UNIVERSITÁ DEGLI STUDI DI MILANO
FACOLTÁ DI MEDICINA E CHIRURGIA
DOTTORATO DI RICERCA IN FISIOLOGIA
SETTORE SCIENTIFICO DISCIPLINARE BIO-09
CICLO XXIII
Tesi di Dottorato di Ricerca
Studio dell’interazione tra le proteine citoscheletriche 4.1R80 e
4.1R135 e la proteina ICln: aspetti molecolari e funzionali.
Dottorando: Dott.ssa
Chiara Zanoni
Matricola: R07659
Tutor: Prof. Giuliano Meyer
Dipartimento di Scienze Biomolecolari e Biotecnologie
Coordinatore: Prof. Paolo Cavallari
Anno Accademico 2009-2010
Indice
1 Riassunto
1
2 Introduzione
5
2.1 La regolazione del volume cellulare
5
2.2 Sensori e modulatori delle variazioni del volume cellulare
8
2.3 L’RVD
17
2.3.1 Meccansimi di trasporto coinvolti nel’RVD
17
2.3.2 La corrente per il Cl- attivata durante l’RVD: ICl,swell
19
2.4 ICln
22
2.4.1 Struttura di ICln
22
2.4.2 La localizzazione e la funzione
28
2.4.3 Interazione di ICln con altre proteine
31
2.5 La proteina 4.1
34
2.5.1 La struttura
34
2.5.2 La funzione e le interazioni
43
3 Scopo del lavoro
47
I
4 Materiali e Metodi
50
4.1 Colture cellulari
50
4.2 Estrazione dell’RNA citoplasmatico
51
4.3 Elettroforesi dell’RNA
53
4.4 Retrotrascrizione (RT) con oligo dT
54
4.5 PCR
54
4.6 Clonaggio del cDNA per la proteina 4.1Rsh e 4.1RII nei vettori, pEYFP-C1, pEYFPN1
56
4.6.1 PCR
58
4.6.2 Elettroforesi del DNA
60
4.6.3 Purificazione dei prodotti di PCR
61
4.6.4 Reazioni di restrizione
61
4.6.5 Purificazione dei prodotti di digestione
63
4.6.6 Ligazione
63
4.7 Mutagenesi del sito di inizio ATG2 presente nel plasmide IRES 4.1 RII
64
4.8 Produzione di batteri competenti
66
4.9 Trasformazione batterica
67
4.10 Miniprep
68
4.11 Maxiprep
69
4.12 Dosaggio acidi nucleici
71
4.13 Trasfezione
71
4.14 FRET, Fluorescence Resonance Energy Transfer
73
4.14.1 Tecnica dell’Acceptor Photobleaching
75
4.14.2 "Sensitized emission"
76
II
4.15 Studi di immunocitochimica
78
4.16 Estrazione proteine totali
79
4.17 Estrazione proteine nucleari e citosoliche
80
4.18 Estrazione proteine totali di membrana
83
4.19 Dodaggio proteico
85
4.20 Coimmunoprecipitazione
85
4.21 Western blot
87
4.22 Esperimenti di patch-clamp
93
4.23 Conte cellulari e curve di crescita
95
4.24 Analisi statistiche
96
5 Risultati
97
5.1 Caratterizzazione delle isoforme di 4.1 espresse in cellule HEK: RT-PCR
97
5.2 Interazione tra ICln e 4.1R
101
5.2.1 Interazione in vivo in cellula. Esperimenti di FRET: interazione tra ICln e 4.1Rsh
101
5.2.2 Studio dell’interazione tra ICln e l’isoforma corta (4.1Rsh) della proteina 4.1
mediante coimmunoprecipitazione.
108
5.2.3 Interazione in vivo in cellula. Esperimenti di FRET: interazione tra ICln e 4.1RII
110
5.2.4 Studio dell’interazione tra ICln e l’isoforma lunga (4.1RII) della proteina 4.1
mediante coimmunoprecipitazione.
112
5.3 Studi di localizzazione subcellulare: nucleo.
115
5.3.1 Studio della localizzazione della proteina 4.1Rsh e di ICln nel nucleo.
115
5.3.2 Studi di immunofluorescenza sulla localizzazione di ICln in cellule HEK 293
Phoenix in cui è overespressa la proteina 4.1Rsh.
120
III
5.3.3 Studi di western blot sui livelli di espressione della proteina ICln in cellule HEK
293 Phoenix transfettate con la proteina 4.1Rsh e sui livelli di espressione totali
della proteina 4.1 in cellule transfettate con ICln.
121
5.3.4 Studi di western blot sui livelli di espressione nel nucleo della proteina ICln in
cellule HEK 293 Phoenix transfettate con la proteina 4.1Rsh e sui livelli di
espressione nucleare della proteina 4.1 in cellule transfettate con ICln.
124
5.3.5 Studio della localizzazione della proteina 4.1RII e di ICln nel compartimento
nucleare.
128
5.3.6 Studi di western blot sui livelli di espressione nel nucleo della proteina ICln in
cellule HEK 293 Phoenix transfettate con la proteina 4.1RII e sui livelli di espressione
nucleare della proteina 4.1 in cellule transfettate con ICln.
131
5.4 Studi di localizzazione subcellulare: membrana.
135
5.4.1 Osservazione delle immagini di FRET della localizzazione in membrana di
entrambe la isoforme: 4.1Rsh e 4.1RII.
135
5.4.2 Studi di western blot sui livelli di espressione della proteina 4.1R in membrana
in cellule HEK 293 Phoenix co-espresse con la proteina ICln.
138
5.5 4.1R e RVD
140
5.5.1 Studi di western blot sulla localizzazione in membrana della proteina 4.1R
sottoposta a uno stimolo ipotonico
141
5.5.2 Significato funzionale dell’interazione tra ICln e 4.1: Esperimenti di patchclamp.
144
5.7 Studi di proliferazione di cellule HEK 293 Phoenix transfettate con 4.1Rsh e
ICln.
148
6 Discussione
150
Bibliografia
163
IV
V
1 Riassunto
ICln riveste un ruolo fondamentale nell’attivazione dell’ICl,swell, la corrente di
cloruro attivata dal rigonfiamento cellulare, (Okada et al., 1997). Lo stress ipotonico
induce una traslocazione di ICln verso la membrana cellulare che accompagna
l’attivazione della corrente ma non è ancora stato chiarito se ICln intervenga come
regolatore o se rappresenti uno dei componenti del canale stesso.
Oltre ad essere coinvolta nel’RVD (Regulatory Volume Decrease), ICln è coinvolta in
numerosi altri processi cellulari come dimostrato dal pattern delle sue interazioni
con altre proteine quali proteine coinvolte nello splicing dell’mRNA (Pu et al., 1999)
e proteine di adesione (integrine) (Larkin et al., 2004). Di particolare interesse per
questo lavoro è l’interazione di ICln con proteine citoscheletriche. E’ noto infatti che
l’ICl,swell è modulata dallo stato del citoscheletro actinico (Lang et al., 1998;
Moustakas et al., 1998) e ICln lega numerose proteine citoscheletriche tra cui la
stessa actina, la miosina e alcune proteine appartenenti alla famiglia delle 4.1, una
famiglia di proteine multifunzione coinvolta nella regolazione del citoscheletro,
della morfologia cellulare e di membrana e della proliferazione cellulare.
E’ già noto che 4.1R e ICln interagiscono: l’interazione è stata appurata tramite le
tecniche del doppio ibrido in lievito, di coimmunoprecipitazione e spettrometria di
massa (Tang et al. 1998; Figeys et al., 2001). In particolare si è visto che il dominio Cterminale di ICln (aminoacidi dal 103 al 237) lega il dominio di 30 kDa C-terminale
(FERM) della proteina 4.1R (80 kDa) (Tang et al., 1998), a livello degli aminocaidi
136-283 (Calinisan et al., 2006). L’utilizzo di queste tecniche, tuttavia, non ha fornito
informazioni né sulla sede dell’interazione, né sulla funzione, aspetto di estremo
interesse, vista la spiccata multifuzionalità di entrambe le proteine.
Su queste basi, in questo lavoro abbiamo studiato in vivo tramite la tecnica del FRET
(Fluorescence Resonance Energy Transfer) l’interazione tra ICln e 4.1R umana, per
studiarne la localizzazione subcellulare e l’evolversi durante l’ipotonia. Sebbene ICln
interagisca con i FERM di tre diverse isoforme di 4.1, l’isoforma R, B e G abbiamo
scelto di focalizzarci sull’ isoforma R.
1
Tale isoforma è il prototipo della famiglia ed è la prima per cui è stata riportata
l’interazione con ICln (Tang et al., 1998) e quella per cui i siti di interazione sono
stati meglio caratterizzati. L’indagine è stata estesa a due tipi di isoforme di 4.1R:
una ad alto peso molecolare, (4.1RII variante lunga) e una a basso peso molecolare
(4.1Rsh, variante corta), che sono clonate da cellule embrionali umane HEK-293
Phoenix. Entrambe le isoforme sono state inserite in vettori per la FRET (pEYFP-N1 e
pEYFP-C1) inoltre abbiamo utilizzato due plasmidi che consentono l’espressione
della proteina ICln come proteina di fusione con il fluoroforo CFP all’ N-terminale o
al C-terminale. Gli esperimenti di FRET sono stati condotti per ogni configurazione
possibile per entrambe le isoforme dando risultati molto diversi a seconda delle
coppie donatore-accettore utilizzate, suggerendo che l’organizzazione spaziale del
complesso 4.1R-ICln sembra essere piuttosto rigidamente orientata nello spazio;
tuttavia possiamo dire che il segnale di FRET più forte l’abbiamo verificato tra la
4.1Rsh e ICln. Questi esperimenti sono stati affiancati da esperimenti di
immunoprecipitazione in cui sono stati utilizzati gli stessi plasmidi in cui sono
inserite le due isoforme della 4.1R (YFP-N1 4.1Rsh o YFP-C1 4.1Rsh; YFP-N1 4.1RII o
YFP-C1 4.1RII) utilizzate per gli esperimenti di FRET e la proteina ICln a cui era legato
un Flag-tag all’estremità C-terminale. Tali esperimenti hanno evidenziato che
entrambe le isoforme 4.1Rsh e 4.1RII interagiscono con ICln. Inoltre i dati di FRET
(utilizzando sia la metodica dell’Acceptor Photobleaching che della Sensitized
emission) indicano che, perlomeno per l’isoforma corta con il YFP fuso all’Nterminale della 4.1Rsh (YFP-4.1Rsh) l’ipotonia aumenta i valori di efficienza di FRET
e quindi presumibilmente l’interazione tra 4.1Rsh e ICln.
Gli esperimenti sulla interazione sono stati affiancati dallo studio dell’influenza
reciproca delle due proteine sulla loro localizzazione subcellulare. I distretti cellulari
su cui abbiamo focalizzato la nostra attenzione sono il nucleo e la membrana.
L’osservazione delle immagini di microscopia confocale ottenute per gli esperimenti
di FRET, gli studi di immunofluorescenza e i western blot hanno evidenziato che
quando la 4.1Rsh è co-espressa con ICln il segnale di entrambe diminuisce
notevolmente nel nucleo. Analogamente alla 4.1Rsh gli esperimenti di western blot
2
ci hanno indicato che quando la proteina 4.1RII e ICln sono co-espresse il loro
segnale nel nucleo diminuisce notevolmente. Gli studi di localizzazione effettuati
sulla membrana hanno dimostrato inoltre che quando è overespressa ICln il segnale
in membrana di entrambe le isoforme della proteina 4.1R diminuisce
significativamente.
Per capire il significato funzionale dell’interazione ICln-4.1R nella regolazione della
corrente per il cloruro ICl,swell abbiamo allestito esperimenti di patch clamp in
configurazione whole cell in cellule HEK per studiare l’effetto della over-espressione
delle due varianti da splicing della 4.1R, 4.1Rsh e 4.1RII sulla corrente attivata dallo
swelling; inoltre abbiamo effettuato studi di western blot sulla localizzazione in
membrana della proteina 4.1R sottoposta ad uno stimolo ipotonico. Dagli
esperimenti di western blot è emerso che l’isoforma lunga diminuisce
significativamente in membrana dopo che è stata sottoposta a stimolo ipotonico
mentre l’isoforma corta ha solo una tendenza alla diminuzione che non risulta però
significativa. Per quanto riguarda l’aspetto funzionale dagli esperimenti di patchclamp abbiamo verificato che le due isoforme 4.1Rsh e 4.1RII sembrano avere un
effetto diverso sulla corrente di cloruro attivata durante l’ipotonia. L’isoforma corta
determina un aumento significativo della ICl,swell a diversi potenziali (+ 100mV,
+80, mV +60 mV e a –100mV, -80mV e -60 mV) rispetto alla condizione di controllo,
mentre per l’isoforma lunga non abbiamo misurato alcun effetto sulla corrente. E’
possibile ipotizzare che la diversa azione della 4.1RII e della 4.1Rsh, sulla corrente
ICl,swell possa dipendere dalla presenza della regione unica U1, l’unica regione che
differenzia la 4.1Rsh da 4.1RII.
Infine abbiamo iniziato una serie di esperimenti volti a valutare se l’overespressione di ICln potesse alterare la proliferazione di cellule over-esprimenti 4.1R.
In questa fase di studio abbiamo scelto di focalizzare la nostra attenzione
sull’isoforma corta (4.1Rsh) della proteina 4.1R e in futuro approfondiremo questo
studio anche per l’isoforma lunga (4.1RII). Ciò che abbiamo osservato è che nella
condizione in cui le cellule sono co-transfettate con 4.1Rsh e ICln i valori a 48h e a
72h sono significativamente maggiori rispetto alla condizione di controllo (cellule
3
transfettate con YFP-4.1Rsh). In conclusione possiamo dire che entrambe le
isoforme 4.1Rsh e 4.1RII interagiscono con ICln; per quanto riguarda l’isoforma
corta abbiamo verificato che l’interazione con la 4.1Rsh aumenta in ipotonia e tale
isoforma ha un effetto diverso sulla corrente rispetto alla 4.1RII in quanto
determina un aumento della ICl,swell a diversi potenziali. Questo differente
comportamento delle due isoforme potrebbe essere dovuto a diversi fattori. Per
quanto riguarda la localizzazione le due proteine sembrano influenzare
reciprocamente la loro localizzazione subcellulare e probabilmente anche la loro
funzione in quanto per entrambe le proteine le differenti funzioni che svolgono in
cellula sono state correlate alle localizzazione (Calinisan et al., 2006; Fürst et al.,
2006).
4
2 Introduzione
2.1 La regolazione del volume cellulare
La membrana plasmatica delle cellule animali è, di norma, permeabile all’acqua e
dotata di bassa rigidità; l’acqua si muove essenzialmente attraverso un processo
diffusivo garantito dalla differenza di osmolarità tra ambiente intracellulare ed
extracellulare (Lang et al., 1998).
Le cellule, oltre a mantenere un volume costante in condizioni basali, sono in grado
di controbilanciare le perturbazioni del proprio volume, che potrebbero alterare lo
stato fisiologico della cellula causando gravi danni e perfino indurre degenerazione
cellulare.
I cambiamenti del volume influenzano numerosi processi cellulari quali i trasporti
ionici (Lang et al., 2007), la maturazione degli eritrociti, la proliferazione e la morte
cellulare, il differenziamento, l’espressione genica l’ipertrofia e l’apoptosi (Lang et
al., 1998a). La migrazione cellulare e i cambiamenti della forma della cellula
richiedono un adattamento del volume cellulare (Lang et al., 1998a). Le variazioni di
volume quindi influenzano il metabolismo cellulare (Lang et al., 1989) e questo
fenomeno è particolarmente evidente nel fegato, dove lo swelling della cellula è
accompagnato da un aumento della sintesi proteica e del glicogeno mentre lo
shrinkage cellulare provoca degradazione proteica e del glicogeno (Häussinger et
al., 1996; Häussinger et al., 1998).
Le variazioni del volume si possono verificare sia in condizioni fisiologiche, come
durante il differenziamento e la divisione cellulare (Hoffman & Simonsen, 1989),
che patologiche, per squilibrio osmotico tra comparto intracellulare ed
extracellulare causato da un allontanamento dalla normale condizione di isoosmolarità, pari a 308 mOsm nei mammiferi, oppure in seguito all’esposizione delle
cellule a soluzioni extracellulari ipotoniche o ipertoniche.
Anche in condizioni fisiologiche alcune cellule sono sottoposte a mezzi cellulari
anisosmotici o ad osmolarità variabile, ad esempio, le cellule epiteliali intestinali e le
5
cellule del sangue che attraversano i capillari intestinali sono esposte ad ipotonicità,
dopo eccessiva introduzione di H2O. Sempre in condizioni fisiologiche,
l’anisosmoticità tra compartimento intra- ed extracellulare può derivare da
alterazioni della composizione del citoplasma, a causa di attività di trasporto che
alterano la concentrazione intracellulare d’acqua o di soluti osmoticamente attivi.
Questa situazione si realizza tipicamente in epiteli assorbenti o secernenti, in
concomitanza con fenomeni di trasporto a carico degli enterociti, delle cellule
tubulari renali o degli epatociti, in relazione all’uptake attivo di soluti, quali zuccheri
od aminoacidi, a carico delle cellule ghiandolari, in relazione a secrezione di fluido o
elettroliti. Infine come abbiamo già visto anche eventi metabolici possono condurre
ad un rapido aumento nel contenuto cellulare di soluti organici, questo può
avvenire nel muscolo scheletrico durante l’esercizio, negli epatociti durante la
glicogenolisi, negli adipociti durante la lipolisi (Hoffmann et al., 2009). Inoltre,
perturbazioni osmotiche si osservano durante la mitosi o la proliferazione cellulare,
il differenziamento cellulare e l’azione ormonale (Hoffmann & Simonsen, 1989).
Numerose sono anche le condizioni patologiche in cui si registra squilibrio osmotico.
Tra queste si trovano ipossia/ischemia (Okada et al., 1997), iponatremia (che
insorge quando le funzioni ormonali e renali sono sbilanciate), ipotermia (con
l’inibizione
della
pompa
Na+K+ATPasi),
incremento
della
concentrazione
extracellulare di K+, acidosi/diabete intracellulare: in tutti questi casi si verifica
swelling cellulare, mentre lo shrinkage della cellula avviene ad esempio durante
ipernatremia (dovuta da eccessivo riassorbimento di Na+ o perdita di acqua),
riduzione della concentrazione di K+ intracellulare iperglicemia e alcalosi (Lang et al.,
1998); (Hoffmann et al., 2009).
Cambiamenti nell’equilibrio osmotico tra i compartimenti intra- ed extracellulare
possono causare gravi danni alle cellule, poiché la permeabilità delle membrane
cellulari all’acqua è sufficientemente alta da permettere un rapido movimento di
questa. Si è evoluto, quindi, un meccanismo regolatorio ubiquitario, che prende il
nome di regolazione del volume cellulare, basato sul fine controllo dei trasporti
ionici finemente controllati (Gschwentner et al., 1995).
6
In condizioni di ipotonicità extracellulare o ipertonicità intracellulare si crea un
flusso di acqua verso il citoplasma che provoca rigonfiamento della cellula, mentre
l’ipertonicità extracellulare o l’ipotonicità intracellulare portano all’uscita di acqua
dalla cellula e quindi ad una riduzione del suo volume.
In caso di rigonfiamento le cellule attivano un meccanismo regolativo chiamato
decremento regolatorio del volume (RVD: Regulatory Volume Decrease) che
permette un recupero del volume cellulare tramite l’uscita di acqua dalla cellula;
mentre in caso di riduzione del volume cellulare il meccanismo attivato dalla cellula
è detto incremento regolatorio del volume (RVI: Regulatory Volume Increase) che
determina un ingresso di acqua in cellula.
Nelle cellule animali, che, differentemente dalle cellule vegetali, sono prive di una
parete esterna, l’attivazione dell’RVD è l’unico meccanismo disponibile per
preservare le funzioni vitali in seguito a rigonfiamento cellulare (Fürst et al., 2002).
Figura 1 Meccanismi di trasporto coinvolti nella regolazione del volume cellulare. RVD = regulatory
volume decrease; RVI = regulatory volume increase. Sono indicati sia meccanismi di trasporto
+
+
+
+
+
elettroneutri (cotrasporto K -Cl , cotrasporto K -Na -2Cl , doppio scambio Na /H , Cl /HCO3 ) che
+
canali ionici per il K e il Cl .
7
2.2 Sensori e modulatori delle variazioni del volume cellulare
Tutti i meccanismi sensori dell’osmolarità nelle cellule di mammifero non sono stati
fino ad oggi chiariti. In questo paragrafo approfondiremo e focalizzeremo
l’attenzione sui diversi sensori e modulatori che intervengono nell’RVD.
Possiamo schematizzare i diversi fattori che intervengono in questo meccanismo:
a-Recettori e proteine di adesione: comprendono integrine, recettori per fattori di
crescita (GFRs), recettori delle citochine e i recettori calcio-sensibili. Le integrine
appartengono ad una famiglia altamente conservata di molecole di adesione
eterodimeriche che connettono la matrice extracellulare con proteine segnale
intracellulari e il citoscheletro. Oltre ad intervenire nella trasduzione del segnale
meccano-sensoriale e nel “signaling” dei fattori di crescita (Aplin et al., 1998) ci
sono numerose evidenze del loro coinvolgimento come sensori del volume cellulare
(Haüssinger et al., 2003) durante lo swelling e lo shrinkage cellulare (Häussinger et
al., 2003; Moeckel et al., 2006).
Secondo Browe e Baumgarten (Browe & Baumgarten, 2003; Browe & Baumgarten,
2004) lo “strech” delle integrine sarebbe seguito dall’attivazione di una cascata di
segnali che coinvolgono FAK; Src, recettori EGF, la PI3K e Rac che portano
all’attivazione dei canali VRAC.
Anche i recettori per i fattori di crescita vengono attivati dallo swelling cellulare e
questo porta all’attivazione di numerosi patways che coinvolgono PI3K-PKB,
MEK1/2-ERK1/2 e un flusso di taurina che porta alla regolazione del volume (Franco
et al., 2004).
b-Canali TRP: i canali TRP (Transient Receptor Potential Channels) sono dei sensori
polimodali di una grande varietà di stimoli chimici e fisici (Nilius et al., 2007;
Pedersen et al., 2005). I TRP interagiscono con molti elementi coinvolti
nell’osmoregolazione tra cui PLCϒ-1 (Van Rossum et al., 2005), PtdIns(4,5)P2,
calmodulina, caveolina1 AQP5, numerose proteine scaffold, proteine chinasi e
8
proteine citoscheletriche (Pedersen et al., 2005; Nilius B et al., 2007). Un esempio
del loro coivolgimento nell’attivazione dello swelling cellulare lo possiamo osservare
nelle cellule epiteliali della ghiandola salivare dove l’interazione TRPV4 e AQP5 è
necessaria per l’attivazione indotta dello swelling (Liu et al., 2006).
Anche se non è ancora stato del tutto chiarito il ruolo dei TRP nella regolazione del
volume, si pensa che essi medino un aumento dell’influsso di calcio in cellula che
successivamente stimola l’RVD con l’attivazione di canali di K+ e Cl- Calcio-attivati
(Numata et al., 2007).
c-Fosfolipasi, Fosfatidilinositoli kinasi e Fosfolipasi C e D: le fosfolipasi (PLA2)
idrolizzano i glicerofosfolipidi con conseguente rilascio di acido arachidonico che è
un importante regolatore di numerosi meccanismi intracellulari; ad esempio puo’
attivare direttamente meccanismi di trasporto per il K+ e osmoliti organici (Ordway
et al., 1991). Anche se non è ancora stato chiarito il meccanismo di regolazione
dell’attività della PLA2 dopo stress osmotico, evidenze farmacologiche in cellule EAT
hanno dimostrato che il rilascio di acido arachidonico indotto da swelling cellulare e
l’RVD implicano l’attivazione delle PLA2 (Thoroed et al., 1997).
Durante gli stress osmotici in numerosi lavori è stata osservata una variazione dei
livelli di molti fosfatidilinositoli (Sbrissa et al., 2005; Van der Kaay et al., 1999;
Hoffmann et al., 2000). In particolare in una grande varietà di cellule dopo stress
osmotico si assiste a variazioni importanti dei livelli di PtdIns(4,5)P2: durante lo
shrinkage cellulare si osserva una aumento dei livelli di questo fosfoinositide mentre
durante lo swelling si verifica una diminuzione (Nielsen et al., 2007). Il
mantenimento dell’omeostasi cellulare dei fosfatidilinositoli è garantito dalla
fosfainositol fosfato chinasi (PIPKs), PIP fosfatasi, e la fosfolipasi C (PLC) (Oude
Weernink et al., 2004; Takenawa et al., 2001); diversi lavori hanno dimostrato come
lo stress osmotico generi una regolazione dell’attività di PIPks (Yamamoto et al.,
2005), PIP fosfatasi e PLC (Nam et al., 2007) che provoca una variazione dei livelli di
PtdIns(4,5)P2. Durante lo stress osmotico i PtdIns(4,5)P2 regolano molti canali ionici
e trasportatori (ad esempio NHE1, VRAC, ENaC) (Voets et al., 2007; Suh et al., 2005;
9
Hilgemann et al., 2001) inoltre hanno un ruolo centrale nella riorganizzazione del
citoscheletro.
Tutti i fosfatidilinositoli fosforilati in posizione 3 sono sintetizzati dalla
fosfatidilinositolo 3 chinasi (PI3K), capace di fosforilare l’inositolo in posizione D3
(Toker et al., 1998). Esistono in letteratura vari lavori che collegano l’attività di
questo enzima con quella dell’RVDC (Regulatory Volume Decrease Channel;
argomento approfondito nel paragrafo successivo). E’ stato ipotizzato per la PI3K un
ruolo come sensore del rigonfiamento cellulare, in quanto la fosforilazione del PI in
posizione 3 dell’anello, per mezzo della PI3K, dipende fortemente dalla curvatura
della membrana, suggerendo che l’attività di questo enzima possa essere regolata
dalla deformazione meccanica della membrana stessa in seguito a variazioni del
volume (Hubner et al., 1998).
Non solo, ma per alcuni PtdIns sono state riportate variazioni a seguito di
alterazione dell’equilibrio osmotico cellulare.
Infatti, il PtdIns(4,5)P2, maggior regolatore dell'organizzazione dell'F-actina, subisce
un decremento in condizioni di cell swelling e al contrario un aumento durante cell
shrinkage (Klausen et al., 2006). Si può ipotizzare pertanto il coinvolgimento di tale
fosfatidilinositolo nei processi di RVD e di RVI, sebbene sia stato anche riportato che
esso non sembra essere implicato nell’attivazione della corrente innescata dallo
swelling (Klaussen et al., 2006).
Non solo, ma anche l'enzima (PIK-five), deputato alla biosintesi del PtdIns(5)P,
introdotto recentemente all'interno della famiglia dei fosfoinositidi, si pensa possa
essere coinvolto nelle risposte cellulari allo stress osmotico (Sbrissa et al., 2002).
A suffragio di questa ipotesi è il risultato ottenuto da esperimenti in fibroblasti e
adipociti 3T3-L1: quando le cellule vengono sottoposte a shock iposmotico, i livelli di
PtdIns(5)P diminuiscono (Sbrissa et al., 2002).
Il PI(3,5)P2 sembra essere un buon candidato come molecola segnale, poichè in
condizioni basali rappresenta solo lo 0,2% dei PtdIns totali di membrana e quindi
piccole variazioni nella sua concentrazione potrebbero costituire un segnale
inequivocabile per la cellula. Recentemente è stato scoperto che la concentrazione
10
di questo lipide aumenta in seguito a stress iperosmotico in cellule di
Saccharomyces cerevisiae (Dove et al., 1997) e si ritiene che possa svolgere un ruolo
importante nella regolazione del volume cellulare (Rudge et al., 2004). Al contrario,
in cellule di scimmia Cos-7 è lo stress iposmotico che stimola la sintesi di
PtdIns(3,5)P2, catalizzata dalla PtdIns3P 5-OH chinasi (Dove et al., 1997).
Anche il PtdIns(3,4,5)P3 è stato messo in relazione agli stress osmotici: la sua
concentrazione in membrana infatti aumenta in seguito a stimolazione con mezzo
ipotonico (Feranchak et al., 1999).
d I livelli di calcio intracellulare: i livelli di calcio intracellulare sembrano influenzare
in modo variabile l’RVD. In alcuni tipi cellulari, tra cui cellule epiteliali intestinali,
cellule SiHa (human cervical cancer cells), e le cellule epiteliali della cornea
(MacLeod et al., 1999; Shen et al., 2001; Uchida et al., 1993) il rigonfiamento
cellulare provoca un aumento della concentrazione di calcio intracellulare mentre in
altri citotipi questo parametro rimane invariato. L’aumento di calcio è dovuto sia
all’ingresso di calcio dall’esterno –via SAC (Strech activated channels), VOC (Voltage
operated canne), SOC (store-operated Ca2+ channel) e lo scambiatore Na+/Ca+ sia al
rilascio dagli stores intracellulari (Hazama et al., 1990). Si ritiene che questo
fenomeno possa sostenere l’RVD in diversi modi: stimolando i canali del K + attivati
dal Ca2+, determinando il rimodellamento del citoscheletro o promuovendo
l’esocitosi di osmoliti (McCarty &O’Neil, 1992).
Prove indirette indicano che il calcio è estruso dalla cellula durante l’RVD, attraverso
la Ca2+/cationi polivalenti (CaR). Quest’ultimo determina un aumento del cAMP
intracellulare proteina G-mediato e alla conseguente attivazione/facilitazione
dell’RVDC (Okada et al., 2006).
e pH intra ed extracellulare: nelle cellule di Ehrlich l’RVD è rallentato in presenza di
soluzioni extracellulari a pH acido. Le variazioni di pH extra- e intra-cellulare in
conseguenza di uno stress ipotonico modulano la corrente di K + volume-sensible,
modificando il numero di canali e anche le proprietà conduttive degli stessi (Duprat
11
et al., 1997). La maggior parte delle cellule risponde al rigonfiamento con un
acidificazione intracellulare che puo’ influenzare il metabolismo, quindi la
concentrazione dei metaboliti e che determina la protonazione degli aminoacidi
carichi negativamente implicati nel bilancio osmotico dell’RVD (Hoffmann &
Dunham, 1995). Le alterazioni del pH possono anche influire sulla selettività di
alcuni canali ionici.
f Protein chinasi e “small GTPasi”: eventi fosforilativi possono essere da un lato
attivatori diretti dei canali coinvolti nell’RVD e dall’altro possono influenzare
l’attività e la conformazione di altre proteine coinvolte nel processo, come ad
esempio proteine citoscheletriche. In differenti citotipi la protein chinasi C (PKC)
può avere effetti stimolanti, inibenti o neutri sui canali attivati dal rigonfiamento.
Lo stress osmotico può attivare inoltre alcune tirosin chinasi (come Pyk2, Fak, Lck o
Syk) e MAP chinasi; quest’ultime prevalentemente in una fase più tardiva della
regolazione del volume (Hoffmann et al., 2009). Anche le proteine che legano il GTP
sono coinvolte nell’RVD e un ruolo piuttosto importante sembra essere svolto dalla
famiglia delle Rho-GTPasi e dai loro effettori, che influenzano la riorganizzazione del
citoscheletro di actina (Tamma et al., 2007; Nilius et al., 2001).
In alcuni tipi cellulari è necessaria una fosforilazione da parte delle chinasi
Ca2+/calmodulina dipendenti per determinare l’attivazione dei canali per il K+ e per il
Cl- coinvolti nell’RVD (Jakab et al., 2002).
Infine, anche la PI3K sembra essere importante per l’attivazione dei canali RVDC
come già discusso nel paragrafo precedente.
g-Il citoscheletro: un ruolo fondamentale nella modulazione dell’RVD sembra essere
svolto dal citoscheletro, infatti in una grande varietà di citotipi il rigonfiamento
determina il rimodellamento del citoscheletro. Inoltre si è osservato che in molti tipi
cellulari l’RVD è inibito dalle citocalasine, che impediscono la polimerizzazione
dell’actina (Lang et al., 1998; Moustakas et al., 1998) e in generale si ritiene che
qualunque tipo di inibizione della riorganizzazione del citoscheletro actinico, sia
12
importante per l’attivazione degli RVDC. Sebbene i meccanismi che legano
riarrangiamento del citoscheletro e attivazione dei canali non siano ancora chiariti
del tutto, nella maggior parte dei tipi cellulari studiati (anche se non in tutti), lo
swelling cellulare è associato a una globale riduzione del contenuto di F-actina
(Pedersen et al., 1999). E’ stato anche proposto è che l’induzione della ICl,swell
richieda la depolimerizzazione di alcuni pull di actina (come quella periferica) ma
non di altri (come quelli di F-actina perinucleare) (Klausen et al., 2006)
Il citoscheletro di actina è di fondamentale importanza per la generazione e il
mantenimento della morfologia cellulare, per l’endocitosi e l’esocitosi, per la
motilità e la divisione cellulare (Stossel et al., 1984), per il controllo dell’azione di
diversi ormoni (Hall et al., 1984) e per la localizzazione specifica di proteine integrali
nella membrana plasmatica (Bennett & Lambert, 1991). Tanti monomeri globulari
(actina G), ciascuno dei quali è costituto da un unico polipeptide del peso
molecolare di circa 42 kDa, formano polimeri, che a loro volta costituiscono i
filamenti di actina. In condizioni fisiologiche l’actina G polimerizza spontaneamente
a costituire filamenti lunghi e rigidi di alcuni mm di lunghezza senza che il processo
richieda energia esterna (Pollard & Cooper, 1986). L’assemblaggio e il
disassemblaggio dei filamenti di actina, così come la loro organizzazione in strutture
funzionali più complesse, sono regolati da numerose proteine che legano l’actina
(ABP: Actin Binding Proteins), come la gelsolina, la profilina e la cofilina, che si
legano alle estremità dei filamenti di actina, impedendo l’aggiunta di nuove
subunità alle estremità neoformate e stabilizzando il filamento di actina stesso. In
cellule umane di melanoma è stato ben dimostrato il coivolgimento delle ABP
nell’RVD, la loro assenza porta infatti all’inibizione dell’RVD tramite l’inattivazione
dei canali per il potassio attivati in seguito al rigonfiamento (Jakab et al., 2002).
L’actina filamentosa (F-actina), molto concentrata a livello submembranario,
sembra essere un buon candidato tra quelli che potrebbero mediare un controllo
nella regolazione del volume cellulare e nelle variazioni morfologiche ad esso
connesse (Mitchinson & Cramer, 1994): il rigonfiamento cellulare porta infatti ad un
rimodellamento del network di actina presente al di sotto della membrana
13
plasmatica. Mediante l’utilizzo di metodiche biochimiche si è potuto evidenziare
come, nel complesso, la struttura dell’actina filamentosa venga disorganizzata
durante il rigonfiamento, processo a cui fa seguito una riorganizzazione
citoscheletrica che coincide temporalmente con l’RVD (Cornet et al., 1993; Hallows
et al., 1996; Mills et al., 1994) e che comporta anche la formazione di segmenti di Factina, sotto forma di spikes, localizzati al di sotto della membrana plasmatica
(Tamma et al., 2007; Nilius et al., 2007).
La riorganizzazione del citoscheletro actinico che coincide con il cambiamento del
volume cellulare sembrerebbe rivestire un ruolo cruciale nel corso dell’RVD,
modulando l’attivazione di diversi meccanismi di trasporto (Kleinzeller & Ziyah,
1990; Mills et al., 1994; Hoffman, 1997; Cantiello, 1997). Tuttavia non si è ancora
compreso pienamente il meccanismo alla base di questi processi.
Si ritiene che il citoscheletro possa essere coinvolto nella regolazione del volume
cellulare in vari modi:
potrebbe funzionare da sensore del volume trasducendo la
deformazione meccanica della membrana, indotta dal rigonfiamento,
ai meccanismi di regolazione del volume cellulare (Jakab et al., 2002).
E’ stato infatti suggerito che il network di filamenti di actina potrebbe
conferire alle invaginazioni presenti normalmente a livello della
membrana plasmatica una resistenza meccanica alla deformazione;
durante il rigonfiamento queste invaginazioni si distenderebbero,
rimodellando in tal modo il citoscheletro e attivando l’RVD (Okada et
al., 1997);
potrebbe modificare direttamente l’attività dei canali RVDC e dei
trasportatori coinvolti nell’RVD, modulando la funzionalità di questi
trasportatori, oltre a garantirne la localizzazione in specifiche regioni
della membrana. E’ stato dimostrato che il citoscheletro può regolare
alcuni canali del Na+ in cellule epiteliali (Cantiello et al.,1991), la
pompa Na+/K+ ATPasi (Nelson & Hammerton, 1989; Cantiello et al.,
14
1997), l’antiporto Na+/H+ (Smith et al., 1991; Watson et al., 1992), lo
scambiatore Cl-/HCO3- (Drenckhahn et al., 1985), il cotrasporto Na+K+-2Cl- (Jorgensen et al., 1984), i canali del Ca2+ nei neuroni (Johnson
& Byerly, 1993; Rosenmund & Westbrook, 1993), alcuni canali del Cltra cui quelli responsabili della ICl,swell e il canale anionico CFTR
(Prat et al., 1995);
potrebbe modulare le vie di attivazione e di conseguenza le
concentrazioni di messaggeri secondari coinvolti nella trasduzione
del segnale durante l’RVD. A tal proposito l’actina sembrerebbe
funzionare come un magazzino intracellulare di Ca2+, rilasciato in
seguito
a
depolimerizzazione
dell’F-actina;
inoltre
il
suo
rimodellamento è strettamente correlato all’ attivazione di numerosi
secondi messaggeri tra cui small GTPasi (Rho- Rac -cdc42) MAPkinasi
e FAk kinasi, per cui è riportato un ruolo nell’attivazione della
ICl,swell.
l’actina submembranaria potrebbe inoltre essere considerata come
una vera e propria barriera fisica per la diffusione, impedendo il
flusso di acqua che si attiva per osmosi, soprattutto nelle cellule
dotate di microvilli (Jakab et al., 2002);
potrebbe regolare l’esocitosi di osmoliti organici e probabilmente di
ATP (Shuba et al., 2000; Van der Wijk et al., 2000);
il rimodellamento del citoscheletro potrebbe creare gradienti
osmotici locali, che potrebbero favorire o impedire lo spostamento di
acqua, e generare forze meccaniche promuovendo l’interazione tra
actina e miosina (Jakab et al., 2002);
le proprietà elettrosmotiche dell’actina sembrerebbero influenzare la
conduzione dei segnali elettrici nel compartimento intracellulare;
una spiegazione alternativa attribuirebbe all’actina la regolazione
dell’inserzione o meno in membrana dei trasportatori contenuti in
vescicole. La riorganizzazione dell’actina osservata durante i
15
cambiamenti del volume cellulare sarebbe cruciale per l’attivazione
e/o inserzione dei trasportatori in membrana. Questo processo
potrebbe essere mediato da proteine che, tramite il legame
all’actina, vanno a regolare la polimerizzazione e l’organizzazione
strutturale della stessa. Almeno per quanto riguarda l’attivazione
della corrente ICl,swell non sembra però essere implicata l’inserzione
di canali per fusione di vescicole con la membrana plasmatica. In
esperimenti di patch-clamp in configurazione whole-cell non si
osserva, inoltre, un incremento della capacità della membrana (Ross
et al., 1994; Jakab et al., 2002);
un’ultima ipotesi sostiene che l’effetto dell’actina sia mediato da
proteine ad essa legate appartenenti alla famiglia delle proteine ERM
(Ezrin/Radixin/Moesin) o delle ABP (Actin Binding Proteins). Le
proteine ERM, in seguito ad interazione dei loro domini PH
(Pleckstrin Homology) con i fosfatidilinositoli di membrana (PtdIns o
PI) permettono l’interazione indiretta tra l’actina e la membrana
plasmatica. L’idrolisi del PtdIns(4,5) ad opera della fosfolipasi C (PLC),
porta ad una depolimerizzazione dell’actina e ad un rilascio delle
ABP; un aumento di PtdIns(4,5), invece, induce la polimerizzazione.
L’idrolisi del PtdIns(4,5) porta alla formazione di due secondi
messaggeri: il diacilglicerolo (DAG) e il PtdIns(1,4,5) entrambi
coinvolti nella regolazione del citoscheletro. Le ERM potrebbero
quindi essere molto importanti nell’accoppiamento tra l’actina
citoscheletrica e la membrana (inclusi i meccanismi di trasporto) e
probabilmente nel processo dell’RVD in quanto è già stato
dimostrato che sono influenzate dale perturbazioni osmotiche
(Ciano-Oliveira et al., 2005; Rasmussen et al., 2008)
16
2.3 L’RVD
Le strategie usate per controbilanciare il rigonfiamento cellulare variano nei diversi
organismi, tessuti e tipi cellulari. Molti organismi unicellulari costruiscono una
parete cellulare per proteggersi da stress osmotici. Negli eucarioti invece
l’attivazione dell’RVD è l’unica difesa disponibile, poiché le loro cellule non possono
sostenere un’elevata pressione idrostatica. I meccanismi messi in atto durante
l’RVD hanno il compito di diminuire l’osmolarità intracellulare, avverrà cosi’ per
osmosi, una fuoriuscita di acqua dalla cellula, in modo da controbilanciare l’acquisto
iniziale e far recuperare alla cellula stessa il volume originario.
Figura 2.
2.3.1 Meccansimi di trasporto coinvolti nel’RVD
Durante l’RVD si attivano diversi meccanismi di trasporto (Fig. 1) che determinano
l’uscita dalla cellula di sostanze osmoticamente (Fig. 2) attive: KCl, osmoliti organici
e, per osmosi, di acqua. Questo meccanismo, opponendosi all’incremento delle
dimensioni cellulari, riporta il volume alle condizioni iniziali.
I meccanismi di trasporto attivati durante l’RVD in diversi tipi di cellule, possono
essere (Hoffmann e Simonsen, 1989):
conduttanze separate per K+e Cl-: sono i principali sistemi di trasporto attivo
durante l’RVD (linfociti, cellule endoteliali, fibroblasti, cellule dell’intestino,
cellule MDCK e cellule del tubulo convoluto prossimale) (McCarty & O’Neil,
1992; Lang et al., 1998). Tra i canali del K+ clonati fino ad oggi, un ruolo nella
17
regolazione del volume è stato attribuito a Kv1.3, Kv1.5 e a minK (Lang et
al., 1998; Stutzin et al., 2006; Hoffmann et al, 2009). Molti studi hanno
stabilito che il canale anionico attivato dal rigonfiamento è aselettivo e
permette il passaggio non solo di Cl-, ma anche di HCO3-, anioni organici e
osmoliti organici neutri; l’identità molecolare di questo canale non è stata
però ancora individuata ed è tuttora fonte di acceso dibattito;
cotrasporto elettroneutro K+-Cl-: è il sistema di trasporto utilizzato più di
frequente, oltre ai canali ionici, per l’efflusso di KCl; sembra essere attivato
preferenzialmente in seguito a rigonfiamento isoosmotico (McCarty &
O’Neil, 1992; Hoffmann et al., 2009);
scambiatori K+/H+ e Cl-/ HCO3- funzionalmente accoppiati tra loro: alcune
cellule come i globuli rossi di Amphiuma perdono KCl utilizzando questo
meccanismo; l’H+ e l’HCO3- entrati in cellula si combinano formando CO2,
che diffondendo all’esterno, non risulta osmoticamente attiva (Hoffmann &
Simonsen, 1989; Lang et al., 1998; Hoffmann et al., 2009);
efflussi diffusionali di anioni organici come la taurina, il sorbitolo e
l’inositolo.
Quest’ultimo
fenomeno
risulta
essere
particolarmente
importante, in quanto esistono casi in cui la concentrazione di Cl- è ridotta e
può raggiungere l’equilibrio prima che il processo di RVD sia completato.
Numerose sono le prove a sostegno del fatto che l’efflusso di questi
osmoliti si realizzi comunque attraverso gli stessi canali anionici attivati
durante l’RVD (Hoffmann et al., 2009).
E’ stato inoltre dimostrato che, durante il rigonfiamento, la cellula va incontro ad
acidosi dovuta all’inibizione del trasportatore Na+/H+ e alla conseguente riduzione
dell’uscita di ioni H+ dal citoplasma all’ambiente extracellulare (MacLeod &
Hamilton, 1996).
Il principale meccanismo coinvolto nell’RVD prevede la contemporanea attivazione
dei canali ionici per K+ e Cl-. Studi condotti in cellule MDCK (Paulmichl et al., 1996)
hanno evidenziato che in seguito all’esposizione delle cellule ad un mezzo ipotonico,
si osserva inizialmente l’attivazione di canali per il K+ con conseguente
18
iperpolarizzazione del potenziale di membrana. La successiva attivazione dei canali
per il cloruro fa sì che a questa iperpolarizzazione seguisse una depolarizzazione.
A dimostrazione dell’importanza della conduttanza per il Cl- nell’omeostasi del
volume cellulare vi è il fatto che essa è presente in tutte le cellule di mammifero
(Burton et al., 1997). E’ stato inoltre provato che l’inibizione della corrente per il Cl ha come conseguenza l’incapacità della cellula di regolare il proprio volume.
2.3.2 La corrente per il Cl- attivata durante l’RVD: ICl,swell
Quando una cellula è esposta ad una soluzione extracellulare ipotonica, si attiva una
corrente anionica determinata dal rigonfiamento cellulare denominata ICl,swell
(Ritter et al., 2003).
Esperimenti di tipo elettrofisiologico non hanno permesso una caratterizzazione
biofisica univoca in tutte le cellule di un singolo canale per il cloruro coinvolto nella
regolazione del volume cellulare, ma sono stati osservati diversi canali per il cloruro,
variabili a seconda dei tipi cellulari esaminati e delle condizioni sperimentali
adottate.
Per spiegare l’eterogeneità riscontrata sono state formulate due ipotesi: la prima è
che diversi tipi cellulari esprimano diversi tipi di RVDC (Regulatory Volume Decrease
Channels/Currents), la seconda è che questi canali siano formati da un differente
assemblaggio di subunità identiche. I diversi fenotipi dipenderebbero dal numero di
subunità assemblate a costituire il canale funzionale in una data situazione.
Maggiore importanza è stata rivolta a questa ultima ipotesi in quanto potrebbe
giustificare la grande varietà di fenotipi osservati (Fürst et al., 2002).
Esperimenti di patch-clamp in configurazione whole-cell hanno mostrato che la
corrente ICl,swell, mediata da questi canali, possiede le seguenti caratteristiche:
rettificazione uscente (Okada, 1997);
veloce cinetica di attivazione (<1ms) (Lang et al., 1998);
lenta inattivazione tempo dipendente a potenziali di +40 mV o superiori
(Ackerman et al., 1994). Anche l’entità dell’inattivazione è variabile a seconda
del tipo cellulare e può inoltre essere modulata da differenti fattori quali: il pH e
la presenza di cationi bivalenti nel mezzo extracellulare. E’ stato proposto che, a
19
livello di singolo canale, questa inattivazione sia dovuta ad un progressivo
decremento nel numero di canali attivi (Okada, 1997);
Ca2+ indipendenza;
indipendenza dal voltaggio (Lang et al., 1998);
selettività prevalentemente anionica (SCN->I->Br->Cl->F->Gluconato) (Hoffmann
et al., 2009);
inibizione ad opera dei comuni bloccanti di canali anionici (SITS, NPPB, DIDS), ma
anche tramite nucleotidi (cAMP, cGMP, ATP) (Tsumura at al., 1996) ed analoghi
nucleosidici (AZT e acyclovir), che inibiscono in modo voltaggio indipendente la
ICl,swell (Meyer & Korbmacher, 1996; Gshwentner et al., 1995);
la possibilità di consentire il passaggio di osmoliti, anioni organici: esistono
prove sostanziali che questo canale medi il trasporto di aminoacidi, quali
taurina, glutammato, glutammina e glicina (Jackson & Strange, 1993; Jackson et
al., 1995).
Attivazione, regolazione e modulazione dell’ICl,swell
L’attivazione di ICl,swell, come ampiamente riportato in letteratura, non sembra
essere legata all’inserzione di canali in membrana in seguito ad esocitosi, in quanto
non è stato osservato un aumento della capacità della membrana in coincidenza
con l’attivazione della corrente. La conduttanza sembrerebbe aumentare per
effetto dell’attivazione di canali preesistenti (Graf et al., 1995; Meyer &
Korbmacher, 1996).
L’applicazione di “stretch” alla membrana non induce l’attivazione del canale; è
pertanto verosimile quindi che l’attivazione richieda l’intervento di secondi
messaggeri. Si sa, però, che l’attivazione della corrente del Cl- non richiede calcio
extracellulare e non è influenzata in modo apprezzabile dal calcio intracellulare
(Ackerman et al., 1994), a differenza di quanto osservato per i canali del K+ coinvolti
nell’RVD. Negli ultimi anni è stato investigato anche il ruolo di altri secondi
messaggeri come Mg++, cAMP, G-protein come cdc42 e Rac, PKC, PKI, ma ad oggi
20
non è ancora emerso un quadro univoco sul ruolo di questi fattori nell’RVD. Il ruolo
di altri secondi messaggeri (Mg++, cAMP, G-protein) non è ancora stato chiarito.
Nel caso della PKC ci sono numerosi pareri controversi: la PKC sembra infatti
svolgere un ruolo attivatorio o inibitorio a seconda del tipo cellulare considerato
(Jakab et al., 2001). In miociti di cane ad esempio, l’attivazione della PKC provoca
una stimolazione degli RVDC (Du et al., 1999), mentre in cellule HeLa i canali RVDC
non sono modulati da fosforilazioni della PKC (Jakab et al., 2001).
Un'altra chinasi che sembra evere un ruolo importante è la PI3K, visto che la sua
inibizione si traduce in una inibizione dell’attivazione della ICl,swell (Yamamoto et
al., 2009; Yamamoto et al., 2008). Recentemente è anche emerso un ruolo per small
GTpasi della famiglia Rho, come Cdc42 e Rac (Klausen et al., 2006; Tamma et al.,
2007) che sono coinvolte nella regolazione della ICl,swell. Tuttavia, la modulazione
di tale corrente da parte del pathway delle Rho GTPasi e del citoscheletro actinico è
piuttosto complessa e controversa. Per quanto riguarda la regolazione dei RVDCs da
parte delle small GTPasi, si ritiene che l’attivazione di tali canali indotta dallo
swelling richieda che la via di signalling associata alle Rho GTPasi sia funzionale, ma
non che ci sia una un’attivazione di tali proteine (Cartone t al., 2002). In altre parole,
è stato proposto che in generale questa via abbia un effetto facilitante sulla
corrente, ma che non sia sufficiente di per sé ad attivare i canali (Klausen et al.,
2006).
Un altro fattore importante è il citoscheletro: sia l’inibizione della polimerizzazione
dell’actina, così come il suo “disassemblamento” blocano l’attivazione del canale.
(Jakab et al., 2001). E’ stato di recente proposto che più che lo stato di
polimerizzazione dell’actina in generale, siano i diversi pools cellulari di F-actina
(corticale, associata alle fibre da stress, perinucleare) ad essere coinvolti nella
regolazione dei RVDCs, probabilmente con effetti diversi (Klausen et al., 2007; Wang
et al., 2005).
L’attivazione di ICl,swell non dipende da variazioni dell’osmolarità, è quindi possibile
che il canale, o una sua proteina accessoria, siano sensibili all’aumento del volume
cellulare. E’ stata avanzata un’ipotesi secondo la quale, in seguito a rigonfiamento
21
cellulare, la membrana, espandendosi, perderebbe una serie di invaginazioni
multiple, portando al riarrangiamento di interazioni tra citoscheletro di F actina,
proteine di membrana, canali del Cl- e proteine accessorie con funzione di “sensori”
del volume; questo riarrangiamento condurrebbe all’attivazione del canale (Okada,
1997).
2.4 ICln
ICln è una proteina multifunzionale, clonata nel 1992 (Paulmichl et al., 1992) dalle
cellule epiteliali MDCK (Madine Darby Canine Kidney), e altamente conservata lungo
la scala evolutiva. Essa è ubiquitariamente espressa in tutte le cellule ed è
fondamentale per la loro sopravvivenza. Infatti, il knockout di ICln effettuato in
nematode, topo e linee cellulari è letale, a dimostrazione del fatto che il suo ruolo è
essenziale per la vitalità dell’embrione già negli stadi precoci (Pu et al., 2000).
Il gene umano di tale proteina è localizzato sul cromosoma 11q13.5-14.1 ed è
controllato da un promotore attivo costitutivamente (Scandella et al., 2000), come
spesso succede per le proteine a funzione house-keeping.
ICln sembra ricoprire svariati ruoli notevolmente diversi tra loro, tra i quali quello di
maggior rilievo, a cui è stata primariamente associata, è il coinvolgimento nei
meccanismi di regolazione del volume cellulare e nell’attivazione dell’ICl,swell. Nel
corso degli anni, tuttavia, nuove importanti funzioni sono state ricondotte alla
proteina, via via che nuovi partners proteici venivano identificati.
2.4.1 Struttura di ICln
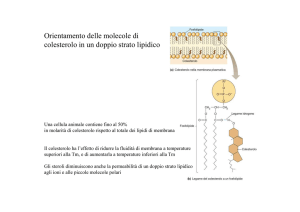
L’isoforma canina di ICln è formata da 235 amminoacidi prevalentemente acidi
(pI=3,8) e con un peso molecolare predetto sulla base della sequenza primaria di
26,5 kDa. Tuttavia, in elettroforesi su gel di poliacrilammide, condotta in condizioni
denaturanti, la proteina migra con una massa apparente di circa 37 kDa. Una simile
discrepanza non è dovuta a modificazioni post-traduzionali, dal momento che ICln
delle cellule eucariotiche, ICln espressa in batterio ed ICln sintetizzata in vitro in
22
lisato di reticolociti di coniglio migrano alla medesima altezza (Buyse et al., 1996;
Fürst et al., 2000). E’ plausibile che il ritardo elettroforetico sia dovuto alla forte
acidità della proteina.
Struttura primaria
La struttura primaria di ICln presenta tre regioni interessanti, denominate Acidic
Domains (AD1, AD2 e AD3); si tratta di porzioni di proteina di lunghezze variabili dai
7 ai 20 residui, a seconda della specie, e composte prevalentemente da
amminoacidi acidi (Fig. 3). E’ ipotizzato che tali domini svolgano un ruolo nelle
interazioni con altre proteine (Emma et al., 1998), come con JBP1 e 4.1.
Figura 3 Struttura primaria di ICln in cellule MDCK.
In giallo sono evidenziati tre Acidic Domains (AD1, AD2, AD3) e in azzurro la sequenza
GXGXG, potenziale sito di interazione con i nucleotidi (Paulmichl et al., 1992).
Mediante esperimenti di mutagenesi è stato possibile identificare nella sequenza
aminoacidica di ICln un potenziale sito di legame per i nucleotidi costituito da tre
residui di glicina alternati ad altri amminoacidi (GxGxGx, regione che si estende
dall’amminoacido 49 all’amminoacido 53 dell’isoforma canina (Fig.3)).
23
Struttura terziaria
Una delle ipotesi sul legame tra ICln e i canali attivati dallo swelling è che ICln
rappresenti il canale stesso o una sua parte (Paulmichl et al., 1996). In accordo con
questa ipotesi e sulla base della sua sequenza aminoacidica, è stata proposta nel
1992 un modello tridimensionale per la proteina ICln inserita in membrana
costituita da un foglietto β composto di quattro putativi filamenti β antiparalleli
(Paulmichl et al., 1992). Secondo quest’ipotesi, si potrebbe formare un omodimero
composto da due molecole di ICln (Fig. 4), le quali, apponendo i rispettivi foglietti β
uno contro l’altro, darebbero luogo ad un canale che attraversa il bilayer lipidico. In
base a questo modello sia l’estremità C-terminale sia quella N-terminale si
affacciano sul lato citoplasmatico. Tale struttura ipotetica è stata modellizzata
attraverso strumenti bioinformatici.
Figura 4 Visualizzazione laterale e dall’alto dell’ipotetico omodimero di ICln. Le catene delle due
molecole sono distinte per colore.
Sulla base di questo modello 3D la sequenza GxGxGx di legame per i nucleotidi,
verrebbe a trovarsi in prossimità dell’imboccatura esterna del putativo canale.
Mutando tale sequenza da GxGxGx a AxGxGx o AxAxAx si riduce notevolmente la
capacità dei nucleotidi di inibire la corrente mediata da ICln (Paulmichl et al., 1992).
24
Ad oggi il modello appena descritto non è stato validato tramite esperimenti
cristallografia o Risonanza Magnetica Nucleare (NMR). Tuttavia è stato possibile
risolvere la struttura dei primi 159 aminoacidi di ICln MDCK in forma idrosolubile,
dato comunque di estremo interesse visto che la proteina ha una localizzazione
prevalentemente citoplasmatica (Krapivinsky et al., 1994).
Esperimenti di Risonanza Magnetica Nucleare (NMR) hanno permesso di risalire alla
struttura tridimensionale di ICln in forma idrosolubile e hanno rivelato che la parte
N-terminale di ICln è organizzata in un PH-domain (Fürst et al., 2005; Schedlbauer et
al., 2003).
Figura 5 Struttura NMR di ICln156 solubile in acqua. a) sovrapposizione di 15 strutture finali di ICln
con relativa rappresentazione a “ribbon plot”; il loop tra β6 e β7 è in color magenta, i β-sheets sono
in ciano, e l’α-elica è in rosso.
Domini PH
Il dominio PH (Pleckstrin Homology) è un modulo di 100-120 residui aminoacidici
appartenente a diverse proteine, tra cui anche ICln.
E’ stato descritto per la prima volta nel 1993 da Haslam (Haslam et al., 1993; Mayer
et al., 1993) come un motivo che si ripete due volte nella plecstrina, una proteina
solubile presente solo in cellule ematopoietiche, dove è una delle proteine più
abbondanti ( 1% delle proteine totali) e costituisce il principale substrato della
protein chinasi C.
I domini PH sono stati evidenziati in centinaia di proteine coinvolte nel traffico
vescicolare, nel riarrangiamento del citoscheletro e nella trasduzione del segnale in
cellula (Lemmon et al., 2002), come le fosfolipasi, le proteine regolate dalle GTPasi,
25
le proteine chinasi, le proteine citoscheletriche e le proteine oncogeniche, dal
lievito ai mammiferi.
Il sequenziamento del genoma umano ha mostrato 252 proteine umane diverse
contenenti almeno un PH-domain, rendendolo così l’undicesimo dominio più
comune nell’uomo (Lemmon et al., 2002).
Grazie all’utilizzo della cristallografia e della NMR, la struttura di questi domini PH è
stata risolta e oggi si conoscono almeno 13 differenti tipi di struttura (Yoon et al.,
1994).
In ogni dominio PH il core strutturale è formato da sette filamenti , organizzati in
due foglietti antiparalleli affacciati l’uno contro l’altro a formare uno stretto barile,
seguiti da un’-elica C-terminale che si accosta all’estremità superiore del barile, in
posizione trasversale rispetto ad esso (Fig. 6). Un foglietto è formato da quattro βstrands (β1-β4), l’altro solo da tre (β5-β7). A due angoli i foglietti sono molto vicini
tra loro, mentre agli altri due angoli i due foglietti sono alla massima distanza.
Figura 6. Topologia dei domini PH. Si possono osservare anche tre Variable Loops (VL).
La parte inferiore è delimitata dai loops 1/2, 3/4 e 6/7, denominati
“Variable Loops”, rispettivamente VL1, VL2 e VL3, i quali sono di lunghezza variabile
a seconda della proteina ed in alcuni casi possono contenere strutture secondarie o
addirittura interi domini (Blomberg et al., 1997). L’alta variabilità di questi loops, sia
per lunghezza sia per composizione aminoacidica, insieme con la loro accessibilità
spaziale, è coerente con l’ipotesi che essi costituiscano un sito di legame per
eventuali ligandi (Lemmon et al., 2002).
26
Il dominio PH di ICln
Per risolvere la struttura di ICln per NMR, si è ricorsi all’utilizzo di un mutante tronco
(ICln-T477) comprendente solo i primi 159 amminoacidi della proteina, dal
momento che la coda C-terminale di ICln WT (Wild Type), dotata di grande mobilità,
rendeva difficile compiere l’assegnazione delle risonanze.
La topologia di ICln-T477, determinata attraverso NMR, corrisponde ad un PHdomain (Fürst et al., 2005; Schedlbauer 2003). Il dominio PH di ICln condivide la
medesima struttura dei PH-domains canonici: un barile composto di filamenti e
sovrastato da un’-elica trasversale.
All’interno dei domini PH, appartenenti alle diverse proteine, l’omologia di
sequenza è molto bassa, dal 7% fino al 30% nei casi migliori (per ICln è solo del 4%),
mentre la struttura tridimensionale è molto ben conservata, suggerendo così che
nel determinare la funzione di una proteina può essere molto più importante la
struttura tridimensionale piuttosto che la sequenza aminoacidica (Lemmon et al.,
2001).
Funzioni dei domini PH
Per i pochi domini PH di cui è stata dimostrata la funzione, si sa che alcuni di essi
sono responsabili della traslocazione di proteine presso la membrana tramite
associazione diretta con specifici fosfoinositidi (Lemmon et al., 2000) come il PtdIns
(3,4,5) e il PtdIns (4,5). Tuttavia questa potrebbe non essere l’unica funzione poichè
solo una piccola frazione dei domini PH, circa il 10%, mostra tale proprietà. Nel
restante 90% l’affinità verso i fosfoinositidi è bassa e meno specifica, suggerendo
così la possibilità che, per lo meno in alcuni casi, questo dominio metta in atto
strategie mirate ad aumentare l’affinità verso i fosfoinositidi, ad esempio tramite la
formazione di oligomeri, oppure che realizzi interazioni con altri ligandi, proteici o
meno (Lemmon et al., 2000). Una seconda funzione che sembra essere comune a
questo tipo di domini è di mediare le interazione proteina-proteina, che giustifica la
loro presenza in proteine coinvolte in vie di signalling (Hemmings et al., 1993;
Baltimore et al., 1993). Il PH domain è inoltre presente in molte proteine legate al
citoscheletro, tra cui la Beta-spectrina e le 4.1 (Saarikangas et al., 2010).
27
2.4.2 La localizzazione e la funzione
-Localizzazione
La proteina ICln è espressa in modo ubiquitario ed ha un alto grado di omologia
nelle diverse specie, a tal punto che l’espressione in oociti di Xenopus delle proteine
provenienti dalle differenti specie genera correnti molto simili (Gschwentner et al.,
1999). A livello subcellulare ICln è prevalentemente distribuita nel citosol in forma
solubile (circa 90%), mentre per il restante 10% è localizzata nella membrana
plasmatica. Condizioni di stress ipotonico possono portare, tuttavia, alla
traslocazione reversibile di ICln dal citoplasma verso la membrana.
In particolare, nei fibroblasti NIH3T3 (Ritter et al., 2003), la riduzione dell’osmolarità
extracellulare porta alla traslocazione di ICln dal citosol alla frazione microsomiale,
così come nelle cellule cardiache di embrione di razza (Musch et al., 1997) e in
miociti cardiaci di ratto (Musch et al., 1998).
La traslocazione è stata dimostrata anche in vivo in cellule HEK-293 tramite
esperimenti di FRET (Rodighiero et al., 2008; Ritter et al., 2003).
Inoltre, trattando i microsomi con il detergente Triton X-100, ICln si solubilizza.
Questo ci permette di ipotizzare l’esistenza di una seppur debole interazione tra la
proteina e il doppio strato lipidico, a conferma della possibilità di una traslocazione
reversibile di ICln verso la membrana plasmatica (Mush et al., 1998).
E’ stato anche proposto, (Paulmichl et al., 1992; Ritter et al., 2003) che la
traslocazione porti ad una vera e proria inserzione di ICln nel bilayer lipidico, ipotesi
supportata dal fatto che la ricostituzione della proteina purificati in bilayer lipidici
artificiale genera una corrente elettrica (Fürst et al., 2000).
-Funzioni
Sebbene la proteina sia stata clonata ormai da molti anni, le sue funzioni non sono
ancora state chiaramente definite e il quadro delle sue attività e andato
ampliandosi nel corso degli anni.
Un metodo molto utile per studiare la funzione delle proteine è realizzare il KnockOut (KO) del gene esprimente la proteina endogena in esame; tuttavia i tentativi di
28
creare animali KO per ICln sono falliti, in quanto il Knock-Out è letale in uno stato
embrionale molto precoce, suggerendo che questa proteina possa avere un ruolo
indispensabile per la vitalità e la funzione della cellula (Pu et al., 2000).
La prima funzione associata ad ICln è stata nel RVD: si ritiene infatti che giochi un
ruolo fondamentale nell’ attivazione della corrente ICl,swell.
L’importanza di ICln nella regolazione del volume cellulare è stata dimostrata
mediante esperimenti con oligodeossinucleotidi antisenso complementari alla
porzione iniziale della regione codificante per ICln (Gschwentner et al., 1995) e con
anticorpi monoclonali anti-ICln (Krapivinsky et al., 1994), che determinano la
soppressione di ICl,swell.
In accordo con questi dati, come si è già detto, l’espressione di ICln in oociti di
Xenopus oltre che in altri tipi cellulari porta all’attivazione di una corrente con
caratteristiche riconducibili a ICl,swell (Paulmichl et al., 1992; Gschwentner et al.,
1994). L’insieme di questi dati ha indotto a supporre che fosse proprio ICln l’entità
molecolare attraverso cui si origina ICl,swell, e che la traslocazione di ICln verso la
membrana coincidesse con la sua inserzione in forma di dimero, forse in
configurazione “beta-barrel”, nella membrana plasmatica funzionando come canale
oppure che permettesse l’attivazione di un canale anionico preesistente, di cui ICln
sarebbe un potenziale regolatore (Buyse et al., 1996; Ritter et al., 2003; Strange et
al., 1998).
Per verificare la correttezza di una di queste due ipotesi, ICln purificata è stata
ricostituita in membrane artificiali microscopiche (mediante esperimenti di tip-dip)
e macroscopiche. Tali esperimenti hanno consentito di dimostrare che la proteina è
effettivamente in grado di traslocare spontaneamente dalla soluzione alla
membrana lipidica generando una corrente ionica (Fürst et al., 2000; Strange et al.,
1998). Tuttavia la relazione tra canale e ICln è ancora oggetto di discussione.
Il pattern d’interazioni proteiche di ICln suggerisce che la proteina, oltre al ruolo di
regolazione del volume cellulare, svolga altre funzioni, non direttamente legate alla
permeazione degli ioni. Più precisamente, è stato proposto,e per lo meno in alcuni
casi dimostrato, un suo coinvolgimento:
29
-nella regolazione della morfologia cellulare (Krapivinsky et al., 1998) soprattutto in
virtù della sua interazione con molecole citoscheletriche come actina, miosina, e
JBP1;
-nello splicing dell’RNA (Pu et al., 1999);
-nell’attivazione delle piastrine (Larkin et al., 2004) e nell’angiogenesi (Li et al.,
2004).
Tra le suddette funzioni quella meglio caratterizzata è quella legata allo splicing
dell’RNA. Sembra infatti che ICln, insieme ad altre due proteine, MEP50 e JBP1 (di
cui si discuterà nel paragrafo successivo), entri a far parte del metilosoma, un
complesso proteico che ha il ruolo di metilare le proteine Sm in modo da favorire la
loro
associazione
con
gli
U
snRNAs
e
la
formazione
delle
snRNPs
(Ribonucleoproteine) (Friesen et al., 2001). La metilazione delle proteine Sm da
parte del metilosoma ne regola l’assemblaggio e quindi la funzionalità.
Una quadro riassuntivo delle funzioni proposte per ICln è riassunto nella figura 7.
Figura 7 Funzioni di ICln proposte. 1) l’interazione con il citoscheletro durante la regolazione del
volume cellulare può essere coinvolta nella trasduzione del segnale, nella traslocazione di ICln verso
la membrana cellulare o nella regolazione di un canale ionico; 2) canale ionico o regolatore di un
canale ionico; 3) coordinazione dell’assemblaggio di piccole ribonucleoproteine ricche di uridina
mediante il complesso survival of motor neurons (SMN) sequestrando e dirigendo le proteine Sm o
LSm al metilosoma; 4) potenziale funzione nella degradazione dell’mRNA; 5) ICln può
30
indirettamente regolare i livelli di espressione dell’mRNA o della proteina tramite i pathways 3) e 4).
(Tratto da Fürst et al., 2006).
2.4.3 Interazione di ICln con altre proteine
Poiché ICln si trova al centro di una complessa rete di interazioni proteiche (Fürst et
al., 2006), è di grande interesse identificare e caratterizzare le molecole con cui essa
interagisce e comprendere come tali legami potrebbero regolare il suo ruolo sia nel
citosol, sia a livello della membrana cellulare.
Vediamo di seguito le varie proteine con cui si è vista interagire, che qui sono state
raggruppate in base alla funzione:
1-Proteine coinvolte nello splicing dell’mRNA
E’ stata scoperta la presenza di ICln nel metilosoma, un insieme funzionale di
proteine deputato alla metilazione di alcune proteine dello spliceosoma, il
complesso di proteine ed RNA il cui compito è partecipare al processo di splicing
dell’RNA messaggero (mRNA), cioè alla rimozione degli introni dal pre-mRNA a
formare l’mRNA maturo (Friesen et al., 2001; Meister et al., 2001; Friesen et al.,
2002)
-
MEP50
Questa proteina, il cui peso molecolare è tra i 45 e i 50 kDa, fa parte del complesso
del metilosoma insieme a JBP1 e ad ICln (Friesen et al., 2001; Friesen et al 2002;
Meister et al 2001), con cui sembra però interagire indirettamente. Sono dirette
invece le sue interazioni con JBP1 e, mediante i WD repeats che MEP50 contiene,
con le proteine dello spliceosoma SmB, SmD2, SmD3, SmE (Friesen et al., 2001).
-
JBP1
Inizialmente non identificata e chiamata IBP72 (ICln Binding Protein, PM=72 kDa), si
è visto che il suo legame con ICln è mediato dall’Acidic Domain 3 (gli ultimi 29 aa in
ICln di ratto) (Krapivinsky et al., 1999; Emma et al., 1998). In seguito, è stata
identificata e denominata JBP1 (Janus Binding Protein 1) o PRMT5 (Protein arginine
Methyltransferase 5), omologa della proteina Skb1 di lievito (Krapivinsky et al.,
1999), la quale lega le PAK-like chinasi ed è implicata nella regolazione della
31
morfologia e del ciclo cellulare (Gilbreth et al., 1998). Più recentemente si è
osservato che essa interagisce con le proteine Sm nei loro domini ricchi di arginina e
di glicina (RG domains) e, dei tre componenti del metilosoma, è quello responsabile
della metilazione delle proteine Sm (Friesen et al., 2001).
-
Proteine Sm
In diverse occasioni sono state evidenziate interazioni di ICln con proteine Sm, in
particolar modo con SmD1, SmD2, SmD3, SmB/B’, SmX5, SmE, SmG (Friesen et al.,
2001; Figeys et al., 2001; Schmarda et al., 2001). E’ stato proposto che ICln sequestri
le proteine Sm appena sintetizzate e le diriga verso il metilosoma (Friesen et al.,
2001; Friesen et al., 2002; Gilbreth et al., 1998). Non solo, ma recentemente è stato
scoperto che anche LSm4, una proteina coinvolta nello splicing e nella degradazione
dell’mRNA, è in grado di interagire con una forma tronca di ICln (ICln159) (Fürst et al
2005).
2-Proteine del citoscheletro
Miosina
Si è riscontrata l’associazione di ICln con una proteina di 17 kDa. E’ stata identificata
come l’isoforma non muscolare della catena leggera della miosina ed il legame non
risulta mediato da alcuno degli Acidic Domains di ICln di ratto (Emma et al., 1998).
Actina
Inizialmente questa proteina è stata identificata come actina mediante esperimenti
di immunoblotting e di immunoprecipitazione (Krapiwinsky et al., 1994). La
possibilità di un’interazione tra ICln e l’actina è stata confermata tramite saggi di
doppio ibrido (Schwartz et al., 1997) e di elettroforesi bidimensionale (Li et al.,
1999), con cui è stato provato un legame con l’F-actina. L’interazione è stata
confermata con esperimenti di immunoprecipitazione in cellue renali CD8 (Tamma
et al., 2006).
32
4.1R
A seguito di esperimenti di doppio ibrido, la parte C-terminale di ICln umana (aa
103-237) risulta in grado di legare l’isoforma eritroide della proteina 4.1 (4.1R),
anche detta Banda 4.1, sugli amminoacidi 162-280, corrispondenti a parte del
dominio FERM (Tang et al., 1998). Di recente è stato inoltre riportato che ICln è
capace anche di legare altri membri della famiglia delle 4.1: la 4.1B e
presumibilmente anche la 4.1G e la 4.1N (Calinisan et al., 2006).
Tale interazione, su cui è stato incentrato il mio lavoro di dottorato, verrà discussa
più ampliamente nel paragrafo successivo, interamente dedicato alle proteine 4.1,
ne verranno approfonditi il ruolo e le funzioni
3)
Proteine di membrana: le integrine
Le integrine sono molecole eterotrimeriche di adesione cellulare composte da
subunità α e β, attraverso cui mediano l’adesione cellula-cellula e cellula-matrice e
coordinano i segnali bidirezionali che attraversano la membrana plasmatica.
Recentemente è stato dimostrato un coinvolgimento delle integrine nella
regolazione dell’osmosi e dei movimenti degli ioni attraverso le membrane cellulari,
in particolare controllano il flusso del calcio, del potassio e del cloro sia
direttamente (Davis et al., 2002; Haussinger et al., 2003) sia indirettamente (Ritter
et al., 2003; Browe et al., 2003; McPhee et al., 1998; Shakibaei et al., 2003; Vom et
al., 2003).
L’integrina αIIbβ3, specifica per le piastrine, è la più studiata ed è stata utilizzata
come modello per la ricerca di eventuali ligandi con cui interagiscono tali proteine
(Larkin et al., 2004) tra di esse è compresa anche ICln.
Dal momento che l’inibizione della ICl,swell attraverso l’Acyclovir induce l’inibizione
delle integrine e dell’aggregazione piastrinica, è stato proposto che ICln svolga un
ruolo rilevante negli eventi di attivazione delle piastrine (Larkin et al., 2004).
33
2.5 La proteina 4.1
2.5.1 La struttura
I membri della famiglia delle proteine adattatrici 4.1 sono espressi in numerosi
tessuti, inclusi molti epiteli dove probabilmente rivestono un importante ruolo nel
mantenimento dell’architettura e della polarità della cellula e nel controllo della
proliferazione cellulare.
Il primo membro della famiglia della proteina 4.1 ad essere individuato è stata la
proteina 4.1 R (chiamata anche banda 4.1), una proteina strutturale multifunzionale
di 80 kDa, individuata inizialmente a livello della membrana degli eritrociti umani,
dove stabilizza il citoscheletro submembranario di actina-spectrina. Per tale motivo
inizialmente si è pensato che i membri della superfamiglia della proteina 4.1
rivestissero un ruolo esclusivamente strutturale.
I membri della famiglia della proteina 4.1 sono stati individuati, in seguito, in molti
tipi cellulari di organismi metazoi, concentrati soprattutto a livello del nucleo e delle
giunzioni cellulari.
Tali
proteine
costituiscono
dei
complessi
multimolecolari
con
proteine
transmembrana e proteine associate alla membrana, importanti per la stabilità
strutturale e la trasduzione del segnale nei siti di contatto tra le cellule (Calinisan et
al., 2006; Diakowski et al., 2006). L’importanza del ruolo strutturale e funzionale
della proteina 4.1 deriva dall’osservazione che una drastica riduzione o la perdita
dell’espressione della proteina determina una diminuzione dei livelli di espressione
e la dislocazione di proteine transmembrana e proteine associate alla membrana,
che normalmente interagiscono con la proteina 4.1 (Nunomura et al., 2009).
Nell’uomo sono stati individuati quattro geni che codificano per quattro distinte
isoforme della proteina 4.1, che prendono il nome dal tessuto in cui sono state
isolate. Questi geni sono altamente complessi: sono localizzati su differenti
cromosomi e presentano diversi patterns di espressione (Kim et al., 1998; Parra et
al., 1998; Peters et al., 1998), contengono diversi esoni e possiedono più di un sito
di inizio per la trascrizione; l’mRNA subisce quindi splicing alternativo generando
34
delle varianti che differiscono per la presenza o meno di alcuni domini e che hanno
peso variabile (ciò è dovuto, molto probabilmente, anche a modificazioni posttraduzionali quali glicosilazione e fosforilazione).
La superfamiglia delle 4.1 include la proteina 4.1R (spesso denominata EPB41:
erythrocyte membrane protein band 4.1), per la quale sono generate un gran
numero di varianti con regolazione tessuto-specifica e sviluppo-specifica (Conboy et
al., 1993); la proteina 4.1N (EBP41-like 1); la proteina 4.1G (EBP41L2); la proteina
4.1B (EBP41L3) (Walensky et al., 1999).
4.1 R (Red blood cell) è espressa soprattutto nei tessuti emopoietici,
polmone, fegato, sistema olfattivo e nel cervello: nel lobo caudato e
occipitale, nel putamen e identificata in modo specifico nelle cellule
granulari del cervelletto e nel giro dentato. Questa proteina è localizzata a
principalmente livello nucleare e della membrana plasmatica (Conboy et al.,
1987; Walensky et al., 1998; Salomao et al., 2008).
4.1N (Neurons) mostra un alto livello di espressione in tutti i neuroni del
cervello ed è localizzata a livello delle sinapsi. È debolmente espressa anche
a livello del polmone, placenta, muscolo scheletrico, pancreas, cuore e rene
(Walensky et al., 1999). La proteina 4.1 stabilizza la plasticità della
membrana neuronale grazie alle interazioni actina-spectrina, con i canali
integrali di membrana e le guanilato chinasi associate alla membrana
(Ramez et al., 2003). 4.1N è espressa in quasi tutti i neuroni centrali e
periferici nei mammiferi ed è stata identificata nei neuroni embrionali nelle
fasi precoci della differenziazione post mitotica. Le zone di contatto
sinaptico sono arricchite di questo omologo (Wallensky et al., 1999).
4.1G (General distribution) è presente in quasi tutti i tessuti dell’organismo
(ad eccezione del rene) e in particolare nel cervello: nell’ippocampo e nel
cervelletto; nel testicolo, con localizzazione a livello citoplasmatico e
perinucleare (Parra et al., 1998).
4.1B (Brain) è localizzata nelle popoloazioni neuronali nel cervello di topo,
specialmente nelle cellule del Purkinje del cervelletto, nelle cellule piramidali
35
della regione ippocampale, nei nuclei talamici e nel bulbo olfattivo (Parra et
al., 2000). Inoltre è stata trovata nel cuore, nei polmoni, nei reni,
nell’intestino e nei testicoli.
Le diverse isoforme che costituiscono la superfamiglia di proteine 4.1 sono formate
da quattro domini strutturali conservati tra le diverse isoforme (Fig.8): all’Nterminale presentano un dominio FERM (4.1 ezrin-radixin-moesin) di 30 kDa, che
media il legame con molte altre proteine, tale dominio è considerato il dominio
“firma” della famiglia; un dominio idrofilico di 16 kDa che possiede un sito di
fosforilazione da parte della PKC; un dominio SAB (spectrin/actin binding domain) di
10 kDa, presente nei geni per 4.1 di tutti i vertebrati (Hoover et al., 2000) e un
dominio di 22-24 kDa al C-terminale (CTD); gli ultimi due domini citati possono
interagire con proteine transmembrana.
Figura 8 Organizzazione dei domini delle proteine 4.1. L’allineamento delle proteine 4.1 mostra la
presenza di tre domini conservati (FERM, SAB e CTD), indicati con sfumature di grigio e di tre regioni
uniche (U1, U2, U3), indicate dai riquadri bianchi. I numeri riportati nei riquadri si riferiscono alla
percentuale di identità dei domini conservati delle proteine 4.1G, 4.1B e 4.1N con i corrispondenti
domini della 4.1R. La rappresentazione della 4.1R mostra una sola regione U3 che è espressa
unicamente nei tessuti epiteliali. Si può notare che il dominio SAB non è conservato nella 4.1N
(Calinisan et al., 2006).
Diversamente dalle proteine 4.1R, 4.1G e 4.1B, la 4.1N è incapace di formare un
complesso ternario con spectrina e actina a causa del ridotto grado di
conservazione del suo dominio SAB.
36
Nei quattro membri della famiglia 4.1 le regioni con più alta omologia sono i domini
FERM, SAB e il dominio CTD. Ciò suggerisce che le proteine 4.1 condividono alcune
funzioni comuni, sono infatti proteine adattatrici associate alla membrana; mentre
la presenza di regioni caratteristiche per ogni 4.1, disseminate tra i domini
conservati, può conferire specifiche proprietà ad ognuna di esse (Gascard et al,
2000).
Figura 9. Schema della struttura delle proteine 4.1. I diversi domini sono evidenziati: CTD carboxyl
terminal domain; FERM - 4.1-ezrin-radixin-moesin domain, SABD -spectrin-actin-binding domain.
Sono inoltre evidenziate le proteine che interagiscono con la proteina 4.1 (Diakowski et al., 2006).
Nell’approfondire le funzioni dei diversi domini farò riferimento soprattutto alla
proteina 4.1R, che oltre a rappresentare un prototipo per la famiglia è anche quella
che è stata oggetto di studio per il mio progetto di dottorato.
Il dominio FERM
Il dominio FERM è situato all’estremità N terminale ed è codificato dagli esoni 4 e 12
del gene per la 4.1R; è responsabile del legame della proteina 4.1R con altre
proteine come ad esempio la glicoforina C, la proteina p55 e la calmodulina
(Diakowski et al., 2006). Il dominio FERM è costituito da tre differenti lobi (N-lobe,
α-lobe e C-lobe) ognuno dei quali contiene una regione specifica per il legame con
la membrana o per una proteina associata alla membrana (Han et al., 2000). Il lobo
N che contiene il sito di legame per la proteina banda 3 è formato da due foglietti β
double-stranded collegati da un α-elica; i foglietti β avvolgono parzialmente l’ α
elica. Il lobo α contiene il sito di legame con la glicoforina C ed è organizzato da 4 αeliche organizzate come uno stretto pacchetto. Infine il lobo C contiene il sito di
legame per la p55, la calmodulina, i PS e la regione di legame per i PIP2; questo lobo
37
è caratterizzato da due foglietti β e da un α-elica, una struttura tipica dei domini PH
(Diakowski et al., 2006). Nella regione centrale del dominio, vicino al punto in cui i
tre lobi si congiungono, ci sono due regioni separate, adibite al legame con la
calmodulina (CaM). Una di queste è composta da una -elica è ed è Ca2+ insensibile;
per quanto riguarda la seconda regione, la presenza di Ca2+ accresce il suo legame
con CaM.
Il legame della 4.1 alla calmodulina porta ad alcune modificazioni nella
disposizione/conformazione dei tre lobi, ne consegue un indebolimento del legame
della proteina 4.1 ad altre proteine che interagiscono con essa. Ad esempio in
presenza di Ca2+ la calmodulina riduce l’affinità della proteina 4.1R per il complesso
actina-spectrina e questo fa diminuire la stabilità meccanica della membrana
(Nunomura et al ., 2006). Tale struttura, nella quale i tre lobi legano tre proteine
associate alla membrana, e la presenza del sito di legame della calmodulina,
permettono al dominio FERM di interagire con le proteine di membrana e di
regolare in modo dinamico la forma cellulare in risposta a cambiamenti
intracellulari di Ca2+.
B
Figura 10 A Immagine cristallografica del dominio FERM (Diakowski et al., 2006); B Diagramma di
Ribbon in cui in verde sono evidenziatele α-eliche, in blu i β-sheets e in giallo i loop (Han et al., 2000)
è possibile inoltre distinguere i tre lobi (N-lobe, α-lobe e C-lobe).
Tra il dominio FERM e il dominio SABD è presente un dominio di 16 KDa (regione
U2) che non ha un’attività di legame con altre proteine ma presenta due residui di
38
Serina (312 e 331) che possono essere fosforilati dalla proteina chinasi A e C
(Manno et al., 2005).
Il dominio SAB
Il dominio SAB è un dominio di 10 KDa codificato dagli esoni 13, 16 e 17 del gene
per la 4.1R. Tale dominio media l’interazione tra spectrina e actina (Correas et al.,
1986), inoltre lega U2AF35 e l’importina α una proteina necessaria per l’import della
proteina 4.1R nel nucleo (Gascard et al., 1999; Luque 2003). La sequenza consenso
minima che è necessaria per l’interazione con spectrina e actina è una sequenza di
21 aminoacidi codificata dall’esone 16 (Diakowski et al., 2006).
La formazione del complesso ternario composto da 4.1R, spectrina e actina regola la
resistenza meccanica e l’elasticità della membrana degli eritrociti (Conboy et al.
1998). Questo dominio contiene inoltre residui quali Serina467 e Tirosina418 che
possono essere fosforilati da diverse chinasi (Manno et al., 2005). La struttura
primaria di questo dominio è altamente conservata in molti vertebrati ad eccezione
della 4.1N che è differente e non lega la spectrina e l’actina (Gimm et al., 2002).
Il dominio CTD
Il dominio CTD, di 22/24 kDa, è codificato dagli esoni 18-21 della 4.1R; i 2 kDa di
differenza tra la proteina 4.1Ra e 4.1Rb sono il risultato di una conversione da
asparagina Asn
502
a Asp502 che avviene durante l’invecchiamento degli eritrociti
(Inaba et al., 1992). Tale dominio interagisce con diverse proteine fra cui recettori di
membrana, tight junction proteins e la proteina nucleare NuMA associata
all’apparato mitotico (Calinisan et al., 2006).
I domini variabili
Oltre ai tre domini altamente conservati, le proteine appartenenti a questa famiglia
contengono le regioni U1, U2 e U3, disseminate tra i domini conservati, che
agiscono da modulatori delle interazioni mediate dalla proteina 4.1 attraverso i
domini conservati e possono conferire specifiche funzioni a ciascuna proteina 4.1.
39
Figura 11 Schema della struttura delle proteine 4.1. I diversi domini sono evidenziati.
E’ stato dimostrato che la regione unica U1 (non presente in tutte le isoforme di
4.1R) modula la traslocazione nel nucleo della proteina 4.1R (Luque et al., 1999).
Questa regione inoltre interagisce con la proteina calmodulina in maniera
strettamente calcio-dipendente e con le proteine associate al centrosoma (Gascard
et al., 2006). La regione unica U1 presenta inoltre un sito di fosforilazione per la
chinasi ciclina dipendente cdc2 il cui livello di fosforilazione varia durante il ciclo
cellulare (Calinisan et al., 2006). La presenza o meno della regione U1 permette di
dividere le proteine 4.1R in due sotto gruppi: isoforme ad alto peso molecolare e
isoforme a basso peso.
Per quanto riguarda le regioni uniche U2 e U3, non sono ancora stati identificati i
partners di legame.
La regione unica U2 contiene un residuo chiave di serina che rappresenta il
substrato primario per la proteina chinasi C (PKC) fosforilazione dipendente
(Calinisan et al., 2006). La fosforilazione di tale serina porta ad una diminuzione
dell’interazione di 4.1R con la proteina transmembrana glicoforina C e con spectrina
e actina (Gascard et al., 2006). Data la conservazione di questo residuo di serina e
dei circostanti aminoacidi in tutte le proteine 4.1, è ipotizzato che la fosforilazione
dipendente da PKC giochi un ruolo chiave nella regolazione della funzione di 4.1G,
4.1N e 4.1B e 4.1R.
Nel corso degli anni sono state identificate numerose isoforme, 7 isoforme
principali più 11 varianti, della proteina 4.1R, espresse sia in eritrociti che in cellule
non eritrocitarie (Parra et al., 1998). Queste isoforme variano nelle dimensioni e
40
mostrano differenze strutturali e funzionali notevoli, e spesso più isoforme sono
espresse all’interno di uno stesso tipo cellulare.
La grande eterogeneità delle isoforme di 4.1 (Fig. 11) è generata dal complesso
splicing alternativo del pre–mRNA di 4.1, dall’uso differente di due siti d’inizio per la
traduzione e da modificazioni post-traduzionali delle proteine 4.1 (Conboy et al.,
1998).
In particolare l’utilizzo di due siti alternativi “splice acceptor” all’estremità 5’
dell’esone 2 genera due popolazioni di RNA di 4.1R: una che include un AUG a
monte (AUG–1) e codifica per isoforme di 4.1R ad alto peso molecolare (135 KD) e
un’altra che salta AUG–1 e codifica 4.1R a partire da un AUG a valle (AUG-2) situato
nell’esone 4 che genera isoforme a basso peso molecolare (80 KD) (Gascard et al.,
1998).
In pratica le isoforme ad alto o basso peso molecolare differiscono per la presenza o
meno della regione U1.
A)
41
B)
Figura 12 Sono mostrate alcune delle isoforme esistenti di 4.1R. In figura A è riportata
l’organizzazione in esoni del gene umano per la 4.1R delle isoforme prodotte dal sito di inizio
trasduzione AUG-1 (isoforme lunghe). All’inizio della figura è rappresentato l’mRNA della 4.1R con gli
esoni alternativi, gli esono costitutivi e gli esoni non codificati. I due siti di inizio trasduzione AUG1 e
AUG2 sono situati all’esone 2 e all’esone 4 rispettivamente. In B sono rappresentate le isoforme
mancanti l’AUG-1, quindi le isoforme corte (Gascard et al., 1998).
Recenti studi hanno dimostrato che l’mRNA della 4.1R che contiene l’AUG1
responsabile della sintesi dell’isoforma lunga (135 kDa), all’interno della sequenza
tra i due siti di inizio della trascrizione (AUG 1 e AUG 2) presenta un elemento IRES
che permette l’uso di un sito interno e quindi la sintesi dell’isoforma corta della 4.1
(80kDa). L’elemento IRES divide la sintesi del secondo cistrone in due sistemi
bicistronici e quindi la sintesi dell’isoforma corta della proteina 4.1R dall’mRNA
dell’isoforma lunga anche a splicing già avvenuto (Lospitao et al., 2008).
42
2.5.2 La funzione e le interazioni
La proteina 4.1R, insieme alla spectrina, conferisce supporto ed elasticità alle
membrane delle cellule animali. Queste due proteine si sono evolute negli
organismi animali per consentire alle cellule di resistere agli stress dovuti ai
movimenti e per organizzare le complesse strutture che coordinano le funzioni
cellulari nei tessuti (Bennet et al., 2001).
Spectrina e 4.1R formano le connessioni tra citoscheletro citoplasmatico e
membrana plasmatica, organizzano la trasmissione funzionale di segnali complessi,
conferiscono elasticità e capacità di resistenza a differenti strutture di membrana,
rafforzano l’adesione cellulare (Bennet et al., 2001).
Le cellule del sangue, in particolare i globuli rossi, necessitano di alcune proteine
per far fronte allo stress che subiscono durante la circolazione. Le cellule muscolari,
sottoposte a ripetute contrazioni, e i neuroni, la cui necessità di trasmissione dei
segnali comporta specifiche organizzazioni e microstrutture, sono esempi di tipi
cellulari in cui spectrina e proteina 4.1R sono abbondanti.
Sembra anche che la proteina 4.1R, così come altri membri della stessa
superfamiglia (in particolare la 4.1B), rivestano anche un importante ruolo nella
soppressione dei tumori e nella regolazione della proliferazione cellulare (Gutman
et al., 2000; Robb et al., 2003; Kuns et al., 2005).
Pertanto sebbene alla famiglia delle proteine 4.1 inizialmente era stato attribuito un
ruolo esclusivamente strutturale nelle cellule, ad oggi ci sono numerose evidenze
del loro coinvolgimento in numerose altre funzioni cellulari tra cui:
partecipano al mantenimento della polarità della cellula (Lamb et al., 1998);
intervengono nel controllo della divisione cellulare (Correas et al., 1991; PerezFerreiro et al., 2004) in quanto partecipano alla formazione del fuso mitotico e
dei poli del fuso interagendo con i microtubuli mitotici (Huang et al., 2004);
intervengono nel mantenimento del volume cellulare in risposta a stress
osmotico (Tang et al., 1998);
hanno un ruolo importante nella proliferazione (Jiang et al., 2005);
43
inoltre intervengono nella morfogenesi cellulare e nella regolazione di alcuni
fattori di trascrizione nel nucleo, dal momento che intervengono nelle maggiori
vie di trasduzione del segnale (Scoles et al., 1998).
Sembra inoltre che alcune isoforme di 4.1 espresse in eritroblasti possano
contribuire in maniera significativa alla funzione e all’architettura nucleare e
centrosomale (Calinisan et al., 2006). Le 4.1 R giocano un ruolo molto importante
per il centrosoma in quanto contribuiscono al mantenimento dell’organizzazione
radiale dei microtubuli (Perez-ferreiro et al., 2004).
Una delle principali funzioni della proteina 4.1R è quella di congiungere i tetrametri
di spectrina e di ancorare i componenti del citoscheletro (Conboy et al., 1993; Lue et
al., 1994), in particolare stabilendo delle interazioni orizzontali tra gli oligomeri di
spectrina e i filamenti di actina (Gascard et al., 2000). Queste proteine, tramite
specifici domini, interagiscono con numerosi partners:
scambiatore anionico AE (Banda 3) mediante il dominio FERM (esone 5);
Glicoforina C (legame ad una superficie carica negativamente di Asp e Glu e
carica positivamente di Lys e Arg, mediante il dominio FERM (esone 8);
richiesto PtdIns(4) come cofattore perché possa avvenire l’interazione);
Ca2+-calmodulina chinasi (CASK);
Fosfatidilinositolo 4-fosfato (PtdIns(4));
Fosfatidilinositolo 4,5-bifosfato (PtdIns(4,5));
Human discs-large (hDlg) (multidomain scaffolding protein con tre domini
PDZ; un dominio SH3 e un dominio guanilate kinase-like (GUK), è una
MAGUK, richiesta per il controllo della crescita e della polarità delle cellule
epiteliali);
p55 (è una proteina che appartiene a proteine del citoscheletro e di segnale
associate alla membrana (MAGUK), presenta un sito di legame HOOK
domain);
ICln;
Ca2+-calmodulina (regola il legame della 4.1 a proteine di membrana e del
citoscheletro);
44
CD44;
Spectrina;
Actina;
Fosfatidilserina;
NCP (Neurexin / CASPR/ Paranodin);
CIF3-p44 (subunità del fattore eucariotico 3 di inizio della traduzione,
costitutivamente espressa in molti tessuti, interagisce con la porzione Cterminale);
NuMA (Nuclear Mitotic Apparatus Protein);
CPAP (Centrosome Protein).
Sebbene la 4.1R sia localizzata nella membrane delle cellule nucleate e degli eritociti
anucleati, molte cellule che possiedono un nucleo presentano appunto una
localizzazione nucleare di questa proteina. Una delle funzioni nucleari di questa
proteina potrebbe dipendere dalla sua capacità di legare fattori di splicing e
regolare così il processamento dell’RNA (Lallena et al., 1998; De Carcer et al., 1995;
Krauss et al., 1997); un altro studio indica che la proteina 4.1 regola la formazione
del fuso mitotico (Mattagajasingh et al., 1999): il dominio C-terminale della 4.1R
lega la proteina NuMA, che forma un complesso molecolare con la dineina,
essenziale per la formazione del fuso mitotico. L’overespressione della 4.1R causa,
però, un’errata localizzazione della proteina NuMA e, di conseguenza, morte
cellulare.
La proteina 4.1R, perciò, è importante non solo a livello della membrana per
l’organizzazione del citoscheletro, ma anche nel nucleo.
Esperimenti di Knock-Out sono stati utili per comprendere la funzione dei geni
codificanti per le 4.1; in particolare il KO del gene 4.1R ha dimostrato l’importanza
di questa isoforma in alcune regioni del cervello, nel giro dentato dell’ippocampo e
nelle cellule granulari del cervelletto, dove c’è una forte espressione dell’mRNA di
tale proteina. Topi mancanti della 4.1R presentano difetti neurocomportamentali
tipici di disfunzioni cerebrali, riguardanti l’equilibrio, la coordinazione e la memoria
(Walensky et al., 1999).
45
Notevole attenzione è stata rivolta allo studio di mutazioni delle proteine del
citoscheletro, alla base di malattie genetiche ereditarie. In particolare, la proteina
4.1R è mutata nell’ellissocitosi ereditaria (HE), caratterizzata da un’anormale forma
degli eritrociti (Conboy et al., 1993); in una forma di HE, con una specifica delezione
del dominio SAB, i difetti eritrocitari sono identici a quelli causati da una completa
assenza della proteina 4.1 (Conboy et al., 1990; Marchesi et al., 1990), a
dimostrazione dell’importanza del dominio SAB per una corretta funzione della 4.1
negli eritrociti. In un’altra famiglia di HE, invece, la mancanza di uno specifico sito di
binding del dominio FERM non provoca sintomi correlati alla mancanza della 4.1,
indicando che questo sito non è di fondamentale importanza per la funzione della
4.1R, che potrebbe, altrimenti, essere svolta da paraloghi della 4.1 in tessuti non
eritroidi (Jons et al., 1992; Conboy et al., 1993). Mutazioni della proteina in esame
sono state ritrovate anche in Drosophila melanogaster e Mus musculus.
46
3 Scopo del lavoro
ICln riveste un ruolo fondamentale nell’attivazione dell’ICl,swell, la corrente di
cloruro attivata dal rigonfiamento cellulare, (Okada et al., 1997). Lo stress ipotonico
induce una traslocazione di ICln verso la membrana cellulare che accompagna
l’attivazione della corrente ma non è ancora stato chiarito se ICln intervenga come
regolatore o se rappresenti uno dei componenti del canale stesso.
Oltre ad essere coinvolta nel’RVD, ICln è coinvolta in numerosi altri processi cellulari
come dimostrato dal pattern delle sue interazioni con altre proteine quali proteine
coinvolte nello splicing dell’mRNA (Pu et al., 1999) e proteine di adesione (integrine)
(Larkin et al., 2004). Di particolare interesse per questo lavoro è l’interazione di ICln
con proteine citoscheletriche. E’ noto infatti che l’ICl,swell è modulata dallo stato
del citoscheletro actinico (Lang et al., 1998; Moustakas et al., 1998) e ICln lega
numerose proteine citoscheletriche tra cui la stessa actina, la miosina e alcune
proteine appartenenti alla famiglia delle 4.1, una famiglia di proteine multifunzione
coinvolta nella regolazione del citoscheletro, della morfologia cellulare e di
membrana e della proliferazione cellulare.
E’ già noto che 4.1R e ICln interagiscono: l’interazione è stata appurata tramite le
tecniche del doppio ibrido in lievito, di coimmunoprecipitazione e spettrometria di
massa (Tang et al. 1998; Figeys et al., 2001). In particolare si è visto che il dominio Cterminale di ICln (aminoacidi dal 103 al 237) lega il lobo C del dominio di 30 kDa
(FERM) della proteina 4.1R (80 kDa) (Tang et al., 1998), a livello degli aminocaidi
136-283 (Calinisan et al., 2006). L’utilizzo di queste tecniche, tuttavia, non ha fornito
informazioni né sulla sede dell’interazione, né sulla funzione, aspetto di estremo
interesse, vista la spiccata multifuzionalità di entrambe le proteine.
In particolare, vista l’importanza di ICln per l’RVD, l’interazione suggerisce un
possibile ruolo, fin’ora non noto, anche per 4.1 nella regolazione del volume
cellulare, come possibile componente del pathway di signalling e/o elemento chiave
per la riorganizzazione del citoscheletro indotta dall’ipotonia. Un altro possibile
47
ruolo per la 4.1 nasce dalla considerazione che garantisce, tra le altre funzioni,
anche il mantenimento dell’associazione tra proteine di membrana e il citoscheletro
di spectrina/actina sottostante. Dato che la 4.1 è in grado di legare sia l’actina che
ICln attraverso due specifici e distinti siti di legame, tale proteina potrebbe agire
come un adattatore proteico che collega lo scheletro actina–spectrina a ICln,
favorendone la traslocazione e il suo ancoraggio alla membrana e l’attivazione della
ICl,swell.
Il citoscheletro è, infatti, uno dei candidati per la trasduzione del segnale durante la
regolazione del volume cellulare. E’ infatti noto che l’organizzazione citoscheletrica
è alterata dalle variazioni di volume (Tamma G. et al, 2007) e molte proteine
trasportatrici regolate dal volume e rispettivi regolatori mostrano interazioni
funzionali con il citoscheletro (Pedersen et al, 2001).
Lo studio delle interazioni tra ICln e proteine associate al citoscheleto potrebbe
chiarire il meccanismo di trasduzione del segnale che porta dal rigonfiamento
cellulare all’attivazione delle conduttanze di membrana responsabili del RVD.
Su queste basi, il primo scopo di questo lavoro è stato di studiare in vivo tramite la
tecnica del FRET (Fluorescence Resonance Energy Transfer) l’interazione tra ICln e
4.1R umana, per studiarne la localizzazione subcellulare e l’evolversi durante
l’ipotonia. Sebbene ICln interagisca con i FERM di tre diverse isoforme di 4.1,
l’isoforma R, B e G abbiamo scelto di focalizzarci sull’ isoforma R. Tale isoforma è il
prototipo della famiglia ed è la prima per cui è stata riportata l’interazione con ICln
(Tange et al., 1998) e quella per cui i siti di interazione sono stati meglio
caratterizzati. L’indagine è stata estesa a due tipi di isoforme di 4.1R: una ad alto
peso molecolare, (4.1R long variante lunga) e una a basso peso molecolare (4.1Rsh,
variante corta), che sono state clonate da cellule embrionali umane HEK-293
Phoenix. Gli esperimenti sulla interazione sono stati affiancati dallo studio
dell’influenza reciproca delle due proteine sulla loro localizzazione subcellulare.
Infine per capire il significato funzionale dell’interazione ICln-4.1R nella regolazione
della corrente per il cloruro ICl,swell abbiamo allestito esperimenti di patch clamp in
48
configurazione whole cell in cellule HEK per studiare l’effetto della over-espressione
delle due varianti da splicing della 4.1R sulla corrente attivata dallo swelling.
49
4 Materiali e Metodi
4.1 Colture cellulari
Gli esperimenti sono stati condotti su cellule HEK 293 Phoenix e HEK 293 T (Human
Epithelial Kidney). Le cellule sono state fatte crescere a 37°C, in presenza di 5% CO 2
in un terreno minimo MEM (Minimum Essential Medium, M5650, Sigma)
complementato con l’aggiunta di siero bovino fetale (FBS, 14-801F, Lonza) al 10%, di
L-glutammina 2 mM, penicillina 280 µM streptomicina 144 µM e 1 mM di acido
piruvico. Le cellule sono seminate in piastre petri e successivamente sottoposte ad
eventuali passaggi di tripsinizzazione. Lavorando sotto cappa a flusso laminare in
condizioni di sterilità, una volta raggiunto l’80-90% di confluenza si preleva il
terreno dalle piastre petri e le cellule vengono lavate con PBS (Phosphate Buffered
Saline) al fine di rimuovere il terreno residuo per evitare l’inibizione dell’azione della
tripsina da parte delle proteine contenute nel siero. Una volta rimosso il PBS, viene
aggiunto PBS contenente 0,05% tripsina-EDTA si lascia agire l’enzima per qualche
minuto, fino a quando le cellule non si staccano dal fondo della piastra. Per bloccare
l’effetto della tripsina si aggiunge un volume opportuno (variabile in base al volume
delle petri in cui le cellule sono coltivate) di terreno. Le cellule vengono poi
seminate nuovamente in altre piastre di volume variabile a seconda del tipo di
esperimento da condurre.
50
Composizione del terreno:
Minimun Essential Medium 435 ml
(M5650 Sigma)
FBS 10%
50 ml
L-glutammina 200 mM
5 ml
Penicillina 100 U/ml
5ml
Streptomicina 0,1 mg/ml
5 ml
Acido piruvico 100 mM
5 ml
Composizione del PBS 1X:
NaCl
136,89 mM
KCl
2,69 mM
Na2HPO4
3,21 mM
KH2PO4
1,47 mM
NaOH
1,00 mM
pH
7,4
4.2 Estrazione dell’RNA citoplasmatico
L’estrazione dell’RNA è stata effettuata utilizzando un kit della Qiagen (RNeasy Maxi
Kit), seguendo un protocollo che ha consentito di estrarre solo RNA citoplasmatico
che nelle cellule animali rappresenta l’85% dell’RNA totale cellulare.
L’RNA veniva estratto da una piastra di cellule HEK 293 T a confluenza.
51
Tutte le soluzioni utilizzate venivano precedentemente trattate con 0,1% DEPC, un
potente, anche se non assoluto, inibitore di RNAsi.
-
La lisi cellulare avveniva direttamente nella Petri aggiungendo 1 ml di Buffer
RLN a 4 °C. Questo buffer conteneva Nonidet® P-40 che lisa la membrana
cellulare.
-
Le cellule lisate venivano staccate con una spatola dalla Petri, messe in un
tubo Eppendorf e incubate in ghiaccio per 5 minuti.
-
Il lisato era centrifugato a 4 °C per 5 minuti a 300-500 x g per rimuovere i
nuclei che restano intatti durante la lisi cellulare, il surnatante era trasferito
in una Falcon da 15 ml Rnase-free.
-
Venivano poi aggiunti 4 ml di Buffer RLT e 2.8 ml d’etanolo (96-100%) al
lisato. Il buffer RLT contiene guanidina isotiocianato, che inibisce le RNAsi
citoplasmatiche. Il buffer e l’etanolo erano aggiunti al surnatante per
rendere ottimale il legame dell’RNA alla membrana di una colonna
cromatografica (fornita con il kit).
-
La miscela veniva quindi posta in questa colonna che era posizionata in una
Falcon da centrifuga da 15 ml e centrifugata per 5 minuti a 3000-5000 x g.
L’eluito era eliminato. L’RNA con dimensioni superiori a 200 bp restava
legato alla colonna.
-
Seguiva la fase di lavaggio in cui 4 ml di Buffer RW1 erano aggiunti alla
Rneasy column che era centrifugata per 5 minuti a 3000-5000 x g. In seguito
erano aggiunti 2,5 ml di Buffer RPE alla RNeasy column che era poi
centrifugata per 2 minuti a 3000-5000 x g. La procedura era ripetuta una
seconda volta.
-
Per l’eluizione 350 μl d’acqua RNasi free erano aggiunti direttamente sulla
membrana della spin-column. Dopo 1 minuto la colonna era centrifugata per
3 minuti a 3000-5000 x g.
52
Composizione buffer RLN
50 mM TrisCl, pH 8.0
140 mM NaCl
1,5 mM MgCl2
0,5% (v/v) Nonidet® P-40 (1,06 g/ml)
Prima dell’utilizzo il Buffer RLN viene raffreddato e mantenuto a 4 °C.
4.3 Elettroforesi dell’RNA
Preparazione del gel 1%
0,5 g d’agarosio erano sciolti in 50 ml di TAE. La miscela era scaldata finché
l’agarosio non si fosse sciolto completamente, dopodichè venivano aggiunti 0,3 μl di
bromuro d’etidio.
La miscela era versata in un’opportuna vaschetta, inserito il pettine per i pozzetti e
lasciata raffreddare.
Preparazione del campione
Veniva preparata una miscela costitutita da:
4 μl RNA
4 μl Formaldeide
1,6 μl Sample Buffer 6X
La miscela era scaldata a 65 °C per 10 minuti. Il campione era poi caricato su gel e
fatto correre a 120 V per 30 minuti.
53
4.4 Retrotrascrizione (RT) con oligo dT
Veniva effettuata utilizzando un kit Invitrogen (Superscript III).
Era inizialmente preparata una miscela la cui composizione era:
Oligo dT (50 mM)*
1 μl
dNTP (10 mM)*
1 μl
RNA
11 μl
Questa miscela veniva preparata in una Eppendorf che era scaldata a 65 °C per 5
minuti (per denaturare eventuali strutture secondarie formate dall’RNA) e
successivamente lasciata in ghiaccio per 1 minuto (per evitare, data l’elevata
temperatura, di inattivare la trascrittasi inversa).
Per la vera e propria reazione di retrotrascrizione venivano aggiunti ad ogni
Eppendorf 7 μl della seguente miscela:
Buffer 5X
4 μl
DTT 0,1M
1 μl
RNAse OUT
1 μl
SuperScript III
1 μl
Seguiva una fase di incubazione a 50 °C per 50 minuti (retrotrascrizione) ed una di
incubazione a 70 °C per 15 minuti (inattivazione della trascrittasi).
4.5 PCR
PCR con Taq Bioline.
Una prima serie di reazioni è stata condotta per individuare quale isoforma di RNA
codificante per la proteina 4.1 fosse presente in cellule HEK.
54
Miscela di reazione per PCR (volume finale 50 μl)
H2O
36,5 μl
Buffer 10X
5 μl
MgCl2 (50mM)
2 μl
dNTP (100 mM)
Bio Taq
1 μl
0,5 μl
Primer senso (50 μM)
1 μl
Primer antisenso (50 μM)
1 μl
Diversi primers sono stati disegnati per le differenti isoforme di 4.1: R, G, B.
Primers per isoforma R:
Senso:
ATGCACTGTAAGGTTTCTTTGTTGG
Tm: 49 °C
Antisenso:
CAGTCTGAAAAACGTGTGATGT
Tm: 46 °C
Dimensioni frammento atteso :735 bp o 840 bp, a seconda delle varianti da splicing.
Primers per isoforma G:
Senso:
GTGCAGTGTAAAGTGACCCTCT
Tm: 50 °C
Antisenso:
GAAGCAAGCCTGTAGAAAGTATGAT
Tm: 49 °C
Dimensioni frammento atteso: 845 bp.
Primers per isoforma B:
Senso:
AAAAGCATGCAGTGGAAAGTGATAC
Tm: 49 °C
Antisenso:
TAAAAGTAGTCTGAAAAATGTATGTGC
Tm: 46 °C
55
Dimensioni del frammento atteso: 852 bp.
Protocollo PCR
95 °C
5 min
denaturazione iniziale
95 °C
45 s
denaturazione
X °C
45 s
X 35
72 °C
1 min 30 s
72 °C
5 min
X = 46 °C
per isoforma B e R
X = 49 °C
per isoforma G
annealing
allungamento
allungamento finale
4.6 Clonaggio del cDNA per la proteina 4.1Rsh e 4.1RII nei vettori, pEYFP-C1,
pEYFP-N1 e pIRES2-EGFP
Il cDNA esprimente le due varianti da splicing 4.1Rsh e 4.1RII della proteina 4.1R è
stato clonato nel vettore YFP sia in posizione N–terminale rispetto alla YFP nel
pEYFP-N1 (Clontech) che in posizione C–terminale nel pEYFP-C1 (Clontech) e nel
vettore pIRES2-EGFP.
56
Figura 13 Mappe del vettore pEYFP-N1 e pEYFP-C1 in cui è evidenziato: 1- il multiple cloning site
(MCS) che contiene diversi siti di restrizione unici del vettore e rappresenta il punto in cui viene
inserito il cDNA; 2- l’EYFP sequenza codificante per la proteina fluorescente localizzata in 3’ rispetto
all’MCS per pEYFP-N1 e in 5’ rispetto al MCS per pEYFP-C1; 3- l’origine di replicazione SV40 e un gene
r
che conferisce resistenza alla neomicina (Neo ) per la selezione (usando il G418) delle cellule
transfettate stabilmente. Un promotore batterico a monte (P) conferisce resistenza alla kanamicina
r
(Kan ) in E. Coli. Il vettore fornisce anche una origine di replicazione pUC19 per la propagazione in
E.Coli e una origine f1 per la produzione di DNA a singola elica.
Figura 14 Mappa del vettore pIRES2-EGFPin cui è evidenziato: 1- il multiple cloning site (MCS) che
contiene diversi siti di restrizione unici del vettore e rappresenta il punto in cui viene inserito il cDNA;
2- la sequenza IRES (Internal Ribosome Entry Site) tra il MCS e la regione EGFP (Enhanced Green
Fluorescent Protein) corrispondente alla regione codificante per la proteina GFP. Questo permette
alle cellule di produrre da un singolo mRNA bicistronico la proteina di interesse (ICln) e la proteina
GFP come proteine separate ; 3- l’origine di replicazione SV40 e un gene che conferisce resistenza
r
alla neomicina (Neo ) per la selezione (usando il G418) delle cellule transfettate stabilmente. Un
r
promotore batterico a monte (P) conferisce resistenza alla kanamicina (Kan ) in E. Coli. Il vettore
fornisce anche una origine di replicazione pUC per la propagazione in E.Coli e una origine f1 per la
produzione di DNA a singola elica.
57
4.6.1 PCR
Per il clonaggio delle varianti dell’isoforma 4.1R è stata utilizzata una Taq
(Advantage) dotata di maggiore attività proofreading rispetto ad una Taq
convenzionale. I primers, specifici per le due varianti della proteina 4.1R, sono stati
disegnati in modo da possedere una sequenza di basi (18-24) che si appaia in modo
specifico alla sequenza codificante per la proteina, una sequenza riconosciuta da
uno specifico enzima di restrizione (Xho I nel caso del primer senso e Pst I nel caso
dell’antisenso). Sono stati utilizzati primers per l’inserimento del cDNA delle due
varianti della 4.1R nei vettori pEYFP-C1 e pEYFP-N1.
Primer utilizzati per l’isoforma 4.1Rsh:
Primers per YFP-N1
Senso: TAG GAC TCG AGA TGC ACT GTA AGG TTT CTT TGT TG
Tm 58 °C
Antisenso: CAAGGAATTCTCTCATCAGCAATCTCGGTCTC
Tm 42 °C
Primers per YFP-C1
Senso: TAG GAC TCG AGG TAT GCA CTG TAA GGT TTC TTT GTT G
Tm 59 °C
Antisenso: CAA GGA ATT CTC ACT CAT CAG CAA TCT CGG TCT C
Tm 59 °C
Primer utilizzati per l’isoforma 4.1RII:
Primers per YFP-N1
Senso : CAAGCTCGAGATGACAACAGAGAAGAGTTTAGTGAC
Antisenso: CAAGGAATTCTCTCATCAGCAATCTCGGTCTC
58
Tm=42
Tm=41.9°C
Primers per YFP-C1
Senso: CAAGCTCGAGCAATGACAACAGAGAAGAGTTTAGTGAC
Tm=46.4°C
Antisenso: CAAGGAATTCTCACTCATCAGCAATCTCGGTCTC
Tm=48°C
Per l’inserimento dell’isoforma corta 4.1Rsh e dell’isoforma lunga 4.1RII nel vettore
pIRES2-EGFP abbiamo utilizzato i primers per YFP-C1 (specifici per le due isoforme)
precedentemente utilizzati per l’inserimento nel vettore pEYFP-C1.
Miscela di reazione per PCR (volume finale 50 µl)
H2O
40,5 µl
Buffer 10X
5 µl
dNTP (10 mM )
1 µl
Primer senso (50 µM)
1 µl
Primer antisenso (50 µM)
1 µl
Pfu polimerasi (…U/µl)
0,5 µl
DNA pSTBlue1-4.1Rsh ( e RII) (3,5 ng/µl)
1 µl
Protocollo PCR
95 °C
1 min
predenaturazione
95 °C
30 sec
denaturazione
55 °C
45 sec
72 °C
3 min
allungamento
72 °C
5 min
allungamento finale
x 32 cicli
annealing
59
4.6.2 Elettroforesi del DNA
Preparazione gel all’ 1% (w/v)
Alla fine della reazione di PCR è stata eseguita la corsa elettroforetica su gel di
agarosio per tutte le miscele.
Il gel è stato preparato sciogliendo 0,4 g di agarosio in polvere in 40 ml di tampone
TAE (Tris Acetato EDTA) 0.5X. Dopo aver scaldato la miscela nel microonde, finché
l’agarosio non è sciolto completamente, vengono aggiunti circa 0.3 μl di bromuro
d’etidio 10 µg/µl.
La miscela viene versata in un’opportuna vaschetta, viene inserito il pettine per i
pozzetti e si attendono circa 30 min per la solidificazione.
Preparazione campione
Al DNA viene aggiunto un appropriato volume di Loading dye 6X in modo che la sua
concentrazione finale nella miscela sia 1X.
Nei pozzetti del gel sono caricati 6 μl di miscela (5µl DNA + 1 µl Loadyng dye) e 7/8µl
di marker di corsa DNA ladder 1kb oppure 15 µl di mass ruler DNA ladder mix
(Fermentas) per la quantificazione del peso molecolare e della quantità del
campione.
Il gel viene ricoperto di TAE 0,5X e sottoposto ad un voltaggio costante di 100 V per
circa 20 min.
Terminata la corsa si osserva il gel al transilluminatore a raggi UV.
Loading dye 6X
0,2% Blu di bromofenolo,
0,2% xilene cyanol FF,
60% glicerolo,
60 mM EDTA
60
TAE 1X:
40 mM
Tris base
20 mM
sodio acetato
1 mM
EDTA
Il tampone è stato portato a pH 8 con acido acetico.
4.6.3 Purificazione dei prodotti di PCR
E’ stato utilizzato il PCR purification kit della Qiagen per purificare i frammenti di
DNA dalla PCR.
Protocollo:
Sono stati uniti i prodotti delle due PCR mantenendo divisi gli N dai C.
Vengono aggiunti 5 volumi di Buffer PB (contenuto nel kit) per ogni volume
del campione di PCR e si mescola con cura.
La miscela viene caricata con una pipetta in una spin column QIAquick e
centrifugata per 1 min. a 13000 rpm. Il flowthrough è eliminato e la
colonnina viene rimessa nel tubo.
Vengono aggiunti 0,75 ml di Buffer PE (contenuto nel kit) alla colonna e si
procede a centrifugazione per 1 min.
Il flowthrough è eliminato ed è ripetuta una seconda centrifugazione per 1
min.
La spin column è quindi inserita in un’eppendorf pulita da 1,5 ml.
Per eluire il DNA,sono aggiunti 30 µl di acqua milliQ nel centro della colonna
QIAquick. Viene quindi atteso un minuto e in seguito essa viene sottoposta a
centrifugazione per 1 min.
4.6.4 Reazioni di restrizione
In passato i clonaggi della 4.1 si sono rivelati difficili, quindi prima di aggiungere gli
enzimi si è proceduto a riscaldare a 65°C per 3 minuti il DNA per aiutare risolvere
eventuali strutture secondarie, poi abbiamo riportato la temperatura a 37°C e
61
abbiamo aggiunto il buffer e gli enzimi. La reazione è stata lasciata per 16 ore circa a
37°C, per assicurarsi che il DNA fosse digerito completamente, in caso contrario si
avrebbero delle gravi difficoltà per la ligazione.
Miscela di restrizione per i frammenti di DNA:
DNA purificato (vedi paragrafo precedente)
28 µl
H2O MilliQ
1 µl
Buffer Y + TANGO 10X
8 µl
XHO I (10U/ µl)
1.5 µl
ECO RI (10U/ µl)
1.5 µl
sono stati aggiunti dopo
denaturazione del DNA
in acqua a 65 °C per 3
min
Miscela di restrizione per il plasmide YFP –N:
pEYFP–N1 (3,26 µg/ µl)
1 µl
H2O MilliQ
36 µl
Buffer Y + TANGO 10X
10 µl
XHO I (10U/ µl)
1.5 µl
ECO RI (10U/ µl)
1.5 µl
sono stati aggiunti dopo
denaturazione del DNA
in acqua a 65 °C per 3
min
Miscela di restrizione per il plasmide YFP – C:
pEYFP–C1 (2,56 µg/ µl)
1,5 µl
H2O MilliQ
36 µl
Buffer Y + TANGO 10X
10 µl
XHO I (10U/ µl)
1.5 µl
ECO RI (10U/ µl)
1.5 µl
62
sono stati aggiunti dopo
denaturazione del DNA
in acqua a 65 °C per 3
min
4.6.5 Purificazione dei prodotti di digestione
E’ stato utilizzato il MIN ELUTE REACTION CLEANUP kit per purificare i prodotti della
digestione.
Protocollo:
Vengono aggiunti 300 µl di Buffer ERC (contenuto nel kit) per ogni volume di
reazione, mescolando con cura.
La miscela viene caricata con una pipetta nella parte superiore di una spin
column e centrifugata per 1min. a 13000rpm. Il flowthrough è eliminato e la
colonnina viene rimessa nel tubo.
Vengono aggiunti 750 µl di Buffer PE alla colonna e si procede a
centrifugazione per 1 min. a 13000 rpm.
Il flowthrough è eliminato ed è ripetuta una seconda centrifugazione per 1
min. sempre alla massima velocità (13000 rpm).
La spin column è quindi inserita in un’eppendorf pulita da 1,5 ml.
Per eluire il DNA sono aggiunti nel centro della colonna 30 µl di H2O milliQ
per i plasmidi e 10 µl di H2O milliQ per i frammenti. Viene quindi atteso un
minuto e in seguito essa è sottoposta a centrifugazione per 1 min.
4.6.6 Ligazione
Per la ligazione (effettuata con il Fast Ligation kit ver2.1 della Takara) sono state
allestite 3 diverse reazioni con diversi rapporti molari plasmide/frammento e una
reazione di controllo, con il solo plasmide, per la valutazione del “background” dato
dal vettore vuoto richiuso:
63
1:2
1:1
2:1
Ø
Frammento
106 ng
53 ng
26 ng
_
Plasmide
120 ng
120 ng
120 ng
120 ng
Tris/ MgCl2
X µl
X µl
X µl
X µl
Volume finale
6 µl
6 µl
6 µl
6 µl
Dove Ø = controllo con il plasmide vuoto
Protocollo:
Si incubano plasmide, frammento e buffer Tris/ MgCl2 (Tris 100 mM, MgCl 2
5 mM, pH 7,6) a 65 °C per 3 min per denaturare eventuali strutture
secondarie
Si scende a 25 °C e si aggiungono 6 µl di T4 DNA ligasi mix (contenente
buffer, ligasi e ATP) e 0,5 µl di ATP 2 mM
Si imposta un gradiente di temperatura - 25 °C per 5 min
- 24 °C per 15 min
- 22 °C per 25 min
- 18 °C per 30 min
- 16 °C per 1 ora
Per la trasformazione sono stati utilizzati 9 µl di ogni miscela di ligazione 1
4.7 Mutagenesi del sito di inizio ATG2 presente nel plasmide IRES 4.1 RII
Per eliminare il secondo sito di inizio trascrizione ATG2 nel cDNA della sequenza
4.1RII nel vettore plasmidico pIRES-4.1RII abbiamo effettuato una mutagenesi sito
specifica. I primers, specifici sono stati disegnati in modo da possedere una
64
sequenza di basi (18-24) che si appaia in modo specifico alla sequenza codificante
per la proteina ma che portano una variazione di una base che deve essere al centro
della sequenza dei primer.
Primer utilizzati:
Senso: CACAGGAACGTGCACTGCAAGGTTTCTTTGTTGG
Tm=79.2°C
Antisenso: GGTTAGTCTTTTGTGTCCTTGCACGTGACGTTCC
Miscela di reazione per PCR (volume finale 50 µl)
H2O
40 µl
Buffer 10X
5 µl
dNTP (10 mM )
1 µl
Primer senso (50 µM)
1 µl
Primer antisenso (50 µM)
1 µl
Pfu polimerasi (…U/µl)
1 µl
DNA pIRES 4.1 RII (40ng/µl)
1 µl
Protocollo PCR
95 °C
30 sec
predenaturazione
95 °C
30 sec
denaturazione
55 °C
1 min
68 °C
8 mine 15 sec min
x 16 cicli
annealing
allungamento
Digestione con Dpn1
Terminata la PCR alla mix viene aggiunto 1 µl dell’enzima di restrizione Dpn1.
Questo enzima va a tagliare e a degradare il DNA metilato, cioè lo stampo che
deriva dal DNA genomico dei batteri. La mix con Dpn1 viene incubata 1h RT.
Per la trasformazione vengono utilizzati 10 µl della miscela proveniente dalla PCR.
65
4.8 Produzione di batteri competenti
Per procedere con la trasformazione batterica è stato necessario rendere i batteri in
grado di assumere il plasmide.
-sono stati versati 5 ml di terreno di coltura liquido LB in una Falcon a cui
successivamente sono stati aggiunti i batteri XL1Blue, conservati a -80°C, grattandoli
con un‟ansa sterile dal glicerolato;
-la Falcon è stata poi incubata a 37°C overnight;
- il giorno successivo sono stati prelevati 2 ml di coltura che sono stati aggiunti a 200
ml di terreno LB; la coltura è stata fatta crescere fino ad ottenere una densità ottica
pari a circa 0,7;
-i batteri sono trasferiti in provette sterili da centrifuga, lasciati in ghiaccio 30 minuti
e poi centrifugati a 3000 rpm con rotore swing out per 15 minuti a 4° C. Il
sovranatante viene eliminato e il pellet risospeso delicatamente in 2 ml (per 200 ml
di coltura di partenza) di buffer RF1 freddo. Sono stati poi raccolti gli 8 ml totali in
una provetta sterile da centrifuga da 15 ml, lasciati in ghiaccio per 30 minuti e
centrifugati a 3000 g per 10 minuti a 4° C e risospesi in 2 ml di buffer RF2.
-sono state quindi aliquotate frazioni da 100 l in eppendorf sterili,
immediatamente congelate in azoto liquido e tenute a- 80° C fino al momento della
trasformazione.
Soluzioni:
RbCl
Acetato
di
RF1
RF2
0.1 M
10 mM
potassio 60 M
KOAc
CaCl2
10 M
75 mM
glicerolo
15% (v/v)
15% (v/v)
MOPS
pH
10 mM
pH
5.8
con
acetico
66
acido pH 6.8 con NaOH
sterilizzazione per filtrazione.
Preparazione delle piastre e del terreno di crescita per E.Coli
Per la preparazione delle piastre su cui far crescere E. coli sono stati sciolti in acqua
20g/l di LB (Luria Broth- Sigma L-3522) + 15 g/l di agar per batteri e sono stati
autoclavati per 20 minuti a 120°C. Una volta raggiunta una temperatura di circa
50ºC è stato aggiunto sotto cappa sterile l'antibiotico (kanamicina 30 g/ml nel caso
dei vettori utilizzati per il FRET o ampicillina 100 g/ml per i vettori pET e pFLAG) e
la soluzione è stata versata in piastre Petri sterili (circa 30 ml per piastra).
Successivamente, dopo la solidificazione dell'agar, le piastre sono state conservate a
4°C in camera fredda fino al momento dell’utilizzo.
Nel caso del terreno liquido, 20 g di LB sono stati sciolti in 1 l d'acqua bidistillata e
autoclavati.
4.9 Trasformazione batterica
La trasformazione batterica consiste nell’inserire DNA esogeno in cellule riceventi.
Affinché ciò avvenga occorre rendere competenti i batteri. Nel corso del mio stage
ho utilizzato batteri precedentemente resi competenti con la tecnica del calcio
cloruro e tenuti congelati a 80°C (per preservare la competenza) fino al momento
della trasformazione.
Protocollo della trasformazione:
Un’aliquota da 100 µl di batteri competenti XL1–Blue è stata rimossa dal
freezer e messa immediatamente in ghiaccio per 5 min.
Una volta accertato che le cellule fossero scongelate si procede con
l’aggiunta di 1 µl di DNA plasmidico alle cellule; si può miscelare
delicatamente utilizzando il puntale.
L’eppendorf viene lasciata in ghiaccio per 15 min.
67
In seguito si sottopongono i batteri ad uno shock termico portandoli da 4 °C
a 42 °C per 45 secondi e infine lasciati per 2 min. in ghiaccio.
Vengono aggiunti 250 µl di LB addizionato con glucosio 20 mM e si lasciano
riposare i batteri a 37 °C per circa 60 min, in modo tale che i batteri che
hanno acquisito il DNA plasmidico abbiano il tempo di produrre l’enzima
necessario per distruggere la kanamicina del terreno.
Infine vengono piastrati su una piastra petri di LB agar contenente
l’antibiotico per la selezione dei cloni trasformati con DNA plasmidico. Dopo
aver atteso che il liquido fosse completamente assorbito, si procedeva a
invertire la piastra ed a incubare overnight a 37 °C.
4.10 Miniprep
Dopo l’incubazione si prelevano dalla piastra i cloni con uno stuzzicadenti sterile e si
inoculano in 2 ml di terreno liquido LB contenente kanamicina 30 µg/ml. Le colture
vengono lasciate a 37°C, in agitazione, per circa 16 ore. Si procede quindi con
l’estrazione plasmidica, secondo il seguente protocollo:
Si prelevano 1,5 ml da ogni tubo di microbiologia e si mettono in una
eppendorf da 2ml
Si centrifuga per 10 min a 10000 rpm. Il surnatante viene scartato.
Viene risospeso il pellet in 100 µl di soluzione I fredda e viene tenuto 5 min a
temperatura ambiente.
Si aggiungono 200 µl di soluzione II , la cui funzione è quella di portare alla
lisi cellulare e alla denaturazione controllata di proteine e DNA genomico. Si
mescola delicatamente per inversione e si lascia agire per 5 min a
temperatura ambiente
Si aggiungono 150 µl di soluzione III, la cui funzione è quella di far precipitare
le proteine e neutralizzare il pH acido della soluzione II, interrompendo
quindi la reazione di denaturazione. Si mescola delicatamente e si lascia
agire 5 min in ghiaccio.
Si centrifuga per 15 min a 13000 rpm.
68
Si trasferiscono 400 µl di surnatante, che dovrebbe contenere il plasmide, in
una nuova eppendorf.
Il DNA plasmidico viene precipitato in etanolo. Si aggiunge 1 ml di etanolo
100% e si vortexa. Il DNA in etanolo viene quindi posto per 20 min a –80 °C,
per facilitare la sua precipitazione.
Si centrifuga per 30 min a 13000 rpm; e il surnatante viene eliminato.
Si lava il pellet con 750 µl di etanolo 70% freddo.
Si centrifuga per 5 min a 13000 rpm.
Si lascia asciugare il pellet per 10 min ed infine si risospende in 20 µl di H 2O
milliQ.
Al termine dell’estrazione, i DNA plasmidici sono stati controllati tramite analisi di
restrizione e corsa su gel di agarosio.
Miscela di digestione
Plasmide
5 µl
Buffer Y + TANGO 10X
4 µl
XHO I
1 µl
ECO RI
1 µl
H2O MilliQ
4 µl
soluzione 1
soluzione 2
soluzione 3
50 mM glucosio
0.2 N NaOH
3 M KOAc
25 mM Tris pH 8
0.1% SDS
1.5% acido acetico glaciale
10 mM EDTA
pH 4.8
4.11 Maxiprep
Per amplificare i plasmidi d’interesse (YFP–N e YFP–C) siamo partiti inoculando un
singolo clone di una piastra contenente batteri trasformati col plasmide d’interesse
in 1 ml di terreno LB addizionato dell’antibiotico kanamicina 30 µg/ml.
69
Il pre-inoculo è stato fatto crescere per 7/8 ore a 37 °C in agitazione e quindi diluito
in 250 ml di LB + antibiotico e fatto crescere overnight a 37 °C con agitazione.
La mattina le cellule batteriche sono state raccolte in un provettone da 250 ml e
centrifugate a 3500 x g, 45 min a 4 °C. Si è proceduti all’estrazione del DNA
plasmidico utilizzando il kit della Nucleobond.
Protocollo:
Il pellet contenuto nei provettoni viene risospeso in 12 ml di buffer S1
freddo.
Si aggiungono 12 ml di buffer S2 a temperatura ambiente e si agita con
cautela il provettone 6-8 volte. Si aspettano poi 5 min a temperatura
ambiente.
Si aggiungono 12 ml di buffer S3 freddo, è il buffer di neutralizzazione:
riporta il PH neutro e rinatura. Si agita nuovamente il provettone 6-8 volte e
poi si aspettano 5 min in ghiaccio.
Il lisato viene chiarificato per filtrazione su carta e applicato alla colonna
contenente (per legare gli acidi nucleici), pre-equilibrata con 6 ml di buffer
N2.
Si procede lavando la colonna con 18 ml di Buffer N3. L’operazione viene
ripetuta una seconda volta.
Il DNA plasmidico viene eluito con 15 ml di Buffer N5.
Per far precipitare il DNA si aggiungono 10 ml di isopropanolo e si agita
utilizzando un vortex.
Si procede a centrifugare con una ultracentrifuga a 15000xg per 30 min a
4°C.
Il DNA eluito è lavato con etanolo al 70% a 4 °C, per eliminare residui di sale
e in seguito centrifugato a 12000 rpm per 10 min a 4 °C.
Una volta che il DNA si è asciugato, si procede a risospendere in 200 µl di
H2O milliQ.
Tutti i buffer sono forniti dal produttore del kit.
70
Il DNA plasmidico estratto, è stato quindi dosato con uno spettrofotometro,
controllato tramite analisi di restrizione e mandato a sequenziare (servizio
sequenziamento esterno Cogentech, Campus IFOM-IEO).
4.12 Dosaggio acidi nucleici
Si usa una cuvetta di quarzo ( trasparente agli UV ), che viene riempita con 350 µl di
TE: (Tris 10 mM, EDTA 1 mM, pH 7,5.
Viene effettuata una prima lettura allo spettrofotometro del solo TE per fare il
bianco: il valore letto verrà sottratto dalla lettura fatta in presenza del DNA.
Successivamente ai 350 µl di TE si aggiungono 5 µl di plasmide direttamente in
provetta, si rimescola e si misura il valore di assorbanza a cui verrà sottratto quello
del bianco.
La lettura viene effettuata a due diverse lunghezze d’onda: 260 nm (per il DNA) e
280 nm (per misurare la concentrazione delle proteine). Il rapporto tra le letture
alle due diverse lunghezze d’onda ci dà un indice della qualità della preparazione
del DNA. Una preparazione ottimale di DNA plasmidico deve avere deve avere un
rapporto di circa 1,9. Il calcolo viene effettuato sapendo che, per un cammino ottico
pari ad 1 cm , una soluzione acquosa di DNA a pH 7–7,5 ad una concentrazione di
50ng/µl ha un’assorbanza pari a 1. La formula utilizzata è la seguente:
[DNA] µg /µl = A260 X 0,05 (µg /µl) x 350/5
4.13 Trasfezione
La transfezione consiste nel trasferimento di molecole di DNA esogeno in cellule
riceventi. Il DNA, una volta transfettato, viene mantenuto per un limitato periodo di
tempo come DNA extra-crosomiale e il prodotto della sua espressione si mantiene
stabile per un determinato periodo, in genere due o tre giorni (transfezione
transiente).
Per
la
transfezione
è
stato
sfruttato
il metodo
del
PEI
(polietilenimmina), una molecola carrier nel cui scheletro sono presenti gruppi
71
amminici facilmente protonabili. A pH neutro essa è dotata di una elevata carica
cationica, che permette di legare il DNA con alta efficienza; questo consente al DNA
di entrare per endocitosi in cellula grazie al legame con i proteoglicani cellulari.
Inoltre il PEI garantisce che i complessi formatisi non vengano poi degradati dalla
cellula.
Il giorno della transfezione (il giorno successivo alla semina delle cellule) vengono
preparate due diverse miscele A e B: la prima composta da DNA e NaCl 150 mM
(tamponato con HEPES 10mM, pH 7,4) e la seconda contenente PEI (jetPEI,
Euroclone) e NaCl 150 mM, Hepes 10 mM, pH 7. Dopo aver lasciato a temperatura
ambiente per 5 minuti, il contenuto delle due Eppendorf è unito, mescolato e
lasciato a temperatura ambiente per 15 minuti. Al termine, la miscela di
transfezione è stata aggiunta goccia a goccia in ciascuna piastra. A distanza di 7-8
ore dalla transfezione, il terreno è stato sostituito con terreno completo fresco.
Composizione delle mix (per la transfezione in piastre da 3 cm):
Mix A
NaCl 150 mM + Hepes
DNA (0,5 µg/µl)
Volume totale Mix A
10 mM, pH 7,4
4 µl (2 µg)
46 µl
50 µl
Mix B
NaCl 150 mM + Hepes
PEI
Volume totale Mix B
10 mM, pH 7
4 µl
46 µl
50 µl
Mix Totale
Volume Mix Totale
100 µl
72
Composizione delle mix (per la transfezione in piastre da 10 cm):
Mix A
NaCl 150 mM + Hepes
DNA (0,5 µg/µl)
Volume totale Mix A
10 mM, pH 7,4
30 µl (15 µg)
220 µl
250 µl
Mix B
NaCl 150 mM + Hepes
PEI
Volume totale Mix B
10 mM, pH 7
30 µl
220 µl
250 µl
Mix Totale
Volume Mix Totale
500 µl
4.14 FRET, Fluorescence Resonance Energy Transfer
Il FRET è una nanotecnologia che può fornire informazioni sulle interazioni proteinaproteina.
La tecnica del FRET è basata sul trasferimento di energia tra due fluorofori che sono
detti donatore e accettore. Tale processo si verifica quando il fluoroforo donatore,
eccitato da luce ad una lunghezza d’onda appropriata trasferisce parte dell’energia
al fluoroforo accettore attraverso un’interazione di tipo dipolo-dipolo. Una delle
condizioni fondamentali affinché ci sia FRET è che i due fluorofori siano a distanza di
pochi nanometri (non oltre 10 nm).
73
Figura 15 Modello schematico della FRET. Nel pannello superiore della figura il donatore (in azzurro)
viene eccitato ma la distanza dall'accettore (in giallo) non consente il passaggio di energia tra le due
molecole. Nel pannello inferiore il donatore viene eccitato ed emette energia che riesce ad eccitare
l'accettore grazie alla stretta vicinanza tra i due fluorofori.
Per generare FRET, accettore e donatore devono avere specifiche caratteristiche.
Una di queste è che lo spettro d’emissione del donatore abbia sufficiente
sovrapposizione con lo spettro di eccitazione dell’accettore.
I due fluorofori da noi utilizzati nell’esperimento sono due proteine derivate dalla
GFP (Green Fluorescence Protein). Noi abbiamo utilizzato come donatore una
variante blu, CFP (Cyan Fluorescence Protein) e come accettore una variante gialla,
YFP (Yellow Fluorescence Protein). CFP/YFP è la coppia di fluorofori più utilizzata nel
FRET per studiare le interazioni proteina–proteina.
Per studiare l’interazione tra due proteine, donatore e accettore vengono fusi
all’estremità delle proteine di interesse e le rispettive proteine di fusione vengono
over-espresse in cellula. Le sequenze proteiche di interesse possono essere inserite
o all’estremità amino–terminale o all’estremità carbossi - terminale delle proteine
fluorescenti. Solo se le due proteine interagiranno direttamente, donatore e
accettore si troveranno sufficientemente vicini da permettere il trasferimento di
energia e generare un segnale di FRET. Nel nostro esperimento la proteina ICln è
stata coniugata al donatore CFP (fornitoci da Prof Hannes Università di Salisburgo)
mentre la proteina 4.1 è stata coniugata all’accettore YFP.
74
4.14.1 Tecnica dell’Acceptor Photobleaching
Una delle tecniche per misurare l’efficienza del FRET è l’ “acceptor photobleaching”
(Kenworthy AK, 2001).
Il metodo si basa sul seguente principio: l’energia trasferita dal donatore
all’accettore è ridotta o eliminata quando l’accettore è sottoposto a “bleaching”,
ovvero alla distruzione delle molecole dell’accettore, eccitando con un laser a
potenza massima per un tempo opportuno.
Questo metodo prevede pertanto tre fasi:
Acquisizione dell’immagine del donatore
Photobleaching dell’accettore
Nuova acquisizione dell’immagine del donatore
Il FRET viene poi calcolato utilizzando il software Image J sulla base della variazione
d’intensità di fluorescenza del donatore secondo la seguente formula:
FRET Eff = Dpost - Dpre/ Dpost
Dove per Dpost si intende l’intensità di fluorescenza del donatore (CFP) dopo
l’“acceptor photobleaching” e per Dpre l’intensità di fluorescenza di CFP prima
dell’“acceptor photobleaching” nell’area sottoposta a photobleaching.
Per intensità di fluorescenza si intende la somma delle intensità di fluorescenza dei
singoli pixel nella regione sottoposta a bleaching. Sono stati considerati per il
calcolo solo quei pixels che, sia nell’immagine del donatore che nell’immagine
dell’accettore, superano una certa soglia di intensità di fluorescenza (20 su 255)
Nel nostro esperimento per l’acquisizione dell’immagine del donatore è stato usato
il microscopio confocale Leica TCS SP2 AOBS dotato di laser Argon. Per l’eccitazione
del donatore è stata utilizzata la linea a 458 nm , mentre per l’eccitazione e il
photobleaching dell’accettore è stata utilizzata la linea a 514 nm. Le rispettive
finestre di emissione sono state impostate in modo da minimizzare il cross – talk tra
i canali CFP e YFP, rispettivamente a 465–505 nm e 525–600 nm. Le immagini sono
state acquisite a zoom 2 e alla risoluzione di 512x512 (line average 2, 8 bit),
posizionando il fuoco in un piano centrale sull’asse z della cellula. Per il
75
photobleaching, dopo aver posto lo zoom a 10X e portato la risoluzione a
1024x1024, sono state effettuate 3 diverse scansioni con il laser al 100% di potenza.
Per gli esperimenti di FRET, la sera prima della trasfezione le cellule sono state
piastrate in una petri da 3,5 cm di diametro con il fondo in vetro, a una
concentrazione di circa 1,5x105 cellule/ml (2 ml per petri).
Gli esperimenti di FRET sono stati effettuati a 24 ore dalla trasfezione delle cellule
HEK-293. Le cellule, tenute in terreno MEM, sono state lavate con la soluzione
isotonica e tenute in questa stessa soluzione per almeno 5 minuti prima di
procedere alla acquisizione delle immagini. Per ogni petri, dopo avere acquisito le
immagini necessarie su più cellule in isotonica, la soluzione è stata cambiata con
una ipotonica, in cui le cellule sono rimaste per 10 minuti, prima di procedere alla
acquisizioni delle immagini su nuove cellule.
4.14.2 "Sensitized emission"
Per verificare una variazione dell’interazione nel tempo tra la proteina ICln e la
proteina 4.1 in ipotonia abbiamo utilizzato la tecnica della Sensitized emission.
Tutte le immagini sono state acquisite a temperatura ambiente mediante
microscopio invertito Leica DM-IRE2 (TCS SP2 AOBS scanhead), dotato di un laser
argon e di un obiettivo di immersione a olio 63X. CFP è stato eccitato con un raggio
laser di 458 nm e la finestra di emissione è stata settata a 465-505 nm; YFP è stato
eccitato con un raggio laser di 514 nm e la finestra di emissione è stata settata a
525-600 nm. In entrambi i casi le immagini sono state acquisite con una risoluzione
di 512x512, line average 2, 8 bit.
La "sensitized emission" (Fsen) è stata calcolata come descritto in van Rheenen et al.
Van RJ et al., 2004). L’entità del cross-talk tra l'accettore ed il donatore è stata
stimata attraverso i parametri α, γ, δ e β. Questi fattori di correzione sono definiti
come:
M Dy
M DAy
M IAy
M DAy
M Dy
M IAy
76
Le y indicano che la fluorescenza misurata è determinata dai campioni che
esprimono solo YFP mediante gli stessi parametri di acquisizione usati negli altri
esperimenti.
M IAc
M Dc
Le c indicano che la fluorescenza misurata è determinata dai campioni che
esprimono solo CFP mediante gli stessi parametri di acquisizione usati negli altri
esperimenti.
Le M rappresentano la fluorescenza (sottratta del background) misurata nelle
regioni d'interesse (ROI) ottenuta nei seguenti modi:
MD = eccitazione del donatore + emissione del donatore;
MIA = eccitazione del donatore + emissione dell'accettore;
MDA = eccitazione dell'accettore + emissione dell'accettore
Il valore della "sensitized emission" (Fsen) è stato ottenuto dalla seguente formula:
Fsen
M IA M D M DA
1
(iii)
Gli indici di N-FRET sono stati calcolati in accordo con Xia e Liu (Xia et al., 2001) nelle
regioni di interesse (ROIs), utilizzando la seguente formula:
77
NFRET
Fsen
M D M DA
(iv)
Le immagini relative all'N-FRET sono state ricavate applicando l'equazione (iv)
direttamente alle immagini di FRET, ottenute eccitando il donatore e raccogliendo
l’emissione dell’accettore.
Soluzione isotonica:
NaCl 90 mM, KCl 5 mM, CaCl2 2 mM, MgCl2 2 mM, glucosio 5 mM, HEPES 10 mM,
mannitolo 90 mM, pH 7,4.
Soluzione ipotonica:
NaCl 90 mM, KCl 5 mM, CaCl2 2 mM, MgCl2 2 mM, glucosio 5 mM, HEPES 10 mM,
mannitolo 10 mM, pH 7,4.
4.15 Studi di immunocitochimica
Le cellule sono state seminate su un vetrino copri oggetto quadrato (2x2 cm 2),
precedentemente deposto sul fondo di ciascuna petri da 3 cm. 24 ore dopo la
semina le cellule (al 50-70% di confluenza) sono state transfettate con il plasmide
pEYFP-C1 4.1Rsh e come controllo con il plasmide pEYFP vuoto.
Dopo 24h dalla transfezione:
si lavano le cellule due volte in PBS 1X con 2 ml per togliere gli eventuali
residui di terreno;
si fissano le cellule aggiungendo in ogni pozzetto 1 ml di formaldeide al 3% in
PBS per 10 minuti a temperatura ambiente;
successivamente si effettuano tre lavaggi con 2 ml di PBS con glicina 0,1 M.
Per permettere la permeabilizzazione delle membrane e quindi l’ingresso
dell’anticorpo primario in cellula, si aggiunge in ciascuna petri 1 ml di PBS
78
addizionato di MgCl2 3mM e Triton 0,1% per 3 minuti a 4°C. Questo processo è
necessario per gli anticorpi che riconoscono proteine intracellulari o proteine di
membrana con epitopi intracellulari.
Dopo permeabilizzazione si lavano i vetrini 3 volte con PBS. Per ridurre un eventuale
legame aspecifico dell’anticorpo, le cellule sono esposte 1 ora a temperatura
ambiente ad una soluzione di blocco contenente BSA al 3%. La BSA compete con
l’anticorpo per i siti di legame. Terminato il blocco le cellule vengono incubate con
anticorpo primario anti-ICln diluito 1:100 in PBS contenente BSA 0,1% over-night a
4°C.
Dopo l’incubazione con anticorpo primario si lavano le cellule tre volte con PBS per
5 minuti. Successivamente vengono esposte all’anticorpo secondario anti-rabbit
diluito 1:400 in PBS contenente BSA 0,1%, coniugato a Cy5, molecola con un
massimo d’eccitazione a λ=650 nm (rosso) ed un massimo di emissione a λ=670 nm
(rosso lontano) per un'ora a temperatura ambiente. Da questo momento si procede
lavorando al buio in quanto gli anticorpi secondari sono fotosensibili.
Si effettuano tre lavaggi in PBS. I vetrini sono stati montati rovesciati su un vetro
porta
oggetto,
in
glicerolo
90%
addizionato
di
DABCO
(1,4-
diazabicyclo[2.2.2]octane), come agente antifading. Successivamente i vetrini sono
contornati con smalto per evitare l’essicazione all’esposizione all’aria. Infine sono
osservati al microscopio confocale Leica (TCS SP2 AOBS), con un obbiettivo 40X a
immersione e le immagini sono state acquisite con il software in dotazione della
Leica, con una risoluzione 1024x1024 (8 bit), zoom 1X, con i seguenti parametri di
eccitazione/emissione: per il Cy5 è stata usata la linea laser 633nm per l’eccitazione
e l’emissione è stata raccolta a 640-720; per il YFP è stata usata la linea laser a
514nm per l’eccitazione e l’emissione è stata raccolta nella finestra 525-590 nm.
4.16 Estrazione proteine totali
A 24 ore dalla transfezione:
si toglie il terreno dalla piastra petri da 3 cm;
79
si effettua un lavaggio con 1 ml di PBS 1X;
si aggiunge 1 ml di PBS 1X;
le cellule vengono staccate utilizzando uno scraper e poi raccolte in
Eppendorf;
la Eppendorf viene centrifugata per 10 minuti a 230 g a 4°C;
il sovranatante viene eliminato.
Il pellet di cellule viene lisato in 70 µl di Buffer di Lisi (addizionato di inibitori delle
proteasi). Successivamente si procede ad un ciclo di congelamento in azoto liquido
e scongelamento, per favorire la lisi delle cellule, seguito da una centrifugata per 5
minuti a 4500 g a 4°C per eliminare i debris cellulari. Dal sovranatante così ottenuto
vengono prelevati 5 µl che serviranno per i dosaggi proteici. Il resto viene
conservato a -80°C fino al momento dell’utilizzo.
Composizione del Buffer di Lisi:
Tris-HCl
20 mM
NaCl
150 mM
EDTA
1 mM
NP40
1%
pH
7
4.17 Estrazione proteine nucleari e citosoliche
A 24h dalla transfezione si effettua un lavaggio in PBS 1X della Petri da 10.
Successivamente si aggiunge 1 ml di PBS, le cellule vengono staccate utilizzando uno
scraper e poi raccolte in Epperdorf.
Si centrifuga a 230 G a 4°C per 10 minuti;
il sovranatante viene eliminato e il pellet di cellule viene risospeso in 200 µl
di Sucrose Buffer con NP-40;
si incuba in ghiaccio per 5 min per lisare le cellule;
80
si effettua una centrifugata a 1500 g per 5 min per pellettare i nuclei.
Si trasferisce il sovranatante (frazione citoplasmatica) in una nuova
Eppendorf. Dal sovranatante così ottenuto vengono prelevati 5 µl che
serviranno per i dosaggi delle proteine citoplasmatiche. Il resto viene
conservato a -80°C fino al momento dell’utilizzo;Il pellet (frazione nucleare)
invece viene risospeso delicatamente in 1 ml di Sucrose Buffer senza NP-40;
si effettua poi una centrifugata per pellettare i nuclei a 1500 g per 5 min e si
elimina il sovranatante;
si risospendono i nuclei in 50 μl di Low Salt Buffer e poi si aggiungono 10 µl
di High Salt Buffer e si agita delicatamente;
si continua ad aggiungere 10 µl di High Salt Buffer fino al raggiungimento del
volume totale (50 μl) oppure fino a quando i nuclei iniziano a rompersi e la
viscosità del campione aumenta.
I campioni vengono poi incubati su una piattaforma rotante a 4°C per 20 min e
successivamente centrifugati a 13000 g per 15 min. Il sovranatante è la frazione
solubile nucleare, viene prelevato e trasferito in una nuova Eppendorf. Dal
sovranatante così ottenuto vengono prelevati 5 µl che serviranno per i dosaggi
proteici. Il resto viene conservato a -80°C fino al momento dell’utilizzo.
Composizione del Sucrose Buffer With NP-40:
Sucrose Buffer W/O NP-40 1 ml
NP-40 (igepal)
5 µl
81
Composizione del Sucrose Buffer W/O NP-40:
Sucrose
1M
3,2 ml
CaCl2
0,1 M
300 µl
MgAc
1M
20 µl
EDTA
250 mM
4 µl
DTT
100 mM
100 µl
PMSF
100 mM
50 µl
H2O
6,326 ml
Composizione del Low Salt Buffer:
HEPES ph 7,9
1M
Glicerolo
200 µl
2,5 ml
MgCl2
1M
15 µl
KCl
1M
200 µl
EDTA
250 mM
8 µl
DTT
100 mM
100 µl
PMSF
100 mM
50 µl
H2O
6,927 ml
82
Composizione del High Salt Buffer:
HEPES pH 7,9
1M
Glicerolo
200 µl
2,5 ml
MgCl2
1M
15 µl
KCl
3M
2,67 ml
EDTA
250 mM
8 µl
DTT
100 mM
100 µl
PMSF
100 mM
50 µl
H2O
4,357 ml
4.18 Estrazione proteine totali di membrana
A 24h dalla transfezione si effettua un lavaggio in PBS 1X della Petri da 10.
Successivamente si aggiunge 1 ml di PBS, le cellule vengono staccate utilizzando uno
scraper e poi raccolte in Epperdorf.
Si centrifuga a 1000g per 10 min a 4°C.
Si trasferisce il sovranatante (frazione citoplasmatica) in una nuova
Eppendorf. Dal sovranatante così ottenuto vengono prelevati 5 µl che
serviranno per i dosaggi delle proteine citoplasmatiche. Il resto viene
conservato a -80°C fino al momento dell’utilizzo; il pellet (frazione di
membrana) invece viene risospeso delicatamente in500ul di Buffer Fosfato
(addizionato con inibitori delle proteasi).
Successivamente si procede a tre cicli di congelamento in azoto liquido e
scongelamento e con una siringa da insulina si siringa il campione 30-40
volte, per favorire la lisi delle cellule
83
Si aggiunge 1 ml di soluzione di saccarosio in ciascun campione.
Si centrifuga a 1000g per 10 min a 4°C. Al termine della centrifugata si
raccoglie il sovranatante lo si trasferisce in una nuova eppendorf e si elimina
il pellet.
Il sovranatante viene centrifugato a 1000g per 30 min a 4°C
Il pellet che si è formato viene lavato risospendendolo per 2 volte in 1ml di
PBS, mescolando bene per inversione e infine centrifugando a 1000g per 10
min a 4°C.
Si elimina il sovranatante e si riso spende il pellet in Buffer di lisi (addizionato di
inibitori delle proteasi). Dal sovranatante così ottenuto vengono prelevati 5 µl che
serviranno per i dosaggi proteici. Il resto viene conservato a -80°C fino al momento
dell’utilizzo.
Soluzioni
Buffer Fosfato
NaH2PO4
8.3 mM
Na2HPO4
11.6mM
pH=7.3
Soluzione di saccarosio 0.5M in 0.02M Tris Buffer pH=7.3
Composizione del Buffer di Lisi:
Tris-HCl
20 mM
NaCl
150 mM
EDTA
1 mM
NP40
1%
pH
7
84
4.19 Dodaggio proteico
Per misurare la concentrazione delle proteine viene utilizzato il metodo di Bradford
(1979). Tale sistema consiste nell’impiego di un colorante costituito da una
soluzione acida di Comassie Brillant Blue G-250 (Sigma) contenuto in un reattivo
formato da acido fosforico ed etanolo (Biorad Protein Assay kit). Il colorante
anionico si lega alle proteine causando una variazione di colore che è possibile
rilevare attraverso una variazione di assorbanza quantificabile a 595 nm. Per il
dosaggio servono 1 ml di reattivo diluito 1:5 in cui viene aggiunto il campione. Il
dosaggio delle proteine viene misurato allo spettrofotometro; per risalire dal valore
di assorbanza alla concentrazione, è necessario allestire una retta di taratura,
servendosi di diluizioni seriali di albumina da siero bovino a concentrazioni note. La
quantificazione dei campioni avviene leggendo l’assorbanza a λ=595 nm. Dopo
essere risalita alla concentrazione proteica è possibile preparare i campioni per
effettuare la corsa elettroforetica.
4.20 Coimmunoprecipitazione
Per effettuare gli esperimenti di coimmunoprecipitazione abbiamo co-transfettato
per ogni caso di interesse tre petri da 10:
pFLAG human ICln
pEYFP-N1 4.1Rsh
pEYFP-C1 4.1Rsh
pFLAG BAP
pFLAG human ICln
pEYFP-N1 4.1Rsh
controllo
pEYFP-C1 4.1Rsh
controllo
pEYFP-N1 4.1 RII
pEYFP-C1 4.1RII
pFLAG BAP
pEYFP-N1 4.1RII
controllo
pEYFP-C1 4.1RII
controllo
85
A 24h dalla transfezione si effettua un lavaggio in PBS 1X delle Petri da 10.
Successivamente si aggiunge 1 ml di PBS, le cellule vengono staccate utilizzando uno
scraper e poi raccolte in Falcon.
Si centrifuga a 1000g per 10 min a 4°C.
Si elimina il sovranatante e il pellet viene risospeso (per 3 petri da 10) in 3 ml
di Buffer di lisi (addizionato con inibitori delle proteasi).
Successivamente si procede a congelare in azoto liquido e scongelare per
favorire la lisi delle cellule.
Si centrifuga a 1000g per 5 minuti a 4°C per eliminare i debris.
Successivamente si aliquota il sovranatante in Eppendorf.
I lisati vengono dosati con metodo Bradford
Si utilizza una resina, l’α – FLAG M2 Affinity Gel (Sigma), costituita da anticorpi
monoclonali α – FLAG coniugati ad agarosio. La resina viene preparata seguendo le
istruzioni del produttore. Si usano 100μl totali di resina che vengono centrifugati a
5000 rpm per 5 minuti per eliminare il sovranatante (glicerolo).
Prima di procedere la resina viene avvinata con il buffer di lisi e nuovamente
pellettata e separata dal surnatante; quindi viene aggiunta ad un volume di
campione corrispondente a 5 mg di proteine totali, secondo il dosaggio ottenuto col
metodo Bradford, e viene lasciata in incubazione a 4°C per 2 ore.
Al termine dell’incubazione la resina viene nuovamente centrifugata a 1000 g per 5
minuti per separare il Flow-through (contenente tutte le proteine non legate alla
resina) dalla resina stessa. Questa viene poi lavata due volte con wash buffer e
nuovamente separata per centrifugazione.
Sucessivamente la resina viene trasferita in colonnine (Biorad) che trattengono la
resina ma lasciano passare la soluzione. La resina viene quindi lavata ulteriormente
con 500 μl di wash buffer per 4 volte, si procede quindi con un ultimo lavaggio con
PBS e infine vengono fatte quattro eluizioni sequenziali con 40 μl di flag peptide 130
μg/ml.
86
Buffer di lisi:
Tris HCl
25 mM
NaCl
150mM
Glicerolo
10%
Triton X100
0.5%
Buffer di lavaggio:
Tris HCl
25 mM
NaCl
150mM
Glicerolo
10%
Triton X100
0.1%
4.21 Western blot
Elettroforesi SDS PAGE
L'elettroforesi su gel di poliacrilammide (PAA) viene condotta in condizioni
denaturanti (SDS-PAGE), utilizzando un metodo di tipo discontinuo, così chiamato
per le differenze di concentrazione e di pH esistenti tra running gel e stacking gel. La
corsa viene effettuata in presenza di sodiododecilsolfato (SDS) e di βmercaptoetanolo. L’SDS è un detergente anionico in grado di legarsi alle proteine,
provocandone la denaturazione. In queste condizioni le proteine assumono una
carica negativa che consente loro di migrare verso il polo positivo, pertanto la
separazione avviene solo in funzione del peso molecolare.I gel di poliacrilammide
per l’SDS-PAGE: sono formati da due fasi distinte, il "running" gel, che permette la
separazione delle proteine sulla base del loro peso molecolare, e lo "stacking" gel,
che impacca le proteine sul fronte di corsa. Nei western blot relativi ai livelli di
espressione totale di ICln sono caricati 10 µg di proteine per campione, mentre negli
esperimenti relativi alla separazione nucleo citosol per i campioni dei nuclei
vengono caricati 20 µg, mentre per il citosol 10 µg. Prima di caricare i campioni, ai
87
preparati è aggiunto Sample Buffer 4X in modo da risultare in concentrazione finale
1X. I campioni sono immediatamente denaturati attraverso bollitura per 3 minuti,
quindi centrifugati per pochi secondi ad alta velocità e caricati nei pozzetti del gel. I
campioni e il marker (Page ruler Prestained Protein Ladder, Fermentas, SM0671)
sono fatti correre in un gel per elettroforesi SDS-PAGE (13% acrilammide). La corsa
è effettuata a voltaggio costante di 120 V.
Soluzioni:
Sample Buffer 4X
Tris-HCl pH 6,8
Buffer di corsa 1X
240 mM Tris-base 25 mM
SDS 8% (w/v)
Glicina 200 mM
Glicerolo 40% (w/v)
SDS 0,1%
Blue di bromo fenolo 0,008% (w/v) pH 8,3
β-mercaptoetanolo 2,5% (w/v)
Stacking gel 4% Running gel 13%
Miscela di acrilammide
bisacrilammide (30% e 0,8%)
H2O bi-distillata
0,87 ml
6,5 ml
4,07 ml
4,52 ml
0,5 M Tris-HCl pH 6,8
1,67 ml
-
1,5 M Tris-HCl pH 8,8
-
3,75 ml
10% SDS
66,7 µl
150 µl
10% APS
33,3 µl
75 µl
6,7 µl
7,5 µl
6,72 ml
15 ml
Temed
Volume finale
88
Trasferimento
Al termine della corsa elettroforetica si elimina lo Stacking Gel e si trasferiscono le
proteine del Running Gel su una membrana di PVDF (polyvinylidene fluoride) per
mezzo di un elettroblotter. Si pre-equilibrano nel Buffer di Trasferimento la
membrana pretrattata (45 secondi in metanolo, lavate in acqua per due minuti), il
gel, due fogli di carta da filtro 3M e due spugnette. La membrana viene poi posta a
contatto con il gel. Membrana e gel vengono messi tra i due fogli di carta da filtro e
il tutto poi posizionato tra le due spugnette (verificando la mancanza di bolle d'aria
che potrebbero compromettere il trasferimento). Dopo aver immerso il tutto nel
Buffer di Trasferimento, il trasferimento avviene tra due elettrodi di platino (con
membrana rivolta all'anodo), con voltaggio di 80V per 120 minuti a 4°C.
Al termine del trasferimento, il gel è colorato con Blue di Coomassie R-205 (Sigma)
0,4% per 20 minuti e poi decolorato per 2 ore con acido acetico al 10% in agitazione
rotante per controllare l’esito della corsa. La membrana invece viene sciacquata in
TBST (Tween 0.1%) per eliminare i residui di buffer di trasferimento.
Composizione del Buffer di Trasferimento:
Metanolo 10%
Tris base
48 mM
Glicina
39 mM
Blocco e incubazione con anticorpo primario e secondario
La membrana è messa a contatto con una soluzione di bloccaggio TBST addizionato
a polvere di latte 5% per un'ora a temperatura ambiente o over night a 4° C (a
seconda della proteina) in agitazione in modo di andare a saturare i siti aspecifici
della membrana cui potrebbe andare a legarsi l'anticorpo. Successivamente la
membrana è incubata con l'anticorpo primario specifico diluito in TBST e polvere di
latte 5%. Al termine la membrana incubata con anti-ICln viene lavata con TBST
addizionato a polvere di latte al 5% per tre volte 10 min, mentre le membrane
89
incubate con anti-GAPDH, anti-Laminina, anti-4.1, anti-Na+ K+ ATPasi, anti GFP, anti
Flag e anti-caderina vengono lavate con TBST. La membrana viene quindi incubata
in agitazione orbitante un'ora con una soluzione 1:20000/10000 dell'anticorpo
secondario corrispondente coniugato a perossidasi di rafano (HRP), diluito in TBST
addizionato con polvere di latte al 5%. Infine si susseguono tre lavaggi da 10 minuti
e due da 5 ciascuno in TBST.
Combinazione proteina–blocco-anticorpo primario–anticorpo secondario:
Proteina
Blocco
Anticorpo Primario
Anticorpo
secondario
ICln
1 h RT milk 5% TBS-T
Anti-ICln (1:1000) O/N Anti-rabbit
4°
(1:20000)
(Pierce)
4.1
1 h RT milk 5% TBS-T
Anti-4.1R (1:1000) O/N Anti-goat
4° (Santa Cruz)
GFP
1 h RT milk 5% TBS-T
Laminina
1 h RT milk 5% TBS-T
Flag
Caderina
(1:10000)
Anti.GFP (1:1000) O/N Anti-rabbit
(1:20000)
4°
(Pierce)
Anti-laminina A/C
Anti-rabbit
(1:1000) O/N 4 °
(1:20000)
(Santa Cruz)
(Pierce)
O/N 4 °C milk 5% TBS- Anti-Flag (1:2000) 1hRT
Anti
mouse
T
(Sigma)
(1:10000) (Pierce)
1 h RT milk 5% TBS-T
Anti-caderina O/N 4°C
Anti-rabbit
(1:1000)
(1:20000)
(Pierce)
Na+ K+ ATPasi O/N 4 °C milk 5% TBS- Anti-Na+ K+ ATPasi 1hRT Anti
T
mouse
(1:20000) (Pierce)
(1:20000) (Millipore)
GAPDH
1 h RT milk 5% TBS-T
Anti-GAPDH (1:10000)
Anti
mouse
(1:10000) (Pierce)
90
Composizione del TBST:
Tris-HCl
15 mM
NaCl
150 mM
Tween
0,1%
pH 8,8
Sviluppo
Il kit utilizzato per lo sviluppo è il kit per chemioluminescenza Immobilon della
Millipore. La membrana viene ricoperta per circa 3 minuti con una soluzione
contenente Luminol, un substrato della perossidasi che, quando viene ossidato in
presenza di H2O2 e in condizioni alcaline, passa ad uno stato eccitato. Il
decadimento dello stato eccitato allo stato fondamentale avviene per un’emissione
di luce che, in seguito a brevi esposizioni (fino a 1 ora circa), può impressionare un
film per autoradiografia sensibile alla luce blu.
Stripping
Dopo lo sviluppo le membrane incubate con un anticorpo prima di essere incubate
nuovamente con un anticorpo primario devono essere strippate per eliminare i
residui di anticorpo legato alla membrana.
Si effettua un lavaggio veloce (1 minuto) in acqua di soluzione;
si lava per 1 minuto con soluzione di Stripping;
si lava con soluzione di Stripping per 40 min;
si effettuano due lavaggi da 5 minuti in acqua da soluzione;
si equilibra la membrana in TBST 0,1%.
Si effettua uno sviluppo di controllo per verificare che tutto l’anticorpo non sia più
legato. La membrana viene messa a contatto con una soluzione di bloccaggio TBST
addizionato a polvere di latte 5% per un'ora, a temperatura ambiente e in
91
agitazione, poi si prosegue l’incubazione della membrana con anticorpo primario e
secondario specifici.
Soluzione di Stripping:
Glicina
0,05 Moli
SDS
1%
pH 2,2 – 2,3 (con HCl)
Colorazione Amido black di membrane in PVDF
Al termine del western blot le membrane vengono messe a colorare con una
soluzione di Amido Black staining solution (Biorad) in agitazione per tre minuti.
Successivamente vengono lavate velocemente con H20 di soluzione, poi vengono
messe in contatto con la soluzione Destain solution per 3 min sempre in agitazione
per decolorare. La membrana è fatta asciugare all’aria per qualche minuto.
Amido Black staining solution:
Metanolo
45%
Acido acetico
10%
H2O
45%
Aggiungere poi 0,1% di Amido Black (Biorad).
Destain solution:
Metanolo
90%
Acido acetico
2%
H2O
8%
92
4.22 Esperimenti di patch-clamp
Per misurare la corrente di Cl- indotta dal rigonfiamento cellulare, è stato utilizzato
il metodo del patch-clamp in configurazione whole-cell che permette, mettendo in
comunicazione la soluzione interna dell’elettrodo con il citoplasma, di effettuare
misure delle correnti che attraversano i canali ionici di tutta la membrana
plasmatica di una singola cellula. L’apparato strumentale utilizzato per gli
esperimenti di patch-clamp, era costituito da: gabbia di Faraday, tavolo
antivibrante, microscopio invertito, amplificatore da patch-clamp [Axopatch 200
Axon Instruments], e relativo holder, micromanipolatore, sistema di perfusione,
convertitore analogico-digitale e computer per immagazzinamento e analisi dati
(programma Pulse Heka, Germania).
Tutte le misure erano effettuate con una frequenza di campionamento di 20 kHz e
filtrate a 5 kHz con un filtro Bessel a 8 poli.
Un sistema di perfusione, permetteva di immettere ad una velocità di circa 5
ml/min una determinata soluzione nel bagno il cui volume era di circa 300 l. La
soluzione contenuta nel bagno era messa a terra con l’ausilio di un elettrodo ad
Ag/AgCl.
Le pipette da patch-clamp erano preparate a partire da capillari in borosilicato
(BRAND, diametro esterno di 1,55 mm, diametro interno di 1,15 mm) con un puller
orizzontale (Sutter Instrument CO Made in U.S.A.Model P-87 Flaming/Brown
Micropipette Puller) ed in modo tale che avessero una resistenza di circa 4-8 M.
Il protocollo sperimentale utilizzato nel corso degli esperimenti prevedeva che il
sigillo fosse realizzato in soluzione ipertonica. Sempre in soluzione ipertonica,
applicando un’ulteriore suzione, si passava alla configurazione whole-cell. A questo
punto si eseguiva un protocollo “I/V” che prevedeva l’invio, alla cellula, di impulsi di
potenziale della durata di 500 ms da –100 a + 100 mV, con incrementi di 20 mV e
da un potenziale di holding di 0 mV. Successivamente veniva sostituita la soluzione
esterna con la soluzione ipotonica e l’incremento della corrente era registrato con
un protocollo “sweeps +40 mV”.
93
Nel corso di tale protocollo, da un potenziale di holding di 0 mV, veniva inviato alla
cellula, ogni 30 secondi e per 400 ms, uno step di potenziale di 40 mV. Dopo 5’
dall’inizio della sostituzione, si eseguiva nuovamente un “I/V”, di seguito altri 8’ con
“step +40 mV” ed infine un ultimo protocollo “I/V”.
Soluzioni utilizzate per gli esperimenti di patch-clamp
Le soluzioni di perfusione del bagno (extracellulare) e della pipetta (intracellulare),
la cui composizione è riportata nelle tabelle 1, 2 e 3, sono state opportunamente
scelte per permettere la misura delle correnti di Cl-.
1)SOLUZIONE PER ELETTRODO
CsCl2
125 mM
MgCl2
5 mM
EGTA
11 mM
HEPES
10 mM
Raffinosio
50 mM
MgATP
pH 7.2
osmolarità 340 mOsm
2 mM
pH 7.4
2)SOLUZIONE IPERTONICA
NaCl
125 mM
MgCl2
2,5 mM
CaCl2
2,5 mM
HEPES
10 mM
Mannitolo
100 mM
osmolarità 390 mOsm
94
3)SOLUZIONE IPOTONICA
NaCl
125 mM
pH 7.4
MgCl2
2,5 mM
osmolarità 270 mOsm
CaCl2
2,5 mM
HEPES
10 mM
4.23 Conte cellulari e curve di crescita
I saggi di conta cellulare sono stati effettuati per i test di proliferazione.
Per i test di proliferazione le cellule sono sono state transfettate in petri da 3 e il
giorno successivo seminate in quantità 5 104 cellule / ml in multiwell da 24.
Ad ogni conta viene eseguito il seguente protocollo:
Prelevare il contenuto di 3 pozzetti e tenerlo in una provetta falcon in
ghiaccio
Lavare i 3 pozzetti con 500 μl di PBS1X e aggiungerlo alla provetta
falcon in ghiaccio
Mettere in ogni pozzetto 100 μl di tripsina e aspettare il distacco delle
cellule dal fondo dei pozzetti
Stoppare la tripsina con 900 μl di terreno fresco per ogni pozzetto
Raccogliere il tutto nella falcon in ghiaccio
Pellettare a 1500 rpm per 10 minuti a 4°C
Eliminare il sovranatante e risospendere il pellet in 50 μl di terreno
fresco
Prelevare 20 μl a cui vengono aggiunti 20 μl di Trypan Blue (0.4% in
PBS)
Tenere 5 minuti in ghiaccio ed effettuare la conta al microscopio
utilizzando la cameretta di burker
95
Le cellule che sono visivamente blu non sono vitali, le altre sì. Questo succede
perché il Trypan Blue contiene un fluorocromo carico negativamente: nelle cellule
con membrana integra non può entrare, ma può entrare laddove la membrana
cellulare è frammentata, ovvero nelle cellule danneggiate.
4.24 Analisi statistiche
I dati sperimentali di patch clamp sono stati espressi come media aritmetica più o
meno l’errore standard (SEM). I dati ottenuti sono stati analizzati attraverso il test
Anova (post test Bonferroni) utilizzando il programma PRISM.
Per quanto riguarda l'analisi statistica dei dati degli esperimenti di FRET e di
Western Blot, tutti i risultati sono stati espressi come media ± errore standard. È
stato applicato il test T di Student per dati non appaiati. Le differenze tra i dati sono
assunte come statisticamente significative quando p < 0,05.
96
5 Risultati
5.1 Caratterizzazione delle isoforme di 4.1 espresse in cellule HEK: RT-PCR e
clonaggio delle isoforme AUG-1 e AUG-2 di 4.1R
Prima di procedere per individuare le isoforme della proteina 4.1 presenti nelle
cellule HEK 293 Phoenix abbiamo realizzato una RT-PCR sull’RNA totale. Questa
procedura permette di avere un arricchimento in mRNA in quanto consente di
eliminare, nel corso del processo d’isolamento, l’RNA con lunghezza minore di 200
nucleotidi (come il 5,8 S rRNA, 5 S rRNA e i tRNA che possono rappresentare fino al
15-20% dell’RNA totale). Inoltre, volendo evitare di isolare RNA non maturo (che
non ha subito ancora il processo di splicing o lo ha subito parzialmente) è stato
eseguito un protocollo che consente l’estrazione del solo RNA citoplasmatico
(permettendo di preservare l’integrità dei nuclei).
Prima di procedere con la retrotrascrizione la qualità dell’RNA è stata controllata
mediante elettroforesi (figura 16).
28 S
18 S
Figura 16 Elettroforesi su gel di agarosio all’1% (w/v)
in TAE dell’RNA estratto da cellule HEK 293T. Sono
evidenti
due
bande
nettamente
distinte
corrispondenti alle bande dell’RNA ribosomiale 18 S
e 28 S.
Il cDNA è stato sottoposto a una prima PCR per caratterizzare quali isoforme della
4.1 (R, G, B) fossero presenti nelle cellule HEK 293T da cui era stato estratto l’RNA.
Sono stati utilizzati quindi i primers specifici per ciascuna isoforma. In parallelo sono
state condotte diverse reazioni di controllo. Una prima reazione di controllo era
97
condotta su un campione di RNA cui non era stata aggiunta la trascrittasi inversa
per, un’eventuale contaminazione da DNA genomico (che comunque poteva essere
esclusa anche sulla base della grandezza del frammento amplificato, dato che i
primers erano stati disegnati in modo che fossero complementari a regioni esoniche
distinte). Altre reazioni di controllo sono state allestite in modo da escludere una
qualsiasi contaminazione da DNA esogeno al momento della PCR. Alle miscele di
reazione complete non veniva, in tal caso, aggiunto il c-DNA (-R, -G, -B in figura 17).
M |- RT | R | -R | G | -G | B |
-B
Figura 17 Elettroforesi su gel di agarosio all’1%
(w/v) in TAE dei prodotti della PCR condotta per
determinare quale isoforma di 4.1 fosse presente
in cellule HEK 293T. Ordine di caricamento: M >
Marker: Mass Ruler DNA Ladder low Range (MBI
Fermentas), RT > controllo (no trascrittasi
inversa), R > isoforma R, -R > controllo (no sDNA),
G > isoforma G, -G > controllo (no sDNA), B >
isoforma B e -B > controllo (no sDNA).
Dall’elettroforesi, avendo ottenuto per le diverse isoforme singole bande
dell’altezza attesa, è risultato evidente che tutte le tre isoforme erano presenti nelle
cellule HEK 293T.
Per clonare i cDNA corrispondenti a isoforme ad alto (che utilizzano l’ATG-1) e basso
(che utilizzano l’ATG-2) peso molecolare della 4.1R abbiamo eseguito due diverse
PCR ciascuna con un diverso primer senso specifico per uno dei due possibili siti di
inizio della trascrizione, (ATG-1, primer senso 1 in figura 18 o ATG-2, primer senso 2
in figura 18), e lo stesso primer antisenso (figura 18). I primer (vedi materiali e
metodi) contenevano anche le sequenze di restrizione necessarie per il clonaggio
nei vettori per il FRET, EYFP-N1 e –C1.
98
A)
B)
kb
2500
2000
1500
M
| primer 1
|
pprimer 2
Figura 18 A) E’ riportato uno schema della sequenza della 4.1R. Sono evidenziati domini altamente
conservati nella famiglia della proteina 4.1 e i punti di innesco dei primers disegnati per determinare
quali varianti da splicing fossero presenti. B) Elettroforesi su gel di agarosio all’1% (w/v) in TAE delle
PCR eseguite con le coppie di primers riportati; M=marker.
Come mostrato in figura 18, in entrambe le PCR l’amplificato corrispondeva ad
un'unica banda, probabilmente la variante più rappresentata in questo tipo
cellulare. Le due varianti da splicing della 4.1R corrispondono alla variante 4.1R135
(isoforma lunga, 4.1RII) e alla variante 4.180 (isoforma corta, 4.1Rsh) (Gascard et al.,
1998) che differiscono per la lunghezza delle 3’-UTR non amplificate nel corso della
reazione di PCR.
99
A)
B)
Figura 19 Sono mostrate alcune delle isoforme esistenti di 4.1R. In figura A è riportata
l’organizzazione in esoni del gene umano per la 4.1R delle isoforme prodotte dal sito di inizio
trasduzione AUG-1 (isoforme lunghe). All’inizio della figura è rappresentato l’mRNA della 4.1R con gli
esoni alternativi, gli esono costitutivi e gli esoni non codificati. I due siti di inizio trasduzione AUG1 e
100
AUG2 sono situati all’esone 2 e all’esone 4 rispettivamente. In B sono rappresentate le isoforme
mancanti l’AUG-1, quindi le isoforme corte (Gascard et al., 1998).
5.2 Interazione tra ICln e 4.1R
Dopo aver clonato per RT-PCR da cellule HEK-293 le due isoforme della proteina
4.1R (4.1Rsh e 4.1RII) abbiamo allestito esperimenti di FRET (Fluorescence
Resonance Energy Transfer, vedi sessione “Materiali e metodi”) per analizzare in
vivo il fenomeno d’interazione tra la proteina ICln e la 4.1R (in particolare per la due
isoforme 4.1RII e 4.1Rsh) e per cercare di chiarire se tale interazione gioca un ruolo
nella traslocazione di pICln dal citosol alla membrana e nell’attivazione dei canali
RVDC.
5.2.1 Interazione in vivo in cellula. Esperimenti di FRET: interazione tra ICln e
4.1Rsh
Per studiare in vivo l’interazione tra la proteina ICln e la proteina 4.1R abbiamo
allestito esperimenti di FRET utilizzando la metodica dell’ Acceptor photobleaching
ampiamente utilizzata in letteratura (Rodighiero et al., 2008). Tale tecnica consente
di valutare la variazione dell’intensità di emissione del donatore ECFP (Enhanced
Cyan Fluorescence Protein) dopo bleaching del fuoroforo accettore YFP (Enhanced
Yellow Fluorescence Protein), ottenuto illuminando una regione del preparato con
un laser ad elevata potenza.
Questi esperimenti sono stati condotti in vivo su cellule transfettate con plasmidi
che consentono l’espressione di proteine di fusione aventi il fluoroforo CFP in N- o
C-terminale rispetto a ICln ed inizialmente con il fluoroforo YFP in N- terminale
rispetto alla proteina 4.1Rsh. Gli esperimenti sono stati condotti stimolando le
cellule per 10 minuti con soluzione isotonica (controllo) o con soluzione ipotonica
(rispetto al mezzo intracellulare) per studiare l’interazione tra le due proteine anche
in presenza di uno di uno stress osmotico.
101
Interazione tra ICln e YFP-4.1Rshort: “Acceptor photobleaching”
Inizialmente gli esperimenti di FRET sono stati condotti per queste possibili coppie:
pEYFP C1 4.1Rsh – pECFP N1 ICln
pEYFP C1 4.1Rsh – pECFP C1 ICln
pEYFP C1 4.1Rsh – pECFP (controllo)
Il vettore pECFP-C1 ha il cDNA codificante per ICln a valle della sequenza
nucleotidica codificante per il fluoroforo CFP (CFP ICln) e il vettore pECFP-N1 ha il
cDNA codificante per ICln a monte della sequenza nucleotidica codificante per il
fluoroforo CFP (ICln-CFP); il vettore pEYFP-C1 ha il cDNA per la 4.1 a valle della
sequenza nucleotidica codificante per il fluoroforo YFP (YFP 4.1Rsh).
Gli esperimenti sono stati condotti per ogni combinazione sia in soluzione isotonica
che ipotonica.
Inizialmente sono stati condotti esperimenti di controllo necessari per valutare il
segnale di fondo, ossia l’efficienza di FRET dovuta all’interazione aspecifica e casuale
tra i due fluorofori in cellula. Nel corso di questi esperimenti le cellule sono state
transfettate con il vettore vuoto pECFP, esprimente il solo CFP, ed il vettore pEYFPC1-4.1. Dopo aver effettuato gli esperimenti di controllo abbiamo allestito gli
esperimenti di FRET per ogni coppia precedentemente illustrata (YFP-4.1Rsh + CFPICln e YFP-4.1Rsh + ICln-CFP).
102
A
B
*
C
#
D
Figura 20 Esperimenti di FRET (acceptor photobleaching) condotti in cellule HEK 293 Phoenix
transfettate con YFP-4.1Rsh e CFP-ICln o ICln-CFP. A) Esempio di immagini ottenute durante un
esperimento. In giallo è evidenziato il segnale emesso dalla molecola YFP, in azzurro quello emesso
dal CFP. I pannelli a sinistra si riferiscono al segnale misurato prima del photobleaching dell’accettore
(YFP), i pannelli a destra si riferiscono all’intensità di fluorescenza rilevata dopo photobleaching
dell’accettore. Nel pannello più a destra (contrassegnato dal simbolo #) è riportata la figura
rappresentante l’efficienza di FRET calcolato nella stessa cellula. B) Immagine rappresentativa del
controllo negativo in cellule transfettate con YFP-4.1Rsh e CFP (vettore senza l’inserto). C)
Istogrammi relativi all’analisi statistica (relativa a più cellule acquisite in tre preparazioni
indipendenti) dei valori di efficienza di FRET misurati in isotonicità e ipotonicità in cellule esprimenti
ICln-CFP + YFP-4.1Rsh e CFP+YFP-4.1Rsh (controllo). D) Analisi statistica (relativa a più cellule
acquisite in tre preparazioni indipendenti) dei valori di efficienza di FRET misurati in isotonicità e
ipotonicità in cellule esprimenti CFP-ICln + YFP-4.1Rsh e CFP + YFP-4.1Rsh (controllo).
In figura 20 è riportata una immagine esemplificativa di un esperimento di FRET e
l’analisi statistica dei valori ottenuti. Negli esperimenti di controllo condotti overesprimendo nelle cellule CFP non legato ad ICln e YFP-4.1Rsh è stata rilevata
un’efficienza di FRET pari a 2.5 1.0% (n=30) (figura 20).
103
Dalle immagini di FRET sembrerebbe che la sede principale dell’interazione tra le
due proteine è il compartimento citosolico.
Per la coppia YFP-4.1Rsh + ICln-CFP dall’analisi statistica abbiamo misurato
un’efficienza di FRET significativamente più alta rispetto ai controlli sia quando le
cellule sono in soluzione iso-osmotica (YFP-4.1Rsh + ICln-CFP: 5.60 ± 0.68, n=29 vs
YFP-4.1Rsh + CFP: 2.5 ± 0.80 n=30) che quando sono poste in soluzione ipoosmotica (YFP-4.1Rsh + ICln-CFP: 4.59 ± 1.44, n=16 vs YFP-4.1Rsh + CFP: 0.99± 0.89
n=22).
Per quanto concerne la coppia YFP-4.1Rsh + CFP-ICln (istogramma figura 20 D)
abbiamo misurato un valore di FRET più alto rispetto a quello misurato negli
esperimenti in cui ICln ha il CFP posto all’estremità C-terminale sia in isotonia (11.3
± 1.1 n=28) che in ipotonia (15.07 ± 1.17 n=20); questi valori inoltre sono
significativamente maggiori rispetto alle condizioni di controllo (rispettivamente
2.50 ± 0.8 n=30 in isotonia e 0.99 ± 0.89 n=20 in ipotonia). Inoltre è interessante
notare che per questa coppia dopo stimolo ipotonico abbiamo misurato un
aumento significativo dell’efficienza di FRET rispetto alla condizione isotonica
suggerendo che l’interazione tra ICln e la 4.1Rsh aumenta quando le cellule sono
sottoposte a uno stimolo ipotonico. Per valutare l’aumento dell’interazione tra le
due proteine, in ipotonicità, abbiamo deciso di utilizzare un’altro tipo di valutazione
dell’efficienza FRET che ci permettesse a differenza dell’Acceptor Photobleaching di
effettuare misure ripetute su una stessa cellula.
Interazione tra ICln e YFP 4.1Rshort ‘Sensitized emission’
La metodica della Sensitized emission consente di arrivare al calcolo di un indice di
N-FRET a partire da quello di FRET, ottenuto eccitando il donatore e misurando
l’emissione dell’accettore. Tale valore viene corretto introducendo una serie di
parametri di correzione, che consentono di eliminare gli eventuali errori derivanti
dalla
sovrapposizione
degli
spettri
dei
due
dettagliatamente nella sezione "Materiali e metodi".
104
fluorofori,
come
descritto
Nei nostri esperimenti le cellule sono state precedentemente co-transfettate in
modo transiente con YFP-4.1Rsh e CFP-ICln e a 24h dalla trasfezione abbiamo poi
acquisito le immagini di tali cellule, al microscopio confocale previo il
mantenimento per almeno 5 minuti in soluzione isotonica. La soluzione
extracellulare è stata quindi sostituita con quella ipotonica e sono state acquisite
nuovamente le immagini a diversi intervalli di tempo fino a 20 minuti dalla
sostituzione. In parallelo sono stati allestiti due controlli transfettando le cellule con
il solo YFP-4.1Rsh o CFP-ICln da solo, la cui analisi permette di ricavare i valori dei
parametri di correzione da utilizzare per il calcolo della N-FRET (vedi sezione
materiali e metodi). Inoltre per valutare l’efficienza del trasferimento energetico
dovuta all’interazione aspecifica e casuale tra i due fluorofori abbiamo allestito
esperimenti in cui abbiamo transfettato le cellule con YFP-4.1Rsh e il vettore vuoto
pECFP, esprimente il solo CFP.
A
B
NFRET HEK T
YFP-4.1sh + CFP-ICln
**
0.5
NFRET
0.4
0.3
ns
0.2
10
C
FP
FP
hy
po
hy
pe
rC
-IC
C
FP
10
hy
po
hy
pe
rC
FP
-IC
ln
ln
0.1
Figura 21 A) Immagini di N-FRET (ottenute come decritto nella sezione materiali e metodi) relative a
cellule HEK 293 Phoenix co-transfetttate con pEYFP-C1-4.1Rsh + pECFP-C1-ICln in basso, e con pEYFPC1-4.1Rsh + CFP (controllo negativo). B) Analisi statistica, mediante il test T di Student, a dati
appaiati, dei valori ricavati dall'analisi di N-FRET. Le differenze tra i dati sono assunte come
statisticamente significative quando p 0,05.
L'analisi statistica dei dati di N-FRET nelle due condizioni è riportata in figura 21 B.
Nel caso delle cellule di controllo (transfetatte con YFP 4.1Rsh e CFP vuoto) si
osserva che il valore di N-FRET è basso sia in isotonia (0.18 ± 0.10 n=9) che dopo lo
105
stimolo ipotonico (0.20 ± 0.009 n=9). Quando le cellule over-esprimono YFP-4.1Rsh
e CFP-ICln già in isotonia il valore di N-FRET è significativamente diverso dal
controllo (0.40 ± 0.012 n=15) inoltre aumenta significativamente quando si passa in
condizioni ipotoniche (0.47 ± 0.013 n=15), dopo 10 minuti dalla sostituzione.
N-FRET HEK T
YFP-4.1sh + CFP-ICln
normalized
N-FRET HEK T
YFP-4.1sh + CFP-ICln
*
0.50
1.2
NFRET norm
NFRET
0.45
0.40
0.35
0.30
1.1
1.0
0.9
-5
0
5
10
15
20
time in hypo (min)
25
-5
0
5
10
Figura 22 Immagine relativa al time course del segnale di N-FRET calcolato in cellule co-transfettate
con YFP-4.1Rsh e CFP-ICln. Il segnale aumenta in modo significativo già a partire da 2.5 minuti dopo
la sostituzione con soluzione ipotonica; tale aumento si mantiene costante nei tempi successivi (t= 5,
10, 15, 20 minuti dopo sostituzione con soluzione ipotonica). * p< 0.05 con t di Student
Il “time course” del segnale di N-FRET calcolato in cellule transfettate con YFP4.1Rsh e CFP-ICln (fig. 22) indica che l’aumento è significativo già a partire da 2,5
minuti dopo lo stimolo ipotonico (0.46 ± 0.013 n=8 vs 0.42 ± 0.02 n=8 in isotonia).
L’N-FRET risulta essere significativamente maggiore rispetto alla condizione di
controllo anche per i tempi successivi fino a 20 minuti dopo la sostituzione con
soluzione ipotonica (N-FRET: 5 min> 0.47 ± 0.012 n=8; 10 min> 0.47 ± 0.014 n=8; 15
min> 0.46± 0.012 n=8; 20 min> 0.46± 0.012 n=8).
Interazione tra ICln e 4.1Rshort-YFP
Abbiamo allestito un’ulteriore serie di esperimenti di FRET utilizzando la tecnica
dell’Acceptor photobleaching per studiare l’interazione tra ICln e pEYFP-N1 4.1Rsh.
In questa configurazione il fluoroforo YFP è stato posto all’estremità C-terminale
della proteina 4.1Rsh.
106
15
time in hypo(min)
20
25
Analogamente a quanto visto per YFP-4.1Rsh, le coppie accettore/donatore
saggiate sono state le seguenti:
pEYFP-N1-4.1Rsh – pECFP-N1-ICln
pEYFP-N1-4.1Rsh – pECFP-C1-ICln
pEYFP-N1-4.1Rsh – pECFP (controllo)
A
B
4.1short-YFP + CFP-ICln
4.1short-YFP + ICln-CFP
ICln-CFP + 4.1sh-YFP
20
CFP-ICln + 4.1sh-YFP
20
ns
ns
p<0.05
10
ICln
5
0
-5
p<0.05
ICln
contr
n=29
ISO
n=30
contr
n=16
CFP + 4.1sh-YFP
(control)
15
FRETeff
FRETeff
15
10
HYPO
-5
ICln
contr
n=29
ISO
CFP + 4.1sh-YFP
(control)
ns
ICln
5
0
n=22
ns
n=30
contr
n=16
n=22
HYPO
Figura 23 Esperimenti di FRET (acceptor photobleaching) condotti in cellule HEK 293 Phoenix
transfettate con 4.1Rsh-YFP e ICln-CFP o CFP-ICln. A) Analisi statistica (relativa a più cellule acquisite
in tre preparazioni indipendenti) dei valori di efficienza di FRET misurati in isotonicità e ipotonicità. in
cellule esprimenti ICln-CFP + 4.1Rsh-YFP e CFP + 4.1Rsh-YFP (controllo B) Istogrammi relativi
all’efficienza di FRET misurata in cellule esprimenti CFP-ICln+ 4.1Rsh-YFP e CFP+ 4.1Rsh-YFP
(controllo).
Dall’analisi statistica risulta che solo nella condizione ICln-CFP (ma non in quella
CFP-ICln) i valori erano significativamente diversi dal controllo con solo CFP. Inoltre,
anche in questa condizione, i valori di FRETeff erano significativamente più bassi
rispetto a quelli ottenuti quando il YFP era fuso all’estremità N-t di 4.1 (isotonia:
4.1Rsh-YFP + ICln-CFP 1.93 ± 0.25, n=12; ipotonia: 1.71 ± 0.14 n=12) e poco distanti
da quelli dei controlli. Infine, non abbiamo riscontrato un aumento significativo
passando dall’isotonia all’ipotonia. Questo suggerisce che l’efficienza della FRET è
molto sensibile alla posizione in cui si trova il fluoroforo e che l’interazione tra le
due proteine è ridotta quando il YFP è posto all’estremità C-t. Per indagare su
questo punto e verificare l’effettiva interazione tra 4.1Rsh e ICln, visti i bassi valori
di FRETeff, abbiamo affiancato esperimenti di coimmunoprecipitazione agli
esperimenti di FRET.
107
5.2.2 Studio dell’interazione tra ICln e l’isoforma corta (4.1Rsh) della proteina 4.1
mediante coimmunoprecipitazione.
Con questa tecnica abbiamo investigato l’interazione della proteina 4.1Rsh con il
fluoroforo fuso al N- o C terminale (gli stessi costrutti utilizzati per gli esperimenti di
FRET) e la proteina ICln a cui era legato un Flag-tag all’etremità C-terminale. Le
cellule sono state co-transfettate con la 4.1Rsh (YFP-4.1Rsh o 4.1Rsh-YFP) e pFlagICln. Abbiamo inoltre allestito un controllo transfettando le cellule con la proteina
4.1Rsh (con il YFP al N- o al C-terminale) e pFlag-BAP (Sigma), Bovine Alkaline
Peroxidase, che si presuppone non interagire con 4.1. A 24h dalla trasfezione le
cellule sono state raccolte e lisate. Il lisato è stato successivamente incubato con
una una resina, l’α – FLAG M2 Affinity Gel (Sigma), costituita da anticorpi
monoclonali α – FLAG coniugati ad agarosio. Le proteine con un tag FLAG si legano
alla resina e con esse le proteine a loro legate. L’eluizione è stata effettuata per
competizione in presenza di un eccesso di un peptide Flag purificato (Sigma).
Riassumendo, i nostri casi sperimentali sono stati i seguenti:
-YFP-4.1Rsh + Flag-ICln
-YFP-4.1Rsh + Flag-Bap (controllo)
e -4.1Rsh-YFP + Flag-ICln
-4.1Rsh-YFP + Flag-Bap (controllo).
I lisati (Lys in figura 24) e gli eluati (tre eluizoni successive, E2, E3 ed E1) sono stati
controllati mediante Western Blot per la presenza negli eluati di ICln (mediante
anticorpi anti-FLAG) e 4.1 (mediante anticorpi anti 4.1 o anti GFP).
In Figura 24 possiamo osservare la lastra autoradiografica, risultante dallo sviluppo
del segnale di chemioluminescenza, relativo alla membrana incubata con anti-4.1,
anti GFP e anti-flag.
108
A
B
Figura 24 Western relativo all’immunoprecipitazione della proteina YFP-4.1Rsh (pannello A) o
4.1Rsh-YFP (pannello B con Flag-ICln), utilizzando una resina coniugata ad agarosio. Le membrane
sono state incubate con anti-4.1 che riconosce la proteina 4.1, con l’anti-GFP che riconosce la
proteina YFP fusa alla proteina 4.1 e l’anti-Flag che riconosce il FLAG fuso alla proteina ICln. In
entrambi i casi la condizione di controllo è stata ottenuta co-transfettando con 4.1Rsh + FLAG-BAP
anziché FLAG-ICln. In entrambi i pannelli: Lys =lisato E2, E3, E1 = eluati successivi. La banda relativa
alla 4.1Rsh è all’altezza di circa 130 kDa mentre la banda relativa ad ICln è a circa 37 kDa.
Nel pannello A della figura 24 osserviamo le lastre autoradiografiche relative al
Western Blot dell’immunoprecipitazione della proteina YFP-4.1Rsh e Flag-ICln e
relativo controllo YFP-4.1Rsh e Flag-BAP. Nella prima corsia è stato caricato il lisato
di controllo (dell’immunoprecipitazione YFP-4.1Rsh + Flag-BAP) e possiamo
osservare la presenza di un segnale sia con l’anti-4.1 che con l’anti-GFP (che prova la
presenza della YFP-4.1Rsh nel lisato di partenza) e con l’anti-Flag (che prova la
presenza della FLAG-BAP, il plasmide di controllo nel lisato di partenza). Nella
seconda, nella terza e nella quarta corsia abbiamo caricato 3 eluati successivi di
questa immunoprecipitazione (E2, E3, E1). Abbiamo caricato diversi eluati in quanto
non sapevamo con quale eluizione le nostre proteine si sarebbero staccate dalla
resina. Per queste corsie abbiamo osservato un segnale solo con l’anti-FLAG; questo
ci indica che negli eluati non è presente la YFP-4.1Rsh ma solo la proteina Flag-BAP
di controllo, come atteso. Nella quinta corsia abbiamo caricato il lisato
109
dell’immunoprecipitazione YFP-4.1Rsh + Flag-ICln. E’ possibile osservare come sia
presente un segnale con tutti e tre gli anticorpi; questo ci indica la presenza nei
lisati di partenza di entrambe le proteine di nostro interesse la proteina YFP-4.1Rsh
e la proteina FLAG-ICln. Nelle rimanenti corsie (sesta, settima e ottava) abbiamo
caricato gli eluati. In questi abbiamo osservato un segnale con l’anti-4.1, con l’antiGFP e anti-Flag. I primi due ci indicano la presenza della proteina YFP-4.1Rsh negli
eluati mentre il segnale con l’anti-FLAG ci indica la co-presenza della proteina ICln.
Questo risultato conferma i dati di FRET e dimostra che la proteina 4.1Rsh con il
fluoroforo posto all’estremità N-terminale lega la proteina ICln fusa con FLAG.
L’immunoprecipitazione è stata effettuata anche per la proteina 4.1Rsh-YFP (Figura
24, pannello B) + Flag-ICln. Come per la proteina YFP-4.1Rsh (YFP al N-terminale
della 4.1Rsh) anche quando il fluoroforo è fuso all’estremità C-terminale della
4.1Rsh abbiamo osservato che la 4.1Rsh coimmunoprecipita con la proteina ICln con
il tag FLAG. Questi esperimenti ci indicano che la proteina 4.1Rsh in fusione con il
YFP o all’N- o al C-terminale interagisce con ICln. E’ pertanto ipotizzabile che il basso
segnale di FRETeff misurato per la 4.1Rsh-YFP non sia dovuto al fatto che le due
proteine non interagiscono ma sia determinato dal fatto che il fluoforo posto al Cterminale della 4.1Rsh non è la configurazione ottimale affinchè avvenga il
trasferimento energetico tra YFP e CFP.
5.2.3 Interazione in vivo in cellula. Esperimenti di FRET: interazione tra ICln e
4.1RII
In parallelo allo studio dell’interazione tra la proteina 4.1Rsh e la proteina ICln
abbiamo effettuato esperimenti di FRET per verificare l’interazione tra l’isoforma
lunga (4.1RII) e ICln in vivo utilizzando, come per l’isoforma corta, la tecnica
dell’Acceptor Photobleaching; gli esperimenti sono stati effettuati, anche in questo
caso, sia in condizioni isotoniche che dopo stimolo ipotonico. Gli esperimenti di
FRET sono stati condotti per queste possibili coppie:
pEYFP C1 4.1RII – pECFP N1 ICln
pEYFP C1 4.1RII – pECFP C1 ICln
110
pEYFP C1 4.1RII – pECFP (controllo) e
pEYFP N1 4.1RII – pECFP N1 ICln
pEYFP N1 4.1RII – pECFP C1 ICln
pEYFP N1 4.1RII – pECFP (controllo)
A
B
C
YFP-4.1long + ICln-CFP
ICln-CFP + YFP-4.1long
20
CFP + YFP-4.1long
(control)
FRETeff
15
10
ns
0
contr
ICln
n=15
n=24
n=15
ISO
n=18
HYPO
E
4.1long-YFP + ICln-CFP
4.1long-YFP + CFP-ICln
20
ICln-CFP + 4.1long-YFP
CFP-ICln + 4.1long-YFP
20
15
10
5
ns
p<0.05
ICln
contr
n=27
n=25
ICln
-5
ISO
10
5
contr
0
n=26
CFP + 4.1lunga-YFP
(control)
15
CFP + 4.1long-YFP
(control)
FRETeff
FRETeff
contr
ICln
-5
D
p<0.05
5
0
n=19
-5
HYPO
ns
ICln
n=15
ISO
ns
contr
ICln
contr
n=25
n=12
n=19
HYPO
Figura 25 A) Esperimenti di FRET (accetor photobleaching) condotti in cellule HEK 293 Phoenix
transfettate con YFP-4.1RII (4.1R-long in figura) o 4.1RII-YFP e CFP-ICln o ICln-CFP. Esempio di
immagini ottenute durante un esperimento con cellule transfettate con CFP-ICln + 4.1RII-YFP (a
111
sinistra) o con CFP + 4.1RII-YFP (controllo negativo), a destra. In giallo è evidenziato il segnale emesso
dal YFP, in azzurro quello emesso dal CFP. B) C) Analisi statistica (relativa a più cellule acquisite in tre
preparazioni indipendenti) dei valori di efficienza di FRET (FRETeff) misurati in isotonia e ipotonia.
Sono riportati gli istogrammi raffiguranti l’efficienza di FRET misurata in cellule esprimenti (pannello
B) CFP-ICln+YFP-4.1RII e CFP+YFP-4.1RII (controllo) in isotonia e in ipotonia. Il pannello C) si riferisce
a cellule esprimenti ICln-CFP + YFP-4.1RII e CFP+YFP-4.1RII (controllo) per entrambe le condizioni
sperimentali. D) si riferisce a cellule esprimenti CFP-ICln + 4.1RII-YFP e i relativi controlli (4.1RII-YFP +
CFP) nel pannello E) abbiamo invece riportato l’analisi statistica dell’efficienza di FRET misurata in
cellule esprimenti ICln-CFP+ 4.1RII-YFP. *p<0.05 con t student
Dall’analisi statistica abbiamo verificato che in ipotonia per la coppia 4.1RII-YFP +
CFP-ICln (figura 25 D) l’efficienza di FRET misurata pur essendo statisticamente
maggiore (2.03 ± 0.46 n=34) rispetto alla condizione di controllo (0.40 ± 0.62 n=19)
è molto basso (e non risulta infatti significativo se viene applicato il test AnovaBonferroni a una via). Per le altre coppie sperimentali (figura 25 B, C, E) l’efficienza
di FRET misurata è bassa e non risulta statisticamente differente rispetto alla
condizione di controllo; in un caso sperimentale YFP-4.1RII + ICln-CFP in ipotonia
l’efficienza di FRET è cosi’ bassa (0.31 ± 0.34 n=15) da risultare essere
significativamente inferiore (solo con il T di student, ma non con Anova-Bonferroni)
rispetto al suo controllo (1.96 ± 0.61 n=18).
5.2.4 Studio dell’interazione tra ICln e l’isoforma lunga (4.1RII) della proteina 4.1
mediante coimmunoprecipitazione.
Per l’isoforma lunga non abbiamo di fatto misurato un efficienza di FRET
significativa in nessuna configurazione testata. Per verificare se la bassa o nulla
FRETeff riflettesse effettivamente una mancanza di interazione, in parallelo agli
esperimenti di FRET abbiamo allestito esperimenti di coimmunoprecipitazione,
come per la 4.1Rsh.
Analogamente a quanto visto per 4.1Rsh i nostri casi sperimentali sono stati:
-YFP-4.1RII + Flag-ICln
-YFP-4.1RII + Flag-Bap (controllo)
e -4.1RII-YFP + Flag-ICln
-4.1RII-YFP + Flag-Bap (controllo).
112
A
B
Figura 26 Western relativo all’immunoprecipitazione della proteina YFP-4.1RII (pannello A) o 4.1RIIYFP (pannello B) con Flag-ICln mediante anticorpo anti-FLAG coniugato ad una resina di sefarosio. Il
controllo è stato effettuato con cellule co-transfettate con YFP-4.1RII+ Flag-BAP anziche FLAG-ICln,
Le membrane sono state incubate con anti-4.1 che riconosce la proteina 4.1, con l’anti-GFP che
riconosce la proteina YFP fusa alla proteina 4.1 e l’anti-Flag che riconosce il FLAG fuso alla proteina
ICln. In entrambi i pannelli Lys = lisato di cellule co-transfettate con YFP-4.1RII+ Flag ICln; E2, E3, E1=
eluati successivi. La banda relativa alla 4.1RII è all’altezza di circa 170 kDa mentre la banda relativa ad
ICln è a circa 37 kDa.
Come si può osservare dalle lastre (figura 26), sia per quanto riguarda YFP-4.1RII che
la proteina 4.1RII-YFP è presente un segnale negli eluati della proteina 4.1RII solo
quando siamo in co-transfezione con FLAG-ICln ma non in caso di co-transfezione
con FLAG-BAP (controllo negativo); con l’anticorpo anti-Flag in questi eluati
abbiamo osservato anche un segnale della proteina FLAG-ICln o FLAG-BAP che ci
conferma la presenza negli eluati delle proteine riconosciute dall’antiFLAG della
resina.
Questi esperimenti dimostrano pertanto che sia YFP-4.1RII che e 4.1RII-YFP
interagiscono con ICln. Il basso segnale di FRET misurato negli esperimenti di
Acceptor Photobleaching potrebbe quindi essere dovuto ad esempio all’interferenza
del dominio U1 (presente nell’isoforma lunga della proteina 4.1) sulla vicinanza tra i
due fluorofori YFP (fuso alla proteina 4.1 all’estremità N- o C-terminale) e il CFP
113
(fuso all’estremità N- o C-terminale della proteina ICln) e non a una mancanza di
interazione tra le due proteine.
Dagli esperimenti di immunopecipitazione sono emersi anche altre osservazioni
interessanti. Dal western blot in figura 26 si vede che per la 4.1RII-YFP negli eluati è
presente anche una banda più bassa (circa 130 kDa, in figura 26) che viene
riconosciuta sia dall’anti-4.1 che dall’anti-GFP e che potrebbe rappresentare la
banda relativa all’isoforma corta (4.1Rsh) in fusione con il YFP. E’ stato infatti
recetemente riportato (Lospitao et al., 2008) che è presente una sequenza IRES tra
ATG1 e l’ATG2 dell’ORF delle 4.1R ad alto peso molecolare che può causare la
traduzione anche di isoforme corte dall’overespressione di isoforme lunghe. Per
studiare
cosa
avvennise
nelle
nostre
condizioni
sperimentali
quando
overesprimiamo la 4.1RII in fusione con YFP, abbiamo effettuato esperimenti di
western blot sui lisati totali di cellule trasfettate con YFP-4.1Rsh, YFP-4.1RII, 4.1RshYFP e 4.1RII-YFP.
Figura 27 Western blot relativo al segnale identificato dall’anticorpo anti-4.1 nei lisati totali di cellule
transfettate nella prima corsia con pEYFPC1-4.1Rsh, nella seconda corsia con pEYFPC1-4.1RII, nella
terza corsia pEYFP-N1-4.1Rsh (4.1Rsh-YFP) e nella quarta con pEYFP-N1-4.1RII (4.1RII-YFP).
Questi esperimenti confermano che soprattutto con la 4.1RII con il YFP fuso
all’estremità C-t è evidenziabile una seconda banda, alla stessa altezza della 4.1RshYFP. Non abbiamo invece riscontrato la presenza significativa di una seconda banda,
quando il YFP era fuso all’estremità C-t di 4.1RII suggerendo che la presenza di un
114
tag voluminoso come la YFP all’etremità N-t potrebbe inibire l’utilizzo della
sequenza interna IRES.
Gli esperimenti di FRET per la 4.1Rsh e per la 4.1RII sono stati effettuati anche su
HEK 293 T (una linea cellulare simile alle Phoenix e utilizzata per gli esperimenti di
patch-clamp) ottenendo risultati del tutto paragonabili (dati non riportati) ai
risultati ottenuti su HEK 293 Phoenix.
5.3 Studi di localizzazione subcellulare: nucleo.
Le immagini di microscopia confocale ottenute per gli esperimenti di FRET hanno
fornito alcune osservazioni interessanti circa la localizzazione delle due proteine in
esame. Due sono i distretti cellulari su cui abbiamo concentrato la nostra
attenzione: il nucleo e la membrana cellulare
5.3.1 Studio della localizzazione della proteina 4.1Rsh e di ICln nel nucleo.
Un esempio delle immagini ottenute co-transfettando le cellule con YFP-4.1Rsh è
riportato nell figura 28.
A)
115
B)
Figura 28 A) Nelle tre immagini in alto è riportata l’acquisizione al microscopio confocale di cellule
HEK-293 Phoenix transfettate contemporaneamente con YFP–4.1Rsh e CFP (senza ICln), sulla destra,
un ingrandimento della localizzazione di 4.1Rsh in queste condizioni (In giallo l’acquisizione nel
canale del YFP, in blu quella relativa al CFP). Nelle tre immagini in basso B) sono riportate le
rispettive acquisizioni al confocale (in giallo YFP, in blu CFP) di cellule HEK-293 Phoenix transfettate
con YFP-4.1Rsh e CFP-ICln (con un ingrandimento sulla destra della localizzazione di 4.1Rsh in queste
condizioni). Le frecce in bianco indicano le cellule in cui il segnale di ICln nel nucleo è minore rispetto
a quello presente nel citoplasma.
Come mostrato in figura 28, la 4.1Rsh over-espressa localizza sia nel citosol sia, in
minor misura, in membrana dove è particolarmente evidente nelle regioni di
contatto tra cellula e cellula. In molte cellule il segnale è evidente anche nel nucleo.
Ciò è in accordo con quanto riportato in letteratura (Conboy et al., 1998). Come è
possibile osservare in figura 28 B quando la 4.1Rsh è co-espressa con la proteina
CFP-ICln il segnale nucleare della 4.1Rsh rispetto alla condizione di controllo
diminuisce notevolmente (figura A).
116
A
B
C
D
Figura 29 In A e B è riportata l’acquisizione al microscopio confocale di cellule HEK 293 Phoenix
transfettate con CFP–ICln e il vettore YFP vuoto (A) o CFP–ICln + YFP-4.1Rsh (B). Nel pannello C e B è
confrontata l’acquisizione al microscopio confocale di cellule HEK–293 transfettate con solo il CFP (D)
o con CFP–ICln (C).
Anche ICln subisce cambiamenti nella localizzazione quando è over–espressa
insieme a 4.1 sh: anche in questo caso c’è, in media, una riduzione del segnale nel
nucleo rispetto a quello citosolico (fig 29).
Over–esprimendo CFP-C1–ICln da solo o insieme al vettore pEYFP-C vuoto, si vede
che la proteina è presente sia nel nucleo che nel citosol, spesso con un rapporto del
segnale nucleo-citoplasma a favore del nucleo (figura 29 A e 29 C). E’ possibile che il
segnale di ICln (proteina citosolubile di piccole dimensioni nel nucleo sia almeno in
parte una conseguenza dell’over–espressione in fusione con il CFP, in quanto è noto
che le GFP, quando sono over- espresse, vanno anche nel nucleo anche se in
condizioni fisiologiche non sono proteine nucleari. Tuttavia, comparando una cellula
che esprime CFP–ICln con una cellula che esprime solo CFP, si può notare un
maggior rapporto del segnale nucleo/citoplasma nel primo caso: ciò suggerisce che
ICln abbia effettivamente localizzazione anche nucleare, indipendentemente da
possibili artefatti dipendenti dalla sua over-espressione come proteina di fusione
con la CFP. In accordo con tale ipotesi, anche esperimenti di immunofluorescenza in
alcuni casi hanno evidenziato un segnale anche a livello del nucleo (Krapivinsky et
117
al., 1994). La localizzazione nucleare di ICln endogena (non in over-espressione) è
del resto evidenziabile anche mediante western blot, come mostrato nei paragrafi
successivi.
Quest’analisi qualitativa è stata accompagnata da un analisi quantitativa. Abbiamo
calcolato infatti il rapporto dell’intensità di fluorescenza nel nucleo rispetto
all’intensità di fluorescenza nel citosol, misurate come valori di fluorescenza media
in ROI (region of interest) selezionate nei distretti cellulari di interesse di una stessa
cellula.
**
Figura 30 Istogrammi relativi all’analisi statistica del rapporto tra l’intensita di fluorescenza del
fluoroforo CFP nel nucleo rispetto al citosol per ogni condizione saggiata negli esperimenti di FRET:
YFP-4.1Rsh + CFP-ICln, YFP-4.1Rsh + ICln-CFP, CFP-ICln + YFP e YFP-4.1Rsh + CFP. N si riferisce al
numero di cellule analizzate.
118
Figura 31 Istogrammi relativi all’analisi statistica del rapporto tra l’intensita di fluorescenza nel
nucleo rispetto al citosol del fluoroforo YFP per ogni condizione saggiata negli esperimenti di FRET:
YFP-4.1Rsh +CFP-ICln, YFP-4.1Rsh + ICln-CFP, CFP-ICln + YFP e YFP-4.1Rsh + CFP. N si riferisce al
numero di cellule analizzate.
Dall’analisi statistica (figure 30 e 31) abbiamo verificato una diminuzione
significativa del segnale di ICln (CFP-ICln e ICln-CFP) nel nucleo quando è coespressa con la proteina YFP-4.1Rsh (Fnucleo/Fcitosol= 0.64 ± 0.05 con CFP-ICln n= 38,
e Fnucleo/Fcitosol= 0.60 ± 0.04 per ICln-CFP n= 28) rispetto alle condizioni di controllo
(CFP-ICln + YFP = 1.44 ± 0.02 n= 34). Il valore di Fnu/Fcyt di CFP-ICln resta sempre
significativamente diverso dal valore del solo CFP, sia in presenza che in assenza di
4.1, suggerendo che la risposta è propria della proteina ICln non dovuta al CFP a cui
è fusa (YFP-4.1Rsh + CFP= 1.14 ± 0.01 n= 31). Analogamente se consideriamo
l’intensità di fluorescenza valutata per il canale del YFP abbiamo verificato una
diminuzione del segnale della YFP-4.1Rsh nel nucleo quando è co-espressa con CFPICln o ICln-CFP (CFP-ICln= 0.06 ± 0.02 n=38; ICln-CFP= 0.06 ± 0.008 n=28 ) rispetto
alle condizioni di controllo, in assenza di ICln (YFP-4.1Rsh + CFP= 0.60 ± 0.007 n=28).
L’analisi conferma anche che la sua ripartizione tra nucleo e citosol differisce da
quella del solo YFP (CFP-ICln + YFP= 1.44 ± 0.027 n=34).
119
5.3.2 Studi di immunofluorescenza sulla localizzazione di ICln in cellule HEK 293
Phoenix in cui è overespressa la proteina 4.1Rsh
L’effetto dell’isoforma corta sulla localizzazione nucleare di ICln potrebbe riflettere
un evento fisiologico oppure, almeno in parte, potrebbe essere dovuto alla
overespressione in grande quantità delle due proteine e alla formazione di un
complesso stabile 4.1–ICln che inibisce la diffusione di ICln (e apparentemente
anche alla 4.1Rsh) nel nucleo.
Per approfondire questo aspetto abbiamo deciso di allestire esperimenti di
immunofluorescenza per verificare se questo fenomeno avvenisse anche per la
proteina ICln endogena.
Le cellule sono state transfettate col plasmide pEYFP-C1-4.1Rsh e a 24h dalla
transfezione le cellule sono state fissate e la localizzazione di ICln endogena valutata
mediante anticorpo anti-ICln seguito da anticorpo secondario (anti-rabbit)
coniugato a Cy5. I campioni sono stati osservati al microscopio confocale.
A
B
C
D)
Figura 32 Immagini di Immunofluorescenza su cellule HEK 293 Phoenix acquisite al microscopio
confocale. Nella figura A osserviamo, in rosso, l’immagine acquisita nel canale del Cy5,
corrispondente ad ICln (anticorpo primario anti-ICln diluito 1:100 e anticorpo secondario anti-rabbit
coniugato a cy5 diluito 1:400 in PBS contenente BSA 0,1%). Le frecce indicano le cellule positive alla
120
trasfezione per 4.1Rsh. L’immagine B mostra invece l’acquisizione nel canale del YFP. In questa
immagine possiamo visualizzare in giallo le cellule che over-esprimono la 4.1Rsh. L’immagine C è
data dalla sovrapposizione delle prime due acquisizioni; D Ingrandimento delle immagini precedenti.
Nel riquadro possiamo osservare le cellule che over-esprimono la proteina 4.1Rsh (in giallo) ed in cui
si osserva una diminuzione della proteina ICln endogena (rosso) nel nucleo rispetto alle cellule prive
di un segnale visibile per la YFP-4.1Rsh.
Nelle immagini acquisite al microscopio confocale possiamo osservare in rosso il
segnale della proteina ICln mentre in giallo la proteina YFP-4.1Rsh over-espressa.
Nella figura 32 la prima acquisizione ci mostra che ICln endogena ha una
localizzazione sia citoplasmatica che nucleare, in accordo con la letteratura
(Krapivinsky et al, 1994). Le frecce indicano le cellule positive alla transfezione con
YFP-4.1Rsh, dove si può notare che il segnale nucleare di ICln è minore rispetto alle
cellule in cui non si riscontra una visibile over-espressione della YFP-4.1Rsh. 4.1Rsh
over-espressa localizza sia nel citosol sia in membrana. In molte cellule il segnale è
evidente anche nel nucleo e ciò è in accordo con quanto riportato in letteratura
(Conboy et al., 1998). Se sovrapponiamo le due immagini, si osserva come le cellule
che presentano una diminuzione nucleare di ICln, siano quelle positive alla
transfezione e che quindi over-esprimono l’ isoforma corta di 4.1. Questi risultati
confermano i dati ottenuti dalle immagini degli esperimenti di FRET e supportano
l’ipotesi che la proteina 4.1Rsh influenzi la localizzazione di ICln.
5.3.3 Studi di western blot sui livelli di espressione della proteina ICln in cellule
HEK 293 Phoenix transfettate con la proteina 4.1Rsh e sui livelli di espressione
totali della proteina 4.1 in cellule transfettate con ICln.
Parallelamente agli esperimenti di immunofluorescenza abbiamo allestito
esperimenti di western blot per confermare il dato su ICln endogena.
Prima di effettuare studi di localizzazione tramite Western blot sulla distribuzione
della proteina ICln e della proteina 4.1Rsh nei compartimenti subcellulari abbiamo
voluto verificare se l’over-espressione della proteina 4.1Rsh influenzasse solo la
distribuzione di ICln in cellula (come visto negli esperimenti di immunocitochimica)
e/o influenzasse anche il suo livello di espressione; e viceversa se l’espressione della
121
proteina ICln influenzasse i livelli totali di espressione della 4.1Rsh. Sono stati quindi
effettuati esperimenti di western blot su preparati di proteine totali.
Per la prima serie di western blot, le cellule HEK 293 Phoenix sono state transfettate
con pEYFP-C1-4.1Rsh o con il vettore pEYFP-C1 senza inserto (controllo).
La membrana è stata incubata con anti-ICln e successivamente con l’anticorpo
secondario specifico, coniugato ad HRP.
In figura 33 sono riportati i risultati di questi esperimenti.
A)
B)
Figura 33 A) Western blot relativo ai livelli di espressione di ICln in preparati di proteine totali di
cellule HEK 293 Phoenix transfettate con pEYFP-C1-4.1Rsh o con pEYFP (controllo). La membrana è
stata incubata con anticorpo primario anti-ICln (1:1000) e anticorpo secondario anti-rabbit
(1:10000). L’immagine è rappresentativa di 4 esperimenti. B) Istogrammi relativi all’analisi
densitometrica del segnale di ICln in cellule transfettate con pEYFP-C1-4.1Rsh e con pEYFP (controllo)
normalizzato per il segnale della GAPDH negli stessi campioni (test: ρ value>0,05).
Al fine di valutare la significatività dell'esperimento, è stata condotta un’analisi
densitometrica delle bande. L’istogramma riportato in figura 33 rappresenta
l’analisi densitometrica usando il software ImageJ (Version 1.37, National Institutes
of Health, USA) condotta sul segnale della proteina ICln opportunamente
normalizzato per il segnale della GAPDH (YFP-4.1Rsh =1,74 ± 0,54 n=4; YFP= 1,79 ±
0,74 n=4) in modo da tener conto di eventuali differenze dovute ad errori o
imprecisioni nel caricamento. Dall’analisi statistica dell’analisi densitometrica
possiamo affermare che la quantità totale di ICln non varia nelle cellule che overesprimono la proteina 4.1Rsh rispetto al controllo.
122
Come accennato in precedenza, abbiamo effettuato anche una seconda serie di
western blot per verificare un possibile effetto della proteina ICln sulla quantità
totale della proteina 4.1. Le cellule sono state transfettate con il vettore plasmidico
pECFP-C1 ICln o con pCFP C1 senza inserto (controllo). A 24h dalla transfezione le
cellule sono state raccolte e lisate con lo stesso protocollo utilizzato negli
esperimenti precedenti.
A)
B)
Figura 34 A) Western blot relativo ai livelli di espressione della proteina 4.1 in preparati di proteine
totali di cellule HEK 293 Phoenix transfettate con pECFP-C1 ICln o con pECFP. La membrana è stata
incubata con anticorpo primario anti-4.1 (1:1000) e anticorpo secondario anti-goat (1:10000).
L’immagine è rappresentativa di 4 esperimenti. B) Istogrammi relativi all’analisi densitometrica del
segnale di 4.1 in cellule transfettate con pECFP-C1 ICln e con pECFP (controllo) normalizzato per il
segnale della tubulina negli stessi campioni (test: ρ value>0,05).
Nei western sono visibili due bande, ad altezza di circa 135 kDa (banda A) e 80 kD
(banda B), come atteso per le isoforme ad alto (4.1R135) e basso (4.1R80) peso
molecolare di 4.1 (Gascard et al., 1998). Per l’analisi densitometrica delle bande
abbiamo normalizzato il segnale della proteina 4.1 rispetto al segnale della tubulina
(marker interno). Dall’analisi statistica (CFP=0,93 ± 0,14 n=4; CFP-C1-ICln=0,99 ±
0.27 n=3) risulta che la quantità totale di 4.1, così come quella delle singole bande
(135 kDa e 80 kDa) non varia nelle cellule che over-esprimono la proteina ICln
rispetto al controllo.
123
5.3.4 Studi di western blot sui livelli di espressione nel nucleo della proteina ICln
in cellule HEK 293 Phoenix transfettate con la proteina 4.1Rsh e sui livelli di
espressione nucleare della proteina 4.1 in cellule transfettate con ICln.
Dopo aver verificato che non ci fossero variazione nei livelli di espressione di ICln, in
overespressione per la 4.1 e della 4.1 in overespressione per ICln abbiamo allestito
due serie sperimentali di studi di localizzazione nucleare per entrambe le proteine.
Sono stati quindi effettuati due serie di western blot speculari:
1) su frazioni citosoliche e nucleari, per indagare la distribuzione in questi due
compartimenti subcellulari di ICln in overespressione per la YFP-4.1Rsh;
2) su frazioni separate di citosol e nuclei, per indagare la distribuzione in questi
due compartimenti della 4.1Rsh in overespressione per CFP-C1-ICln.
Nel primo caso le cellule sono state transfettate con il vettore plasmidico pEYFP-C14.1Rsh (scelto perché già utilizzato negli esperimenti di immunofluorescenza) o con
pEYFP-C1 senza inserto (controllo). A 24h dalla transfezione le cellule sono state
raccolte e abbiamo separato le proteine nucleari dalle proteine citosoliche; tutte le
frazioni nucleari di campioni diversi sono poi state caricate su uno stesso gel per
l’analisi densitometrica. Lo stesso si è fatto per le relative frazioni citosoliche.
A)
B)
Figura 35 – A) Western blot effettuato sulle preparazioni proteiche nucleari (lastra in alto) e le
corrispondenti frazioni citosoliche (lastra in basso) di cellule HEK 293 Phoenix. In entrambi i western
124
blot nelle prime tre corsie sono stati caricati i campioni provenienti dalle cellule di controllo
(transfettate con pEYFP) mentre nelle ultime 3 corsie i campioni provenienti dalle cellule transfettate
con la proteina YFP-4.1Rsh. Le membrane sono state incubate entrambe con anticorpo primario antiICln (1:1000) e anticorpo secondario anti-rabbit (1:10000). B) Istogrammi relativi all’analisi
densitometrica del segnale nucleare di ICln normalizzato per il segnale citosolico di ICln (t test p<
0.05).
L’immagine 35 è rappresentativa di 7 campioni indipendenti.
Nella prima lastra della figura 35 possiamo osservare una diminuzione del segnale
nel nucleo della proteina ICln in cellule transfettate con la proteina 4.1Rsh (ultime
tre corsie). Per l’analisi densitometrica delle bande abbiamo normalizzato il segnale
della proteina ICln nucleare rispetto al segnale citosolico. L’analisi statistica
conferma che quando è overespressa la proteina 4.1Rsh il segnale di ICln nel nucleo
diminuisce significativamente rispetto alla condizione di controllo.
Per poter effettuare una analisi statistica del segnale nucleare di ICln, inizialmente
avevamo scelto come fattore di normalizzazione per il caricamento la proteina
laminina, una proteina sub-membranaria nucleare.
Figura 36 Western blot relativo alla membrana degli estratti proteici nucleari. Abbiamo utilizzato
l’anticorpo primario anti-laminina diluito in TBST 1:1000 e successivamente l’anticorpo secondario
anti-rabbit 1:20000 diluito in TBST. Nelle prime tre corsie abbiamo i campioni delle cellule di
controllo mentre nelle restanti corsie abbiamo caricato i campioni delle cellule transfettate col
plasmide pEYFP-C1 4.1Rsh.
Dalla lastra in figura 36 è possibile tuttavia notare un forte aumento del segnale
della laminina nei campioni corrispondenti alle cellule transfettate con 4.1Rsh
rispetto ai controlli. La proteina laminina sembra quindi essere up-regolata dalla
4.1Rsh, come confermato dall’analisi densitometrica (YFP= 0.082 ± 0.03, YFP4.1Rsh= 0.244 ± 0.027, n=3). Abbiamo pertanto preferito non utilizzare la laminina
nel dubbio che non fosse un valido fattore di normalizzazione interno. La
125
normalizzazione è stata fatta fatta sia rispetto al segnale di ICln citosolico (vedi
sopra) che rispetto al segnale originato dalla colorazione della membrana in PVFD
con il metodo dell’amido Black, che è un indice della quantità proteica totale
trasferita su membrana. Per calcolare questo valore di normalizzazione abbiamo
considerato il segnale dell’intera corsia.
A)
B
Figura 37 A) (In basso) Membrana in PVDF colorata con Amido Black. Per normalizzare il segnale di
ICln è stata considerata l’intensità corrispondente all’intera corsia sulla membrana in PVDF. B)
Istogrammi relativi all’analisi densitometrica del segnale nucleare (A in alto) di ICln normalizzato per
il segnale della membrana colorata con Amido Black (t test: ρ value < 0,01).
Il risultato di questa analisi è riportato nell’istogramma in figura 37 B (YFP-4.1Rsh=
0,67 ± 0,062 n=7; YFP= 1,43 ± 0,16 n=7). Anche con questo secondo tipo di analisi
densitometrica si osserva una diminuzione significativa nella quantità della proteina
ICln nel nucleo nelle cellule che over-esprimono la 4.1Rsh rispetto al controllo,
confermando i dati ottenuti dalle immagini di FRET e di immunofluorescenza.
Nella seconda serie sperimentale abbiamo voluto verificare cosa succedeva alla
distribuzione nucleo/citosol della proteina 4.1Rsh, overespressa con un vettore
pIRES-4.1Rsh che ci permette di seguire esclusivamente questa isoforma via
western blot, quando overesprimiamo la porteina ICln. Il vettore bicistronico pIRES,
che esprime EGFP e 4.1 come proteine separate (vedi materiali metodi) permette
sia di overesprimere la proteina senza tag aggiuntivi, che potrebbero interferire con
126
il suo funzionamento, che di monitorare l’efficienza di trasfezione ad un
microscopio a fluorescenza. Le cellule sono state co-transfettate con il vettore
pIRES-4.1Rsh e pECFP-C1-ICln e con pECFP-C1 senza inserto (controllo). A 48h dalla
transfezione le cellule sono state raccolte e abbiamo separato le proteine nucleari
dalle proteine citosoliche. Per ogni campione sono stati caricati 20 µg di proteine
della frazione nucleare.
A)
B)
Figura 38 – A) Western blot effettuato sulle preparazioni proteiche nucleari (lastra in alto) di cellule
HEK 293 Phoenix. In entrambi i western blot nelle prime quattro corsie sono stati caricati i campioni
provenienti dalle cellule transfettate con pECFP-C1-ICln mentre nelle ultime 4 corsie i campioni
provenienti dalle cellule di controllo (transfettate con pECFP) (tutte le cellule sono in cotrasfezione
con pIRES 2EGFP-4.1Rsh). B) Istogrammi relativi all’analisi densitometrica del segnale di 4.1Rsh in
cellule transfettate con pECFP-C1 ICln e con pECFP (controllo) normalizzato per il segnale della
laminina negli stessi campioni (test: ρ value<0,05).
La lastra in figura 38 mostra una diminuzione del segnale della proteina 4.1Rsh nelle
frazioni nucleari di cellule transfettate con CFP-ICln (prime quattro corsie). Tale dato
è stato confermato dall’analisi statistica dell’analisi densitometrica effettuata sul
segnale delle bande della proteina 4.1Rsh (CFP-C1-ICln = 0.36 ± 0.08 n=4; CFP = 1.7
4 ± 0.41, n=4) normalizzato per il segnale della laminina (nostro marker interno).
Per accertarci della qualità della procedura di separazione nucleo/citosol utilizzata
per questi esperimenti, abbiamo caricato nuovamente i campioni provenienti dalle
separazioni nucleo/citosol e incubato le membrane con un anticorpo diretto verso
una proteina prevalentemente citosolica quale la GAPDH (Glyceraldehyde 3phosphate dehydrogenase) e dopo avere sottoposto le membrane ad un protocollo
127
di “stripping” le abbiamo incubate nuovamente con un anticorpo anti-laminina
ampliamente utilizzata come marker del nucleo (Grunewald et al., 2007). Per ogni
campione sono stati caricati 10 µg di proteine.
Figura 39 Immagine relativa a Western blot di arricchimento. Nella lastra autoradiografica in alto
osserviamo il segnale della laminina solo nelle frazioni nucleari, mentre nella lastra in basso è
presente il segnale della GAPDH solo nelle frazioni citosoliche.
Nelle lastre autoradiografiche della figura 39 possiamo osservare che nelle prime sei
corsie dove sono stati caricati i campioni provenienti dalle frazioni citosoliche è
presente il segnale per la GAPDH, marker del citosol ma non il segnale della
laminina invece presente nelle ultime sei corsie dove sono stati caricati gli estratti
nucleari. Ciò conferma che la contaminazione tra le due frazioni durante la
separazione è estramemante ridotta.
5.3.5 Studio della localizzazione della proteina 4.1RII e di ICln nel compartimento
nucleare.
Come per l’isoforma corta, abbiamo analizzato le immagini provenienti dagli
esperimenti di FRET delle cellule transfettate con la proteina YFP-4.1RII e CFP-ICln
per valutare i rapporti nucleo/citosol delle due proteine.
128
A)
4.1RII + CFP
4.1RII + CFP ICln
B)
CFP ICln + YFP
CFP ICln + YFP 4.1RII
Figura 40 Nelle due immagini in alto A) è riportata l’acquisizione al microscopio confocale di cellule
HEK-293 Phoenix transfettate contemporaneamente con YFP–4.1RII e CFP (senza ICln), (a sinistra), e
sulla destra l’immagine relativa a cellule transfettate contemporaneamente con YFP–4.1RII e CFPICln. Nelle due immagini in basso B) a sinistra è riportata l’acquisizione al microscopio confocale di
cellule HEK-293 Phoenix transfettate contemporaneamente con YFP e CFP-ICln e a destra l’immagine
di cellule HEK-293 Phoenix transfettate con YFP-4.1Rsh e CFP-ICln (In giallo l’acquisizione nel canale
del YFP, in blu quella relativa al CFP).
Come riportato in figura 40, la proteina 4.1RII in fusione con il fluoroforo YFP
localizza prevalentemente nel citosol e in membrana ma il segnale nucleare è
estremamente basso; sia in presenza sia in assenza di ICln. Per quanto riguarda la
proteina ICln dall’immagine 40 B osserviamo che sembra esserci solo una debole
riduzione del suo segnale nucleare in presenza della proteina 4.1RII rispetto alla
condizione di controllo. L’analisi quantitativa del rapporto dell’intensità di
fluorescenza tra Fnucleo/Fcitosol per entrambi i canali di acquisizione (CFP e YFP)
129
conferma la bassa presenza nucleare di YFP-4.1RII, che non viene influenzata dalla
over-espressione di ICln.
Figura 41 Istogrammi relativi all’analisi statistica dei rapporti Intensita di fluorescenza nel nucleo/
citosol (Fnucleo/Fcitosol) del fluoroforo CFP (a sinistra) e YFP (a destra) (per ogni condizione saggiata
negli esperimenti di FRET): YFP-4.1RII +CFP-ICln, YFP-4.1RII + ICln-CFP, CFP-ICln + YFP e YFP-4.1RII +
CFP.
Per quanto riguarda ICln il risultato dell’analisi è ambiguo: si ha una diminuizione
significativa di Fnucleo/Fcitosol solo nel caso di CFP-ICln (Fnucleo/Fcitosol=1.00 ± 0.06
n=28) rispetto al controllo con YFP senza la 4.1 (Fnucleo/Fcitosol =1.44 ± 0.027 n=34).
Per ICln-CFP non abbiamo osservato una riduzione significativa del segnale nel
nucleo.
Per verificare la presenza di 4.1 nel nucleo anche in assenza di over-espressione
abbiamo effettuato esperimenti di Western blot per studiare la presenza delle 4.1R
endogene in frazioni nucleari e citplasmatiche di HEK 293 Phoenix.
130
Figura 42 Immagine relativa alla lastra autoradiografica del western blot relativo ad una separazione
nucleo/citosol di cellule HEK 293 Phoenix (sono stati caricati 40 ug di proteine in entrambe le corsie).
Il wetern è stato eseguito utilizzando anti-4.1 come anticorpo primario.
Nella lastra in figura 42 osserviamo nella frazione nucleare e in quella citosolica la
presenza di entrambe le isoforme della proteina 4.1R ad alto (circa135 kDa) e basso
(circa 80 kDa) peso molecolare presenti in questo tipo cellulare.
5.3.6 Studi di western blot sui livelli di espressione nel nucleo della proteina ICln
in cellule HEK 293 Phoenix transfettate con la proteina 4.1RII e sui livelli di
espressione nucleare della proteina 4.1 in cellule transfettate con ICln.
Per effettuare gli esperimenti di localizzazione dell’isoforma lunga abbiamo scelto di
over-esprimere la 4.1RII tramite un vettore IRES che ci permette di esprimere la
proteina 4.1 separata dalla proteina GFP (mentre la GFP ci permette di valutare
l’efficienza di transfezione). Prima di effettuare gli esperimenti di localizzazione
abbiamo deciso di silenziare per mutagenesi sito specifica il secondo sito di inizio
trascrizione ATG2 presente nella sequenza di tale isoforma per evitare che quando
over-esprimiamo la 4.1RII venga in contemporanea prodotta la 4.1Rsh.
Figura 43 Organizzazione in esoni del gene umano per la 4.1R.
131
Figura 44 Lastra riguardante l’overespressione in cellula delle due isoforme 4.1Rsh (prima corsia) e
4.1RII (seconda corsia) (overespresse in un vettore IRES) e l’isoforma 4.1RII mutata nell’ATG2 (terza
corsia).
Come si può vedere dalla lastra in figura 44 nella prima corsia dove abbiamo
caricato 10 µg del lisato totale di cellule transfettate con pIRES-4.1Rsh c’è una
banda all’altezza di circa 80 kDa, come atteso nella seconda corsia abbiamo caricato
il lisato totale di cellule transfettate con pIRES-4.1RII, in questa corsia vediamo la
presenza di una banda alta a 135 kDa corrispondente all’isoforma lunga (come
atteso) e una seconda banda più bassa che presumibilmente corrisponde alla 4.1Rsh
che viene prodotta tramite l’utilizzo della sequenza IRES presente tra ATG1 e ATG2,
come riportato anche da altri (Lospitao et al., 2008). A riprova di questa ipotesi, nel
lisato di cellule transfettate con il vettore pIRES-4.1RII ATG2 mut, in cui l’ATG2 è
stato mutato in GTG, la banda a 80 kDa non è più visibile, indicando che non viene
più prodotta la 4.1Rsh ma solo l’isoforma lunga.
Come per l’isoforma corta per effettuare studi di localizzazione subcellulare nel
nucleo e nel citosol per le proteine ICln e 4.1RII abbiamo allestito due serie
sperimentali di western blot:
1) su frazioni separate di citosol e nuclei, per indagare la distribuzione di ICln in
questi due compartimenti quando è over-espressa la 4.1RII ATG2 mut.
2) su frazioni separate di citosol e nuclei, per indagare la distribuzione della
4.1RII quando è over-espressa la proteina CFP-C1 ICln.
I risultati ottenuti per la prima serie sperimentale sono riportati in figura 45.
132
A)
B)
Figura 45 A) Western blot effettuato sulle preparazioni proteiche nucleari (lastra in alto) e le
corrispondenti frazioni citosoliche (lastra in basso) di cellule HEK 293 Phoenix. In entrambi i western
blot nelle prime quattro corsie sono stati caricati i campioni provenienti dalle cellule di controllo
(transfettate con pIRES-GFP) mentre nelle ultime quattro corsie i campioni provenienti dalle cellule
transfettate con la proteina pIRES 4.1RII ATG2mut. Per ogni campione sono stati caricati 20 µg di
proteine della frazione nucleare e 10 µg della frazione citosolica Le membrane sono state incubate
entrambe con anticorpo primario anti-ICln. B) Istogrammi relativi all’analisi densitometrica del
segnale nucleare di ICln normalizzato per il segnale citosolico di ICln (t test: ρ value < 0,05).
Nella prima lastra in alto mostrata della figura 45 possiamo osservare una
diminuzione del segnale nel nucleo della proteina ICln in cellule transfettate con la
proteina 4.1RII (con ATG2 mutato) (ultime quattro corsie); l’osservazione è
confermata dall’analisi densitometrica delle bande effettuata normalizzando il
segnale di ICln nucleare rispetto al segnale citosolico. Quando è over-espressa la
proteina 4.1RII ATG2mut il segnale di ICln nel nucleo (pIRES-4.1RII ATG2mut= 0,17 ±
0,04) diminuisce significativamente rispetto alla condizione di controllo (pIRES2EGFP= 0,43 ± 0,07).
Come per i Western blot in cui abbiamo over-espresso l’isoforma corta il segnale di
ICln nel nucleo è stato normalizzato anche per il segnale originato dalla colorazione
della membrana in PVFD con il metodo dell’amido Black (fig. 46).
133
A)
B)
ICln nucleo/membrana
Figura 46 A) (In basso) Membrana in PVDF colorata con Amido Black. Per normalizzare il segnale di
ICln è stata considerata l’intensità corrispondente all’intera corsia sulla membrana in PVDF. B)
Istogrammi relativi all’analisi densitometrica del segnale nucleare (A in alto) di ICln normalizzato per
il segnale della membrana colorata con Amido Black (t test: ρ value < 0,01).
Anche in questo caso si osserva una diminuzione significativa nella frazione nucleare
di ICln quando overesprimiamo la 4.1ATG2 mut (pIRES-2EGFP= 0,34 ± 0,03; pIRES4.1RII ATG2 mut= 0,13 ± 0,03).
Nella seconda serie sperimentale abbiamo voluto verificare la distribuzione
nucleo/citosol della proteina 4.1RIIATG2 mut overespressa con un vettore IRES in
co-espressione per la proteina ICln espressa come proteina di fusione con il CFP
posto alla sua estremità C-terminale. Analogamente a quanto fatto per 4.1Rsh,
abbiamo scelto di lavorare in overespressione della proteina 4.1RII ATG2 mut per
assicurarci di studiare l’effetto della proteina ICln in modo specifico per questa
isoforma.
134
A)
B)
Figura 47 A) Western blot effettuato sulle preparazioni proteiche nucleari (lastra in alto) di cellule
HEK 293 Phoenix. Nelle prime quattro corsie sono stati caricati i campioni provenienti dalle cellule
transfettate con pECFP-C1 (controllo) mentre nelle ultime 4 corsie i campioni provenienti dalle
cellule transfettate con pECFP-C1 ICln (tutte le cellule sono in co-transfezione con pIRES-4.1RIIATG2
mut). Inoltre è riportata la lastra riguardante il segnale della laminina nelle stesse estrazioni nucleari.
Per ogni campione sono stati caricati 40 µg di proteine della frazione nucleare. B) Istogrammi relativi
all’analisi densitometrica del segnale di 4.1RIIATG2mut in cellule transfettate con pECFP-C1-ICln e
con pECFP (controllo) normalizzato per il segnale della laminina negli stessi campioni (test: ρ value<
0,05).
Il segnale della proteina 4.1RII è stato normalizzato per il segnale della laminina.
Dall’analisi statistica abbiamo verificato che in presenza di ICln il segnale nel nucleo
della proteina 4.1RII diminuisce significativamente rispetto alla condizione di
controllo (CFP= 0,70 ± 0,22; CFP-ICln= 0,027 ± 0,004) come avviene per l’isoforma
corta.
5.4 Studi di localizzazione subcellulare: membrana.
5.4.1 Osservazione delle immagini di FRET della localizzazione in membrana di
entrambe la isoforme: 4.1Rsh e 4.1RII.
Oltre agli gli studi di localizzazione nucleare abbiamo analizzato la localizzazione in
membrana della proteina 4.1R in presenza di ICln (Diakowski et al., 2006). Entrambe
le isoforme 4.1Rsh e 4.1RII hanno l’esone 5 che codifica per una regione importante
per il legame con la membrana (Gascard et al., 1998).
135
A) CFP+4.1Rsh
B) CFP-ICln+4.1Rsh
Figura 48 Nell’immagine A) è riportata l’acquisizione al microscopio confocale di cellule HEK-293
Phoenix co-transfettate con YFP–4.1Rsh e CFP (senza ICln) come indicato dalla freccia si osserva un
segnale della 4.1Rsh in membrana, l’immagine B) è relativa a cellule transfettate
contemporaneamente con YFP–4.1Rsh e CFP-ICln. In questa immagine si nota come il segnale in
membrana della 4.1Rsh diminuisce notevolmente.
Figura 49 Immagini acquisite al microscopio confocale del segnale relativo al YFP (eccitazione 514
nm, finestra di acquisizione 525-600 nm, immagini A e B) e del CFP (eccitazione 485 nm, finestra di
acquisizione 465-505 nm,immagini C e D) di cellule HEK–293 Phoenix transfettate con 4.1Rsh-YFP e
CFP (A e C) o 4.1Rsh e CFP-ICln (B e D). Le immagini sono state ottenute come ”Max Projection” di 50
136
(A) o 40 (B) sezioni confocali delle cellule. Nell’immagine in A sono visibili numerosi filopodi, che
sono invece meno evidenti e ridotti nelle cellule in B, in cui è over-espressa anche ICln.
Dalle immagini della 4.1 in overespressione ottenute durante gli esperimenti di
FRET riportate in figura 48 osserviamo nell’immagine A che il segnale della 4.1Rsh
quando è co-transfettata con il CFP (vuoto) è presente nel nucleo, nel citoplasma e
in membrana (freccia bianca) mentre quando la 4.1Rsh è coespressa con ICln il suo
segnale in membrana diminuisce visibilmente.
Inoltre abbiamo notato (figura 49 A) che over–esprimendo 4.1Rsh le cellule hanno
la tendenza ad aumentare notevolmente il numero di filopodi emessi. Questo
fenomeno sembra essere inibito dalla cotransfezione con ICln (figura 49 B).
La presenza di ICln sembra, quindi, influenzare la localizzazione in membrana
dell’isoforma corta della proteina 4.1Rsh.
Lo stesso fenomeno è presente anche nel caso dell’isoforma lunga (Fig 50).
A) 4.1RII + CFP
B) 4.1RII + CFP ICln
Figura 50 Nell’immagine A) è riportata l’acquisizione al microscopio confocale di cellule HEK-293
Phoenix co-transfettate con YFP–4.1RII e CFP (senza ICln) come indicato dalla freccia si osserva un
segnale della 4.1RII in membrana, l’immagine B) è relativa a cellule transfettate
contemporaneamente con YFP–4.1RII e CFP-ICln. In questa immagine si nota come il segnale in
membrana della 4.1RII diminuisce notevolmente in presenza di ICln.
Osservando le immagini di FRET possiamo vedere che l’isoforma lunga quando la
proteina 4.1 è espressa con il CFP (controllo) localizza sia nel citosol che in
membrana dove è particolarmente evidente nelle regioni di contatto tra cellula e
cellula, mentre quando viene co-espressa con ICln il segnale in membrana
diminuisce notevolmente. Abbiamo inoltre notato (figura 51) che come per la
137
proteina 4.1Rsh over–esprimendo 4.1RII le cellule hanno la tendenza ad aumentare
il numero di filopodi emessi. Questo fenomeno come abbiamo già osservato per
l’altra isoforma sembra essere inibito dalla cotransfezione con ICln (figura 51 B).
Figura 51 Immagini acquisite al microscopio confocale del segnale relativo al YFP (eccitazione 514
nm, finestra di acquisizione 525-600 nm) e del CFP (eccitazione 485 nm, finestra di acquisizione 465505 nm,immagini C e D) di cellule HEK–293 Phoenix transfettate con YFP–4.1RII e CFP (A e C) o YFP–
4.1RII e CFP-ICln (B e D). Le immagini sono state ottenute come ”Max Projection” di 50 (A) o 40 (B)
sezioni confocali delle cellule. Nell’immagine in A sono visibili numerosi filopodi, che sono invece
meno evidenti e ridotti nelle cellule in B, in cui è over-espresso anche ICln.
5.4.2 Studi di western blot sui livelli di espressione della proteina 4.1R in
membrana in cellule HEK 293 Phoenix co-espresse con la proteina ICln.
Dall’osservazione delle immagini in over-espressione di YFP-4.1RII e di YFP-4.1Rsh
abbiamo visto che entrambe le isoforme della proteina 4.1 (4.1Rsh e 4.1RII) in
presenza di ICln tendono a diminuire il loro segnale in membrana. Per confermare
quest’osservazione abbiamo effettuato esperimenti di western blot sulla 4.1R
endogena in frazioni di membrane totali. Le cellule (HEK-293 Phoenix) sono state
transfettate con pCFP-C1-ICln o con il solo CFP (controllo). A 24h dalla trasfezione le
cellule
sono
state
raccolte
e
abbiamo
membrane/citosol.
138
effettuato
una
separazione
Figura 52 A) Western blot effettuato sulle preparazioni di membrana di cellule transfettate con CFPC-ICln o il solo CFP; A e B si riferiscono alla banda alta e alla banda bassa della proteina 4.1
endogena. E’ mostrata la lastra riguardante il segnale della caderina per cui abbiamo normalizzato il
segnale della 4.1. Per ogni campione sono stati caricati 40 µg di proteine. B) C) D) Analisi
densitometrica del segnale di membrana della 4.1R endogena (pannello B somma della bande A+B,
pannello C segnale della banda bassa e pannello D segnale della banda alta) di cellule transfettate
con pECFP-C1 ICln e con pECFP (controllo) normalizzato per il segnale della caderina (test: ρ
value<0,05).
La lastra in alto in figura 52 A mostra il segnale della proteina 4.1 endogena nelle
frazioni di membrana derivanti da cellule transfettate con CFP-C1-ICln (prime
quattro corsie) e CFP (ultime quattro). Possiamo osservare la presenza di due bande
una a circa 135 kDa corrispondente all’isoforma lunga della proteina 4.1 (4.1RII)
banda A e una più bassa a circa 80 kDa corrispondente all’isoforma corta della 4.1R
(4.1Rsh) banda B. In presenza di ICln entrambi i segnali (banda A e banda B) della
proteina 4.1 diminuiscono notevolmente. Il dato è stato confermato dall’analisi
densitometrica, effettuata normalizzando il segnale della proteina 4.1R per il
segnale della caderina (proteina marker di membrana). L’over-espressione con ICln
causa una diminuzione significativa del segnale di membrana di entrambe le
isoforme di 4.1R (per la 4.1Rsh> CFP-C1 ICln= 0.39 ± 0.13 rispetto alla condizione di
controllo 4.1Rsh> CFP= 1.28 ± 0.21, per la 4.1RII> CFP-C1 ICln 0.26 ± 0.07 vs
139
controllo 4.1RII> CFP= 1.21 ± 0.22). Questo dato conferma quanto osservato nelle
immagini di 4.1R over-espressa in fusione con YFP.
La qualita delle preparazioni di membrane totali è stata controllata mediante
western blot utilizzando anticorpi specifici per marker citosolici (GAPDH) o di
membrana (Caderina).
Figura 53 Immagine relativa a Western blot di arricchimento. Nella lastra autoradiografica in alto
osserviamo il segnale della GAPDH solo nelle preparazioni citosoliche, mentre nella lastra in basso è
presente il segnale della caderina solo nelle estrazioni di membrana. Per ogni campione sono stati
caricati 20 µg di proteine sia per le frazioni di membrana che per i citosol.
Come mostrato in figura 53 possiamo osservare che il segnale per la GAPDH, marker
del citosol è presente solo nelle frazioni citosoliche mentre la caderina è fortemente
arricchita nell frazioni di membrana.
5.5 4.1R e RVD
E’ noto che la proteina ICln è coinvolta nella regolazione del volume cellulare e in
ipotonia la proteina trasloca dal citosol alla membrana (Musch et al., 1997). Durante
l’RVD (Decremento regolatorio del volume cellulare) (Fürst et al., 2002) anche il
citoscheletro subisce numerosi riarrangiamenti (Rasmussen et al., 2008). Ci siamo
pertanto chiesti quale fosse il comportamento della 4.1R sottoposta ad uno stress
ipotonico, visto che è una proteina adattatrice associata alla membrana che
connette elementi del citoscheletro a proteine associate alla membrana (Gascard et
al., 2000). In particolare, ci siamo soffermati su due aspetti principali:
1) poiché un aumento della espressione di ICln sembra causare una
diminuzione della frazione di membrana di 4.1R, abbiamo voluto verificare
se ci fossero alterazioni nella quantità di di 4.1R in membrana indotte
dall’ipotonia.
140
2) Abbiamo voluto verificare quale fosse l’effetto dell’over-espressione delle
due isoforme di 4.1R sull’attivazione della ICl,swell.
5.5.1 Studi di western blot sulla localizzazione in membrana della proteina 4.1R
sottoposta a uno stimolo ipotonico
Per indagare sul primo punto, abbiamo allestito esperimenti di western blot per
verificare l’effetto dello stress osmotico sulla presenza della 4.1R in membrana. Gli
esperimenti sono stati effettuati sia utilizzando cellule non trasfettate, seguendo le
4.1R endogene, sia su cellule transfettate con 4.1Rsh o 4.1RII, per studiare il
comportamento delle singole varianti.
In entrambi i casi, per massimizzare gli effetti di uno shock osmotico le cellule (HEK293 Phoenix) sono state trattate con una soluzione ipotonica o lievemente
ipertonica (condizione di controllo) per 5 minuti, prima di procedere con la
separazione membrane totali/citosol.
Figura 54 A) Western relativo al segnale in membrana della proteina 4.1 endogena in preparati di
frazioni di membrana di cellule trattate con soluzione ipotonica (prime 4 corsie) o con soluzione
141
+ +
ipertonica. La lastra in basso rappresenta il segnale della Na K ATPasi negli stessi campioni. Per ogni
campione sono stati caricati 70 µg di proteine. B) C) Istogrammi relativi all’analisi densitometrica del
segnale in membrana della 4.1RII (B) e del segnale della 4.1Rsh (C) di cellule trattate con soluzione
+ +
ipotonica o soluzione ipertonica normalizzato per il segnale della Na K ATPasi negli stessi campioni
(test: ρ value<0,05).
I risultati relativi alle 4.1R endogene sono riportati in figura 54. L’analisi
densitometrica è stata effettuata sui segnali della banda alta (relativa alla 4.1RII) e
della banda bassa (4.1Rsh) separatamente, normalizzandoli per quello della Na+-K+ATPasi. Dall’analisi statistica (figura 54 B C) risulta che il segnale in membrana della
proteina 4.1RII (IPO= 0,10 ± 0,03) diminuisce significativamente in ipotonia rispetto
alla condizione di controllo (IPO=0,46 ± 0,13). L’isoforma corta invece ha una
tendenza alla diminuzione in condizioni ipotoniche (IPO= 0,25 ±0,089) ma
dall’analisi statistica non risulta essere significativa (IPER=0,62 ± 0,25).
Gli stessi esperimenti di western blot sono stati effettuati anche su cellule
precedentemente transfettate con pIRES-4.1RII ATG2mut o con pIRES-4.1Rsh. Dopo
48h dalla trasfezione le cellule sono state trattate con soluzione ipotonica o
ipertonica prima di procedere con la separazione membrane/citosol.
Figura 55 A) Western blot relativo al segnale in membrana della proteina 4.1RII in preparati di
frazioni di membrane totali di cellule HEK-293 Phoenix transfettate con pIRES-4.1RIIATG2mut e
trattate con soluzione ipotonica (prime 4 corsie) o con soluzione ipertonica (ultime quattro). A destra
142
è riportato l’istogramma relativo all’analisi densitometrica del segnale della 4.1RII normalizzato per
+ +
la Na K ATPasi. Per ogni campione sono stati caricati 50 µg di proteine delle frazioni di membrana B)
Western relativo al segnale in membrana della proteina 4.1Rsh in preparati di frazioni di membrana
di cellule transfettate con pIRES-4.1Rsh e trattate con soluzione ipotonica o con soluzione ipertonica.
A destra è riportato l’istogramma relativo all’analisi densitometrica del segnale in membrana della
+ +
4.1Rsh normalizzato per la Na K ATPasi (T test: ns = ρ value>0,05, * = p<0.05).
Analogamente a quanto riscontrato
per le 4.1R endogene, dall’analisi
densitometrica di questi western blot (figura 55) è emerso che, l’isoforma lunga
della proteina 4.1R diminuisce significativamente (IPO= 0,53 ± 0,07; IPER= 1,10 ±
0,1) in membrana dopo che è stata sottoposta a stimolo ipotonico, mentre
l’isoforma corta ha solo una tendenza a diminuire ma non risulta significativa (IPO=
0,437 ± 0,10; IPER= 0,62 ± 0,14).
In parallelo a questi studi abbiamo effettuato esperimenti di colocalizzazione, al
momento da considerarsi solo preliminari. Per queste prove abbiamo transfettato
transientemente cellule HEK 293 Phoenix con YFP-C-4.1Rsh + CFPmem (proteina di
membrana) o con YFP-C-4.1RII + CFPmem.
A 24h dalla trasfezione abbiamo acquisito le immagini delle cellule al microscopio
confocale, previo il mantenimento per almeno 5 minuti in soluzione ipertonica.
Infine, la soluzione extracellulare è stata sostituita con quella ipotonica e sono state
acquisite nuovamente le immagini a 5 e a 10 minuti dalla sostituzione.
In parallelo abbiamo allestito dei controlli transfettando le cellule con CFPmem +
YFPmem due proteine di membrana non funzionalmente interessate nell’ipotonia
che permettono di valutare aspetti aspecifici dovuti allo stress ipotonico. La
quantificazione della colocalizzazione è stata calcolata utilizzando due indici di
colocalizzazione: PC (coefficiente di Pearson) che tiene conto della media
dell’intensità dei pixel per ognuno dei due canali, CFP e YFP, a cui è sottratta
l’intensità dei pixel iniziale; l’intervallo del coefficiente di Pearson va da -1 a 1 dove
un valore di -1 indica la totale mancanza di sovrapposizione tra i pixel dell’immagine
mentre 1 indica la perfetta sovrapposizione dei pixel nell’immagine e OC
(coefficiente di overlapping), che a differenza del PC non sottrae l’intensità dei pixel
iniziale; il valore che si ottiene è in un intervallo tra 0 e 1. L’analisi statistica è stata
143
fatta sulla variazione percentuale dei coefficienti di colocalizzazione(ΔPC% o ΔOC%)
indotta dall’ipotonia rispetto a alla condizione di ipertonia.
I primi dati ottenuti nell’ambito di un singolo esperimento, e pertanto da
considerarsi solo preliminari, sembrano in accordo con gli esperimeinti di western
blot. Sia nei controlli (CFPmem + YFPmem) che nei campioni trasfettati con 4.1, gli
indici di colocalizzazione sono diminuiti dopo stimolazione ipotonica. Tuttavia la
diminuzione del coefficiente PC ottenuto per le cellule transfettate con CFPmem +
YFP-C-4.1Rsh o YFP-C-4.1RII (ΔPC%= -16,2 % ± 3.3 %, n=5 e ΔPC%=-17% ± 3.1 %,
n=3 rispettivamente) è significativamente maggiore rispetto ai controlli (CFPmem +
YFPmem: ΔPC%= -5.3 % ± 1.7 %, n=5). Nel caso del coefficiente OC, la diminuzione è
risultata diversa dal controllo nel caso della isoforma lunga (CFPmem + YFP C-4.1RII
ΔOC%= -14.1 % ± 2.0 %, n=3 vs CFPmem + YFPmem: ΔOC%= -4.2 % ± 3.1, n=5), ma
non per la corta (CFPmem + YFP-C-4.1Rsh: ΔPC%=-11 % ± 3.2 %, n=5).
Sia gli esperimenti di colocalizzazione che gli esperimenti di Western blot
suggeriscono che entrambe le isoforme tendono a diminuire in membrana dopo
uno stimolo ipotonico e che tale tendenza è più marcata per la 4.1RII.
5.5.2 Significato funzionale dell’interazione tra ICln e 4.1: Esperimenti di patchclamp.
Al fine di analizzare le conseguenze funzionali dell’interazione tra le proteine ICln e
la 4.1R, e il ruolo svolto nella modulazione della corrente per il cloruro ICl,swell
attivata durante il rigonfiamento cellulare, sono stati condotti esperimenti di patchclamp in configurazione whole-cell.
Le cellule HEK 293T sono state transfettate con le sequenze codificanti per le due
varianti da splicing della 4.1R, 4.1RII ATG2 mut (variante lunga) e 4.1Rsh (variante
corta), clonate nel vettore pIRES-2-EGFP, che consente la contemporanea
espressione della proteina d’interesse e della GFP, per selezionare le cellule
transfettate. Inoltre sono stati condotti esperimenti di controllo su cellule
transfettate con il vettore vuoto pIRES-2-EGFP.
144
Le cellule erano inizialmente incubate in una soluzione ipertonica; dopo avere
raggiunto la configurazione whole-cell, la stabilità del sigillo veniva controllata per
circa 2 minuti al termine dei quali veniva registrata la corrente imponendo gradini
di potenziale della durata di 500 ms che variavano da -100 mV a +100 mV
(protocollo I-V) con un intervallo di 20 mV. In un secondo tempo la soluzione del
bagno in cui erano mantenute le cellule era sostituita con una soluzione ipotonica
per determinare gli effetti indotti dal rigonfiamento cellulare sull’attivazione nel
tempo e sull’entità della corrente. Dopo l’aggiunta della soluzione ipotonica la
corrente era misurata per 10 minuti, facendo delle registrazioni con il protocollo I-V
ad intervalli di tempo di 2 minuti e 30 secondi.
Nei grafici di figura 56 sono riportate le relazioni densità di corrente-voltaggio
registrate in cellule HEK 293T. La capacità di membrana non variava in modo
significativo nel corso dell’esperimento (anche dopo esposizione alla soluzione
ipotonica).
Nelle cellule di controllo, transfettate con il vettore pIRES-2-EGFP-EGFP (nel cui
multiple clonig site è stato inserito l’ORF della EGFP), (Figura 56) si osserva, dopo la
sostituzione con la soluzione ipotonica, l’attivazione di una corrente per il cloruro
che presenta una rettificazione uscente, come atteso per la ICl,swell. Sebbene la
curva corrente-voltaggio della corrente dopo 10 minuti dalla sostituzione con
soluzione ipotonica non sia statisticamente differente dalla corrente registrata in
ipertonica (Anova a due via, Bonferroni post-Test); a +80 mV e a + 100 mV la
densità di corrente (+25.31 ± 14.76 pA/pF (n=6) e +33.9 ± 19.82 pA/pF (n=6),
rispettivamente) è statisticamente maggiore (p<0.05, T student) rispetto alla
densità di corrente misurata a +80 mV e +100 mV in soluzione ipertonica (4.34 ±
1.06 pA/pF (n=7) e 5.38 ± 1.33 pA/pF (n=7), rispettivamente).
145
A)
B)
60
iper GFP
ipo 2.5' GFP
ipo 5' GFP
ipo 7' 30'' GFP
ipo 10' GFP
***
d(pA/pF)
50
*
40
30
20
10
-125
-100
-75
-50
-25
25
50
75
-10 )
100
125
V(mV)
-20
-30
Figura 56 Corrente ICl,swell generata in cellule HEK 293T transfettate con il vettore pIRES-2-EGFP. A)
Tracciati di corrente registrati con il protocollo I-V in presenza di una soluzione ipertonica e 10’ dopo
la sostituzione con la soluzione extracellulare ipotonica. B) Relazione d/V: in ordinata sono riporati i
valori della corrente normalizzata per la capacità di membrana (densità di corrente, espressa in
pA/pF) e in ascissa il potenziale imposto nel corso dell’esperimento, espresso in mV). La corrente è
stata registrata in soluzione ipertonica e a diversi tempi dalla sostituzione con la soluzione ipotonica).
I dati sono riportati come medie ± S.E.M. *= p<0.05 rispetto al controllo (t test p<0.05).
Nelle cellule transfettate con il plasmide codificante per la proteina 4.1RIIATG2mut (pIRES-2-EGFP-4.1RII ATG2 mut) la corrente registrata in presenza di
soluzioni ipertoniche, così come in ipotonia non è differente dalla corrente
registrata in cellule transfettate con il vettore vuoto (figura 57). In presenza di
soluzioni ipertoniche la densità di corrente è pari a +7.44 ± 2.35 pA/pF (n=8) e a 4.98 ± 1.66 pA/pF (n=8) a + e –100 mV rispettivamente ed è + 23.55 ± 8.75 pA/pF
(n=8) e -15.52 ± 5.8 pA/pF (n=8) dopo 10’ dalla sostituzione con la soluzione
ipotonica.
146
Nel caso invece di cellule transfettate con il plasmide codificante per la proteina
4.1Rsh (pIRES-2-EGFP-4.1Rsh), già dopo 5’ dalla sostituzione con soluzione ipotonica
a +100 mV abbiamo registrato una corrente +48.82 ± 14.49 pA/pF (n=6)
significativamente maggiore rispetto quella misurata in condizione di controllo 7.09
± 0.85 pA/pF (n=6). Dopo 10’ la sostituzione con la soluzione ipotonica abbiamo
notato un incremento della densità di corrente a + 100mV +80 mV +60 mV e a –
100mV -80mV e -60 mV (densità di corrente a +100 mV è paria a +107.22 ± 25.55
pA/pF (n=6), a +80 mV è pari a +82.46 ± 19.55 pA/pF (n=6), a + 60 mV è pari a
+58.33 ± 13.62 pA/pF (n=6), a -100 mV è pari a -64.21 ± 14.02 pA/pF (n=6), a -80 mV
11.78 ± -54.24 pA/pF (n=6) e a -60 mV è pari a -41.16 ± 9.27 pA/pF (n=6))
significativo rispetto al controllo in ipertonia (p<0.05).
***
150
hypo 10' GFP n=8
hypo 10' 4.1sh n=6
hypo 10' 4.1RII ATG2 mut n=8
***
100
50
pA/pF
-150
***
-100
-50
50
100
150
-50
*** **
*
-100
mV
Figura 57 Correnti ICl,swell in cellule HEK 293T transfettate con il vettore pIRES-2-EGFP (controllo),
con il vettore pIRES-2-EGFP 4.1Rsh o con con il vettore pIRES-2-EGFP 4.1RII ATG2 mut. Relazione d/V
(densità di corrente, espressa in pA/pF, e voltaggio, espresso in mV) registrata in cellule HEK 293T
dopo 10’ dalla sostituzione con soluzione ipotonica (I dati sono riportati come medie ± S.E.M.
*=p<0.05; **=p<0.01 ***=p<0.001 (t test).
147
5.7 Studi di proliferazione di cellule HEK 293 Phoenix transfettate con
4.1Rsh e ICln.
I dati esposti precedentemente indicano che l’overespressione di ICln determina
una alterazione della localizzazione di 4.1R. Poiché la sua maggiore o minore
presenza in membrana e nel nucleo è stata messa in relazione all’attività antiproliferativa di 4.1R (Kuns et al., 2005) abbiamo di recente iniziato una serie di
esperimenti volti a valutare se l’over-espressione di ICln potesse alterare la
proliferazione di cellule over-esprimenti 4.1R. In questa fase di studio abbiamo
scelto di focalizzare la nostra attenzione sull’isoforma corta (4.1Rsh) della proteina
4.1R e in futuro approfondiremo questo studio anche per l’isoforma lunga (4.1RII). I
test di proliferazione sono stati eseguiti su cellule HEK 293 Phoenix co-transfettate
con CFP-ICln e YFP-4.1Rsh o con YFP-4.1Rsh + CFP (controllo). Le cellule a 24 ore
dalla trasfezione sono state seminate in pozzetti da 24 (5 104 cellule/pozzetto) e
contate al tempo 0 (momento della semina) alle 24h, 48h, 72h dalla semina
utilizzando un emocitometro. Il test di esclusione del Trypan blue è stato utilizzato
per discriminare le cellule morte dalle vive.
*
200
*
150
ns
YFP-4.1Rsh
YFP-4.1Rsh+CFP-ICln
100
n=4 n=4
n=4 n=4
t=
72
n=4 n=4
t=
48
0
t=
24
50
Figura 58 Istogrammi relativi agli studi di proliferazione di 4 serie sperimentali indipendenti. Per ogni
prova indipendente il valore per le cellule transfettate con YFP-4.1Rsh + CFP-ICln è stato calcolato in
rapporto percentuale rispetto alla condizione di controllo (cellule transfettate con YFP.4.1Rsh). *p<
0.05 (t test).
Dall’analisi statistica (figura 58) risulta che nella condizione in cui le cellule sono cotransfettate con 4.1Rsh e ICln i valori a 48h e a 72h erano significativamente
148
maggiori (alle 48h: 176,72 ± 20,99; alle 72h: 158,26 ± 4,67) rispetto alla condizione
di controllo (in cui la cellule sono co-transfettate con YFP-4.1Rsh e CFP vuoto, il
valore delle cellule contate è stato posto a 100%).
Per assicurarci che la transfezione si sia mantenuta elevata per tutta la durata
dell’esperimento
abbiamo
effettuato
un
western
blot
di
controllo
dell’overespressione di ICln e della 4.1Rsh.
Figura 59 Western blot di controllo dell’overespressione di CFP-ICln e della proteina YFP-4.1Rsh.
Nella lastra A la membrana è stata incubata con l’anti-4.1 che riconosce la proteina 4.1 endogena e
l’overespressa mentre nella lastra B la membrana è stata incubata con l’anti ICln che riconosce sia la
proteina ICln endogena che overespressa. Per ogni campione sono stati caricati 5 µg di proteine
totali.
Nelle lastre autoradiografiche riportate nella figura 59A possiamo osservare che il
segnale della proteina 4.1Rsh overespressa al tempo 0 che si mantiene poi costante
alle 72h nelle cellule transfettate con YFP-4.1Rsh + CFP (seconda e terza corsia) e
YFP-4.1Rsh + CFP-ICln (quinta e sesta corsia) mentre si nota un debole segnale delle
proteine 4.1R endogene nelle cellule non transfettate come atteso (si osservano
due bande relative alle due isoforme ad alto e basso peso mlecolare (135 kDa e 80
kDa). Viceversa nel pannello B della figura 59 si può osservare un segnale ad una
altezza corrispondenete alla proteina di fusione CFP- ICln (intorno ai 50 kDa) al
tempo 0h e alle 72h solo nelle cellule transfettate con CFP-ICln + YFP-4.1Rsh (quinta
e sesta corsia). In tutte le corsie si nota anche il segnale della proteina ICln
endogena a circa 37kDa.
149
6 Discussione
Durante l’RVD si attiva una corrente di cloruro denominata ICl,swell e la proteina
ICln riveste un ruolo di primaria importanza nella sua attivazione anche se non è
ancora stato chiarito se interviene come regolatore del canale o è un componente
molecolare del canale stesso (Fürst et al., 2006). La corrente ICl,swell è modulata
dallo stato del citoscheletro actinico (Lang et al., 1998; Moustakas et al., 1998) e
ICln interagisce con componenti citoscheletriche tra cui l’actina (Krapiwinsky et al.,
1994; Emma et al., 1998), la miosina (Emma et al., 1998) e due componenti
appartenenti alla famiglia delle 4.1 l’isoforma R e B (Tang et al., 1998; Calinisan et
al., 2006), anche se ICln sembra essere in grado di legare la regione FERM di tutte le
4.1 (Gascard et al., 2006).
Fino ad oggi l’interazione è stata studiata con diversi metodi tra cui saggio di doppio
ibrido in lievito e immunoprecipitazione (Tang et al., 1998) che hanno stabilito che
la proteina ICln interagisce con il suo dominio C-terminale (aa dal 103 al 237) e il
lobo C del dominio FERM delle 4.1 (Gascard et al., 2006) ma non hanno chiarito il
significato di tale interazione.
Interazione tra ICln e la 4.1R
In questo lavoro abbiamo clonato due isoforme di 4.1R da cellule HEK per studiare
la loro interazione con la proteina ICln umana. Abbiamo scelto di focalizzarci sulle
isoforme di 4.1R in quanto è la prima per cui è stata individuata l’interazione con
ICln e quella per cui le regioni di interazione sono state meglio definite. Inoltre la
4.1R rappresenta il membro più conosciuto e meglio caratterizzato della famiglia, di
cui spesso è utilizzata come prototipo. Dell’isoforma R esistono diverse varianti da
splicing
(Gascard
et
al.,
1998)
e
di
regola
molte
sono
espresse
contemporaneamente in un unico tipo cellulare (Gascard et al., 1998). La sintesi
delle diverse varianti di 4.1R è stata approfondita soprattutto nei globuli rossi, dove
si ritiene che le forme lunghe siano tipiche delle fasi iniziali di sviluppo degli
150
eritrociti (Nunomura et al., 2009), mentre le forme corte sono considerate tipiche
degli eritrociti maturi. I dati ad oggi disponibili hanno fin’ora riguardato solo le
interazioni di ICln con varianti a basso peso molecolare di 4.1R o singoli domini di
4.1R (Tang et al., 1998; Gascard et al., 1998). Per questo motivo abbiamo voluto
includere nel nostro progetto anche varianti ad alto peso molecolare, argomento
potenzialmente interessante in quanto correlato al differenziamento cellulare. Le
isoforme utilizzate per questo lavoro sono state clonate da cellule umane HEK-293,
eseguendo due diverse PCR ciascuna con un diverso primer senso specifico per uno
dei due possibili siti di inizio della trascrizione, e lo stesso primer antisenso. Le due
isoforme isolate, 4.1RII, isoforma lunga e 4.1Rsh a basso peso molecolare (che
presumibilmente rappresentano anche quelle presenti in maggiore quantità nelle
cellule) sono risultate identiche se non per la presenza o meno dei primi 209
aminoacidi iniziali corrispondenti alla regione U1, presente solo nelle varianti ad
alto peso molecolare e assente nelle varianti corte che iniziano direttamente con la
regione FERM. Le due varianti da splicing individuate sono corrispondenti alla
4.1R135, (4.1RII) e alla 4.1R80, (4.1Rsh) che sono tra le varianti da splicing
maggiormente espresse anche in altri tipi cellulari (Gascard et al., 1998). Ad
esempio, negli eritroblasti la 4.1R135 (4.1RII) risulta essere il 36% delle isoforme la
cui trascrizione parte dall’AUG-1 e la 4.1R80 (4.1Rsh) è il 63% delle isoforme
prodotte a partire dal secondo AUG di inizio trascrizione (Gascard et al., 1998). Le
due isoforme hanno entrambe l’esone 16 che è fondamentale per l’interazione con
actina e spectrina (Correas et al., 1986) e per la localizzazione nucleare (Gascard et
al., 1999) e l’esone 5 che codifica per una regione responsabile del legame con la
membrana (Gascard et al 1998). La presenza di queste regioni è in accordo con la
localizzazione che abbiamo riscontrato over-esprimendo le proteine in fusione con il
YFP. Gli esperimenti di FRET sono stati condotti per ogni configurazione possibile
per entrambe le isoforme (vedi paragrafo 5.2) dando risultati molto diversi a
seconda delle coppie donatore-accettore utilizzate, suggerendo che l’organizzazione
spaziale del complesso 4.1R-ICln sembra essere piuttosto rigidamente orientata
nello spazio. I dati riguardanti l’isoforma corta validano le notizie disponibili sui siti
151
di interazioni tra le due proteine. Per quanto riguarda l’isoforma corta 4.1Rsh,
abbiamo visto che quando il CFP si trova all’estremità C-terminale di ICln l’efficienza
di FRET diminuisce. Poichè ICln lega la 4.1 con la sua estremità C-terminale (Tang et
al., 1998) è possibile che il fluoroforo in questa posizione interferisca con
l’interazione tra le due proteine a causa del suo ingombro sterico oppure che
posizionare il CFP all’estremità C-t di ICln sia sufficiente ad allontanare donatore e
accettore quanto basta per inibire il trasferimento energetico. Analogamente,
quando il YFP si trova al C-terminale della 4.1Rsh abbiamo misurato un’ efficienza di
FRET molto bassa. A differenza dei saggi di immunoprecipitazione, il FRET risente
fortemente della vicinanza tra i fluorofori, che è esattamente il motivo per cui viene
utilizzato per discriminare tra interazioni indirette (come ad es. tra proteine facenti
parte di uno stesso complesso proteico) e interazioni dirette. I nostri dati di FRET
con YFP-4.1Rsh, i saggi di doppio ibrido e, soprattutto, quelli di interazione in vitro,
(Tang et al., 1998) indicano che nel caso di 4.1R e ICln si tratta di una interazione
diretta. Poiché i dati di immunoprecipitazione confermano che nel nostro caso non
si tratta di una assenza di interazione, è probabile che il basso FRETeff con 4.1RshYFP sia soprattutto una conseguenza dell’allontanamento di donatore e accettore.
Quando il YFP è fuso all’estremità N-t di 4.1Rsh, viene infatti a trovarsi legato
direttamente al FERM, il dominio di interazione di ICln, mentre quando è legato al
C-t viene a trovarsi all’estremità opposta. Anche i dati sulla isoforma lunga
suggeriscono una rigida organizzazione spaziale del complesso. La FRETeff è infatti
risultata nulla per tutte le coppie 4.1-ICln utlizzate, anche quelle in cui è stata
utilizzata la variante con il YFP N-t, che nel caso delle variante corta aveva dato un
buon segnale di FRETeff. Entrambe le proteine di fusione YFP-4.1RII e 4.1RII-YFP
tuttavia coimmunoprecipitano con ICln. Non si può escludere a priori che la perdita
del segnale di FRET possa dipendere da una diversa organizzazione del complesso
ICln-4.1R (e forse altri parner), in cui le due proteine vengono a trovarsi più distanti.
Tuttavia sembra ragionevole attendersi che, così come per la variante corta priva di
U1, anche nel caso di 4.1RII l’interazione tra ICln e 4.1R si mantenga diretta. E’
pertanto probabile che la FRETeff rifletta soprattutto una maggiore distanza dei
152
fluorofori. Nel caso di 4.1RII-YFP possono valere le stesse considerazioni già fatte
per 4.1Rsh-YFP, che dimostra chiaramente come lo spostamento della YFP al C-t
della proteina si traduca in una perdita pressochè totale del segnale di FRET (figura
25). Per quanto riguarda invece YFP-4.1RII, la bassa efficienza di FRET potrebbe
dipendere dalla presenza della regione U1, caratterizzante la 4.1RII; il dominio
FERM della 4.1RII, interagirebbe con la porzione C-terminale di ICln, ma i due
fluorofori YFP (legato a monte della regione U1) e CFP verrebbero distanziati dalla
presenza della regione U1, interposta tra fluoroforo e sito di interazione (il FERM).
Le due proteine potrebbero interagire, come indica la co-immuoprecipitazione, ma
la distanza tra i fluorofori non consentirebbe un trasferimento energetico efficace.
E’ possibile che a questo fatto si sommi anche una effettiva ridotta interazione di
ICln con l’isoforma lunga. Sebbene non abbiamo effettuato degli esperimenti
quantitativi di immunoprecipitazione i saggi di immunoprecipitazione con 4.1RII-YFP
forniscono comunque un dato interessante in tal senso. Come già accennato in
precedenza, dai western blot di figura 26, è evidente che quando viene overespressa la 4.1RII-YFP viene contemporaneamente espressa l’isoforma corta, che
viene riconosciuta sia dall’anti-4.1 (figura 26) che dall’anti-GFP (figura 26), in quanto
la proteina prodotta a partire dall’ATG2 presente nell’ORF della 4.1RII-YFP ha
anch’essa il YFP fuso al C-terminale. Tale dato è in accordo con quanto riscontrato
anche da altri (Lospitao et al., 2008), e si è ipotizzato che possa dipendere dalla
presenza di una sequenza IRES tra ATG1 e ATG2, entrambi presenti nell’ORF delle
isoforme lunghe. Abbiamo riscontrato che questo tipo di comportamento è molto
attenuato (se non assente) nel caso della YFP-4.1RII (fig 26), come se la presenza di
un tag C-terminale interferisse con l’utilizzo della sequenza IRES responsabile della
traduzione della isoforma corta. Come mostrato nella corsia corrispondente ai lisati
cellulari di fig 26 B, la quantità prodotta di isoforma corta resta comunque inferiore
rispetto alla lunga. Tuttavia negli eluati della immunoprecipitazione di YFP-4.1RII,
con FLAG-ICln, entrambe le isoforme vengono arricchite e compaiono in quantità
paragonabile, nonostante l’isoforma corta addirittura non appaia nei lisati di
partenza. Questo suggerisce che l’isoforma corta possa rappresentare un
153
competitore più forte della lunga per il legame con ICln, che legherebbe quindi con
maggiore affinità. Il problema legato alla produzioni di varianti corte anche overesprimendo i soli ORF delle lunghe non sembrerebbe alterare il quadro ottenuto
dagli esperimenti di FRET, né le informazioni riguardo la loro localizzazione. Per
quanto riguarda gli esperimenti con YFP-4.1RII infatti, la sintesi di isoforma corta
sembra molto bassa se non assente, come già discusso. Inoltre l’eventuale quota di
isoforma corta prodotta con l’over-espresione sarebbe comunque priva di YFP e
quindi non contribuirebbe né alla localizzazione né alla FRETeff, se non, in questo
ultimo
caso,
competendo
con
YFP-RII
per
il
legame
con
ICln.
L’immunoprecipitazione ci conferma tuttavia che questa competizione, se esiste, è
estremamente ridotta: non ci sono bande all’altezza attesa per 4.1Rsh negli eluati, a
differenza di quanto ottenuto con 4.1RII-YFP (fig. 26), in accordo con una bassa o
nulla produzione di 4.1Rsh da YFP-4.1RII. Per quanto riguarda 4.1RII-YFP, il risultato
ottenuto negli esperimenti di FRET non è presumibilmente alterato dalla simultanea
espressione delle due isoforme perché l’efficienza di FRET che abbiamo misurato in
cellule transfettate con 4.1Rsh-YFP era già di per sé non significativa. Inoltre le
immagini in fluorescenza di 4.1RII-YFP indicano una localizzazione diversa da
4.1Rsh-YFP, dove in genere è presente anche un segnale nucleare (fig. 48), a riprova
dell’ipotesi che le due proteine non sono prodotte in uguale quantità e la forma
lunga resta comunque predominante.
I dati di FRET (utilizzando sia la metodica dell’Acceptor Photobleaching che della
Sensitized emission) indicano che, perlomeno per l’isoforma corta con il YFP fuso
all’N-terminale della 4.1Rsh (YFP-4.1Rsh) l’ ipotonia aumenta dai valori di efficienza
di FRET e quindi presumibilmente l’interazione tra 4.1Rsh e ICln. I dati ottenuti con
N-FRET ci dicono inoltre che l’aumento dell’interazione tra ICln e la 4.1Rsh avviene
con tempistiche compatibili con la traslocazione di ICln dal citosol alla membrana
(Tamma et al., 2007; Ritter et al., 2003; Rodighiero et al., 2008), che viene indotta
dall’ipotonia, e con l’attivazione della ICl,swell (fig. 56) (Tamma et al., 2007). In
esperimenti di N-FRET è stato dimostrato che la traslocazione di ICln in membrana
aumenta significativamente dopo 10 minuti e fino a 20 minuti dallo stimolo
154
ipotonico e negli esperimenti di N-FRET sull’interazione tra ICln e la 4.1Rsh abbiamo
visto che dopo uno stimolo ipotonico l’interazione aumenta significativamente già
dopo 2 minuti e si mantiene costante fino a 20 minuti dopo la sostituzione con
soluzione ipotonica.
Ruolo dell’interazione tra ICln e 4.1R nell’ipotonia
Dagli esperimenti di FRET abbiamo verificato che l’interazione tra la 4.1Rsh e ICln
aumenta in ipotonia. Per valutare se l’interazione tra le due proteine fosse coinvolta
nell’attivazione della corrente ICl,swell (Tamma et al., 2007) abbiamo allestito
esperimenti di patch-clamp in configurazione whole cell. Da questi esperimenti
abbiamo verificato che le due isoforme 4.1R135 e 4.1R80 sembrano avere un effetto
diverso sulla corrente di cloruro attivata durante l’ipotonia. L’isoforma corta
determina un aumento significativo della ICl,swell a diversi potenziali (+ 100mV,
+80mV, +60 mV e a –100mV, -80mV, e -60 mV) rispetto alla condizione di controllo,
mentre per l’isoforma lunga non abbiamo misurato alcun effetto sulla corrente. Una
differenza nelle funzioni e nel comportamento tra le due varianti di 4.1R è stata
riportata anche da altri. Le due isoforme ad esempio hanno affinità di legame
diverso per molte proteine di membrana (Nunomura et al., 2009) e questo influisce
sulle rispettive funzioni. E’ possibile ipotizzare che la diversa azione della 4.1RII e
della 4.1Rsh, sulla corrente ICl,swell possa dipendere dalla presenza della regione
unica U1, l’unica regione che differenzia la 4.1Rsh da 4.1RII. Questa regione al pari
delle altre regioni U2 e U3, è meno conservata tra le diverse isoforme rispetto ai
domini FERM, SAB, CTD e per tale motivo si ritiene che U1, insieme alle altre regioni
uniche U2 e U3, possa conferire specifiche funzioni regolatorie ad ogni 4.1. E’ stato
dimostrato che U1 modula la traslocazione nucleare di 4.1R (Gascard et al., 1998;
Gascard et al., 1999; Luque et al., 1999), interagisce con una proteina centrosomale
(Hung et al., 2000) e possiede inoltre un sito di fosforilazione da parte della chinasi
ciclina dipendente cdc2 (Huang et al., 2005) inoltre il livello di fosforilazione delle
4.1R ad alto peso molecolare varia durante il ciclo cellulare. Le 4.1R HMW (high
molecular weight) inoltre interagiscono con la calmodulina in modo strettamente
155
Ca2+ dipendente a differenza della 4.1Rsh che è CaM Ca2+ indipendente(Leclerc &
Vetter, 1998).
I dati in nostro possesso non ci permettono di chiarire il meccanismo con cui 4.1Rsh
interviene nell’attivazione della corrente. Anche per altre 4.1 è stato riportato un
effetto diretto su correnti di Na+ Cl- e K+ (Rivera et al., 2006) con implicazioni
importanti per alcune patologie cardiache, come la sindrome del QT lungo (Stagg et
al., 2008) e per la conduzione nervosa (Shen et al., 2000; Douyard et al., 2007). Per
alcuni casi è stato proposto che 4.1R regolasse l’espressione in membrana di questi
sitemi di trasporto (Binda et al., 2002) e/o la loro funzione. Per quanto riguarda la
ICl,swell il discorso è complesso in quanto non è ancora stato identificato il canale
vero e proprio, canale per cui lo stesso ICln rappresenta un candidato. Una
possibilità è che l’ipotonia causi una traslocazione di ICln verso la membrana, dove
aumenta l’interazione con 4.1 che ne facilita l’inserzione in membrana (nel caso in
cui ICln sia una parte del canale) o la vicinanza con un canale di cui ICln funge da
regolatore. Tuttavia, i dati di microscopia e i western blot indicano che un aumento
della quantità di ICln determina una diminuzione della presenza in membrana di 4.1
Ciò suggerisce che la traslocazione di ICln verso la regione di membrana porti al
legame di 4.1R, causando il suo distacco dalla membrana o inibendone comunque la
associazione, un evento che presumibilmente partecipa al riarrangiamento del
citoscheletro sottomembranario che accompagna l’attivazione della ICl,swell, e che
coincide tra l’altro con un aumento dell’interazione tra ICln e l’actina (Tamma et al,
2007).
Il complesso formato tra ICln e 4.1R (e forse anche da altri partner) sembra di fatto
sequestrarle entrambe nel citosol, inibendone la presenza negli altri ambiti subcellulari (membrana, per lo meno per quanto riguarda la 4.1R nel nucleo), con
possibili conseguenze sulle loro funzioni. E’ probabile che questo rappresenti un
evento chiave per la complessa rete di eventi che accoppia lo stimolo ipotonico alla
attivazione dei canali RVDC. In questo senso, è possibile che l’effetto della sola
variante corta di 4.1 (ma non della lunga) sulla attivazione della corrente sia legato
ad un maggior legame di questa isoforma con ICln, come suggerito dagli
156
esperimenti di FRET e di coimmunoprecipitazione. Un effetto maggiore della
isoforma corta su ICln è suggerito anche dal fatto che i rapporti nucleo/citosol
analizzati dalle immagini di immunofluorescenza indicano che YFP-4.1Rsh sembra
avere un impatto maggiore sulla localizzazione nucleare di ICln rispetto alla
isoforma lunga (YFP-4.1RII).
Anche l’aumento transiente di calcio intracellulare che avviene nelle fasi iniziali
dell’RVD (Weskamp et al., 2000; Light et al., 2003) contribuirebbe all’inibizione
dell’associazione di 4.1R con la membrana. Il calcio infatti favorisce il distacco di 4.1
dalla membrana, sia legandosi direttamente sia regolando il legame della
calmodulina al FERM e, soprattutto alla regione U1, se presente (Diakowski et al.,
2006). In effetti gli esperimenti di western blot e di colocalizzazione suggeriscono
proprio che l’ipotonia causi una dissociazione della 4.1 dalla membrana, che
sembrerebbe più marcata per l’isoforma lunga rispetto alla corta. E’ possibile che
questo rifletta la maggiore sensibilità delle isoforme ad alto peso molecolare al
calcio.
In questo quadro estremamente complesso possiamo affermare che l’interazione
tra la proteina ICln e la proteina 4.1R (soprattutto le isoforme corte) è coinvolta
nella complessa via di signaling che porta all’attivazione della corrente ICl,swell ma
non possiamo ancora stabilire a che livello intervenga in tale pathway.
Localizzazione
L’isoforma corta della proteina 4.1Rsh localizza nel nucleo, in membrana e nel
citosol.
Ciò è in accordo con quanto riportato in letteratura (Conboy et al., 1998). Un’analisi
comparativa della composizione in esoni del cDNA della 4.1 ha evidenziato che il
segnale di localizzazione nucleare è dato dall’ esone 16, che è presente in entrambe
le nostre isoforme. L’esone 5 (anch’esso presente nelle nostre isoforme), al
contrario, conferisce una localizzazione citosolica in quanto codifica per una
sequenza idrofobica e ricca di leucine che è molto simile alla sequenza dei segnali di
esportazione dal nucleo. Nel caso di simultanea espressione degli esoni 5 e 16
157
l’effetto della localizzazione nucleare determinata dall’esone 16 sembra
predominare sull’effetto inibente indotto dall’espressione dell’esone 5 (Parra et al.,
1998). L’isoforma lunga localizza prevalentemente in membrana e nel citosol e la
presenza nucleare sembra ridotta (anche se non assente) rispetto alla corta (come
dimostrato dalle immagini ottenute con YFP-4.1RII), in accordo con dati presenti in
letteratura che indicano che la presenza della regione U1 ostacola l’ingresso nel
nucleo dell’isoforma lunga (Luque et al., 1999). Nel nostro lavoro abbiamo visto che
quando la 4.1RII è in fusione con il YFP è presente in quantità molto minore nel
nucleo rispetto all’isoforma corta. E’ tuttavia vero che i western blot fatti sulla
proteina endogena (vedi figura 42) indicano che sia le isoforme lunghe che le corte
identificate dal nostro anticorpo (e che quindi contengono presumibimente l’esone
16) localizzano anche nel nucleo, in proporzione non uguale, ma comunque
paragonabile. Nel dubbio che la contemporanea presenza della regione U1 e del
fluoroforo possa ulteriormente ostacolare l’import nel nucleo di tale proteina gli
esperimenti di western blot sulla localizzazione della proteina 4.1RII sono stati
effettuati over-esprimendo tale isoforma mendiante un vettore IRES che permette
di esprimere la 4.1RII separata dalla GFP. Lavorare in over-espressione per una delle
due isoforme ci ha permesso di studiare negli esperimenti di localizzazione il
comportamento specifico di una o dell’altra isoforma.
Sia l’analisi delle immagini degli esperimenti di FRET che gli esperimenti di western
blot hanno dimostrato che quando ICln è in coespressione con la proteina 4.1Rsh
diminuisce considerevolemente la quota di 4.1R associata alla membrana, e
entrambe diminuiscono nel nucleo.
Per quanto riguarda il primo punto è importante sottolineare che le ripercussioni
della diminuzione di 4.1 in membrana possono essere estremamente importanti per
la cellula. La presenza di 4.1R in questa sede è infatti considerata fondamentale per
la corretta struttura e rigidità di membrana, come provato dal fatto che la sua
ridotta espressione sia associata ad elliptocitosi, una malattia dei globuli rossi (dove
le 4.1R sono particolarmente presenti) che comporta l’indebolimento del
citoscheletro submembranario che genera deformazione della morfologia
158
dell’eritrocita, questo comporta fragilità degli eritrociti e l’incapacità di rispondere
agli stress meccanici (Gallagher, 2004). Senza arrivare a situazioni estreme come
queste, è comunque probabile che una alterazione della distribuzione della 4.1R
abbia importanti conseguenze anche sulle vie di signalling associate alla membrana
in cui la proteina è coinvolta (Bretscher et al., 2002; Sherman and Gutmann, 2001).
E’ possibile che l’inibizione della associazione di 4.1R con la membrana sia una
conseguenza diretta dell’interazione tra le due proteine. ICln infatti lega 4.1R in una
regione del FERM (la parte C-t) dove legano anche altri fattori critici (come ad
esempio la CaM) per la regolazione del suo legame con la membrana (Nunomura et
al., 2009), mentre gli altri due lobi del FERM sono quelli che prevalentemente
legano altre proteine di membrana, come lo scambiatore anioco e la glicoforina C. A
tale proposito è interessante notare che la regione del FERM interessata al legame
con ICln è rappresentata da un PH domain, che ICln legherebbe con la sua porzione
C-terminale (Gascard et al., 2006). In un complesso di questo tipo il PH domain di
4.1 (importante per il legame con il PIP2 che ne regola l’associazione con la
membrana e il citoscheletro (Gascard et al., 2000) verrebbe a trovarsi mascherato o
anche solo affiancato dal PH domain di ICln, con possibili importanti conseguenze
sul pattern di interazioni della 4.1.
Per quanto riguarda la localizzazione nucleare è difficile dire se sia ICln a indurre
l’uscita dal nucleo della 4.1Rsh (fungendo da proteina shuttle) o se avvenga
l’opposto, anche perché la funzione di ICln nel compartimento nucleare è ancora
oscura e poco indagata.
ICln oltre ad essere implicata nelle RVD è coinvolta in numerose altre funzioni
cellulari. Una di queste si basa sulla sua interazione con PRMT5 (Skb1) (con la
regione AD3) e Mep50 (Pu et al., 1999, Friesen et al,. 2001, 2002, Meister et al.,
2001) per formare il metilosoma. Sebbene il metilosoma svolga la sua funzione
principalmente nel citosol, dove si occupa della metilazione delle proteine Sm
(Friesen et al., 2001), l’interazione tra le proteine in questione sembra avvenire
anchea livello nucleare. PRMT5 e Mep50 sono due proteine che oltre a partecipare
159
alla formazione del metilosoma sono in grado di traslocare nel nucleo per formare
un nuovo complesso con la proteina AJUBA (Hou et al., 2008); tale complesso è
reclutato da fattori trascrizionali (SNAIL) e si comporta da repressore trascrizionale
per numerosi geni tra cui ad esempio ST7 and NM23; inoltre promuove lo stato
tumorale in cellule NIH 3T3 (Pal et al., 2004). PRMT5 interviene inoltre nella
regolazione della trascrizione attraverso la metilazione degli istoni (Zhao et al.,
2009). Per ICln non è stato ancora chiarita la sua funzione nel nucleo ma come per
PRMT5 la sua traslocazione nel nucleo (o nel nostro caso dal nucleo) potrebbe avere
importanti conseguenze sulle sue funzioni tra cui la regolazione dello splicing
dell’RNA. Infatti è già stato dimostrato che ICln facendo parte del metilosoma va a
regolare l’assemblaggio dello spliceosoma (Grimmler et al., 2005). E’ inoltre
importante sottolineare che recentemente è stato riportato che PRMT5, la cui
funzione è spesso stata associata ad ICln (Guderian et al., 2010) è in grado di legare
oltre ad ICln anche varianti di 4.1, come la B (Jiang et al., 2004). Questo suggerisce
che PRMT5 possa rappresentare un ulteriore componente del complesso 4.1R-ICln,
un aspetto che meriterebbe di essere approfondito.
Per quanto riguarda un possibile effetto legato alle funzioni della proteina 4.1, ci
sono numerose evidenze del suo coinvolgimento nella regolazione della
proliferazione cellulare. La perdita negli stadi precoci della proteina 4.1R è un
evento comune nella patogenesi di meningiomi (Robb et al., 2003). In letteratura
inoltre ci sono numerosi lavori che hanno dimostrato che la 4.1B e la proteina
merlin si comportano come dei regolatori negativi della crescita in diversi tipi di
cancro inclusi i meningiomi (Gutman et al., 2000; Kuns et al., 2005). Robb e
collaboratori hanno verificato che anche la proteina 4.1R viene inattivata (a livello
della proteine e del DNA) come la 4.1B e la proteina merlin nei meningiomi
sporadici. La localizzazione delle 4.1 sembra importante per questo aspetto. La
proteina 4.1R, 4.1B e merlin si associano con CD44 e si concentrano presso la
membrana cellulare durante l’arresto della crescita suggerendo che il segnale
regolatorio della crescita ha inizio dalla membrana (Bretscher et al., 2002; Sherman
and Gutmann, 2001). Inoltre per la proteina 4.1N è stato dimostrato che la sua
160
traslocazione nel nucleo in PC12 differenziate provoca l’arresto della crescita
cellulare (Ye et al., 2000; Calinisan et al., 2006).
Nel nostro lavoro abbiamo visto che quando ICln è overespressa 4.1R diminuisce in
membrana e nel nucleo facendo di ICln un possibile regolatore negativo di 4.1R
contrastandone l’effetto anti-proliferativo. Un legame tra ICln e lo stato
proliferativo delle cellule, sebben poco studiato, è già stato proposto. Nel caso delle
cellule ECs (Li et al., 2004), è stato infatti riportato che l’overespressione di ICln
stimola la proliferazione cellulare, favorendo l’angiogenesi. Tornando a 4.1, i dati,
pur preliminari, sull’effetto delle coespressione di ICln con 4.1 (fig 58) sembrano
validare questa ipotesi e confermano l’importanza che l’interazione tra le due
proteine può avere anche in ambiti diversi dalla regolazione del volume
161
162
Bibliografia
ACKERMAN M.J., WICKMAN K.D., CLAPHAM D.E. (1994). -Hypotonicity activates a native
chloride current in Xenopus oocytes. J. Gen. Physiol. 103(2):153-79.
APLIN AE., HOWE A., ALAHARI SK., JULIANO RL. (1998) -Signal transduction and signal
modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell
adhesion molecules, and selectins. Pharmacol Rev 50: 197–263.
BALTIMORE D, MAYER BJ, REN R, CLARK KL (1993). A putative modular domain present in
diverse signaling proteins". Cell 73 (4): 629–630.
BENNETT V, BAINES AJ (2001) - Spectrin and Ankyrin-Based Pathways: Metazoan Inventions
for Integrating Cells Into Tissues Physiol. Rev. 81: 1353-1392
BENNETT V., LAMBERT S. (1991) -The spectrin skeleton: from red cells to brain- J Clin Invest
87:1483-1489.
BINDA AV, KABBANI N, LIN R, LEVENSON R. (2002) - D2 and D3 dopamine receptor cell
surface localization mediated by interaction with protein 4.1N. Mol Pharmacol. 62(3):50713
BLOMBERG N., NILGES M. (1997). -Functional diversity of PH domains: an exhaustive
modelling study Fold. Des 2:343-55.
BRETSCHER, A., EDWARDS, K., FEHON, R.G. (2002) - ERM proteins and merlin: integrators at
the cell cortex. Nat. Rev. Mol. Cell. Biol. 3, 586–599.
BROWE DM, BAUMGARTEN CM.(2003). -Stretch of _1 integrin activates an outwardly
rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol 122:
689–702
163
BROWE DM, BAUMGARTEN CM. (2004). -Angiotensin II (AT1) receptors and NADPH oxidase
regulate Cl_ current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen
Physiol 124: 273–287.
BUYSE G, DEGREEF C, RAEYMAEKERS L, DROOGMANS G, NILIUS B, EGGERMONT J (1996) The ubiquitously expressed pICln protein forms homodimeric complexes in vitro- Biochem
Biophys Res Commun 218:822.
BURTON L, TAYLOR DL (1997) -Traction forces of cytokinesis measured with optically
modified elastic substrata- Nature 30:385, 450-454.
CALINISAN V, GRAVEM D, CHEN RP, BRITTIN S, MOHANDAS N ET AL. 2006. New insights into
potential functions for the protein 4.1 superfamily of proteins in kidney epithelium Front
Biosci. 11:1646-66
CANTIELLO HF (1995) -Role of the actin cytoskeleton on epithelial Na+ channel regulationKidney 48:970-984.
CANTIELLO HF, STOWN JL, PRAT AG, AUSIELLO DA (1991) -Actin filaments regulate epithelial
Na+ channel activity- Am J Physiol 261:882-888.
CARTON I, TROUET D, HERMANS D, BARTH H, AKTORIES K ET AL. (2002) - RhoA exerts a
permissive effect on volume-regulated anion channels in vascular endothelial cells Am. J.
Physiol Cell Physiol 283:C115-C125.
CIANO-OLIVEIRA C, LODYGA M, FAN L, SZASZI K, HOSOYA H, ROTSTEIN OD, KAPUS A. (2005)
-Is myosin light-chain phosphorylation a regulatory signal for the osmotic activation of the
Na_-K_-2Cl_ cotransporter? Am J Physiol Cell Physiol 289: C68–C81.
CONBOY JG (1993) -Structure, function, and molecular genetics of erythroid membrane
skeletal protein 4.1 in normal and abnormal red blood cells- Semin Hematol 30(1):58-73.
164
CONBOY JG, CHASIS JA, WINARDI R, TCHEMIA G, KAN YW, MOHANDAS N (1993) -An
isoform-specific mutation in the protein 4.1 gene results in heredeitary elliptocytosis and
complete deficiency of protein 4.1 in erythrocytes but not in nonerythroid cells- J Clin Invest
91:77-82.
CONBOY J, MARCHESI S, KIM R, AGRE P, KAN YW, MOHANDAS N (1990) -Molecular analysis
of insertion/deletion mutations in protein 4.1 in elliptocytosis. Determination of molecular
genetics origins of rearrangements- J Clin Invest 86:524-530.
CORNET M, LAMBERT IH, HOFFMANN EK (1993) -Relation between cytoskeleton, hypoosmotic treatment and volume regulation in Ehrlich ascites tumor cells- J Membr Biol
131:55-66.
CORREAS I, SPEICHER DW, MARCHESI VT (1986) Structure of the spectrin-actin binding site
of erythrocyte protein 4.1. J Biol Chem 261: 13362-13366
CORREAS I, LETO TL, SPEICHER DW, MARCHESI VT. (1986) - Identification of the functional
site of erythrocyte protein 4.1 involved in spectrin-actin associations. J Biol Chem 261:3310.
CORREAS I (1991): Characterization of isoforms of protein 4.1 present in the nucleus.
Biochem J 279, 581-585
CRAMER LP, MITCHISON TJ, THERIOR J A (1994) -Actin-dependent motile forces and cell
motility- Cell Biol 6:82-6.
DAVIS MJ, WU X, NURKIEWICZ TR, KAWASAKI J, GUI P ET AL. (2002) - Regulation of ion
channels by integrins Cell Biochem. Biophys. 36:41-66.
DIAKOWSKI W, GRZYBEK M AND SIKORSKI AF (2006) -Protein 4.1, a component of the
erythrocyte membrane skeleton and its related homologue proteins forming the protein
4.1/FERM superfamily Folia Histochemica et Cytobiologica. 44:231-248
165
DOUGLAS B. LIGHT, ANDREW J ATTWOOD, CORRYN SIEGEL AND NICOLE L. BAUMANN
(2003) - Cell swelling increases intracellular calcium in Necturus erythrocyte Journal of Cell
Science 116: 101-10
DOUYARD J, SHEN L, HUGANIR RL, RUBIO ME (2007) - Differential neuronal and glial
expression of GluR1 AMPA receptor subunit and the scaffolding proteins SAP97 and 4.1N
during rat cerebellar development. J Comp Neurol. 502(1):141-56.
DOVE SK, COOKE FT, DOUGLAS MR, SAYERS LG, PARKER PJ, MICHELL RH (1997) - Osmotic
stress activates phosphatidylinositol-3,5-bisphosphate synthesis Nature 390:187-92.
DRENCKHAHN D, SCHLUTER K, ALLEN DP, BENNETT V (1985) -Colocalization of band 3 with
ankyrin and spectrin at the basal membrane of intercalated cells in the rat kidney- Science
230:1287-1289.
DU XY, SOROTA S, (1999) – Protein kinase C stimulates swelling-induced chloride current in
canine atrial cells. Pflugers Arch 437: 227-234.
DUPRAT F., LESAGE F, FINK M, REYES R, HEURTEAUX C AND LAZDUNSKI M (1997) -TASK, a
human background K+ channel to sense external pH variations near physiological pH The
EMBO Journal 16, 5464 – 5471.
EMMA F, SANCHEZ-OLEA R, STRANGE K. (1998) -Characterization of pI(Cln) binding proteins:
identification of p17 and assessment of the role of acidic domains in mediating proteinprotein interactions Biochim. Biophys. Acta 1404:321-8.
FERANCHAK AP, ROMAN RM, DOCTOR RB, SALTER KD, TOKER A, FITZ JG (1999) - The lipid
products of phosphoinositide 3-kinase contribute to regulation of cholangiocyte ATP and
chloride transport J. Biol. Chem. 274:30979-86.
FIGEYS D, MCBROOM LD, MORAN MF. (2001). - Mass spectrometry for the study of proteinprotein Interactions Methods 24:230-9
166
FRANCO R, LEZAMA R, ORDAZ B, PASANTES-MORALES H. (2004) -Epidermal growth factor
receptor is activated by hyposmolarity and is an early signal modulating osmolyte efflux
pathways in Swiss 3T3 fibroblasts. Pflu¨ gers Arch 447: 830–839.
FRIESEN WJ, WYCE A, PAUSHKIN S, ABEL L, RAPPSILBER J ET AL. (2002) -A novel WD repeat
protein component of the methylosome binds Sm proteins J. Biol. Chem. 277:8243-7.
FRIESEN WJ, PAUSHKIN S, WYCE A, MASSENET S, PESIRIDIS GS ET AL. (2001) -The
methylosome, a 20S complex containing JBP1 and pICln, produces dimethylargininemodified Sm proteins Mol. Cell Biol. 21:8289-300.
FÜRST J, BOTTA G, SAINO S, DOPINTO S, GANDINI R ET AL. (2006).-The ICln interactome
Acta Physiol (Oxf) 187:43-9.
FÜRST J, SCHEDLBAUER A, GANDINI R, GARAVAGLIA ML, SAINO S ET AL. (2005). ICln159
folds into a pleckstrin homology domain-like structure. Interaction with kinases and the
splicing factor LSm4 J. Biol. Chem. 280:31276-84.
FÜRST J, GSCHWENTNER M, RITTER M, JAKAB M, MEYER M, GARAVAGLIA ML, BAZZINI C,
RODIGHIERO S, MEYER G, EICHMULLER S, WOLL E, PAULMICHL M (2002). -Molecular and
functional aspects of anionic channel activated during regulatory volume decrease in
mammalian cells- Cell Physiol Biochem 12:235-258.
FÜRST J, BAZZINI C, JAKAB M, MEYER G, KONIG M ET AL. (2000). -Functional reconstitution
of ICln in lipid bilayers Pflugers Arch. 440:100-15.
GALLAGHER PG. (2004) - Hereditary elliptocytosis: spectrin and protein 4.1R. Semin
Hematol. 41(2):142-64.
GASCARD P, LEE G, COULOMBEL L, AUFFRAY I, LUM M, PARRA M, CONBOY JG, MOHANDAS
N, CHASIS JA (1998) -Characterization of multiple isoforms of protein 4.1R espressed during
erythroid terminal differentiation- Blood 92,4404-4414.
167
GASCARD P, NUNOMURA W, LEE G, WALENSKY LD, KRAUSS SW, TAKAKUWA Y, CHASIS JA,
MOHANDAS N, CONBOY JG (1999). -Deciphering the nuclear import pathway for the
cytoskeletal red cell protein 4.1R. Mol Biol Cell 10: 1783-1798
GASCARD P, MOHANDAS N (2000) -New insights into functions of erythroid proteins in
nonerythroid cells- Current Opinion in Hematology 7:123-129.
GILBRETH M, YANG P, BARTHOLOMEUSZ G, PIMENTAL RA, KANSRA S ET AL. (1998). Negative regulation of mitosis in fission yeast by the shk1 interacting protein skb1 and its
human homolog, Skb1Hs Proc. Natl. Acad. Sci. U. S. A 95:14781-6.
GIMM JA, AN X, NUNOMURA W, MOHANDAS N (2002) -Functional characterization of
spectrin-actin binding domains in 4.1 family of proteins. Biochemistry 41: 7275-7282
GRAF J., RUPNIK M., ZUPANCIC G., ZOREC R. (1995). -Osmotic swelling of hepatocytes
increases membrane conductance but not membrane capacitance. Biophys. J. 68: 13591363.
GRIMMLER, M., BAUER, L., NOUSIAINEN, M. ET AL. (2005).- Phosphorylation regulates the
activity of the SMN complex during assembly of spliceosomal U snRNPs. EMBO Rep 6, 70–
76.
GSCHWENTNER M, FÜRST J, RITTER M., BAZZINI C, WOLL E, DIENSTL A, JAKAB M, KONIG M.,
SCANDELLA E, RUDZI J, BOTTA’ G, MEYER G, LANG F, DEETJEN P AND PAULMICHL M (1999) ICln an anion channel-forming protein associated with cell volume regulation- Experimental
Physiology 84:1023-1031.
GSCHWENTNER M, NAGL UO, WOLL E, SCHMARDA A, RITTER M, PAULMICHL M (1995). Antisense oligonucleotides suppress cell-volume-induced activation of chloride channelsPflügers Arch 430:464-470.
GUDERIAN G, PETER C, WIESNER J, SICKMANN A, SCHULZE-OSTHOFF K, FISCHER U,
GRIMMLER M. (2010) - RioK1, a new interactor of protein arginine methyltransferase 5
168
(PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and
substrate specificity. J Biol Chem. (Epub ahead of print).
GUTMANN, D.H., DONAHOE, J., PERRY, A., LEMKE, N., GORSE, K., KITTINIYOM, K., REMPEL,
S.A., GUTIERREZ, J.A., NEWSHAM, I.F. (2000) - Loss of DAL-1, a protein 4.1-related tumor
suppressor, is an important early event in the pathogenesis of meningiomas. Hum. Mol.
Genet. 9, 1495–1500.
HALL PF (1984) -The role of the cytoskeleton in hormone action- Cell Biol 62:653-65.
HALLOWS KR, LAW FY, PACKMAN C H, KNAUF PA (1996) -Changes in distribution, and
surface morphology during HL-60 cell volume regulation- J Cell Physiol 167:6071.cytoskeletal actin content, F-actin
HAN BG, NUNOMURA W, TAKAKUWA Y, MOHANDAS N, JAP BK (2000). -Protein 4.1R core
domain structure and insights into regulation of cytoskeletal organization. Nature Struct
Biol 7: 871- 875
HASLAM RJ, KOIDE HB, HEMMINGS BA. (1993). -Pleckstrin domain homology Nature
363:309-10.
HÄUSSINGER D, KURZ AK, WETTSTEIN M, GRAF D, VOM DS, SCHLIESS F. (2003). Involvement of integrins and Src in tauroursodeoxycholate- induced and swelling-induced
choleresis. Gastroenterology 124: 1476–1487.
HÄUSSINGER D, SCHLIESS F, KIRCHEIS G. (2002) - Pathogenesis of hepatic
encephalopathy. Journal of Gastroenterology and Hepatology 17, Issue Supplement
s3: S256–S259.
HÄUSSINGER H, (1996) - The role of cellular hydration in the regulation of cell function
Biochem. J. 313: 697-710.
169
HAZAMA A, OKADA Y. (1999). -Biphasic rises in cytosolic free Ca2_ in association with
activation of K_ and Cl_ conductance during the regulatory volume decrease in cultured
human epithelial cells. Pflu¨ gers Arch 416: 710–714.
HEMMINGS BA, HASLAM RJ, KOIDE HB (1993). "Pleckstrin domain homology". Nature 363
(6427): 309–310.
HILGEMANN DW, FENG S, NASUHOGLU C. (2001). -The complex and intriguing lives of PIP2
with ion channels and transporters. Sci STKE 2001: RE19.
HOFFMANN ELSE K., LAMBERT IAN H., AND PEDERSEN STINE F. (2009). -Physiology of Cell
Volume Regulation in Vertebrates Physiol Rev 89: 193–277.
HOFFMANN EK. (2000) Intracellular signalling involved in volume regulatory decrease. Cell
Physiol Biochem 10: 273–288.
HOFFMANN EK, DUNHAM PB. (1995). -Membrane mechanisms and intracellular signalling
in cell volume regulation. Int Rev Cytol 161:173–262.
HOFFMANN E.K., SIMONSEN L.O (1989). -Membrane mechanisms in volume and pH
regulation in vertebrate cells. Physiol. Rev. 69(2): 315-82.
HOU Z, PENG H, AYYANATHAN K, YAN KP, LANGER EM, LONGMORE GD, RAUSCHER F (2008)
-The LIM Protein AJUBA Recruits Protein Arginine Methyltransferase 5 To Mediate SNAILDependent Transcriptional Repression MOLECULAR AND CELLULAR BIOLOGY Vol. 28,No. 10:
3198–3207
HUANG S. C, E. S. LIU, S. H. CHAN, I. D. MUNAGALA, H. T. CHO, R. JAGADEESWARAN, E. J.
BENZ JR (2005) Mitotic regulation of protein 4.1R involves phosphorylation by cdc2 kinase.
Mol Biol Cell 16, 117-127
170
HUANG SC, JAGADEESWARAN R, LIU ES AND BENZ E (2004) -Protein 4.1R, a Microtubuleassociated Protein Involved in Microtubule Aster Assembly in Mammalian Mitotic Extract
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 33: 34595–34602
HUBNER S, COUVILLON AD, KAS JA, BANKAITIS VA, VEGNERS R ET AL. (1998) - Enhancement
of phosphoinositide 3-kinase (PI 3-kinase) activity by membrane curvature and inositolphospholipid-binding peptides Eur. J. Biochem. 258:846-53.
HUNG LY, TANG CJ, TANG TK (2000) -Protein 4.1R-135 interacts with a novel centrosomal
protein (CPAP) which is associated with the gamma-tubulin complex- Mol Cell Biol 20,78137825.
INABA M, GUPTA KC, KUWABARA M, TAKAHASHI T, BENZ EJ JR, MAEDE Y (1992) Deamidation of human erythrocyte protein 4.1: possible role in aging. Blood 79: 3355-3361
JAKAB M, FÜRST J, GSCHWENTNER M, BOTTÀ G, GARAVAGLIA ML, BAZZINI C, RODIGHIERO
S, MEYER G, EICHMUELLER S, WÖLL E, CHWATAL S, RITTER M, PAULMICHL M. (2002). Mechanisms sensing and modulating signals arising from cell swelling. Cell Physiol Biochem.
12(5-6):235-58.
JACKSON P.S., STRANGE K. (1993). -Volume-sensitive anion channels mediate swellingactivated inositol and taurine efflux. Am. J. Physiol. 265(6 Pt 1):C1489-500.
JACKSON P.S., CHURCHWELL K., BALLATORI N., BOYLER J.L., STRANGE K. (1995). -Swelling
activeted anion conductance in skate hepatocytes: regolation by cell Cl- and ATP. Am. J.
Physiol. 270:C57-C66.
JIANG W, ME ROEMER & IF NEWSHAM (2005):The tumor suppressor DAL-1/4.1B modulates
protein arginine Nmethyltransferase 5 activity in a substrate-specific manner. Biochem
Biophys Res Commun 329, 522-530
171
JORGENSEN PL, PETERSEN J, REES WD (1984) -Identification of a Na+,K+, Cl- cotransport
protein of Mr 34 000 from kidney by photolabeling with [3H]bumethanide. The protein is
associated with cytoskeleton components- Biochim Biophys Acta 8;775:105-110.
JOHNSON BD, BYERLY L (1993) -A cytoskeletal mechanism for Ca2+ channel metabolic
dependence and inactivation by intracellular Ca2+- Neuron 10:797-804.
KLAUSEN TK, HOUGAARD C, HOFFMANN EK, PEDERSEN SF (2006) - Cholesterol modulates
the volume-regulated anion current in Ehrlich-Lettre ascites cells via effects on Rho and Factin Am. J. Physiol Cell Physiol 291:C757-C771.
KUNS R, KISSIL JK, NEWSHAM IF, JACKS T, GUTMANN DH AND SHERMAN LS (2005) - Protein
4.1B expression is induced in mammary epithelial cells during pregnancy and regulates their
proliferation Oncogene 24, 6502–6515.
KLEINZELLER A, BOOZ GW, MILLS JW, ZIYADEH FN (1990) -pCMBS- induced swelling of
dogfish (Squalus acanthias) rectal gland cells: role of the Na+-K+ATPase and the
cytoskeleton- Biochim Biophys Acta 11;1025(1):21-31.
KRAPIVINSKY GB, ACKERMAN, MJ, GORDON, EA, KRAPIVINSKY, LD, CLAPHAM, DE (1994) Molecular characterization of a swelling-induced chloride conductance regulatory protein,
pICln- Cell 76:3,439-448.
KRAPIVINSKY G, PU W, WICKMAN K, KRAPIVINSKY L, CLAPHAM DE. (1998). -pICln binds to a
mammalian homolog of a yeast protein involved in regulation of cell morphology J. Biol.
Chem. 273:10811-4.
KRAUSS SW, CHASIS JA, ROGERS C, MOHANDAS N, KROCKMALNIC G, PANMAN S (1997) Structural protein 4.1 is located in mammalian centrosome- Proc Natl Acad Sci USA
94:7297-7302.
172
KRAUSS SW, LARABELL CA, LOCKETT S, GASCARD P, PENMAN S, MOHANDAS N, CHASIS JA
(1997) -Structural protein 4.1 in the nucleus of human cells: dynamic rearrangements during
cell division- J Cell Biol 137:275-289.
LALLENA MJ, MARTINEZ C, VALCARCEL J, CORREAS I (1998) -Functional association of
nuclear protein 4.1 with pre-mRNA splicing factors- J Cell Sci 111:1963-1971.
LAMB RS, RE WARD, L SCHWEIZER & RG FEHON (1998). -Drosophila coracle, a member of
the protein 4.1 superfamily, has essential structural functions in the septate junctions and
developmental functions in embryonic and adult epithelial cells. Mol Biol Cell 9, 3505-3519
LANG F., FOLLER M., LANG K., RITTER M., VERENINOV A., SZABO I., HUBER SM., GULBINS E.
(2007). -Cell volume regulatory ion channels in cell proliferation and cell death. Methods
Enzymol 428: 209–225
LANG F., RITTER M., GAMPER N., HUBER S., FILLON S., TANNEUR V., LEPPLE-WIENHUES A.,
SZABO I., GULBINS E. (2000). -Cell volume in the regulation of cell proliferation and
apoptotic cell death. Cell Physiol Biochem 10: 417–428.
LANG F., BUSH G.L., RITTER L., VOLKI H., WLADEGGER S., GULBINS E., HAUSSINGER D.
(1998a). -Functional significance of cell volume regulatory mechanisms. Physiolog. Rew.
78(1):247-306.
LANG F, STEHLE T, HAUSSINGER D (1998b) -Water, K+, H+, lactate and glucose fluxes during
cell-volume regulation in perfused rat liver- Pflügers Arch 413:209-216.
LARKIN D, MURPHY D, REILLY DF, CAHILL M, SATTLER E ET AL. (2004). ICln, a novel integrin
alphaIIbbeta3-associated protein, functionally regulates platelet activation J. Biol. Chem.
279:27286-93.
LECLERC E, VETTER S (1998) -Characterization of a calcium-dependent calmodulin-binding
domain in the 135 kD human protein 4.1 isoform- Eur J Biochem 258,567-571.
173
LEMMON MA, FERGUSON KM. (2001). Molecular determinants in pleckstrin homology
domains that allow specific recognition of phosphoinositides. Biochem. Soc. Trans. 29:37784.
LEMMON MA, FERGUSON KM, ABRAMS CS. (2002). -Pleckstrin homology domains and the
cytoskeleton FEBS Lett. 513:71-6.
LEPPLE-WIENHUES A., SZABO I., LAUN T.,KABA N.K.,GULBINS E.,LANG F. (1998).The tyrosine
kinase p56lck mediates activation of swelling-induced chloride channels in lymphocytes. J.
Cell. Biol. 141(1):281-6.
LI Y, TAO G, NAGASAWA H, TAZAWA H, KOBAYASHI A ET AL. (1999).-pI(Cln) and cytosolic Factin constitute a heteromeric complex with a constant molecular mass in rat skeletal
muscles J. Biochem. 126:643-9.
LI, HUI, YONEKURA, HIDETO, KIM, CHUL-HEE, SAKURAI, SHIGERU, YAMAMOTO, YASUHIKO,
TAKIYA, TOSHIYUKI, FUTO, SATOSHI, WATANABE, TAKUO AND YAMAMOTO, HIROSHI (2004)
'Possible Participation of pICln in the Regulation of Angiogenesis Through Alternative
Splicing of Vascular Endothelial Growth Factor Receptor mRNAs', Endothelium, 11:5, 293 —
300.
LI S, ARMSTRONG CM, BERTIN N, GE H, MILSTEIN S ET AL. (2004). -A map of the interactome
network of the metazoan C. elegans Science 303:540-3.
LIU X, BANDYOPADHYAY BB, NAKAMOTO T, SINGH BB, LIEDTKE W, MELVIN JE, AMBUDKAR
IS. (2006). -A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of
AQP5 and TRPV4 in regulation of cell volume recovery. J Biol Chem 281: 15485-15495.
LOSPITAO E, PÉREZ-FERREIRO CM, GOSÁLBEZ A, ALONSO M AND CORREAS I (2008) . An
internal ribosome entry site element directs the synthesis of the 80 kDa isoforms of protein
4.1R BMC Biology 2008, 6:51 doi:10.1186/1741-7007-6-51
174
LUE RA, MARFATIS SM, BRANTON D, CHISHTL AH (1994) -Cloning and characterization of
hdlg: the human homologue of the Drosophila discs large tumor suppressor binds to protein
4.1- Proc Natl Acad Sci USA 91:9818-9822.
LUQUE CM, PEREZ-FERREIRO CM, PEREZ-GONZALES A, ENGLMEIER L, KOFFA MD, CORREAS I
(2003) -An alternative domain containing a leucine-rich sequence regulates nuclear
cytoplasmic localization of protein 4.1R. J Biol Chem 278: 2686-2691
LUQUE CM, LALLENA MJ, PEREZ-FERREIRO CM, DE ISIDORO Y, DE CARCER G, ALONSO MA,
CORREAS I (1999) -The N-terminal 209-aa domain of high molecular weight 4.1R isoforms
abrogates 4.1R targeting to the nucleus- Proc Natl Acad Sci USA 96,14925-14930.
MCCARTY N.A., O’NEIL R.G. (1992) -Calcium signaling in cell volume regulation.Physiol. Rev.
72(4): 1037-71, Review.
MACLEOD RJ, HAMILTON JR. (1999). -Ca2_/calmodulin kinase II and decreases in
intracellular pH are required to activate K_ channels after substantial swelling in villus
epithelial cells. J Membr Biol 172: 59–66.
MACLEOD RJ, HAMILTON JR (1996) -Activation of Na+ / H+ exchange is required for
regulatory volume decrease after modest “physiological” volume increases in jejunal villus
epithelial cells- J Biol Chem 271(38):23138-45.
MANNO S, TAKAKUWA Y, MOHANDAS N (2005). -Modulation of erythrocyte membrane
mechanical function by protein 4.1 phosphorylation. J Biol Chem 280: 7581-7587
MARCHESI SL, CONBOY J, AGRE P, LETAINGER JT, MARCHESI VT, SPEICHER DW, MOHANDAS
N (1990) -Insertion/deletion mutations in protein 4.1 in elliptocytosis. Biochemical
identification of rearrangements in the spectrin/actin binding domain and functional
characterizations- J Clin Invest 86:516-523.
175
MATTAGAJASINGH SN, HUANG SC, HARTENSTEIN JS, SNYDER M, MARCHESI VT, BENZ EJ
(1999) -A non erythroid isoform of protein 4.1R interacts with the nuclear mitotic apparatus
(NuMA) protein- J Cell Biol 145:29-43.
MAYER BJ, REN R, CLARK KL, BALTIMORE D. (1993). -A putative modular domain present in
diverse signaling proteins Cell 73:629-30.
MCCARTY N.A., O’NEIL R.G. (1992) Calcium signaling in cell volume regulation. Physiol. Rev.
72(4): 1037-1061.
MCPHEE JC, DANG YL, DAVIDSON N, LESTER HA. (1998). -Evidence for a functional
interaction between integrins and G protein-activated inward rectifier K+ channels J. Biol.
Chem. 273:34696-702
MEISTER G, EGGERT C, BUHLER D, BRAHMS H, KAMBACH C, FISCHER U. (2001). Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP
assembly factor pICln Curr. Biol. 11:1990-4.
MEYER K, KORBMACHER C (1996) -Cell swelling activates ATP – dependent voltage-gated
chloride channels in M-1 mouse cortical colletting duct cells- J Gen Physiol 108:177-193.
MILLS JW, MANDEL LJ (1994) -Cytoskeletal regulation of membrane transport events- Faseb
J 8(14):1161-5.
MITCHINSON TJ, CRAMER LP (1996) -Actin-based cell motility and cell locomotion – Cell
9;84(3):371-9.
MOECKEL GW, ZHANG L, CHEN X, ROSSINI M, ZENT R, POZZI A. (2006) Role of integrin
alpha1beta1 in the regulation of renal medullary osmolyte concentration. Am J Physiol
Renal Physiol 290: F223–F231.
MOUSTAKAS A, THEODOROPOULOS PA, GRAVANIS A, HAUSSINGER D, STOURNARAS C
(1998) -The cytoskeleton in cell volume regulation- Contrib Nephrol 123:121-134.
176
MUSCH MW, LUER CA, DAVIS-AMARAL EM, GOLDSTEIN L (1997) -Hypotonic stress induces
translocation of the osmolyte channel protein pICln in embryonic skate (Raja eglanteria)
heart- J Exp Zool 15;277(6):460-3.
MUSCH MW, DAVIS-AMARAL EM, VANDENBURGH HH, GOLDSTEIN L (1998) -Hypotonicity
stimulates translocation of ICln in neonatal rat cardiac myocytes- Pflügers Arch Eur J Physiol
436(3):415-422.
NAM JH, LEE HS, NGUYEN YH, KANG TM, LEE SW, KIM HY, KIM SJ, EARM YE, KIM SJ. (2007). Mechanosensitive activation of K_ channel via phospholipase C-induced depletion of
phosphatidylinositol 4,5-bisphosphate in B lymphocytes. J Physiol 582: 977–990.
NELSON WJ, HAMMERTON RW (1989) -A membrane-cytoskeletal complex containing Na+K+-ATPase, ankyrin, and fodrin in Madin-Darby canine kidney (MDCK) cells: implications for
the biogenesis of epithelial cell polarity- J Cell Biol 108(3):893-902.
NIELSEN DK, JENSEN AK, HARBAK H, CHRISTENSEN SC, SIMONSEN LO. (2007). -Cell content
of phosphatidylinositol (4,5)bisphosphate in Ehrlich mouse ascites tumour cells in response
to cell volume perturbations in anisotonic and in isosmotic media. J Physiol 582: 1027–1036.
NILIUS B, OWSIANIK G, VOETS T, PETERS JA. (2007).Transient receptor potential cation
channels in disease. Physiol Rev 87: 165–217.
NILIUS B, DROOGMANS G (2001) - Ion Channels and Their Functional Role in Vascular
Endothelium Physiol. Rev. 81: 1415-1459.
NUMATA T, SHIMIZU T, OKADA Y. (2007). -TRPM7 is a stretch- and swelling-activated cation
channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol
292: C460–C467.
NUNOMURA W, PARRA M, HEBIGUCHI, SAWADA K, MOHANDAS N, TAKAKUWA Y (2009) –
Marked difference in membrane-protein-binding properties of the two isoforms of protein
4.1R expressed at early stages of erythroid differentiation. Biochem 417: 141-148
177
NUNOMURA W, TAKAKUWA Y (2006) Regulation of protein 4.1R interactions with
membrane proteins by Ca2+ and calmodulin. Front Biosci 11:1522-1539
OKADA Y. (2006). - Cell volume-sensitive chloride channels: phenotypic properties and
molecular identity Contrib. Nephrol. 152:9-24
OKADA Y (1997). -Volume expansion-sensing outward-rectifer Cl- channel: fresh start to the
molecular identity and volume sensor- Am. J. Physiol. 273:C755-C789.
ORDWAY RW, SINGER JJ, WALSH JV JR. (1991). -Direct regulation of ion channels by fatty
acids. Trends Neurosci 14: 96–100.
OUDE WEERNINK PA, SCHMIDT M, JAKOBS KH. (2004). -Regulation and cellular roles of
phosphoinositide 5-kinases. Eur J Pharmacol 500: 87–99.
PAL, S., S. N. VISHWANATH, H. ERDJUMENT-BROMAGE, P. TEMPST, AND S. SIF (2004) Human SWI/SNF associated PRMT5 methylates histone H3 arginine 8 and negatively
regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 24:9630–
9645.
PAL, S., R. YUN, A. DATTA, L. LACOMIS, H. ERDJUMENT-BROMAGE, J. KUMAR, P. TEMPST,
AND S. SIF. (2003) - mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is
involved in transcriptional repression of the Myc target gene cad. Mol. Cell. Biol. 23:7475–
7487.
PARRA M, GASCARD P, WALENSKY LD, GIMM JA, BLACKSHAW S, CHAN N, TAKAKUWA Y,
BERGER T, LEE G, CHASIS JA, SNYDER SH, MOHANDAS N, CONBOY JG . (2000). -Molecular
and functional characterization of protein 4.1B, a novel member of the protein 4.1 family
with high level, focal expression in brain. J Biol Chem 275: 3247-3255
178
PARRA M, GASCARD P, WALENSKY LD, SNYDER SH, MOHANDAS N, CONBOY JG (1998) Cloning and characterization of 4.1G (EPB41L2), a new member of the skeletal protein 4.1
(EPB41) gene family- Genomics 15;49(2):298-306.
PAULMICHL M, GESCHWENTNER M, SUSANNA A, SCHMARDA A, LAICH A, NAGL UO,
ELLEMUNTER H, DEETJEN P, FRICK J (1996) -ICln: a chloride channel paramount for cell
volume regulation- J Allergy Clin Immunol 98(5 Pt 2):S98-101.
PAULMICHL M, LI Y, WICKMAN K, ACKERMAN M, PERALTA E, CLAPHAM D. (1992). -New
mammalian chloride channel identified by expression cloning Nature. 356:238-41.
PEDERSEN SF, OWSIANIK G, NILIUS B. (2005). -TRP channels: an overview. Cell Calcium 38:
233–252.
PEDERSEN SF, MILLS J W AND HOFFMANN EK (1999) - Role of the F-actin Cytoskeleton in
the RVD and RVI Processes in Ehrlich Ascites Tumor Cells Experimental Cell Research
252: 63-74.
PEREZ-FERREIRO CM, I VERNOS & I CORREAS. (2004). -Protein 4.1R regulates interphase
microtubule organization at the centrosome. J Cell Sci 117, 6197-6206.
POLLARD TD, COOPER JA (1986) -Actin and actin-binding proteins. A critical evaluation of
mechanisms and functions- Annu Rev Biochem 55:987-1035.
PRAT AG, XIAO YF, AUSIELLO DA, CANTIELLO HF (1995) -cAMP-independent regulation of
CFTR by the actin cytoskeleton- Am J Physiol 268:C1552-61.
PU WT, WICKMAN K, CLAPHAM DE. (2000). -ICln is essential for cellular and early embryonic
viability J. Biol. Chem. 275:12363-6.
PU WT, KRAPIVINSKY GB, KRAPIVINSKY L, CLAPHAM DE. (1999). -pICln inhibits snRNP
biogenesis by binding core spliceosomal proteins Mol. Cell Biol. 19:4113-20.
179
RAMEZ M, BLOT-CHABAUD M, CLUZEAUD F, CHANAN S, PATTERSON M, WALENSKY LD,
MARFATIA S, BAINES AJ, CHASIS JA, CONBOY JG, MOHANDAS N, GASCARD P (2003) -Distinct
distribution of specific members of protein 4.1 gene family in the mouse nephron. Kidney Int
63: 1321-1337
RASMUSSEN M, ALEXANDER RT, DARBORG BV, MOBJERG N, HOFFMANN EK, KAPUS A,
PEDERSEN SF. (2008). -Osmotic cell shrinkage activates ezrin/radixin/moesin (ERM)
proteins: activation mechanisms and physiological implications. Am J Physiol Cell Physiol
294: C197–C212.
RITTER M, RAVASIO A, JAKAB M, CHWATAL S, FÜRST J, LAIGH A, GSCHWENTNER M,
SIGNORELLI S, BURTSCHER C, EICHMULLER S AND PAULMICHL M (2003) -Cell swelling
stimulates cytosol to membrane transposition of ICln- J Biol Chem 12;278(50):50163-74.
RIVERA A, DE FRANCESCHINI L, PETERS LL, GASCARD P, MOHANDAS N, BRUGNARA C, (2006)
– Effect of complete protein 4.1R deficiency on ion transport properties of murine
erythrocytes Am J Physiol Cell Physiol 291: C880-C886
ROBB VA, LI W, GASCARD P, PERRY A, MOHANDAS N, GUTMANNA DH (2003) - Identification
of a third Protein 4.1 tumor suppressor, Protein 4.1R, in meningioma pathogenesis
Neurobiology of Disease 13 (2003) 191–202
RODIGHIERO S, BAZZINI C, RITTER M, FÜRST J, BOTTA G, MEYER G, PAULMICHL M (2008)Fixation, Mounting and Sealing with Nail Polish of Cell Specimens Lead to Incorrect FRET
Measurements using Acceptor Photobleaching Cell Physiol Biochem; 21:489-498
ROSENMUND C, WESTBROOK GL (1993) -Calcium-induced actin depolymerization reduces
NMDA channel activity- Neuron 10(5):805-14.
ROSS AF, OLEYNIKOW Y, KISLAUSKIS EH, TANEJA KL, SINGER RH (1997) -Characterization of
a beta-actin mRNA zipcode-binding protein- Mol Cell Biol 17(4):2158-65.
180
RUDGE SA, ANDERSON DM, EMR SD (2004) - Vacuole size control: regulation of
PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific
phosphatase Mol. Biol. Cell 15:24-36.
SAARIKANGAS J, ZHAO H, LAPPALAINEN P (2010) - Regulation of the actin cytoskeletonplasma membrane interplay by phosphoinositides. Physiol Rev.;90:259-89.
SALOMAO M, ZHANG X, YANG Y, LEE S, HARTWIG JK, CHASISJA, MOHANDAS N, AND AN X
(2008) - Protein 4.1R-dependent multiprotein complex: New insights into the structural
organization of the red blood cell membrane PNAS 105: 8026–8031
SBRISSA D, SHISHEVA A. (2005). -Acquisition of unprecedented phosphatidylinositol 3,5bisphosphate rise in hyperosmotically stressed 3T3–L1 adipocytes, mediated by ArPIKfyvePIKfyve pathway. J Biol Chem 280: 7883–7889.
SBRISSA D, IKONOMOV OC, DEEB R, SHISHEVA A (2002) - Phosphatidylinositol 5-phosphate
biosynthesis is linked to PIKfyve and is involved in osmotic response pathway in mammalian
cells J. Biol. Chem. 277:47276-84.
SCANDELLA E, NAGL UO, OEHL B, BERGMANN F, GSCHWENTNER M ET AL. (2000). -The
promoter for constitutive expression of the human ICln gene CLNS1A J. Biol. Chem.
275:15613-20.
SCOLES DR, HUYNH DP, MORCOS PA, COULSELL ER, ROBINSON NG, TAMANOI F, PULST SM
(1998) -Neurofibromatosis 2 tumour suppressor schwannomin interacts with betaII-spectrinNat Genet 18(4):354-9.
SHAKIBAEI M, MOBASHERI A. (2003). -Beta1-integrins co-localize with Na, K-ATPase,
epithelial sodium Channels (ENaC) and voltage activated calcium channels (VACC) in
mechanoreceptor complexes of mouse limb-bud chondrocytes Histol. Histopathol. 18:34351
181
SCHEDLBAUER A, KONTAXIS G, KONIG M, FÜRST J, JAKAB M ET AL. (2003). -Sequencespecific resonance assignments of ICln, an ion channel cloned from epithelial cells J. Biomol.
NMR 27:399-400.
SCHMARDA A, FRESSER F, GSCHWENTNER M, FÜRST J, RITTER M ET AL. (2001). Determination of protein protein interactions of ICIn by the yeast two-hybrid system Cell
Physiol Biochem. 11:55-60
SCHWARTZ RS, RYBICKI AC, NAGEL RL (1997) -Molecular cloning and expression of a chloride
channel-associated protein pICln in human young red blood cells: association with actinBiochem J 327(609–616).
SHEN L, LIANG F, WALENSKY LD, HUGANIR RL. (2000) - Regulation of AMPA receptor GluR1
subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J Neurosci.
20(21):7932-40.
SHEN MR, CHOU CY, BROWNING JA, WILKINS RJ, ELLORY JC. (2001). -Human cervical cancer
cells use Ca2_ signalling, protein tyrosine phosphorylation and MAP kinase in regulatory
volume decrease. J Physiol 537: 347–362.
SHERMAN, L.S., GUTMANN, D.H. (2001) - Merlin: hanging tumor suppression on the Rac.
Trends Cell Biol. 11, 442–444.
SHUBA YM, PREVARSKAYA N, LEMONNIER L, VAN COPPENOLLE F, KOSTYUK P. G., MAUROY
B., AND SKRYMA R (2000) - Volume-regulated chloride conductance in the LNCaP human
prostate cancer cell line Am J Physiol Cell Physiol 279: C1144-C1154.
SMITH PR, SACCOMANI G, JOE EH, ANGELIDES KJ, BENOS DJ (1991) -Amiloride-sensitive
sodium channel is linked to the cytoskeleton in renal epithelial cells- Proc Natl 15:6971-5.
182
STAGG M A; CARTER E, SIEDLECKA U; SOPPA G K; MEAD F; BENNETT P; TAYLOR-HARRIS P;
PINDER J C; YACOUB M; BAINES A; TERRACCIANO C (2008) - Cytoskeletal Protein 4.1R
Affects Sodium Current in Cardiomyocytes from Transgenic Mice with Prolonged QT Interval
Circulation. 2008;118:S_341
STOSSEL TP (1984) -Contribution of actin to the structure of the cytoplasmic matrix- J Cell
Biol 99(1 Pt 2):15s-21s.
STRANGE K. (1998) - Molecular identity of the outwardly rectifying, swelling-activated anion
channel: time to reevaluate pICln. J Gen Physiol. 111:617-22.
STRANGE K., EMMA F., JACKSON P.S. (1996). -Cellular and molecular physiology of volumesensitive anion channels. Am. J. Physiol. 270(3 Pt 1):C711-30.
STUTZIN A, HOFFMANN EK. (2006) - Swelling-activated ion channels: functional regulation
in cell-swelling, proliferation and apoptosis. Acta Physiol 187: 27–42.
SUH BC, HILLE B. (2005). -Regulation of ion channels by phosphatidylinositol 4,5bisphosphate. Curr Opin Neurobiol 15: 370–378.
TAMMA G, PROCINO P, STRAFINO A, BONONI E, MEYER G, PAULMICHL M, FORMOSO V,
SVELTO M AND VALENTI G (2007) - Hypotonicity Induces Aquaporin-2 Internalization and
Cytosol-to-MembraneTranslocation of ICln in Renal Cells Endocrinology 148:1118-1130.
TAMMA G; PROCINO G; SVELTO; VALENTI (2006) Hypotonicity causes actin reorganization
and recruitment of the actin-binding ERM protein moesin in membrane protrusion in
collecting duct principal cells Am J Cells Physiol 292: C1476-C1484
TANG CJ, TANG TK. (1998). -The 30-kD domain of protein 4.1 mediates its binding to the
carboxyl terminus of pICln, a protein involved in cellular volume regulation Blood 92:1442-7
183
TAKENAWA T, ITOH T. (2001). -Phosphoinositides, key molecules for regulation of actin
cytoskeletal organization and membrane traffic from the plasma membrane. Biochim
Biophys Acta 1533: 190– 206.
THOROED SM, LAURITZEN L, LAMBERT IH, HANSEN HS, HOFFMANN EK. (1997). Cell swelling
activates phospholipase A2 in Ehrlich ascites tumor cells. J Membr Biol 160: 47–58,
TINEL H, KINNE-SAFFRAN E, KINNE RK. (2000). -Calcium signalling during RVD of kidney
cells. Cell Physiol Biochem 10: 297–302.
TOKER A. (1998) - The synthesis and cellular roles of phosphatidylinositol 4,5-bisphosphate
Curr. Opin. Cell Biol. 10:254-61.
UCHIDA S, YAMAUCHI A, PRESTON AS, KWON HM, HANDLER JS. (1993). -Medium tonicity
regulates expression of the Na_- and Cl_-dependent betaine transporter in Madin-Darby
canine kidney cells by increasing transcription of the transporter gene. J Clin Invest 91:
1604–1607.
VAN DER KAAY J, BECK M, GRAY A, DOWNES CP. (1999).-Distinct phosphatidylinositol 3kinase lipid products accumulate upon oxidative and osmotic stress and lead to different
cellular responses. J Biol Chem 274: 35963–35968.
VAN DER WIJK T, TOMASSEN S, DE JONGE HR,. TILLY BC (2000) - Signalling Mechanisms
Involved in Volume Regulation of Intestinal Epithelial Cells Cell Physiol Biochem;10:289-29
VAN RJ, LANGESLAG M, JALINK K. (2004) - Correcting confocal acquisition to optimize
imaging of fluorescence resonance energy transfer by sensitized emission Biophys. J.
86:2517-29
VAN ROSSUM DB, PATTERSON RL, SHARMA S, BARROW RK, KORNBERG M, GILL DL, SNYDER
SH. (2005). -Phospholipase Cgamma1 controls surface expression of TRPC3 through an
intermolecular PH domain. Nature 434: 99–104.
184
VOETS T, NILIUS B. (2007). -Modulation of TRPs by PIPs. J Physiol 582: 939–944.
VOM DS, SCHLIESS F, REISSMANN R, GORG B, WEIERGRABER O ET AL. (2003). -Involvement
of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver J. Biol.
Chem. 278:27088 95
WALENSKY LD, BLACKSHAW S, LIAO D, WATKINS CC, WEIER HU, PARRA M, HUGANIR RL,
CONBOY JG, MOHANDAS N, SNYDER SH (1999) -A novel neuron-enriched homolog of the
erythrocyte membrane cytoskeletal protein 4.1. J Neurosci 19: 6457-6467
WANG GX, DAI YP, BONGALON S, HATTON WJ, MURRAY K ET AL (2005) - Hypotonic
activation of volume-sensitive outwardly rectifying anion channels (VSOACs) requires
coordinated remodeling of subcortical and perinuclear actin filaments J. Membr. Biol.
208:15-26.
WATSON AJ, LEVINE S, DONOWITZ M, MONTROSE M H (1992) - Serum regulates Na+/H+
exchange in Caco-2 cells by a mechanism which is dependent on F-actin- J Biol Chem 15;
267(2):956-62.
WESKAMP M, SEIDL W AND GRISSMER S (2000) - Characterization of the Increase in [Ca2+] i
During Hypotonic Shock and the Involvement of Ca2+-activated K+ Channels in the Regulatory
Volume Decrease in Human Osteoblast-like Cells Journal of Membrane Biology 178, Number
1, 11-20.
XIA Z, LIU Y. (2001) - Reliable and global measurement of fluorescence resonance energy
transfer using fluorescence microscopes Biophys. J. 81:2395-402
YAMAMOTO M, CHEN MZ, WANG YJ, SUN HQ, WEI Y, MARTINEZ M, YIN HL. (2006). Hypertonic stress increases PI(4,5)P2 levels by activating PIP5KIbeta. J Biol Chem 281:
32630-32638.
185
YAMAMOTO S, ICHISHIMA K, EHARA T (2009) -- Reduced volume-regulated outwardly
rectifying anion channel activity in ventricular myocyte of type 1 diabetic mice. J Physiol Sci.
59:87-96.
YAMAMOTO S, ICHISHIMA K, EHARA T (2008) Regulation of volume-regulated outwardly
rectifying anion channels by phosphatidylinositol 3,4,5-trisphosphate in mouse ventricular
cells. Biomed Res. 29:307-15.
YE K, KJ HURT, FY WU, M FANG, HR LUO, JJ HONG, S BLACKSHAW, CD FERRIS & SH SNYDER
(2000) Pike: A nuclear gtpase that enhances PI3kinase activity and is regulated by protein
4.1N. Cell 103, 919-930
YOON HS, HAJDUK PJ, PETROS AM, OLEJNICZAK ET, MEADOWS RP, FESIK SW. (1994). Solution structure of a pleckstrin-homology domain Nature 369:672-5.
ZHAO Q, RANK G, TAN YT, LI H, MORITZ R L, SIMPSON RJ, CERRUTI L, CURTIS D J, PATEL D J,
ALLIS C D, CUNNINGHAM J JANE S (2009) - PRMT5-mediated methylation of histone H4R3
recruits DNMT3A, coupling histone and DNA methylation in gene silencing NATURE
STRUCTURAL & MOLECULAR BIOLOGY VOLUME 16: 304-311
186