Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
SINOSSI DEL PROTOCOLLO
Gilead Sciences, Inc.
199 East Blaine Street
Seattle, WA 98102, USA
Titolo dello studio:
Studio di Fase 3, randomizzato, in doppio cieco, controllato con placebo
per valutare l’efficacia e la sicurezza di Idelalisib (GS-1101) in
combinazione con bendamustina e rituximab per linfomi non-Hodgkin
indolenti trattati in precedenza
Centri di studio
programmati:
Circa 120 centri in tutto il mondo
Numero IND:
101254
Numero EudraCT:
2012-004034-42
Obiettivo primario: •
Obiettivi
secondari:
RISERVATO
Valutare l’effetto dell’aggiunta di idelalisib a bendamustina/rituximab
(B/R) sulla sopravvivenza senza progressione (progression-free
survival, PFS) nei soggetti affetti da linfoma non Hodgkin indolente
trattato in precedenza (indolent non-Hodgkin lymphoma, iNHL)
•
Valutare l’effetto dell’aggiunta di idelalisib a
bendamustina/rituximab B/R sull’insorgenza, sulla grandezza e sulla
durata del controllo del tumore
•
Valutare l’effetto dell’aggiunta di idelalisib a
bendamustina/rituximab B/R sui parametri del benessere del
soggetto, ivi compresi sopravvivenza globale (Overall survival, OS),
qualità della vita correlata alla salute (health-related quality of life,
HRQL) e performance status
•
Valutare gli effetti dell’aggiunta di idelalisib a
bendamustina/rituximab B/R sui biomarcatori associati alla malattia
•
Caratterizzare l’effetto di bendamustina/rituximab sull’esposizione a
idelalisib attraverso la valutazione delle concentrazioni di idelalisib
nel plasma col passare del tempo
•
Descrivere il profilo di sicurezza osservato con l’aggiunta di
idelalisib a bendamustina/rituximab B/R
•
Stimare l’utilizzo delle risorse sanitarie associate all’aggiunta di
idelalisib a bendamustina/rituximab B/R
Pagina 1
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
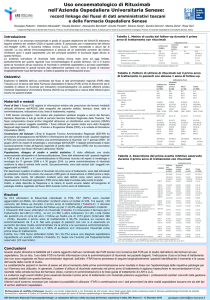
Disegno dello
studio:
Version 2.2(VHP)
Studio di fase 3, multicentrico, a 2 bracci, randomizzato, in doppio cieco,
controllato con placebo e a gruppi paralleli
Piano dello studio
Randomized,
Double-Blind
Terapia combinata,
iniziale,
randomizzata,
in doppio
cieco
Initial
Combination
Therapy
Continuing
Study Drug
Terapia
continuativa
con il farmaco in
(GS-1101/Placebo)
Therapy
studio (GS-1101/Placebo)
2
Braccio A
Arm
A
N = 300
N=300
2 Day 1,2
B,B,90
1, 2 every
ogni 2828
giorni
90mg/m
mg/mGiorni
daysper
for 44cicli
cycles
GS-1101, 150 mg 2 volte al dì
GS-1101, 150 mg BID
Terapia
dopo la
Post-Study
Therapy
conclusione
2 2
R,R,
375
mg/m
281,
giorni
per
cicli for 4 cycles
375
mg/mogni
Day
every
284 days
Terapia
continuativa
fino a progressione
Screening
Arm B B
Braccio
N=150
N = 150
Investigators’
Choice
Scelta degli
Commercial
/investigational
sperimentatori
2
2 Day 1,2
B,B,9090mg/m
1, 2 ogni
2828
giorni
mg/mGiorni
every
daysper
for44cicli
cycles
Placebo150
BIDmg 2 volte al dì
Placebo
2
2 Day
R,R,375
281,giorni
cicli for 4 cycles
375mg/m
mg/mogni
everyper
284days
Stratificazione
e
Stratification
and
randomizzazione
2:1
2: 1 Randomization
Progressione
della
iNHL Disease
malattia
iNHL
Progression
Abbreviazioni: B=bendamustina, BID=2 volte al dì, iNHL=linfoma non Hodgkin indolente, R=rituximab
Gruppo di trattamento
•
Braccio A: idelalisib + bendamustine+ rituximab
Braccio B: Placebo + bendamustine+ rituximab
Randomizzazione e stratificazione
•
Assegnazione 2:1 al braccio A rispetto al braccio B con implementazione
tramite un sistema interattivo di risposta Web (Interactive web response
system, IWRS)
•
Randomizzazione centralizzata per blocchi fissi con assegnazione dei
soggetti entro gli 8 strati, come stabilito dall’intersezione dei 3 fattori di
stratificazione binari:
o Tipo di tumore: Linfoma follicolare (LF) rispetto ad altri
o Carico tumorale: Elevato 1 vs basso
o Tempo trascorso dall’ultima terapia pregressa per iNHL: <18 mesi vs
≥18 mesi
1
Un carico tumorale elevato comprende la presenza di uno qualsiasi dei seguenti, come rilevato dalla tomografia
computerizzata (TAC) o dalla risonanza magnetica (RM): ≥3 sedi linfonodali con ≥1 linfonodo di dimensione più
lunga (DL) pari a≥3 cm, ≥1 massa linfonodale o extralinfonodale con DL pari a ≥7 cm, splenomegalia con
dimensione verticale più lunga (DVL) >10 cm, versamento pleurico o ascite peritoneale (a prescindere dal
contenuto cellulare) o compressione degli organi adiacenti (es. epidurale, ureterale, gastrointestinale, orbitale,
ecc.). Le sedi linfonodali sono collo (es. preauricolare, sottomandibolare, cervicale, sopraclavicolare), mediastino
(es. paratracheale, ilare), ascellare, epitrocleare, addominale (es. mesenterica, splenica, ilare, portale, celiaca),
retroperitoneale (paraortica), pelvica (iliaca comune, iliaca esterna, inguinale, femorale) o altra.
RISERVATO
Pagina 2
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
Numero
programmato di
soggetti:
Un totale di ~450 soggetti
(~300 soggetti nel braccio A e 150 soggetti nel braccio B)
Popolazione target
Il target di popolazione comprende soggetti adulti con iNHL ricorrente
trattato in precedenza, ivi compresi linfoma follicolare (LF), linfoma
linfocitico a piccole cellule (CLL), linfoma marginale (MZL) o linfoma
linfoplasmacitoide (LPL)/macroglobulinemia di Waldenström (WM),
che soffrano di linfoadenopatia misurabile, necessitino di terapia per il
iNHL, siano stati sottoposti in precedenza a una terapia con anticorpi
anti-CD20 e a chemioterapia e che soffrano di iNHL non refrattario al
bendamustina.
Criteri di inclusione
Diagnosi e criteri
principali di idoneità: Per essere idonei a partecipare a questo studio, i soggetti devono
soddisfare i criteri di inclusione elencati di seguito:
1) Maschio o femmina ≥18 anni di età.
2) Diagnosi confermata istologicamente di iNHL a cellule B, con
sottotipo istologico limitato alle seguenti condizioni in base ai
criteri stabiliti dalla classificazione dei tumori del tessuto
ematopoietico e linfoide da parte dell’Organizzazione Mondiale
della Sanità (OMS) del 2008.
a) Linfoma follicolare (LF) di grado 1, 2 o 3a
b) Linfoma linfocitico a piccole cellule (SLL) con calcolo assoluto
dei linfociti <5 x 109/l al momento della diagnosi
c) Linfoma linfoplasmacitoide/macroglobulinemia di Waldenström
(LPL/WM)
d) Linfoma con sede marginale (MZL) (splenica, linfonodale o
extralinfonodale)
3) iNHL linfonodale o extralinfonodale recidivante, misurabile tramite
radiografia (stabilito dalla presenza di ≥1 lesione misurante ≥2,0 cm
nel diametro più lungo, [DL] e ≥1,0 cm nel diametro perpendicolare
più lungo [DPL], come valutato mediante TAC o RM).
4) Trattamento pregresso per malignità linfoide comprendente
≥1 regime contenente un anticorpo terapeutico anti-CD20
(rituximab, ofatumumab, GA-101) somministrato per ≥2 dosi di
trattamento con anticorpi.
5) Trattamento pregresso per malignità linfoide comprendente ≥1
regime contenente chemioterapia terapeutica (es. agente alchilante)
somministrato per ≥2 cicli di trattamento.
Nota: il trattamento pregresso potrà essere stato somministrato come
singoli agenti o come componenti di terapie combinate. Soggetti che
abbiano ricevuto altri tipi di chemioterapia disponibili in commercio
(compreso bendamustina), immunoterapia o terapie sperimentali non
RISERVATO
Pagina 3
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
escluse. Ciascuna applicazione terapeutica ripetuta ma separata, dello
stesso agente singolo o combinato, è considerata un regime
indipendente.
6) Interruzione di tutte le terapie (comprese radioterapia,
chemioterapia, immunoterapia o terapia sperimentale) per il
trattamento del cancro ≥ 6 settimane prima della randomizzazione.
Nota: i soggetti possono ricevere corticosteroidi per gestire i
sintomi di iNHL.
7) Qualsiasi effetto tossico acuto di qualsiasi terapia antitumorale
precedente dovrà essere risolto al grado ≤1 prima della
randomizzazione (ad eccezione dell’alopecia [Grado 1 o 2
consentito], neurotossicità [Grado 1 o 2 consentito] oppure
parametri del midollo osseo [Grado 1 o 2 consentito]).
8) Punteggio dello stato di performance secondo Karnofsky: ≥60.
9) Dati di laboratorio richiesti al basale (entro 4 settimane prima
dell’inizio della terapia in studio) come mostrato nella tabella in
basso. Nota: considerare la conferma dei valori fuori norma per
stabilire se l’anomalia sia reale o artificiale. I valori dovranno
essere ottenuti entro il periodo di screening e, in generale, devono
essere quanto più recenti possibile.
Valori di laboratorio richiesti per lo screening
Sistema organico
Ematopoietico
Parametro
ANC
Piastrine
≥1,0 x 10 /l
≥75 x 109/la
Emoglobina
≥80 g/l (8,0 g/dl o 4,9 mmol/l)a
≤1,5 x limite superiore alla norma (ULN) (se non elevata a
causa della sindrome di Gilbert)
Bilirubina sierica totale
Epatico
Renale
Gravidanza
Valore richiesto
ALT sierica
AST sierica
eC Cr b
β-HCGc
HIV
HBV
9 a
≤2,0 x ULN
>40 ml/min
Negativo
Anticorpi anti-HIV assenti
HbsAg negativo e anticorpi anti-HBc negativid
Infezione
RNA virale negativo (se gli anticorpi anti-HCV sono
HCV
positivi)
a Neutropenia digrado ≥3, trombocitopenia o anemia sono consentite se l’anomalia è correlata al coinvolgimento
del midollo osseo con il iNHL (come documentato dalla biopsia/aspirato del midollo osseo ottenuto dall’ultima
terapia pregressa).
b Come calcolato dalla formula di Cockcroft-Gault
c Soltanto per donne in età fertile; il β-HCG urinario o sierico deve essere negativo durante lo screening e la
randomizzazione (Visita 2)
d I soggetti falsi positivi per gli anticorpi anti-HBc (a causa della somministrazione di Ig per via endovenosa)
potranno esser arruolati se l’HBV-DNA non è rilevabile mediante PCR quantitativa. [Arnold 2010, Benton
2012]
RISERVATO
Pagina 4
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
Abbreviazioni: βHCG= beta-gonadotropina corionica umana, ALT= alanina aminotransferasi, AST= aspartato
aminotransferasi, DNA= acido desossiribonucleico, eC Cr = clearance stimata della creatinina, anticorpi anti-HBc=
anticorpi anti-core dell’epatite B, HBsAg= antigene di superficie dell’epatite B, HBV= virus dell’epatite B, HCV=
virus dell’epatite C, HIV= virus dell’immunodeficienza umana , Ig= immunoglobulina, PCR= reazione a catena
della polimerasi, RNA= acido ribonucleico, ULN= limite superiore alla norma
10) Per i soggetti di sesso femminile in età fertile, disponibilità a
utilizzare un metodo contraccettivo raccomandato dal protocollo dalla
visita di screening (Visita 1), per l’intera durata dello studio e per 30
giorni dopo l’ultima dose di bendamustina o di idelalisib oppure per
>12 mesi dall’ultima dose di rituximab (a seconda di quale sia stato
somministrato per ultimo). Nota: un soggetto di sesso femminile è
considerato fertile fatto salvo il caso in cui abbia subito
un’isterectomia, legatura bilaterale delle tube oppure ooforectomia
bilaterale oppure in caso di insufficienza ovarica clinicamente
documentata (con livelli di estradiolo sierico e dell’ormone
follicolo-stimolante [FSH] entro i valori normali post-menopausa e
βHCG urinario o sierico negativo) oppure quando il soggetto sia in
menopausa (età ≥55 anni con amenorrea da ≥6 mesi).
11) Per i soggetti di sesso maschile fertili che abbiano rapporti sessuali
con soggetti di sesso femminile in età fertile, disponibilità a utilizzare
un metodo contraccettivo raccomandato dal protocollo dalla
randomizzazione (Visita 2), per l’intera durata dello studio e fino a >
6 mesi dopo l’ultima dose di bendamustina o del farmaco in studio (a
seconda di quale sia stato somministrato per ultimo) e dovrà astenersi
dalla donazione del seme dalla randomizzazione (Visita 2), per
l’intera durata dello studio e per 6 mesi dopo l’ultima dose di
bendamustina o del farmaco in studio (a seconda di quale sia stato
somministrato per ultimo). Nota: un soggetto di sesso maschile è
considerato fertile, fatto salvo il caso in cui abbia subito una
vasectomia bilaterale con aspermia documentata oppure
orchiectomia bilaterale oppure soppressione testicolare continua
con una formulazione depot di un antagonista dell’ormone di
rilascio dell’ormone luteinizzante (es. goserelin acetato
[Zoladex®]), leuprolina acetato [Lupron®]), o triptorelina pamoato
[Trelstar®]).
12) A giudizio dello sperimentatore, la partecipazione al protocollo offre
un rapporto rischio-beneficio accettabile considerato lo stato attuale
del iNHL, la condizione medica e i potenziali benefici e rischi di
trattamenti alternativi disponibili per i soggetti affetti da iNHL.
13) Disponibilità a rispettare le visite programmate, il piano di
somministrazione del farmaco, gli studi di imaging, gli esami di
laboratorio e altre procedure e restrizioni dello studio. Nota: devono
essere presi in considerazione fattori geografici, familiari, sociali o
psicologici che possano precludere un’adeguata partecipazione allo
RISERVATO
Pagina 5
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
studio.
14) Evidenza del modulo di consenso informato firmato personalmente
che indichi che il soggetto è a conoscenza della natura neoplastica
della malattia e che è stato informato sulle procedure da seguire, sulla
natura sperimentale della terapia, sulle alternative, sui potenziali
benefici e i possibili effetti collaterali, potenziali rischi e disagi e su
altri aspetti relativi alla partecipazione allo studio.
Criteri di esclusione
I pazienti che rispondono a uno dei seguenti criteri di esclusione non sono
idonei a partecipare al presente studio.
1) Linfoma del sistema nervoso centrale o leptomeningeale noto. Nota:
non è richiesta documentazione di imaging su assenza o presenza di
malattia centrale.
2) Trasformazione istologica nota in un linfoma aggressivo Nota: non è
richiesta documentazione bioptica su assenza o presenza di
trasformazione.
3) Presenza nota di sindrome mielodisplastica.
4) Anamnesi di malignità non linfoide, ad eccezione dei seguenti casi:
carcinoma della pelle a cellule squamose o a cellule basali locale
trattato adeguatamente, carcinoma in situ della cervice, tumore
superficiale della vescica, tumore alla prostata asintomatico, senza
malattia metastatica nota e senza necessità di terapia o richiedente
solo terapia ormonale e con antigene prostatico specifico nella norma
per ≥1 anno prima della randomizzazione, altro cancro di Stadio 1 o 2
trattato adeguatamente in completa remissione o qualsiasi altro cancro
in completa remissione da ≥ 5 anni.
5) Ipersensibilità nota o intolleranza a uno qualsiasi dei principi attivi o
eccipienti contenuti nelle formulazioni di idelalisib, o rituximab.
6) Conferme di infezione virale, micotica o batterica sistemica in atto al
momento della randomizzazione (Visita 2). Nota: i soggetti con
infezioni micotiche localizzate della pelle o delle unghie sono idonei.
I soggetti possono essere attualmente sottoposti a regimi profilattici
antivirali o antibatterici (es. profilassi anti-pneumocisti) a
discrezione dello sperimentatore. Per i soggetti che presentano un
rischio sostanziale di infezioni (es. influenza) che possono essere
prevenute mediante immunizzazione, si dovrebbe considerare
l’eventualità di un vaccino prima di iniziare la terapia del
protocollo.
7) Lesione epatica farmaco-indotta in atto, epatite C cronica attiva
(HCV), epatite B cronica attiva (HBV), epatopatia alcolica,
steatoepatite non alcolica, cirrosi biliare primitiva, ostruzione
RISERVATO
Pagina 6
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
extraepatica dovuta a colelitiasi, cirrosi epatica o ipertensione portale.
8) Polmonite farmaco-indotta in atto.
9) Malattia intestinale infiammatoria in atto.
10) Dipendenza da alcol o droghe in corso.
11) Gravidanza e allattamento.
12) Anamnesi di pregresso trapianto allogenico di organo solido o di
cellule staminali del midollo osseo.
13) Terapia immunosoppressiva diversa da corticosteroidi in corso. Nota:
i soggetti possono utilizzare corticosteroidi sistemici, per inalazione,
topici o enterici come terapia per le manifestazioni del iNHL, di
condizioni di comorbilità o anemia autoimmune e/o
trombocitopenia. Durante la partecipazione allo studio, i soggetti
possono ricevere corticosteroidi per via sistemica o altri
corticosteroidi come pretrattamento per le infusioni di rituximab o
come opportuno per condizioni di comorbilità emergenti dal
trattamento.
14) In un soggetto con anamnesi di terapia pregressa con bendamustina,
un intervallo di tempo dall’ultima dose di bendamustina alla
successiva progressione del iNHL di < 6 mesi.
15) Anamnesi di pregressa terapia con qualsiasi inibitore dell’AKT
(serina/treonina chinasi), tirosin-chinasi di Bruton (BTK), Janus
chinasi (JAK), bersaglio della rapamicina nei mammiferi (mTOR),
fosfatidilinositolo-3-chinasi (PI3K) (compreso GS-1101) o tirosinchinasi della milza (SYK).
16) Partecipazione contemporanea a un’altra sperimentazione clinica
terapeutica.
17) Partecipazione pregressa a una sperimentazione clinica conidelalisib .
18) Patologia clinicamente significativa pregressa o in atto, condizione
medica, anamnesi chirurgica, esiti fisici, esiti di elettrocardiogramma
(ECG) o anomalie dei valori di laboratorio che, secondo il parere
dello sperimentatore, potrebbero influenzare negativamente la
sicurezza del soggetto o compromettere la valutazione dei risultati
dello studio.
19) Pazienti che hanno ricevuto un vacciono per la febbre giallaentro 30
giorni prima della randomizzazione (Visita 2)
20) Pazienti che si sono sottoposti a grandi interventi chirurgici entro 30
giorni prima delal randomizzazione (Visita 2)
RISERVATO
Pagina 7
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Procedure dello
studio/
Frequenza:
Version 2.2(VHP)
I soggetti saranno randomizzati con un rapporto di 2:1 al braccio A o al
braccio B dello studio. I soggetti assumeranno idelalisib o placebo per via
orale, due volte al dì (BID) continuativamente. Ai soggetti sarà
somministrato rituximab al Giorno 1 di ciascun ciclo di 28 giorni del
ciclo fino alla fine del Ciclo -6. Ai soggetti verrà somministrato
bendamustina ai Giorni 1 e 2 per ciascun ciclo di 28 giorni a partire dal
Ciclo 1 e concludendo la somministrazione almeno al Ciclo 4 e non oltre
il Ciclo 6.
Le visite cliniche/di laboratorio avranno luogo ogni 2 settimane fino alla
Settimana 24 e, successivamente, ogni 12 settimane. I soggetti che
continueranno il trattamento in studio dopo la Settimana 48 saranno
sottoposti a visite cliniche ogni 12 settimane. I soggetti saranno valutati in
merito alla sicurezza ad ogni visita clinica.
I soggetti saranno valutati per lo stato della malattia iNHL attraverso
esami obiettivi e di laboratorio a ogni visita clinica e mediante TAC o
RM al basale e successivamente ogni 12 settimane.
Farmaco in studio,
dose e modalità di
somministrazione:
• Idelalisib, 150 mg/dose 2 volte al dì o placebo corrispondente 2
volte al dì assunti per via orale a partire dal Giorno 1 e
somministrati successivamente in modo continuativo.
•
Rituximab: 375 mg/m2 per via endovenosa al Giorno 1 di ciascun
ciclo di trattamento di 28 giorni (per un totale di 6 cicli, in base alla
tolleranza)
•
Bendamustina: 90 mg/m2/dose per via endovenosa al Giorno 1 e 2 di
ciascun ciclo di trattamento di 28 giorni (4-6 cicli, in base alla
tolleranza)
Idelalisib/placebo saranno assunto continuativamente fino al ritiro del
Durata del/i
farmaco/i in studio: soggetto dallo studio, alla progressione definitiva del iNHL, alla tossicità
intollerabile correlata al farmaco in studio, all’instaurarsi di una
gravidanza, alla non aderenza sostanziale alle procedure dello studio o
all’interruzione dello studio, a seconda di quale si verifica per primo.
Rituximab sarà assunto continuativamente fino a un massimo di 6
infusioni, al ritiro del soggetto dallo studio, alla progressione definitiva
del iNHL, alla tossicità intollerabile correlata a rituximab, all’instaurarsi
di una gravidanza, alla non aderenza sostanziale alle procedure dello
studio o all’interruzione dello studio, a seconda di quale si verifica per
primo.
Bendamustina sarà assunto continuativamente fino a un massimo di 8-12
infusioni, al ritiro del soggetto dallo studio, alla progressione definitiva
del iNHL, alla tossicità intollerabile correlata a bendamustina,
all’instaurarsi di una gravidanza, alla non aderenza sostanziale alle
procedure dello studio o all’interruzione dello studio, a seconda di quale
RISERVATO
Pagina 8
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
si verifica per primo.
I soggetti proseguiranno con il trattamento specificato nel protocollo per
ciascun farmaco in studio (idelalisib/placebo, bendamustina o rituximab)
che continua a essere tollerato, anche se altri farmaci in studio deve
essere sospeso in modo permanente a causa di tossicità specifica del
farmaco.
Criteri di
valutazione:
Endpoint primario
•
Sopravvivenza senza progressione della malattia (PFS), definita
come l’intervallo fra la randomizzazione e l’evento che si verifica
per primo fra i seguenti: prima documentazione della progressione
definitiva della malattia iNHL o decesso per qualsiasi causa; la
progressione definitiva della malattia iNHL è basata sui criteri
standard.
Endpoint secondari e terziari
Quattro endpoint sono designati come endpoint secondari per i quali
saranno eseguiti test sequenziali per controllare la percentuale di errore di
Tipo 1. Gli endpoint secondari saranno la percentuale di risposta
completa (Complete Response, CR), percentuale di risposta complessiva
(Overall Response Rate, ORR), percentuale di risposta linfonodale e
sopravvivenza globale (Overall Survival, OS). Tutti gli altri endpoint
saranno considerati terziari.
Controllo del tumore
•
Percentuale di risposta complessiva (ORR): definita come la
percentuale di soggetti che raggiungono una risposta completa (CR) o
parziale (PR) (oppure una risposta parziale molto buona [Very Good
Partial Response, VGPR] o una risposta secondaria [Minor Response,
MR] per i pazienti con LPL/WM)
•
RISERVATO
Percentuale di risposta nella sede linfonodale: definita come la
percentuale dei soggetti che ottengono una diminuzione maggiore
del 50%rispetto al basale nella somma dei prodotti dei diametri
perpendicolari più lunghi (SPD) dei linfonodi di riferimento
•
Percentuale di risposta completa (Complete Response, CR): definita
come percentuale dei soggetti che ottengono una risposta completa
•
Tempo alla risposta (Time To Response, TTR): definito come
intervallo dalla randomizzazione alla prima documentazione di CR o
PR (o VGPR o MR per pazienti con LPL/WM)
•
Durata della risposta (Duration Of Response, DOR): definita come
Pagina 9
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
intervallo dalla prima documentazione di CR o PR (o VGPR o MR
per pazienti con LPL/WM) fino alla prima documentazione di
progressione definitiva della malattia o il decesso per qualsiasi causa,
a seconda di quale si verifica per primo
Benessere del paziente
•
Sopravvivenza globale (Overall survival, OS): definita come il tempo
dalla randomizzazione all decesso per qualsiasi causa.
•
•
Variazione rispetto al basale nei punteggi dei sintomi e dei domini di
HRQL in base alla Valutazione funzionale della terapia
antitumorale: Linfoma (FACT-Lym) definito come variazione
rispetto al basale e il tempo di incrementi o decrementi definitivi di
10%, 20% e 40% rispetto al basale; tempo all’incremento definitivo
(migliore del basale dell’importo specificato) è l’intervallo fra la
randomizzazione e il primo punto temporale in cui la misura HRQL
è sostanzialmente migliore che al basale (incluso quel punto
temporale e i punti temporali successivi) in un soggetto, il cui ultimo
punteggio HRQL sia migliore che al basale; e il tempo al
decremento HRQL definitivo (peggiore che al basale dell’importo
specifico) è l’intervallo fra la randomizzazione e il primo evento che
si verifica fra il decesso o il primo punto temporale in cui la misura
di HRQL sia sostanzialmente peggiore rispetto al basale (incluso
quel punto temporale nonché i punti temporali successivi) in un
soggetto nel quale l’ultimo punteggio HRQL sia peggiore che al
basale
Variazioni rispetto al basale nello stato di performance secondo
Karnofsky
Marcatori farmacodinamici dell’attività e della resistenza al farmaco
•
Variazioni delle concentrazioni plasmatiche delle chemochine e
citochine associate alla malattia
•
Variazioni nei recettori dei linfociti B correlati alla malattia nel
sangue periferico
•
Variazioni nei subset di cellule mieloidi e linfoidi nel sangue
periferico
Esposizione
•
RISERVATO
Somministrazione del farmaco in studio come valutata dai dati relativi
alle prescrizione e conformità come valutata mediante la
Pagina 10
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
quantificazione del farmaco utilizzato e non utilizzato
•
Concentrazioni plasmatiche minime (pre-dose) e di picco (campioni
di 1,5 ore) di idelalisib come valutate mediante metodo bioanalitico
convalidato
Sicurezza
•
Il profilo di sicurezza complessivo di ciascun regime di trattamento in
studio caratterizzato da tipo, frequenza, gravità, tempo
dell’insorgenza, durata e relazione con la terapia in studio di qualsiasi
evento avverso o anomalia nei test laboratorio, eventi avversi gravi o
eventi avversi che portano all’interruzione del trattamento in studio
Farmacoeconomia
Metodi statistici:
•
Cambiamento nello stato di salute: definito come variazione dei
punteggi per singolo item e salute complessiva, come valutati usando
la misura dell’utilità EuroQoL a cinque dimensioni (EQ-5D)
•
Misure delle risorse sanitarie, compreso l’utilizzo delle risorse, i costi
totali e le misure del costo per unità di beneficio (es. costo per un
ulteriore mese senza progressione, costo per anno di vita adeguato
alla qualità)
Metodi di analisi
Sarà definita l’analisi appropriata delle serie di dati. La serie di analisi per
intent-to-treat (ITT) sarà utilizzata nelle analisi dell’endpoint di efficacia
primario, PFS. La serie di analisi ITT comprenderà i dati di tutti i soggetti
che sono randomizzati, con l’assegnazione al farmaco in studio designata
secondo la randomizzazione iniziale, indipendentemente da se i soggetti
ricevono qualsiasi farmaco in studio o ricevono un regime diverso da
quello al quale sono stati randomizzati. La serie di analisi sulla sicurezza
comprenderà i dati dei soggetti che ricevono ≥1 dose di farmaco/i in
studio, con assegnazione del regime di trattamento (braccio A o braccio
B) designato in base al regime effettivamente ricevuto. Saranno utilizzate
anche altre serie di analisi [per protocollo (PP) e serie di analisi di
farmacodinamica/farmacocinetica] per determinate analisi.
Le caratteristiche dei soggetti e risultati dello studio saranno descritti e
sintetizzati per braccio di trattamento e valutazione per la serie di analisi
pertinenti. Le sintesi descrittive saranno preparate in modo da mostrare la
dimensione del campione, la media, la deviazione standard, gli intervalli
di confidenza (IC) del 95% sulla media, la mediana, il valore minimo e
massimo per le variabili continue e i calcoli, le percentuali, e gli IC del
95% sulla percentuale per le variabili categoriche.
Un comitato di revisione indipendente (Independent review commitee,
RISERVATO
Pagina 11
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
IRC) verificherà i dati radiografici e i dati clinici pertinenti al fine di
fornire la valutazione degli esperti in merito ai cambiamenti nello stato
del tumore. I risultati dell’IRC saranno considerati primari per le analisi
della PFS e di altri endpoint di controllo del tumore.
Per l’analisi di efficacia primaria, si adotterà un set di analisi ITT
utilizzando i metodi di Kaplan-Meier e il test log-rank stratificato per
confrontare la differenza nella PFS tra i bracci di trattamento. Saranno
presentati mediane, intervalli, percentuali dei soggetti senza progressione
a 24 e 48, 72 e 96, 120 e 144 settimane dalla randomizzazione (in base
alle stime di Kaplan-Meier), rapporti di rischio e i corrispondenti IC 95%
(calcolati usando un modello di regressione dei rischi proporzionale di
Cox).
Gli endpoint secondari saranno la percentuale di risposta complessiva,
percentuale di risposta linfonodale e sopravvivenza globale. L’ipotesi di
efficacia primaria relativa alla PFS deve essere rifiutata al livello di
significatività bilaterale di 0,05 prima che vengano verificate le ipotesi di
efficacia per questi endpoint di efficacia secondari. I 4 endpoint secondari
saranno verificati in sequenza al livello di significatività bilaterale di 0,05
nell’ordine elencato. Se non viene rigettata un’ipotesi nulla, il test
sequenziale formale verrà interrotto e sarà citata solo la significatività
nominale per i restanti endpoint secondari. Per gli endpoint secondari o
terziari relativi al controllo del tumore, al benessere del paziente e ai
biomarcatori, le analisi verranno eseguite in base alle serie di analisi ITT,
risposta, valutabilità e farmacodinamica, come opportuno. Gli endpoint di
efficacia time-to-event saranno analizzati in maniera simile alla PFS. Le
variabili categoriche saranno confrontate usando il test di CochranMantel-Haenszel adeguato per i fattori di stratificazione. Gli endpoint
continui saranno valutati usando l’analisi della covarianza (ANCOVA)
con valori basali e fattori di stratificazione come covarianti. Le variazioni
rispetto al basale nei parametri HRQL e nello stato di performance
saranno confrontati fra i gruppi di trattamento usando il test dei ranghi
segnati di Wilcoxon, considerando lo stato di progressione della malattia
dei soggetti.
In base alla serie di analisi sulla sicurezza, saranno descritte e riassunte le
informazioni relative alla somministrazione del trattamento in studio, alla
conformità del farmaco in studio, alle variabili di sicurezza e alle terapie
successive alla fine dello studio. Usando i dati derivanti dalla serie di
analisi farmacocinetiche, saranno anche descritte e riassunte le
concentrazioni plasmatiche di idelalisib.
Calcolo della dimensione dei campioni
In base ai dati derivanti da studi precedenti, è ragionevole presumere che
la somministrazione di B/R a soggetti con iNHL trattati in precedenza nel
braccio B di questa sperimentazione produca una PFS mediana di ~20
RISERVATO
Pagina 12
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
mesi. Un miglioramento della PFS mediana dai 20 ai 30 mesi dovuta
all’aggiunta di idelalisib a B/R nel braccio A dello studio
corrisponderebbe a un rapporto di beneficio di 1,5 (rapporto di rischio
0,667).
Si presume che i tempi della PFS siano esponenzialmente
distribuiti in ciascuno dei 2 bracci. Con un rapporto di
rischio pari a 1 nell’ipotesi nulla dell’assenza di differenza
fra i 2 bracci di trattamento e un rapporto di rischio di
0,667 nell’ipotesi alternativa di superiorità della
combinazione contenente GS-1101, 267 eventi
(progressioni del iNHL, trasformazioni o decessi) devono
raggiungere un potere di 0,88 in base a un test log-rank
con un livello di significatività bilaterale di 0,05.
Presumendo inoltre un periodo programmato di
arruolamento di 21 mesi (con circa la metà dei pazienti
arruolati durante il 60% iniziale del periodo di
arruolamento e la restante metà dei soggetti arruolata
durante l’ultimo 40% del periodo di arruolamento), un
periodo di follow-up minimo di 30 mesi e la previsione
che fino al 25% dei soggetti sarà perso al follow-up, sono
stati arruolati 450 soggetti (300 soggetti nel braccio A e
150 soggetti nel braccio B in base a un rapporto di
randomizzazione di 2:1) al fine di ottenere un numero
previsto di eventi entro la fine del periodo di follow-up
minimo programmato di 30 mesi.
Questo studio sarà condotto ai sensi delle direttive delle Buone pratiche cliniche (Good Clinical
Practices, GCP) compresa l’archiviazione dei documenti essenziali.
RISERVATO
Pagina 13
27Jun2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
Version 2.2(VHP)
Programma delle procedure dello studio
Periodo
Visita
Settimana
Giorno di studio
Screening
1
2 3 4
-4
0 2
Trattamento
5
4
6 7
6
8
8
9 10 11 12
10
12
Entro
1 2 15 29 30 43 57 58 71 85
28 giorni
86
13 14 15 16
14
16
18
Follow-up
17
18
19
22
20
24
21
36
+22
Lungo
30 giorni
periodo
Fine
Ogni 12 dello
Entro
A
99 113 114 127 141 142 155 169 253 settimane studio più di 30 più di 5
giorni
anni
±2 ±3 +1 ±3 ±3 +1 ±3 ±3 ±3
±7
20
Finestra per la visita
-- +1 ±2 ±2a +1 ±1 ±2a +1 ±2 ±3a +1
Consenso informato
X
Anamnesi medica
X
Valutazione CIRS
X
Virologia sierica
X
β-HCG (donne in età fertile)
X
X
X
X
X
X
X
X
X
Xe
Xe
Coagulazione
X
Analisi delle urine
X
ECG a 12 derivazioni
X
Analisi genotipica e dell’espressione genica
X
X
IWRS
X
X
X
X
X
X
X
X
X
X
X
FACT-Lym/EQ-5D
X
X
X
X
X
X
X
X
X
X
Eventi avversi
X
X
X
X
X
X
X
X
X
X
X
Farmaci concomitanti
X
X
X
X
X
X
X
X
X
X
X
X
Stato di performance
X
X
X
X
X
X
X
X
X
X
X
Saturazione dell’ossigeno (mediante pulsossimetria)
X
X
X
X
X
X
X
X
X
X
X
Esame obiettivo (compresi linfonodi, fegato, milza)
X
X
X
X
X
X
X
X
X
X
X
Analisi ematochimiche
X
X
Xa X
Xa X
Xa X
Xa X
Xa X
Xa X
X
X
X
Ig sieriche
X
X
X
X
X
X
X
X
X
X
Citometria di flusso
X
X
X
X
X
X
X
X
X
X
Cellule circolanti
X
X
X
X
X
X
X
X
X
X
Biomarcatori
X
X
X
X
X
X
X
X
X
X
Somministrazione in clinica di idelalisib/placebo)
X
X
X
X
X
X
Premedicazione
X X
X X
X X
X
X
X X
X
X
Somministrazione di rituximab
X
X
X
X
X
X
Somministrazione di bendamustina
X X
X X
X X
X
X
Xd Xd
Xd Xd
Valutazione della gravità/durata della reazione
X
X
all’infusione
Farmacocinetica di idelalisib
X
X
X
X
Rendicontazione/dispensazione di idelalisib/placebo
X
X
X
X
X
X
Valutazione radiologica (TAC/RM)
Xb
X
X
X
Xb
Xb
Biopsia del midollo osseo
Xb
Xc
Xc Xc
Xc
Xc
SPEP/IFE (solo soggetti con LPL/WM)
X
X
X
X
X
X
Terapia iNHL post-studio/follow-up sul lungo periodo
X
a
Per i soggetti affetti da mielosoppressione di Grado ≥3 o aumenti dei valori di ALT/AST, potrebbe essere opportuno eseguire le analisi ematochimiche con una frequenza maggiore (es.
settimanale)
b
Allo screening, sarà possibile eseguire una TAC o RM entro le 6 settimane precedenti alla randomizzazione; il follow-up dello stato del tumore post-terapia a intervalli . Le valutazioni di
TAC/RM continueranno finché non saranno stati osservati ~267 eventi nella popolazione in studio.
c
Al basale, da eseguirsi a discrezione dello sperimentatore per stabilire la misura dell’interessamento del iNHL e la cellularità del midollo osseo. Dopo il basale, da eseguirsi per confermare la RC;
se il soggetto non dovesse soddisfare i criteri CR oppure se soddisfa altri criteri CR, ma dalla biopsia del midollo osseo al basale non risultasse alcun iNHL, non sarà necessario ottenere una biopsia
del midollo osseo di follow-up.
RISERVATO
Pagina 14
27 Jun 2013
Idelalisib
Studio GS-US-313-0125
Gilead Sciences, Inc.
d
Version 2.2(VHP)
A discrezione dello sperimentatore, in base a un precedente trattamento con bendamustina e alla tollerabilità, i cicli 5 e 6 di bendamustina potrebbero essere omessi.
e
A partire dalla Visita 20, l’esame della β-HCG deve essere effettuato ogni 6 settimane (±5 giorni) in seguito a ciascuna visita (solo per donne in età fertile)
Abbreviazioni: βHCG= beta-gonadotropina corionica umana, CIRS= scala di valutazione della malattia cronica, CR= risposta completa, TAC= tomografia assiale computerizzata, ECG=
elettrocardiogramma, EQ5D= EuroQoL a cinque dimensioni, FACT-Lym= valutazione funzionale della terapia antitumorale del linfoma, IFE= immunoelettroforesi, Ig= immunoglobulina, iNHL=
linfoma non-Hodgkin indolente, IWRS= sistema interattivo di risposta Web, LPL/WM= linfoma linfoplasmacitoide/ macroglobulinemia di Waldenström, RM= risonanza magnetica, SPEP= elettroforesi
sieroproteica
RISERVATO
Pagina 15
27 Jun 2013