(La chimica farmaceutica ti sta provocando un’ulcera?
è la lezione che fa per te!)
Questa
FARMACI ANTIULCERA
INTRODUZIONE
L’epitelio secretore dello stomaco contiene tre tipi di cellule: caliciformi, che secernono mucina;
peptiche, che secernono il pepsinogeno precursore inattivo dell’enzima proteolitico pepsina; infine le
cellule ossintiche o parietali, che secernono acido cloridrico.
La secrezione della maggior parte del succo gastrico è stimolata dal cibo presente nello stomaco.
Nell’uomo in genere nell’arco delle ventiquattro ore vengono secreti 2-3 litri di succo gastrico, che
contiene mucina, elettroliti ed enzimi. La pepsina è il principale enzima proteolitico secreto dalla mucosa
gastrica, che elaborando contemporaneamente acido cloridrico crea nel succo gastrico il pH necessario
(pH = 1,2-1,6) affinchè la pepsina risulti attiva.
In vitro il succo gastrico idrolizza completamente le proteine ad amminoacidi, mentre durante la
digestione nello stomaco la trasformazione si arresta a frammenti polipeptidici più piccoli detti peptoni.
L’attività gastrica ha la funzione di attivare la pepsina, di inibire l’amilasi salivare, di rigonfiare le
fibre collagene presenti nel cibo e di stimolare la secrezione del pancreas. E’ da tenere presente che il
pH della secrezione gastrica influenza la natura della flora batterica presente nello stomaco.
Durante la fase primaria o nervosa della secrezione gastrica il succo è ricco sia in pepsina che in
acido cloridrico. La fase secondaria della secrezione è iniziata dalla presenza di cibo nello stomaco, che
causa la liberazione dalla regione pilorica di un polipeptide detto gastrina, che sembra agisca sulle cellule
ossintiche dimodochè la secrezione nella fase secondaria è ricca in acido cloridrico ma povera in
pepsina.
La massima secrezione di succo gastrico richiede la stimolazione sia da parte del vago che da
parte della gastrina. Anche l’istamina, presente in quantità considerevoli nella mucosa gastrica, stimola la
secrezione delle cellule ossintiche. La gastrina, analogamente all’istamina ed all’acetilcolina (ACh), è una
delle sostanze fisiologiche coinvolte nella stimolazione della secrezione gastrica.
La secrezione di mucina protegge di norma la mucosa stessa dall’essere digerita dai succhi
gastrici. Tuttavia in caso di erosione della mucosa, l’area danneggiata è soggetta ad ulteriori attacchi
poiché manca della usuale protezione e si forma così l’ulcera peptica.
L’iperattività vagale sembra essere un importante fattore causale dell’ulcera peptica; questa
pertanto può nascere da una stimolazione dei centri vagali ipotalamici provocata da lesioni o indotta da
farmaci. Le ulcere peptiche talvolta si possono produrre in seguito ad un trauma grave, specialmente ad
una ustione; l’eccessiva produzione di succo gastrico in queste circostanze è probabilmente dovuta alla
messa in libertà di istamina da parte del tessuto danneggiato.
Le ulcere possono formarsi nello stomaco e nel duodeno. Le ulcere gastriche si instaurano
quando vi è un’eccessiva secrezione di succo gastrico a stomaco vuoto, particolarmente nei pazienti
soggetti a stati emotivi ed ansiosi. Le ulcere duodenali si formano quando la secrezione gastrica è
sovrabbondante e lo svuotamento dello stomaco è troppo rapido. Come risultato si ha che una grande
quantità di succo gastrico acido entra nel duodeno, l’eccesso di acido non viene neutralizzato dalle
secrezioni duodenali e la pepsina attiva erode la mucosa duodenale. Anche l’ulcera duodenale si
instaura comunemente nei soggetti con una predisposizione alle preoccupazioni.
Sebbene la patogenesi dell’ulcera peptica non sia stata ancora completamente compresa, sono
stati riconosciuti tre fattori causali principali: l’aumento della secrezione di acido cloridrico, una difesa
FARMACI ANTIULCERA
inadeguata della mucosa contro l’acido gastrico e l’infezione indotta dal bacillo gram-negativo
Helicobacter pylori.
Ai fini della presente trattazione possiamo individuare le seguenti categorie di farmaci usati nella
prevenzione e nella cura dell’ulcera:
♦ Farmaci Antiacidi
♦ H2 Antagonisti
♦ Inibitori della Pompa Protonica
♦ Farmaci Vari (antimuscarinici, antigastrinici, proteggenti della mucosa)
♦ Farmaci Antimicrobici
Ø Farmaci Antiacidi
Gli antiacidi sono quei farmaci che vengono usati per neutralizzare l’acidità gastrica nelle dispepsie
ipercloridriche, nell’ulcera gastrica e nelle altre condizioni morbose che spostano il pH del succo
gastrico a valori < 1,2-1,6.
L’antiacido in realtà ha solo effetto sintomatico, nel senso che elimina gli effetti prodotti
dall’aumento di acidità senza eliminare la causa che lo produce.
Nonostante le numerose specialità reperibili in commercio, a tutt’oggi non esiste ancora un
antiacido ideale. Tra gli effetti indesiderati presentati da questi farmaci sono da mettere in risalto i
seguenti
Ø possibilità di provocare alcalosi sistemica allorquando la loro azione non è localizzata al tratto
gastroenterico soltanto
Ø azione sulla peristalsi intestinale con possibilità di provocare stipsi ovvero diarrea
Ø formazione da parte di alcuni antiacidi di anidride carbonica con conseguente dilatazione della
parete gastrica e notevoli eruttazioni gassose
Ø il “ritorno acido” dovuto ad una iperneutralizzazione della acidità gastrica con conseguente ripresa
dell’attività secretoria da parte della mucosa gastrica che può portare il pH nello stomaco a valori
anche più bassi di quello di partenza.
Un antiacido ideale dovrebbe
• elevare il pH gastrico nell’intorno ottimale di 3,5-4,5; se infatti si supera il valore di 4,5 si possono
avere fenomeni fermentativi dovuti ad un accrescimento della flora batterica e ad un aumento della
secrezione gastrica dovuto ad un incremento di istamina favorito dall’elevato pH. Con pH più alti di 7 si
ha il “ritorno acido”
• avere azione inattivante sulla pepsina (attiva al pH ottimale di 1,6) al fine di ridurre la proteolisi,
sebbene questa non possa essere mai completamente annullata in quanto nel succo gastrico è presente
un altro enzima proteolitico, la catepsina, che è attiva fino a pH 5.
In base all’azione esercitata sull’organismo gli antiacidi si possono suddividere in sistemici e non
sistemici. I primi vengono assorbiti a livello dell’intestino e passando in circolo possono provocare
alcalosi sistemica (spostamento del pH del sangue verso valori più basici del normale). I secondi non
vengono assorbiti, agiscono solo a livello del tratto gastroenterico e non provocano quindi alcalosi
sistemica.
Un tipico esempio di antiacido sistemico è il bicarbonato di sodio. Questo composto ha azione
immediata sull’acidità (con il conseguente vantaggio di dare immediato sollievo ai bruciori provocati
2
FARMACI ANTIULCERA
dall’iperacidità), ma viene assorbito, supera la mucosa gastrica, entra in circolo e può produrre alcalosi
sistemica; inoltre l’azione neutralizzante dell’acidità è seguita da sviluppo di gas (Rompicapo: dare una
spiegazione chimica di questo fenomeno!) con eruttazioni gassose e dilatazione della parete gastrica
(con possibilità quindi di aggravare un’ulcera conclamata fino ad avere perforazione della parete stessa);
ed ancora può portare il pH a valori superiori a 7 e quindi provocare il ritorno acido.
Non va usato come antidoto nell’ingestione accidentale o volontaria (a scopo suicida) di acidi; in
tali casi va usato l’ossido di magnesio che non sviluppa gas (Suggerimento per lo studente: accertarsi
di saper dimostrare questa affermazione attraverso equazioni chimiche appropriate).
Tra gli antiacidi non sistemici si annoverano ossidi, idrossidi, sali inorganici e sali organici del
calcio, del magnesio, del bismuto e dell’alluminio. Ricordiamo per semplicità solo il magaldrato
(Riopan®), costituito da alluminio magnesio idrossido solfato [Al5Mg10(OH) 31(SO4)2] idrato, che trova
largo impiego come antiacido ed antiulcera.
•
H2 Antagonisti
Ø Introduzione
I recettori H2 si trovano principalmente nelle cellule parietali, che sono localizzate nella mucosa del
fondo e del corpo dello stomaco. I recettori H2 controllano la secrezione acida gastrica in risposta
all’agonista endogeno, l’istamina. Si tratta di recettori accoppiati ad una proteina G (GPCRs), il che
significa che il messaggio dell’istamina è comunicato tramite livelli elevati di cAMP.
Quandi i recettori H2 sono stimolati, gli aumentati livelli di cAMP stimolano un enzima
contenente un gruppo tiolico (SH) e chiamato H+ , K+-ATPasi (noto più comunemente come la
pompa protonica) a secernere attivamente acido cloridrico.
I recettori H2 si trovano anche nella muscolatura liscia vascolare e bronchiale, ma gli H2 antagonisi
hanno effetti molto limitati in questi distretti.
Oltre all’istamina, anche la gastrina e l’acetilcolina (ACh) possono stimolare la secrezione gastrica
delle cellule parietali. L’azione dell’ACh è mediata dal sistema del vago, la gastrina è un ormone
rilasciato dalle cellule gastriche antrali G. L’istamina invece è rilasciata dalle cellule enterocromaffinosimili presenti nella mucosa gastrica.
Sia la gastrina che l’ACh possono stimolare le cellule enterocromaffino-simili a rilasciare istamina,
che a sua volta stimola la secrezione gastrica interagendo con i recettori H2 delle cellule parietali.
Ø Effetti Farmacologici degli H 2 Antagonisti
Gli H2 antagonisti inibiscono la secrezione acida gastrica indotta dall’istamina in maniera dosedipendente e competitiva. Inibiscono anche la secrezione acida gastrica mediata dalla gastrina e, in
misura minore, quella mediata dall’ACh.
Gli H2 antagonisti inibiscono la secrezione acida basale e notturna, come pure quella stimolata dal
cibo.
Gli H2 antagonisti riducono la secrezione del fattore intrinseco (ma non in misura tale da influire
sull’assorbimento della vitamina B12).
•
Struttura degli H2 Antagonisti
3
FARMACI ANTIULCERA
A differenza degli H1 antagonisti, gli H2 antagonisti presentano una struttura essenzialmente idrofila
ed “istamino-simile”.
A differenza degli H1 antagonisti (che devono essere cationici per poter competere con l’istamina
per il sito anionico sul recettore H1 costituito da un residuo ASP), i bloccanti H2 non possono avere
natura cationica nella stessa zona della molecola in cui è cationica l’istamina. Vedremo presto come sia
un catione in una differente area moleculare che permette all’antagonista di competere con
l’istamina per l’ancoraggio al sito anionico del residuo ASP.
Potenti antagonisti del recettore H2 possono essere ottenuti attraverso la modificazione della
struttura dell’istamina. Questo approccio per l’ottenimento di potenti H1 antagonisti ha invece prodotto
soltanto agonisti meno potenti o addirittura composti inattivi.
• Ciò ci dice che, sebbene sia gli H1 che gli H2 antagonisti inibiscano il recettore dell’istamina, essi
esplicano tale azione legandosi a siti diversi del recettore. Per legarsi allo stesso sito dovrebbero avere
strutture simili.
• Mentre gli H1 antagonisti si legano allostericamente al recettore H1, si ritiene che gli H2 antagonisti si
leghino all’effettivo sito attivo del recettore H2, dove normalmente si lega l’agonista istamina.
Ø Relazioni Struttura-Attività per gli H2 Antagonisti
R1
R2
H N
τ
R3
N
π
Un anello aromatico (imidazolo nell’istamina ed in uno dei farmaci anti-H2 in commercio) è
fondamentale per il legame di questi ligandi con il recettore, probabilmente perché consente interazioni
di van der Waals (π stacking).
Come sapete, l’agonista istamina ha un anello imidazolico. L’azoto basico π del tautomero
Nτ H dell’istamina (che è il tautomero che viene riconosciuto per primo dai recettori) è inizialmente
coinvolto in un’importante interazione ione-dipolo con il residuo anionico ASP186 del recettore H2.
[Rompicapo: perché l’azoto π del tautomero N τ H dell’istamina è più basico dell’azoto τ ?]
Anche la presenza di un gruppo funzionale basico associato con l’anello aromatico è un aspetto
molto importante della struttura di un H2 antagonisa. Un gruppo basico è necessario per generare un
catione che possa poi ancorarsi al recettore H2 mediante un legame ione-ione con il residuo ASP98
(lo stesso che lega l’azoto carico positivamente della catena laterale dell’istamina).
L’anello imidazolico, presente nell’agonista istamina ed in uno degli antagonisti (cimetidina), non
è essenziale in sé e per sé, ma lo sono le sue caratteristiche chimiche (aromaticità, azoto basico).
Sebbene abbiamo usato un anello imidazolico per rappresentare la struttura generale degli anti-H2,
vedremo come anche altri sistemi aromatici siano in grado di fornire una buona attività antiistaminica al
recettore H2 fintanto che posseggono un gruppo funzionale basico incorporato nell’anello o ad esso
attaccato come sostituente. Di nuovo, il gruppo basico è essenziale perché si pensa che si protoni a
pH 7.4 ed ancori l’antagonista al sito anionico ASP 98 sul recettore H2.
In altre parole, il sistema aromatico degli H2 antagonisti deve essere isosterico con l’anello
imidazolico basico per avere un’efficace azione antagonista:
• Se è presente l’anello imidazolico, la sua struttura deve essere tale da stabilizzare e mantenere la
forma tautomerica Nτ H necessaria per il riconoscimento da parte del recettore.
4
FARMACI ANTIULCERA
• Non vogliamo che si stabilisca un equilibrio tautomerico che possa portare alla forma N π H
capace di stimolare il recettore. Ciò che vogliamo è un effetto antagonista!
Ben due dei tre gruppi “R” del farmacoforo H2 antagonista servono a stabilizzare la struttura in
questa importantissima forma tautomerica N τH. Essi sono:
v R1
– Questo piccolo gruppo ad effetto +I spinge elettroni verso l’anello lungo i legami σ,
determinando lo spostamento dei doppi legami “tautomerizzabili” nella direzione del
flusso di elettroni.
– Poichè N π è costretto a formare un doppio legame, Nτ dovrà prendere l’idrogeno.
– In altre parole, un piccolo gruppo alchilico in posizione R1 promuove il tautomero Nτ H
a spese del tautomero N π H. Questi composti sono quindi “bloccati” nella forma
necessaria per il riconoscimento da parte del recettore ed al tempo stesso impossibilitati
a dare il tautomero richiesto per l’attivazione del recettore. Essi legano fortemente il
recettore H2, ma non possono stimolarlo. E questo è proprio ciò che vogliamo da un
antagonista H2!
– CH3 è il gruppo più comune in questa posizione. Gruppi di maggiori dimensioni
portano ad una diminuzione della potenza dell’antagonista, probabilmente a causa di
impedimenti sterici con il recettore.
v R2
– Per mantenere la struttura dell’inibitore nella forma tautomera Nτ H, è necessario che il
flusso di elettroni si muova nella giusta direzione. Si richiede quindi un gruppo
elettronattrattore (gruppo -I) dall’altra parte dell’anello rispetto al gruppo +I R1. In tal
modo R1 e R2 cooperano nello stabilire il corretto flusso elettronico lungo l’anello
imidazolico. Il risultato finale è la stabilizzazione del tautomero desiderato Nτ H e alta
affinità per il recettore H2.
– C’è un requisito sterico importante per questo sostituente. R2 deve essere equivalente
come dimensioni ad una catena di quattro atomi di carbonio (n-butile). Ciò permette di
mantenere la distanza opportuna tra l’anello aromatico ed il gruppo R3 (entrambi
importanti per il binding al recettore).
– Il gruppo R2 che si ritrova in tutti gli antagonisti H2 utilizzati in terapia è il gruppo
etiltiometilico (-CH2-S-CH2-CH2-)
♦Lo zolfo ha effetto -I rispetto al carbonio, ed attira elettroni dal gruppo CH2
che lo separa dall’anello aromatico. Questo richiamo di elettroni si trasmette
all’anello promuovendo il tautomero Nτ H.
♦Lo zolfo ha grosso modo le stesse dimensioni di un CH2, per cui il gruppo
etiltiometilico rispetta il requisito sterico, corrispondendo ad una catena di
quattro atomi di carbonio.
♦[Rompicapo 1: Se R2 deve avere le dimensioni di un gruppo n-butilico,
perché non si usa direttamente questo gruppo come spaziatore tra l’anello e
R3?]
♦[Rompicapo 2: Perché la posizione ottimale per l’atomo di zolfo è tra il
primo ed il secondo gruppo CH2 della catena?]
5
FARMACI ANTIULCERA
v R3
– Questa porzione terminale deve essere polare ma non-cationica. Questa è la zona
molecolare in cui si trova l’azoto cationico dell’istamina (l’agonista fisiologico). Se si
formasse un catione in questa parte della molecola, avremmo attività stimolante e non H2
antagonista.
– E’ stato riportato in letteratura che questo segmento strutturale non ionizzato degli H2
antagonisti potrebbe in effetti legarsi ai residui ASP186 e ARG257 (o TYR182) che
solitamente legano l’anello imidazolico dell’istamina quando questo agonista interagisce
con i siti H2.
Concediamoci un “time out” per cercare di capire come il binding di un antagonista al recettore
H2 differisce da quello dell’agonista istamina. Da quanto detto finora, possiamo pensare che, rispetto
all’istamina, gli H2 antagonisti facciano una “capriola” a livello del recettore. Il gruppo funzionale basico
dell’anello aromatico degli H2 antagonisti (nella forma cationica protonata) lega il residuo ASP98 che
normalmente lega la catena laterale dell’istamina. Analogamente, la catena laterale dell’antagonista si
lega all’area recettoriale [ASP 186 e ARG257 (o TYR182)] che originariamente interagisce con gli atomi di
azoto dell’anello imidazolico dell’istamina. Metaforicamente, sia l’agonista (istamina) che l’antagonista
usano lo stesso letto (sebbene non contemporaneamente!), ma mentre uno dorme con la testa sul
cuscino, l’altro dorme con i piedi sul cuscino e la testa in fondo al letto.
R3 (continua)
– Riprendiamo ora la nostra discussione della chimica del gruppo R3. Occorsero un
paio di tentativi ai chimici farmaceutici per arrivare a capire che questo gruppo deve
rimanere non ionizzato, dal momento che normalmente si è portati a pensare che se nella
struttura dell’agonista è necessario un catione, questo sia di norma richiesto anche nella
struttura dell’antagonista e nella stessa posizione relativa. Ma non in questo caso!
– Pensando quindi che un catione fosse essenziale nella struttura degli H2 antagonisti, i
chimici farmaceutici dapprima provarono come gruppo R3 il gruppo guanidinico, che
però non risultò idoneo per la sua eccessiva basicità. Presenta un pKa di 13 ed a pH
7.4 esiste quasi esclusivamente come il suo acido coniugato (catione).
[Suggerimento per gli studenti: Assicurarsi di essere in grado di provare questa
N C NH 2
N C NH 2
H
H
NH
pKa - 13
NH 2
base molto forte
acido molto debole
affermazione usando l’equazione di Henderson-Hasselbach!!]
[Rompicapo: Perché la guanidina è una base così forte? (Meditate, gente!!)]
♦L’equazione
di Henderson-Hasselbach ci dice che, a pH 7.4, il rapporto tra forma
ionizzata e forma non-ionizzata è di circa 106/1. Inizialmente, si pensò che ciò fosse
un dato positivo in quanto questa specie fortemente cationica appariva in grado di
competere con il catione dell’istamina (che ha un pKa di 9.4 ed è meno ionizzata
della guanidina a pH 7.4 ). Si pensò erroneamente che, una volta legata al recettore
H2 attraverso questo legame ione-ione molto forte, il resto della struttura potesse
6
FARMACI ANTIULCERA
fornire l’attività antagonista desiderata. Ma si vide ben presto che questi composti
erano ancora H2 agonisti proprio a causa del loro gruppo cationico.
– Successivamente i chimici scelsero il gruppo tioureico come sostituente R3.
Considerando che è una versione (isostere) non ionizzabile della guanidina, pensarono
che la tiourea era quel che ci voleva per ottenere una potente azione anti H2. Ed
avevano ragione!
N
H
C
N
H
S
Tiourea (isostere della guanidina)
♦L’elettronegatività
dello zolfo elimina il carattere basico dei due atomi di azoto, così
che a pH 7.4 non si forma alcun acido coniugato. Questo gruppo è essenzialmente
neutro (e non ionizzato) a pH 7.4, determinando una potente azione H2 antagonista.
♦Sfortunatamente, questo gruppo tioureico si dimostrò tossico provocando
agranulocitosi. Di conseguenza questo composto non ha mai raggiunto il mercato.
– Mettendo a frutto l’esperienza maturata con la tiourea si giunse allo sviluppo del primo
H2 antagonista efficace sull’uomo: la cimetidina (Tagamet). Semplicemente i chimici
usarono un altro gruppo con forte effetto elettronattrattore, ma privo di zolfo, per
eliminare la basicità del gruppo guanidinico ed evitare la formazione di specie cationiche
in questa parte della molecola.
– Due gruppi di questo tipo vennero subito alla mente: il gruppo cianoguanidinico e
quello del 1,1-diaminonitroetilene. Entrambi hanno prodotto H2 antagonisti molto
N
H
C
N
N
H
C
N
H
C
N
H
NO2
C
N
cianoguanidina
1,1-diamminonitroetilene
potenti e non tossici.
– Si ritiene che in entrambi questi gruppi i due NH agiscano come donatori di legame a
idrogeno nei riguardi di residui (ASP and ARG or TYR) del recettore H2. Tali legami a
idrogeno sono molto importanti per mantenere l’antagonista sul recettore H2 e nel
forzarlo ad assumere una conformazione che disaccoppia il recettore dalla proteina G.
– Gli H2 antagonisti contengono nella loro struttura molti gruppi funzionali polari che li
rendono idrofili. Se si aggiunge un gruppo CH3 all’azoto terminale di queste “guanidine
modificate”, si ottiene un lieve aumento della lipofilia, che sembra favorire l’antagonismo
H2. Tuttavia questo sostituente deve essere piccolo ed in pratica solo il CH3 viene
utilizzato in alcuni dei farmaci antiulcera in commercio.
Ø H2 Antagonisti di Impiego Terapeutico
H3C
S
HN
H
N
N
H
N
N
Cimetidina
C
CH 3
N
H3C
O
N
S
H
N
CH 3
H
N
NO 2
Ranitidina
NH
CH 3
H
H
7
FARMACI ANTIULCERA
Indicazioni: ulcera duodenale; ulcera gastrica benigna; riflusso gastro-esofageo (RGE); condizioni di
ipersecrezione patologica; prevenzione di emorragie del tratto gastrointestinale superiore.
Forme Farmaceutiche: compresse; capsule (Nizatidina); polvere per sospensione orale (Famotidina);
sciroppi (Ranitidina); soluzioni per iniezione ed infusione.
Cimetidina (Tagamet)
Note SAR
• L’anello aromatico imidazolico ha carattere basico
• R1 = CH3: gruppo +I che favorisce il tautomero N τ H richiesto per il riconoscimento da parte del
recettore H2 ma non per l’attivazione del recettore
R2 = etiltiometile: gruppo -I che favorisce la forma tautomerica N τ H; isosterico con la catena butilica
•
che garantisce la separazione ottimale tra l’anello aromatico e R3
• R3 = cianoguanidina: polare, ma neutra. 100% di forma non-ionizzata a pH 7.4, il che assicura un
effetto antagonista puro, senza alcuna azione agonista
• N-CH3 aumenta il carattere lipofilo, con conseguente aumento di attività
Interazioni Farmaco-Farmaco
• La Cimetidina inibisce il citocromo P450 (CYP450)
• Provoca un aumento significativo della durata di azione di altri farmaci cosomministrati che vengono
metabolizzati dal CYP450
• L’anello imidazolico è implicato in questo tipo di attività. L’atomo di azoto dell’imidazolo
complessa il Fe+2 dell’eme che è associato all’enzima CYP450 .
• Anche il CYP3A4 ed altre isoforme dell’enzima vengono inibite, aumentando il rischio di interazioni
farmaco-farmaco.
Altri Effetti Indesiderati
• Effetti collaterali antiandrogenici possono causare ginecomastia ed impotenza
• Ha maggiore propensione rispetto ad altri H2 antagonisti a provocare effetti sul SNC (e.g.
confusione mentale)
• Gli antiacidi riducono l’assorbimento po della Cimetidina. Dal momento che i pazienti assumono sia
antiacidi che H2 antagonisti per il trattamento di RGE o altri disturbi gastrici, questo è un aspetto da
considerare attentamente
8
FARMACI ANTIULCERA
• Nel caso di cosomministrazione, antiacidi e H2 antagonisti dovrebbero essere assunti a distanza di
almeno un’ora gli uni dagli altri.
Ranitidina (Zantac)
Note SAR
• L’anello aromatico del furano con la catena laterale basica dimetilamminometilica è considerato
isostere dell’imidazolo
Presenta tra Ar and R3 una catena stericamente equivalente ad un butile ma con effetto
•
elettronattrattore
• R3 è un gruppo diamminonitroetilenico: polare, ma non-ionizzabile
• Vantaggi della Ranitidina rispetto alla Cimetidina:
• La potenza è aumentata di 4-10X
• La catena laterale basica permette la formazione di sali idrosolubili
• Non ha un anello imidazolico, per cui non inibisce CYP 450
Famotidina (Pepcid )
Note SAR
• Il gruppo guanidinico sull’anello tiazolico fornisce il centro basico necessario in questi composti
• Non ha il gruppo metilico sull’azoto terminale
• La potenza è 40-60X quella della Cimetidina e 9-15X quella della Ranitidina
• La Famotidina ha un indice terapeutico analogo a quello della Ranitidina
• Non ha l’imidazolo, quindi non inibisce CYP450 .
• Minori interazioni farmaco-farmaco rispetto alla Cimetidina
Nizatidina (Axid )
Note SAR
• La Nizatidina presenta l’anello aromatico tiazolico della Famotidina ed i gruppi
dimetilamminometilico e diamminonitroetilenico della Ranitidina
• Possiede attività comparabile a quella della Ranitidina, ma migliore biodisponibilità dopo
somministrazione orale
• La sua potenza è 5-18X quella della Cimetidina
Non inibisce CYP 450
•
• E’ un’ammina terziaria, perciò può formare sali idrosolubili
9
FARMACI ANTIULCERA
Sintesi della cimetidina
H3 C
O
EtOOC
H3 C
NH2
CHO
+ 2
Cl
H
N
1. Na/NH 3 liq.
N
H3 C
N
2. HCl
EtOOC
etile 2-cloroacetato
formamide
NH2
HS
etile 5-metilimidazol4-carbossilato
4-idrossimetil-5-metilimidazolo cloridrato
NC
H
N
H3 C
SCH3
N
SCH3
2 HCl
N
S
H2 N
HCl
HOCH2
HCl
cisteamina cloridrato
H
N
N-cianoimido-S,S-dimetilditiocarbonato
4-(2-aminoetil)-tiometil-5-metilimidazolo dicloridrato
N
CH3 S
CN
N
H
H3 C
S
H
N
N
CH3 NH2
N
CH3 NH
CN
H3 C
H
N
N
S
N
H
cimetidina
Sintesi della ranitidina
condensazione
(CH 3) 2NH
HCl + (CH 2O)x +
paraformaldeide
OH
di Mannich
O
furfurolo
(CH 3) 2N
5-dimetilaminometilfurfurolo
O2N
HS
NH2
HCl
(CH 3) 2N
SCH3
NHCH 3
S
O
cisteamina cloridrato
OH
O
NH2
N-metil-1-metiltio2-nitroetenamina
NO 2
(CH 3) 2N
S
O
ranitidina
N
H
N
H
CH3
10
FARMACI ANTIULCERA
♦ Inibitori della Pompa Protonica (PPI)
Ø Introduzione
Indipendentemente dal tipo di stimolo che ha indotto la secrezione acida gastrica (istamina,
gastrina o ACh), il solo modo per l’acido di raggiungere lo spazio extracitoplasmatico a partire dalle
cellule ossintiche è grazie all’azione dell’enzima H+ , K+ -ATPasi, anche noto come pompa protonica
(PP). Questa pompa è stimolata a secernere acido dal cAMP, prodotto dall’azione dell’istamina sul
recettore H2 accoppiato alla proteina G. Anche il Ca+2 può stimolare la secrezione acida, e la
concentrazione intracellulare di questo ione aumenta quando la gastrina e l’ACh agiscono sui loro
recettori.
In presenza di H2 antagonisti l’istamina non riesce a stimolare la pompa protonica perché rilasci
acido cloridrico nello stomaco, ma la gastrina e l’ACh sono ancora attive. Perciò, per bloccare
completamente la secrezione di acido gastrico, a prescindere dall’origine dello stimolo chimico,
bisogna inibire la pompa stessa, perché questo è l’ultimo stadio nel processo di secrezione acida.
Oltre a bloccare la secrezione acida indotta dai tre mediatori sopra ricordati, gli inibitori della
pompa protonica (PPI) fermano anche la secrezione basale di acido gastrico, risultando quindi agenti
terapeutici potenti e ad ampio raggio nel trattamento dell’ulcera gastrica e del riflusso gastroesofageo
(RGE).
Dal momento che i PPI bloccano anche la secrezione acida indotta dalla gastrina, aumenta la
secrezione della gastrina stessa (meccanismo feed-back), portando a ipergastrinemia. Gli abnormi livelli
di gastrina possono provocare iperplasia delle cellule enterocromaffino-simili del fondo gastrico che
contengono istamina. Fortunatamente nell’uomo, a differenza di quanto avviene in alcune specie animali,
non si è osservata la ulteriore evoluzione di questa iperplasia in carcinomi gastrici.
Ø Chimica della Pompa Protonica
La H+, K+-ATPasi è una proteina di elevato peso molecolare composta da due subunità, la
subunità catalitica alfa ed una subunità regolatoria beta (che è glicosilata). La subunità alfa possiede dieci
domini transmembranari e contiene un totale di 28 residui di cisteina (CYS), uno o due dei quali sono
critici per il legame degli inibitori (PPI).
Diversamente dagli H2 antagonisti, i quali interagiscono competitivamente e reversibilmente con il
recettore H2, i PPI formano un legame covalente (in particolare, un legame disolfuro) con l’enzima H+,
K+ -ATPasi, portando ad inibizione irreversibile.
v Determinando l’inattivazione completa e totale del sito recettoriale (in questo caso l’enzima
H+, K+ -ATPasi), i farmaci che agiscono in modo irreversibile sono agenti terapeutici molto
potenti. L’enzima non è capace di recuperare dalla sua interazione irreversibile con l’inibitore
perché non può idrolizzare il legame covalente che si è formato con il farmaco e l’organismo
deve pertanto sintetizzare nuovo enzima “partendo da zero” o, per usare l’esatta terminologia
scientifica, de novo. Ma la sintesi delle proteine richiede tempo, ed è per questo motivo che
tutti i farmaci che agiscono irreversibilmente hanno una durata d’azione molto lunga.
11
FARMACI ANTIULCERA
Ma quale dei residui di CYS della H+,K + -ATPasi è quello importante ai fini del legame covalente
con l’inibitore? In effetti due residui CYS potrebbero rappresentare siti potenziali di interazione con
questi farmaci.
v Esistono evidenze sperimentali che inducono a ritenere che ciascuno di tali residui possa
essere “quello” che si lega irreversibilmente all’inibitore compromettendo l’attività della pompa
protonica; si pensa generalmente che la CYS813 possa essere più probabilmente coinvolta, ma
è stato anche proposto che due molecole di inibitore possano legarsi simultaneamente
all’enzima, una a livello della CYS813 e l’altra in corrispondenza della CYS 822.
Oltre ai residui di CYS, residui critici anionici alle posizioni 820 e 824 sono importanti per
rafforzare l’interazione tra l’enzima e l’inibitore come pure per posizionare correttamente il farmaco ai
fini del suo legame irreversibile con il residuo di CYS. Ovviamente l’amminoacido anionico sarà GLU o
ASP, ma mentre in posizione 820 c’è effettivamente un residuo di GLU, non sappiamo ancora qule
residuo occupi la posizione 824.
Ø Chimica degli Inibitori della Pompa Protonica
Come già detto i PPI formano un ponte disolfuro irreversibile con la CYS 813 (e/o CYS 822)
dell’enzima H+ , K+ -ATPasi, agendo esclusivamente sulla porzione extracitoplasmatica della proteina.
Un ponte disolfuro non è altro che un legame covalente tra due atomi di zolfo: uno di questi è
rappresentato dal gruppo SH del residuo CYS, mentre l’altro atomo di zolfo deve essere presente nella
struttura dell’inibitore.
Ciò detto, non può sorprendere che la porzione farmacoforica dei PPI sia il 2-piridilmetilsolfinilbenzimidazolo. (Il termine solfinil indica la presenza di un gruppo S=O). Ma per potersi legare
al gruppo SH della CYS, il sulfinile deve essere attivato attraverso una serie di reazioni che inizia con la
protonazione dell’azoto piridinico.
R1
R3
R2
N
N
Struttura 2-Piridilmetilsolfinilbenzimidazolica
R4
S
2-Piridilmetil
O
Sulfinil
N
H
Benzimidazolo
La successiva conversione nella forma solfenamidica attiva è rapida, e grazie alla sua elevata
reattività la reazione irreversibile con il residuo di CYS dell’enzima è di fatto garantita.
Acquisiti questi concetti, diamo ora un’occhiata alla chimica dell’intero processo. L’esempio di
inibitore della pompa protonica riportato nello schema seguente è l’omeprazolo, che è stato il primo di
questi farmaci ad entrare in terapia.
12
FARMACI ANTIULCERA
MECCANISMO DI ATTIVAZIONE DEGLI INIBITORI DELLA POMPA PROTONICA
(OMEPRAZOLO)
MeO
MeO
Me
Me
MeO
Me
Me
H+
N
Me
N
N
H
N
S
N
O
N
MeO
Omeprazolo
S
H
O
N
MeO
H
Me
H
N
O
N
MeO
Forma protonata attivata
S
H
Intermedio spiro labile
H2O
MeO
MeO
Me
Me
Me
Me
N
Me
Me
N
S
N
MeO
N
N
N
H2O
H
S OH
N
H
MeO
N
N
S OH
OH
H
MeO
OMe
Solfenamide
Acido solfenico
Enz−SH
Formazione
del ponte
disolfuro
MeO
Me
Me
S
N
N
NH
S
Enz
Enz−SH
OMe
Enzima inattivato
Molto interessante è il meccanismo con cui l’omeprazolo (analogamente agli altri farmaci PPI) è in
grado di giungere a livello della pompa protonica gastrica e di esplicare il suo effetto (vedi Figura 1).
13
FARMACI ANTIULCERA
L’omeprazolo è una base debole con un discreto grado di lipofilia e capace quindi di attraversare le
membrane cellulari. Esso viene somministrato per os in forma di capsule resistenti all’acidità gastrica, è
assorbito a livello intestinale e, attraverso la circolazione sistemica, giunge nella cellula gastrica. Appena
diffonde nel canalicolo secretore, dato il bassissimo pH presente, si trasforma in un composto protonato
carico non più in grado di attraversare la membrana cellulare. In questo modo il farmaco rimane
intrappolato nel canalicolo secretore dove la sua concentrazione arriva ad essere fino a 1000 volte
superiore a quella plasmatica. Il farmaco protonato va incontro ad un’ulteriore conversione acida in
solfenamide capace di interagire con i gruppi CYS della pompa protonica.
Poiché il legame del farmaco è irreversibile, l’attività della pompa riprende solo dopo che nuove
molecole di enzima sono state sintetizzate ed inserite nella membrana; nel frattempo la secrezione
acida gastrica dovuta a questo enzima si blocca completamente.
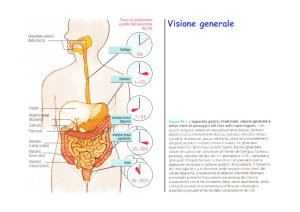
VEDI DIAPOSITIVA 32
Figura 1. Schema della relazione tra instabilità in acido, assorbimento intestinale e trasformazione acida
dei PPI
Ø Metabolismo degli Inibitori della Pompa Protonica (PPI)
Gli inibitori della pompa protonica si legano saldamente alle proteine seriche e sono estesamente
metabolizzati dagli enzimi della famiglia CYP450.
Le isoforme 3A4 e 2C19 sono particolarmente importanti per la conversione di questi farmaci in
metaboliti inattivi. Alcuni composti, in particolare l’omeprazolo, non solo vengono metabolizzati dalle
isoforme del CYP, ma le inibiscono anche. L’omeprazolo ad esempio inibisce il CYP2C19 ed il CYP 2C8.
Ciò ovviamente apre la strada a possibili interazioni farmaco-farmaco, per cui i PPI devono essere usati
con cautela in associazione con altri farmaci che sono metabolizzati da queste stesse isoforme, quali:
♦ ansiolitici benzodiazepinici (e.g., diazepam, triazolam, ecc.)
♦ anticoagulanti del tipo del warfarin
♦ ciclosporina
♦ anticonvulsivanti a struttura idantoinica (e.g., fenitoina, mefenitoina, ecc.)
♦ agenti ipoglicemizzanti a struttura solfonilureica (e.g., tolazamide, ecc.)
14
FARMACI ANTIULCERA
La degradazione metabolica dell’omeprazolo è mostrata nella pagina seguente. Oltre al
metabolismo ossidativo catalizzato dalle isoforme del CYP450, si può avere metabolismo riduttivo a
carico del gruppo solfinile con formazione di un tioetere.
Nuovi PPI che non hanno un gruppo metossilico al C 5 dell’anello benzimidazolico (e.g.,
pantoprazolo, rabeprazolo) vengono metabolizzati dalla isoforma CYP2C19 in altre posizioni. In
particolare:
♦il pantoprazolo subisce O-dealchilazione CYP2C19-mediata al gruppo OCH3 legato al
nucleo piridinico.
♦anche il rabeprazolo è O-dealchilato dal CYP 2C19 al gruppo OCH3 terminale della catena
laterale. Il gruppo alcolico terminale che si forma da questa O-dealchilazione è
ulteriormente ossidato ad aldeide (via alcol deidrogenasi) e successivamente ad acido
carbossilico (via aldeide ossidasi).
Gli schemi che illustrano il destino metabolico del pantoprazolo e del rabeprazolo seguono quello
relativo all’omeprazolo.
Alcune popolazioni asiatiche che esprimono bassi livelli dell’isoforma CYP 2C19 rischiano di
andare incontro ad effetti tossici con l’uso dell’omeprazolo. Mentre solo il 3-5% dei Caucasici
ricadono in questa classe, il 15-22% di Cinesi e Giapponesi esprimono bassi livelli di questo enzima.
Sebbene anche altri PPI siano metabolizzati dal CYP 2C19, il massimo rischio di tossicità si ha con
l’omeprazolo, senza dubbio perché questo farmaco è anche capace di inibire questa isoforma.
Il succo di pompelmo inibisce il CYP 3A4 e può perciò interferire con il metabolismo dei PPI,
portando ad un aumento del tempo di semivita plasmatica.
Ø Interazioni Farmaco-Farmaco Dovute alla Acloridria Indotta dai PPI
I PPI sono farmaci progettati per indurre acloridria (mancanza di succo gastrico), la quale però
può interferire con l’assorbimento di farmaci cosomministrati. Sono state identificate o fortemente
sospettate interazioni farmaco-farmaco tra i PPI ed i seguenti farmaci:
♦cefalosporine, chetoconazolo, indinavir, vitamina B12 (assorbimento ridotto)
♦claritromicina, digossina (assorbimento aumentato).
Ø Sintesi dei PPI
Si riporta di seguito a titolo di esempio la sintesi chimica dell’omeprazolo.
15
FARMACI ANTIULCERA
METABOLISMO DELL’OMEPRAZOLO
MeO
MeO
Me
Me
Me
N
N
S
O
CYP 2C19
N
O-dealchilazione
N
N
Me O
O
H
Omeprazolo
Fenolo (inattivo)
ossidazione
riduzione
MeO
MeO
Me
Me
Me
Me
N
N
O
N
S
O
N
N
Me O
S
N
HO
H
CYP3A4
Me
N
MeO
H
Solfone (inattivo)
S
H
Tioetere (inattivo)
METABOLISMO DEL PANTOPRAZOLO
MeO
HO
OMe
N
N
F2 HCO
S
N
H
Pantoprazolo
O
OMe
CYP 2C19
N
O-dealchilazione
N
F2 HCO
S
O
N
H
Fenolo (inattivo)
16
FARMACI ANTIULCERA
METABOLISMO DEL RABEPRAZOLO
OMe
O
OH
O
OMe
OMe
CYP 2C19
N
O-dealchilazione
N
N
N
S
O
N
S
O
N
ossidazione
N
OMe
O
H
O
S
Aldeide (inattiva)
Aldeide
ossidasi
ossidazione
riduzione
OMe
O
OMe
O
O
OMe OH
OMe
N
N
N
S
N
S
O
N
O
N
Alcol (inattivo)
ossidazione
N
S
H
Rabeprazolo
N
OMe H
Alcol
deidrogenasi
N
H
CYP3A4
O
O
O
N
N
H
H
Solfone (inattivo)
Tioetere (inattivo)
H
Acido carbossilico (inattivo)
17
FARMACI ANTIULCERA
SINTESI DELL’OMEPRAZOLO
H3C
OCH 3
CH3
N
OCH3
(CH 3CO) 2O
H3C
CH3
CH3
NaOH
O
N
H3 C
CH 3
OCH3
CH3
OH
N
O
O
SOCl2
SK
NH2
CH3 O
OCH 3
S
N
OC2H 5
NH2
(potassio etil
xantogenato)
H3C
SH
N
H
CH3O
CH3
N
Cl
NaOH
H3 C
OCH3
CH3
N
N
S
NH
CH3 O
O
O
O
OH
OCH3
H3 C
CH3
Cl
N
MCPBA
N
(acido meta cloroperbenzoico)
S
NH
CH3 O
Omeprazolo
♦ Farmaci Vari
Sono noti anche farmaci antiulcera che operano con meccanismi diversi da quelli finora
considerati.
La pirenzepina, chimicamente il 5,11-diidro-11-[(4-metil-1-piperazinil)acetil]-6H-pirido [2,3b][1,4]benzodiazepin-6-one, disponibile come dicloridrato, è un antimuscarinico selettivo che si lega in
maniera preferenziale ai recettori della mucosa gastrica riducendo la secrezione sia dell’acido cloridrico
che della pepsina.
La proglumide è un inibitore della gastrina e pertanto riduce il volume, l’acidità ed il potere
proteolitico del succo gastrico come pure la peristalsi gastroduodenale. Oltre all’azione antigastrinica la
proglumide possiede proprietà trofiche per la mucosa gastrica e duodenale, azione gastroprotettiva ed
antispasmodica. Trova impiego nel trattamento delle ulcere gastriche e duodenali, nelle gastriti e
18
FARMACI ANTIULCERA
gastroduodeniti ed anche nella prevenzione e nel trattamento dei disturbi gastrici provocati da farmaci.
E’ un composto ottimamente tollerato e praticamente atossico.
Il gefarnato o geranil farnesilacetato è un antiulceroso ad azione protettiva e trofica sulla mucosa
gastrica e duodenale con meccanismo analogo a quello fisiologico. E’ usato nella cura dell’ulcera
gastroduodenale, delle gastriti e delle enterocoliti. Non ha controindicazioni; può provocare fatti allergici
cutanei di tipo pruriginoso.
E’ noto che le prostaglandine sono coinvolte nella conservazione dell’integrità dell’epitelio
gastrico e che la loro somministrazione preventiva protegge la mucosa gastrica degli animali da vari
stimoli ulcerogenici. Come farmaci per il trattamento dell’ulcera peptica sono stati sviluppati vari
analoghi delle prostaglandine dotati di maggiore stabilità e di attività per via orale. Uno di questi, il
misoprostolo (analogo sintetico della prostaglandina E1, PGE1) è usato come agente antisecretorio
gastrico con effetti protettivi sulla mucosa gastroduodenale.
O
H
N
O
N
N
COOH
N
HN
O
O
N
Pirenzepina
N
CH 3
CH3
CH3
Proglumide
CH3
O
H3C
CH3
O
CH3
CH 3
Gefarnato
O
COOCH 3
H3C
OH
HO
Misoprostolo
♦ Farmaci Antimicrobici
19
FARMACI ANTIULCERA
La terapia ottimale dei pazienti con ulcera peptica (sia gastrica che duodenale) infetti da
Helicobacter pylori richiede il trattamento antimicrobico. L’eradicazione di H. pylori induce la rapida
guarigione delle ulcere peptiche attive e basse frequenze di ricaduta. E’ possibile una efficace
eradicazione di H. pylori con varie associazioni di farmaci antimicrobici. Per esempio, un regime di
scelta attuale fondato sull’efficacia (tasso di eradicazione del 90% circa) e sul costo è un ciclo di due
settimane di terapia triplice con bismuto, metronidazolo e tetraciclina, cui spesso si aggiunge un
antisecretivo. Regimi di seconda linea sono rappresentati da associazioni di due farmaci antimicrobici
(metronidazolo e amossicillina) con un antisecretivo (omeprazolo).
N
O2 N
N
CH 3
OH
Metronidazolo
20