitore mTOR
(87)
III
54
22%
2%
Everolimus
Inibitore mTOR
(88)
II
19
36%
11%
Inibitore CDK4 e 6
(90)
I
10
80%
70%
Inibitore PI3Kd
(92)
I
8
75%
nd
Inibitore Btk
(94)
I
4
50%
0%
Mab anti CD20
(95)
II
15
27%
13%
Mab anti
CD3/CD19
(97)
I
7
43%
14%
Flavopiridolo**
Cal-101
PCI-32765
GA-101
Blinatumomab
ORR = tasso di risposte globali; CR = tasso di risposte complete; IMiD = farmaco immunomodulatore; mAb = anticorpo monoclonale; nd = non disponibile.
*in associazione a rituximab; **In associazione a fludarabina e rituximab.
TABELLA 3 - Nuovi farmaci a bersaglio molecolare attualmente in corso di sperimentazione nel MCL.
39
40
Seminari di Ematologia Oncologica
c) gli inibitori della cascata di trasduzione del
segnale del recettore delle cellule B (BCR),
come l’enzastaurina (inibitore della PKCβ: proteina chinasi Cβ) (93) e il PCI-32765 (inibitore della Btk: tirosina chinasi di Bruton) (94);
d) tra i nuovi anticorpi monoclonali sono interessanti i primi dati relativi al GA101, un anticorpo anti-CD20 umanizzato di seconda
generazione (95) e dal blinatumomab, un anticorpo monoclonale bispecifico antiCD19/anti-CD3, che agisce reclutando linfociti T citotossici sulla superficie delle cellule
B di linfoma (BiTE: Bi-specific T-cell-engaging)
(96, 97).
n CONCLUSIONI
La passata decade ha registrato considerevoli
successi nel campo del MCL, legati ad una forte integrazione tra biologia, ricerca traslazionale e ricerca clinica.
È stato possibile superare almeno in parte le problematiche relative alla rarità di questo tipo istologico, grazie allo sviluppo di fruttuose sinergie
internazionali tra cui la più significativa è rappresentata dallo European Mantle Cell Lymphoma
Network. I principali elementi di progresso possono essere così riassunti:
a) sono stati chiariti alcuni aspetti essenziali della patogenesi molecolare di questa patologia
(98);
b) la miglior conoscenza biologica ha consentito di identificare alcune nuove molecole
potenzialmente sfruttabili a livello clinico che
sono attualmente oggetto di attiva sperimentazione e alcune delle quali sono già impiegate nella pratica clinica (77);
c) sono stati sviluppati degli efficaci marcatori
prognostici, in particolare lo score clinico MIPI
e la valutazione della malattia minima residua
in PCR, attualmente utilizzati nell’ambito di
svariati studi clinici (34, 39, 40);
d) sono state definite le strategie terapeutiche
ottimali per la terapia di prima linea in particolare nel paziente giovane (71).
Grazie a questi importanti successi la prognosi
del MCL è decisamente migliorata negli ultimi
anni (5). È importante notare che tale spinta pro-
pulsiva non sembra essersi affatto esaurita: gli
studi attualmente in corso vedranno una sempre maggiore integrazione dei nuovi farmaci biologici nel contesto dei programmi terapeutici
disponibili e un sempre maggiore ricorso ad
approcci risk-adapted. È quindi lecito attendersi un ulteriore significativo miglioramento della
sopravvivenza globale sia nei soggetti giovani sia
in quelli più anziani. Il più importante dei fattori
capaci di condizionare la velocità di questo processo in una patologia comunque non comune
sarà la volontà degli onco-ematologi di inserire
i pazienti con MCL all’interno di studi clinici controllati soprattutto di tipo multicentrico.
Ringraziamenti:
Gli autori ringraziano Antonella Fiorillo e la
Dott.ssa Odetta Antico per il supporto segretariale e iconografico.
n BIBLIOGRAFIA
1. O’Connor OA. Mantle cell lymphoma: identifying novel molecular targets in growth and survival pathways.
Hematology Am Soc Hematol Educ Program. 2007;
270-6.
2. Tumori in Italia - Rapporto 2006: dati di incidenza e
mortalità dei Registri Tumori generali, 1998-2002
(http://www.registri-tumori.it/incidenza19982002/gruppi.html).
3. Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK,
Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the
International Lymphoma Study Group. Blood. 1994;
84: 1361-92.
4. Nickenig C, Dreyling M, Hoster E, Pfreundschuh M,
Trumper L, Reiser M, et al. Combined cyclophosphamide, vincristine, doxorubicin, and prednisone
(CHOP) improves response rates but not survival and
has lower hematologic toxicity compared with combined mitoxantrone, chlorambucil, and prednisone
(MCP) in follicular and mantle cell lymphomas:
results of a prospective randomized trial of the
German Low-Grade Lymphoma Study Group. Cancer.
2006; 107: 1014-22.
5. Herrmann A, Hoster E, Zwingers T, Brittinger G,
Engelhard M, Meusers P, et al. Improvement of overall survival in advanced stage mantle cell lymphoma.
J Clin Oncol. 2009; 27: 511-8.
6. Hummel M, Tamaru J, Kalvelage B, Stein H. Mantle
cell (previously centrocytic) lymphomas express VH
genes with no or very little somatic mutations like the
Linfoma mantellare
physiologic cells of the follicle mantle. Blood. 1994;
84: 403-7.
7. Fisher RI, Dahlberg S, Nathwani BN, Banks PM, Miller
TP, Grogan TM. A clinical analysis of two indolent lymphoma entities: mantle cell lymphoma and marginal
zone lymphoma (including the mucosa-associated
lymphoid tissue and monocytoid B-cell subcategories): a Southwest Oncology Group study. Blood.
1995; 85: 1075-82.
8. Palumbo GA, Parrinello N, Fargione G, Cardillo K,
Chiarenza A, Berretta S, et al. CD200 expression may
help in differential diagnosis between mantle cell lymphoma and B-cell chronic lymphocytic leukemia. Leuk
Res. 2009; 33: 1212-6.
9. Orchard J, Garand R, Davis Z, Babbage G, Sahota
S, Matutes E, et al. A subset of t(11;14) lymphoma
with mantle cell features displays mutated IgVH genes
and includes patients with good prognosis, nonnodal
disease. Blood. 2003; 101: 4975-81.
10. Kienle D, Kröber A, Katzenberger T, Ott G, Leupolt
E, Barth TF, et al. VH mutation status and VDJ
rearrangement structure in mantle cell lymphoma: correlation with genomic aberrations, clinical characteristics, and outcome. Blood. 2003; 102: 3003-9.
11. Thelander EF, Rosenquist R. Molecular genetic characterization reveals new subsets of mantle cell lymphoma. Leuk Lymphoma. 2008; 49: 1042-9.
12. Hadzidimitriou A, Agathagelidis A, Murray F, DelfauLarue MH, Darzentas N, Navarro Lopez A, et al.
Sequence-Based evidence for antigen selection in
mantle cell lymphoma: remarkable immunoglobulin
gene repertoire biases, stereotyped antigen-binding
sites and recurrent hypermutations in certain subsets
(Abstract). Blood. 2009; 114: 1933.
13. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer.
2007; 7: 750-62.
14. Rosenwald A, Wright G, Wiestner A, Chan WC,
Connors JM, Campo E, et al. The proliferation gene
expression signature is a quantitative integrator of
oncogenic events that predicts survival in mantle cell
lymphoma. Cancer Cell. 2003; 3: 185-97.
15. Fu K, Weisenburger DD, Greiner TC, Dave S, Wright
G, Rosenwald A, et al. Cyclin D1-negative mantle cell
lymphoma: a clinicopathologic study based on gene
expression profiling. Blood. 2005; 106: 4315-21.
16. Pinyol M, Hernandez L, Cazorla M, Balbín M, Jares
P, Fernandez PL, et al. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas.
Blood. 1997; 89: 272-80.
17. Hernández L, Beà S, Pinyol M, Ott G, Katzenberger
T, Rosenwald A, et al. CDK4 and MDM2 gene alterations mainly occur in highly proliferative and aggressive mantle cell lymphomas with wild-type INK4a/ARF
locus. Cancer Res. 2005; 65: 2199-206.
18. Stilgenbauer S, Winkler D, Ott G, Schaffner C, Leupolt
E, Bentz M, et al. Molecular characterization of 11q
deletions points to a pathogenic role of the ATM gene
in mantle cell lymphoma. Blood. 1999; 94: 3262-4.
19. Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni
M, Pavan A, et al. Distinct functional significance of
Akt and mTOR constitutive activation in mantle cell
lymphoma. Blood. 2008; 111: 5142-51.
20. Rizzatti EG, Falcão RP, Panepucci RA, Proto-Siqueira
R, Anselmo-Lima WT, Okamoto OK, et al. Gene
expression profiling of mantle cell lymphoma cells
reveals aberrant expression of genes from the PI3KAKT, WNT and TGFbeta signalling pathways. Br J
Haematol. 2005; 130: 516-26.
21. Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo
GR, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;
13: 944-51.
22. Fu L, Lin-Lee YC, Pham LV, Tamayo A, Yoshimura L,
Ford RJ. Constitutive NF-kappaB and NFAT activation leads to stimulation of the BLyS survival pathway in aggressive B-cell lymphomas. Blood. 2006;
107: 4540-8.
23. Fernàndez V, Salamero O, Espinet B, Solé F, Royo C,
Navarro A, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma.
Cancer Res. 2010; 70: 1408-18.
24. Velders GA, Kluin-Nelemans JC, De Boer CJ,
Hermans J, Noordijk EM, Schuuring E, et al. Mantlecell lymphoma: a population-based clinical study. J
Clin Oncol. 1996; 14: 1269-74.
25. Argatoff LH, Connors JM, Klasa RJ, Horsman DE,
Gascoyne RD. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood. 1997; 89: 2067-78.
26. Schrader C, Meusers P, Brittinger G, Teymoortash A,
Siebmann JU, Janssen D, et al. Topoisomerase IIalpha
expression in mantle cell lymphoma: a marker of cell
proliferation and a prognostic factor for clinical outcome. Leukemia. 2004; 18: 1200-6.
27. Bosch F, López-Guillermo A, Campo E, Ribera JM,
Conde E, Piris MA, et al. Mantle cell lymphoma: presenting features, response to therapy, and prognostic factors. Cancer. 1998; 82: 567-75.
28. Samaha H, Dumontet C, Ketterer N, Moullet I,
Thieblemont C, Bouafia F, et al. Mantle cell lymphoma:
a retrospective study of 121 cases. Leukemia. 1998;
12: 1281-7.
29. Tiemann M, Schrader C, Klapper W, Dreyling MH,
Campo E, Norton A, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients
with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J
Haematol. 2005; 131: 29-38.
30. No authors listed. A predictive model for aggressive
non-Hodgkin’s lymphoma. The International NonHodgkin’s Lymphoma Prognostic Factors Project. N
Engl J Med. 1993; 329: 987-94.
41
42
Seminari di Ematologia Oncologica
31. Solal-Céligny P, Roy P, Colombat P, White J,
Armitage JO, Arranz-Saez R, et al. Follicular lymphoma international prognostic index. Blood. 2004;
104: 1258-65.
32. Federico M, Bellei M, Marcheselli L, Luminari S,
Lopez-Guillermo A, Vitolo U, et al. Follicular lymphoma
international prognostic index 2: a new prognostic
index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009; 27: 4555-62.
33. Møller MB, Pedersen NT, Christensen BE. Mantle cell
lymphoma: prognostic capacity of the Follicular
Lymphoma International Prognostic Index. Br J
Haematol. 2006; 133: 43-9.
34. Hoster E, Dreyling M, Klapper W, Gisselbrecht C, van
Hoof A, Kluin-Nelemans HC, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008; 111: 558-65.
35. Weisenburger DD, Vose JM, Greiner TC, Lynch JC,
Chan WC, Bierman PJ, et al. Mantle cell lymphoma.
A clinicopathologic study of 68 cases from the
Nebraska Lymphoma Study Group. Am J Hematol.
2000; 64: 190-6.
36. Räty R, Franssila K, Joensuu H, Teerenhovi L,
Elonen E. Ki-67 expression level, histological subtype,
and the International Prognostic Index as outcome
predictors in mantle cell lymphoma. Eur J Haematol.
2002; 69: 11-20.
37. Corradini P, Ladetto M, Zallio F, Astolfi M, Rizzo E,
Sametti S, et al. Long-term follow-up of indolent lymphoma patients treated with high-dose sequential
chemotherapy and autografting: evidence that durable
molecular and clinical remission frequently can be
attained only in follicular subtypes. J Clin Oncol. 2004;
22: 1460-8.
38. Pott C, Schrader C, Gesk S, Harder L, Tiemann M,
Raff T, et al. Quantitative assessment of molecular
remission after high-dose therapy with autologous
stem cell transplantation predicts long-term remission
in mantle cell lymphoma. Blood. 2006; 107: 2271-8.
39. Andersen NS, Pedersen LB, Laurell A, Elonen E,
Kolstad A, Boesen AM, et al. Pre-emptive treatment
with rituximab of molecular relapse after autologous
stem cell transplantation in mantle cell lymphoma. J
Clin Oncol. 2009; 27: 4365-70.
40. Pott C, Hoster E, Delfau-Larue MH, Beldjord K,
Böttcher S, Asnafi V, et al. Molecular remission is an
independent predictor of clinical outcome in patients
with mantle cell lymphoma after combined
immunochemotherapy: a European MCL intergroup
study. Blood. 2010; 115: 3215-23.
41. Magni M, Di Nicola M, Devizzi L, Matteucci P,
Lombardi F, Gandola L, et al. Successful in vivo purging of CD34-containing peripheral blood harvests in
mantle cell and indolent lymphoma: evidence for a
role of both chemotherapy and rituximab infusion.
Blood. 2000; 96: 864-9.
42. Brugger W, Hirsch J, Grünebach F, Repp R, Brossart
P, Vogel W, et al. Rituximab consolidation after highdose chemotherapy and autologous blood stem cell
transplantation in follicular and mantle cell lymphoma:
a prospective, multicenter phase II study. Ann Oncol.
2004; 15: 1691-8.
43. Geisler CH, Kolstad A, Laurell A, Andersen NS,
Pedersen LB, Jerkeman M, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivopurged stem cell rescue: a nonrandomized phase 2
multicenter study by the Nordic Lymphoma Group.
Blood. 2008; 112: 2687-93.
44. Andersen NS, Donovan JW, Borus JS, Poor CM,
Neuberg D, Aster JC, et al. Failure of immunologic
purging in mantle cell lymphoma assessed by polymerase chain reaction detection of minimal residual
disease. Blood. 1997; 90: 4212-21.
45. Gianni AM, Magni M, Martelli M, Di Nicola M, CarloStella C, Pilotti S, et al. Long-term remission in mantle cell lymphoma following high-dose sequential
chemotherapy and in vivo rituximab-purged stem cell
autografting (R-HDS regimen). Blood. 2003; 102:
749-55.
46. Ladetto M, Magni M, Pagliano G, De Marco F, Drandi
D, Ricca I, et al. Rituximab induces effective clearance of minimal residual disease in molecular relapses of mantle cell lymphoma. Biol Blood Marrow
Transplant. 2006; 12: 1270-6.
47. Martin P, Chadburn A, Christos P, Furman R, Ruan
J, Joyce MA, et al. Intensive treatment strategies may
not provide superior outcomes in mantle cell lymphoma: overall survival exceeding 7 years with standard therapies. Ann Oncol. 2008; 19: 1327-30.
48. Martin P, Chadburn A, Christos P, Weil K, Furman
RR, Ruan J, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;
27: 1209-13.
49. Oken MM, Creech RH, Tormey DC, Horton J, Davis
TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am
J Clin Oncol. 1982; 5: 649-55.
50. Balducci L, Yates J. General guidelines for the management of older patients with cancer. Oncology.
2000; 14: 221-7.
51. Lenz G, Dreyling M, Hoster E, Wörmann B, Dührsen
U, Metzner B, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves
response and time to treatment failure, but not longterm outcome in patients with previously untreated mantle cell lymphoma: results of a prospective
randomized trial of the German Low Grade
Lymphoma Study Group (GLSG). J Clin Oncol.
2005; 23: 1984-92.
52. Hermine O, Hoster E, Walewski J, Ribrag V, Brousse
N, Thieblemont C, et al. Alternating courses of 3x
Linfoma mantellare
CHOP and 3x DHAP plus Rituximab followed by a
high dose ARA-C containing myeloablative regimen
and autologous stem cell transplantation (ASCT) is
superior to 6 courses CHOP plus Rituximab followed
by myeloablative radiochemotherapy and ASCT in
mantle cell lymphoma: results of the MCL younger
trial of the European Mantle Cell Lymphoma Network
(MCL net) (Abstract). Blood. 2010; 116: 110.
53. Lefrère F, Delmer A, Levy V, Delarue R, Varet B, Hermine
O. Sequential chemotherapy regimens followed by
high-dose therapy with stem cell transplantation in
mantle cell lymphoma: an update of a prospective
study. Haematologica. 2004; 89: 1275-6.
54. Dreyling M, Lenz G, Hoster E, Van Hoof A,
Gisselbrecht C, Schmits R, et al. Early consolidation
by myeloablative radiochemotherapy followed by
autologous stem cell transplantation in first remission
significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood. 2005;
105: 2677-84.
55. Dreyling MH, Hoster E, Van Hoof A, Metzner B,
Gisselbrecht C, Pfreundschuh M, et al. Early consolidation with myeloablative radiochemotherapy followed by autologous stem cell transplantation in first
remission in mantle cell lymphoma: long term follow
up of a randomized trial of the (Abstract). Blood. 2008;
112: 769.
56. Delarue R, Haioun C, Ribrag V, Brice P, Delmer A, Tilly
H, et al. RCHOP and RDHAP followed by autologous
stem cell transplantation (ASCT) in mantle cell lymphoma (MCL): final results of a phase II study from
the GELA (Abstract). Blood. 2008; 112: 581.
57. Magni M, Di Nicola M, Carlo-Stella C, Matteucci P,
Devizzi L, Tarella C, et al. High-dose sequential
chemotherapy and in vivo rituximab-purged stem cell
autografting in mantle cell lymphoma: a 10-year
update of the R-HDS regimen. Bone Marrow
Transplant. 2009; 43: 509-11.
58. Romaguera JE, Fayad L, Rodriguez MA, Broglio KR,
Hagemeister FB, Pro B, et al. High rate of durable
remissions after treatment of newly diagnosed
aggressive mantle-cell lymphoma with rituximab
plus hyper-CVAD alternating with rituximab plus highdose methotrexate and cytarabine. J Clin Oncol. 2005;
23: 7013-23.
59. Romaguera JE, Fayad LE, McLaughlin P, Pro B,
Rodriguez A, Wang M, et al. Phase I trial of bortezomib in combination with rituximab-HyperCVAD
alternating with rituximab, methotrexate and cytarabine for untreated aggressive mantle cell lymphoma.
Br J Haematol. 2010; 151: 47-53.
60. Epner EM, Unger J, Miller T, Rimzsa L, Spier C,
Leblanc M, et al. A multi center trial of hyperCVAD +
Rituxan in patients with newly diagnosed mantle cell
lymphoma (Abstract). Blood. 2007; 110: 387.
61. Lefrère F, Delmer A, Suzan F, Levy V, Belanger C,
Djabarri M, et al. Sequential chemotherapy by CHOP
and DHAP regimens followed by high-dose therapy
with stem cell transplantation induces a high rate of
complete response and improves event-free survival
in mantle cell lymphoma: a prospective study.
Leukemia. 2002; 16: 587-93.
62. Forstpointner R, Dreyling M, Repp R, Hermann S,
Hänel A, Metzner B, et al. The addition of rituximab
to a combination of fludarabine, cyclophosphamide,
mitoxantrone (FCM) significantly increases the
response rate and prolongs survival as compared with
FCM alone in patients with relapsed and refractory
follicular and mantle cell lymphomas: results of a
prospective randomized study of the German LowGrade Lymphoma Study Group. Blood. 2004; 104:
3064-71.
63. de Guibert S, Jaccard A, Bernard M, Turlure P,
Bordessoule D, Lamy T. Rituximab and DHAP followed by intensive therapy with autologous stem-cell
transplantation as first-line therapy for mantle cell lymphoma. Haematologica. 2006; 91: 425-6.
64. Meusers P, Engelhard M, Bartels H, Binder T, Fülle
HH, Görg K, et al. Multicentre randomized therapeutic trial for advanced centrocytic lymphoma: anthracycline does not improve the prognosis. Hematol
Oncol. 1989; 7: 365-80.
65. Zucca E, Roggero E, Pinotti G, Pedrinis E, Cappella
C, Venco A, et al. Patterns of survival in mantle cell
lymphoma. Ann Oncol. 1995; 6: 257-62.
66. Ghielmini M, Schmitz SF, Cogliatti S, Bertoni F, Waltzer
U, Fey MF, et al. Effect of single-agent rituximab given at the standard schedule or as prolonged treatment in patients with mantle cell lymphoma: a study
of the Swiss Group for Clinical Cancer Research
(SAKK). J Clin Oncol. 2005; 23: 705-11.
67. Rummel MJ, Niederle N, Maschmeyer G, Banat A, von
Gruenhagen U, Losem C, et al. Bendamustine plus
Rituximab is superior in respect of progression free
survival and CR rate when compared to CHOP plus
Rituximab as first-line treatment of patients with
advanced follicular, indolent, and mantle cell lymphomas: final results of a randomized phase III study
of the StiL (Study Group Indolent Lymphomas,
Germany) (Abstract). Blood. 2009; 114: 405.
68. Unterhalt M, Herrmann R, Tiemann M, Parwaresch R,
Stein H, Trümper L, et al. Prednimustine, mitoxantrone
(PmM) vs cyclophosphamide, vincristine, prednisone
(COP) for the treatment of advanced low-grade nonHodgkin’s lymphoma. German Low-Grade Lymphoma
Study Group. Leukemia. 1996; 10: 836-43.
69. Forstpointner R, Unterhalt M, Dreyling M, Böck HP,
Repp R, Wandt H, et al. Maintenance therapy with
rituximab leads to a significant prolongation of
response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide,
and mitoxantrone (R-FCM) in patients with recurring
and refractory follicular and mantle cell lymphomas:
43
44
Seminari di Ematologia Oncologica
Results of a prospective randomized study of the
German Low Grade Lymphoma Study Group (GLSG).
Blood. 2006; 108: 4003-8.
70. Smith MR, Zhang L, Gordon LI, Foran J, Kahl B,
Gascoyne RD, et al. Horning phase II study of RCHOP followed by 90Y-Ibritumomab Tiuxetan in
untreated mantle cell lymphoma: eastern cooperative
oncology group study E1499 (Abstract). Blood.
2007; 110: 389.
71. Dreyling M, Weigert O, Hiddemann W; European MCL
Network. Current treatment standards and future
strategies in mantle cell lymphoma. Ann Oncol.
2008;19: iv41-4.
72. Khouri IF, Lee MS, Saliba RM, Jun G, Fayad L, Younes
A, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma.
J Clin Oncol. 2003; 21: 4407-12.
73. Tam CS, Bassett R, Ledesma C, Korbling M, Alousi
A, Hosing C, et al. Mature results of the M. D.
Anderson Cancer Center risk-adapted transplantation
strategy in mantle cell lymphoma. Blood. 2009; 113:
4144-52.
74. Rummel MJ, Al-Batran SE, Kim SZ, Welslau M, Hecker
R, Kofahl-Krause D, et al. Bendamustine plus rituximab
is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol. 2005; 23: 3383-9.
75. Rodríguez J, Gutierrez A, Palacios A, Navarrete M,
Blancas I, Alarcón J, et al. Rituximab, gemcitabine and
oxaliplatin: an effective regimen in patients with refractory and relapsing mantle cell lymphoma. Leuk
Lymphoma. 2007; 48: 2172-8.
76. Wang M, Oki Y, Pro B, Romaguera JE, Rodriguez MA,
Samaniego F, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantl e cell lymphoma. J Clin Oncol. 2009; 27:
5213-8.
77. Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell
lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;
117: 26-38.
78. Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B,
Robertson MJ, de Vos S, et al. Multicenter phase II
study of bortezomib in patients with relapsed or
refractory mantle cell lymphoma. J Clin Oncol. 2006;
24: 4867-74.
79. Martínez N, Camacho FI, Algara P, Rodríguez A,
Dopazo A, Ruíz-Ballesteros E, et al. The molecular
signature of mantle cell lymphoma reveals multiple
signals favoring cell survival. Cancer Res. 2003; 63:
8226-32.
80. Weigert O, Pastore A, Rieken M, Lang N, Hiddemann
W, Dreyling M. Sequence-dependent synergy of the
proteasome inhibitor bortezomib and cytarabine in
mantle cell lymphoma. Leukemia. 2007; 21: 524-8.
81. Weigert O, Unterhalt M, Hiddemann W, Dreyling M.
Mantle cell lymphoma: state-of-the-art management
and future perspective. Leuk Lymphoma. 2009; 50:
1937-50.
82. Ruan J, Martin P, Furman RR, Lee SM, Cheung K,
Vose JM, et al. Bortezomib Plus CHOP-Rituximab for
Previously Untreated Diffuse Large B-Cell Lymphoma
and Mantle Cell Lymphoma. J Clin Oncol. 2010 Dec
28. (Epub ahead of print).
83. Kaufmann H, Raderer M, Wöhrer S, Püspök A, Bankier
A, Zielinski C, et al. Antitumor activity of rituximab plus
thalidomide in patients with relapsed/refractory mantle cell lymphoma. Blood. 2004; 104: 2269-71.
84. Habermann TM, Lossos IS, Justice G, Vose JM,
Wiernik PH, McBride K, et al. Lenalidomide oral
monotherapy produces a high response rate in
patients with relapsed or refractory mantle cell lymphoma. Br J Haematol. 2009; 145: 344-9.
85. Zinzani PL, Witzig TE, Vose JM, Reeder CB, Buckstein
R, Haioun C, et al. Confirmation of the efficacy and
safety of Lenalidomide oral monotherapy in patients
with relapsed or refractory mantle-cell lymphoma:
results of an International study (NHL-003) [Abstract].
Blood. Nov 2008; 112: 262.
86. Witzig TE, Geyer SM, Ghobrial I, Inwards DJ,
Fonseca R, Kurtin P, et al. Phase II trial of single-agent
temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005; 23: 5347-56.
87. Hess G, Herbrecht R, Romaguera J, Verhoef G,
Crump M, Gisselbrecht C, et al. Phase III study to
evaluate temsirolimus compared with investigator’s
choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009; 27:
3822-9.
88. Witzig TE, Reeder CB, Laplant BR, Gupta M,
Johnston PB, Micallef IN, et al. A phase II trial of the
oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011; 25: 341-7.
89. Renner C, Zinzani PL, Gressin R, Klingbiel D, Favet
L, Hitz F, et al. A multi-center phase II study (SAKK
36/06) of single agent everolimus (RAD001) in
patients with relapsed or refractory mantle cell lymphoma (Abstract). Blood. 2010; 116: 2803.
90. Lin TS, Blum KA, Fischer DB, Mitchell SM, Ruppert
AS, Porcu P, et al. Flavopiridol, fludarabine, and rituximab in mantle cell lymphoma and indolent B-cell
lymphoproliferative disorders. J Clin Oncol. 2010; 28:
418-23.
91. Lannutti BJ, Meadows SA, Herman SE, Kashishian A,
Steiner B, Johnson AJ, et al. CAL-101, a p110{delta}
selective phosphatidylinositol-3-kinase inhibitor for the
treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011; 117: 591-4.
92. Brown J, Byrd J, Furman R, Flinn I, Benson D, Coutre
D, et al. Clinical activity in a phase I study of CAL-101,
an isoform-selective inhibitor of phosphatidylinositol 3Kinase P110 delta, in patients with B-Cell malignancies (Abstract). Haematologica. 2010; 95 (Suppl. 2): 466.
93. Morschhauser F, Seymour JF, Kluin-Nelemans HC,
Linfoma mantellare
Grigg A, Wolf M, Pfreundschuh M, et al. A phase II
study of enzastaurin, a protein kinase C beta inhibitor,
in patients with relapsed or refractory mantle cell lymphoma. Ann Oncol. 2008; 19: 247-53.
94. Advani R, Sharman JP, Smith SM, Pollyea DA, Boyd
TE, Grant BW, et al. Effect of Btk inhibitor PCI-32765
monotherapy on responses in patients with relapsed
aggressive NHL: Evidence of antitumor activity from
a phase I study (Abstract). J Clin Oncol. 2010: 8012.
95. Cartron G, Thieblemont C, Solal-Celigny P,
Morschhhauser F, Haioun C, Bouabdallah R, et al.
Promising efficacy with the new anti-CD20 antibody
GA101 in heavily pre-treated NHL patients – first results
from a phase II study in patients with relapsed/refractory DLBCL and MCL (Abstract). Blood. 2010; 116: 2878.
96. Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler
M, Knop S, et al. Tumor regression in cancer patients
by very low doses of a T cell-engaging antibody.
Science. 2008; 321: 974-7.
97. Viardot A, Goebeler M, Scheele JS, Zugmaier G,
Noppeney R, Knop S, et al. Treatment of patients with
non-hodgkin lymphoma (NHL) with CD19/CD3 bispecific antibody Blinatumomab (MT103): double-step
dose increase to continuous infusion of 60 µg/m2/d
is tolerable and highly effective (Abstract). Blood. 2010;
116: 2880.
98. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer.
2007; 7: 750-62.
45
47
Linfoma linfoblastico
dell’adulto
STEFANO LUMINARI1, ALESSANDRA DONDI1, GINO SANTINI2
1
Dipartimento di Oncologia ed Ematologia, Università di Modena e Reggio Emilia;
Divisione di Ematologia Ematologia I, Azienda Ospedaliera San Martino, Genova
2
n INTRODUZIONE
Il linfoma linfoblastico (LBL) costituisce, insieme
alla leucemia linfoblastica acuta (LLA), il gruppo
dei linfomi/leucemie dei precursori linfocitari (1).
Con il termine di LBL si fa generalmente riferimento a malattie con interessamento prevalente dei
linfonodi. La distinzione tra LBL e LLA è puramente clinica, generalmente arbitraria e basata sulla
percentuale dei blasti midollari che nel caso dei
LBL non dovrebbe superare il 25% (2). Da un punto di vista istomorfologico invece il LBL non è
distinguibile dalla LLA e in entrambi i casi la malattia può originare da un precursore della linea cellulare T o B. Anche da un punto di vista terapeutico i trattamenti del LBL e della LLA sono simili, basati sull’utilizzo di trattamenti polichemioterapici sequenziali ed intensivi. Pur in assenza di
un razionale ben documentato sull’esistenza di
chiare differenze biologiche tra LBL e LLA esiste
tuttavia un piccolo gruppo di pazienti con T-LBL,
che peraltro rappresenta la maggioranza dei LBL,
Stefano Luminari
per i quali si apprezzano maggiori differenze di
presentazione e di andamento clinico rispetto alla
controparte leucemica e per i quali è più verosimile mantenere una distinzione tra forma linfomatosa e leucemica.
Il trattamento dei LBL non si discosta molto da
quello della LLA, e deve prevedere programmi di
chemioterapia intensificata diversi da quanto previsto per i linfomi non-Hodgkin (LNH) di tipo periferico. La risposta completa ai trattamenti iniziali è in genere buona aggirandosi attorno all’8090%. Il problema maggiore della gestione di un
paziente con LBL è tuttavia l’elevato rischio di recidiva che determina una sopravvivenza globale del
50-60% a 5 anni di osservazione.
Oggetto di questo lavoro di revisione è quello di
fornire un inquadramento generale dei LBL dell’adulto. Verranno descritti gli aspetti epidemiologici, biologici, clinici e le principali conoscenze
sui trattamenti di questa rara patologia.
n EPIDEMIOLOGIA
Parole chiave: linfoma linfoblastico, chemioterapia, linfoma dell’adulto, linfoma dei precursori
Indirizzo per la corrispondenza
Dr. Stefano Luminari
Ricercatore Universitario, Oncologia medica
Dipartimento di Oncologia ed Ematologia
Università di Modena e Reggio Emilia
C/O Centro Oncologico Modenese
Via del pozzo, 71 - 41124 Modena
E-mail: [email protected]
Il LBL rappresenta una malattia molto rara nei soggetti adulti (con un’incidenza inferiore al 2% dei
linfomi non-Hodgkin), e colpisce prevalentemente gli adolescenti e i giovani adulti di sesso
maschile. L’incidenza di LBL/LLA presenta un
andamento bimodale con un primo picco compreso tra i 2 e i 5 anni, in cui si registrano tra i 4
e i 5 casi per 100.000 bambini ed un secondo picco che inizia dopo i 50 anni di età e raggiunge
un tasso annuo di circa 2 casi per 100.000 per-
48
Seminari di Ematologia Oncologica
sone (3). Relativamente ai pazienti adulti il tasso
registrato negli Stati Uniti per i soggetti di età compresa tra i 25 e i 50 anni è di 0,4-0,6 casi per
100.000, e aumenta di 2-3 volte nelle persone oltre
i 60 anni.
Complessivamente il fenotipo T è il più comune,
rappresentando oltre l’80%-90% di tutte le forme di LBL. Generalmente il rischio di ammalarsi
di LBL è più elevato nei soggetti di razza bianca
rispetto ai neri e nei maschi rispetto alle femmine. La maggiore prevalenza di LBL nei maschi è
osservata in particolare nei casi a fenotipo T(4).
In Italia i dati di incidenza sono sovrapponibili a
quelli osservati per gli altri Paesi occidentali con
un tasso complessivo di 1,6 casi/100.000 per i
maschi e di 0,9 casi/100.000 per le femmine.
Complessivamente, in base ai dati prodotti dal
registro tumori della provincia di Modena, si stima che in Italia, nel corso del 2011, verranno diagnosticati circa 700 nuovi casi di LBL/LLA.
Attualmente non sono noti fattori eziologici o di
rischio specifici per i linfomi/leucemia linfoblastici e in particolare per il LBL. Nei casi pediatrici
un ruolo importante è sicuramente svolto da fattori genetici che invece sono meno od affatto noti
nell’adulto. Relativamente ai fattori ambientali, è
noto che il rischio di LLA è aumentato per effetto dell’esposizione a radiazioni ionizzanti (3). Il
rischio di LLA può inoltre aumentare in seguito ad
esposizione a sostanze chimiche quali il benzene o ad agenti chemioterapici, in particolare agli
alchilanti, agli inibitori delle topoisomerasi II e, più
raramente, alle antracicline (5, 6). Infine mancano evidenze dirette sul possibile ruolo eziologico
degli agenti virali con l’unica eccezione rappresentata dai rari casi di leucemia/linfoma a cellule T dell’adulto associati ad infezione da virus
HTLV1. Quanto sopra riportato potrebbe essere
ragionevolmente vero anche per il LBL dell’adulto, anche se mancano dimostrazioni dirette sull’argomento.
n ANATOMIA PATOLOGICA
ED IMMUNOFENOTIPO
Come già detto non esistono caratteristiche citologiche, fenotipiche e biologiche che consentano di distinguere un LBL da una LLA. Vale tutta-
via l’osservazione di casi relativamente più differenziati per i LBL rispetto alle LLA (T-LBL timici,
corticali o midollari, e LBL a cellule B più mature). La diagnosi morfologica di LBL e LLA è tuttora basata sul sistema classificativo FAB (French
American British) che distingue gli elementi cellulari in tre classi: L1, L2, L3. In base agli studi
immunofenotipici più recenti la distinzione tra L1
e L2 ha perso molto significato; si conferma invece l’importanza dell’identificazione dei casi con
morfologia L3 per la correlazione di questo citotipo con il linfoma di Burkitt (7).
Nel LBL a cellule T, che rappresenta la maggioranza dei LBL dell’adulto, la morfologia è quella
del corrispondente linfoma a cellule B e della LLA
(in genere FAB L1). Le cellule, di grandezza media
o piccola, hanno rima citoplasmatica per lo più
sottile, con positività focale paranucleare delle reazioni per la fosfatasi acida; il nucleo è rotondo od
ovoidale oppure variamente convoluto, con cromatina finemente dispersa, nucleoli piccoli e poco
appariscenti o anche assenti. L’aspetto a nucleo
convoluto può essere presente solo in poche cellule oppure in più dell’85%: tali casi vengono considerati un sottotipo di T-LBL. Un’ulteriore variante del T-LBL è costituita dai casi a cellule grandi
e pleomorfe, simili a quelle della LLA FAB L2, con
nucleo convoluto e uno o più grandi nucleoli.
L’attività mitotica è elevata in tutte le varianti di
LBL. La proliferazione linfomatosa, di tipo diffuso, spesso cancella del tutto l’architettura linfonodale, talora coinvolge solo aree T paracorticali e interfollicolari. Si possono osservare aree di
iperplasia di cellule epitelioidi a cielo stellato, a
distribuzione focale. Capsula, tessuti molli perilinfonodali, pareti vasali sono abitualmente infiltrati. In circa il 15% dei casi alle cellule linfomatose sono frammisti granulociti eosinofili e plasmacellule. Nel timo, frequentemente interessato, solo
i reperti immunofenotipici permettono di distinguere il LBL dal timoma (1, 7, 8).
Nel raro linfoma linfoblastico B (B-LBL) i linfoblasti hanno una grandezza media, sottile orletto citoplasmatico debolmente basofilo, nucleo rotondo
oppure convoluto, a rete cromatinica fine e con
distinta membrana; i nucleoli sono piccoli e poco
appariscenti. I fini caratteri morfologici sono meglio
apprezzabili nelle apposizione che nelle sezioni.
Nel 10% circa dei casi i linfoblasti presentano
Linfoma linfoblastico dell’adulto
nuclei più grandi, spesso convoluti, nucleoli prominenti, simili a quelli delle leucemie linfatiche acute di tipo L2: la presenza di una struttura cromatinica finemente dispersa è l’unico reperto che
consenta di distinguere il linfoblasti di un LBL dalle cellule dei linfomi indifferenziati non-Burkitt e
da alcuni linfomi a grandi cellule B. La sub-classificazione morfologica dei LBL in sottotipi a
nucleo convoluto, non convoluto, pleomorfo, non
ha valore clinico. Negli organi infiltrati i linfoblasti
appaiono uniformemente stipati. La struttura linfonodale è del tutto cancellata. Capsula e tessuti peri-linfonodali sono infiltrati e la presenza di
macrofagi può talora dar luogo ad immagini a cielo stellato. Nei linfoblasti si osserva intensa positività citoplasmatica granulare delle reazioni citochimiche per il PAS, l’ATPasi e la 5-nucleotidasi.
Le reazioni per la fosfatasi acida e per le esterasi non specifiche sono quasi sempre negative, mai
di tipo focale. La diagnosi differenziale deve tenere presente la variante blastica del linfoma mantellare, le cui cellule tuttavia mostrano positività
per la ciclina D1 e per la traslocazione cromosomica t(11;14) (1, 7).
Il LBL è facilmente studiabile con analisi citofluorimetrica di cellule in sospensione ottenibili dal
sangue periferico o midollare e/o da tessuto. Una
prima distinzione deve essere posta tra LBL a cellule B o T(9). Le caratteristiche fenotipiche dei TLBL sono più complesse rispetto alle forme B. In
genere i pazienti con T-LBL esprimono TdT e CD7,
quest’ultimo presente nel 95-100% dei casi, variabilmente associati con altri antigeni quali il CD2,
CD1, CD3, CD4 a seconda della fase di differenziazione cellulare. In una minoranza di casi è
descritta l’espressione di CD10. L’espressione del
CD7 è attualmente considerata un requisito
essenziale per porre diagnosi di T-LBL; tuttavia,
poichè CD7 può essere espresso anche da alcuni casi di B-LBL/LLA e da alcune leucemie mieloidi, è necessario dimostrare una coespressione con altri marcatori T (CD2, CD3 e CD5). Altre
molecole variabilmente espresse sono CD99 e
CD34. Nel 29-48% dei casi vi è espressione
nucleare di TAL-1 (10). Per il T-LBL esistono alcuni sottotipi fenotipici: la forma pre-T che rappresenta il 7% dei casi dell’adulto, la forma timica
che costituisce il 13% circa dei casi ed è associata all’espressione di CD1a e la forma a cellu-
le T mature che rappresenta il 7% dei casi e presenta espressione di CD3. I T-LBL possono essere ulteriormente classificati in base alla tipologia
di riarrangiamento del T-cell receptor.
Tra i pochi casi a fenotipo B sono identificabili ulteriori sottotipi in analogia a quanto descritto per
le B-LLA. La forma più frequente, diagnosticata
nel 50% dei casi è il cosiddetto common-LBL,
caratterizzato dall’espressione di TdT, CD19 e
CD10. In una percentuale inferiore pari a circa
l’11% la diagnosi è di pro-B-LBL (altrimenti
descritto come pre-pre-B o early pre-B), in cui la
trasformazione neoplastica avviene in una fase più
precoce dell’ontogenesi linfocitaria, caratterizzata dalla presenza di riarrangiamento del gene delle catene pesanti delle immunoglobulina (IgH) ma
dall’assenza di espressione di linea B; in questi
casi è comunque presente l’espressione di TdT
e di HLA-DR. La presenza di immunoglobuline
citoplasmatiche in un paziente con caratteristiche
di common-LBL identifica i rari casi di pre-B-LBL.
In un 4% dei casi è possibile porre diagnosi di
LBL a cellule B mature, caratterizzato dall’espressione di TdT e di antigeni di superficie B maturi
quali il CD19, CD20, CD22, e CD23; questo sottotipo in particolare corrisponde più frequentemente alla forma linfomatosa. Espressioni antigeniche aberranti sono infine descritte in una percentuale variabile dal 30 al 45% dei casi pediatrici e, in misura minore, dell’adulto. Tra gli antigeni aberranti quelli mieloidi sono più frequenti
degli antigeni della linea T.
Nonostante siano state descritte numerose
varianti fenotipiche sia per i T-LBL che per i B, non
esistono attualmente dati che siano utilizzabili
come fattore prognostico in termini di sopravvivenza. Si è solo osservato che più frequentemente i casi di T-LBL presentano fenotipi più maturi
(corticali e midollari) rispetto alle forme T-ALL che
sono più spesso pro-T e pre-T (11).
n ANOMALIE GENETICHE
E CARIOTIPICHE
Sia nei LBL a cellule B che nelle forme T i geni
delle immunoglobuline e del T-cell receptor (TCR)
risultano riarrangiati e la presenza del riarrangiamento è documentabile con diverse metodiche
49
50
Seminari di Ematologia Oncologica
di biologia molecolare ed utilizzabile in fase di diagnosi e in corso di trattamento per monitorare la
risposta alle cure. A differenza dei casi pediatrici per i quali sono descritte numerose varianti
genetiche in base alle quali i pazienti sono divisi
in gruppi ad alto, intermedio o basso rischio, per
i LBL dell’adulto, molto più rari, alterazioni cromosomiche aggiuntive sono presenti nella maggioranza dei casi, ma ognuna è ripetuta in meno
del 10% dei casi rendendone impossibile una verifica del ruolo prognostico (12, 13). Nella LLA così
come nel LBL l’alterazione cromosomica più frequente è rappresentata dal cromosoma
Philadelphia (Ph), risultato della traslocazione
t(9;22) tipica della leucemia mieloide cronica
(LMC). I casi Ph+ presentano un fenotipo B, hanno età più avanzata e si manifestano con conta
leucocitaria più elevata. La maggior parte dei
pazienti adulti presenta il trascritto p190 del gene
di fusione BCR-ABL prodotto dalla traslocazione t(9;22) diversamente dai casi di LMC che presentano il più comune trascritto p210, presente
nel 25% dei LBL. È stato suggerito che l’espressione di p190 identifichi i casi di de novo LBL differenziandoli dai casi che si manifestano nella fase
blastica della LMC.
In uno studio in 304 pazienti con LBL condotto
per identificare ulteriori lesioni geniche presenti in
aggiunta al cromosoma Ph, sono state identificate frequenti delezioni del gene Ikaros (14). In particolare le alterazioni di Ikaros, un fattore di trascrizione fondamentale per l’ontogenesi B-linfocitaria, erano descritte solo nei casi di LBL pediatrico e dell’adulto e mai nei casi di LMC. Anche
nei casi non esorditi de novo tuttavia il gene Ikaros
risultava deleto nel passaggio alla fase blastica
della malattia (14). Lo stesso gene Ikaros nelle sue
diverse isoforme sembra avere un ruolo importante nel determinare la resistenza ai trattamenti con inibitori delle tirosinochinasi (15). Oltre al cromosoma Ph nei B-LBL sono frequenti i riarrangiamenti del gene MLL1 sul cromosoma 11q23,
descritti nel 10% dei casi. L’alterazione cromosomica più frequente è rappresentata dalla traslocazione t(4;11) in cui il gene MLL1 viene fuso
con il gene AF-4. Anche per i casi con t(4;11) è
descritta una prognosi più infausta rispetto agli
altri casi Ph- (13, 16).
Dal punto di vista genico, T-ALL/LBL mostra pres-
soché sempre il riarrangiamento monoclonale dei
geni del TCR. In circa il 20% dei casi c’è simultanea presenza di riarrangiamento dei geni delle
Ig. Un cariotipo anormale è presente in circa il 5070% dei casi. Le più comuni anomalie citogenetiche coinvolgono i loci del TCR: i loci delle catene alfa e delta al 14q11.2, il locus beta al 7q35 e
il locus gamma al 7p14-15 che possono avere
svariati geni partners nella traslocazione. Nella
maggioranza dei casi queste traslocazioni portano a disregolazione di geni di trascrizione presenti nella regione partner della traslocazione che vengono giustapposti a geni regolatori dei loci del
TCR traslocati. Tra i geni più comunemente coinvolti vi sono i fattori di trascrizione HOX11 (TLX1)
(10q24) (nel 7% dei bambini e nel 30% degli adulti) e HOX11L2 (TLX3) (5q35) presente nel 20% dei
bambini e nel 10-15% degli adulti. Altri fattori di
trascrizione coinvolti nelle traslocazioni sono:
MYC, TAL1, RBTN1, RBTN2 e LYL1 (1, 16).
Tra le delezioni presenti, la più importante è la
del(9p) che determina la perdita del gene soppressore CDKN2A. Infine, in circa il 50% dei casi si
osservano mutazioni che coinvolgono il gene
NOTCH 1 che codifica per una proteina fondamentale per l’iniziale maturazione del linfocita T.
La presenza di queste mutazioni missense nel
gene portano come conseguenza all’aumento dell’emivita della proteina NOTCH1. Secondo i dati
di uno studio recente, la mutazione del gene
NOTCH1 sembra associata ad una ridotta sopravvivenza nei pazienti adulti e non nei bambini affetti da T-ALL (17).
Analisi genomiche più complesse condotte su
ampie casistiche di LBL/LLA confermano di fatto quanto già noto relativamente alla presenza di
multiple alterazioni geniche ampliando il numero
di loci coinvolti. Alcuni studi hanno mostrato la
presenza di lesioni genetiche nascoste nella pressoché totalità dei casi di LBL/LLA dell’adolescente e dell’adulto, in analogia a quanto osservato
nei casi di LLA pediatrici (18).
Più recentemente, studi sui profili di espressione
genica mediante tecniche di DNA micro-array hanno evidenziato alcune signatures geniche che corrispondono a specifici stadi di maturazione che
si osservano durante lo sviluppo dei linfociti normali (19). In particolare per i T-LBL: LYL1 signature corrisponde allo stadio di pro-T, HOX11+
Linfoma linfoblastico dell’adulto
signature allo stadio di corticale early, e TAL1+
signature allo stadio corticale late. Studi clinici indicano che il gruppo di pazienti con signature di tipo
HOX11+ sembra avere una prognosi relativamente favorevole.
Molti altri studi, sempre basati su GEP, hanno
identificato svariati fattori prognostici genici ma
nella maggior parte dei casi gli studi sono stati
condotti su casistica pediatrica di T-ALL. Nel
2006 e 2007 sono stati pubblicati dati di particolare interesse che differenzierebbero la T-ALL
dal T-LBL ed il gene-profile identificherebbe possibili e potenziali targets terapeutici differenziati (20-23).
Tuttavia poco o nulla è stato fatto su casistiche
adulte, soprattutto se il campo si restringe al LBL.
Una recente recensione sul T-LBL sottolinea che
le caratteristiche cliniche, immunofenotipiche e la
risposta alla terapia od al trapianto, come più
avanti riportato, sembrerebbero riflettere il confronto tra malattie simili, una localizzata (T-LBL)
ed una sistemica (T-LLA) (23). Tuttavia, la mancanza di dati sulla malattia residua minima
(MRM) e la scarsità di notizie sul profilo genico,
e su quanto questi possano incidere sull’andamento della malattia, sulla risposta alla terapia, o
su una potenziale differenza tra le due entità, implicano che questo argomento resta completamente da chiarire.
N
Fenotipo
Età
Sesso
Sintomi
Bulky
Sedi EN
Hb
Stadio
T
B
Null
NV
Mediana
Range
M
B
(>10 cm)
1+
<10 g/dl
I-Ie
II-IIe
III-IV
n ASPETTI CLINICI
Al momento attuale la clinica rappresenta il principale criterio diagnostico per identificare i casi
di LBL rispetto alla più comune LLA. La maggior
parte dei paziente con ALL si presentano all’esordio della malattia con i tipici sintomi e i segni clinici e di laboratorio dell’insufficienza midollare. Nei
pazienti con LBL non potendo essere presente
per definizione un marcato infiltrato midollare questo quadro clinico è meno frequente e prevalgono i sintomi d’organo (2). Gli organi più frequentemente coinvolti sono i linfonodi, il fegato e la milza. Il coinvolgimento del SNC è anche frequente (24). Tra i diversi sottotipi di LLA/LBL dell’adulto la forma che si manifesta più frequentemente
come linfoma è il T-LBL. Il T-LBL si evidenzia spesso a livello mediastinico anteriore (timico) anche
se può coinvolgere ogni sede linfonodale o extranodale (cute, tonsilla, fegato, milza, sistema nervoso centrale e testicolo). Comune è anche un
coinvolgimento della pleura.
Anche nei casi di LBL il coinvolgimento midollare è comune ma tende a comparire durante il corso della malattia (fino al 50% dei casi), spesso con
rapida progressione sino alla fase leucemica che
può costituire l’ evento finale. La malattia si presenta in oltre il 50% dei casi in stadio avanzato
(Tabella 1).
Morel et al. (47)
Sweetenham et al. (48)
Sweetenham et al. (28)
54
85
2
13
0
33
15-76
65
46
61
48
11
10
33
57
214
55
23
2
20
27
16-57
71
39
30
5
17
70
119
68
25
2
5
26
14-65
70
32
8
27
62
Legenda: N: Numero; EN: Extranodale; Hb: Emoglobina; NV: Non valutabile.
TABELLA 1 - Caratteristiche cliniche dei pazienti con LBL (%).
51
52
Seminari di Ematologia Oncologica
I pazienti con LBL sono comunemente stadiati
con il sistema di Ann Arbor utilizzato per gli altri
LNH. Rispetto ad altri sistemi di stadiazione come
quello proposto da Murphy (2) per i casi pediatrici, la stadiazione di Ann Arbor sembra fornire
nel paziente adulto le migliori informazioni ai fini
prognostici. Lo stadio di un paziente con LBL
deve essere definito con un approccio classico,
ovvero con esami di laboratorio, biopsia osteomidollare e TAC con mezzo di contrasto. Visto il
frequente coinvolgimento del SNC è inoltre raccomandato lo studio radiologico dell’encefalo e
l’esecuzione di un esame liquorale. L’esame
liquorale, in particolare, deve essere sempre completato con analisi citologica e citofluorimetrica
alla ricerca di piccole popolazioni linfocitarie clonali.
Nonostante il LBL sia una neoplasia ad alta attività proliferativa e ad elevato metabolismo non vi
sono attualmente dati sufficienti per suggerire di
utilizzare la PET con FDG anche in questa patologia, se non nell’ambito di studi controllati.
n SOPRAVVIVENZA
E FATTORI PROGNOSTICI
Complessivamente con i trattamenti più moderni la sopravvivenza dei pazienti con LBL si attesta attorno al 50-60% a 5 anni (26, 27).
Ad oggi non esiste un sistema di stratificazione prognostica specifica per i LBL anche se la possibilità di identificare correttamente il rischio individuale dei pazienti potrebbe avere importanti risvolti nell’impostazione del programma terapeutico.
Fattori comunemente descritti come associati ad
una prognosi più infausta sono l’età con un limite a 30 anni, la presenza di stadio III-IV, l’interessamento midollare, il coinvolgimento del sistema
nervoso centrale (SNC), la presentazione leucemica, la presenza di sintomi B e di valori elevati
di LDH (25). Ulteriori fattori prognostici sfavorevoli sono rappresentati da un punteggio superiore a 2 dell’indice prognostico internazionale IPI e
dal tempo impiegato per raggiungere una remissione completa.
Gli studi più recenti condotti su casistiche ampie
e utilizzando trattamenti moderni hanno messo
in discussione il ruolo prognostico dei parame-
tri clinici e demografici precedentemente descritti (26, 27). Anche lo studio europeo randomizzato sul trapianto di midollo non ha dimostrato
l’esistenza di fattori predittivi dell’outcome nel
LBL dell’adulto, non confermando tra l’altro il
ruolo prognostico dell’indice prognostico internazionale (IPI) (28).
La definizione di categoria ad alto rischio che rimane di fondamentale importanza per impostare un
programma di cura adeguato dovrà necessariamente integrare le attuali conoscenze cliniche con
i più moderni dati biologici.
Le maggiori novità sulla definizione della prognosi dei LBL sono attese dagli studi sulla MRM. La
presenza di riarrangiamento dei geni delle immunoglobuline o del T-cell receptor, o la presenza
del cromosoma Ph offrono infatti strumenti molto importanti per poter definire con elevata sensibilità la qualità della risposta ai trattamenti e per
poter monitorare l’andamento della malattia
durante la fase di follow-up (29).
Gli studi sulla MRM riguardano soprattutto i casi
di LLA pediatrica e concordano nell’identificare
l’ottenimento della risposta molecolare quale
importante fattore prognostico indipendente. In
particolare in uno studio condotto dal gruppo
GMALL i pazienti con MRM hanno fatto osservare un tempo mediano alla recidiva di 9,5 mesi,
mentre solo il 6% dei pazienti con negativizzazione della MRM sono recidivati (30). I dati ottenuti
per la LLA potrebbero essere estesi anche ai LBL
tuttavia in questi casi è opportuno considerare la
possibilità di false negatività legate alla potenziale assenza di infiltrato midollare.
n TERAPIA NELL’ADULTO
La rarità del LBL ha molto limitato lo studio della malattia e a tutt’oggi sono disponibili solo poche
pubblicazioni ed in genere riferite a pochi pazienti. In una serie iniziale di 95 pazienti riportati da
Nathwani et al. (31), la sopravvivenza mediana a
lungo termine è stata di soli 17 mesi e solo il 15%
dei pazienti erano vivi a 30 mesi.
Tuttavia è necessario precisare che i giovani ed
adulti trattati negli anni ’60 e fino agli anni ‘70 hanno ricevuto sicuramente trattamenti inadeguati. La
chemioterapia (CT) cosiddetta ciclica, non inten-
Linfoma linfoblastico dell’adulto
siva e non sequenziale, usata in quegli anni e simile a quella utilizzata nei linfomi non-Hodgkin, ha
mostrato risultati deludenti anche nei bambini con
LBL, con sopravvivenza di circa il 10% a 5 anni
(32).
Tra il 1970 ed il 1980, nei bambini furono utilizzati trattamenti più intensivi con un miglioramento significativo dell’outcome (2, 33). Questi includevano regimi induttivi di tipo intensivo, profilassi del SNC, terapia di consolidamento e terapia
di mantenimento. Risultati molto stimolanti furono riportati da Wollner et al. (34) in giovani trattati con una CT sequenziale intensificata, il regime LSA2L2.
Questo ottenne in un gruppo di pazienti con linfoma aggressivo in stadio avanzato, il 40% dei
quali era costituito da LBL una probabilità di
sopravvivenza globale (OS) del 76%, con 25 mesi
di mediana di osservazione. In considerazione di
ciò l’adozione di schemi simili, sequenziali e/o più
aggressivi, fu estesa anche agli adulti. Lo stesso
autore (35), utilizzando uno schema variato della
durata di 3 anni, riporta nel linfoma linfoblastico
del bambino in stadio III-IV una OS ed una sopravvivenza libera da eventi (EFS) pari rispettivamente al 90% e all’85%. Tuttavia, come descritto da
Hvidzala et al. (36) rimaneva un gruppo a prognosi infausta rappresentato dai casi pediatrici in stadio IV; la sopravvivenza libera da malattia (DFS)
di questo sottogruppo di pazienti risultava infatti inferiore al 20%.
Molti studi sul LBL dell’adulto hanno usato
schemi simili a quelli utilizzati nella ALL. Il gruppo di studio di Stanford (37) pubblicò i risultati di
uno studio pilota intensivo in 14 LBL con risultati buoni in termini di risposta (100%) ma con una
serie di problemi legati a frequente ricaduta nel
SNC. Lo schema fu successivamente modificato includendo una fase induttiva CHOP-like, e
aggiungendo la somministrazione settimanale di
vincristina ed asparaginasi, la profilassi del SNC,
programmando 4 blocchi di consolidamento e
terapia di mantenimento, il tutto per la durata di
1 anno. Nei 44 pazienti trattati la risposta fu del
100% (risposta completa, (RC): 95%; risposta parziale, (RP): 5%), con una OS ed un tempo libero
da recidiva (FFS) del 56%. Anche in questa esperienza si confemava il limite della terapia nei
pazienti in IV stadio, i quali continuavano a rica-
dere durante e al termine terapia ottenendo una
OS a 3 anni del 20%. Oltre alla diagnosi in stadio IV altri fattori prognostici sfavorevoli erano la
presenza di coinvolgimento midollare o del SNC,
e l’aumento >150% della LDH.
Slater et al. (25) hanno pubblicato la loro esperienza su 51 pazienti con LBL trattati con 5 schemi successivi LLA-like.
I pazienti con >25% di infiltrato midollare o con
blasti circolanti vennero classificati come LBL
leucemici, gli altri come non leucemici. La RC
fu del 78%, 18 pazienti ricaddero o morirono per
complicazioni, e la sopravvivenza a 5 anni fu del
45%.
Tuttavia, nel LBL non leucemico la DFS fu di circa il 75%. Non vi fu differenza tra questi pazienti e pazienti con LLA in termini di OS (circa 50%).
Fattori negativi per la sopravvivenza furono l’età
>30 anni, globuli bianchi >50.000 mm3, il non ottenimento della RC o una RC ottenuta dopo 4 settimane (25).
Mazza et al. (38) riportarono, in 64 pazienti adulti, una RC del 38% con una terapia CHOP-like
ed una RC del 50% con lo schema LSA2L2. La
RC fu del 77% in stadio I-II e del 35% in stadio
III-IV. La sopravvivenza degli stadi III-IV fu tra il
10% ed il 20%.
Levine et al. (39) avevano ottenuto in 15 LBL, con
uno schema LSA2L2 modificato, una RC del 73%,
con una DFS di circa il 50%.
Bernasconi et al. (40) riportarono i dati su 31
pazienti adulti, trattati con uno schema LLA-like.
Il trattamento ottenne una RC del 77%, con
sopravvivenza libera da recidiva (RFS) del 50%
a 18 mesi, ed OS a 4 anni del 35%. Gli stadi IV
mostrarono una prognosi significativamente peggiore nei confronti degli stadi iniziali.
Il Gruppo tedesco ha pubblicato i risultati di due
schemi terapeutici utilizzati nel T-LBL ma anche
nella LLA (26). In particolare, sono stati definiti TLBL i pazienti con un infiltrato midollare ≤25%.
La terapia nel LBL, raccomandata per circa 1
anno, includeva una induzione a 8 farmaci, con
chemio-radioterapia (CT/RT) cranica profilattica e
radioterapia (RT) mediastinica, seguite da terapia
di consolidamento e da terapia reinduttiva. In termini di risposta, 42/45 pazienti (93%) ottennero
una RC e 2 pazienti (4%) una RP. La RC fu del
100% nei pazienti in stadio I-III e del 89% in
53
54
Seminari di Ematologia Oncologica
pazienti in stadio IV. Il 36% dei pazienti ricadde,
con una OS e DFS a 7 anni del 51% e del 62%
rispettivamente. La ripresa di malattia mediastinica fu l’evento più frequente. Non vi furono fattori prognostici negativi individuabili.
L’osservazione finale è che è possibile ottenere
un’alta percentuale di RC con schemi disegnati
per la LLA ma che è anche auspicabile una terapia più intensificata e che sia estesa l’indicazione al trapianto con cellule staminali.
Nel 2004 il gruppo di Houston ha pubblicato i risultati dello hyper-CVAD (27). Con due schemi simili della durata di circa 3 anni, vennero trattati 33
pazienti: 80% erano T-LBL, 70% erano in IV stadio, 70% avevano coinvolgimento mediastinico,
9% coinvolgimento del SNC, e 15% un infiltrato
midollare < al 25%. Il 91% dei pazienti ottenne una
RC ed il 9% una RP. Con una mediana di osservazione di 13 mesi, il 30% dei pazienti ricadde.
Le curve attuariali del T-LBL mostrano una probabilità di sopravvivenza libera da progressione
(PFS) e OS del 62% e del 67%, rispettivamente.
Nessun fattore potenzialmente prognostico ha
modificato l’outcome. Le osservazioni finali sono
che l’aggiunta di antracicline liposomiali allo schema originale non migliora l’outcome e che appare auspicabile l’utilizzo di anticorpi monoclonali e/o
analoghi delle purine.
Il dato che colpisce in questa prima fase di
osservazione è che l’evoluzione terapeutica
abbia decisamente aumentato la possibilità di
RC ma che una serie di fattori prognostici molto variabili ed incerti, probabilmente per l’esiguità dei pazienti trattati, ed in particolare lo stadio avanzato (IV), pregiudichino l’outcome. Il
fenomeno della ricaduta, contenuta negli ultimi
studi attorno al 30%-35%, sembra poi indipendente dal fatto che il paziente sia in RC ed in
terapia di mantenimento. L’altra osservazione è
che la durata della terapia oscilla tra 1 e 3 anni
e la durata del trattamento pregiudica la qualità di vita del paziente.
Ruolo della radioterapia
Nonostante il LBL sia una malattia radiosensibile, il trattamento localizzato è stato abbandonato come procedura di routine da quando si sono
iniziati ad utilizzare schemi polichemioterapici
Studio
Anno
N. pazienti
DFS (%)
Autotrapianto in prima RC
Milpied et al. (49)
Santini et al. (50)
Baro et al. (51)
Morel et al. (47)
Verdonck et al. (52)
Sweetenham et al. (48)
Jost et al. (53)
Zinzani et al. (54)
Bouabdallah et al. (55)
Conde et al. (56)
Sweetenham et al. (28)
Levine et al. (41)
N. tot di pz e mediana di DFS (range)
1989
1991
1992
1992
1992
1994
1995
1996
1998
1999
2001
2003
13
18
14
5
9
105
17
10
16
58
31
47
343
68
74
77
60
67
63
31
70
62
76
55
44
65 (31-77)
Allo-trapianto in prima RC
Milpied et al. (49)
Bouabdallah et al. (55)
Sweetenham et al. (28)
Levine et al. (41)
N. tot di pz e mediana di DFS (range)
1989
1998
2001
2003
12
11
12
24
59
68
91
58
39
63 (39-91)
Da: Aljurf M and Zaidi SZ. (modificata) (57).
TABELLA 2
Linfoma linfoblastico dell’adulto
intensificati, principalmente per cercare di limitare la tossicità dei trattamenti.
Ad oggi tuttavia non vi sono sufficienti dati per
poter confermare o meno se l’aggiunta della radioterapia nei pazienti con lenta risposta a livello di
localizzazioni bulky incrementi l’efficacia dei trattamenti, con l’unica eccezione per la malattia
mediastinica dove la RT è ancora raccomandata (26). L’uso della radioterapia è attualmente utilizzato come misura profilattica della malattia cerebrale in combinazione con gli schemi BFM (irradiazione del cranio con 12Gy). Vi è invece indicazione alla radioterapia encefalica nei rari casi
di localizzazioni documentate al SNC.
Trapianto con cellule staminali
Benché la terapia intensificata o ALL-like abbia
modificato la possibilità di ottenimento della RC
e l’outcome del LBL dell’adulto, la sopravvivenza è ancora breve in molte serie di pazienti, collocandosi attorno al 50-60%, e molti pazienti ricadono, anche nel corso della terapia, e per un
periodo di circa 3 anni. Come conseguenza, la
terapia ad alto dosaggio con trapianto autologo
(ABMT) o allogenico è stata utilizzata per consolidare la prima RC dopo CT convenzionale.
I dati disponibili in letteratura suggeriscono che
una terapia di induzione-consolidamento seguita da trapianto autologo o allogenico possano
migliorare l’outcome del LBL in termini di DFS. È
tuttavia ancora poco chiaro se e quali pazienti,
definibili a maggiore rischio alla diagnosi, debbano necessariamente utilizzare questa terapia.
La tabella 2 mostra un sommario di dati recenti
sul trapianto di midollo e/o cellule staminali periferiche. Dall’analisi di questi studi, emerge che
l’autotrapianto mostra un trend favorevole in termini di DFS nei confronti della CT convenzionale, ma questa procedura non sembra migliorare
statisticamente la OS. Questo dato emerge dall’unico studio randomizzato del Gruppo Europeo
che ha confrontato CT convenzionale ed ABMT
in 65 pazienti con LBL in risposta dopo una induzione-consolidamento convenzionale (28).
I dati degni di nota di quest’ultimo studio, che
ricalcano a grandi linee quanto riportato negli studi di fase II, sono che:
- i pazienti trattati con CT convenzionale ricadono per circa 3 anni mentre quelli sottoposti a
ABMT ricadono, in genere, nel primo anno (RFS
55% vs 24%, p=0.06);
- la OS è simile (ABMT, 56%; CT, 45%; p=0.70)
poiché i pazienti ricaduti dopo CT convenzionale possono utilizzare l’ABMT come salvataggio;
- non sono emersi fattori prognostici sfavorevoli
che possano indicare un sub-set di pazienti in cui
sia suggeribile l’ABMT;
- la sopravvivenza in accordo all’IPI aggiustato per
fascia d’età mostra una differenza statistica a sfavore dei pazienti con 3 fattori prognostici sfavorevoli alla diagnosi (OS <10%; p=0.01);
- la sopravvivenza di 12 pazienti avviati al trapianto allogenico è del 58%, quindi sovrapponibile a
quanto osservato con la procedura autotrapiantologica. Il trapianto allogenico utilizzato in prima
RC è caratterizzato da una DFS simile a quanto
osservato con il trapianto autologo. Questo non
è legato ad una ricaduta analoga delle due procedure, ma alla elevata mortalità legata alla procedura allotrapiantologica, a fronte di una minore ricaduta di malattia (41). Studi successivi, pubblicati dallo EBMT Group (42), dal Gruppo francese (43), e da Gruppi di studio giapponesi e
canadesi (44, 45) non hanno apportato modifiche
di contenuto a quanto detto sopra.
In sintesi l’ABMT è caratterizzato da ricadute più
frequenti nei confronti del trapianto allogenico e
quest’ultimo da un maggior numero di decessi
legati alla procedura. Ne consegue che i risultati
a lungo termine sono sovrapponibili. Per questo
motivo, in molti centri, il trapianto allogenico viene prosposto in alternativa, ove possibile, all’ABMT
qualora quest’ultimo sia ritenuto opportuno.
Ovviamente, la prognosi dei casi trapiantati oltre
la prima RC od in presenza di malattia, argomento qui non discusso, è peggiorativa rispetto a quella dei pazienti trapiantati in prima RC.
Terapia della recidiva
La recidiva nei pazienti con LBL coinvolge nella
maggior parte dei casi le sedi coinvolte inizialmente ma possono comparire localizzazioni al SNC
o midollari. I pazienti in recidiva sono generalmente trattati in maniera analoga ai pazienti con recidiva di LLA ad alto rischio e con trattamenti sistemici a più farmaci.
L’outcome del paziente con LBL in recidiva è in
genere molto sfavorevole e inversamente corre-
55
56
Seminari di Ematologia Oncologica
lato con la durata della risposta alla prima linea
di trattamento. I pazienti con B-LBL tendono ad
avere una maggiore probabilità di ottenere una
seconda remissione rispetto ai T-LBL.
Nuovi farmaci
Lo sviluppo recente di nuovi farmaci antitumorali
non-mielosoppressori e non cross-resistenti con
i trattamenti convenzionali ha apportato importanti possibilità di miglioramento nel campo delle
malattie linfoproliferative. Limitatamente ai casi di
LBL/LLA tuttavia le novità possibili sono abbastanza limitate (46). Migliori prospettive sembrano offerte dalle piccole molecole, prevalentemente ad attività anti tirosinochinasica e dagli anticorpi monoclonali. Tra gli inibitori delle tirosinochinasi, il farmaco imatinib è utilizzabile nei casi di LBL portatori di cromosoma Ph. L’efficacia e la buona tollerabilità dell’aggiunta di imatinib o dasatinib alla
terapia citotossica sono dimostrate per i casi di LLA
Ph+ ma i dati relativi ai LBL sono molto scarsi.
Oltre agli inibitori delle tirosinochinasi classici sono
attualmente in fase di sviluppo clinico nei LBL gli
inibitori di src, un gruppo di tirosinochinasi non
recettoriali e gli inibitori dell’angiogenesi.
Tra gli anticorpi monoclonali il rituximab fa già parte dei farmaci utlizzati nei casi a fenotipo B e a
maggiore grado di differenziazione, dal momento che l’espressione di CD20 è acquisita in una
fase relativamente tardiva dello sviluppo dei precursori linfocitari. L’aggiunta di rituximab non
determina un aumento della tossicità come
osservato nei LNH B periferici.
Una dimostrazione, chiara e metodologicamente corretta, di aumento dell’efficacia dei trattamenti con l’aggiunta di rituximab non è attualmente
disponibile. In ogni caso prima di decidere se
aggiungere rituximab al trattamento citotossico è
sempre indicato documentare l’espressione di
CD20 sulla biopsia diagnostica.
Target più interessanti del CD20 per lo sviluppo
di nuovi anticorpi monoclonali per i LBL sono rappresentati da antigeni di superficie più precoci,
quali il CD19 e il CD22.
Per entrambe le proteine sono in fase di sviluppo più o meno avanzata anticorpi monoclonali
nudi o coniugati con immunotossine. I risultati di
efficacia non sono attualmente disponibili e
dovranno essere rivalutati nei prossimi anni.
n CONCLUSIONI
Il LBL rappresenta una rara forma di LNH e in base
alle conoscenze attuali non è distinguibile dalla
più comune LLA. La malattia ha un decorso estremamente aggressivo e necessita di un trattamento immediato con programmi intensivi.
Nonostante la rarità della malattia ed il grande
numero di approcci utilizzati nel trattamento del
LBL dell’adulto, si possono fare alcune considerazioni:
- i trattamenti più intensivi, o LLA-like di ultima
generazione, sembrano superiori ai programmi
LNH-like;
- induzioni-consolidamenti di breve durata necessitano di mantenimento per ridurre il rischio di ricaduta;
- è necessaria una CT profilattica intratecale per
ridurre il rischio di ripresa a livello del SNC;
- il trapianto autologo ed allogenico sono stati utilizzati come consolidamento dopo una fase di
induzione-consolidamento convenzionale. Le
procedure sembrano migliorare la DFS ma non
la sopravvivenza. Il trapianto allogenico è ancora caratterizzato da un’elevata mortalità e questo
fa sì che la OS e la DFS ottenute da trapianto
autologo ed allogenico siano uguali;
- la CT ha una durata di 1-3 anni. La procedura trapiantologica viene sviluppata nell’arco di 4-6 mesi:
questo sembra migliorare la qualità di vita dei
pazienti. In genere, per il rischio peri-procedurale,
si preferisce l’utilizzo del trapianto autologo come
prima scelta e del trapianto allogenico come seconda scelta, quindi come salvataggio;
- non sappiamo, a tutt’oggi, quale terapia sia realmente più indicata poiché manca completamente la definizione di alto rischio nel LBL dell’adulto.
Dal momento che non sono disponibili a tutti
oggi modelli prognostici clinici e/o biologicimolecolari efficaci e validati per valutare l’outcome del LBL e stabilire quali pazienti possano o debbano accedere alle terapie intensificate di tipo trapiantologico, è necessario continuare a condurre programmi di ricerca con
l’obbiettivo di raccogliere casistiche ampie ed
omogenee per cercare di identificare parametri predittivi di alto rischio a evoluzione sfavorevole e definire con approccio moderno l’effetto dei trattamenti sistemici.
Linfoma linfoblastico dell’adulto
n BIBLIOGRAFIA
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA,
Stein H, Thiele J, Vardiman JW. WHO Classification of
tumours of haematopoietic and lymphoid tissues, fourth
edition. IARC WHO Classification of Tumours, No 2 ed
2008.
2. Murphy SB. Childhood non-Hodgkin’s lymphoma. The
New England journal of medicine. 1978; 299: 1446-8.
3. Faderl S, Jeha S, Kantarjian HM. The biology and therapy of adult acute lymphoblastic leukemia. Cancer.
2003; 98: 1337-54.
4. Smith OP, Hann I. Pathology of leukemia. Pediatric
Oncology. 3rd ed. London: Arnold 2004; 83-100.
5. Pui CH, Ribeiro RC, Hancock ML, Rivera GK, Evans
WE, Raimondi SC, et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Eng J Med. 1991; 325: 16827.
6. Zuna J, Cave H, Eckert C, Szczepanski T, Meyer C,
Mejstrikova E, et al. Childhood secondary ALL after ALL
treatment. Leukemia. 2007; 21: 1431-5.
7. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton
DA, Gralnick HR, et al. Proposals for the classification
of the acute leukaemias. French-American-British
(FAB) co-operative group. Br J Haematol. 1976; 33:
451-8.
8. Sander CA, Jaffe ES, Gebhardt FC, Yano T, Medeiros
LJ. Mediastinal lymphoblastic lymphoma with an immature B-cell immunophenotype. Am J Surg Pathol. 1992;
16: 300-5.
9. Riley RS, Massey D, Jackson-Cook C, Idowu M,
Romagnoli G. Immunophenotypic analysis of acute
lymphocytic leukemia. Hematol/Oncol Clin. 2002; 16:
245-99.
10. Chetty R, Pulford K, Jones M, Mathieu-Mahul D, Close
P, Hussein S, et al. SCL/Tal-1 expression in T-acute lymphoblastic leukemia: an immunohistochemical and
genotypic study. Human pathology. 1995; 26: 994-8.
11. Lewis RE, Cruse JM, Sanders CM, Webb RN, Tillman
BF, Beason KL, et al. The immunophenotype of preTALL/LBL revisited. Exp Mol Pathol. 2006; 81: 162-5.
12. Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical
significance of cytogenetic abnormalities in adult acute
lymphoblastic leukemia. Blood. 1998 ; 91: 3995-4019.
13. Moorman AV, Harrison CJ, Buck GA, Richards SM,
Secker-Walker LM, Martineau M, et al. Karyotype is an
independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data
from patients treated on the Medical Research Council
(MRC) UKALLXII/Eastern Cooperative Oncology Group
(ECOG) 2993 trial. Blood. 2007; 109: 3189-97.
14. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton
J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is
characterized by the deletion of Ikaros. Nature. 2008;
453: 110-4.
15. Iacobucci I, Lonetti A, Messa F, Cilloni D, Arruga F,
Ottaviani E, et al. Expression of spliced oncogenic
Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine
kinase inhibitors: implications for a new mechanism of
resistance. Blood. 2008; 112: 3847-55.
16. Mancini M, Scappaticci D, Cimino G, Nanni M,
Derme V, Elia L, et al. A comprehensive genetic classification of adult acute lymphoblastic leukemia (ALL):
analysis of the GIMEMA 0496 protocol. Blood. 2005;
105: 3434-41.
17. Zhu YM, Zhao WL, Fu JF, Shi JY, Pan Q, Hu J, et al.
NOTCH1 mutations in T-cell acute lymphoblastic
leukemia: prognostic significance and implication in
multifactorial leukemogenesis. Clin Cancer Res. 2006;
12: 3043-9.
18. Paulsson K, Cazier JB, Macdougall F, Stevens J,
Stasevich I, Vrcelj N, et al. Microdeletions are a general feature of adult and adolescent acute lymphoblastic leukemia: unexpected similarities with
pediatric disease. Proceedings of the National
Academy of Sciences of the United States of America.
2008; 105: 6708-13.
19. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard
C, Raimondi SC, et al. Gene expression signatures
define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002; 1: 75-87.
20. Lin YW, Aplan PD. Gene expression profiling of precursor T-cell lymphoblastic leukemia/lymphoma identifies oncogenic pathways that are potential therapeutic targets. Leukemia. 2007; 21: 1276-84.
21. Martinez-Delgado B, Melendez B, Cuadros M, Alvarez
J, Castrillo JM, Ruiz De La Parte A, et al. Expression
profiling of T-cell lymphomas differentiates peripheral
and lymphoblastic lymphomas and defines survival
related genes. Clin Cancer Res. 2004; 10: 4971-82.
22. Raetz EA, Perkins SL, Bhojwani D, Smock K, Philip M,
Carroll WL, et al. Gene expression profiling reveals
intrinsic differences between T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma.
Pediatr Blood Cancer. 2006; 47: 130-40.
23. Uyttebroeck A, Vanhentenrijk V, Hagemeijer A, Boeckx
N, Renard M, Wlodarska I, et al. Is there a difference
in childhood T-cell acute lymphoblastic leukaemia and
T-cell lymphoblastic lymphoma? Leuk Lymphoma.
2007; 48: 1745-54.
24. A clinical evaluation of the International Lymphoma
Study Group classification of non-Hodgkin’s lymphoma.
The Non-Hodgkin’s Lymphoma Classification Project.
Blood. 1997; 89: 3909-18.
25. Slater DE, Mertelsmann R, Koziner B, Higgins C,
McKenzie S, Schauer P, et al. Lymphoblastic lymphoma
in adults. J Clin Oncol. 1986; 4: 57-67.
26. Hoelzer D, Gokbuget N, Digel W, Faak T, Kneba M,
Reutzel R, et al. Outcome of adult patients with T-lymphoblastic lymphoma treated according to protocols
for acute lymphoblastic leukemia. Blood. 2002; 99:
4379-85.
57
58
Seminari di Ematologia Oncologica
27. Thomas DA, O’Brien S, Cortes J, Giles FJ, Faderl S,
Verstovsek S, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood. 2004; 104:
1624-30.
28. Sweetenham JW, Santini G, Qian W, Guelfi M, Schmitz
N, Simnett S, et al. High-dose therapy and autologous
stem-cell transplantation versus conventional-dose
consolidation/maintenance therapy as postremission
therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European
Group for Blood and Marrow Transplantation and the
United Kingdom Lymphoma Group. J Clin Oncol. 2001;
19: 2927-36.
29. Bailey LC, Lange BJ, Rheingold SR, Bunin NJ. Bone
marrow relapse in paediatric acute lymphoblastic leukaemia. Lancet Oncol. 2008; 9: 873-83.
30. Bruggemann M, Raff T, Flohr T, Gokbuget N, Nakao
M, Droese J, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;
107: 1116-23.
31. Nathwani BN, Diamond LW, Winberg CD, Kim H,
Bearman RM, Glick JH, et al. Lymphoblastic lymphoma: a clinicopathologic study of 95 patients.
Cancer. 1981; 48: 2347-57.
32. Murphy SB. Management of childhood non-Hodgkin’s
lymphoma. Cancer Treat Rep. 1977; 61: 1161-73.
33. Aur RJ, Hustu HO, Simone JV, Pratt CB, Pinkel D.
Therapy of localized and regional lymphosarcoma of
childhood. Cancer. 1971; 27: 1328-31.
34. Wollner N, Exelby PR, Lieberman PH. Non-Hodgkin’s
lymphoma in children: a progress report on the original patients treated with the LSA2-L2 protocol.
Cancer. 1979; 44: 1990-9.
35. Mora J, Filippa DA, Qin J, Wollner N. Lymphoblastic
lymphoma of childhood and the LSA2-L2 protocol: the
30-year experience at Memorial-Sloan-Kettering
Cancer Center. Cancer. 2003; 98: 1283-91.
36. Hvizdala EV, Berard C, Callihan T, Falletta J, Sabio H,
Shuster JJ, et al. Lymphoblastic lymphoma in children
- a randomized trial comparing LSA2-L2 with the ACOP+ therapeutic regimen: a Pediatric Oncology Group
Study. J Clin Oncol. 1988; 6: 26-33.
37 Coleman CN, Picozzi VJ, Jr., Cox RS, McWhirter K,
Weiss LM, Cohen JR, et al. Treatment of lymphoblastic lymphoma in adults. J Clin Oncol. 1986; 4: 162837.
38. Mazza P, Bertini M, Macchi S, Lauria F, Pileri S, Rivano
MT, et al. Lymphoblastic lymphoma in adolescents and
adults. Clinical, pathological and prognostic evaluation.
Eur J Cancer Clin Oncol. 1986; 22: 1503-10.
39. Levine AM, Forman SJ, Meyer PR, Koehler SC,
Liebman H, Paganini-Hill A, et al. Successful therapy
of convoluted T-lymphoblastic lymphoma in the adult.
Blood. 1983; 61: 92-8.
40. Bernasconi C, Brusamolino E, Lazzarino M, Morra E,
Pagnucco G, Orlandi E. Lymphoblastic lymphoma in
adult patients: clinicopathological features and
response to intensive multiagent chemotherapy analogous to that used in acute lymphoblastic leukemia.
Ann Oncol. 1990; 1: 141-6.
41. Levine JE, Harris RE, Loberiza FR, Jr., Armitage JO,
Vose JM, Van Besien K, et al. A comparison of allogeneic and autologous bone marrow transplantation for lymphoblastic lymphoma. Blood. 2003; 101:
2476-82.
42. Peniket AJ, Ruiz de Elvira MC, Taghipour G,
Cordonnier C, Gluckman E, de Witte T, et al. An
EBMT registry matched study of allogeneic stem cell
transplants for lymphoma: allogeneic transplantation
is associated with a lower relapse rate but a higher
procedure-related mortality rate than autologous
transplantation. Bone Marrow Transplant. 2003; 31:
667-78.
43. Le Gouill S, Lepretre S, Briere J, Morel P, Bouabdallah
R, Raffoux E, et al. Adult lymphoblastic lymphoma: a
retrospective analysis of 92 patients under 61 years
included in the LNH87/93 trials. Leukemia. 2003; 17:
2220-4.
44. Kim SW, Tanimoto TE, Hirabayashi N, Goto S, Kami
M, Yoshioka S, et al. Myeloablative allogeneic
hematopoietic stem cell transplantation for nonHodgkin lymphoma: a nationwide survey in Japan.
Blood. 2006; 108: 382-9.
45. Song KW, Barnett MJ, Gascoyne RD, Chhanabhai M,
Forrest DL, Hogge DE, et al. Primary therapy for adults
with T-cell lymphoblastic lymphoma with hematopoietic stem-cell transplantation results in favorable outcomes. Ann Oncol. 2007; 18: 535-40.
46. Larson RA. Three new drugs for acute lymphoblastic
leukemia: nelarabine, clofarabine, and forodesine.
[Abstract]. Sem Oncol. 2007; 34 (Suppl 5): S13-20.
47. Morel P, Lepage E, Brice P, Dupriez B, D’Agay MF,
Fenaux P, et al. Prognosis and treatment of lymphoblastic lymphoma in adults: a report on 80 patients. J Clin
Oncol. 1992; 10: 1078-85.
48. Sweetenham JW, Liberti G, Pearce R, Taghipour G,
Santini G, Goldstone AH. High-dose therapy and autologous bone marrow transplantation for adult patients
with lymphoblastic lymphoma: results of the European
Group for Bone Marrow Transplantation. J Clin Oncol.
1994; 12: 1358-65.
49. Milpied N, Ifrah N, Kuentz M, Maraninchi D, Colombat
P, Blaise D, et al. Bone marrow transplantation for adult
poor prognosis lymphoblastic lymphoma in first complete remission. Br J Haemat. 1989; 73: 82-7.
50. Santini G, Coser P, Chisesi T, Porcellini A, Sertoli R,
Contu A, et al. Autologous bone marrow transplantation for advanced stage adult lymphoblastic lymphoma
in first complete remission. Report of the NonHodgkin’s Lymphoma Cooperative Study Group
(NHLCSG). Ann Oncol. 1991; (Suppl 2) 9: 181-5.
51. Baro J, Richard C, Sierra J, Garcia-Conde J, Garcia
Larana J, Rocha E, et al. Autologous bone marrow
Linfoma linfoblastico dell’adulto
transplantation in 22 adult patients with lymphoblastic lymphoma responsive to conventional
dose chemotherapy. Bone Marrow Transplant.
1992; 10: 33-8.
52. Verdonck LF, Dekker AW, de Gast GC, Lokhorst HM,
Nieuwenhuis HK. Autologous bone marrow transplantation for adult poor-risk lymphoblastic lymphoma in
first remission. J Clin Oncol. 1992; 10: 644-6.
53. Jost LM, Jacky E, Dommann-Scherrer C, Honegger
HP, Maurer R, Sauter C, et al. Short-term weekly
chemotherapy followed by high-dose therapy with
autologous bone marrow transplantation for lymphoblastic and Burkitt’s lymphomas in adult patients.
Ann Oncol. 1995; 6: 445-51.
54. Zinzani PL, Bendandi M, Visani G, Gherlinzoni F, Frezza
G, Merla E, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk
Lymphoma. 1996; 23: 577-82.
55. Bouabdallah R, Xerri L, Bardou VJ, Stoppa AM, Blaise
D, Sainty D, et al. Role of induction chemotherapy and
bone marrow transplantation in adult lymphoblastic
lymphoma: a report on 62 patients from a single center. Ann Oncol. 1998; 9: 619-25.
56. Conde E, Zuazu J, Lopez-Guillermo A. Autologous
stem cell trasplant (ASCT) for lymphoblastic lymphoma
(LBL). [Abstract]. Blood. 1999; 94 (Suppl 1): 477a.
57. Aljurf M, Zaidi SZ. Chemotherapy and hematopoietic
stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol
Blood Marrow Transplant. 2005; 11: 739-54.
59
61
Linfomi non Hodgkin
T/NK
ANNALISA PELI, GIUSEPPE ROSSI
Struttura Complessa di Ematologia, Spedali Civili, Brescia
n INTRODUZIONE
I linfomi T comprendono numerose entità patologiche spesso rare e tuttora solo parzialmente
conosciute. Una loro trattazione approfondita è
al di fuori dello scopo del presente lavoro, nel quale verranno trattati in particolare gli aspetti clinico patologici rilevanti delle entità relativamente più
frequenti nell’ambito dei linfomi T sistemici, cioè
i linfomi T periferici, non altrimenti specificati, il linfoma anaplastico, il linfoma angioimmunoblastico e i linfomi T/NK.
n CARATTERISTICHE GENERALI
Le neoplasie a cellule T/NK sono patologie clonali dei linfociti T o NK, maturi o immaturi, a diversi livelli di differenziazione. Nella più recente classificazione della World Health Organization (WHO)
del 2008 i linfomi T e NK sono considerati insieme per la stretta correlazione tra i due subsets di
linfociti, in termini sia di caratteristiche immunofenotipiche sia di funzione.
Parole chiave: linfomi T periferici, caratteristiche clinico-patologiche, terapia
Indirizzo per la corrispondenza
Dr. Giuseppe Rossi
Struttura Complessa di Ematologia
Spedali Civili di Brescia
Piazzale Spedali Civili, 1 - 25100 Brescia
E-mail: [email protected]
Giuseppe Rossi
La classificazione WHO delle neoplasie linfoidi si
basa sulle caratteristiche biologiche (morfologia,
immunofenotipo, genetica, biologia molecolare),
sulla presentazione clinica e sulla correlazione tra
ciascuna entità e la propria controparte normale, considerato che le neoplasie linfoidi sembrano ricapitolare i diversi stadi differenziativi della
linfopoiesi stessa.
Le neoplasie che derivano dai precursori dei linfociti T (dal progenitore staminale fino a timociti
midollari) sono raggruppate sotto il nome di TLymphoblastic Lymphoma/Leukemia; mentre le
neoplasie che derivano dalle cellule T mature
(post-timiche) e dalle cellule NK sono raggruppate sotto il nome di Peripheral (mature) T-cell
(PTCLs) and NK cell lymphomas/leukemias. La
categoria dei PTCLs comprende un gruppo molto eterogeneo di entità tra cui è possibile distinguere le forme “specificate”, con caratteristiche
clinico-patologiche ben distinte, dalle forme “non
altrimenti specificate”, fra le quali sono verosimilmente comprese malattie diverse ma non ancora sufficientemente caratterizzate.
La lista completa dei linfomi a cellule T/NK mature secondo la terza e la quarta classificazione
WHO, pubblicate rispettivamente nel 2001 e nel
2008, è riportata in tabella 1 (1). Nella classificazione WHO, pubblicata nel 2001, i PTCLs erano
stati suddivisi in diverse categorie: le entità ad
espressione prevalentemente leucemica/disseminata, extranodale, cutanea e linfonodale (4). Tale
suddivisione non è stata mantenuta dalla classificazione WHO del 2008 per la frequente sovrapposizione tra le diverse categorie (1). Inoltre, come
62
Seminari di Ematologia Oncologica
indicato in tabella 1, nella classificazione del 2008
sono state riconosciute nuove entità ed entità
provvisorie.
La classificazione WHO comprende le forme extranodali cutanee, fra cui la micosi fungoide, che è
l’entità largamente più frequente fra i linfomi T. Essa
si presenta prevalentemente in stadio iniziale e a
livello esclusivamente cutaneo dove tende a
rimanere durante gran parte del suo decorso clinico. Per questo la sua gestione, così come quella degli altri linfomi T cutanei, è per molto tempo
di tipo esclusivamente dermatologico nella maggior parte dei pazienti. Di conseguenza esiste una
classificazione EORTC dei linfomi cutanei (2), internazionalmente accettata e utilizzata, solo parzialmente integrata nella classificazione WHO, ed esiste un sistema di stadiazione specifico della micosi fungoide (3), che prescinde dai criteri classifi-
cativi di Ann Arbor utilizzati nei linfomi. Il ruolo dell’ematologo è marginale e limitato alle fasi terminali di queste patologie, quali la sindrome di Sezary,
mentre sarebbe auspicabile una maggiore collaborazione per migliorare risultati terapeutici tuttora insoddisfacenti. La gestione dei più rari PTCL
è invece prettamente emato-oncologica.
I PTCLs presentano le caratteristiche immunofenotipiche e genetiche dei linfociti T post-timici o
delle cellule NK mature (1, 5). Ad esempio, la leucemia prolinfocitica T (T-PLL) deriva dalle cellule
T naive allo stadio maturativo intermedio tra il timocita corticale e il timocita naive della zona midollare; i linfomi T extranodali derivano dai linfociti T
citotossici; i linfociti T γδ danno origine al linfoma
epatosplenico (hepatosplenic T cell lymphoma,
HSTL) mentre i linfociti T αβ dell’epitelio intestinale danno origine al linfoma T enteropatico
Classificazione WHO del 20014
Classificazione WHO del 20081
Entità ad espressione leucemica/disseminata
Leucemia Prolinfocitica T
Leucemia a grandi linfociti granulari T
Leucemia a cellule NK, aggressiva
Leucemia/linfoma a cellule T dell’adulto (HTLV1 positiva)
Leucemia Prolinfocitica T
Leucemia a grandi linfociti granulari T
Disordini linfoproliferativi cronici dei linfociti NKa
Leucemia a cellule NK, aggressiva
Leucemia/linfoma a cellule T dell’adulto (HTLV1 positiva)
Malattie sistemiche linfoproliferative a cellule T, EBV+,
del bambinoa
Entità ad espressione extranodale
Linfoma extranodale a cellule NK/T, di tipo nasale
Linfoma T Enteropatico
Linfoma T Epatosplenico
Linfoma T sottocutaneo, simil-panniculitico
Linfoma extranodale a cellule NK/T, di tipo nasale
Linfoma T Enteropatico
Linfoma T Epatosplenico
Linfoma T sottocutaneo, simil-panniculitico
Entità ad espressione cutanea
Micosi Fungoide
Sindrome di Sezary
Disordini Linfoproliferativi a cellule T CD30+,
primitivi cutanei
Micosi Fungoide
Sindrome di Sezary
Disordini Linfoproliferativi a cellule T CD30+, primitivi cutanei
Linfomi a cellule γδ, primitivi cutaneia
Entità ad espressione nodale
Linfoma a cellule T periferiche, non altrimenti specificato
Linfoma T angioimmunoblastico
Linfoma a grandi cellule anaplastiche
Linfoma a cellule T periferiche, non altrimenti specificato
Linfoma T angioimmunoblastico
Linfoma a grandi cellule anaplastiche, ALK positivoa
Linfoma a grandi cellule anaplastiche, ALK negativob
γδ, gamma delta; ALK, tirosin chinasi del linfoma anaplastico; EBV +, virus di Epstein Barr; HTLV1, virus dei linfociti T dell’uomo; NK, cellule natural killer.
Indicati come nuova entità; bIndicati come entità provvisoria.
a
TABELLA 1 - Confronto tra le classificazioni della WHO dei linfomi a cellule T/NK mature.
Linfomi non Hodgkin T/NK
CD3 CD4 CD8 CD7 CD5 CD2 TIA1 GrB Per CD30 CD25 CD56 CD16 CD57 BCL6 CD10 EBV EMA
ATLL
+
+
-
-
+
+
-
-
-/+
++
-
-
-
-
-
-
-
ENK/T,
+c
Nasal Type
-
-/+
-
-
+
+
+
-
-
+
-
-
-
-
+
-
EATL
+
-
-/+
+
-
+
+
+
-/+
-/+
-/+
-
-
-
-
-
-/+
HSTL
+
-
+/-
+
-
+
+
-
-
-
+
-
-
-
-
-
-
MF/SS
+
+
-/+
-/+
+/-
+
-
-
-
-
-
-
-
-
-
-
-
AITL
+
+
-
+
+
+
-
-
-
-
-
-
-
+/-
+/-
-
-
+/- -/+
-/+
-/+
+
-
-
-/+
-
-
-
-
-
-
-
-
ALCL, ALK+-/+ -/+ -/+
-/+
-/+
-/+
+
+
++
++
+/-
-
-
+
-
-
++
ALCL, ALK- +/- +/- -/+
-/+
+/-
+/-
+/-
+/-
++
++
+/-
-
-
-
-
-
+\
PTCL-NOS +
AITL, linfoma T angioimmunoblastico; ALCL, linfoma anaplastico a grandi cellule; ALK, anaplastic lymphoma kinase; ATLL, leucemia/linfoma T “adulta”; EATL,
linfoma T associate a enteropatia; EBV +, Epstein Barr virus; ENK/T linfoma T/NK extranodale “nasal type”, HSTL, linfoma T epatosplenico; HTLV1,” human
T lymphotropic virus-1”; MF/SS, Micosi fungoide/syndrome di Sezary; NK, natural killer; PTCL-NOS, linfoma T periferico, non altrimenti specificato; GrB granzyme B; Pr, perforina.
TABELLA 2 - Caratteristiche immunofenotipiche dei linfomi derivanti da cellule T/NK mature.
(Enteropathy Associated T-cell lymphomas, EATL)
e infine la leucemia/linfoma a cellule T adulte
(ATLL) deriva da un particolare subset di T CD4+
con fenotipo regolatorio (CD25+FoxP3+). Le
caratteristiche immunofenotipiche dei PTCLs
sono riportate in tabella 2 (1). L’immunoistochimica
generalmente mostra l’espressione di molecole
associate alle cellule T ma il fenotipo è aberrante in circa l’80% dei casi (6, 7).
Le alterazioni del cariotipo non rappresentano un
criterio classificativo; il cariotipo è aberrante in oltre
l’80% dei casi di PTCLs e spesso è complesso
(8). Sono state individuate alterazioni specifiche
del cariotipo nel linfoma anaplastico a grandi cellule (ALCL), associato a traslocazioni che coinvolgono il gene ALK (anaplastic lymphoma kinase)
sul cromosoma 5 e nel linfoma T epatosplenico
associato all’isocromosoma 7q (1). Lo studio del
riarrangiamento dei geni che codificano per il TCR
ne mostra un riarrangiamento clonale, anche se
non in tutti i casi di PTCL (9).
n EPIDEMIOLOGIA E FATTORI
DI RISCHIO
I dati epidemiologici relativi ai PTCLs derivano
dall’International Peripheral T-Cell Lymphoma
Project, un ampio studio multicentrico, internazionale, retrospettivo che ha coinvolto 1153 pazienti da 22 centri (10). La frequenza delle diverse entità raggruppate sotto il nome di PTCLs secondo
questo studio è riportata in figura 1 (10). In ordine di frequenza, i sottotipi più comuni sono i
PTCL, non altrimenti specificati (NOS 25,9%), il
linfoma T angioimmunoblastico (18,5%) e i linfomi T/NK extranodali, di tipo nasale (10,4%). Tra
le entità più frequenti seguono l’ATLL (9,6%), il linfoma a grandi cellule anaplastiche ALK positivo
(6,6%), ALK negativo (5,5%) e il linfoma enteropatico (4,7%). Tutte le restanti entità specified non
rappresentano più del 2% di tutti i PTCLs. Nello
studio erano state incluse anche altre entità non
specificate (1,8%), classificate non correttamente come PTCLs nel 10,4% dei casi e, in seguito
alle revisioni eseguite dall’International TCL
Project, risultate essere linfomi di Hodgkin (3%),
linfomi a cellule B (1,4%) altri linfomi (2,3%) o, nel
restante 3.6% dei casi, non classificabili per difficoltà tecniche o inadeguatezza del materiale istologico (10).
In generale, i linfomi T/NK periferici rappresentano circa il 12% dei linfomi non Hodgkin (10). La
loro incidenza varia a seconda della razza e della area geografica: in Occidente rappresentano il
15-20% dei linfomi aggressivi e il 5-10% dei lin-
63
64
Seminari di Ematologia Oncologica
1,7%
1,4%
0,9%
2,5%
n PTCL, NOS
n E NK/TCL
n ALCL, ALK+
n EATL
n HSTL
n PTCLs non classificabili
12,2%
25,9%
4,7%
5,5%
n AITL
n ATLL
n ALCL, ALKn Primary cutaneous ALCL
n SPTCL
n Altri disordini
Modificato da (10).
18,5%
6,6%
AITL, linfoma T angioimmunoblastico; ALCL, linfoma T a grandi cellule anaplastiche; ATLL, linfoma/leucemia a cellule Tdell’adulto; E
NK/TCL linfoma T/NK extranodale, di tipo nasale; EATL linfoma T entoropatico; HSTL linfoma T epatosplenico; PTCL, NOS linfoma T periferico, non altrimenti specificato; SPTCL linfoma T sottocutaneo similpanniculitico.
9,6%
10,4%
FIGURA 1 - Distribuzione dei diversi sottotipi di PTCLs secondo International T Cell Lymphoma Project.
ne dei PTCLs non sono del tutto note. Una maggiore esposizione e suscettibilità genica ad agenti patogeni quali l’HTLV1 e il virus di Epstein Barr
si associano all’elevata incidenza di ATLL e linfomi T/NK EBV-correlati in Asia rispetto all’Europa
e al Nord America (11). È riportato che nelle regio-
fomi non Hodgkin; mentre in Asia il 15-20% di tutti i linfomi non Hodgkin (10). L’incidenza dei principali sottotipi nel Nord America, in Europa e in
Asia secondo l’International PTCLs Project (10),
è riportata in tabella 3. Le cause biologiche delle differenze geografiche/razziali nella distribuzio-
PTCL-NOS
Linfoma T angioimmunoblastico
ALCL, ALK+
ALCL, ALK NK/TCL
ATLL
Linfoma T Enteropatico
Linfoma T Epatosplenico
ALCL primitivo cutaneo
Linfoma T sottocutaneo, simil panniculitico
Linfomi T non classificabili
Nord America (%)
Europa (%)
Asia (%)
34,4
16,0
16,0
7,8
5,1
2,0
5,8
3,0
5,4
1,3
2,3
34,4
28,7
6,4
9,4
4,3
1,0
9,1
2,3
0,8
0,5
3,3
22,4
17,9
3,2
2,6
22,4
25,0
1,9
0,2
0,7
1,3
2,4
ALCL, linfoma a grandi cellule analplastiche; ATLL, leucemia/linfoma a cellule T dell’adulto; NK/TCL, linfomi T/NK; PTCL-NOS, linfomi T periferici, non altrimenti specificati.
TABELLA 3 - Distribuzione geografica dei principali linfomi T/NK periferici secondo l’International Peripheral T-cell Lymphoma
Project (10).
Linfomi non Hodgkin T/NK
n PROGNOSI
ni del Giappone dove l’infezione da HTLV1 è endemica (prevalenza dell’infezione 8-10%), il rischio
di sviluppare una ATLL è del 6,9% per i maschi
sieropositivi e del 2,9% per le femmine (1). Le cause del riscontro di una maggiore incidenza del linfoma angioimmunoblastico in Europa e del linfoma a grandi cellule anaplastiche ALK positivo nel
Nord America non sono ancora note (10). È nota
invece una correlazione tra il linfoma enteropatico e l’enteropatia da glutine, associata agli aplotipi HLA DQ2 e HLA DQ8, diffusi nella popolazione del Nord Europa, dove il linfoma enteropatico
è più diffuso (10, 12).
Linfoma a grandi cellule anaplastiche, ALK+
Linfoma a grandi cellule anaplastiche, ALK –
Linfomi T/NK
Linfomi T periferici, non altrimenti specificati
Linfoma angioimmunoblastico
Leucemia/linfoma a cellule T dell’adulto
100
90
Overall survival (%)
I PTCLs rientrano nella categoria dei linfomi
aggressivi. L’overall survival (OS) dei PTCLs è
riportata in figura 2 e in tabella 4 (10). Nessuno
dei linfomi T sistemici, ad eccezione del rarissimo linfoma T sottocutaneo simil-panniculitico e
del ALCL ALK-positivo, ha sopravvivenze a 10
anni superiori al 50%. In particolare, la prognosi
più infausta è dei pazienti affetti da linfoma epatosplenico (OS 7%, failure free survival (FFS) 0%),
da ATLL (OS 14%, FFS 12%) e da linfoma enteropatico (OS 20%, FFS 4%) (10). Anche i più fre-
80
70
FIGURA 2
60
50
40
30
20
10
0
P<.001
1
2
3
4
5
6
7
8
9 10 11 12 13 14 15 16 17 18
Tempo (anni)
OS a 5 anni
FFS a 5 anni
Diagnosi
%
IPI 0/1
IPI 4/5
%
IPI 0/1
IPI 4/5
PTCL, NOS
Linfoma angioimmunoblastico
NK/TCL, nasale
NK/TCL, extranasale
ATLL
ALCL, ALK+
ALCL, ALK Linfoma enteropatico
ALCL, primitivo cutaneo
Linfoma epatosplenico
Linfoma sottocutaneo, simil panniculitico
32
32
42
9
14
70
49
20
90
7
64
50
56
57
17
28
90
74
29
100
0
60
11
25
0
20
7
33
13
14
NA
0
0
20
18
29
6
12
60
36
4
55
0
24
33
34
53
21
26
80
62
7
62
0
30
6
16
0
20
0
25
13
14
NA
0
0
IPI, International Prognostic Index; PTCL, NOS linfoma T periferico, non altrimenti specificato; TCL, linfoma T; ALCL, linfoma a grandi cellule anaplastiche;
NA, non applicabile; OS = overall survival; FFS = failure free survival
TABELLA 4 - Sopravvivenza dei pazienti affetti da PTCLs in base all’istotipo e al’IPI secondo l’International Peripheral T Cell
Lymphoma Project (10).
65
66
Seminari di Ematologia Oncologica
Fattore prognostico
Tipo di PTCLs
Bibliografia
Istotipo
Tutte le entità
(10), (14), (15), (16)
Tutte le entità
ALCL
ATLL
NK/T cell lymphoma
(14)
(15), (17)
(18)
(19)
PTCL/NOS
(15), (20)
PTCL/NOS, AITL
(7), (21)
Indice prognostico koreano
NK/T cell lymphoma
(22)
Indice prognostico NK
NK/T cell lymphoma
(23)
Virus di Epstein Barr
PTCL, NOS
NK/T cell lymphoma
(7), (24), (25)
(26), (27)
Indice proliferativo (Ki67)
PTCL, NOS
(7)
Derivazione cellulare
PTCL, NOS
(7)
Attivazione di NFkB
PTCL, NOS
(21), (28)
International Prognostic Index (IPI)
PIT
Bologna score
AITL, linfoma T angioimmunoblastico; ALCL, linfoma a grandi cellule anaplastiche; ATLL, leucemia/linfoma a cellule T dell’adulto; NK, natural killer; PTCLNOS, linfomi T periferici, non altrimenti specificati.
TABELLA 5 - Fattori prognostici e score nei PTCLs.
quenti PTCL, NOS e angioimmunoblastico sono
tra i più aggressivi con un relapse free survival e
overall survival a 5 anni intorno al 30%. Il linfoma a grandi cellule anaplastiche ALK positivo ha
una prognosi significativamente migliore rispetto
all’ALK negativo (OS 70% vs 49% rispettivamente); mentre quando ha una localizzazione cutanea
primitiva, il linfoma anaplastico ha una prognosi
ancora migliore. (OS 90%) (10). Per quanto riguarda i linfomi T/NK, l’OS a 5 anni delle forme a localizzazione esclusivamente nasale è pari al 42%
rispetto alle forme extranasali che hanno una prognosi nettamente più sfavorevole (OS 9%) (10).
Da questi dati risulta quindi che:
1) l’istotipo è uno dei principali fattori prognostici;
2) le forme nodali vanno considerate entità cliniche distinte dalle forme extranodali, in particolare cutanee, caratterizzate da una prognosi significativamente più favorevole;
3) nell’ambito dei PTCLs nodali, l’ALCL va distinto dalle altre entità così come i linfomi ALCL
ALK+ vanno distinti dai ALCL ALK-.
I fattori prognostici e gli score elaborati per la stratificazione del rischio sono riportati in tabella 5 (13).
Tra i fattori prognostici, l’International Prognostic
Index (IPI), elaborato per i linfomi non Hodgkin
aggressivi, può essere utile nella stratificazione del
rischio anche dei pazienti affetti da PTCLs e da
NK/TCL anche se molti pazienti hanno una prognosi sfavorevole nonostante un IPI score basso (10). Le categorie in cui l’IPI è di scarsa utilità, dai risultati del International T Cell Lymphoma
Project, sono l’ATLL, il linfoma T enteropatico, il
linfoma T epatosplenico e il linfoma T/NK extranasale (10).
n ENTITÀ CLINICO-PATOLOGICHE
SPECIFICHE
Linfoma T periferico, non altrimenti
specificato (PTCL-NOS)
I PTCL-NOS sono il sottogruppo di PTCLs più
comune, rappresentando circa il 30% dei PTCLs.
La maggior parte dei pazienti sono adulti; l’età
mediana di presentazione è la settima decade. Il
rapporto maschi/femmine è 2/1 (29).
Il riscontro di adenopatie diffuse rappresenta
l’esordio più frequente e i sintomi B sono presen-
Linfomi non Hodgkin T/NK
ti nel 45% dei casi, una percentuale nettamente superiore alla media dei linfomi non-Hodgkin;
talvolta si possono associare eosinofilia, prurito
e più raramente una sindrome emofagocitica (29,
30). Il 65% dei pazienti si presenta già in stadio
avanzato di malattia con infiltrati nel midollo
osseo, fegato, milza e in altre sedi extranodali.
Talvolta è possibile riscontrare cellule blastiche
anche nel sangue periferico, anche se la presentazione leucemica è un evento raro. Le sedi extranodali più frequentemente coinvolte sono la cute
e il tratto gastrointestinale (5, 31). Dal punto di
vista istologico, gli infiltrati sono diffusi o paracorticali con sovvertimento della normale architettura del linfonodo stesso (1, 5). Le caratteristiche citologiche sono variabili, da quadri polimorfi a quadri monomorfi. Caratteristiche morfologiche frequenti sono la presenza di cellule di
media/grossa taglia, con citoplasma chiaro,
nucleo irregolare, nucleoli prominenti e molte figure mitotiche. Sono di frequente osservazione
un’intensa vascolarizzazione e un background
infiammatorio formato da eosinofili, linfociti, plasmacellule, grandi cellule B e clusters di istiociti epitelioidi (1, 5). Questi ultimi sono particolarmente numerosi nella variante linfoepitelioide (o
linfoma di Lennert), che consiste di piccoli linfociti T citotossici, più spesso CD8 positivi (1, 5).
È stata recentemente descritta anche la variante follicolare, nella quale le cellule neoplastiche
infiltrano i follicoli linfonodali dando origine ad un
pattern di crescita che ricorda un linfoma B follicolare. Per quanto riguarda invece le sedi extranodali, a livello cutaneo le cellule linfomatose tendono ad infiltrare il derma e il tessuto sottocutaneo, dando origine a noduli che spesso vanno
incontro ad ulcerazione; a livello splenico invece il pattern di crescita varia da noduli solitari o
multipli a livello della polpa bianca ad un’infiltrazione predominante della polpa rossa (1, 5).
I PTCL-NOS sono spesso caratterizzati da un
fenotipo T aberrante con una ridotta espressione del CD5 e CD7 (1). Nella maggior parte dei casi,
soprattutto nelle forme nodali, sono CD4+ CD8(1, 5). La catena beta del T-cell receptor (TCR) ( F1)
è espressa, a differenza dei linfomi a cellule T γδ
e dei linfomi NK (1, 5). Il CD52 è assente nel 60%
dei casi con metodiche immunoistochimiche su
sezioni da materiale in paraffina, ma questo dato,
che ha anche implicazioni terapeutiche vista la
disponibilità di alemtuzumab, un anticorpo monoclonale IgG1 umanizzato specifico per CD52, non
è costante in diversi lavori. Con la citometria a flusso su materiale fresco infatti, la positività del CD52
è stata riportata fino al 90-100% dei casi di PTCLNOS (1, 32-34). Il CD30 può essere espresso,
eccezionalmente con il CD15; ciononostante il
profilo immunofenotipico globale e la morfologia
consentono una diagnosi differenziale dal Linfoma
a grandi cellule T anaplastico e dal linfoma di
Hodgkin (1). L’indice proliferativo è spesso elevato con un’espressione di Ki67 superiore al 70%
e quindi associato ad una prognosi più severa (1).
Nella maggior parte dei casi il riarrangiamento delle sequenze geniche che sovrintendono alla produzione del TCR è clonale (1, 35). Il cariotipo è
spesso complesso, con alterazioni diverse da
quelle osservabili nei AITL e ALCL (1, 5). In particolare addizioni cromosomiche ricorrenti sono
osservabili a carico dei cromosomi 7q (coinvolgendo le chinasi ciclino-dipendenti 6), 8q (coinvolgendo il gene MYC), 17q e 22q; mentre le delezioni sono osservabili a carico di diversi cromosomi (1, 5). È stato riportato che delezioni del cromosoma 5q, 10q e 12q sono associate ad una
prognosi migliore (5, 36). Il virus di Epstein Barr
è integrato nel genoma delle cellule neoplastiche
solo raramente (1, 5).
Anche gli studi del profilo di espressione genica
(GEP, gene expression profiling) hanno confermato l’eterogeneità della categoria PTCL, NOS (1,
5). Rispetto ai linfociti T normali, i PTCL, NOS sono
caratterizzati da una down-regolazione dell’espressione dei geni che regolano la proliferazione, l’apoptosi, l’adesione cellulare ed il rimodellamento della matrice extracellulare. L’overespressione del PDGFR- è stata identificata dagli
studi di GEP e confermata da studi di immunoistochimica. Essa può rappresentare un target terapeutico per inibitori delle tirosinochinasi (1, 5). In
effetti un recente studio di fase II con dasatinib
in pazienti con linfoma recidivato o refrattario ha
mostrato una sensibilità elettiva del farmaco nei
linfomi T periferici rispetto ad altri istotipi (37).
Alcuni lavori riportano che può esserci una upregolazione o una down-regolazione dei geni del
pathway di NF-kB, con possibili differenze in termini di prognosi. In particolare sembra che bas-
67
68
Seminari di Ematologia Oncologica
si livelli di molecole correlate al pathway di NFkB
o l’attivazione di tale pathway siano fattori prognostici favorevoli, con una mediana di sopravvivenza globale di 25 mesi (range 0-124 mesi) contro una mediana di 12 mesi (range 0-19 mesi)
(p=0,032) (28, 29, 39).
I PTCL-NOS sono caratterizzati da una scarsa
risposta alla terapia e da frequenti recidive con
sopravvivenza a 5 anni molto bassa (20-30%) (10).
I fattori prognostici relativi ai PTCL, NOS sono
elencati in tabella 5. I più importanti sono lo stadio di presentazione e l’IPI. Sulla base di un’analisi retrospettiva di 385 pazienti affetti da PTCL,
NOS, l’Intergruppo Italiano Linfomi ha elaborato
un nuovo modello prognostico (the Prognostic
Index for PTCL unspecified, PIT) (20). Il modello
prognostico include il coinvolgimento midollare
oltre all’età, al performance status e alla LDH, considerandolo un fattore prognostico sfavorevole. Il
PIT è risultato leggermente più efficace dell’IPI nello stratificare i pazienti affetti da PTCL, NOS (logrank 66,79 vs 55,94) ed è stato proposto come
strumento di riferimento (13, 20).
Dal punto di vista clinico un fattore prognostico
altamente significativo che sta emergendo recentemente, in generale nei PTCL, è rappresentato
dal numero assoluto di linfociti circolanti all’esordio di malattia. Una conta linfocitaria inferiore a
1000/mm3, presente nel 37% dei pazienti, si associa infatti ad una sopravvivenza mediana di un
mese, rispetto a 59 mesi nei pazienti con linfociti > a 1000/mm3 (40) Anche in recidiva il numero
di linfociti circolanti è l’unico fattore prognostico
indipendente, insieme al performance status, che
ha dimostrato un effetto negativo sulla sopravvivenza dei pazienti (41). Ciò suggerisce un ruolo
negativo dello stato di immunodepressione dei
pazienti, che risulta particolarmente frequente nei
PTCL e consente di interpretare anche la generale tendenza degli stessi a sviluppare infezioni
opportunistiche, talora simili a quelle dei pazienti HIV sieropositivi, in misura nettamente maggiore rispetto ai linfomi di derivazione B linfocitaria.
Linfoma anaplastico a grandi cellule,
ALK positivo
La definizione di “linfoma anaplastico a grandi cellule” è stata per la prima volta utilizzata da Stein
nel 1985 per indicare un linfoma costituito da gran-
di cellule linfoidi anaplastiche, intensamente CD30
positive e con tendenza ad un pattern di crescita coesiva e sinusoidale (15). Studi di immunoistochimica e genetica hanno successivamente
consentito di restringere la definizione ai linfomi a
cellule T o null, ovvero a cellule che hanno perso
l’espressione degli antigeni T ma che presentano
un riarrangiamento clonale del TCR (15). Già nella terza edizione della classificazione WHO, il linfoma primitivo sistemico (ALCL) è stato considerato come entità distinta dal linfoma anaplastico
primitivo cutaneo (Primary Cutaneous ALCL), sulla base di differenze immunofenotipiche e cliniche.
Nel 1994, l’identificazione di una traslocazione cromosomica, t (2;5) (p23;q35) ha messo in evidenza l'eterogeneità della categoria degli ALCL sistemici (2). Nella più recente WHO (2008) il ALCL ALK
positivo è riconosciuto come entità distinta (1); inoltre si stanno accumulando evidenze per le quali
anche il ALCL ALK negativo andrebbe separato
dagli altri PTCLs, ma ad oggi non sono ancora state identificate caratteristiche sufficientemente
distintive ed esso viene considerato per ora come
una entità provvisoria (1).
I ALCLs sono rari; rappresentano circa il 3% dei
LNH, il 12% dei PTCLs e il 10-20% circa dei linfomi in età pediatrica. Mediante l’impiego di anticorpi anti-ALK, l’espressione di ALK è dimostrabile nel 50-85% degli ALCLs sistemici (15).
Il picco di massima incidenza dell’ALCL ALK positivo è in età pediatrica e giovane-adulta, mentre
il picco di incidenza dell’ALCL ALK negativo è in
età avanzata (età mediana 58 anni) (1, 5, 15).
Il riscontro di adenopatie superficiali è la più comune presentazione clinica, anche se è frequente
anche il coinvolgimento di sedi extranodali quali la cute, il midollo osseo e i tessuti molli, soprattutto per il ALCL ALK positivo. Sintomi sistemici, in particolare la febbre, sono comuni. Oltre la
metà dei pazienti esordisce in stadio avanzato (III
o IV) di malattia (5).
- ALCL ALK positivo. Diversi studi hanno dimostrato che i pazienti affetti da ALCL ALK positivo hanno una prognosi più favorevole (OS a 5
anni, 70-80%) rispetto ai pazienti affetti da ALCL
ALK negativo o da altri PTCLs aggressivi a parità di trattamento con regimi di chemioterapia contenenti antracicline; questo potrebbe essere in parte dovuto anche alla giovane età dei pazienti stes-
Linfomi non Hodgkin T/NK
si (15, 42-45). Il ALCL ALK+ è un’entità autonoma dal punto di vista nosografico perché caratterizzata a livello biologico dal riarrangiamento del
gene ALK sul cromosoma 2p23 (1, 5, 46).
Diverse traslocazioni possono coinvolgere il gene
ALK; la più comune è la t(2;5) (p23;q35).
Tale traslocazione porta alla fusione del gene ALK
posto sul cromosoma 2 con il gene Nucleofosmina (NPM) posto sul cromosoma 5; ne risulta
un gene di fusione che codifica per una proteina
chimerica di 80-kDa NPM-ALK, dotata di un’attività tirosinochinasica intrinseca. La cascata
intracellulare che deriva dall’attivazione di ALK
dipende dal tipo di traslocazione (1, 5, 46). Diversi
studi molecolari hanno dimostrato il ruolo dell’oncogene NPM-ALK nel processo di trasformazione neoplastica (46, 47). Studi in vitro hanno dimostrato che proteine chimeriche ALK costitutivamente attivate inducono la trasformazione, la proliferazione e la sopravvivenza cellulare. Il processo di oncogenesi è mediato dall’attivazione di
cascate di segnale intracellulare tra cui il pathway
di JAK3-STAT3, ERK e PI3-Akt (46, 47).
Da un punto di vista morfologico esistono almeno 5 varianti, non correlate alle varianti geniche di
ALK: comune, a piccole cellule, linfoistiocitica,
Hodgkin-like e composita. Tutte le varianti contengono le cosiddette hallmark cells, cellule patognomoniche, caratterizzate da un nucleo eccentrico
e reniforme e una regione di Golgi eosinofila. La
proteina ALK può essere riscontrata sia nel nucleo
sia nel citoplasma delle hallmark cells (1, 5).
Per quanto riguarda l’immunofenotipo, le cellule
neoplastiche sono uniformemente CD30 positive
sia in superficie sia sulla regione di Golgi; frequente è anche la positività di EMA (epithelial membrane antigen) e di markers citossici (TIA-1, perforina e granzyme B). Le cellule neoplastiche mostrano un fenotipo T aberrante con un’espressione
difettiva di molti antigeni T e spesso mostrano un
apparente fenotipo null. In quest’ultimo caso l’origine T cellulare del ALCL ALK positivo è dimostrabile dal riarrangiamento clonale delle catene β e
γ del TCR. L’espressione di CD3 è assente nel 7580% dei casi, così come l’espressione di CD5 e
CD7 e CD8; al contrario marcatori come CD2, CD4
e CD45 sono espressi in molti casi (1, 5).
- ALCL ALK negativo. Come già accennato, nella
più recente classificazione WHO, l’ALCL ALK nega-
tivo è considerato un’entità provvisoria da distinguere sia dall’ALCL ALK positivo sia dai PTCL,
NOS. Il meccanismo di oncogenesi non è noto (1).
La prognosi è significativamente peggiore rispetto all’ALCL ALK positivo (OS a 5 anni, 30-51%) ma
migliore rispetto ai PTCL-NOS (10, 15, 29).
Dal punto di vista della morfologia, l’ALCL ALK
negativo è simile all’ALCL ALK positivo nella
variante comune, anche se le hallmark cells sono
di maggiori dimensioni e più pleomorfe. Non sono
state identificate varianti istologiche specifiche (5).
L’immunofenotipo dell’ALCL ALK negativo è
simile all’ALCL ALK positivo. La diagnosi differenziale tra ALCL ALK negativo e PTCL-NOS può
essere difficoltosa: i PTCL-NOS mostrano
un’espressione del CD30 a diversa intensità in una
minoranza di casi mentre il ALCL ALK negativo
è intensamente CD30 positivo; la perdita di marcatori di superficie della linea T è evento più raro
nei PTCL-NOS così come l’espressione di EMA.
Da un punto di vista pratico, la classificazione
WHO raccomanda che la diagnosi di ALCL ALK
negativo sia posta solo se la morfologia e l’immunofenotipo sono simili ai casi ALK positivi e in
assenza dell’overespressione di ALK (1, 5, 29).
Linfoma T angioimmunoblastico
Il linfoma T angioimmunoblastico (Angioimmunoblastic T cell Lymphoma, AITL) è una delle entità più comuni tra i PTCLs in Occidente; rappresenta infatti il 25-30% dei casi di PTCLs in Europa
(Tabella 3) (10).
L’incidenza è maggiore in età avanzata (età
mediana, 60 anni). Clinicamente, i pazienti si presentano con adenopatie superficiali, epatosplenomegalia, sintomi B, in particolare febbre e calo
ponderale e in oltre la metà dei casi sono presenti un rash cutaneo e artralgie. Gli esami di
laboratorio spesso mostrano un’ipergammaglobulinemia policlonale e un’anemia emolitica con
test di Coombs diretto positivo. Circa l’80% dei
pazienti esordisce in stadio avanzato di malattia
(stadio III o IV) con coinvolgimento di sedi extranodali quali milza, midollo osseo, cute, fegato e
polmone.
La prognosi è sfavorevole con una mediana di
sopravvivenza inferiore a 3 anni, anche se da alcuni studi emerge che il 30% dei pazienti affetti da
AITL sono long-term survivors (5, 48).
69
70
Seminari di Ematologia Oncologica
Le seguenti caratteristiche anatomo-patologiche
fanno dell’AITL un’entità distinta e ben definita tra
i PTCLs:
- un diffuso infiltrato polimorfo costituito da cellule neoplastiche T di media taglia con abbondante citoplasma chiaro, piccoli linfociti, istiociti o cellule epitelioidi, immunoblasti, eosinofili e plasmacellule;
- intensa vascolarizzazione;
- proliferazione perivascolare di cellule follicolari
dendritiche (follicular dendritic cells, FDC);
- la presenza di grandi cellule B ad abito blastico, spesso EBV positive, che ricordano morfologicamente la cellula di Reed-Sternberg.
Il background reattivo è spesso predominante
rispetto alla componente neoplastica (49). Le
diverse componenti cellulari si distribuiscono in
modo da creare tre diversi quadri morfologici o
pattern architetturali:
- con follicoli iperplastici (pattern I, poco frequente);
- con follicoli depleti (pattern II);
- senza follicoli (pattern III).
La progressiva perdita dei follicoli corrisponde ad
un progressivo incremento della componente neoplastica e rappresenta quindi uno stadio più avanzato di malattia (50, 51).
Le cellule neoplastiche nell’AITL sono linfociti T
maturi CD4+ CD8- ed esprimono la maggior
parte degli antigeni pan-T (Tabella 2) (1). È stato
recentemente dimostrato che la controparte normale dell’AITL è un particolare subset di linfociti
T helper follicolari (follicular helper T cells, TFH)
(50, 52, 53). I TFH sono fisiologicamente situati
al limite tra la zona del mantello e i centri germinativi dove interagiscono con le cellule B inducendo l’espressione di AID (activation-induced
deaminase), importante nella differenziazione dei
linfociti B. In effetti la maggior parte dei casi di
AITL esprime CD10 e BCL6, tipicamente associati ai centri germinativi ed alle TFH cells, e
CXCL13, chemochina che favorisce l’espansione delle cellule B, la differenziazione in plasmacellule e l’ipergammaglobulinemia (50, 52, 53). Ad
ulteriore conferma, recentemente studi di GEP
hanno mostrato un profilo di espressione genica
simile tra le TFH e le cellule neoplastiche
dell’AITL. Inoltre, gli studi di GEP hanno confermato la predominanza della componente cellu-
lare reattiva nel milieu cellulare. In sostanza, le cellule non neoplastiche coinvolte nella risposta
immune umorale rappresentano la principale
caratteristica distintiva dell’AITL (54).
Il VEGF (vascular endothelial growth factor) è altamente espresso nell’AITL, è responsabile della tipica intensa vascolarizzazione e potrebbe rappresentare un possibile target terapeutico (5).
Le alterazioni molecolari che sottendono alla trasformazione neoplastica delle TFH non sono ad
oggi note. Recenti studi di citogenetica hanno individuato tra le alterazioni più frequenti la trisomia
dei cromosomi 3, 5 e 21, la perdita di 6q e del
cromosoma X (55).
Linfoma T/NK extranodale (nasal type)
Le entità definite extranodal NK/T cell lymphoma,
nasal type (NK/TCLs) rappresentano circa il 5-10%
dei LNH registrati soprattutto in Estremo Oriente
e nel Sud- Centro America, mentre rappresentano un’entità rara nelle popolazioni Occidentali (10).
Nella classificazione REAL, i NK/TCLs erano chiamati “linfomi angiocentrici” per il caratteristico pattern di crescita angioinvasivo, con distruzione
vascolare e necrosi. Da un punto di vista anatomo-patologico è facilmente riconoscibile un’invasione dell’albero vascolare da parte delle cellule
neoplastiche con conseguente occlusione vascolare, ischemia e necrosi tissutale (29).
La 3a edizione della WHO ha sostituito il termine
linfomi angiocentrici con il termine extranodal NK/T
cell lymphoma, nasal type per i seguenti motivi (2):
- la definizione NK/T è stata utilizzata per indicare che nella maggior parte dei casi la cellula neoplastica deriva da cellule NK [CD2+, CD56+,
cCD3 +, EBV+] ma che in rari casi, pur con le
stesse caratteristiche cliniche e morfologiche,
la cellula neoplastica può esprimere un fenotipo T (EBV+ CD56- cytotoxic T cell);
- il termine nasal-type è stato utilizzato in considerazione del fatto che la maggior parte dei casi
si presenta a livello della cavità nasale e delle
strutture anatomiche ad essa associate.
Per quanto riguarda l’immunofenotipo, le cellule
neoplastiche esprimono fenotipo NK e sono tipicamente CD2+. Il marcatore di superficie CD3 è
negativo mentre è positivo il cCD3 ; questo pattern è tipico di neoplasie di derivazione NK.
Molecole citossiche quali granzyme B, perforine
Linfomi non Hodgkin T/NK
e TIA-1 sono anch’esse espresse. Il CD56 è un
utile marcatore NK ma non è un marcatore specifico degli NK/TCLs e può espresso anche da
altri PTCLs (Tabella 2). Infine il virus di Epstein Barr
(EBV) è quasi sempre dimostrabile nelle cellule
neoplastiche mediante tecniche di ibridizzazione
in situ (ISH) (1).
Il NK/TCLs è diagnosticato più spesso in età adulta; l’età mediana alla diagnosi è nella quinta decade, con un rapporto maschi/femmine di 3/1. Il linfoma si localizza dapprima a livello delle cavità
nasali, del nasofaringe, e può diffondere a livello
delle orbite e del palato duro. La disseminazione
a livello della cute, del tratto gastrointestinale e
degli organi genitali, è generalmente un evento tardivo nella storia naturale della malattia. Nella maggior parte dei casi non c’è, almeno in fase iniziale, un coinvolgimento del sistema nervoso centrale e del midollo osseo (57). È osservabile una
pancitopenia talora secondaria ad emofagocitosi, se non è presente un coinvolgimento midollare (56).
Non esiste ad oggi un sistema di stadiazione standardizzato. La stadiazione secondo Ann Arbor non
è adatta agli NK/TCLs in quanto lo stadio I può
includere sia pazienti con un malattia localizzata
solo alle cavità nasali sia pazienti con malattia che
invade anche le strutture adiacenti (58). Per le forme ad esordio esclusivamente nasale è raccomandato l’impiego del sistema di stadiazione - T
che si basa sull’estensione locale del tumore (57):
- T1 indica un tumore confinato alle cavità
nasali;
- T2 indica un’estensione a livello mascellare, dei
seni etmoidali e del palato duro;
- T3 l’estensione alla parte posteriore dei seni
etmoidali, ai seni sfenoidali, alle orbite, alle guance o allo spazio buccinatorio superiore;
- T4 indica un’estensione allo spazio buccinatorio inferiore, alla fossa infratemporale o al nasofaringe.
L’utilità dell’International Prognostic Index (IPI) nella stratificazione prognostica dei pazienti affetti da
NK/TCLs è controversa (10, 22, 23, 36, 27). La
presenza del DNA dell’EBV nel sangue è stata
recentemente segnalata come un fattore prognostico nettamente sfavorevole (59). I livelli plasmatici della viremia all’esordio correlano infatti con
la risposta alla terapia e con la sopravvivenza e
la persistenza dell EBV DNA nel plasma dopo terapia è strettamente correlato alla probabilità di recidiva (60).
Linfoma T enteropatico
I linfomi gastrointestinali primitivi rappresentano
il 4-12% di tutti i linfomi non Hodgkin e l’1-4%
di tutte le neoplasie gastrointestinali (10, 61). I linfomi gastrointestinali a cellule T sono una rara entità e l’unica entità clinico-patologica ben definita
è il linfoma T enteropatico (EATL, entheropatyassociated T cell lymphoma) che presenta tuttavia caratteristiche peculiari e una frequenza in
aumento recente e merita perciò una breve trattazione separata. L’età mediana di insorgenza è
57 anni (28-82 anni) (61). Il quadro clinico di presentazione più comune è rappresentato da
malassorbimento, dolori addominali e talvolta quadri di perforazione intestinale in pazienti con una
storia di celiachia (61). Talvolta la diagnosi di celiachia avviene in seguito alla diagnosi di EATL.
Come già riportato, esiste una correlazione tra
EATL e celiachia (10, 12, 61). Nei pazienti affetti
da celiachia infatti l’ingestione di glutine determina un’infiammazione cronica della mucosa del piccolo intestino e, in una piccola percentuale di tali
pazienti (2-5%) non si osserva un miglioramento
nonostante una dieta priva di glutine. I pazienti
resistenti possono presentare un’espansione clonale dei linfociti intraepiteliali con un fenotipo aberrante. L’OS a 5 anni è pari a 50-58% e la principale causa di morte è proprio l’EATL (61).
La sede di insorgenza del linfoma è più spesso
il digiuno e l’ileo, anche se qualsiasi parte del tratto gastrointestinale può essere coinvolta (1, 61).
Nella maggior parte dei pazienti il linfoma è multifocale, dà origine ad ulcere, noduli, placche (più
raramente a grosse masse) e spesso infiltra il
mesentere e i linfonodi mesenterici.
Da un punto di vista anatomo-patologico, le cellule neoplastiche sono spesso di dimensioni
medio-grandi, con nucleo arrotondato o angolato, nucleoli prominenti e abbondante citoplasma.
È spesso presente un infiltrato infiammatorio costituito da eosinofili e istiociti. La mucosa intestinale adiacente all’EATL spesso presenta un’atrofia
dei villi, iperplasia delle cripte, incremento dei linfociti, plasmacellule nella lamina propria e una linfocitosi anche intraepiteliale. Le cellule neoplasti-
71
72
Seminari di Ematologia Oncologica
che sono CD3+, CD4-, CD8+/-, CD5-, CD7+ e
contengono granuli citotossici (1).
La prognosi dell’EATL trattato con chemioterapia convenzionale è rapidamente infausta (OS a
5 anni 20%) (10). Le opzioni di trattamento includevano storicamente l’approccio chirurgico con
o senza regimi di chemioterapia contenenti
antracicline o meno frequentemente chemioterapia ad alte dosi seguita da trapianto di cellule
staminali autologhe (62, 63). È stato recentemente riportato da Sieniawski et al. (61) un nuovo regime di trattamento comprendente chemioterapia
ad alte dosi con IEV/MTX (ifosfamide, vincristina, etoposide/methotrexate) e successivo trapianto di cellule staminali autologhe. L’OS a 5 anni
dei 26 pazienti arruolati nello studio è risultata pari
al 60% e la progression free survival pari al 52%,
quindi significativamente migliore rispetto alla prognosi dei pazienti trattati con chemioterapia convenzionale (p=0,003 e p=0,01 rispettivamente per
OS e PFS). Il profilo di tossicità si è rivelato inoltre accettabile (61).
n TERAPIA
Il trattamento dei PTCLs rappresenta ad oggi
un’area controversa per la rarità di tali patologie,
la difficoltà a formulare una diagnosi istopatologica rapida e definitiva, il decorso variabile di ciascuna entità e la scarsità di trials clinici randomizzati. Le strategie di trattamento non sono quindi
ben definite e derivano dai principi di trattamento dei linfomi B.
Terapia di prima linea
Molti studi riguardano in generale i PTCL e solo
recentemente sono stati condotti studi rivolti specificamente a sottotipi istologici dei PTCL, che
descriveremo separatamente. Il trial clinico randomizzato che all’inizio degli anni ’90 aveva identificato il regime di chemioterapia CHOP come
il regime di trattamento standard per i linfomi a
grandi cellule includeva anche i PTCLs, che nell’era pre-Rituximab, ricevevano quindi lo stesso trattamento dei pazienti affetti da linfoma a
grandi cellule B (64). I risultati poco incoraggianti dell’impiego del regime CHOP o dei regimi contenenti antracicline hanno successivamente
portato diversi gruppi ad investigare l’impiego di
approcci alternativi e, tra questi, i regimi contenenti platino quali l’ESHAP o l’HyperCVAD (65,
66). In uno studio di fase II su 58 pazienti di età
>60 anni trattati con regime ESHAP, il gruppo
GELA ha riportato solo il 33% di remissioni complete (67). In un altro studio di fase II condotto
su pazienti di età <60 anni trattati con regimi più
intensivi (simili ai protocolli impiegati nei pazienti pediatrici affetti da linfoma di Burkitt), lo stesso gruppo GELA ha riportato un tasso di remissioni complete del 51% e una mediana di
sopravvivenza libera da eventi di 6 mesi (68).
Ulteriori tentativi sono stati fatti con molte altre
associazioni, per esempio con gemcitabina e
etoposide, in piccoli studi di fase II con pochi
pazienti, che non hanno portato a miglioramenti significativi (69). Lo studio più ampio sul trattamento dei quattro principali istotipi dei PTCL,
pubblicato recentemente dal German HighGrade NHL Study Group ha analizzato 343
pazienti trattati nell’ambito degli studi prospettici del gruppo, confermandone la prognosi
insoddisfacente con sopravvivenza libera da
eventi (EFS) inferiore al 50% per tutti gli istotipi
ad eccezione dell’ALCL ALK positivo (Figura 4)
(70). I risultati dello schema CHOP sono stati
migliorati significativamente solo nel sottogruppo di pazienti di età < a 60 anni e con LDH superiore alla norma, nei quali l’aggiunta di etoposide (CHOEP) ha portato la EFS a tre anni dal 51%
al 75%. Tuttavia schemi di terapia ancora più
intensificati (megaCHOEP), utilizzati dallo stesso gruppo, non hanno migliorato ulteriormente
i risultati (70). A dimostrazione della peculiare
risposta terapeutica dei PTCL, rispetto ad altri
linfomi, è la pari efficacia rispetto a CHOP ottenibile con un regime di somministrazione continua ambulatoriale di ciclofosfamide orale e
prednisone (71).
Alla domanda se esista un rituximab anche per i
linfomi T la risposta è purtroppo ancora no. I trials
in cui alemtuzumab è stato associato allo schema CHOP hanno dato tassi di risposta del 6570% in prima linea con tuttavia un alto tasso di
recidive e una sopravvivenza a un anno inferiore
al 50% (72, 73). La tossicità ematologica e soprattutto infettiva è stata considerevole. Discorso analogo vale per l’associazione CHOP con denileu-
Linfomi non Hodgkin T/NK
kin-diftitox (74). Solo nell’istotipo ALCL risultati
preliminari molto interessanti ottenuti con l’immunoconiugato brentuximab-vedotin, un’associazione fra l’anticorpo monoclonale anti-CD30 e la tossina del fuso mitotico, fanno ritenere aperta l’era
dell’immunochemioterapia
Per quanto riguardo l’impiego del trapianto di cellule staminali, è riportato che il trapianto autologo garantisce un controllo di malattia a lungo termine in circa il 50-70% dei pazienti, quando eseguito in uno stato di prima remissione e in presenza di una malattia chemio sensibile (75, 76).
Il tasso di recidive/progressioni durante il trattamento resta però elevato e non è chiaro se i buoni risultati derivino da una selezione dei pazienti o da una reale maggiore efficacia terapeutica,
visto i risultati sovrapponibili fra CHOP e autotrapianto in uno studio caso-controllo del gruppo GELA (77). Il trapianto allogenico da donatore HLA-identico sembra offrire, grazie all’effetto
graft vs lymphoma, un miglior controllo a lungo
termine della malattia a prezzo di una maggiore
tossicità (78). La sua superiorità in prima linea
rispetto al trapianto autologo è in corso di valutazione in uno studio controllato multicentrico
Italiano in cui le procedure trapiantologiche
seguono un programma di chemioterapia iniziale a dose standard associato ad alemtuzumab e
ad un consolidamento con chemioterapia a dose
intensificata (79).
Terapia di seconda linea
Certamente in una situazione clinica così difficile come il linfoma T che non ha risposto o è recidivato dopo una terapia di prima linea le procedure trapiantologiche vanno considerate di prima
scelta. Fra queste il trapianto allogenico anche a
intensità di condizionamento ridotta, laddove possibile, va preferito al trapianto autologo, essendo in grado di ottenere una sopravvivenza a lungo termine del 50% in diverse casistiche, nei
pazienti, certamente selezionati, che sono riusciti a ricevere questo trattamento. Al di là della scelta fra trapianto autologo e allogenico, che dipende soprattutto dalla tipologia di paziente, va sottolineato che il ricorso al trapianto dovrebbe essere il più precoce possibile, senza attendere che
recidive plurime rendano la malattia completamente refrattaria anche alla terapia ad alte dosi.
Nuovi farmaci
Al di fuori delle opzioni trapiantologiche o in preparazione ad esse, si stanno esplorando nuove
opzioni terapeutiche e nuovi farmaci. Un elenco
certamente non esauriente delle principali classi
e dei target terapeutici è riportato in tabella 6. Per
alcuni di questi sono stati completati importanti
studi di fase II su un numero di pazienti adeguato (80).
- Pralatrexate (PDX; 10-propargyl 10-deazaaminopterin). Il pralatrexate è un nuovo antifolato, ed
è l’unico farmaco approvato nel 2009 dalla Food
and Drug Administration per il trattamento dei
PTCLs recidivati o refrattari. Si differenzia strutturalmente dal methotrexate (MTX) per la presenza di un gruppo propargile in posizione 10.
Tale struttura favorisce l’internalizzazione del farmaco nella cellula (10 volte maggiore rispetto al
MTX), garantendo un maggior effetto antitumorale. Il meccanismo d’azione è poi simile a quello del MTX (81).
Lo studio PROPEL (Pralatrexate in relapsed or
refractory Peripheral T Cell Lymphoma) (82) condotto in pazienti affetti da PTCLs recidivati o resistenti, incluse tutte le entità compresa la micosi
fungoide e i NK/TCLs, ha arruolato 115 pazienti,
già sottoposti ad una mediana di 3 linee di trattamento (1-12); di questi, inoltre, 18 erano già stati sottoposti anche ad autotrapianto e il 63% era
refrattario all’ultima linea di terapia (82). Lo schema di trattamento comprendeva la somministrazione di PDX 30 mg/m2/settimana a cicli di 6 settimane con una settimana di pausa tra ogni ciclo
fino a progressione o tossicità. Il tasso di risposta in 109 pazienti valutabili è stato del 29% (32
di 109), di cui 12 risposte complete (11%) e 20
risposte parziali (18%) (82). I più comuni eventi
avversi di grado 3/4 sono stati i seguenti: trombocitopenia (32%), mucosite (22%), neutropenia
(22%) e anemia (18%) (82). La mucosite è il problema clinico principale, può essere prevista
dosando i livelli di acido metilmalonico e omocisteina, e deve essere prevenuta con la somministrazione continua di acido folico e vitamina B12.
La conclusione degli autori è stata che il pralatrexate ha indotto risposte durature in PTCL recidivati o refrattari, indipendentemente dall’età, dal
sottotipo istologico e dal numero di linee precedenti di trattamento (82). Sulla base di tali risul-
73
74
Seminari di Ematologia Oncologica
Classe
Agente
Target
Antifolati
Pralatrexate
RFC-1
Inibitori delle istone deacetilasi
Vorinostat
Romidepsina
Belinostat
Panobinostat
Plitidepsin
Deacetilasi degli istoni e
di proteine non istoniche
Bortezomib
PR-171 (carfilzomib)
Vari
Analoghi purinici
Gemcitabina
Forodesina
Clofarabina
Sintesi delle purine
Inibitori di mTOR
Temsirolimus
Everolimus
Target della rapamicina
Immunomodulatori
Lenalidomide
Vari
Anticorpi monoclonali
Alemtuzumab
Anti CD30
AntiCD4
CD52
CD30
CD4
Imatinib, Dasatinib
PDGFR-alfa,
Syc
Tipifarnib
Ras
Brentuximab-vedotin
Denileukin diftitox
CD30+tubulina
Recettore di IL-2
Inibitori del proteasoma
Inibitori delle tirosin-chinasi
Inibitori delle farnesil-transferasi
Immunoconiugati e tossine di fusione
TABELLA 6 - Agenti terapeutici con bersaglio specifico in sperimentazione per la terapia dei PTCLs.
tati, sono in corso studi di associazione del PDX
con altri farmaci citotossici e biologici, tra cui la
gemcitabina, il bortezomib e gli inibitori delle istone deacetilasi, e studi in cui PDX viene dato come
terapia di mantenimento in pazienti responsivi a
CHOP (83).
- Inibitori delle istone-deacetilasi (HDAC). Uno studio internazionale multicentrico analogo al PROPEL per numero e caratteristiche dei pazienti ha
valutato l’efficacia di romidepsina, un HDAC inibitore ciclico, con attività pleitropa su molti
pathways intracellulari, già approvato negli Stati
Uniti per l’uso nei linfomi T cutanei recidivati o
refrattari (84). I risultati sono stati riportati in via
preliminare. Il farmaco è somministrato alla dose
di 14 mg/m 2 per via endovenosa lenta nei giorni
1, 8, 15 di cicli di 28 giorni fino a progressione di
malattia per almeno 6 cicli. Il tasso di risposta globale (ORR) è stato del 26%, con RC nel 13% di
130 pazienti. La tossicità di grado 3-4 è stata
soprattutto di tipo ematologico e non si sono verificati i temuti eventi avversi cardiaci legati al potenziale allungamento del QT (84).
Risultati simili sono stati riportati per altri farmaci
quali il belinostat, un pan-inibitore delle HDAC, e
il plitidepsin, un farmaco di origine marina, somministrato alla dose di 3,2 mg/m2 ev con una schedula simile alla Romidepsina.
- Immunoconiugati - anti CD30. Il CD30 è un
membro della famiglia dei recettori del TNF ed
è espresso da diverse neoplasie ematologiche
quali i linfomi di Hodgkin, gli ALCL sistemici e
cutanei e alcuni casi di micosi fungoide trasformata. Esistono diversi anticorpi monoclonali
antiCD30. L’anticorpo monoclonale SGN-30
aveva dimostrato un profilo di attività promettente verso il ALCL con un tasso di risposta parziale del 17% e senza tossicità (85). Tuttavia il reale progresso terapeutico è stato ottenuto quando questo anticorpo è stato coniugato, attraver-
Linfomi non Hodgkin T/NK
so un linker proteico sensibile alla rottura da parte delle proteasi lisosomiali, con aurastatina, una
tossina specifica per l’apparato microtubulare del
fuso mitotico. Il farmaco che ne è derivato, brentuximab-vedotin (SGN-35) si è dimostrato molto più attivo. Recentemente sono stati riportati i
dati eccezionali dello studio di fase I (86) che sono
stati ampiamente confermati nello studio di fase
2. Alla dose di 1,8 mg/m2 ev ogni 21 giorni, l’86%
di 58 pazienti con ACLC recidivato o refrattario
(62% dei casi) ha ottenuto una risposta obiettiva e il 53% una RC, indipendentemente dalla
positività di ALK (87). La tollerabilità è stata buona; una neuropatia periferica di grado 3, prevalentemente sensitiva, si è riscontrata nel 13% dei
pazienti dopo in media 16 settimane di trattamento, con un profilo di tossicità simile agli alcaloidi della vinca. La PFS è stata significativamente maggiore rispetto all’ultima linea di trattamento ricevuta (41 vs 26 settimane; P<0.02). Alla luce
di questi dati brentuximab vedotin può a ragione essere considerato un fondamentale progresso nella terapia dei linfomi che esprimono CD30,
quali l’ALCL e il linfoma di Hodgkin. Nell’ALCL
esso verrà testato in prima linea e in associazione ad altri farmaci, per migliorare ulteriormente
i già ottimi risultati.
- Inibitori delle tirosinochinasi. Nel sottogruppo
ALK-positivo degli ALCL, caratterizzato dalla traslocazione t(2;5) e dalla conseguente formazione
di una proteina di fusione ad attività tirosinochinasi intrinseca, sembra promettente anche il crizotinib, un TK-inibitore specifico per ALK che si
è dimostrato in grado di indurre risposte obiettive in alcuni casi di malattia avanzata e refrattaria
(88), come in altre neoplasie caratterizzate dallo
stesso meccanismo di patogenesi molecolare (89).
il tasso di recidiva è intorno al 50% e la maggior
parte delle recidive è locale e avviene entro il primo anno dal termine della radioterapia. In considerazione dell’elevato rischio di recidiva di malattia, non solo nelle forme sistemiche ma anche nelle forme localizzate, è utile associare regimi di chemioterapia, anche prima dell’inizio della radioterapia. Tra i regimi impiegati, lo schema CHOP e
i regimi CHOP-like contenenti antracicline non
hanno dato risultati soddisfacenti (92) per l’elevata incidenza di chemioresistenza dovuta anche
all’espressione del gene della multi-drug resistance (MDR) ABCB1 che codifica per la P-glicoproteina nelle cellule neoplastiche (93). La L-asparaginasi (L-Asp) è in grado di per sé di ridurre l’attività NK nelle cellule normali, non risente dei meccanismi di farmaco-resistenza tipo MDR e causa apoptosi in linee cellulari NK e in cellule di leucemia/linfoma NK in vitro (94). In effetti si ottengono buone risposte sia con l’impiego della L-Asp
da sola che in combinazione con prednisone e
vincristina (tasso di risposta fino al 50% dei
NK/TCLs) (95). In uno studio del GELA, Jaccard
et al. (96) hanno combinato L-Asp con MTX a alte
dosi e con desametasone ottenendo risposte
eccellenti in pazienti recidivati e refrattari, con ORR
del 79% e RC del 63% e una sopravvivenza globale di oltre 12 mesi in una categoria di pazienti
particolarmente difficile. Uno studio giapponese
(97) ha ideato uno schema contenente L-Asp in
combinazione con steroide, methotrexate, ifosfamide ed etoposide (SMILE). L’esperienza iniziale
con questo protocollo è molto favorevole: l’ORR
è pari a 67% con un tasso di RC del 50% in
pazienti con malattia disseminata o recidivata/resistente.
Terapia dei linfomi T/NK extranodali
(nasal-type)
Da un punto di vista terapeutico i NK/TCLs meritano di essere distinti dagli altri PTCLs. Il trattamento delle forme localizzate comprende la
radioterapia. Sono state utilizzate dosi variabili da
30 a 60 Gy ma la dose ottimale dovrebbe essere non inferiore a 55 Gy, cioè molto più alta di quelle efficaci in altri tipi di linfoma. Il tasso di risposta è di oltre l’80% e circa il 70% dei pazienti ottiene la remissione completa (90, 91). Ciononostante,
n CONCLUSIONI
I linfomi T sistemici restano una patologia particolarmente difficile per l’ematologo. Le loro
peculiarità biologiche, la relativa rarità, la particolare refrattarietà ad approcci terapeutici efficaci in
altri tipi di linfomi possono scoraggiare il clinico.
Tuttavia recentemente sono iniziati sforzi collaborativi internazionali, fra i quali l’International T-Cell
Lymphoma Project e lo studio registrativo prospettico T-cell project, che possono rappresentare la
75
76
Seminari di Ematologia Oncologica
base indispensabile per ottenere in futuro progressi decisivi. Una conoscenza più approfondita dei
meccanismi biologici e dei quadri istopatologici
caratteristici delle diverse entità di linfomi T/NK
consentirà verosimilmente di ottenere, su più vasta
scala, quei risultati che già oggi si iniziano ad ottenere in alcuni gruppi di pazienti. L’esempio dell’immunoconiugato anti-CD30/aurastatina nel linfoma anaplastico, è paradigmatico di come una
caratteristica biologica distintiva possa essere
specificamente sfruttata per massimizzare il differenziale fra efficacia e tossicità della terapia.
Nell’attesa che questi progressi si traducano in
armi terapeutiche concretamente utilizzabili
dobbiamo continuare a fare il miglior uso delle
terapie disponibili. Il ricorso a procedure trapiantologiche precocemente durante il decorso del
linfoma rappresenta un’indicazione unanime, e
vedremo presto se ciò debba essere fatto in prima o in seconda remissione e se con cellule staminali autologhe o allogeniche. Anche la chemioterapia convenzionale non dovrebbe limitarsi
sempre e comunque allo schema CHOP, essendovi evidenze di possibili miglioramenti in sottogruppi di pazienti con l’aggiunta o l’utilizzo di
altri farmaci come l’etoposide nei giovani a prognosi sfavorevole e l’asparaginasi nei linfomi
T/NK. Certamente il suggerimento resta quello
di inserire il più possibile i pazienti in studi clinici controllati, nei quali possano venire verificati i reali vantaggi dei nuovi farmaci, quali pralatrexate, inibitori delle istone-deacetilasi e delle
tirosin-chinasi, e molti altri, che stanno percorrendo il lungo iter degli studi di fase I-II e poi di
fase III necessario a farli entrare a pieno titolo,
da soli o in combinazione, nello scarso armamentario terapeutico oggi disponibile per i linfomi T sistemici.
n BIBLIOGRAFIA
1. Swerdlow SH, Campo E, Harris NL, et al. WHO
classification of tumors of haematopoietic and lymphoid
tissues, edn 4. Lyon, France: International Agency nfor
Research on Cancer. 2008.
2. Jaffe ES, Harris NL, Stein H, et al. World Health
Organization Classification of Tumors. Pathology and
genetics of tumors of haematopoietic and lymphoid
tissues. Lyon, France. IARC Press. 2001.
3. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC
classification for cutaneous lymphomas. Blood. 2005;
105: 3768-85.
4. Olsen E, Vonderheid E, Pimpinelli N, et al. Revision to
the staging and classification of mycosis fungoides and
sezary syndrome: a proposal of international society
of cutaneous lymphomas (ISCL) and the cutaneous
lymphoma task force of the European Organization of
Research and Treatment of Cancer (EORTC). Blood.
2007; 110: 1713-22.
5. de Leval L, Gaulard P. Pathobiology and molecular
profiling of peripheral T-cell lymphomas. Hematology
Am Soc Hematol Educ Program. 2008; 272-9.
6. de Leval L, Gaulard P. Pathology and biology of
peripheral T-cell lymphomas. Histopatology. 2011; 58:
49-68.
7. Went A, Agostinelli C, Gallamini A, et al. Marker
expression in peripheral T-cell lymphoma: a proposed
clinical-pathologic prognostic score. J Clin Oncol. 2006;
24: 2472-9.
8. Zettl A, Rudiger T, Konrad MA, et al. Genomic
profiling of peripheral T cell lymphoma, unspecified, and
anaplastic large T cell lymphoma delineates novel
recurrent chromosomal alterations. Am J Pathol.
2004; 164: 1837.48.
9. Rudiger T, Weisenburger DD, Anderson JR et al.
Peripheral T-Cell Lymphoma (excluding anaplastic
large-cell lymphoma): results from the non Hodgkin’s
Lymphoma Classification Project. Ann Oncol. 2002; 13:
140-149.
10. Vose J, Armitage J, Weisenburger D. International
peripheral T-cell and natural/t-cell lymphoma study:
pathology findings and clinical outcome. J Clin Oncol.
2008; 26: 4124-30.
11. Rudiger T, Weisenburger DD, Anderson JR, et al.
Peripheral T cell lymphoma (including anaplastic largecell lymphoma): Results from the Non Hodgkin’s
Lymphoma Classification Project. Ann Oncol. 2002;
13: 140-9.
12. Deleeuw RJ, Zettl A, Klinker E, et al. Whole genome
analysis and HLA genotyping fo entheropathy-type T
cell lymphoma reveales two distinct lymphoma
subtypes. Gastroenterology. 2007; 132: 1902-11.
13. Piccaluga PP, Agostinelli C, Gazzola A, et al. Prognostic
markers in peripheral T cell lymphoma. Curr Hematol
Malign Rep. 2010; 5: 222-8.
14. A clinical evaluation of the International Lymphoma
Study Group classification of non Hodgkin’s
Lymphoma. The Non Hodgkin’s Lymphoma
Classification Project. Blood. 1997; 89: 3909-18.
15. Savage KJ, Harris NL, Vose JM et al. ALK - anaplastic
large cell lymphoma is clinically and immunophenotypically different from both ALK+ALCL and
peripheral T cell lymphoma, not otherwise specified:
report from the International Peripheral T Cell
Lymphoma Project. Blood. 2008; 111: 5496-504.
16. Lopez-Guillermo A, Cid J, Salar A, et al. Peripheral T
Linfomi non Hodgkin T/NK
cell lymphomas: initial features, natural history and
prognostic factors in a series of 174 patients
diagnosed to the REAL Classification. Ann Oncol.
1998, 9: 849-55.
17. Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma:
clinico-pathological findings and outcome. Blood
1999; 93. 2697-706.
18. Suzumiya J, Ohshima K, Tamura K, et al. The
International Prognostic Index predicts outcome in
aggressive adult T-cell leukaemia/lymphoma: analysis
of 126 patients from the International Peripheral T Cell
Lymphoma Project. Ann Oncol. 2009; 20: 715-20.
19. Au WY, Weisenburger DD, Intragumtornchai T, et al.
Clinical difference between nasal and extranasal
natural killer/T cell lymphoma: a study of 136 cases
from the International T Cell Lymphoma Project. Blood.
2009; 113: 3931-7.
20. Gallamini A, Stelitano C, Calvi R, et al. Peripheral T cell
lymphoma, unspecified (PTCL-U): a new prognostic
model from a retrospective multicentric clinical study.
Blood. 2004; 103: 2474-9.
21. Briones J, Moga E, Espinosa I, et al. Bcl-10 protein
highly correlates with the expression of phosphorylated
p65 NK-kappaB in peripheral T cell lymphomas and
os associated with clinical outcome. Histopatology.
2009; 54: 478-85.
22. Lee J, Suh C, Park YH, et al. Extranodal natural killer
T cell lymphoma, nasal type: a prognostic model from
a retrospective multicenter study. J Clin Oncol. 2006;
24: 612-8.
23. Suzuki R, Suzimiya J, Yamagughi M, et al. Prognostic
factors for mature killer (NK) cell neoplasms: aggressive
NK cell leukaemia and extranodal NK cell lymphoma,
nasal type. Ann Oncol. 2010; 21: 1032-40.
24. Kluin PM, Feller A, Gaulard P, et al. Peripheral T/NK
cell lymphoma: a report og IXth Workshop of the
European Association for Haematopathology.
Histopathology. 2001; 38: 250-70.
25. Dupuis J, Emile JF, Mounier N, et al. Prognostic
significance of Epstein Barr virus in nodal peripheral
T cell Lymphoma, unspecified : A Group d’Etude des
Lymphomes de d’Adulte (GELA) study. Blood. 2006;
108: 4136-69.
26. Au WY, Pang A, Choy C, et al. Quantification of
circulating Epstein Barr virus (EBV) DNA in the
diagnosis and monitoring of natural killer cell and EBVpositive lymphomas in immunocompetent patients.
Blood. 2004; 104: 243-9.
27. Lee J, Suh C, Huh J, et al. Effect of positive bone
marrow EBV in situ hybridization in staging and survival
of localized extranodal natural killer/T cell lymphoma,
nasal type. Clin Cancer Res. 2007; 13: 3250-4.
28. Martinez-Delgado B, Cuadros M, Honrado E, et al.
Differential expression of NF-kappaB pathway genes
among peripheral T cell lymphomas. Leukemia. 2005;
19: 2254-63.
29. O’Leary H, Savage K. The spectrum of peripheral T cell
lymphomas. Curr Opin Hematol. 2009; 16: 292-8.
30. Lopez-Guillermo A, Cid J, Salar A, et al. Peripheral T
cell lymphomas: initial features, natural history, and
prognostic factors in a series of 174 patients diagnosed
according to the REAL Classification. Ann Oncol. 1998;
9: 849-55.
31. Gisselbrecht C, Gaulrd P, Lepage E, et al. Prognostic
significance of T cell phenotype in aggressive non
Hodgkin’s lymphomas. Groupe d’Etudes des
Lymphomes de l’Adulte (GELA). Blood. 1998; 92:
76-82.
32. Chang ST, Lu CL, Chuang SS. CD52 expression in nonmycotic T and NK/T cell lymphomas. Leuk Lymphoma.
2007; 48: 117-21.
33. Piccaluga PP, Ascani S, Agostinelli C, et al. Expression
of CD52 in peropheral T cell lymphoma.
Haematologica. 2007; 92: 566-7.
34. Rodig SJ, Abramson JS, Pinkus GS, et al.
Heterogeneous CD52 expression among hematologic
neoplasms: implications for the use of Alemtuzumab
(CAMPATH-1H). Clin Cancer Res. 2006; 12: 7174-9.
35. Rizvi MA, Evens AM, Tallman MS, et al. T cell non
Hodgkin’s lymphoma. Blood. 2006; 107: 1255-64.
36. Zettl A, Rudiger T, Konrad MA, et al. Genomic
profiling of peripheral T cell lymphoma, unspecified, and
anaplastic large T cell lymphoma delineates novel
recurrent chromosomal alterations. Am J Pathol.
2004; 164: 1837-48.
37. Basem M. William, Maribeth Hohenstein, Fausto R, et
al. Phase I/II study of dasatinib in relapsed or refractory
non-Hodgkin’s lymphoma (NHL). Blood. 2010; 116 (21):
Abs 288.
38. Martinez-Delgado B, Mendelez B, Cuadros M, et al.
Expression profiling of T-cell lymphomas differentiates
peripheral and lymphoblastic lymphomas and defines
survival related genes. Clin Cancer Res. 2004; 10:
4971-82.
39. Ballestrer B, Ramuz O, Gisselbrecht C, et al. Gene
expression profiling identifies molecular subgroups
among nodal peripheral T cell lymphomas. Oncogene
2006; 25: 1560-71.
40. Beltran B, Morales D, Quinones P, et al. Lymphopenia
predicts survival in peripheral T cell lymphoma,
unspecified. [Abstract]. Blood. 2009; 114 (22): 1930.
41. Choi DR, Yoon DH, Ahn JH, et al. Significance of
absolute lymphocyte count at relapse as a prognostic
factor in patients with t cell non Hodgkin’s lymphoma.
[Abstract]. Blood 2009; 114 (22): 2939.
42. Salaverria I, Bea S, Lopez-Guillermo, A et al.
Genomic profiling reveals different genetic aberrations
in systemic ALK-positive and ALK-negative anplastic
large cell lymphomas. Br J Haematol. 2008; 140:
516-26.
43. Gascoyne RD, Aoun P, Wu D, et al. Prognostic
significance of anaplastic lymphoma kinase (ALK)
protein expression in adults with anaplastic large cell
lymphoma. Blood. 1999; 93: 3913-21.
77
78
Seminari di Ematologia Oncologica
44. Falini B, Bigerna B, Fittozzi M, et al. ALK expression
defines a distinct group of T/null lymphomas (‘ALK
lymphomas’) with a wide morphological spectrum. Am
J Path. 1998; 153: 875-86.
45. Suzuki R, Kagami Y, Takeuchi K, et al. Prognostic
significance of CD56 expression for ALK-positive and
ALK-negative anaplastic large cell lymphoma of T/null
cell phenotype. Blood. 2000; 96: 2993-3000.
46. Amin HM, Lai R. Pathobiology of ALK+ anaplastic largecell lymphoma. Blood 2007; 110: 2259-953.
47. Chiarle R, Voena C, Ambrogio C, et al. The anaplastic
lymphoma kinase in the pathogenesis of cancer. Nat
Rev Cancer. 2008; 8: 11-23.
48. Mourad N, Mounier N, Briere J, et al. Clinical, biologic
and pathologic features in 157 patients with
angioimmunoblastic T-cell lymphoma treated within the
Groupe d’Etude des Lymphomes de l’Adulte (GELA)
trials. Blood. 2008; 111: 4463-70.
49. Willenbrock K, Renne C, Gaulard P, et al. In
angioimmunoblastic T cell lymphoma, neoplastic T cells
may be a minor population. A molecular single cell and
immunohistochemical study. Virchows Arch. 2005; 446:
15-20.
50. Attygalle A, Al-jehani R, Diss TC, et al. Neoplastic T
cells in angioimmunoblastic T cell lymphoma express
CD10. Blood. 2002; 99: 627-33.
51. Attygalle AD, Kyriakou C, Dupui J, et al. Histologic
evolution of angioimmunoblastic T cell lymphoma in
consecutibe biopsies: clinical correlation and insights
into natural history and disease progression. Am J Surg
Pathol. 2007; 31: 1077-88.
52. Vinuesa CG, Tangye SG, Moser B, et al. Follicular B
helper T cells in antibody responses and autoimmunity.
Nat Rev Immunol. 2005; 5: 853-65.
53. Ree HJ, Kadin ME, Kikuchi M, et al. Bcl-6 expression
in reactive follicular hyperplasia, follicular lymphoma,
and angioimmunoblastic T cell lymphoma with
hyperplastic germinal centers: heterogeity of
intrafollicular T cells and their altered distribution in the
pathogenesis of angioimmunoblastic T cell lymphoma.
Hum Pathol. 1999; 30: 403-11.
54. de Leval L, Rickman DS, Thielen C, et al. The gene
expression profile of nodal peripheral T cell lymphoma
demonstrates
a
molecular
link
between
angioimmunoblastic T cell lymphoma and follicular
helper T cells. Blood. 2007; 109: 4952-63.
55. Nelson M, Horsman DE, Weisenburger DD, et al.
Cytogenetic abnormalities and clinical correlations in
peripheral T cell lymphoma. Br J Haematol. 2008; 141:
461-9.
56. Liang R. Advances in the management and monitoring
of extranodal NK/T cell lymphoma, nasal type. Br J
Haematol. 2009; 147: 13-21.
57. Wong KF, Chan JK, Cheung MM, et al. Bone marrow
involvment by nasal NK cell lymphomaat diagnosis is
uncommon. Am J Clin Pathol. 2001; 115: 266-70.
58. Ooi GC, Chim CS, Liang R, et al. Nasal T/NK cell
lymphoma: CT and MR imaging features of new clinicopathological entity. Am J Roenterol. 174; 1141-45.
59. Suzuki R, Yamaguchi M, Izutsu K, et al. Prospective
measurement of EBV DNA in plasma and peripheral
blood mononuclear cells of Extranodal NK/T cell
Lymphoma, Nasal Type. Blood. 2009; 114 (22): abs 135.
60. Jaccard A, Gachard N, Coppo P, et al. A prospective
phase II trial of an L-Asparaginase containing
regimen in patients witn refractory or relapsing
Extranodal NK/T cell Lymphoma. [Abstract]. Blood.
2008; 112 (11): 579.
61. Sieniawski M, Angamuthu N, Boyd K, et al. Evaluation
of enteropathy-associated T cell lymphoma comparing
standard therapies with a novel regimen including
autologous stem cell transplantation. Blood. 2010; 115:
3664-70.
62. Daum S, Ullrich R, Heise W, et al. Intestinal non
Hodgkin’s lymphoma: a multicenter prospective
clinical study from the german Study Group on
Intestinal non Hodgkin’s Lymphoma. J Clin Oncol.
2003; 21: 2740-6.
63. Rongey C, Micallef I, Smyrk T, et al. Successful
treatment of entheropathy-associated T cell lymphoma
with autologous stem cell trasnplantation. Dig Dis Sci.
2006, 51: 1082-6.
64. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison
of a standard regimen (CHOP) with three intensive
chenotherapy regimens for advanced non-Hodgkin’s
lymphoma. N Engl J Med. 1993; 328: 1992-006.
65. Di Venuti G, Nawgiri R, Foss F. Denileukin diftitos and
hyper-CVAD in the treatment of human T cell
lymphotropic virus associated acute T cell
leucemia/lymphoma. Clin Lymphoma. 2003; 4: 176-8.
66. Karakas T, Bergmann L, Stutte HJ, et al. Peripheral T
cell lymphomas respond well to vincristine, adriamycin,
cyclophosphamide, prednisone and etoposide (VACPE)
and have similar outcome as high-grade B cell
lymphomas, Leuk Lymphoma. 1996; 24: 121-9.
67. Bouabdallah R, Delmer A, Xerri L, et al. ESHAP
chemotherapy regimen and 13-cis-reinoic acid in
elderly patients with intreated poor-prognosis peripheral
T cell lymphoma: a GELA phase II trial of feasibility and
efficacy. [Abstract]. Ann of Oncol. 2005; 16 (Suppl. 5):
v131 (abs.#322).
68. Delmer A, Mounier N, Gaulard P, et al. Intensified
induction therapy with etoposide (VP16) and high dose
cytarabina (AraC) in patients aged less than 60 years
with peripheral T-cell/NK lymphoma: Preliminary results
of the phase II GELA study LNH98 T7. [Abstract]. Proc
Am Soc Clin Oncol. 2003; 22: 591 (#2375).
69. Kim JG, Sohn SK, Chae YS, et al. Cancer Chemother
Pharmacol. 2006; 58: 35-39.
70. Schmitz N, Trumper L, Ziepert M, et al. Treatment and
prognosis of mature T cell and Nk cell lymphoma: an
analysis of patients with T cell lymphoma treated in
studies of the German high-grade Non-Hodgkin
Lymphoma Study Group. Blood. 2010; 116: 3418-25.
Linfomi non Hodgkin T/NK
71. Tucci A, Cerqui E, Ungari M, et al. Continuous oral
cyclophosphamide and prednisolone as valuable
treatment option for peripheral T cell lymphoma.
[Abstract]. Br J Haematol. 2011; 152: 113-6.
72. Gallamini A, Zaja F, Patti C, et al. Alemtuzumab
(Campath-1H) and CHOP chemotherapy as first-line
treatment of peripheral T-cell lymphoma: results of a
GITIL (Gruppo Italiano Terapie Innovative nei Linfomi)
prospective multicenter trial. Blood. 2007; 110:
2316-32.
73. Kim JG, Sohn SK, Chae YS, et al. Alemtuzumab plus
CHOP as front-line chemotherapy for patients with
peripheral T-cell lymphomas: a phase II study. Cancer
Chemother Pharmacol. 2007; 60: 129-34.
74. Foss FM. Enhancing existing approaches to Periheral
T Cell Lymphomas. Semin Hematol. 2010; 47: 8-10.
75. Reimer P, Rüdiger T, Geissinger E, et al. Autologous
stem-cell transplantation as first-line therapy in
peripheral T-cell lymphomas: results of a prospective
multicenter study. J Clin Oncol. 2009; 2: 106-13.
76. Rodríguez J, Conde E, Gutiérrez A, et al. Frontline
autologous stem cell transplantation in high-risk
peripheral T-cell lymphoma: a prospective study
from The Gel-Tamo Study Group. Eur J Haematol.
2007; 79: 32-8.
77. Mounier N, Gisselbrecht C, Brière J, et al. All aggressive
lymphoma subtypes do not share similar outcome after
front-line autotransplantation: a matched-control analysis
by the Groupe d'Etude des Lymphomes de l'Adulte
(GELA). Ann Oncol. 2004; 15: 1790-7.
78. Corradini P, Dodero A, Zallio F, et al. Graft-versuslymphoma effect in relapsed peripheral T-cell nonHodgkin’s lymphomas after reduced-intensity
conditioning followed by allogeneic transplantation of
hematopoietic cells. J Clin Oncol. 2004; 22: 2172-6.
79. Dodero A, Spina F, Narni F, et al. Allogeneic stem cell
transplantation (Allo-SCT) follonig a reduced intensity
conditioning (RIC) regimen in relapsed Peripheral T cell
Lymphomas (PTCL): results at 4 years of median follow
up. [Abstract]. Blood. 2009; 114 (22): 875.
80. Zain MJ, O’Connor O. Targeted treatment and new
agents in peripheral T cell lymphoma. Int J Hematol.
2010; 92: 33-44.
81. Sirotnak FM, DeGraw JI, Moccio DM, et al. New folte
analogs of the 10-deaza-aminopterin series: basis for
structural design and biochemical and pharmacologic
properties. Cancer Chemother Pharmacol. 1984; 12:
18-25.
82. O’Connor O, Pro B, Pinter-Brown LL. Results of the
pivotal, multicenter, phase II study of pralatrexate in
patients with relapsed or refractory peripheral T cell
lymphoma (PTCL). [Abstract]. J Clin Oncol. 2009; 27:
15s (8561).
83. Horwitz S, Vose J, O’Connor O. A phase 1/2 A open
label study of Pralatrexate and Gemcitabine in patients
with relapsed or refractory lymphoproliferative
malignancies. ASH 2010, [abstract] 1674.
84. Coiffier B, Pro B, Prince M, et al. Final results from a
pivotal, multicenter, international, open-label, phase 2
study of Romidepsin in progressive or relapsed
Peripheral T Cell Lymphoma (PTCL) following prior
systemic therapy. [Abstract]. Blood. 2010; 116 (21): 114.
85. Bartlett NY, Younes A, Carabasi MH. A phase I
multidose study of SGN-30 immunotherapy in patients
with refractory or recurrent CD30+ hematologic
malignancies. Blood. 2008; 111: 1848-54.
86. Younes A, Bartlet NL, Leonard JP, et al. Brentuximab
vedotin (SGN-35) for relapsed CD30-positive
lymphomas. N Engl J Med. 2010; 363: 1812-21.
87. Shustov RA, advani R, Brice P, et al. Complete
remission with Brentuximb Vedotin (SNG-35) in patients
with relapsed or refractory Sistemic Anaplastic Large
cell Lymphoma. [Abstract]. Blood. 116 (21): 961.
88. Gambacorti-Passerini CB, Dilda I, Giudici G, et al.
Clinical activity of Crizotinib in advanced,
chemoresistance ALK+ lymphoma patient. [Abstract].
Blood. 2010; 116: 2877.
89. Butrynski JE, D'Adamo DR, Hornick JL, et al. Crizotinib
in ALK-rearranged inflammatory myofibroblastic tumor.
N Engl J Med. 2010; 363: 1727-33.
90. Huang ML, Jiang Y, Liu WP, et al. Early or up-front
radiotherapy improved survival of localized extranodal
T/NK-cell lymphoma, nasal type in the upper
aerodigestive tract. Intern J Radiat Oncol Biol Phys.
2008; 70: 166-74.
91. Wang B, Lu JJ, et al. Combined chemotherapy and
external beam radiation for stage IE and IIE natural killer
T-cell lymphoma of nasal cavity. Leuk Lymph. 2007;
48: 396-402.
92. Suzuki R, Takeuchi K, Oshima K, et al. Extranodal NK/T
cell lymphoma: diagnosis and treatment cues. Heamtol
Oncol. 2008: 26: 66-72.
93. Yamaguchi M, Kita K, Miwa H, et al. Frequent
expression of P-glycoprotein/MRD1 by nasal T-cell
lymphoma cells. Cancer. 1995; 76: 2351-6.
94. Jaccard A, Petit B, Girault S, et al. L-Asparaginase
based treatment of 15 western patients with extranodal
NK/T cell lymphoma and leukemia and a review of the
literature. Ann Oncol. 2009; 20: 110-6.
95. Yong W, Zheng W, Zhu J, et al. L-Asparaginase in the
treatment of refractory and relapsed extranodal NK/T
cell lymphoma, nasal type. Ann Hematol. 2009; 88:
647-52.
96. Jaccard A, Gachard N, Marin B, et al. Efficacy of Lasparaginase with methotrexate and dexamethasone
(AspaMetDex regimen) in patients with refractory or
relapsing extranodal NK/T-cell lymphoma, a phase 2
study. Blood. 2011; 117: 1834-9.
97. Yamaguchi M, Suzuki R, Kwong YL, et al. Phase I trial
of dexamethasone, methotrexate, ifosfamide, Lasparaginase and etoposide (SMILE) chemotherapy for
advanced stage, relapsed or refractory extranodal
natural killer (NK)/T cell lymphoma and leucemia.
Cancer Sci. 2008; 99: 1016-20.
79