
UNIVERSITÀ DEGLI STUDI DI TORINO
FACOLTÀ DI MEDICINA E CHIRURGIA
LAUREA IN TECNICO SANITARIO DI
LABORATORIO BIOMEDICO
TESI DI LAUREA
Candidato: ELEONORA TESTA
Relatore: Dott.ssa CRISTIANA LO NIGRO
ANNO ACCADEMICO 2008/2009.
EXEMESTANE E POLIMORFISMI DEL CYP19A1
NEL CARCINOMA MAMMARIO OPERATO
EXEMESTANE AND CYP19A1 POLYMORPHISMS
IN EARLY BREAST CANCER
2
1. INTRODUZIONE
5
1.1 ANATOMIA DELLA MAMMELLA
5
1.2 IL TUMORE DELLA MAMMELLA
6
1.3 FARMACOGENETICA E FARMACOGENOMICA
13
1.4 I POLIMORFISMI
14
1.5 GLI INIBITORI DELL’AROMATASI
15
1.6 TAMOXIFENE ED EXEMESTANE
18
1.7 L’ALTERAZIONE DEL PATHWAY PROLIFERATIVO
19
2. SCOPO DEL LAVORO
25
2.1 POLIMORFISMI DEL GENE CYP19A1
25
2.2 POLIMORFISMI NEL GENE TP53 ED MDM2
26
2.3 POLIMORFISMI DEI GENI GLUTATIONE-S-TRANSFERASI
26
3. MATERIALI E METODI
28
3.1 ESTRAZIONE AUTOMATICA DI DNA
28
3.2 QUANTIFICAZIONE MEDIANTE NANODROP
29
3.3 PCR
31
3.4 IL SEQUENZIAMENTO
37
3.5 GENESCAN
45
3.6 ALLELIC DISCRIMINATION
47
3
3.7 PCR-AGAROSIO
50
3.8 STRUMENTI STATISTICI PER LA VALUTAZIONE DEI DATI
52
4. RISULTATI
55
4.1 RAPPRESENTAZIONE MEDIANTE BOX-PLOT
55
4.2 I DUE GRUPPI DI PAZIENTI A CONFRONTO
57
4.3 ANALISI GLOBALE DEI GENOTIPI IDENTIFICATI
66
4.4 ANALISI STATISTICA DEI DATI
67
5. CONCLUSIONI
70
6. BIBLIOGRAFIA
76
4
INTRODUZIONE
ANATOMIA DELLA MAMMELLA
La mammella è un organo pari e simmetrico, posto nella regione anteriore del torace tra il
terzo e sesto spazio intercostale. La componente ghiandolare è costituita da 15-20 lobi
ognuno dei quali sboccia verso il capezzolo attraverso un dotto galattoforo. Queste
strutture sono immerse all’interno di una componente di tessuto adiposo, mentre la
componente fibrosa di sostegno si porta in profondità e divide il parenchima ghiandolare in
lobi e lobuli. Ogni lobulo comprende gli alveoli che fungono da unità secernenti e che sono
rivestiti da epitelio semplice. I dotti hanno un calibro progressivamente crescente: dotti
alveolari, lobulari ed infine i galattofori. Ogni lobulo ha il suo dotto galattoforo che sbocca
lateralmente al capezzolo in un’ampolla che ha la capacità di accumulare il secreto
prodotto. L’epitelio da cubico semplice dei dotti alveolari diventa pluristratificato non
cheratinizzato nei dotti galattofori.
Schema della mammella normale.
5
IL TUMORE DELLA MAMMELLA
EPIDEMIOLOGIA
Il tumore alla mammella colpisce entrambi i sessi anche se nell’uomo ha un’incidenza di
100 volte inferiore. Nei Paesi Industrializzati, inoltre, rappresenta il tumore più frequente
nelle donne. In Italia ci sono circa 262 casi/100.000 ogni anno in donne con età > 65 anni e
117.6 morti/100.000 ogni anno. Ciò significa che una donna su 14 manifesta un carcinoma
mammario nel corso di una vita media normale.
Colpisce maggiormente le donne in età adulta con due picchi di insorgenza: tra i 40 e i 50
anni e dopo i 70 anni.
La prevenzione e la diagnosi precoce hanno comunque ridotto di molto la mortalità (per
esempio tra il 1993 e il 1998 si è registrato un sensibile regresso della mortalità (-9,3%).
FATTORI DI RISCHIO
L’insorgenza della neoplasia si verifica secondo un meccanismo multi-step. Nel suo
sviluppo concorrono sia una componente genetica che una componente ambientale volta a
facilitare il fenotipo legato alla mutazione genetica.
Gli studi su base epidemiologica hanno dimostrato che diversi fattori possono aumentare il
rischio di tumore alla mammella:
•
Età e sesso: raramente, se non in alcuni casi familiari colpisce donne < 25 anni.
L’incidenza aumenta nel corso della vita e l’età media alla diagnosi è di 64 anni.
•
Razza: sono maggiormente colpite le donne bianche. Un maggior numero di casi di
carcinoma alla mammella è diagnosticato nelle donne nere di età inferiore ai 40
anni rispetto alle donne bianche; più frequentemente però queste presentano i
recettori ormonali negativi e hanno differenti tipi di mutazioni sporadiche di p53.
Le donne caucasiche generalmente presentano la più alta incidenza di carcinoma
alla mammella.
6
•
Aumentata esposizione agli estrogeni ed al progesterone: menarca precoce (prima
degli 11-12 anni), menopausa tardiva (dopo i 55 anni), nulliparità o terapia
ormonale sostitutiva.
•
Anamnesi personale di precedente carcinoma alla mammella, all’endometrio o
all’ovaio: probabilmente a causa dei condivisi fattori di rischio ormonali per questi
tumori.
•
Anamnesi personale di iperplasia atipica o carcinoma in situ.
•
Storia familiare di carcinoma mammario: il rischio per una donna di sviluppare il
cancro è maggiore se la propria madre, sorella, figlia o altri membri femminili della
famiglia, ad esempio cugine, ne sono state colpite, specie se in giovane età.
•
Modificazioni genetiche: mutazioni geni BRCA1/ BRCA2
•
Radiazioni: le donne sottoposte a radiazioni durante l'infanzia, specialmente per il
trattamento del morbo di Hodgkin, hanno maggiori probabilità di sviluppare un
tumore della mammella nel corso della vita. Secondo alcuni studi, più bassa è l'età
in cui si è ricevuta la terapia radiologica, più tale rischio aumenta.
•
Dieta: elevato consumo di grassi saturi ed alcool.
LA CLASSIFICAZIONE DEL TUMORE ALLA MAMMELLA
Distinguere i vari tipi di neoplasia della mammella può esser utile nel determinare il
miglior approccio terapeutico.
Il carcinoma duttale e il carcinoma lobulare sono i più comuni. Come dice il nome,
colpiscono rispettivamente le cellule duttali e le ghiandole deputate alla produzione di
latte.
In entrambi i casi la neoplasia può rimanere localizzata (carcinoma in situ) oppure
estendersi al di fuori del sito originario del tumore e metastatizzare in altre parti del corpo
attraverso il sangue o il sistema linfatico (cancro alla mammella invasivo).
7
Esistono altre forme che originano dal tessuto connettivo fibroso, capezzolo, areola. In
alcuni casi non hanno origine all’interno della mammella ma sono metastasi di neoplasie
con sede in altre parti del corpo, come ad esempio colon, polmoni, linfoma non-Hodgkin,
melanoma.
Una buona classificazione deve tener conto dello stato dei recettori ormonali, in particolare
quelli per gli estrogeni e progesterone. Il recettore è una struttura proteica situata sulla
parte extracellulare di una cellula bersaglio, che legandosi in modo specifico ad un ligando,
media una risposta biologica. Le cellule normali della mammella ed alcune cellule di
carcinoma mammario possiedono i recettori per gli ormoni femminili. Gli ormoni mandano
segnali alle cellule affinché venga aumentata la crescita cellulare. Il cancro che presenta
questi recettori è definito ormono-responsivo ed in particolare può esser ER+ (recettore
positivo per l’estrogeno) oppure PR+ (recettore positivo per il progesterone). Nel suo
trattamento sono usati farmaci bloccanti gli ormoni (tamoxifene) in modo da rallentare la
progressione del tumore. Il cancro non ormono-responsivo non può invece avvalersi della
terapia ormonale in quanto i recettori ormonali non sono presenti.
Conoscere se nelle cellule neoplastiche è presente una overespressione del gene HER2 può
infine esser un’informazione utile ai fini della classificazione e del trattamento. Il gene
HER-2 codifica per una glicoproteina di 185 kDa appartenente alla famiglia di tipo I dei
recettori per fattori di crescita, tra cui il recettore per l’EGF, c-erbB3 e c-erbB4. La
fosforilazione del dominio intracellulare tirosin-chinasico provoca l’attivazione di un
segnale intracellulare che porta all’accensione di diversi geni coinvolti nella proliferazione
e nella crescita cellulare.
Casi di HER2 negativo, ER- e PR- sono definiti tripli negativi e la neoplasia tende ad esser
più aggressiva.
IL TRATTAMENTO DEL TUMORE ALLA MAMMELLA
Avendo oggi a disposizione diversi metodi terapeutici è necessario pianificare un
programma di trattamento. La terapia va scelta in base alla dimensione, alla localizzazione
del tumore all’interno della mammella, alle metastasi e ai risultati degli esami diagnostici
eseguiti. Pertanto le modalità di intervento dipendono in modo stretto dalla stadiazione del
tumore.
stadio: si basa sulla classificazione TNM e con esso intendiamo l’estensione della
neoplasia. (Frederick L. 2007)
8
•
Stadio 0: chiamato anche carcinoma non invasivo. Segnala un rischio elevato di
sviluppare un cancro ad entrambe le mammelle, ma non è detto che questo tumore
evolva in cancro invasivo.
•
Stadio I - II: identificano forme precoci di carcinoma mammario invasivo. Nello
stadio I il tumore ha un diametro di circa 2 cm e non è esteso oltre la mammella. Lo
stadio II invece comprende diversi livelli: tumore con un diametro di 2 cm con
invasone dei linfonodi ascellari, tumore con un diametro di 2-5 cm con o senza
invasione dei linfonodi ascellari, tumore che misura più di 5 cm di diametro ma
senza invasione linfonodale. In questi casi si interviene con terapia chirurgica
seguita da radioterapia e da una terapia adiuvante con chemioterapia o inibitori
dell’aromatasi per prevenire il rischio di una recidiva o lo sviluppo di metastasi.
•
Stadio III: definito anche tumore localmente avanzato. Il diametro è maggiore di 5
cm con estensione ai linfonodi ed ai tessuti adiacenti. Si interviene con intervento
chirurgico e radioterapia alla mammella e alla zona ascellare. I cicli di
chemioterapia possono precedere (chemioterapia neoadiuvante) o seguire
(chemioterapia adiuvante) il trattamento chirurgico.
•
Stadio IV ovvero carcinoma mammario metastatico: diffusione al cavo ascellare
delle cellule neoplastiche e attraverso il sistema linfatico estensione anche ad altri
organi. Le metastasi si riscontrano con più frequenza in ossa, polmone e fegato. La
chirurgia mira a trattare il tumore alla mammella. La chemioterapia o la
radioterapia mirano a tenere sotto controllo le metastasi. (Simona Lambertini 2004)
I tipi di trattamento del carcinoma mammario possono essere locali o sistemici. I primi
consistono nell’asportare le cellule cancerose di una determinata zona. La chirurgia e la
radioterapia, ad esempio, sono trattamenti locali. Le terapie sistemiche eliminano le cellule
cancerose diffuse in tutto l'organismo. La chemioterapia e l'endocrinoterapia, ad esempio,
sono trattamenti sistemici. Può esser utilizzata una sola forma di trattamento o una
combinazione di questi, praticati uno alla volta o in successione. L’integrazione di varie
forme di trattamento dei tumori è definita terapia integrata o multidisciplinare. In generale
possiamo
parlare
di
una
terapia
finalizzata
alla
guarigione
o
palliativa.
L’intervento chirurgico: è la prima linea di trattamento nel caso di tumore della
mammella. Viene chiamata chirurgia demolitiva l'operazione di asportazione della
mammella (o di una consistente porzione di tessuto mammario). Un'operazione che asporta
9
il tumore ma non l'intera mammella è invece denominato intervento di chirurgia
conservativa. Di solito, sono seguite da sedute di radioterapia allo scopo di distruggere
ogni cellula cancerosa residua.
Radioterapia: è un tipo di terapia oncologica loco-regionale basata sull'utilizzo di
radiazioni ad alta energia nell’ordine dei MeV (raggi X, alfa, beta, gamma), in grado di
distruggere le cellule tumorali. Le radiazioni danneggiano il DNA impedendo alla cellula
di replicarsi e causandone la morte. La terapia è diretta specialmente contro le cellule
tumorali poiché si replicano più attivamente. Tuttavia alcune cellule normali possono
essere uccise, soprattutto quelle ad elevata velocità di replicazione. La radiazione può
provenire dall'esterno del corpo mediante l’uso di apposite strumentazioni chiamate
acceleratori lineari (LINAC) capaci di generare un fascio di elettroni, raggi X o raggi
GAMMA; oppure può provenire da materiali radioattivi collocati direttamente all'interno
del tumore (brachiterapia). La radioterapia viene spesso utilizzata in associazione alla
chemioterapia, per ridurre la dimensione di un tumore prima della chirurgia o per
distruggere eventuali cellule tumorali rimaste dopo un intervento chirurgico. Solo in alcuni
casi di tumori localizzati è impiegata come trattamento principale.
Chemioterapia: è una terapia farmacologia sistemica, in grado di distruggere le cellule
tumorali con diversi meccanismi d'azione che bloccano la crescita cellulare e la sintesi del
DNA.
Un chemioterapico ideale dovrebbe uccider solo le cellule neoplastiche senza danneggiare
le cellule normali. In realtà vengono danneggiate anche le cellule normali in fase di
replicazione. L’azione citotossica che ne deriva è proporzionale al tempo di contatto e non
alla dose di farmaco somministrata.
Attualmente può portare a guarigione definitiva solo di alcuni tipi di tumore. Per molte
altre neoplasie fornisce una terapia palliativa consentendo un prolungamento delle
aspettative di vita.
Uno dei maggiori problemi che si riscontrano è relativo alla resistenza ai farmaci. Essa può
essere primaria se compare fin dal primo contatto. Si parla di resistenza acquisita se
compare verso un farmaco precedentemente efficace nei confronti della neoplasia.
I farmaci più usati sono le antracicline (adriamicina, epirubicina), gli agenti alchilanti
(ciclofosfamide),
antimetaboliti
(5-fluoruracile,
methotrexate),
alcaloidi
vegetali
10
(vincristina). Nei vari programmi di terapia, possono essere combinati fra loro: si parla in
questo caso di polichemioterapia.
Esistono diverse linee di azione della chemioterapia.
1. Chemioterapia adiuvante: eseguita in aggiunta alla terapia primaria ottenuta con la
chirurgia, serve per eliminare le cellule neoplastiche che si sono diffuse dalla sede
principale del tumore ad altri distretti all’interno del corpo. Viene impiegata in
pazienti nelle quali non è stato possibile evidenziare con i comuni esami
strumentali (scintigrafia ossea, RX torace, ecografia) metastasi a distanza, ma la
presenza di fattori prognostici sfavorevoli (istologicamente non differenziati,
invasione vascolare o linfatica, coinvolgimento del cavo ascellare), fa aumentare la
probabilità che queste siano presenti sotto forma di micrometastasi non
documentabili
quindi
con
gli
esami
strumentali.
L’alta
possibilità
di
micrometastasi a distanza è dovuta al fatto che fin dalle prime fasi il tumore alla
mammella sembra avere una diffusione sistemica. Questo tipo di terapia permette
di aumentare il tasso di sopravvivenza e l’intervallo libero da malattia. Numerosi
studi randomizzati hanno dimostrato come essa determini un significativo aumento
della sopravvivenza, in particolare nelle pazienti in premenopausa, mentre gli
stessi risultati non sono mai stati confermati nelle pazienti in postmenopausa.
(Simona Lambertini 2004) (M. Luisa Brandi 1997)
2. Neo adiuvante: il trattamento viene effettuato prima dell’intervento chirurgico
affinché venga ridotta la massa tumorale e vengano eseguiti interventi meno
demolitivi. Terapia di elezione in pazienti con carcinoma mammario localmente
avanzato, in cui si cerca di sostituire la mastectomia con un intervento chirurgico
conservativo. (Simona Lambertini 2004) Oltre alla diminuzione del volume
tumorale, la terapia neo adiuvante ha l’obiettivo di eliminare precocemente le
micrometastasi occulte. (M. Luisa Brandi 1997)
3. Palliativa: in questo caso i farmaci chemioterapici vengono impiegati al fine di
controllare l’evoluzione del tumore. Viene somministrata per migliorare la qualità
di vita del paziente riducendone i sintomi e per ottenere un beneficio in termini di
sopravvivenza.
11
Terapia ormonale: oggi la terapia ormonale ha assunto un ruolo chiave nella cura di molti
tipi di tumore in quanto le conoscenze scientifiche sul ruolo degli ormoni sessuali si sono
notevolmente perfezionate nel corso degli anni. Molte cellule tumorali richiedono gli
estrogeni per la propria proliferazione. I farmaci impiegati, agiscono interferendo con
l’attività di questi ormoni in due modi: impedendo alla cellula tumorale di utilizzare gli
ormoni prodotti (farmaci antiestrogeni) oppure inibendo la produzione degli estrogeni
stessi (inibitori dell’aromatasi). (Simona Lambertini 2004) Nella terapia ormonale gioca
pertanto un ruolo fondamentale l’espressione del recettore per gli estrogeni (ER): maggiore
è il contenuto e maggiori sono le possibilità di un successo terapeutico.
Esistono due tipologie di ER: ERα e ERβ. Sono prodotti da geni diversi ma hanno una
struttura simile ed entrambi funzionano come fattori trascrizionali nucleari dopo essere
stati attivati dai rispettivi ligandi (Fuqua SA 2004). ERα regola attraverso proteine
coregolatrici (AIB1 E SCR3) la trascrizione di geni responsivi all’estrogeno (importanti
per la proliferazione cellulare), l’inibizione dell’apoptosi, la stimolazione della
metastatizzazione e la promozione dell’angiogenesi. (McKenna NJ 1998) Meno conosciute
sono le funzioni di ERβ che pare avere un attività antagonista a quella di ERα sulla crescita
della neoplasia. (McInerney EM 1998). Quando i recettori sono attivati dal tamoxifene, che
modula selettivamente ER, alti livelli di ERβ aiutano ad inibire la crescita tumorale.
Grazie alle terapie adiuvanti endocrine come ad esempio il tamoxifene, il rischio di
recidiva dopo il trattamento chirurgico primario si riduce del 30-35%. Nella malattia
metastatica il tasso di risposte obiettive varia dal 30-35% al 65-75%.
Il tamoxifene considerato per molto tempo l’antiestrogeno per eccellenza, ha però
evidenziato come la sua somministrazione prolungata sia la causa di un’aumentata
incidenza di carcinomi dell’endometrio. Negli ultimi anni è stato introdotto l’uso di una
nuova classe di farmaci: gli inibitori dell’aromatasi (formestane, anastrozolo, letrozolo ed
exemestane) che sembrano avere un rischio minore nel determinare una nuova neoplasia.
(Simona Lambertini 2004) (Dino Amadori 2005)
12
FARMACOGENETICA E FARMACOGENOMICA
La farmacogenetica è una disciplina che si occupa di studiare le basi scientifiche della
variabilità interindividuale delle risposte ai farmaci. Grazie agli studi sui meccanismi
d’azione dei farmaci, all’individuazione di nuovi bersagli e all’identificazione del ruolo dei
fattori genetici in relazione alla risposta alla terapia, si potrebbe arrivare a programmare
l’intervento terapeutico in modo da colpire selettivamente le patologie neoplastiche. Si
ridurrebbe in questo modo la variabilità interindividuale della tollerabilità nei confronti dei
chemioterapici. Alla base della regolazione a livello individuale della farmacocinetica e
della farmacodinamica dei farmaci, ci sono i fattori genetici. Le analisi genetiche
permettono pertanto di predire in quale misura un trattamento sarà efficace e tollerato dal
paziente. Uno dei punti critici nel trattamento mediante chemioterapici consiste nel loro
indice terapeutico. Sebbene l’attività sia prevalentemente rivolta verso le cellule tumorali,
si verificano inevitabilmente anche fenomeni di tossicità nei confronti dei tessuti sani.
Mutazioni del bersaglio o fenomeni di down regulation sono la causa del fallimento di
alcuni farmaci volti ad inibire vie metaboliche determinanti per la sopravvivenza delle
cellule neoplastiche. Il sequenziamento del genoma, ha permesso la scoperta di varianti
alleliche alla base della regolazione del metabolismo dei farmaci che potrebbero essere
correlate con la loro chemiosensibilità piuttosto che con la loro tolleranza. (Danesi R
2001).
La farmacogenomica analizza l’intero genoma individuando le alterazioni geniche
responsabili delle risposte terapeutiche che non sono spiegabili con il metodo
farmacogenetico e individuando nuovi bersagli per la terapia con chemioterapici. La
classificazione della variabilità genica comprende le mutazioni geniche (delezioni,
inserzioni poco frequenti, inaspettate e che influenzano negativamente la funzione
cellulare) e le varianti delle sequenze geniche (gli SNPs) che sono presenti rispetto alle
prime con frequenza maggiore nella popolazione. Se gli SNPs interessano gli esoni il
risultato sarà il cambiamento della sequenza aminoacidica, se interessano gli introni
causano l’inserzione di siti alternativi di splicing, se invece coinvolgono la porzione
regolatoria dei geni verrà alterata l’espressione genica. Con la scoperta della correlazione
tra i vari profili di espressione dovuti a mutazioni o SNPs, è stato possibile scegliere in
modo più razionale i farmaci da utilizzare nel trattamento.
13
In particolare l’azione dei chemioterapici viene influenzata da: attivazione e inattivazione
metabolica (CYP450 e UTG), espressione dei bersagli farmacologici (EGFR), integrità
sistemi di trasduzzione che promuovono o inibiscono l’apoptosi (p53, bcl2), sistemi di
riparazione del DNA (ERCC1), attività dei trasportatori che portano i farmaci al di fuori
delle cellule (trasportatori ABC).
Le varianti genetiche coinvolte nel metabolismo dei farmaci possono esser associate a
fenotipi metabolizzatori lenti o veloci. Nel caso di un fenotipo metabolizzatore veloce
responsabile dell’inattivazione dei farmaci, esso è associato ad una ridotta esposizione
delle cellule al farmaco. Pertanto la tollerabilità è alta ma la sua attività scarsa, al contrario
degli enzimi del metabolismo dei profarmaci (CYP 450) in cui un’elevata attività risulterà
con una tollerabilità minore ma con una maggiore attività antineoplastica. (Dino Amadori
2005)
I POLIMORFISMI
Il termine polimorfismo indica, per definizione, l'esistenza in una popolazione di più di un
allele per un dato locus con frequenza superiore all'1%.
Un polimorfismo a singolo nucleotide (SNP) è un polimorfismo che si presenta tra
individui della stessa specie, caratterizzato da una differenza a carico di un unico
nucleotide.
Gli SNPs possono interessare una regione codificante di un gene, una regione non
codificante o una regione intergenica. Per quanto riguarda i primi, non necessariamente
cambiano la sequenza aminoacidica della proteina prodotta. Uno SNP che genera in tutte le
sue forme la stessa sequenza aminoacidica è detto sinonimo (mutazione silente), se invece
la sequenza prodotta è diversa allora gli SNPs sono definiti non sinonimi. Questi ultimi
vanno ancora distinti in missenso (l’aminoacido codificato è diverso dall’originale) o non
senso (il risultato è la formazione di un codone di stop). Gli SNPs che coinvolgono
sequenze non codificanti possono presentar effetti negativi sullo splicing o su legami con i
fattori di trascrizione.
Nel genoma umano ci sono circa 10 milioni di polimorfismi. Si trovano in media ogni 100300 coppie di basi all’interno dei 3 miliardi di coppie di basi del genoma, sebbene la loro
densità varia tra le regioni. Nonostante la maggior parte degli SNPs sia silente, essi hanno
14
conseguenze molto importanti sulla suscettibilità individuale ad alcune neoplasie. Per
esempio, il trattamento e l’outcome clinico delle pazienti affette da carcinoma alla
mammella, secondo alcuni studi, potrebbe dipendere dal genotipo degli enzimi
metabolizzatori. In letteratura esistono studi che evidenziano l’associazione tra gli SNPs
del gene CYP2D6 ed il successo terapeutico con tamoxifene: in particolare essi
evidenziano come il polimorfismo nel gene CYP2D6 sia associato con una ridotta efficacia
dell’enzima nel metabolizzare il farmaco. Al contrario il polimorfismo del gene CYP2C19
sembra associato ad una maggiore efficacia del tamoxifene. ( Lim HS 2007) (Schroth W
2007)
GLI INIBITORI DELL’AROMATASI
Il passaggio finale nella biosintesi degli estrogeni è l’aromatizzazione degli androgeni
(androstenedione e testosterone) in estrogeni (estrone ed estradiolo). Questa conversione,
nelle donne in premenopausa avviene nell’ovaio mentre nelle donne in postmenopausa
avviene nei tessuti periferici (pelle, muscoli e fegato).
La sintesi dell’estrogeno nel tessuto tumorale avviene attraverso la trasformazione di
estrone solfato in estrone ed estradiolo ed attraverso l’aromatizzazione di estrogeni
intratumorali (Pasqualini JR 2002). Questo dimostra come l’inibizione dell’aromatasi può
rappresentare uno strumento di riduzione della spinta proliferativa del tumore, mediante il
controllo della produzione degli estrogeni.
15
Il primo ad essere utilizzato in campo clinico fu l’aminoglutetimide. Oggi vengono
utilizzati inibitori dell’aromatasi di terza generazione (anastrozolo, letrozolo ed
exemestane) che si sono dimostrati più specifici ed efficaci rispetto ai precedenti
(inibizione dell’enzima pari al 97-99%). (Lonning PE 2002) Classificati secondo le loro
strutture ed i loro meccanismi d’azione possono essere distinti in 2 gruppi. Anastrozolo e
letrozolo sono inibitori non steroidei che legano reversibilmente l’enzima. L’exemestane
lega irreversibilmente il sito attivo dell’aromatasi competendo con il ligando. (Dino
Amadori 2005)
EXEMESTANE:
Struttura chimica dell’exemestane.
E’ un inibitore steroideo dell’aromatasi. Mima l’androstenedione e agisce come falso
substrato per l’enzima, causando la sua inattivazione (inibizione suicida). Appartiene agli
inibitori di classe I (insieme a formestane, plomestane, atamestane) che legano in modo
irreversibile l’aromatasi impedendogli di svolgere il compito di conversione degli
androgeni in estrogeni. In particolare la percentuale di androgeni soppressi varia da un
85% per l’estradiolo al 95% per l’estrone. Indicato per il trattamento adiuvante nelle donne
in postmenopausa con recettori ER+ nel cancro alla mammella iniziale, che hanno ricevuto
2-3 anni di terapia con tamoxifene e sono poi passate all’exemestane come completamento
del trattamento ormonale adiuvante. Utilizzato anche nel trattamento del cancro alla
mammella avanzato, in donne in postmenopausa nelle quali la neoplasia è progredita
nonostante trattamento con tamoxifene. (Stephen Neidle 2008)
16
L’aromatasi catalizza la conversione dell’ androstenedione in estrone.
ANTIESTROGENI
TAMOXIFENE:
Formula chimica del tamoxifene.
Fino a poco tempo fa considerato come il trattamento di elezione per il tumore alla
mammella. Si ritiene che agisca come inibitore dell’azione del recettore degli estrogeni nel
tessuto mammario. Composto non steroideo, somministrato nelle donne in premenopausa
in quanto compete con gli effetti degli estrogeni. In donne ipoestrogeniche possiede una
debole azione estrogenica sufficiente a mantenere una buona densità ossea e a prevenire
malattie cardiocircolatorie in quanto riduce i livelli di colesterolo circolante. Nei dosaggi
normalmente usati è in grado di ridurre il colesterolo di circa il 12% e la frazione LDL di
circa il 20%. Studi in vitro dimostrano che il tamoxifene agisce a livello del recettore degli
estrogeni provocando un’alterazione nella sua conformazione con conseguente danno a
livello della trascrizione dell’RNA e riduzione della proliferazione cellulare. La sua attività
antiproliferativa può esser misurata dai fattori di crescita: inibisce la secrezione dell’αTGF
17
e dell’EGF (coinvolti nella promozione del tumore) mentre stimola il βTGF (nell’inibitore
della crescita tumorale). Inizialmente non erano sconosciuti gli effetti collaterali e il
tamoxifene veniva largamente impiegato anche negli stadi iniziali del tumore mammario.
Recenti studi hanno evidenziato una correlazione tra l’uso di questo farmaco e l’insorgenza
del tumore all’endometrio e altre neoplasie. Tra gli effetti collaterali sono state descritte
anche severe alterazioni oculari, alcune irreversibili.
Il 60% delle pazienti con tumore alla mammella con lesioni ER-α positivo inizialmente
traggono beneficio dalla terapia con tamoxifene ma molte vanno incontro a ricaduta.
TAMOXIFENE ED EXEMESTANE
La terapia ormonale del carcinoma alla mammella presenta un profilo tossicologico più
favorevole rispetto a quello della chemioterapia. Per poter usufruire di questo tipo di
terapia è necessario avere una positività per i recettori ormonali, che non tutte le donne con
neoplasia mammaria presentano.
Per circa 40 anni, il farmaco di elezione nella terapia ormonale come trattamento adiuvante
era il tamoxifene, rimpiazzato recentemente dagli inibitori dell’aromatasi (anastrozolo,
letrozolo ed exemestane). La soppressione degli ormoni circolanti con gli inibitori
dell’aromatasi di terza generazione è circa del 95%-98%. (Geisler J 1998).
Abitudinariamente l’exemestane viene somministrato in sequenza dopo 3 anni di
tamoxifene, al fine di ottenere una terapia con una durata complessiva di 5 anni. Infatti, è
dimostrato come il rischio di ricaduta nelle donne con tumore alla mammella operato
persista dopo 5 anni di trattamento adiuvante con tamoxifene, sebbene non è indicato
prolungare oltre i 5 anni la terapia adiuvante con questo farmaco.
Inoltre, nelle pazienti ormono-responsive il rischio di ricaduta è più basso nei primi 5 anni
dopo terapia adiuvante rispetto alle pazienti non ormono-responsive ma, nonostante ciò,
rimane costante senza diminuire per 15 anni.
Il tamoxifene, sembra esser correlato ad una sovra regolazione dei recettori della tiroxina
chinasi (HER2, EGFR) implicati nella resistenza primaria o acquisita al farmaco stesso.
(Osborne CK 2003). Inoltre, le pazienti dimostrano un rischio aumentato per emorragie
vaginali, tumore all’endometrio, e trombosi venose. L’exemestane, invece, sembra essere
ben tollerato ed inoltre si associa ad un diminuito rischio per eventi trombotici (p=0.004),
18
iperplasia endometriale (p=0.0001), polipi uterini (p=0.0001) e severi eventi ginecologici
(p=0.0002). Nonostante ciò, gli inibitori dell’aromatasi possono provocare alterazioni del
metabolismo osseo sebbene tra tutti l’exemestane abbia effetti meno dannosi a 12 mesi di
trattamento.
L’ALTERAZIONE DEL PATHWAY PROLIFERATIVO
Le cellule cancerose hanno una capacità proliferativa incontrollata in quanto non rispettano
le fasi del ciclo cellulare e sono pertanto indipendenti da stimoli proliferativi extracellulari. Il ciclo cellulare è diviso in varie fasi rigorosamente regolate, che portano alla
duplicazione e alla trasmissione del materiale genetico da una cellula madre alle cellule
figlie. Il DNA deve essere stato precedentemente duplicato, in modo che le cellule figlie
ricevano lo stesso corredo cromosomico.
La fase G0 definisce una situazione nella quale le cellule sono quiescenti
temporaneamente, o in maniera indefinita per tutto il ciclo. Nella fase G1 la cellula è
sottoposta ad una serie di stimoli da parte dell’ambiente extracellulare. La fase S permette
la sintesi del DNA. L’intervallo tra il completamento della sintesi e la mitosi vera e propria
è chiamata G2. Infine la fase M è contrassegnata dalla formazione del fuso mitotico, dalla
divisione dei cromatidi e dalla divisione cellulare. (Sharpless NE 2001)
Esistono punti chiamati di controllo, checkpoint, che servono per monitorare l’integrità del
DNA. Posti in posizioni strategiche nelle fasi del ciclo, al fine di impedire la proliferazione
delle cellule danneggiate o mutate che porterebbero ad un accumulo di danni cellulari. Tra
i vari checkpoint, il più importante è quello nella fase G1.
La cellula normale, per passare dalla fase di quiescenza alla fase G1 ha bisogno di stimoli
esterni ai quali risponde mediante un sistema di interazione ligando-recettore ad attività
tirosino-chinasica. Si realizza così una cascata di fosforilazioni intracellulari che si
traducono con l’attivazione e l’espressione di proteine chiamate cicline. Queste ultime,
legano la CDK per formare complessi attivati. Nella fase G1, le cicline D incrementano la
loro espressione e formano complessi con la CDK4 e CDK6 che sono regolate da 2
famiglie di inibitori: p21 e p27 e dalla famiglia che comprende p16INK4a e p19ARF. I
complessi ciclina/CDK4 e ciclica/CDK6 una volta attivati fosforilano la proteina Rb che
19
libera il fattore EF2. Il fattore EF2 in forma libera può così legare sequenze di DNA e
trascrivere i geni richiesti per superare il ceckpoint in G1. (Ashcroft M 1998)
In caso di danno al DNA, Rb sequestra i fattori proteici di trascrizione EF2 bloccando la
cellula nel punto di restrizione. In questo pathway biomolecolare sono fondamentali il gene
oncosoppressore p53 e la sua unità regolatoria Mdm2.
Le cellule cancerose, tendono a rimanere nel ciclo in quanto sono in grado di evadere il
punto di controllo in fase G1, continuando a proliferare in modo incontrollato. Questa
situazione si può verificare in seguito a mutazioni a carico di p53, Rb, cicline, CDK o dei
loro inibitori. Anche una sovra-espressione della ciclica D, può portare ad una crescita
incontrollata in quanto promuove la fosforilazione di Rb ed il rilascio di EF2.
Di seguito sono brevemente spiegati i geni presi in considerazione in questo studio.
p53: È un proteina di 393 aminoacidi, con un peso molecolare di 53 kDa. Codificata dal
gene TP53 presente sul braccio corto del cromosoma 17, in particolare in posizione
17p13.1, una regione frequentemente deleta nei carcinomi. Possiamo distinguere 3 domini:
N-terminale denominato dominio di trascrizione-attivazione (TAD) che attiva fattori di
trascrizione; un dominio legante il DNA (DNA-binding core domain DBD), che contiene
ioni zinco e residui di arginina; un dominio c-terminale di omo-oligomerizzazione (OD).
Agisce come fattore di trascrizione, regolando la trascrizione di geni critici per il controllo
del ciclo cellulare. Avendo il compito di controllare l’omeostasi dell’organismo è espressa
costitutivamente, ma in condizioni normali la sua concentrazione è molto bassa poiché
viene velocemente degradata. (Kubbutat MHG 1998) (Meek DW 1998). Tuttavia, in caso
di danno al DNA viene stabilizzata e la sua concentrazione aumenta. I meccanismi di
azione di questa proteina sono sostanzialmente due: attiva la trascrizione di geni che
bloccano la progressione del ciclo cellulare e reprime i geni indispensabili per la
sopravvivenza cellulare con apoptosi della cellula stessa. Strutturalmente la proteina p53
presenta molti siti di fosforilazione dove possono agire le protein-kinasi, quando attivate a
seguito di situazioni di stress (Jayaraman L. 1999) (Shieh Sy 1997). La fosforilazione porta
a modificazioni nella conformazione della proteina che impediscono l’interazione con
mdm2. (Bottger V 1999) (Kamijo T 1998) Tra i meccanismi che stabilizzano la proteina,
un ruolo importante è svolto dalla proteina p14ARF (Stott FJ 1998) (Bates S 1998), la cui
sintesi è promossa da diversi fattori di trascrizione tra cui E2F1, MYC, E1A. (Zindy F.
1998) (Honda R 1999) La p14ARF lega mdm2 ed è in grado di impedire la degradazione
20
di p53 (Weber JD 1999) (Hsieh JK 1999). Anche la pRB è in grado di legare la p53 e di
impedirne la degradazione, con un meccanismo però un po’ diverso in quanto non
impedisce il legame tra le due proteine ma solamente l’azione di p53. (Hanahan D 2000)
Per quanto riguarda la prima delle conseguenze dell’attivazione di p53, ossia l’arresto del
ciclo cellulare, la proteina è in grado di attivare la p21WAF30 che lega le CDK e ne
impedisce l’attività. Il ciclo cellulare si arresta in corrispondenza del checkpoint al termine
della fase G1. Infatti, l’inibizione delle CDK impedisce la fosforilazione della proteina Rb
che non può rilasciare il fattore di trascrizione E2F, responsabile dell’attivazione di alcuni
geni indispensabili affinché la proteina entri nella fase S del ciclo cellulare. La cellula non
potendo duplicare il DNA non può pertanto trasmettere la mutazione alle generazioni
successive. Per quanto riguarda poi l’induzione del processo apoptotico, le modalità con
cui viene indotto non sono ancora oggi del tutto chiare, ma si pensa che questo processo
comporti l’attivazione di processi ossidoriduttivi e la lisi dei mitocondri.
Mutazioni del gene TP53 sono state descritte in quasi tutti i tipi di tumore ed in particolare
nel 25% dei tumori alla mammella. (Olivier M 2001) Al giorno d’oggi se ne conoscono
circa 20.000, suddivise in diverse categorie: delezione di uno o di entrambi gli alleli;
troncamento della proteina da parte di mutazioni puntiformi che generano un segnale
prematuro di stop nella traduzione dell’mRNA; mutazioni missenso che sono il tipo più
comune con una frequenza del 75% e che consistono in una sostituzione di uno o più
aminoacidi nella sequenza.
21
Pathways di p53.
Mdm2: la minute doble murine 2 è una proteina di 491 aminoacidi con un peso
molecolare di 90 kDa. Il gene che codifica per questa proteina si trova sul cromosoma 12
in posizione 12q15. Presenta una localizzazione nucleare e citoplasmatica ed è presente in
quantità ridotta o è del tutto assente nelle cellule p53 negative. Possiede un dominio Nterminale, un dominio acidico centrale (la cui fosforilazione pare importante per la
regolazione delle funzioni della proteina) ed infine un dominio RING C-terminale, che
conferisce alla proteina l’attività E3 ubiquitina ligasi. Questa funzione promuove la
degradazione proteasoma dipendente di p53 e regola la sua attività mediante un
meccanismo di feed-back negativo (il legame avviene con il dominio N-terminale di
transattivazione). Infatti l’mdm2 inibisce le funzioni di apoptosi e di blocco della
proliferazione in fase G1, proprie della proteina p53.
Inoltre, facilita la degradazione della proteina Rb (retinoblastoma) nei proteasomi
dipendenti e pertanto una sua overespressione contribuisce allo sviluppo del cancro dovuto
alla destabilizzazione di Rb.
Mdm2 funziona in due siti: a livello genico riduce la trasformazione di p53; a livello
proteico lega il prodotto di p53, ne riduce la sua attività e media a sua esportazione
nucleare, aumentandone la degradazione proteosomiale e l’ubiquitinazione. In presenza di
22
danno al DNA, come già detto precedentemente, il gene p53 viene indotto, con un
incremento del suo prodotto proteico. La successiva fosforilazione rende la proteina p53
attiva con aumento della sua emivita e con l’inattivazione della proteina Mdm2 attraverso
p19arf.
Polimorfismi a singolo nucleotide sono stati identificati nel promotore di Mdm2 e hanno
mostrato una maggiore affinità per l’attivatore trascrizionale sp1 con un conseguente
aumento dei livelli di RNA e di proteina che attenuano il pathway di p53. Le mutazioni di
questa proteina sono insolite e solitamente quelle coinvolte nella cancerogenesi sono
mutazioni puntiformi. E’ coinvolta soprattutto nello sviluppo dei tumori nei tessuti molli,
osteosarcomi o carcinoma esofageo. (Uhrinova S 2005) (Vassilev LT 2004) (Lorusso
2005) (Meister A 1983)
Glutatione-S-transferasi: enzima coinvolto nella resistenza a diverse famiglie di
chemioterapici. Agisce coniugando diverse sostanze al glutatione ridotto in modo che
possano essere eliminati dall’organismo. Il tripeptide γ-glutamilcisteinglicina o glutatione
(GSH) è pertanto molto importante nell’omeostasi redox intracellulare ed esiste in forma
ridotta (GSH) ed in forma ossidata (GSSG). (Kalyanaraman B. 1996) (Briviba K. 1999)
(Hayes J.D. 1995)
Le GST vengono suddivise in 3 grandi famiglie: citosoliche o solubili suddivise in base
all’omologia della sequenza aminoacidica. Le mitocondriali e le microsomiali o MAPEG
coinvolte soprattutto nel matabolismo degli ecosanoidi.
Tutte hanno la comune funzione di detossificare le sostanze dannose. L’azione di questo
enzima porta inevitabilmente ad una diminuzione di GSH intracellulare con un inevitabile
aumento del GSSG, aumento molto tossico perché porta alla formazione di ponti disolfuro
nelle proteine cellulari. (Pemple S.E 1996) (Sheehan D.001) (Frova C. 2006) (Philpot RM.
1991)
Per quanto riguarda la correlazione tra questo enzima ed i tumori, le GST sono coinvolte
nel processo di prevenzione del processo carcinogenico mediante inattivazione o
detossificazione del composto elettrofilico di derivazione carcinogenica, ma come detto
precedentemente sono coinvolte anche nel processo di insorgenza delle farmacoresistenze.
Tra le varie forme isoenzimatiche spicca come importanza la GSTP1-1 presente in molti
tumori solidi, overespressa nel caso di forme resistenti ai farmaci.
23
Citocromo p450: i citocromi sono proteine vettori di elettroni che permettono
l'utilizzazione dell'ossigeno a livello cellulare. La famiglia del citocromo P450 è una
superfamiglia enzimatica di emoproteine appartenente agli enzimi di fase 1, coinvolti nel
metabolismo dei farmaci. Nell’uomo sono stati sinora identificati più di 63 geni codificanti
per isoforme del citocromo P450, di cui 57 geni completi e 5 pseudogeni, divisi in 18
famiglie e 43 sottofamiglie, espressi nel fegato ed in altri tessuti come il tratto
gastrointestinale, i reni, i polmoni, la cute ed il sistema nervoso centrale. (Gaetano Crepaldi
2002)
Sono i maggiori attori coinvolti nella detossificazione dell'organismo, essendo in grado di
agire su un gran numero di differenti substrati, sia esogeni (farmaci e tossine di origine
esterna) che endogeni (prodotti di scarto dell'organismo).
Le reazioni catalizzate dalle isoforme del citocromo P450 sono svariate. La più comune è
una classica reazione da monossigenasi: il trasferimento di un atomo di ossigeno
dall'ossigeno molecolare ad un substrato organico, con riduzione del secondo atomo di
ossigeno ad acqua:
RH + O2 + 2H+ + 2e– → ROH + H2O
Questi enzimi si ritrovano principalmente legati alle membrane del reticolo endoplasmatico
liscio ed alla membrana mitocondriale interna tramite la regione N-terminale idrofobica, in
particolare nella frazione microsomiale delle cellule epatiche.
Le funzioni svolte dal citocromo P450 nell’uomo sono di ossidazione ed eliminazione di
sostanze endogene, come la bilirubina derivante dal metabolismo dell’emoglobina, e di
sostanze esogene, come inquinanti e farmaci, ma comprendono anche la regolazione dei
livelli di concentrazione degli ormoni steroidei, come gli estrogeni ed il testosterone, la
biosintesi del colesterolo ed il metabolismo della vitamina D.
Per quanto riguarda la regolazione della concentrazione degli ormoni steroidei, il
citocromo implicato è il CYP19A1. Costituito da nove esoni. La regione a monte di questo
gene è piuttosto complessa e contiene più promotori. La trascrizione può avere inizio a
livello dei diversi promotori con la produzione di 7 diversi prodotti, tutti codificanti però
per il CYP19A1.
24
SCOPO DEL LAVORO
Lo scopo dello studio presentato in questa tesi è valutare la possibile correlazione tra gli
SNPs nel gene CYP19A1 e l’outcome clinico in pazienti affette da carcinoma mammario
operate e trattate con exemestane. Inoltre, lo studio è stato esteso all’analisi di SNPs in
p53, MDM2 e nei geni GST al fine di:
•
identificare
ulteriori
determinanti
molecolari
per
la
resistenza/sensibilità
all’exemestane;
•
determinare il ruolo di markers CYP19A1 in associazione ad un pannello di
markers candidati in p53, MDM2, e nei geni GST;
•
predire l’outcome alla terapia in un gruppo di pazienti omogenee;
•
generare un algoritmo multigenico di risposta al trattamento basato sulla
combinazione di questi markers.
POLIMORFISMI DEL GENE CYP19A1
Sono noti vari polimorfismi di CYP19A1 coinvolti nella regolazione dell’attività
dell’aromatasi attraverso la stabilizzazione dell’mRNA, l’aumento della trascrizione o la
regolazione post-traduzionale della sua espressione. I polimorfismi che caratterizzano geni
coinvolti nel metabolismo degli estrogeni potrebbero alterare la possibilità di avere una
corretta soppressione estrogenica mediata dal farmaco. In particolare, il target CYP19A1
presenta dei polimorfismi che influenzano l’attività enzimatica e quindi la risposta
tumorale agli inibitori delle aromatasi.
Particolare importanza ha il polimorfismo C1588T localizzato a livello della regione
3’UTR non tradotta. Alcuni studi, hanno dimostrato che l’allele C è associato ad una scarsa
soppressione pituitaria durante la stimolazione ovarica. I pazienti con genotipo CC
necessitano di un numero di giorni maggiore per ottenere una soppressione pituitaria,
25
rispetto ai pazienti con genotipo TT. L’allele C, infatti, porta alla formazione di una
quantità minore di enzima aromatasi ed è pertanto è associato con una riduzione
statisticamente significativa dei livelli di estradiolo e di estrone riducendo il rapporto
estradiolo/testosterone ed estrone/androstenedione. Al contrario, l’allele T è associato con
alti livelli di estradiolo ed estrone.
In particolare, in questo lavoro sono state analizzate due regioni del gene CYP19A1: una
nel gene 3’UTR (C1558T) dell’esone 10 ed una nell’introne 4 (IVS4[TCT]+/-). (Alison M.
Dunning 2004)
POLIMORFISMI NEL GENE TP53 ED MDM2
Di spiccata importanza in p53 è il polimorfismo Arg72Pro; in particolare, le proteine che
contengono l’Arginina sembrano essere più efficaci nell’induzione dell’apoptosi di quanto
non lo siano quelle che presentano la variante Prolina. (Dumont P 2004) (Thomas M
1999). Pertanto, alcuni studi suggeriscono come l’omozigosi Arg/Arg induca l’apoptosi
meglio di quanto non faccia il genotipo omozigote per Pro/Pro. Per quanto riguarda la
correlazione tra polimorfismo e terapia adiuvante, la sopravvivenza libera da malattia nelle
pazienti con genotipo Pro/Pro è inferiore rispetto alle omozigoti Arg/Arg. (Tatsuya
Toyama 2007)
Per quanto riguarda i polimorfismi in Mdm2 invece è stato preso in considerazione lo
SNP309 T/G localizzato a livello della regione del promotore. In letteratura è riportato
come l’omozigosi G/G sia correlata con una minor sopravvivenza rispetto all’omozigosi
T/T. (Manner Gareth 2006)
POLIMORFISMI DEI GENI GLUTATIONE-S-TRANSFERASI
Abbiamo analizzato in particolare il polimorfismo di GSTP1 nell’esone 5 (A313G) in cui
un’Isoleucina è sostituita con una Valina (I105V). Questo codone che subisce la mutazione
si trova nel sito legante di GSTP1. Questo polimorfismo è associato secondo alcuni studi
ad una consistente diminuizione dell’attività dell’enzima. La forma Val/Val ha un 60% di
sopravvivenza in più rispetto alla variante Ile/Ile. Infatti, alti livelli di GST dati dalla forma
26
Ile/Ile sono correlati ad una minor sopravvivenza delle pazienti proprio perché l’enzima
GST è molto più efficace nella sua azione ed elimina velocemente il chemioterapico.
Il polimorfismo presente nel gene Glutatione S-transferasi mu, M1 (GSTM1) è dovuto ad
una delezione del gene stesso. Caratterizzato dalla delezione della maggior parte della
regione codificante del gene, determina una perdita di funzionalità dell’enzima. Anche il
polimorfismo nel gene Glutatione S-transferasi theta, T1 (GSTT1) origina dalla delezione
della maggior parte della regione codificante del gene e determina anch’esso una perdita di
funzionalità dell’enzima. (Kathleen M. Egan 2004) (Mingfang Zhao 2001)
27
MATERIALI E METODI
Le caratteristiche cliniche delle pazienti affette da carcinoma mammario ER positivo
operate e trattate con exemestane arruolate in questo studio, sono riportate nella tabella
seguente:
CARATTERISTICHE DELLE PAZIENTI
n
PAZIENTI
106
ETA' MEDIA (RANGE)
62,6 (48,0 85,7)
ER+ (n° PAZIENTI)
106
NON RICADUTE
101
FOLLOW UP DALLA CHIRURGIA (MESI)
RICADUTE
50.5 (10-87)
5
TEMPO LIBERO DA MALATTIA
26,2(13-63)
Il DNA è stato ottenuto mediante l’estrazione da linfociti di sangue periferico intero,
prelevato in provette vacutainer da 6 ml contenenti anticoagulante EDTA. Il consenso,
approvato dal comitato etico, è stato ottenuto prima di iniziare l’arruolamento e tutte le
pazienti hanno fornito un consenso informato scritto di adesione allo studio.
ESTRAZIONE AUTOMATICA DI DNA
Per velocizzare e facilitare l’analisi è stata eseguita l’estrazione del DNA mediante il BIO
ROBOT EZ1 (DNA Blood Kit QUIAGEN). Da 200 µl di sangue si ottengono 200 µl di
estratto di DNA, con una concertazione di circa 20ng/µl.
28
L’estrazione degli acidi nucleici è il primo passo nelle applicazioni di biologia molecolare.
Il procedimento si suddivide in tre fasi: la lisi cellulare, la deproteinizzazione del campione
lisato e la precipitazione dell’acido nucleico. La particolarità dell’estrattore automatico da
noi utilizzato è l’adsorbimento del DNA su microsfere (biglie) di silice magnetiche.
Il DNA estratto è stato conservato a -20°C fino al momento della genotipizzazione.
QUANTIFICAZIONE MEDIANTE NANODROP
La quantificazone del DNA è stata effettuata misurando l’assorbanza del campione a 260 e
280 nm utilizzando lo spettrofotometro Nanodrop ND-1000. Lo strumento utilizza soltanto
1µl di campione per la lettura e permette quindi di riservare più materiale per le successive
analisi. Inoltre, non richiede l’uso di cuvette o capillari, perché il campione viene pipettato
direttamente sulla superficie di misura. Non sono nemmeno necessarie diluizioni del
campione: ciò consente di eseguire misure più rapide ed in modo più semplice. Bastano
infatti meno di 30 secondi per effettuare la lettura e prepararsi alla successiva. La tensione
superficiale serve per mantenere sulla colonna il campione liquido mentre viene effettuata la
29
misurazione tramite due fibre ottiche. Lo spettro e la relativa analisi vengono visualizzati
sullo schermo del PC ed archiviati in esso.
Esempio di misurazione della concentrazione di DNA al Nanodrop
Prima di eseguire la determinazione dei campioni, è importante eseguire una misura del
bianco.
Per determinare la purezza dell'acido nucleico vengono utilizzati i seguenti rapporti:
•
260/280= indice della contaminazione da proteine. Per in DNA il rapporto deve
essere 1.6-1.8; rapporti inferiori indicano una contaminazione da proteine.
•
260/230= indice della contaminazione da fenoli (solventi); il valore ottimale di
questo rapporto e di circa 2.2: rapporti inferiori indicano contaminazione da
solventi.
E' possibile utilizzare anche la lunghezza d'onda di 320nm come background: a questa
lunghezza d'onda non assorbono né acidi nucleici né proteine quindi l'assorbanza dovrebbe
essere zero. Il valore di assorbanza a 320nm deve essere sottratto agli altri valori di
assorbanza.
30
Eempio di rappresentazione delle misure effettuate al Nanodrop
PCR
La PCR (Polymerase Chain Reaction) è una tecnica di biologia molecolare che consente
l’amplificazione di frammenti di acidi nucleici dei quali si conoscano le sequenze
nucleotidiche iniziali e terminali. L'amplificazione mediante PCR consente di ottenere
molto rapidamente in vitro la quantità di materiale genetico necessaria per le successive
applicazioni.
Esistono numerose variabili che condizionano la reazione di amplificazione. Innanzitutto
l’enzima Taq DNA polimerasi, che è capace di resistere a temperature molto elevate
(97°C) ed è in grado di funzionare fino da temperature di 65°C. La sua concentrazione può
oscillare tra 1 e 5 unità per 100µl di reazione in quanto con concentrazioni troppo elevate si
possono accumulare prodotti di reazione aspecifici: se, invece, la concentrazione
dell’enzima è troppo bassa, difficilmente si raggiungono rese ottimali di amplificato.
La concentrazione ottimale dei primer è circa 0,75-1µM: concentrazioni elevate possono
promuovere l’accumulo di prodotti aspecifici, che determinano un notevole calo in resa dei
prodotti desiderati.
I deossiribonucleotidi trifosfati devono essere ad una concentrazione di circa 0,2 mM, ed è
consigliabile utilizzare per i quattro nucleotidi la stessa concentrazione, questo infatti
minimizza gli errori di incorporazione.
31
Essenziale per la PCR è lo ione magnesio (Mg2+), utilizzato in concentrazione variabile tra
0,05 e 5 mM (mediamente 1,5 mM), che influenza l’attività dell’enzima aumentando la
temperatura di denaturazione del DNA bersaglio, condiziona l’attacco dei primer
stabilizzando l’ibrido molecolare e forma complessi solubili con i dNTPs che sono i veri
substrati riconosciuti dalla DNA polimerasi.
Esistono 3 fasi nella reazione di PCR:
Denaturazione: il DNA a doppia elica è denaturato alla temperatura di circa 95°C ed è
convertito in DNA a singola catena;
Annealing: i primer oligonucleotidici complementari alle due estremità 3' della sequenza
da amplificare ibridano con i due filamenti denaturati ad una temperatura che è
orientativamente 5 °C più bassa della Tm ( temperatura di Melting = 4 (G+C ) + 2 (A + T))
dei primer stessi;
Estensione: i primer oligonucleotidici, in presenza dei quattro deossinucleotidi trifosfati e
di una DNA polimerasi, vengono estesi ognuno in direzione dell'altro ma su due diverse
catene complementari portando alla sintesi di due molecole di DNA a doppia elica copie
della regione bersaglio delimitata dagli inneschi. (Innis M. A. 1990)
Ciascuna delle tre fasi della reazione di amplificazione è caratterizzata da strette condizioni
termodinamiche (temperatura e tempo di ciascuna fase), dalle quali dipende la riuscita del
ciclo di amplificazione. Si deve tenere presente che temperature eccessive determinano la
perdita di attività della polimerasi: la Taq polimerasi ha infatti un’emivita che diminuisce
progressivamente con l’aumentare della temperatura (> 2 ore a 92,5°C, 40 minuti a 95°C e
5 minuti a 97,5°C).
Si deve evitare l’uso di primer che presentino complementarità all’estremo 3’, perché in tal
caso si possono formare dei dimeri tra loro che riducono la resa del prodotto desiderato; si
devono inoltre evitare primer con sequenze palindrome e con strutture secondarie estese.
Una considerazione generale è che i primer stessi devono essere sufficientemente
complessi affinché la probabilità di ibridare sequenze diverse da quella voluta sia
estremamente bassa.
Una conseguenza dell’uso di una temperatura di annealing troppo bassa è che uno o
entrambi i primer possono ibridarsi con sequenze diverse da quella bersaglio
32
(amplificazione non specifica) determinando quindi un calo in resa del prodotto desiderato;
al contrario una temperatura più alta determina una riduzione dell’ibridazione dei primer
sul DNA bersaglio e quindi della resa stessa.
Il numero di cicli di amplificazione è un fattore condizionante la resa di amplificazione.
Tale numero dipende da vari parametri, e principalmente dalla quantità di DNA bersaglio
di partenza.
In generale condizioni che incrementano la resa di amplificazione portano ad una ridotta
specificità di amplificazione cioè a rischi di amplificazioni non specifiche. Le condizioni
ottimali di reazione sono quelle che assicurano un bilanciamento tra queste due opposte
tendenze.
Schema delle fasi di una PCR:
1. Denaturazione
2. Annealing
3. Allungamento
4. Termine del ciclo
33
L’elettroforesi con l’amplificato su gel di agarosio e’ un metodo semplice e veloce che
permette di separare, e quindi identificare, frammenti di DNA amplificati in base al loro
peso molecolare. I frammenti migrano, nel campo elettrico che attraversa il gel, dal polo
negativo a quello positivo, in funzione delle cariche elettriche conferitegli dai gruppi
fosfato. La velocità di migrazione dipende: 1) dalle dimensioni dei frammenti; 2) dalla
percentuale dell’agarosio nel gel; 3) dal voltaggio applicato. Frammenti lineari più piccoli
migrano più velocemente rispetto a quelli più grandi.
Esempio di gel di controllo.
Le reazioni di PCR per l’amplificazione della regione contenente gli SNPs nei singoli geni
sono state così allestite:
TABELLA PRIMER UTILIZZATI
Geni analizzati
Primer F
Primer R
CYP19A1
5'GTCTGGAACACTA 5'ATGCCATGGGCCACT
3’UTR_F2 e R2
GAGAAGGCTGGTC
GAGTGTTCAC3'
AGTAGC3'
CYP19A1deltaTCT ACCAGGAGTTCTCC CAAAAAAGGCACATTC
_F e R
TGACC
ATAGAC
P53EX4F2-R2
CAATGGTTCACTGA CTGTCCCAGAATGCAA
AGACCC
GA
MDM2 SNP309F e CGGGAGTTCAGGG
AGCAAGTCGGTGCTTAC
R
CTG
TAAAGGT
GSTP1 I105V F e R CCAGTGACTGTGTG CAACCCTGGTGCAGAT
GSTT1 del F e R
GSTM1 del F e R
TTGATC
GCTC
TTCCTTACTGGTCC
TCACCGGATCATGGCCA
TCACATC
GCA
GAACTCCCTGAAA
GTTGGGCTCAAATATAC
AGCTAAAGC
GGTGG
34
La mix per la PCR dei vari geni è stata così allestita:
Mix PCR
conc finale
stock
Volume x 1 camp.
dNTPs
200 µM
2 mM
1 µl
buffer
1X
10X
1 µl
MgCl2
1.5 mM
25 mM
0.6 µl
primer F+R
0.75 µM
5 µM
1 µl
TaqGold
0.25U
5 U/µl
0.05 µl
H2O
fino a 10.5 µl (0.5 µl in + per evaporazione) 4.85 µl
DNA
30 ng
20 ng/µl
2 µl
Il numero dei cicli e le temperature delle varie fasi nel termociclatore sono state
personalizzate in base ai geni da amplificare:
CYP19A1 3’UTR
Cicli di temperatura:
95°C 10’
95°C 30’’
60°C 30’’
35 cicli
72°C 40’’
72°C 15’
4°C
10’
CYP deltaTCT
Cicli di temperatura:
95°C 10’
94°C 25’’
55°C 20’’
10 cicli
72°C 30’’
90°C 25’’
55°C 20’’
25 cicli
72°C 30’’
72°C 15’
4°C
10’
35
p53EX4
Cicli di temperatura:
95°C 10’
95°C 1’
55°C 1’
40 cicli
72°C 1’
72°C 7’
4°C 10’
MDM2 SNP309
Cicli di temperatura:
95°C 10’
95°C 30’’
60°C 30’’
35 cicli
72°C 40’’
72°C 15’
4°C
10’
GSTP1 I105V
Cicli di temperatura:
95°C 10’
95°C 30’’
60°C 30’’
35 cicli
72°C 40’’
72°C 15’
4°C
10’
GSTT1
Cicli di temperatura:
95°C 10’
95°C 30’’
60°C 30’’
35 cicli
72°C 40’’
72°C 7’
4°C 10’
36
GSTM1 del F e R
Cicli di temperatura:
95°C 10’
95°C 30’’
60°C 30’’
35 cicli
72°C 40’’
72°C 7’
4°C 10’
Per il controllo della corretta riuscita della PCR abbiamo corso 4 µl di amplificato su gel di
agarosio al 2%.
Schema delle metodiche utilizzate per lo studio dei vari geni:
Gene
Variante
Metodica
CYP19A1 TCT (+/-)
primer marcato 6-Fam e sequenziatore- gene
scan
CYP19A1 3’UTR T/C
Allelic discrimination
p53 esone
4
Sequenziamento
GSTT1
ins/del
PCR-agarosio
GSTM1
ins/del
PCR-agarosio
GSTP1
MDM2
Sequenziamento con primer F
A/G
Sequenziamento con primer R
IL SEQUENZIAMENTO
La purificazione della reazione di PCR per il sequenziamento è stata eseguita con il kit
EXO-SAP. Il DNA, per la buona riuscita del sequenziamento deve essere pulito dai
nucleotidi, primer e sali che potrebbero inficiare il risultato. Il metodo EXO-SAP si basa su
una
reazione
enzimatica
mediante
esonucleasi
I
e
fosfatasi
alcalina.
Gli enzimi degradano rispettivamente primer e dNTPs, ma non rimuovono sali o altri
prodotti secondari della PCR.
37
I campioni sono stati purificati da residui di primer e dNTPs con 2 µl di EXO-SAP in
termociclatore per 20 min a 37°C e 5 min a 90°C.
Le fasi per ottenere il prodotto finale da far correre sul sequenziatore automatico sono:
1. PCR
2. purificazione del prodotto di sequenza
3. PCR di sequenza
4. purificazione del prodotto di sequenza
5. corsa sul sequenziatore
La PCR di sequenza è simile ad una normale PCR, nonostante esistano alcune sostanziali
differenze: nella reazione viene usato solo un primer così che l’amplificazione del prodotto
risulta lineare e non esponenziale. Inoltre, vengono usati dei dideossinucleotidi oltre ad i
normali deossinucleotidi. La particolarità di questi nucleotidi è nel fatto che interrompono
la reazione quando vengono incorporati. La TAQ polimerasi da utilizzare dovrà pertanto
avere come caratteristica una bassa discriminazione tra dNTP e ddNTP: se si accorgesse
che il ddNTP non è il dNTP perfetto, potrebbe non incorporarlo.
Elettroferogramma (traccia) di una porzione di sequenza di DNA
T=Timina A=Adenina G=Guanina C=Citosina.
Il sequenziamento del DNA è la determinazione dell'ordine dei diversi nucleotidi
(Adenina, Citosina, Guanina e Timina) che costituiscono l'acido nucleico.
38
Sono state ideate diverse strategie per ottenere la sequenza nucleotidica del DNA. I primi
metodi, tra cui quello ideato da Allan Maxam e Walter Gilbert nel 1973, (Proc Natl Acad
Sci U S A. 1973) erano piuttosto complicati; una svolta si ebbe nel 1975 con la prima
pubblicazione di una strategia enzimatica ancora oggi diffusissima, sviluppata da Frederick
Sanger, il cosiddetto metodo dei terminatori di catena, chain termination method o metodo
Sanger, dal nome del suo scopritore (Sanger F. 1975) (Sanger F. 1977) che ricevette per
questo il suo secondo premio Nobel. Un'altra strategia inizialmente molto popolare ed
utilizzata fu sviluppata dagli stessi Maxam e Gilbert nel 1977 ed è conosciuta sotto il nome
di metodo di Maxam e Gilbert.
Più recentemente sono stati sviluppati nuovi metodi caratterizzati dalla capacità di
sequenziare molti frammenti di DNA contemporaneamente (anche se con efficienza
minore in termini di numero di basi sequenziate per frammento) aprendo una nuova era del
sequenziamento. Queste metodiche vanno sotto il nome di sequenziamento ad elevato
parallelismo.
METODO SANGER
T=Timina A=Adenina G=Guanina C=Citosina
Il metodo Sanger è un metodo cosiddetto enzimatico, poiché richiede l'utilizzo di un
enzima; il principio della tecnica sviluppata da Sanger si basa sull'utilizzo di nucleotidi
39
modificati (dideossitrifosfato, ddNTPs) per interrompere la reazione di sintesi in posizioni
specifiche. I nucleotidi dideossitrifosfato sono molecole artificiali corrispondenti ai
nucleotidi naturali, ma si differenziano per l'assenza del gruppo idrossilico (-OH) sul
carbonio 2' e 3' della molecola. I dideossinucleotidi, a causa della loro conformazione,
impediscono che un altro nucleotide si leghi ad essi, in quanto non si possono formare
legami fosfodiesterici.
Confronto tra deossiadenosina (sopra) e dideossiadenosina (sotto).
I dideossinucleotidi devono essere marcati (radioattivamente o per fluorescenza) in modo
da poter visualizzare le bande dei frammenti di DNA neosintetizzato dopo aver effettuato
l'elettroforesi.
Il campione di DNA da sequenziare viene diviso in quattro reazioni separate, ognuna delle
quali contiene la DNA polimerasi e tutti e 4 i deossiribonucleotidi (dATP, dCTP, dGTP,
dTTP). Ad ognuna di queste reazioni viene poi aggiunto solo uno dei quattro nucleotidi
dideossi (ddATP, ddCTP, ddGTP, ddTTP) in quantità stechiometricamente inferiore per
permettere un’elongazione del filamento sufficiente per l'analisi. L'incorporazione di un
dideossinucleotide lungo il filamento di DNA in estensione ne causa la terminazione prima
del raggiungimento della fine della sequenza di DNA stampo; questo dà origine ad una
serie di frammenti di DNA di lunghezza diversa interrotti in corrispondenza
dell'incorporazione del dideossinucleotide.
I frammenti generati da queste reazioni, vengono poi fatti correre su gel di poliacrilammide
che permette la separazione dei vari frammenti con una risoluzione di un nucleotide.
Ognuna delle 4 reazioni è corsa su pozzetti vicini, successivamente le bande sono
40
visualizzate su lastra autoradiografica o sotto luce UV, e la sequenza viene letta
direttamente sulla lastra o sul gel, a seconda del tipo di marcatura dei dideossinucleotidi.
Basandosi su questa procedura, la metodica è stata affinata per facilitare la reazione, e con
l'avvento dell'automatismo la reazione di sequenziamento è diventata molto più veloce.
Attualmente è possibile effettuare, anziché quattro reazioni distinte per ogni nucleotide
modificato, una sola reazione utilizzando i 4 ddNTPs marcati fluorescentemente in modo
diverso tra loro ed utilizzando lettori ottici appropriati. In questo modo ogni filamento di
DNA emetterà una luce di colore diverso in base al nucleotide (ddNTP) con il quale
terminerà.
SEQUENZIATORE AUTOMATICO
Il sequenziatore automatico permette di eseguire, coniugando a ciascun ddNTP un diverso
marcatore fluorescente, quattro reazioni di sequenziamento in un unico tubo da saggio.
Rivestono particolare importanza i terminatori BigDye, sistemi a trasferimento di energia a
singola molecola, costituiti da accettore e donatore. Presenta il vantaggio di emettere un
segnale omogeneo, basso rumore di fondo, luminosità maggiore, facilità interpretativa per
A e G attigue.
Le emissioni fluorescenti vengono captate da un rilevatore e le informazioni vengono
integrate e trasformate in picchi di colore diverso, con aree proporzionali all’intensità di
emissione.
I sequenziatori automatici possono essere a gel o a capillare, quello da noi utilizzato
appartiene alla seconda categoria. Il capillare è caricato del polimero di corsa (POP4 o
POP7) e i frammenti marcati di DNA vengono rivelati man mano che corrono lungo il
capillare.
41
Il sequenziatore automatico utilizzato per eseguire l’analisi dei geni è AB 3100 della ditta
APPLIED BIOSYSTEMS. Il Big Dye da noi utilizzato è il 3.1, migliore rispetto alla
versione 1.1 per le sequenze lunghe e con migliore risoluzione soprattutto nella parte
finale.
Volendo riassumere il principio del sequenziatore automatico, il prodotto di PCR viene
sottoposto ad elettroforesi all’interno di un capillare. Un raggio laser colpisce il capillare
eccitando la fluorescenza dei fluorocromi che lo attraversano e che marcano i frammenti di
DNA. Ciascuno dei quattro diversi fluorocromi emette una diversa lunghezza d’onda. Una
cellula fotoelettrica rileva sequenza, tipo ed intensità, delle varie emissioni luminose ed il
tutto viene registrato in forma grafica. La sequenza dei picchi corrisponde alla sequenza
dei nucleotidi, il colore del picco alla base azotata rilevata. Di norma il sistema interpreta
automaticamente l’elettroferogramma, sebbene possa comparire una N quando non riesce a
dare interpretazione. In ogni caso è possibile inserire a mano la base azotata mancante,
rifacendosi se è possibile alla sequenza del filamento reverse.
42
Reazione di sequenza:
Mix Sequenza
Volume x 1 camp.
Sequencing Buffer 5X
2 µl
Ready Reaction Mix V3.1
1 µl
Primer 5 µM
1 µl
H2O
fino a 10.5 µl
4.5 µl
2 µl
Prodotto di PCR purificato
Cicli di temperatura:
96°C 1’
96°C 10’’
60°C 3’
4°C
25 cicli
∞
Purificazione della reazione di sequenza dai ddNTPs fluorescenti e dai sali
•
95 µl di Precipitation Solution (NaAc 3M 33µl ; Et-oh 694µl; H2O 272µl)
•
Centrifugare a 4000 rpm per 20 min. a temperatura ambiente.
•
Aggiungere 150 µl di etanolo al 70%
•
Centrifugare a 4000 rpm per 5 min. a temperatura ambiente.
•
Centrifugare a 1000 rpm per 1 min con la piastra in posizione invertita
•
Risospendere in 15 µl di HiDi-Formamide
•
Lasciare a temperatura ambiente x 10 minuti (o in ghiaccio)
•
Denaturare per 2 min. a 95°C e mettere in ghiaccio per 5 min.
•
Trasferire tutto il campione in piastra da sequenza.
43
Gene MDM2 esempio di eterozigoti T/G
Gene MDM2 esempio di omozigosi T/T
44
Gene MDM2 esempio di omozigosi G/G
GENESCAN
L'analisi dei frammenti di DNA tramite genescan viene utilizzata per determinare la
presenza di mutazioni, polimorfismi e siti di restrizione in una sequenza genica. A
differenza del DNA sequencing, prevede l'utilizzo di primers fluorocromati nella reazione
standard di PCR in modo tale che il frammento di interesse viene subito visualizzato e
quindi analizzato. Usando questa tecnica si può contemporaneamente, molto rapidamente,
distinguere tra un prodotto di PCR normale e uno mutato o si può individuare un
polimorfismo che implichi variazione nella lunghezza del frammento analizzato.
.La presenza del polimorfismo TCT ins/del nell’introne 4 del gene CYP è stata analizzata
utilizzando il programma GeneScan Analysis version 3 (sullo strumento AB 3100 della
ditta APPLIED BIOSYSTEMS); il primer forward è stato marcato con una tag
fluorescente 6-Fam (Applera).
Ogni frammento viene confrontato con uno standard interno, nel nostro caso abbiamo
utilizzato il LIZ 500 (Perkin-Elmer). La particolarità di questa sequenza è che presenta i
primer marcati anziché i nucleotidi. L’elettroferogramma presenterà picchi di due colori
diversi, nel nostro caso il blu per indicare il campione e l’arancione per indicare lo
standard. Un eterozigote sarà costituito da due picchi a lunghezze d’onda diverse, un
45
omozigote un unico picco d’altezza doppia rispetto all’eterozigote. Questa metodica è stata
utilizzata per l’analisi dell’inserzione/delezione CYP delta TCT con le seguenti condizioni:
•
dye set G5 (filtro), FAM (fluorocromo campione) e ROX (fluorocromo orange per
il marcatore interno)
•
1 µl PCR diluito 1:10
•
0.15 µl liz 500 (standard)
•
13 µl formammide
•
Denaturazione 95x5’
CYP delta TCT Esempio di campione omozigote per l’inserzione (sopra) e di campione
eterozigote (sotto)
46
Programma GeneScan Analysis version 3. In arancione il marker, in blu il campione.
ALLELIC DISCRIMINATION
La discriminazione allelica è un metodo utile nella discriminazione di forme diverse di uno
stesso gene, che differiscono per una sostituzione di base, una delezione o un’inserzione.
Automatizzata e veloce, combina la metodica PCR con quella dell’identificazione in tempo
reale di prodotti di reazione fluorescenti; per questo può essere usata come screening di
differenze alleliche, presenza di mutazioni in diversi individui, oppure anche per
identificare mutazioni ancora ignote.
Una sonda complementare viene tagliata dall’attività 3’esonucleasica della Taq DNA
polimerasi
solo
se
inclusa
nell’amplificazione.
La
sonda
contiene,
come
nell’amplificazione real time normale, un quencher ed un reporter. Se la sonda rimane
intatta durante la reazione, non ci sarà emissione, quando invece la sonda verrà inclusa
nell’amplicone il quencher ed il reporter verranno separati grazie all’azione esonucleasica
47
della Taq polimerasi, generando fluorescenza. Una sonda complementare rimarrà ben
appaiata durante l’annealing di ogni ciclo di reazione di PCR risultando efficientemente
tagliata e rilasciando quindi il fluoroforo che potrà emettere fluorescenza. Quando è
presente una mutazione puntiforme la sonda non sarà ibridizzata e tagliata con la stessa
efficienza, quindi la fluorescenza emessa sarà notevolmente inferiore.
In un sistema biallelico ci saranno due diverse sonde marcate nella stessa reazione.
Ciascuna sarà pertanto marcata da un diverso fluoroforo, FAM e VIC. La prima sonda
presenterà il fluorocromo FAM posizionato all’estremità 5’(massimo di eccitazione a 500510 nm, massimo di emissione a 540 nm, MGB all’estremità 3’). La seconda sonda
presenterà il fluorocromo VIC posizionato all’estremità 5’(massimo di eccitazione a 515 530 nm, massimo di emissione a 540 - 560 nm, MGB all’estremità 3’). Naturalmente una
sarà complementare ad un allele mentre l’altra all’altro. Durante la fase di annealing le
sonde andranno ciascuna ad ibridizzarsi, con la massima efficienza nella competizione, al
proprio filamento specifico. L’aumento di fluorescenza alla lunghezza d’onda specifica di
uno dei due fluorofori significa omozigosi per quell’allele, mentre l’aumento
contemporaneo ad entrambe le lunghezze d’onda significa eterozigoti.
Nonostante il mismatch sia costituito da una singola sostituzione di base, influisce
notevolmente sulla temperatura di melting della sonda e dunque sulla sua capacità di
appaiarsi. Nella reazione vengono utilizzate due sonde che competono rendendo ancora più
restrittive la condizioni di appaiamento anche per un singolo mismatch. Perché la Taq
possa tagliarla la parte 5’ della sonda deve essere almeno in parte denaturata (necessita di
una forca di tre basi). La totale dissociazione in sonde che portano un mismatch avviene
molto più velocemente che in quelle perfettamente complementari, diminuendo così
l’efficienza con cui vengono tagliate.
A differenza dell’amplificazione real time non viene utilizzato cDNA retro trascritto da
RNA ma si parte da DNA.
Per poter eseguire l’analisi sono stati utilizzati dei saggi precasted (Applied) che
contengono le 2 sonde e i primer. La Genotipyng master mix contiene la DNA polimerasi,
i dNTPs, il tampone di reazione ed il Passive Reference.
Per l’analisi del CYP19A1 3’UTR T/C e del polimorfismo nell’esone 4 di p53 è stata
utilizzata questa metodica sullo strumento Abi Prism 7000 Sequence Detection System.
48
Per quanto riguarda la reazione, le condizioni utilizzate sono le seguenti:
Volume x 1 camp.
Master MIX (2x)
12.5 µl
Assay (20x)
1.25 µl
H2O
9.25 µl
Il volume di ogni singola reazione è di 25 µl.
Per ottenere il prodotto finale sono necessarie 3 fasi:
1. Pre-Read (serve per la determinazione della fluorescenza di fondo): 1’ a 60°C
2. Real time: 50°C 2’
95°C 10’ (fase di attivazione della Taq Polimerasi)
95°C 15” secondi
60°C 1’
40 cicli
3. Post-Read (lettura della flourescenza finale): 1’ a 60°C
In “Result-Allelic Discrimination” si ottiene, in presenza dei tre diversi genotipi, un grafico
simile a quello riportato. Sull’asse delle ordinate sono riportati i valori di fluorescenza
relativa ad un allele e sull’asse delle ascisse i dati per l’altro allele. Si determinano dei
raggruppamenti (clusters) di segnali di fluorescenza:
Omozigoti (allele XX)
Eterozigoti (entrambi gli alleli X e Y: both)
Omozigoti (allele YY)
NTD (campioni bianchi contenenti l’acqua)
49
Esempio di allelic discrimination visualizzato mediante rappresentazione grafica.
PCR-AGAROSIO
Per determinare le inserzioni/delezioni nei geni GSTT1 e GSTM1 è stata eseguita una PCR
(con le condizioni già citate precedentemente) con successiva analisi mediante gel di
agarosio al 2%. La regione di inserzione/delezione è localizzata a livello del primer. In
caso di delezione il primer non riuscirà a legare la sequenza di DNA ed ad amplificarlo: su
gel di agarosio non si vedrà nessuna banda. In caso di inserzione il primer riuscirà invece a
legare lo strand ed ad amplificare la regione: su gel di agarosio verrà visualizzata una
banda. E’ possibile una visualizzazione diretta, mediante colorazione con il colorante
fluorescente bromuro di etidio ed esame in luce ultravioletta (UV) al transilluminatore. I
gel di agarosio hanno un potere di risoluzione minore rispetto a quelli di poliacrilamide,
ma una gamma di separazione molto più ampia (globalmente, da 100 bp a 50 kb; lo
specifico intervallo di risoluzione varia a seconda della concentrazione di agarosio).
50
Le molecole lineari di dsDNA migrano a velocità inversamente proporzionali al logaritmo
in base 10 del numero di paia di basi.
Le molecole più grandi sono sottoposte a maggiori forze di attrito e inoltre incontrano
maggiore ostacolo nel trovare una via attraverso i pori del gel rispetto alle molecole più
piccole.
Successivamente la corsa elettroforetica è stata letta e valutata su strumentazione
VERSADOC 1000, sistema ad alta sensibilità, risoluzione ed uniformità per l’acquisizione
e l’analisi delle immagini di campioni chemiluminescenti, fluorescenti a singolo o multiplo
canale, chemifluorescenti e visibili.
Il sistema è così composto:
•
Telecamera CCD digitale ad alta sensibilità ed alta risoluzione
•
Transilluminatore a lampade ultraviolette e in luce bianca.
•
Epi-illuminatore a LED Blu, Verdi, Rossi e bianchi al fine di eccitare in maniera
differenziale e ad alta intensità qualsiasi tipo di fluoroforo o cromogeno.
•
Il sistema VersaDoc è inoltre dotato di camera oscura integrata a completa tenuta di
luce per l’acquisizione di campioni chemiluminescenti.
•
Stazione computer, software Quantity One, per l’acquisizione delle immagini.
51
Analisi dei geni GSTT1 e GSTM1 mediante analisi della PCR su gel di agarosio.
STRUMENTI STATISTICI PER LA VALUTAZIONE DEI DATI
RAPPRESENTAZIONE MEDIANTE BOX-PLOT
Al fine di rendere più completa l’analisi dei dati ottenuti, abbiamo pensato di visualizzare
alcune caratteristiche delle pazienti da un punto di vista statistico. La rappresentazione
grafica che abbiamo deciso di utilizzare è il box-plot. Questo modello, è costituito dal
disegno su un piano cartesiano di un rettangolo, i cui estremi sono il primo e terzo quartile
(in statistica, i quartili ripartiscono una distribuzione di dati in 4 parti di pari frequenze),
tagliato da una linea all'altezza della mediana (si definisce come mediana il valore assunto
dalle unità statistiche che si trovano nel mezzo della distribuzione). Il minimo della
distribuzione viene indicato con (Q0), mentre il massimo con (Q4). Abitualmente vengono
aggiunte due righe corrispondenti ai valori distanti 1,5 volte la distanza interquartile, a
partire rispettivamente dal primo e dal terzo quartile.
52
TEST DI FISHER
Il test esatto di Fisher è un test per la verifica d'ipotesi utilizzato nell'ambito della statistica
non parametrica in situazioni con due variabili nominali (divise ciascuna in due sole
categorie) e campioni piccoli. Usato per verificare se i dati dicotomici di due campioni
riassunti in una tabella di contingenza 2x2 siano compatibili con l'ipotesi nulla (H0) che le
popolazioni di origine dei due campioni abbiano la stessa suddivisione dicotomica e che le
differenze osservate con i dati campionari siano dovute semplicemente al caso.
Per descrivere il test di Fisher è utile introdurre la seguente notazione, nella quale le lettere
a, b, c e d indicano i valori nelle celle e n è la somma totale.
53
La tabella di contingenza viene costruita così:
si malattia
no malattia
Si esposizione
a
b
a+b
no esposizione
c
d
c+d
a+c
b+d
n
Ronald Fisher dimostrò che la probabilità di ottenere tali valori (vincolati alle somme di
riga e colonna realmente osservati) segue la variabile casuale ipergeometrica ed è pari a:
Questa formula dà le probabilità esatte di osservare i valori a, b, c, d (dati a+b, a+c, c+d,
b+d) qualora fosse vera l'ipotesi nulla sopra enunciata.
54
RISULTATI
RAPPRESENTAZIONE MEDIANTE BOX-PLOT
Mediante il box-plot abbiamo rappresentato l’età delle pazienti al momento della chirurgia,
paragonando i due gruppi: le pazienti con e le pazienti senza ricaduta.
90
età
80
70
60
50
40
non ricadute
ricadute
Come si può notare sopra, la maggior parte delle pazienti senza ricaduta ha un’età
compresa tra i 48/68 anni; tra le pazienti con ricaduta, la maggior parte si concentra tra
un’età di 48/75 anni. Questo concorda con il fatto che, il tumore alla mammella, come
esposto nella parte introduttiva, presenta 2 picchi di insorgenza: a 40/50 anni e verso i 70
anni.
55
La tabella seguente dimostra come i due gruppi di pazienti prese in esame non abbiano
differenze significative da questo punto di vista.
NON
RICADUTE
RICADUTE
Number of values
103
5
Minimum
48
51,1
25% Percentile
57
56,7
Median
62,8
66,9
75% Percentile
67,4
75,05
Maximum
85,7
78,8
Mean
62,38
66,08
Std. Deviation
7,944
10,35
Std. Error
0,7828
4,628
Interessante è invece la distribuzione in base ai mesi che intercorrono tra chirurgia e
ricaduta. Nella maggior parte delle pazienti che presentano ricaduta, questa si riscontra,
infatti, nei primi anni.
80
età
60
40
20
0
mesi tra chirurgia e ricaduta
56
I DUE GRUPPI DI PAZIENTI A CONFRONTO
Di seguito, sono riportati gli istogrammi che riassumono in modo schematico i risultati
ottenuti dallo studio di genotipizzazione. I grafici sono stati costruiti mettendo a confronto
le pazienti non ricadute con quelle ricadute ed è stato creato un istogramma per ogni gene.
I dati che abbiamo ottenuto sono interessanti, nonostante il gruppo delle pazienti con
ricaduta sia limitato (solo 5 pazienti). Per la maggior parte dei geni, infatti, tutte le pazienti
con ricaduta presentano lo stesso genotipo; dato il piccolo numero degli eventi osservati,
non viene raggiunta mai la significatività statistica tra le differenze, ma in certi casi è
chiaramente evidenziabile una tendenza in tal senso. Meno evidenti, invece, sono le
differenze in p53, GSTT1 e GSTM1.
CYP19_3’UTR: Questo gene è importante, poiché coinvolto nella regolazione del
livello degli estrogeni. La determinazione del genotipo è stata eseguita con il metodo
dell’allelic discrimination su strumento per real-time PCR; la figura sotto riportata
rappresenta un esempio di distribuzione allelica. Dal momento che in polimorfismo è in
una zona del gene non tradotta in proteina, le due varianti (C e T) non codificano per un
diverso aminoacido.
57
La maggior parte delle pazienti senza ricaduta presenta la variante in eterozigosi (C/T). La
distribuzione dei due genotipi omozigoti (C/C e T/T) sembra invece essere piuttosto
omogenea. Tutte le pazienti con ricaduta presentano genotipo C/T.
CYP19 IVS4[tct]: la presenza della delezione e/o dell’inserzione della tripletta TCT è
stata analizzata mediante un’analisi di gene scan su sequenziatore automatico.
Nell’immagine sotto riportata, sono rappresentate rispettivamente la condizione di
omozigote ins/ins e di eterozigote ins/del (picchi blu; in arancione sono riportati i picchi a
250 e a 300 bp del marcatore di peso molecolare 500-liz).
58
Tutte le pazienti con ricaduta presentano tutte un genotipo eterozigote ins/del, mentre
questo è presente solo nel 47% delle pazienti che non ricadono.
p53: l’analisi del polimorfismo Arg/Pro (G/C) al codine 72 del gene p53 è stata effettuata
mediante sequenza diretta con sequenziatore automatico. Abbiamo riscontrato i 3 genotipi
pazienti possibili: gli omozigoti G/G (Arginina/Arginina), C/C (Prolina/Prolina) ed la
condizione di eterozigosi C/G
omozigote C/C
59
eterozigote C/G
omozigote G/G
Tra il gruppo delle pazienti non ricadute, prevale il genotipo G/G o C/G. Solo 9 pazienti
presentano la forma C/C. Nessuna tra le pazienti ricadute presenta la variante C/C. Non
sono comunque emerse differenze statisticamente significative tra i due gruppi.
60
MDM2: La ricerca della variabile T/G è stata effettuata mediante sequenziamento
diretto.
eterozigote T/G
omozigote T/T
61
omozigote G/G
Questo SNP si trova a livello del primo introne del gene Mdm2 in una regione, pertanto,
non codificante. Nonostante ciò, la forma G sembra essere correlata con una prognosi
sfavorevole in quanto è associata con una maggior attenuazione di p53; tutte e cinque le
pazienti appartenenti al gruppo di ricadute presentano la variante G in eterozigosi. Poche
pazienti senza ricaduta e nessuna di quelle ricadute presentano il genotipo G/G.
GST: lo SNP in GSTP1 si è rivelato interessante in quanto il genotipo G/G (Valina),
correlato in letteratura ad una prognosi migliore in termini di sopravvivenza (60% in più
rispetto alle pazienti ile/ile), non è stato evidenziato in nessuna delle pazienti con ricaduta,
che presentano tutte il genotipo omozigote A/A (Isoleucina). Tra le pazienti senza ricaduta
62
abbiamo la seguente distribuzione: il 62% A/A, 26% A/G, 12% G/G. Di seguito sono
riportati esempi di sequenza dello SNP.
Nel primo pannello è forma riportato un eterozigote A/G, nel secondo un omozigote A/A e
nel terzo un omozigote G/G.
63
Per quanto riguarda lo studio della delezione/inserzione del gene GSTT1 e del gene
GSTM1, non si sono riscontrate significative differenze tra i due gruppi di pazienti; la
distribuzione è risultata, infatti, piuttosto omogenea.
Qui sopra è riportata l’immagine dell’elettroforesi degli amplificati con PCR corsi su gel di
agarosio con cui abbiamo analizzato questi polimorfismi. La presenza della banda a circa
450 bp del gene GSTT1 e di circa 300 bp del gene GSTM1 rappresenta il genotipo ins/ins
o ins/del, mentre l’assenza della banda dell’amplificato identifica il genotipo del/del in
omozigosi (assenza di entrambi gli alleli del gene in analisi).
Di seguito sono riportate le distribuzioni alleliche dei geni della famiglia GSTP1, GSTT1 e
GSTM1 nelle pazienti da noi analizzate.
64
65
ANALISI GLOBALE DEI GENOTIPI IDENTIFICATI
In conclusione, analizzando i risultati relativi ai singoli geni, non sono state messe in
evidenza differenze statisticamente significative tra i due gruppi di pazienti. Tuttavia, è
possibile notare la non casualità di certe associazioni di polimorfismi quando si analizzano
globalmente i vari geni.
In particolare:
•
quasi tutte le pazienti che presentano il genotipo C/T al 3’UTR del gene CYP19A1
presentano la forma ins/del dello SNP
IVS4[tct] dello stesso gene e, tutte le
pazienti, tranne una, omozigoti per la delezione nell’introne, presentano genotipo
C/C.
•
la maggior parte delle pazienti con genotipo eterozigote A/G per il polimorfismo
del gene GSTP1 sono ins/ins per il gene GSTT1; inoltre, la maggior parte delle
pazienti con genotipo ins/ins per il gene GSTM1 presentano sono delete in
omozigosi anche nel gene GSTT1.
66
ANALISI STATISTICA DEI DATI
Al momento di effettuare l’analisi statistica dei risultati ottenuti dallo studio di
genotipizzazione del gruppo di pazienti incluse nello studio retrospettivo, abbiamo preso in
considerazione l’ODD RATIO (o rapporto incrociato) ed il test del CHI-QUADRATO. A
causa del limitato numero di pazienti incluse nello studio, iniziato nel 2003, e soprattutto di
quelle che presentavano ricaduta clinica al momento dell’analisi dei dati, non è stato
possibile utilizzare questi test. Abbiamo invece utilizzato il test di Fisher, alternativa al test
del CHI-QUADRATO, che è utilizzabile anche con dati < 5 e con un numero totale di
osservazioni < 30.
Per poter adattare la tabella che si utilizza nel test di Fisher (vedi materiali e metodi a pag.
54) alla nostra statistica è stato necessario accorpare la condizione di eterozigote ad uno dei
due gruppi di omozigoti. Dopo un’analisi della letteratura, per ogni singolo gene abbiamo
incluso la condizione di eterozigosi al gruppo dei genotipi presentati il polimorfismo in
oggetto in omozigosi.
67
a
b
c
d
MDM2
5
63
0
38
p53
3
49
2
52
GSTT1
3
77
2
24
GSTM1
3
48
2
53
GSTP1
5
88
0
12
CYP19IVS4
5
71
0
27
CYP19UTR
5
78
0
23
68
I risultati dell’analisi mediante il test di Fisher sono riportati nella tabella che segue:
RISULTATI DEL TEST DI FISHER
CYP19_IVS4
0,211
CYP19_3'UTR
1,32
p53
0,312
MDM2
0,103
GSTT1
0,263
GSTM1
0,305
GSTP1
1,15
I risultati ottenuti, tuttavia, non sono quindi statisticamente significativi, in quanto il test
può essere considerato significativo quando la probabilità che l’associazione analizzata sia
casuale è <0.05 (5%).
69
CONCLUSIONI
Nello studio sono state prese in esame 106 pazienti con carcinoma mammario operato: di
queste, 5 sono ricadute. Tutte le pazienti presentavano i recettori ormonali positivi ER+,
condizione necessaria affinché potessero essere arruolate nello studio, in quanto tutte sono
state sottoposte a terapia adiuvante con exemestane. Questo farmaco è utilizzato nella
terapia ormonale insieme agli altri inibitori dell’aromatasi di terza generazione. Agisce
come falso substrato per l’enzima aromatasi legandolo irreversibilmente ed impedendo
così la conversione degli androgeni (androstenedione e testosterone) in estrogeni (estrone
ed estradiolo). Di recente è stato introdotto come farmaco d’elezione nel trattamento
adiuvante in donne in postmenopausa, sostituendo il tamoxifene. A differenza di
quest’ultimo, infatti, l’exemestane ha meno effetti collaterali e sembra esser ben tollerato.
Nello studio abbiamo cercato di evidenziare la relazione tra i polimorfismi o SNPs nel
gene CYP19A1 e l’outcome clinico delle pazienti, estendendo poi l’analisi anche ad altri
geni coinvolti nel metabolismo, nella resistenza ai farmaci e nella probabilità di ricaduta:
p53, MDM2 e la famiglia dei geni GST.
Il gene CYP19A1 fa parte di una famiglia di citocromi impegnati nel metabolismo
dei farmaci ed è pertanto importante per la detossificazione dell’organismo. Inoltre,
questo enzima, ha il compito di regolare la concentrazione degli ormoni steroidei.
Le regioni del gene analizzate sono 2: la regione 3’UTR (C1558T) dell’esone 10 e
l’introne 4 (IVS4[TCT]+/-). Il primo si trova in una zona non tradotta in proteina, le
due varianti non codificano pertanto per un diverso amminoacido.
Il gene TP53 codifica per la proteina p53. Questa, agisce come cofattore di
trascrizione e modula l’espressione di alcuni geni implicati nella regolazione del
ciclo cellulare. Nonostante venga espressa costitutivamente, i livelli di p53, in
condizioni normali, sono bassi poiché la proteina viene degradata velocemente. In
caso di danno al DNA la sua concentrazione aumenta e ciò, provoca l’espressione
di geni che inibiscono la progressione del ciclo cellulare ed inducono l’apoptosi.
Più dettagliatamente, la proteina attiva la p21WAF30 che lega le CDK
impedendone l’attività. In questo modo, la cellula danneggiata non può trasmettere
la mutazione alle generazioni successive. Lo SNPs preso in esame è il Arg72Pro,
localizzato a livello dell’esone 4.
70
Il gene MDM2 codifica per una proteina che regola, mediante un meccanismo di
feedback negativo, la proteina p53 inibendo la funzione di apoptosi propria di
questo gene. In caso di danno al DNA, i livelli di p53 aumentano come detto
precedentemente, mentre la proteina mdm2 viene inattivata attraverso p19arf. Gli
SNPs localizzati a livello del promotore correlano con una maggiore affinità per
l’attivatore trascrizionale Sp1, che aumenta i livelli di Mdm2 attenuando il pathway
di p53. Abbiamo preso in considerazione lo SNP309 T/G, localizzato in una zona
non codificante (nel primo introne) a livello del promotore.
La famiglia GST, infine, codifica per enzimi coinvolti nella resistenza a diversi
chemioterapici. La modalità di azione consiste nel coniugare le sostanze con il
glutatione ridotto: in questo modo, vengono espulse dall’organismo le sostanze
tossiche. Sebbene sia importante per il processo di detossificazione, è anche
implicato nell’insorgenza delle farmacoresistenze. Il polimorfismo studiato è
localizzato nell’esone 5 di GSTP1 (A313G) in cui una Isoleucina è sostituita con
una Valina (I105V). In particolare, il codone che subisce la mutazione si trova nel
sito legante di GSTP1. Abbiamo preso in considerazione, inoltre, inserzioni e
delezioni nelle forme GSTM1 (Glutatione S-transferasi mu) e GSTT1 (Glutatione
S-transferasi theta).
La scelta dei polimorfismi da analizzare e le informazioni sulle modalità di
genotipizzazione derivano da un’accurata analisi degli studi riportati in letteratura. Le
metodiche utilizzate sono state: il sequenziamento per i geni p53, MDM2, GSTP1; l’analisi
di gene scan per il CYP19A1 (IVS4[TCT]+/-); la discriminazione allelica su strumento real
time PCR per CYP19A1 3’UTR (C1558T); le inserzioni-delezioni in GSTM1 e GSTT1
sono state determinate più semplicemente mediante PCR ed elettroforesi su gel d’agarosio.
Il DNA per le analisi è stato ricavato mediante estrattore automatico da linfociti di sangue
periferico intero.
I risultati ottenuti si sono dimostrati interessanti e si è notata una correlazione con quanto
riportato in letteratura. L’analisi statistica ha confermato che i due gruppi in cui sono state
suddivise le pazienti (gruppo 1 non ricadute e gruppo 2 ricadute) erano omogenee
nonostante il gruppo delle pazienti con ricaduta era limitato a 5 casi. Mediante
rappresentazione con box-plot, abbiamo rappresentato due caratteristiche delle pazienti:
l’età alla chirurgia ed i mesi intercorsi tra chirurgia e ricaduta. Nel primo caso abbiamo
71
ottenuto che in media le pazienti senza ricaduta avevano un’età compresa tra 48/68 anni,
mentre le pazienti con ricaduta un’età tra 48/75 anni. Molto probabilmente, se il secondo
gruppo fosse più numeroso i due range tenderebbero ad essere ancora più sovrapponibili.
Per quanto riguarda la seconda caratteristica, invece, quasi tutte le pazienti appartenenti al
secondo gruppo, sono ricadute nei primi anni. Solo una paziente ha presentato la ricaduta
dopo 63 mesi.
I dati sono stati rappresentati mediante un istogramma, mettendo a confronto le pazienti
non ricadute con le pazienti ricadute. Tutte le pazienti con ricaduta presentano lo stesso
genotipo per molti geni. In p53, GSTT1, GSTM1 la distribuzione appare più omogenea.
Analizzando i geni uno alla volta:
CYP19_3’UTR: tutte le pazienti ricadute e 50 delle pazienti non ricadute
presentano la variante C/T. Le restanti presentano: l’omozigosi T/T (23) e
l’omozigosi C/C (28). Come riportato in letteratura, l’allele C porta ad una quantità
minore di enzima aromatasi e si associa ad una riduzione dei livelli di estradiolo ed
estrone riducendo il rapporto estradiolo/testosterone ed estrone/androstenedione.
(Alison M. 2004). Gli SNPs portano a variazioni del livello di estrogeni, come già
detto precedentemente, ma non sembrano coinvolti in un rischio aumentato di
insorgenza di tumore alla mammella. Possono però avere una correlazione con la
risposta al farmaco in quanto l’exemestane ha come bersaglio proprio questo
enzima.
CYP19 IVS4[tct]: il 100% delle pazienti con ricaduta e 47 tra pazienti senza,
presenta il genotipo eterozigote ins/del. Le restanti: 27 hanno l’ins/ins e 24 hanno la
forma del/del.
p53: 3 casi del primo gruppo e 40 casi appartenenti al secondo gruppo presentano
la forma eterozigote C/G. Solo 9 pazienti tra le non ricadute hanno la forma C/C
mentre ben 52 pazienti tra le non ricadute e 2 tra le ricadute presentano la forma
omozigote G/G. La letteratura, riporta che le proteine contenenti Arginina
(genotipo G/G) inducono con maggior efficacia l’apoptosi nelle cellule
danneggiate. In questo modo, la sopravvivenza libera da malattia è maggiore
rispetto
alla
forma
pro/pro.
(Dumont
P
2004)
(Tatsuya Toyama 2007). Nel nostro specifico caso, abbiamo evidenziato che solo
72
poche pazienti tra le non ricadute presentano effettivamente l’omozigosi
prolina/prolina ma altrettanto non è stato dimostrato per il gruppo delle ricadute,
dove ci saremo aspettati un numero maggiore di casi, essendo questa condizione
correlata con una minor sopravvivenza. Con l’ampliamento dello studio, ci
aspettiamo di vedere un aumento, in questo secondo gruppo di pazienti, della
variante sfavorevole.
MDM2: la distribuzione si è rivelata interessante sopratutto per il gruppo delle
ricadute dove il 100% presenta la forma eterozigote. Tra le pazienti non ricadute la
distribuzione è stata le seguente: 51 casi T/G, 38 T/T, 12 G/G. La forma omozigote
G, rivelatasi rara nel primo gruppo di pazienti, in letteratura viene descritta come
caratteristica sfavorevole ai fini della sopravvivenza. Si associa, infatti, con una
maggiore attenuazione di p53, condizione che porta anche ad un aumentato rischio
di insorgenza del tumore stesso poiché si correla ad una minore risposta i p53 al
danno provocato dai chemioterapici. La regolazione del grado di espressione di
mdm2 avviene a livello degli estrogeni, in particolare è indotta da ER ALFA. Gli
estrogeni e gli ormoni tiroidei legano il promotore di mdm2 attivando la sua
trascrizione. Se lo SNP, localizzato proprio in questa regione, si presenta nella
forma G/G porta ad un aumento di affinità tra mdm2 promotore e Sp1. Questa
variante accelera la formazione del tumore, infatti, alcuni studi sostengono che una
cura per le donne con questo tipo di genotipo, potrebbe essere quella di ridurre il
segnale estrogenico. (Manner Gareth L. 2006)
GST: tutte le pazienti con ricaduta presentano la forma omozigote A/A. La
distribuzione nelle pazienti non ricadute è la seguente: 62 A/A 26 A/G e 12 G/G.
La letteratura correla con i dati ottenuti, infatti la forma val/val (G/G) che nessuna
delle pazienti ricadute presenta, ha una prognosi in termini di sopravvivenza molto
più favorevole rispetto alla variante Ile/Ile (A/A), ed essa è circa del 60%. La
motivazione è da ricercarsi nel fatto che questa forma porta alla formazione di
livelli più bassi di GST che eliminano meno efficacemente il farmaco, lasciandolo
svolgere la sua azione nel migliore dei modi. Dallo studio di “Gong Yang el al”
(Gong Yang 2004) è emerso che la prognosi migliore propria della variante val/val
è risultata indipendente dall’età alla diagnosi a dagli altri fattori di sopravvivenza
relativi. Nello stesso studio compare come essa sia presente con maggiore
73
frequenza nelle donne con stadio avanzato di malattia. Nonostante ciò, in letteratura
abbiamo trovato uno studio che dimostra il contrario, vale a dire che il genotipo
val/val è correlato con una prognosi sfavorevole in termini di sopravvivenza.
(Goode EL 2002). In realtà il dato è influenzato dal fatto che alle pazienti è stato
diagnosticato il tumore quasi due anni prima dell’arruolamento. Spostando
l’attenzione sui risultati ottenuti in GSTM1 la distribuzione appare più omogenea,
senza sostanziali differenze. La delezione si trova con frequenza maggiore nelle
non ricadute, rispetto al gruppo delle ricadute dove prevale l’inserzione. Le
differenze sono comunque molto lievi. Le stesse considerazioni si possono fare
anche per il gene GSTT1 dove però prevale le delezione nelle pazienti con ricadute,
mentre l’inserzione in quelle senza ricaduta. In letteratura esistono diversi studi, in
alcuni casi discordanti, se variazioni in questi geni siano implicati o meno
nell’aumento del rischio di tumore. Alcuni dimostrano come la forma deleta porti
alla perdita di parte di gene e sia pertanto responsabile di un funzionamento non
ottimale dell’enzima. (Kathleen M. 2004) (Gong Yang, 2004).
Non sempre è possibile fare un paragone in senso stretto tra i dati ottenuti dallo studio e
quelli relativi a studi precedenti trovati in letteratura. In alcuni casi, cambiano le condizioni
alla base dello studio: i criteri di campionamento, l’importanza data ai fattori di
sopravvivenza relativi, le caratteristiche del gruppo di pazienti studiato, il numero delle
pazienti arruolate.
L’arruolamento di altre pazienti nello studio, l’allungamento del periodo di follow up con
la possibilità di evidenziare nuove ricadute cliniche ed il conseguente del numero di
campioni su cui effettuare l’analisi di genotipizzazione potrebbe mettere in evidenza tra i
due gruppi di pazienti, differenze, ora solo evidenziate come tendenza, in termini oggettivi
e statisticamente significative con i test adatti. Al momento le analisi statistiche eseguite
non hanno dato, come già detto in precedenza, dei risultati significativi. I dati sono
comunque stati analizzati statisticamente mediante il test di Fisher. Per poter costruire la
tabella abbiamo dovuto accorpare la condizione di eterozigote ad uno dei due gruppi di
omozigoti. Il test, può essere considerato significativo solo dal momento che la probabilità
che sia vera l’ipotesi H0 “le differenze osservate tra i due gruppi di pazienti sono dovute
semplicemente al caso” è <0.05 (5%).
74
I risultati ottenuti sono comunque i seguenti:
•
CYP19_IVS4
0,211
•
CYP19_3'UTR
1,32
•
p53
0,312
•
MDM2
0,103
•
GSTT1
0,263
•
GSTM1
0,305
•
GSTP1
1,15
Nonostante ciò, sono comunque necessari ancora altri studi per poter dimostrare con
certezza una relazione tra gli SNPs e l’efficacia della terapia ormonale con exemestane. Lo
studio che abbiamo effettuato è uno studio retrospettivo: sono state genotipizzate delle
pazienti con tumore alla mammella operato e trattate con exemestane, già in follow up,
alcune da tempo. In realtà, lo scopo dello studio è poter identificare dei marcatori in modo
che la ricaduta possa essere diagnosticata prima di essere clinicamente evidente. Lo studio
dei marcatori è uno studio piuttosto recente ma che ha dato risultati di notevole
importanza, nonostante ancora solo per pochi tumori sono noti dei marcatori specifici e
sensibili. Mediante la realizzazione di questo pannello di geni, si potrebbe adattare e
personalizzare la terapia in base al profilo genico proprio di ogni paziente e sapere quale
efficacia aspettarsi dalla terapia ormonale con exemestane. Infatti, come dimostrano i
risultati, alcuni genotipi del CYP19A1 presentano, con chiara tendenza, una minor
possibilità di successo del farmaco. Varianti in altri geni, come ad esempio la forma C/C in
p53, G/G in Mdm2, A/A in GSTP1, correlano con una prognosi sfavorevole in termini di
sopravvivenza. Poter predire in senso prospettico, attraverso studi inizialmente retrospettivi
come quelloqui presentato, l’outcome clinico delle pazienti affette da carcinoma
mammario potrebbe dare dei grandi benefici per lo sviluppo della clinica oncologica.
75
BIBLIOGRAFIA
Alison M. Dunning, Mitch Dowsett, Catherine S. Healey, Louise Tee, Robert N.
Luben, Elizabeth Folkerd, Karen L. Novik, Livia Kelemen, Saeko Ogata, Paul D. P.
Pharoah, Douglas F. Easton, N. E. Day, Bruce A. J. Ponder Polymorphisms
Associated With Circulating Sex Hormone Levels in Postmenopausal Women.
Journal of the National Cancer Institute, Vol. 96, No. 12, June 16, 2004.
Ashcroft M and Vousden KH. Regulation of p53 stability. Oncogene; 18: 7637-43;
1998.
Bates S, Philips AC, Clark Pa, Stott F, Peters G, Ludwig RL. Vousden KH.
p14ARF links the tumor suppressor RB and p53, Nature; 10;395 (6698): 124-125;
Sep 1998.
Bottger V, Bottger A, Garcia-Echeverria C, Ramos YF, van der Eb AJ, Jochemsen
AG, Lane DP, Comparative study of the p53-mdm2 and p53-MDMX interfaces,
Oncogene; 7; 18 (1): 189-199; Jan 1999.
Briviba K.; Klotz, L.O.; Sies, H.; Defenses against peroxynitrite, Methods
Enzymol.; 301, 301-311; 1999.
Danesi R, De Braud F, Fogli S et al Pharmacogenetic determinants of anti-cancer
drug activity and toxicity. Trends Pharmacol Sci; 22: 420-426; 2001.
Dino Amadori, Carlo Maria Croce. Terapia molecolare in oncologia. Editore
Poletto, ottobre; 2005.
Dumont P, Leu JI, Della Pietra AC 3rd, George DL, Murphy M. The codon 72
polymorphic variants of p53 have markedly different apoptotic potential. Int J
Cancer. Jan 10, 2004.
76
Frederick L. Greene, American Joint Committee on Cancer, Frederick L Greene,
Sprinter Link. AJCC. Atlante per la stadiazione dei tumori maligni. Pubblicato da
Springer; 8-9; 2007.
Frova C.; Glutathione transferases in the genomics era: new insights and
perspectives, Biomol. Eng.; 23, 149-169; 2006.
Fuqua SA, Schif R. The biology of estrogen receptors. In: Harris JR, Lippman ME,
Morrow M et al (EDS). Disease of the breast. Lippincot Williams & Wilkins,
Filadelfia, 3 edizione 2004.
Gaetano Crepaldi, Aldo Baritussio Trattato di medicina interna Pubblicato da
PICCIN; 3763-64; 2002.
Geisler J, King N, Anker G, et al. In vivo inhibition of aromatization by
Exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast
cancer patients. Clin Cancer Res.; 4: 2089-93; 1998.
Gong Yang, Xiao-Ou Shu, Zhi-Xian Ruan, Qiu-Yin Cai, Fan Jin, Yu-Tang
Gao,Wei Zheng, Genetic Polymorphisms in Glutathione-S-Transferase Genes
(GSTM1, GSTT1, GSTP1) and Survival after Chemotherapy for Invasive Breast
Carcinoma. American Cancer Society 2004.
Goode EL, Dunning AM, Kuschel B, et al. Effect of germ-line genetic variation on
breast cancer survival in a populationbased study. Cancer Res.; 62:3052-3057;
2002.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell.; 7; 100(1):57-70; Jan
2000.
Hayes J.D.; Pulford, D.J. The glutathione S-transferase supergene family:
regulation of GST and the contribution of the isoenzymes to cancer
chemoprotection and drug resistance, Crit. Rev. Biochem. Mol. Biol.; 30, 445-600;
1995.
77
Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase
activity of Mdm2 for tumor suppressor p53, EMBO J.; 4; 18(1):22-27; Jan 1999.
Hsieh JK, Chan FS, O’Connor DJ, Mittnacht S, Zhong S, Lu X. RB regulates the
stability and the apoptotic functions of p53 via MDM2, Mol. Cell.; 3 (2): 181-193;
Feb 1999.
Innis, M. A., Gelfand, D. H., Sninsky, J. J., and White, T. J. PCR protocols: a
guide to methods and applications. New York: Academic Press, 1990.
Jayaraman L and Prives C. Covalent and noncovalent modifiers of the p53 protein,
Cell Mol. Life Sci; 55(1): 76-87; Jan 1999.
Kalyanaraman B.; Karoui H.; Singh, R.J.; Felix, C.C. Detection of thiyl radical
adducts formed durino hydroxyl radical- and peroxynitrite-mediated oxidation of
thiols-a high resolution ESR spin-trapping study at Q-band, Anal. Biochem.; 241,
75-81; 1996.
Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF and Sherr CJ. Functional
and physical interactions of the ARF tumor suppressor with p53 and Mdm2, Proc.
Natl. Acad. Sci. USA; 7; 95 (14): 8292-8297; Jul 1998.
Kathleen M. Egan, Qiuyin Cai1, Xiao-Ou Shu, Fan Jin, Tian-Li Zhu, Qi Dai, YuTang Gao and Wei Zheng, Genetic Polymorphisms in GSTM1, GSTP1, and GSTT1
and the Risk for Breast Cancer Results from the Shanghai Breast Cancer Study and
Meta-Analys. Biomarkers & Prevention, February 2004.
Kubbutat MHG, Vousden KH. Keeping an old friend under control: regulation of
p53 stability. Mol Med. Today; 4: 250-256; 1998.
Lim HS, Ju Lee H, Seok Lee K et al. Clinical implications of CYP2D6 genotypes
predictive of tamoxifen pharmacokinetics in metastatic Breast cancer. J Clin
Oncol.; 25: 3837-3845; 2007.
78
Lonning PE. overview of aromatase inhibitors and inactivators in the treatment of
advanced breast cancer. In: Miller WR, Ingle JR(EDS), Endocrine therapy in
breast cancer. Dekker, Basel; 2002.
Lorusso, Cinieri, Cova, Silvestris Nuovi farmaci biomolecolari in Oncologia
Medica- Un manuale per il clinico Volume I Ragusa Grafica Moderna; 26-29 4586; 2005.
M. Luisa Brandi, a cura di E.V. Cosmi, J.A. Pinotti et al. Parathyroid diseases.
Pubblicato da SEE Editrice Firenze; 152-153- 243-304; 1997.
Manner Gareth L. Bond Kim M. Hirshfield Tomas Kirchhoff Gabriella Alexe
Elisabeth E. Bond Harlan Robins Frank Bartel Helge Taubert Peter Wuerl William
Hait Deborah Toppmeyer Kenneth Offit and Arnold J. Levine, MDM2 SNP309
Accelerates Tumor Formation in a Gender-Specific and Hormone-Dependent.
Cancer Res 2006.
McInerney EM, Weis KE, Sun J et al. trascription attivation by the human estrogen
receptor subtype β (ERβ) studied with Erβ and Erα receptor chimeras.
Endocrinology; 139: 4513-4522; 1998.
McKenna NJ, Nawaz Z, Tsay SY et al. Distinct steadystate nuclear recerptor
coregulator complexes exist in vivo. Procl Natl Acad Sci USA; 95:11697-11702;
1998.
Meek DW. Multisite phosphorylation and the intregration of stress signals at p53,
Cell Signal.; 10 (3): 159-166; Mar 1998.
Meister A.; Anderson, M.E.; Glutathione, Ann. Rev. Biochem.; 52, 711-760; 1983.
Mingfang Zhao, Ryan Lewis, Deborah R. Gustafson, Wan-Qing Wen, James R.
Cerhan and Wei Zheng, No Apparent Association of GSTP1 A313G Polymorphism
with Breast Cancer Risk among Postmenopausal Iowa Women, Cancer
Epidemiology, Cancer Epidemiology, Biomarkers & Prevention. December 2001.
79
Olivier M, Hainaut P. TP53 mutation patterns in breast cancers: searching for
clues of environmental carcinogenesis. Semin Cancer Biol.; 11 (5): 353-60; Oct
2001.
Osborne CK, Bardou V, Hopp TA, et al. Role of the estrogen receptor coactivator
AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl
Cancer Inst.; 95:353-61; 2003.
Pasqualini JR, Chetrite GS. The selective estrogen enzyme modulators (SEEM) in
breast cancer. In: Pasqualini JR (ED), Breast cancer prognosis, treatment and
prevention. Dekker, Basel, 2002.
Pemple S.E.; Wardle, A.F.; Taylor, J.B.; Glutathione S-transferase class kappa:
classification by cloning of rat mitochondrial GST and identification of a human
homologue, Biochem.; 319, 749-754; 1996.
Philpot RM. Characterization of cytochrome P450 in extrahepatic tissues. Meth
Enzymology; 206: 623-631; 1991.
Proc Natl Acad Sci U S A. The Nucleotide Sequence of the lac Operator, Walter
Gilbert and Allan Maxam.; 70(12 Pt 1-2): 3581–3584; December 1973.
Sanger F, Coulson AR. A rapid method for determining sequences in DNA by
primed synthesis with DNA polymerase. J Mol Biol.; 25;94(3):441-448; May 1975.
Sanger F, S. Nicklen, and A. R. Coulson. DNA sequencing with chain-terminating
inhibitors, Proc Natl Acad Sci U S A.; 74(12): 5463–5467; December 1977.
Schroth W, Antoniadou L, fritz P et al. Breast cancer treatment outcome with
adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. Jclin
Oncol.; 25: 5187-5193; 2007.
Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH; AGuirre AJ, Wu
EA, Corner JW, DePinho RA. Loss of 16INK4a with retention of p19arf
predisposes mice to tumorigenesis. Nature; 413:86-91; 2001.
80
Sheehan D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution
of glutathione transferases: implications for classification of non-mammalian
members of an ancient enzyme superfamily, Biochem. J.;360, 1-16; 2001.
Shieh Sy, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of
p53 alleviates inhibition by MDM2, Cell.;31; 91(3): 325-334; Oct 1997.
Simona Lambertini. Capire il cancro. Pubblicato da Alpha Test; 94-103-104 2004.
Stephen Neidle. Cancer Drug Design and Discovery. Edition: illustrated Pubblicato
da Academic Press; 108-109; 2008.
Stott FJ, Bates S, James MC; McConnell BB, Starborg M, Brookes S, Palmero I,
Ryan K, Hara E; Vousden KH, Peters G. The alternative product from the human
CDKN2A locus, P14(ARF), partecipates in a regulatory feedback loop with p53
and MDM2, EMBO J.; 1; 17 (17): 5001-5014; Sep 1998.
Tatsuya Toyama, Zhenhuan Zhang, Mariko Nishio, Maho Hamaguchi, Naoto
Kondo, Hirotaka Iwase, Hiroji Iwata, Satoru Takahashi, Hiroko Yamashita and
Yoshitaka Fujii, Association of TP53 codon 72 polymorphism and the outcome of
adjuvant therapy in breast cancer patients, Breast Cancer Research 2007.
Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two
polymorphic variants of wild-type p53 differ biochemically and biologically.
Molecular and Cellular Biology, Feb. 1999.
Uhrinova S, Uhrin D, Powers H, et al. Structure of free MDM2 N-terminal domain
reveals conformational adjustments that accompany p53-binding J. Mol. Biol.; 350
(3): 587–98; 2005.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N,
Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53
pathway by small-molecule antagonists of MDM2. Science; 6: 844–848; 2004.
81
Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters
Mdm2 and activates p53. Nat. Cell. Biol.; 1 (1): 20-26; May 1999.
Zindy F, Eischen CM, Randle DH, KamijoT, Cleveland JL, Sherr CJ, Roussel MF.
Myc signling via the ARF tumor suppressor reguletes p53-dependent apoptosis and
immortalization, Genes Dev.;1; 12 (15): 2424-2433; Aug 1998.
82
83

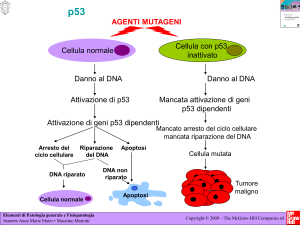







![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)


