Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa
DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W,
Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber
DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ.
Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer
N Engl J Med 2010;363:1693-703.
Commento a cura di:
Giampaolo Tortora
Azienda Ospedaliera Universitaria Integrata
Università di Verona
[email protected]
Introduzione e background
Un obiettivo dell’oncologia moderna è selezionare i pazienti in base alla presenza nel loro
tumore di geni/proteine che identificano sottogruppi specifici, utilizzando poi tali informazioni
come fattori prognostici, per la scelta del trattamento mirato e come fattori predittivi di risposta
ad esso e, infine, come bersagli di nuovi farmaci da sviluppare. Nei tumori del polmone non a
piccole cellule (NSCLC), soprattutto con istologia adenocarcinoma, dopo le alterazioni in EGFR e Kras sono state individuate quelle in MET e, infine, in ALK (anaplastic lymphoma kinase) ampliando
così la possibilità di trattamento con farmaci mirati (1).
Il gene ALK codifica per una proteina con funzione di recettore tirosin-chinasico e ha potere
trasformante in seguito a una mutazione o, più frequentemente, ad un riarrangiamento
cromosomico con formazione di un gene di fusione con diversi altri geni (2). In circa il 60% dei
linfomi non-Hodgkin anaplastici a grandi cellule si reperta la traslocazione 2;5, che consiste nella
fusione della porzione 3’ del gene ALK, posizionato sul cromosoma 2, con il 5’ della nucleofosmina
(NPM), posizionato sul cromosoma 5 (2). Mutazioni attivanti o traslocazioni di ALK sono state
identificate anche in altri tipi di neoplasia, tra cui il neuroblastoma, il tumore miofibroblastico
infiammatorio e, appunto, il NSCLC. Recentemente è stato identificato un prodotto di fusione tra
ALK e il gene Echinoderm microtubule-associated protein-like (EML4) (3). La fusione EML4-ALK è
una piccola inversione all’interno del cromosoma 2p che codifica una proteina con porzione Nterminale di EML4 e dominio catalitico intracellulare di ALK che possiede un’attività chinasica
costitutiva. Sono state identificate diverse e distinte varianti di EML4-ALK, dovute a breakpoint in
diversi esoni di EML4, ma tutte dotate di potere trasformante in vitro. Altri partner di fusione di
ALK più rari, come KIF5B e TFG, sono stati segnalati in pazienti con NSCLC (2).
I primi studi hanno documentato che il riarrangiamento EML4-ALK: a) è presente nel 7% dei
pazienti giapponesi con NSCLC mentre è ancora più raro, circa 2-5%, nei pazienti non asiatici; b)
che esso prevale in pazienti con adenocarcinoma e storia di non fumatori o modesti fumatori (3, 4).
1
Studi preclinici su oltre 600 linee cellulari derivate da NSCLC umani, hanno mostrato che
l’inibizione di ALK con inibitori specifici come TAE684 e PF-02.341.066, attualmente in sviluppo
clinico, causa blocco della proliferazione cellulare e apoptosi (5, 7).
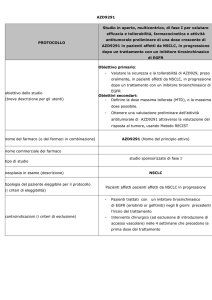
Il farmaco impiegato nello studio in esame è il PF-02.341.066, denominato crizotinib (Pfizer), un
inibitore orale, ATP-competitivo, selettivo per i recettori tirosin chinasici ALK e MET, in grado di
inibire la fosforilazione della tirosina di ALK a concentrazioni nano molari (5, 7).
Lo studio
Coorte di pazienti e analisi
Lo studio condotto da Kwak e coll. è un open-label multicentrico di fase 1 per valutare il
profilo di tossicità e di efficacia di crizotinib che, originariamente, era stato disegnato come un
classico dose-escalation di Fase I per pazienti con tumori solidi refrattari alla terapia standard.
Tuttavia, una volta riscontrato un drammatico miglioramento della sintomatologia dopo le prime
somministrazioni di crizotinib in 2 pazienti con NSCLC e un riarrangiamento di ALK, si è deciso di
riservare lo studio solo a una coorte di pazienti con tali caratteristiche, che è quindi stata ampliata.
A tale scopo da agosto 2008 a febbraio 2010 è stato condotto un vastissimo screening mediante
FISH per la fusione di ALK su circa 1500 pazienti con NSCLC. Sono risultati positivi e sono stati
arruolati nello studio 82 pazienti (circa 5% dei pazienti analizzati) che avevano un Performance
status ECOG 0-2. Nessun limite è stato posto al numero di trattamenti precedenti praticati.
Tumori inclusi in paraffina sono stati valutati in modo prospettico e considerati FISH positivi se più
del 15% delle cellule tumorali mostravano un segnale che indicava riarrangiamento di ALK. Degli
82 campioni, 70 (85%) sono stati confermati FISH-positivi da un laboratorio centralizzato. A
posteriori, in un sottogruppo di campioni tumorali, sono state eseguite anche RT-PCR per
specifiche fusioni EML4-ALK e analisi immunoistochimica (IHC) per valutare la proteina ALK.
Le dosi di crizotinib nel dose-finding sono state progressivamente incrementate da 50
mg/die a 300 mg due volte/die (bid). Dagli studi preclinici risulta che la concentrazione
antitumorale efficace di crizotinib è 120 ng/ml. Poiché la dose di 200 mg bid consente di
raggiungere e superare tale concentrazione plasmatica e la tossicità dose-limitante (DLT), l’astenia,
compariva a dosi di 300 mg bid, si è deciso di considerare 250 mg bid come Massima Dose
Tollerata, utilizzandola in tutti i pazienti, in cicli di 28 giorni, fino a progressione di malattia (PD) o
effetti collaterali intollerabili.
Tutti hanno praticato esami strumentali di valutazione basale e post trattamento dopo aver
ricevuto 1 ciclo di crizotinib e dopo ogni 2 cicli. I pazienti che avevano ottenuto una risposta
parziale (PR) secondo i criteri RECIST venivano sottoposti a scansioni di conferma dopo 4
settimane dalla risposta documentata.
Risultati principali e attività
Un primo risultato è stato la dimostrazione, che conferma osservazioni precedenti, che gli
82 pazienti con NSCLC ALK- positivi (provenienti da USA, Australia e Corea) erano accomunati dalle
seguenti caratteristiche:
• adenocarcinomi, spesso con cellule ad anello con castone;
• età mediamente più giovane di quella attesa in NSCLC;
• condizione di fumatori o fumatori moderati (≤ 10 pacchetti-anno), eccetto 5 pazienti con
una storia di più di 10 pacchetti-anno, 3 dei quali erano forti fumatori.
Degli 82 pazienti, 76 pazienti (94%) avevano già praticato almeno 1 linea di terapia e 34 avevano
ricevuto almeno 3 linee. Solo in 6 pazienti crizotinib è stato usato come prima linea (Tabella 1).
2
Tabella 1. Caratteristiche degli 82 pazienti in studio.
Utilizzando i criteri RECIST, il tasso di Risposta (Figura 1) ottenuto negli 82 pazienti FISH-positivi
per ALK ,è stato di :
• 1 risposta completa (CR) e 46 risposte parziali (PR), per un tasso di risposta complessivo del
57% (95% CI)
• 27 (33%) stabilizzazioni di malattia (SD). In realtà 5 pazienti avevano ottenuto una PR ma,
avendo praticato in ritardo la TAC di conferma, sono stati classificati come SD. Il tasso di
controllo della malattia dopo 8 settimane è stato quindi dell'87%.
• 6 (7%) progressioni di malattia (PD). In 2 casi si era avuto un aumento di malattia di oltre il
20% dopo 2 cicli di terapia, 3 pazienti avevano una SD sulle lesioni target ma un
contemporaneo sviluppo di nuove lesioni, 1 aveva regressione del tumore del 25% alla TAC
dopo il I ciclo ma un successivo aumento di oltre il 20% al momento della ristadiazione.
3
•
2 pazienti non sono stati valutati per la risposta per un brusco deterioramento clinico
causato da complicanze acute della malattia (trombosi e polmonare acuta emorragia).
Figura 1. Risposta dei pazienti con tumori ALK-positivi trattati con crizotinib.
Le barre indicano la variazione percentuale tumorale rispetto al basale.
Tre pazienti non sono inclusi: 1 è stato valutato clinicamente come PR, anche se la risposta è stata
principalmente nelle zone di malattia non misurabili, per cui è stato classificato come SD; 2 pazienti
con brusco peggioramento clinico non sono stati valutati.
Quattro pazienti hanno avuto una completa risoluzione delle lesioni bersaglio ma sono stati
classificati come PR, sulla base della stabilità nelle lesioni non-target. Otto pazienti avevano
riduzione del tumore di più del 30%, ma sono stati classificati come SD perché le scansioni di
conferma non erano disponibili al momento del cut-off dei dati (5 pazienti) o perchè la
ristadiazione è stata eseguita 6 settimane dopo l'inizio di crizotinib (3 pazienti).
La linea tratteggiata indica una riduzione del tumore del 30% rispetto al basale, la diminuzione
percentuale minima che costituisce una risposta parziale, in base alla risposta nei Criteri RECIST.
Un sottogruppo di pazienti non selezionati è stato valutato anche con 18FDG-PET, e alcuni,
sempre non selezionati, anche con scansioni esploratorie con 18FLT-PET dopo 1 ciclo di
trattamento. Le PET hanno confermato l’impressione clinica che l’inibizione di ALK avviene molto
rapidamente.
Analisi molecolari
Poiché il test FISH “break-apart” rileva perturbazioni del locus ALK ma non conferma che il
gene partner di fusione sia proprio EML4, è stata usata la Real Time-PCR per analizzare i punti di
interruzione dei pazienti con riarrangiamenti di ALK FISH-positivi. Si è constatato che esistono
diversi prodotti di fusione EML4-ALK, dovuti a breakpoints in diversi punti di EML4 e che la RT-PCR
non era in grado di confermare sempre tali trascritti, suggerendo che EML4 potrebbe non essere il
partner di fusione di ALK in questi pazienti.
4
L’analisi di MET in 33 tumori non ha rilevato alcuna alterazione in questo gene, né sono state
riscontrate mutazioni in EGFR.
Tossicità
Gli eventi avversi negli 82 pazienti trattati principalmente con 250 mg bid, sono stati
moderati, consistendo prevalentemente di nausea e diarrea di grado 1. Lievi disturbi visivi sono
stati riportati da 34 pazienti (41%) e miglioravano con il proseguire della terapia. Gli aumenti delle
transaminasi osservati erano solitamente di grado 1 o 2, e, con l’eccezione di 1 paziente, i rari casi
di grado 3 e 4 sono in regrediti alla cessazione di crizotinib, consentendo di riprendere il
trattamento con un dosaggio inferiore (Tabella 2).
Tabella 2. Eventi avversi negli 82 pazienti in studio.
Durata del trattamento e PFS
Degli 82 pazienti trattati, 63 (77%) hanno continuato a ricevere crizotinib dopo il cut-off dei
dati con una durata media del trattamento di 6,4 mesi e follow-up ancora in corso. 19 pazienti
hanno interrotto il trattamento: 13 per PD, 1 per eventi avversi correlati al crizotinib, 1 per eventi
avversi indipendenti da crizotinib, 2 per morte dovuta a cause non connesse al trattamento e 2 per
altri motivi (Figura 2).
5
Figura 2. Durata del trattamento per 82 pazienti in studio.
Le barre blu indicano i pazienti che stanno continuando a ricevere crizotinib dopo il cut-off dei dati.
Le barre rosse indicano 19 pazienti che hanno interrotto il trattamento. Nel pannello B si vede la
curva di Kaplan-Meier con la stima della PFS e le 2 curve (sopra e sotto) con i limiti di confidenza
Hall-Wellner del 95%.
6
Lo studio non è stato pianificato per avere come end-point la PFS e gli effetti osservati nei pazienti
con riarrangiamento ALK sono stati inattesi rispetto alla stesura del protocollo, quindi nello studio
non c’era nessun obiettivo statisticamente predeterminato per i pazienti ALK positivi. Inoltre, i
pazienti ALK-positivi che ricevevano crizotinib avevano una storia eterogenea di trattamenti
precedenti. Considerate tali limitazioni, e con un follow-up mediano di PFS di 6,4 mesi (95% CI), è
stata stimata una probabilità di PFS a 6 mesi del 72% (95% CI).
Commento
I tumori EML4-ALK-positivi costituiscono una nuova entità tra gli adenocarcinomi del polmone
caratterizzati da giovane età, istologia acinare, nessuna o rare mutazioni in EGFR, KRAS e TP53, e
una espressione di TTF-1, tutte caratteristiche che correlano con l’essere non fumatori o modesti
fumatori (8, 9). Il riarrangiamento EML4-ALK sembra essere specifico perché nello studio di Kwak e
coll. i campioni FISH-negativi e i tessuti polmonari normali non esprimevano proteina ALK.
Benché manchi un gruppo controllo, aspetti molto rilevanti di questo studio sono stati
certamente:
• il tasso di risposta complessivo, pari al 57% (PR e CR confermate), e di stabilizzazione della
malattia, pari al 33% (incluse alcune PR non confermate) con una probabilità stimata di PFS a 6
mesi del 72% a follow-up ancora in corso. Poiché la maggioranza dei pazienti era stata già
pretrattata con più linee di terapia è particolarmente significativo il confronto di attività con la
tipica risposta attesa dalla chemioterapia di II linea, pari a circa 10% (10) , e se si considera che
una metanalisi in pazienti trattati con polichemioterapia di II linea ha mostrato una mediana di
PFS di 14 settimane e un tasso di PFS a 6 mesi del 27,2% (11). Pertanto, l'inibizione mirata di
ALK in pazienti genotipizzati può portare a miglioramento della sopravvivenza rispetto alla
chemioterapia convenzionale. Pazienti senza anomalie di ALK, inclusi anche alcuni con
neoplasie diverse da NSCLC, non hanno ottenuto alcuna risposta al trattamento.
• L’entità e la velocità della risposta clinica, paragonabili con quanto si osserva con inibitori
tirosin-chinasici di EGFR in pazienti con EGFR mutato (12). Ciò significa che i tumori ALK-positivi
costituiscono un altro sottogruppo di NSCLC con meccanismo oncogene-dipendente altamente
sensibile alla terapia mirata.
• La tossicità relativamente modesta, prevalentemente gastrointestinale di grado 1 o 2. E’
confortante notare che gli stessi modesti effetti collaterali sono stati descritti anche da
Butrinsky e coll. nello studio condotto su pazienti affetti da tumore miofibroblastico
infiammatorio e trattati con la stessa dose di 250 mg bid, pubblicato sullo stesso numero del
NEJM. (13)
• La grande perseveranza ed efficienza nel condurre un vasto pre-screening in tempo reale,
considerata la relativa rarità di ALK riarrangiato (circa il 5% dei pazienti), incorporando uno
studio basato sul genotipo nello studio clinico di fase 1 originario.
Nonostante non sia possibile determinare l'efficacia della chemioterapia standard nei
pazienti ALK-positivi che non hanno praticato crizotinib, dati preliminari in un piccolo numero di
pazienti studiati retrospettivamente suggeriscono che ALK non condiziona la risposta alla
chemioterapia. Infatti una chemioterapia a base di platino produce una risposta simile sia in
pazienti ALK positivi sia in quelli senza riarrangiamento di ALK o mutazione di EGFR (8)
Poiché crizotinib inibisce anche MET e 33 tumori ALK positivi non hanno mostrato
amplificazione di MET, è evidente che l’efficacia di crizotinib in questi pazienti non può essere
ascritta alla sua attività anti-MET. Aspetto interessante è il fatto che nessuno dei pazienti ALK7
positivi aveva anche una mutazione in EGFR nonostante fossero pazienti con adenocarcinoma e
storia di non fumatori, caratteristiche cliniche tipiche dei pazienti con mutazioni EGFR. Nello studio
di Kwak e coll. nei 29 campioni analizzati non è stato possibile correlare la presenza del breakpoint
EML4-ALK con storia di fumo o RR, presumibilmente per l’esiguità del campione. Tuttavia è fuori
discussione che in pazienti con adenocarcinoma e una storia di non fumatori o modesti fumatori, i
driver genetici per la crescita tumorale comprendono EGFR nel 50% dei casi e ALK in un altro 20%,
indicando che una categorizzazione genetica molecolare è fondamentale per guidare la scelta della
terapia appropriata, molto meglio se eseguita in modo prospettico. Come si fa notare
nell’editoriale di accompagnamento del lavoro, a soli 2 anni dal primo studio che ha descritto il
riarrangiamento EML4-ALK (3) è stato dimostrato con lo studio di Kwak e coll. che l'inibizione di
ALK riduce le dimensioni tumorali in pazienti con NSCLC (comunicato già all’ASCO 2009), poi, dopo
solo 3 anni, è partito uno studio di fase 3 di registrazione per l’impiego di crizotinib in pazienti ALKpositivi. Diversamente, ci sono voluti circa 10 anni tra lo studio negativo di fase 3 con inibitori di
EGFR in pazienti non-genotipizzati e gli studi randomizzati di Mok e coll. che ha provato
inequivocabilmente l'efficacia di gefitinib rispetto alla chemioterapia di I linea in pazienti con EGFR
mutato (15).
E’ interessante notare che nello studio di Mok e coll. è stata arricchita la popolazione dei pazienti
con mutazioni EGFR selezionando adenocarcinomi e non- o modesti fumatori, caratteristiche che
però non garantiscono una risposta al gefitinib. Anche nello studio di Kwak e coll. diversi pazienti
erano forti fumatori e nonostante ciò avevano ALK-riorganizzato e hanno risposto al crizotinib.
Ora dovrà essere provato se crizotinib darà risposte altrettanto forti in I linea e se un
approccio combinato con altri farmaci o altri inibitori potrebbe essere più vantaggioso.
Per esempio, studi preclinici con un altro inibitore di ALK, TAE684, hanno dimostrato che la coattivazione di HER2 può renderlo inefficace, e che, in questi casi, la combinazione di TAE684 con
un inibitore di EGFR e HER2 ha un forte effetto antitumorale (6).
Considerando un tasso medio di positività per il riassetto ALK di circa il 5%, si può stimare
che, nei soli Stati Uniti, circa 10.000 pazienti con NSCLC sono potenzialmente candidabili ogni anno
a essere trattati con crizotinib. Considerata la rilevanza assunta da EGFR, K-RAS e ALK dovrebbe
essere presa in considerazione una genotipizzazione di routine per arrivare realmente a una
terapia personalizzata.
Un aspetto importante che dovrà essere attentamente considerato è che, in analogia a
quanto avviene già con piccole molecole come gli anti-EGFR ed imatinib, anche con crizotinib
possono comparire mutazioni di farmaco-resistenza. Infatti, sebbene Kwak e coll. non affrontino
questo problema, è possibile che i loro pazienti ALK positivi che hanno avuto una risposta limitata
possano aver sviluppato mutazioni prima o durante il trattamento con crizotinib. A tale proposito,
sullo stesso numero di NEJM, uno studio di Choi et al. (16) già descrive le mutazioni in EML4-ALK
che conferiscono resistenza a crizotinib. Per esempio essi riportano alcune mutazioni, tra cui la
L1196M, in un paziente che aveva inizialmente avuto una forte risposta clinica al crizotinib.
L1196M è una mutazione del residuo critico gatekeeper che preverrebbe il legame di crizotinib ad
ALK, in analogia a quanto avviene con le mutazioni T790M nell’esone 20 dell’EGFR e T315I nelle
leucemie con Bcr/Abl. Effettivamente studi sperimentali hanno mostrato che inserendo alcune
mutazioni in EML4-ALK in cellule di NSCLC si può conferire resistenza a crizotinib a cellule
precedentemente sensibili. Ciò richiama la necessità di sviluppare altri farmaci anti EML4-ALK che
superino le mutazioni di resistenza al crizotinib (17, 18).
Una incoraggiante conclusione da trarre comunque è che, ancora una volta, il rapporto di
collaborazione tra scienziati di base e clinici può produrre risultati molto importanti per la cura dei
tumori.
8
Bibliografia
1. Pao W, Iafrate AJ, Su Z. Genetically informed lung cancer medicine. J Pathol. 223:230-240, 2011.
Epub 2010 Oct 25.
2. Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in
development and disease. Biochem J 2009;420:345-61.
3. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature 2007;448:561-6.
4. Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics
in a subset of lung cancers. J Thorac Oncol 2008;3(1):13–7.
5. Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a
novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic
large-cell lymphoma. Mol Cancer Ther 2007;6:3314-22.
6 Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase
inhibitor in lung cancer. Clin Cancer Res 14:4275-83, 2008
7. McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase
may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008;68:3389-95.
8. Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with nonsmall-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009; 27:4247-53.
9. Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic
types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115:1723-33.
10. Gridelli C, Ardizzoni A, Ciardiello F, et al. Second-line treatment of advanced
non-small cell lung cancer. J Thorac Oncol 2008;3:430-40.
11. Di Maio M, Chiodini P, Georgoulias V, et al. Meta-analysis of single-agent chemotherapy
compared with combination chemotherapy as second-line treatment of advanced non-small-cell
lung cancer. J Clin Oncol 2009;27:1836-43.
12. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nature Reviews Cancer 10: 760-774, 2010
13. Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory
myofibroblastic tumor. N Engl J Med 2010;363:1727-33.
14. Hallberg B. and Palmer, R.H. Crizotinib-Latest Champion in the Cancer Wars? N Engl J Med
363: (18), 1760-1762, 2010
15. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin–paclitaxel in pulmonary
adenocarcinoma. N Engl J Med 2009;361:947-57.
16. Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance
to ALK inhibitors. N Engl J Med 2010;363:1734-9.
17. Lu L, Ghose AK, Quail MR, et al. ALK mutants in the kinase domain exhibit altered kinase
activity and differential sensitivity to small molecule ALK inhibitors. Biochemistry 2009;48: 3600-9.
18. Zhang S, Wang F, Keats F, et al. AP26113, a potent ALK inhibitor, overcomes mutations in
EML4-ALK that confer resistance to PF-02341066 (PF1066). In: Proceedings of the 101st Annual
Meeting of the American Association for Cancer Research, Washington, DC, April 17–21, 2010, :LB298. abstract.
9