Ciclo dell’acido citrico (Krebs)
Durante la via glicolitica che avviene nel citosol il glucosio viene convertito in piruvato. In
condizioni aerobiche tramite la decarbossilazione ossidativa del piruvato si forma acetil coenzima A
(acetil CoA). L’acetil CoA viene poi completamente ossidato a CO2 nel ciclo dell’acido citrico
tramite una serie di reazioni conosciute come ciclo degli acidi tricarbossilici o ciclo di Krebs. Il
ciclo di Krebs è la via finale comune per l’ossidazione delle molecole di sostanze nutrienti
riconducibili a tre gruppi essenziali rappresentati da aminoacidi, acidi grassi e carboidrati. Negli
eucarioti le reazioni del ciclo di Krebs hanno luogo nei mitocondri.
In questa via metabolica ossidativa, gli elettroni vengono estratti da intermedi del metabolismo dei
grassi, dei carboidrati e delle proteine ed utilizzati per la produzione della maggior parte dei
nucleotidi ridotti, responsabili, a loro volta, della sintesi di adenosina trifosfato (ATP) nella catena
di trasporto degli elettroni. Il ciclo di Krebs non utilizza direttamente l’ossigeno per alcuna delle sue
reazioni, tuttavia necessita del metabolismo ossidativo dei mitocondri per la riossidazione dei
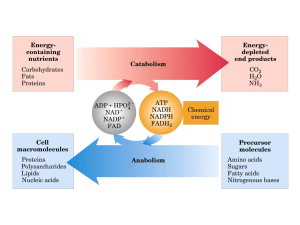
nucleotidi ridotti. Il ciclo di Krebs ha due importanti funzioni: produrre energia e fornire precursori
per le biosintesi.
Il substrato di partenza per il ciclo di Krebs è l’acetil-Coenzima A (acetil-CoA), un prodotto
comune dell’ossidazione dei carboidrati, degli acidi grassi e degli aminoacidi. L’acetil-CoA viene
ossidato per permettere la riduzione dei coenzimi coinvolti nelle quattro reazioni di ossidazione del
ciclo. In particolare, queste reazioni generano tre molecole di nicotinamide adenina dinucleotide
ridotto (NADH) ed una molecola di flavin adenina dinucleotide ridotto (FADH2) che, mediante la
catena di trasporto degli elettroni accoppiata alla fosforilazione ossidativa, rilasceranno l’energia
libera necessaria alla sintesi di ATP. Inoltre, nel ciclo di Krebs si forma direttamente un legame ad
alta energia di una molecola di guanosina trifosfato (GTP), mediante una reazione di fosforilazione
a livello del substrato. La maggior parte della CO2 prodotta dall’organismo deriva dalle reazioni di
questa via metabolica.
L’acetil-CoA, che inizia il ciclo di Krebs, deriva da tre principali precursori metabolici. Nella
glicolisi, i carboidrati sono convertiti a piruvato, che entra nel mitocondrio e viene decarbossilato
ossidativamente ad acetil-CoA dal complesso multienzimatico della piruvato deidrogenasi. I
trigliceridi sono convertiti ad acidi grassi liberi che, nella cellula, sono trasportati nel mitocondrio
ed ossidati ad acetil-CoA. Infine, molti aminoacidi derivati dall’idrolisi delle proteine sono
convertiti in acetil-CoA o in altri intermedi del ciclo di Krebs.
1
La formazione di acetil CoA avviene nella matrice mitocondriale e rappresenta il legame tra la
glicolisi ed il ciclo di Krebs, la reazione irreversibile è catalizzata dal complesso multienzimatico
della piruvato deidrogenasi.
Piruvato + CoA + NAD+
acetil CoA + CO2 + NADH
L’enzima piruvato deidrogenasi è un complesso formato da tre tipi di enzimi: piruvato deidrogenasi
(E1), diidrolipoil transacetilasi (E2), diidrolipoil deidrogenasi (E3). I cofattori catalitici necessari per
questi enzimi sono rispettivamente la tiamina pirofosfato (TPP) coinvolto nella decarbossilazione
ossidativa del piruvato, la lipoamide coinvolto nel trasferimento del gruppo acetile al CoA, ed il
FAD coinvolto nella rigenerazione della forma ossidata della lipoamide. L’acido lipoico è
covalentemente legato alla proteina mediante un legame amidico con il gruppo -aminico di un
residuo di lisina, per formare un lungo braccio flessibile in grado di legare e trasferire i substrati ai
diversi siti attivi. Dopo la formazione della acetildiidrolipoamide ad opera dell’enzima E1, il braccio
flessibile di E2 sposta il gruppo acetildiidrolipoamidico al sito di legame del CoA, dove si forma
acetil-CoA. La diidrolipoamide ridotta si sposta su E3 dove viene riossidata a lipoamide. Il FADH2
è successivamente riossidato a FAD dal NAD+, producendo NADH e rigenerando l’enzima attivo.
Gli arsenicati trivalenti (es arsenito inorganico) sono considerati composti in grado di reagire
con i gruppi sulfidrici. In tal modo inibiscono numerosi enzimi in quanto reagiscono con
ligandi biologici contenenti gruppi –SH disponibili, come ad esempio la lipoamide.
Il ciclo dell’acido citrico inizia con la condensazione di una unità a quattro atomi di carbonio,
l’ossalacetato, con una unità a due atomi di carbonio, il gruppo acetile dell’acetil CoA.
L’ossalacetato reagisce con acetil CoA e con acqua generando citrato e CoA. La reazione è
catalizzata dalla citrato sintasi e avviene tramite una condensazione alcolica seguita da una idrolisi.
Ossalacetato + acetil CoA + H2O
citrato + CoA + H+
Successivamente il citrato viene isomerizzato a isocitrato per permettere all’unità a sei atomi di
carbonio di subire una decarbossilazione ossidativa. L’isomerizzazione del citrato avviene tramite
una tappa di deidratazione seguita da una di idratazione che porta ad uno scambio di posizione tra
un atomo di idrogeno ed un gruppo ossidrilico. L’enzima che catalizza entrambe le tappe è
chiamato aconitasi, in quanto il cis-aconitato è un intermedio della reazione. L’aconitasi contiene
atomi di ferro che sono complessati con quattro solfuri inorganici e quattro atomi di zolfo di
2
altrettanti residui di cisteina. Questo complesso ferro-zolfo si lega al citrato e partecipa alla
deidratazione e reidratazione del substrato (citrato) legato all’enzima (aconitasi).
Citrato cis-aconitato isocitrato
Il fluoroacetato, isolato originariamente dalle piante, è un potente veleno. Esso viene attivato come
fluoroacetil-CoA e si condensa con l’ossalacetato per formare fluorocitrato. Il 2-fluorocitrato è un
potente inibitore dell’aconitasi e causa l’inibizione del ciclo di Krebs che può portare alla morte. Il
fluoroacetato è un esempio di “substrato suicida”, cioè un composto che non è di per sé tossico, ma
che viene metabolicamente trasformato in un derivato tossico. Pertanto, la cellula commette una
sorta di suicidio, trasformando un composto innocuo in un veleno letale. Processi analoghi sono
responsabili della trasformazione di agenti ambientali pro-cancerogeni in composti cancerogeni che
inducono mutazioni del DNA.
A questo punto del ciclo iniziano le quattro reazioni di ossido-riduzione, la decarbossilazione
ossidativa dell’isocitrato è catalizzata dalla isocitrato deidrogenasi. L’intermedio di questa reazione
è l’ossalsuccinato, quando questo composto è legato all’enzima perde CO2 e forma chetoglutarato.
Isocitrato + NAD+ ossalsuccinato -chetoglutarato + CO2 + NADH
chetoglutarato deidrogenasi che porta alla formazione del succinil CoA
-chetoglutarato + NAD+ + CoA succinil CoA + CO2 + NADH
Il legame tioestere tra il succinato ed il CoA è un legame ad alta energia e la scissione del legame
tioestere del succinil CoA è accoppiata alla fosforilazione della guanosina difosfato con formazione
di GTP. Questa rappresenta l’unica tappa del ciclo in cui si ha la produzione diretta di un legame
fosforico ad alta energia. Il GTP viene utilizzato come donatore di gruppi fosforici nella sintesi
proteica e nei processi di trasduzione di segnali extracellulari. La reazione è catalizzata dalla
succinil CoA sintetasi
Succinil CoA + Pi + GDP succinato + GTP + CoA
3
La fase finale del ciclo prevede la rigenerazione dell’ossalacetato. Il succinato viene convertito in
ossalacetato in tre tappe: una ossidazione, una idratazione ed una seconda reazione di ossidazione.
L’ossalacetato, cosi rigenerato, può essere utilizzato per un altro giro del ciclo, mentre l’energia
viene conservata sotto forma di FADH2 e di NADH. Il succinato è ossidato a fumarato dalla
succinato deidrogenasi, questo enzima come l’aconitasi è una proteina ferro-zolfo. La succinato
deidrogenasi contiene tre tipi diversi di centri ferro-zolfo: 2Fe-2S, 3Fe-4S, 4Fe-4S. La succinato
deidrogenasi è parte integrale della membrana interna dei mitocondri ed è direttamente legata alla
catena di trasporto degli elettroni. Infatti, il FADH2 prodotto dall’ossidazione del succinato non si
dissocia dall’enzima come accade invece al NADH prodotto nelle altre reazioni di ossidazione. Due
elettroni vengono trasferiti dal FADH2 ai centri Fe-S dell’enzima. L’accettore finale di questi
elettroni è l’ossigeno molecolare come si vedrà più dettagliatamente nella fosforilazione ossidativa.
Succinato + E-FAD fumarato + E-FADH2
L’idratazione del fumarato, catalizzata dalla fumarasi, porta alla formazione del malato che viene
successivamente ossidato ad ossalacetato tramite la malato deidrogenasi
Fumarato + H2O Malato + NAD+ ossalacetato + NADH + H+
Il ciclo dell’acido citrico è la via principale di degradazione per la generazione di ATP e nello
stesso tempo è in grado di produrre intermedi per i processi di biosintesi. Per esempio, la
maggioranza degli atomi di carbonio della porfirina deriva dal succinil-CoA e molti degli
aminoacidi derivano dall’-chetoglutarato e dall’ossalacetato.
La velocità del ciclo è finemente regolata dalla necessità di ATP della cellula. La disponibilità di
NAD+ e di FAD segnala che la carica energetica è bassa. La sintesi del citrato dall’ossalacetato e
dall’acetil CoA è un importante punto di controllo del ciclo. L’ATP è un inibitore allosterico della
citrato sintasi. L’effetto dell’ATP è quello di aumentare la KM dell’enzima per l’acetil CoA. Quando
i livelli di ATP aumentano, la frazione di enzima saturato con acetil CoA diminuisce e si forma
meno citrato.
4
Fosforilazione ossidativa
L’ossidazione delle sostanze nutritive è un processo essenziale negli esseri viventi. Nelle forme di
vita superiore le sostanze alimentari, rappresentate principalmente da carboidrati e lipidi, sono
metabolizzate a biossido di carbonio, che rappresenta la forma completamente ossidata del
carbonio, e ad acqua la forma completamente ridotta dell’ossigeno. Nel processo della
fosforilazione ossidativa, gli elettroni sono rimossi dai substrati nutritivi, trasferiti a coenzimi
ossido-riducenti (redox) e infine, in una sequenza di reazioni redox, all’ossigeno. L’energia fornita
da questa catena di reazioni ossidative è conservata sotto forma di un composto fosforilato ad alta
energia, l’adenosina trifosfato (ATP), che può essere in seguito utilizzato per compiere lavoro,
come nel trasporto di metaboliti e nelle biosintesi.
L’ACCOPPIAMENTO DELL’OSSIDAZIONE ALLA SINTESI DI ATP
Prerogativa dei sistemi viventi è il trasferimento d’energia da una molecola all’altra, assicurando
che l’energia stessa, invece che essere dissipata come calore, venga almeno in parte conservata in
una forma chimica ed utilizzata per alimentare le reazioni non spontanee delle biosintesi. La
maggior parte dell’energia ottenuta dall’ossidazione dei substrati metabolici è cosi incanalata nella
sintesi di ATP, la molecola universalmente utilizzata dai sistemi viventi per il trasferimento
d’energia, indicata anche come moneta di scambio dell’energia metabolica, dato che è utilizzata in
numerosissime reazioni che richiedono energia. L’ATP è composto da una base purinica, l’adenina,
uno zucchero a cinque atomi di carbonio, il ribosio, e dai gruppi fosforici , , e .
NAD+, FAD E FMN SONO I PRINCIPALI COENZIMI REDOX
I principali coenzimi redox coinvolti nel trasferimento dell’energia dai substrati sottoposti ad
ossidazione all’ATP sono: il nicotinamide-adenina-dinucleotide (NAD+), il flavina-adeninadinucleotide (FAD) e il flavina-mononucleotide (FMN). Il trasferimento di elettroni dai carboidrati
e dai grassi alla forma ossidata di questi coenzimi, li trasforma nelle corrispondenti forme ridotte:
NADH, FADH2 e FMNH2.
Il NADH ed il FADH2 formati nella glicolisi, nell’ossidazione degli acidi grassi e nel ciclo di Krebs
sono molecole ricche di energia, in quanto entrambe contengono una coppia di elettroni con un
elevato potenziale di trasferimento. Quando questi elettroni vengono donati all’ossigeno molecolare
viene liberata una grande quantità di energia libera. La fosforilazione ossidativa è il processo in cui
viene formato ATP, mentre gli elettroni sono trasferiti dal NADH oppure dal FADH2 all’ossigeno,
5
attraverso una serie di trasportatori di elettroni. Questa rappresenta la principale fonte di ATP negli
organismi aerobici. La fosforilazione ossidativa genera 32 delle 36 molecole di ATP che vengono
formate quando il glucosio è ossidato completamente a CO2 ed acqua.
La fosforilazione ossidativa ha luogo su strutture respiratorie localizzate nella membrana interna dei
mitocondri. Il ciclo dell’acido citrico e la via di ossidazione degli acidi grassi, che producono la
maggior parte del NADH e del FADH2, avviene nella matrice mitocondriale. L’ossidazione di una
molecola di NADH rende 3 molecole di ATP, mentre quella del FADH2 solo due molecole di ATP.
L’ossidazione e la fosforilazione sono processi accoppiati.
Le strutture respiratorie contengono numerosi trasportatori di elettroni, come i citocromi. Il
trasferimento, tappa dopo tappa, degli elettroni dal NADH e dal FADH2 all’O2 attraverso questi
trasportatori produce la contemporanea translocazione di protoni al di fuori della matrice
mitocondriale. In questo modo viene generata una forza motrice protonica, costituita da un
gradiente di pH e da un potenziale transmembrana. L’ATP viene sintetizzato quando i protoni
ritornano nella matrice mitocondriale attraverso un complesso enzimatico. Quindi, l’ossidazione e
la fosforilazione sono processi accoppiati da un gradiente protonico creato attraverso la membrana
interna dei mitocondri.
I mitocondri sono organelli di forma ovoidale costituiti da due sistemi di membrane: una membrana
esterna ed una membrana interna molto estesa e ripiegata. Quindi, nei mitocondri esistono due
compartimenti: lo spazio intermembrana, tra la membrana interna e quella esterna, e la matrice, lo
spazio delimitato dalla membrana interna. La fosforilazione ossidativa ha luogo nella membrana
mitocondriale interna, mentre la maggior parte delle reazioni del ciclo dell’acido citrico e della via
di ossidazione degli acidi grassi avvengono nella matrice. La membrana esterna è permeabile alla
maggior parte delle molecole piccole ed agli ioni, in quanto contiene molte copie della proteina
porina, una proteina transmembrana che forma un poro. Al contrario la membrana interna è
impermeabile a quasi tutti gli ioni ed alle molecole polari. La membrana interna è attraversata da
specifiche proteine trasportatrici, come quella per l’ADP e per gli acidi grassi a lunga catena.
IL SISTEMA MITOCONDRIALE DI TRASPORTO DEGLI ELETTRONI
L’intero sistema di trasporto degli elettroni, noto anche come catena di trasporto degli elettroni o
catena respiratoria, è localizzato nella membrana mitocondriale interna, e consiste di diversi grandi
complessi proteici e di due piccole componenti indipendenti: l’ubichinone e il citocromo c. Gli
elettroni attraversano questo sistema in una sequenza che li porta dai coenzimi ridotti all’ossigeno, e
6
lungo la quale i cambiamenti di energia libera alimentano il trasporto di protoni dalla matrice allo
spazio intermembrana.
Vi sono quattro siti d’ingresso per gli elettroni nella catena respiratoria: uno per il NADH
(Complesso I) e tre per il FADH2 (Complesso II). Queste vie confluiscono a livello della piccola
molecola lipofila dell’ubichinone (coenzima Q10), all’inizio della porzione comune della catena di
trasporto, che consiste nel Complesso III, del citocromo c e del Complesso IV. Solo tre complessi
(Complesso I, III, IV) sono in grado di pompare protoni nello spazio intermembrana. L’accettore
finale degli elettroni, al termine della catena, è l’ossigeno molecolare che viene ridotto ad acqua.
Per ogni due moli di elettroni trasportati attraverso i Complessi I, III, e IV, è pompato un numero di
protoni approssimativamente sufficiente per la sintesi di una mole di ATP. In particolare, se gli
elettroni (due moli) provengono dal NADH, vengono sintetizzate tre moli di ATP, mentre si
ottengono due sole moli di ATP se gli elettroni (due moli) provengono dal FADH2, in quanto questi
ultimi entrano nel Complesso II, a valle del Complesso I.
I COMPLESSI I E II CONTENGONO FLAVOPROTEINE
Il Complesso I, è una flavoproteina contenente FMN. Il Complesso I è responsabile
dell’ossidazione del NADH mitocondriale e del trasferimento degli elettroni, attraverso il FMN e i
complessi ferro-zolfo, all’ubichinone, fornendo cosi energia sufficiente, sottoforma di gradiente di
protoni, per la sintesi di una mole di ATP.
L’UBICHINONE (COENZIMAQ10) TRASFERISCE GLI ELETTRONI AL COMPLESSO III
L’ubichinone che si trova in tutti i sistemi viventi, è una piccola molecola liposolubile localizzata
nella membrana interna dei mitocondri di cellule vegetali ed animali, e nella membrana
citoplasmatica dei batteri. L’ubichinone delle cellule dei mammiferi contiene una coda
idrocarburica di 10 unità isopreniche, e viene perciò definito Coenzima Q10. E’ in grado di
diffondere lungo la membrana mitocondriale interna dove accetta elettroni dalle flavoproteine e li
trasferisce al Complesso III (QH2-citocromo c reduttasi).
Il COMPLESSO III (UBICHINONE QH2)-CITOCROMO C REDUTTASI
L’ubichinone incanala il flusso di elettroni provenienti dalle flavoproteine dei Complessi I e II verso
il Complesso III, un oligomero di circa otto peptidi contenente il citocromo b, un centro ferro-zolfo
e il citocromo c1. Questo complesso enzimatico (ubichinone-citocromo c reduttasi) ossida
l’ubichinone e riduce il citocromo c. Il trasporto di due elettroni al citocromo c fornisce energia
libera sufficiente per la sintesi di una mole di ATP.
7
IL CITOCROMO c
Il citocromo c è una piccola proteina contenente eme, che si trova associata debolmente alla
superficie esterna della membrana mitocondriale interna e trasporta elettroni dal complesso III al
complesso IV. La proteina si lega a questi complessi con legami elettrostatici che coinvolgono
diversi residui di lisina esposti sulla superficie della molecola. La riduzione, ad opera del citocromo
c1, del Ferricitocromo c (Fe3+) a Ferrrocitocromo c (Fe2+), induce un cambiamento nella
conformazione tridimensionale di quest’ultimo, che favorisce il trasferimento di elettroni al
citocromo a del Complesso IV.
IL COMPLESSSO IV
Il complesso IV, noto come citocromo c ossidasi, ossida il citocromo c e trasferisce gli elettroni,
attraverso i citocromi a e a3, all’ossigeno che viene ridotto. Il rame è un componente comune a
questo enzima ed ad altre ossidasi, alcuni veleni come l’azide ed il cianuro (discussi in seguito)
reagiscono con il rame ed inibiscono questo enzima. Come per i complessi I e III, il complesso
citocromo ossidasi pompa protoni all’esterno del mitocondrio fornendo energia libera per la sintesi
di un’ulteriore mole di ATP per ogni due moli di elettroni trasferiti.
Il GRADIENTE DI PROTONI CHE DIRIGE LA SINTESI DI ATP
Secondo l’ipotesi chemio-osmotica il trasporto mitocondriale di elettroni è accoppiato alla sintesi di
ATP attraverso un gradiente di protoni. Durante il trasporto di elettroni, i protoni sono pompati
dalla matrice nello spazio intermembrana, creando un potenziale elettrochimico attraverso la
membrana interna. L’esterno del mitocondrio diventa così più acido e più carico positivamente
rispetto alla matrice. La sintesi di ATP, una reazione non spontanea, è sostenuta dal ritorno dei
protoni, in favore del gradiente elettrochimico, nella matrice attraverso l’ATP-sintasi. Il
mitocondrio assomiglia quindi ad unna “batteria” ad ATP in cui l’energia per la sintesi di ATP è
immagazzinata sotto forma di gradiente di protoni. Per funzionare, richiede l’integrità del sistema di
membrane interne, che devono essere impermeabili ai protoni tranne che in corrispondenza del
complesso ATP- sintasi.
8
Il CONTROLLO RESPIRATORIO E’ IL MECCCANISMO PER IL QUALE L’OSSIDAZIONE E
LA
FOSFORILAZIONE
SONO
ACCOPPIATE
E
CONTROLLATE
DALLA
CONCENTRAZIONE MITOCONDRIALE DI ADP
In condizioni normali, i processi di ossidazione e fosforilazione sono strettamente accoppiati: i
substrati sono ossidati, gli elettroni trasportati, e l’ossigeno consumato solo quando c’è richiesta di
sintesi di ATP. Perciò nei mitocondri a riposo, la velocità di consumo di ossigeno è bassa. Questa
può essere fortemente aumentata dall’aggiunta di ADP. Il consumo di ossigeno diminuisce
nuovamente quando la concentrazione di ADP si abbassa a seguito della sua trasformazione in
ATP. I mitocondri sono definiti “disaccoppiati” quando la membrana mitocondriale interna perde la
sua integrità strutturale: in questa circostanza i protoni possono diffondere attraverso la membrana
lungo vie improduttive, che non interessano il sistema dell’ATP-sintasi. Questo evento si verifica
quando i mitocondri isolati sono trattati con detergenti blandi, che introducono dei pori nella
membrana interna. Il meccanismo del controllo respiratorio dipende probabilmente dal legame di
ADP e Pi al complesso dell’ATP-sintasi: in assenza di ADP e Pi, i protoni non possono entrare nel
mitocondrio attraverso questo complesso. Il gradiente elettrochimico raggiunge allora un valore
elevato e per un meccanismo di retroazione la catena di trasporto di elettroni ed il consumo di
ossigeno sono inibiti.
LE SOSTANZE DISACCOPPIANTI
I disaccoppianti della fosforilazione ossidativa dissipano il gradiente di protoni favorendone il
reflusso nella matrice mitocondriale scavalcando il sistema dell’ATP-sintasi. Questo evento agisce
da stimolo della respirazione mitocondriale poiché il sistema effettua un tentativo, per quanto futile
di ripristinare il gradiente di protoni, ossidando sempre più substrati e pompando sempre più protoni
fuori dalla matrice. Un disaccoppiante tipico è il 2,4-dinitrofenolo (DNP), altri comuni
disaccopianti sono sostanze conservanti ed antimicrobiche come il pentaclorofenolo e il p-cresolo
GLI INIBITORI DEL METABOLISMO OSSIDATIVO
Gli inibitori del trasporto degli elettroni interrompono il flusso di elettroni lungo la catena
respiratoria agendo selettivamente sui complessi I, III o IV. Il risultato è sempre il blocco delle
pompe protoniche, della sintesi di ATP e del consumo di ossigeno. Molti di questi inibitori sono
veleni, quali ad esempio il rotenone che è un insetticida che inibisce il complesso I del sistema di
trasporto degli elettroni. Poiché il malato e il lattato sono ossidati dal NAD, il rotenone inibirà la
loro ossidazione. Al contrario, l’ossidazione di substrati che determinano la formazione di FADH2
9
sarà possibile anche in presenza di rotenone, in quanto il complesso I non viene interessato e gli
elettroni possono essere donati all’ubichinone attraverso il complesso II.
L’antimicina A, inibendo il complesso III, impedisce il trasferimento di elettroni dai complessi I o
II al citocromo c.
Sia il cianuro che il monossido di carbonio si legano, inibendolo, al complesso IV. Le cellule
rispondono all’avvelenamento da cianuro o da monossido di carbonio passando ad un metabolismo
anaerobico, che causa acidosi e da ultimo morte, se non si prendono immediate contromisure,
l’avvelenamento da monossido di carbonio può essere trattato con la somministrazione di ossigeno.
10
Processi di biotrasformazione (detossificazione)
Tutti gli organismi viventi sono continuamente esposti a composti xenobiotici di origine naturale o
sintetica. Questi possono essere escreti come tali attraverso la bile, le urine, le feci o il sudore
oppure possono essere assorbiti e subire delle modificazioni nell’organismo. La velocità con cui i
composti esogeni sono eliminati dall’organismo dipende in primo luogo dalla loro idrosolubilità. I
composti lipofili, infatti, tendono ad essere riassorbiti nell’intestino e nei tubuli renali data la loro
capacità di diffusione attraverso le membrane cellulari. Gli organismi superiori, perciò, hanno
sviluppato dei meccanismi di difesa per convertire i composti lipofili in metaboliti più idrosolubili.
Tali processi biochimici vengono detti processi di biotrasformazione.
Le reazioni enzimatiche deputate all’eliminazione di xenobiotici vengono distinte in reazioni di fase
I e reazioni di fase II. Le reazioni di fase I consistono in ossidazioni, riduzioni ed idrolisi: aggiunta
di gruppi funzionali, -SH, -OH, -NH2, -COOH, nella molecola del composto che viene
metabolizzato. Le reazioni di fase II consistono in processi di coniugazione e sintesi: il composto
esogeno, o un suo metabolita derivante dalle reazioni di fase I, si combina con una molecola
endogena formando un coniugato maggiormente solubile in acqua. Gli enzimi ed i sistemi
enzimatici che catalizzano le reazioni di biotrasformazione sono localizzati soprattutto a livello
epatico: è il fegato, infatti, che riceve e processa le sostanze estranee assorbite per via
gastrointestinale prima che queste possano raggiungere gli altri organi e tessuti.
Localizzzazione subcellulare degli enzimi.
Il metabolismo epatico dei composti estranei all’organismo avviene ad opera di svariati sistemi
enzimatici. Questi possono agire su una estrema varietà di farmaci e di tossici con struttura chimica
diversa che entrano nell’organismo per ingestione, inalazione, iniezione o attraverso la cute. Gli
enzimi di fase I (quelli che aggiungono o espongono gruppi funzionali) hanno principalmente sede a
livello del reticolo endoplasmatico, una rete di canali interconnessi presenti nel citoplasma della
maggior parte delle cellule. Si tratta di enzimi di membrana, dal momento che il reticolo
endoplasmatico è fondamentalmente una membrana composta di lipidi e proteine. La localizzazione
in una matrice lipoproteica costituisce un elemento di grande importanza in quanto i substrati
lipofili degli enzimi metabolizzanti si distribuiscono preferenzialmente nelle membrane lipidiche. In
seguito a omogenizzazione di un campione di fegato si ha la rottura del reticolo endoplasmatico e i
frammenti delle membrane si aggregano a formare microvescicole. A queste si dà comunemente il
nome di microsomi. In laboratorio, il loro isolamento avviene mediante centrifugazione
11
differenziale dell’omogenato di fegato. Il sopranatante che si ottiene dalla centrifugazione
dell’omogenato a 9000 X g (per eliminare nuclei, mitocondri e lisosomi) viene a sua volta
centrifugato a 105.000 g. Il materiale che precipita è fortemente arricchito in microsomi. La
rimanente frazione (sopranatante) contiene numerosi enzimi solubili e prende il nome di citosol. Gli
enzimi di biotrasformazione vengono definiti citoplasmatici o microsomiali per indicare la loro
specifica localizzazione subcellulare. Nel citosol, ad esempio, sono presenti molti degli enzimi che
intervengono nella fase II di biotrasformazione. In genere l’azione degli enzimi di fase I e II risulta
“positiva”: rendono più solubili gli xenobiotici favorendo il loro allontanamento dall’organismo e
limitandone l’eventuale azione tossica. In altri casi, però, i metaboliti che si formano in seguito a
biotrasformazione risultano più tossici del composto di partenza (es. pesticidi,…) oppure si formano
intermedi altamente instabili e reattivi. In questo caso i processi di biotrasformazione vengono
indicati come bioattivazioni.
Reazioni di fase I:
Le biotrasformazioni di fase I avvengono attraverso due sistemi enzimatici ossidativi: il sistema del
citocromo P-450 e il sistema delle monoossigenasi microsomiali (aminoossidasi a funzione mista).
In ogni caso si ha l’addizione di gruppi idrossilici nella molecola del substrato.
Citocromo P-450
I più importanti sistemi enzimatici implicati nelle reazioni di fase I sono le monoossigenasi
contenenti il citocromo P-450. Questo è in realtà un sistema comprendente due enzimi, la NADPHcitocromo P-450 reduttasi più lo stesso citocromo P-450, enzima contenente eme. Questi fattori
sono incorporati nella matrice fosfolipidica del reticolo endoplasmatico. I fosfolipidi facilitano
l’interazione tra i due enzimi. La NADPH-citocromo P-450 reduttasi, che utilizza il NADPH come
cofattore, è una flavoproteina capace di trasferire uno o due elettroni al citocromo P-450.
Ciclo catalitico del citocromo P-450: nelle reazioni catalizzate dal citocromo P-450 il substrato
(RH) si combina con la forma ossidata del citocromo (Fe3+) a formare il complesso substratocitocromo. Questo riceve in seguito un elettrone dal NADPH (attraverso la NADPH-citocromo P450 reduttasi): il ferro del gruppo eme viene ridotto a Fe2+. Il complesso substrato-citocromo ridotto
reagisce con l’ossigeno molecolare e riceve un altro elettrone dal NADPH. I due elettroni vengono
trasferiti all’ossigeno: ne derivano forme molto reattive ed instabili. Il substrato, intanto, si dissocia
dal citocromo P-450, rigenerando la forma ossidata dello stesso. Il monossido di carbonio è un
12
potente inibitore delle reazioni catalizzate dal citocromo P-450 in quanto compete con l’ossigeno
per il legame con la forma ridotta dell’enzima.
Esempi di reazioni catalizzate dal citocromo P-450 sono:
l’idrossilazione alifatica del carbonio o -1
RCH2CH2CH3RCH2CHOHCH3
l’idrossilazione aromatica
R-Ar R-Ar-OH
L’ossidazione dello zolfo e dell’azoto
R-S-R R-SO-R
Sebbene sia classificato come ossigenasi, il citocromo P-450, in alcuni casi, catalizza anche la
riduzione di nitro e azo gruppi. Ciò avviene solo a basse tensioni di ossigeno: i substrati xenobiotici
accettano uno o due elettroni e si riducono; l’ossigeno agisce come inibitore di queste reazioni in
quanto compete per gli equivalenti di riduzione.
Monoossigenasi microsomiali FAD-dipendenti: sono enzimi ossidativi che intervengono nelle
biotrasformazioni di fase I. In origine indicati come “aminoossidasi a funzione mista”, si tratta di
flavoproteine del reticolo endoplasmatico capaci di ossidare funzioni azotate nucleofile utilizzando
NADPH e O2. Susbrati della monoossigenasi flavinica sono le amine, composti solforati (solfuri,
tioeteri, tioli,…) ed organofosforici. Il substrato fisiologico è la cisteamina che viene ossidata a
cistamina. La regolazione di tale enzima avviene con modalità differenti rispetto al citocromo P450. Non è chiaro il suo ruolo in altri processi di biotrasformazione e nella tossicità degli
xenobiotici.
Reazioni di fase II:
Le biotrasformazioni di fase II hanno carattere biosintetico, avvengono mediante attivazione di
cofattori o del substrato. I cofattori, in genere, sono attivati direttamente o indirettamente dall’ATP,
la possibilità che tali reazioni avvengano è legata, quindi, allo stato energetico dell’organo o del
tessuto.
Glucuroniltransferasi: la coniugazione con acido glucuronico è una delle principali reazioni di fase
II: consente la conversione di sostanze esogene o endogene in metaboliti (glucuronidi) polari e
idrosolubili, che vengono eliminati con la bile o con le urine. L’enzima che catalizza la reazione è la
uridin difosfato glucuroniltrasferasi (UDP glucuronil trasferasi): consente l’interazione tra un
13
nucleotide ad alta energia (l’acido-UDP glucuronico) ed il gruppo funzionale della molecola
accettrice (substrato). L’attività risiede nel reticolo endoplasmatico di numerosi tessuti, a differenza
di quanto avviene per la maggior parte degli enzimi di fase II citosolici. La localizzazione a livello
della membrana microsomiale è importante in quanto consente all’enzima di avere accesso diretto
ai prodotti che si formano nella fase I per azione della citocromo P-450 microsomiale. Il fegato è il
principale organo sede di coniugazione con acido glucuronico, ma una discreta attività si riscontra
anche nel rene, nell’intestino, nella cute, nel cervello e nella milza. Si configura dunque l’esistenza
di un sistema microsomiale altamente integrato che ha la capacità di sequestrare i composti più
lipofili, aggiungere determinati gruppi funzionali al fine di coniugarli con un accettare fortemente
polare come l’acido glucuronico. Esistono diverse forme di UDP-glucuroniltransferasi. Questa
eterogeneità spiega in parte le differenze dell’attività enzimatica nei confronti dei diversi substrati.
Tra i numerosi gruppi funzionali presenti in sostanze esogene o endogene che vengono coniugate
con l’acido glucuronico troviamo: alcoli alifatici e aromatici, acidi carbossilici, amine aromatiche e
alifatiche ed inoltre gruppi sulfidrici liberi. Da queste sostanze si formano rispettivamente O-, N- e
S- glucuronidi. Durante il processo di coniugazione si forma un gruppo carbossilico che si trova in
gran parte in forma ionizzato a pH fisiologico. Questo gruppo è in grado di promuovere
l’eliminazione della molecola coniugata non solo per la idrosolubilità che conferisce ad essa ma
anche perché viene “riconosciuto” dal sistema di trasporto degli anioni organici presente a livello
renale e biliare. I glucuronidi sono dunque escreti con la bile o con le urine. Alcuni glucuronidi
possono rappresentare un veicolo per il trasporto di composti reattivi dal fegato a tessuti bersaglio.
L’esempio più noto è quello dei glucuronidi delle N-idrossiarilamine che sono stati chiamati in
causa per spiegare l’induzione di tumori della vescica a seguito dell’esposizione a 2-naftilamina, 4aminobifenile e composti analoghi. Nel fegato queste arilamine vanno incontro a N-idrossilazione
con conseguente formazione di glucuronidi N-idrossi-arilaminici che passano nel rene e si
concentrano nell’urina presente in vescica. Tali glucuronidi sono instabili a pH acido e vengono
quindi idrolizzati con formazione dell’agente cancerogeno N-idrossilamina.
Solfotransferasi:
Nei mammiferi, un altro processo importante di coniugazione dei gruppi idrossilici è la
solfoconiugazione. Tale reazione è catalizzata dalle solfotransferasi, enzimi solubili contenuti
principalmente nel fegato, nel rene, nel tratto intestinale e nei polmoni. La loro principale funzione
è quella di trasferire un solfato inorganico al gruppo idrossilico di fenoli ed alcoli alifatici.
14
N-Acetil transferasi
Nella maggior parte delle specie animali, la principale via di biotrasformazione delle arilamine è
l’acetilazione della funzione aminica. Substrati per le N-acetil transferasi possono essere, a titolo di
esempio, amine aromatiche primarie, idrazine, idrazidi, sulfonamidi e alcune amine alifatiche
primarie.
Coniugazione con aminoacidi
Gli xenobiotici contenenti un gruppo carbossilico possono essere metabolizzati mediante
coniugazione con un aminoacido. Ciò porta alla formazione di un legame peptidico tra il carbossile
dello xenobiotico ed il gruppo amminico dell’aminoacido. Comuni substrati per questo tipo di
coniugazione sono gli acidi carbossilici aromatici, gli acidi arilacetici e gli acidi acrilici
arilsostituiti. Una reazione molto frequente è quella con la glicina.
Glutatione S-transferasi
Le glutatione S-transferasi (GSTs) intervengono nella tappa iniziale del processo di coniugazione
che porta alla sintesi degli acidi mercapturici. Sono presenti nel citoplasma e in minor misura nella
frazione microsomiale. Si tratta di enzimi ubiquitari con prevalente attività nel fegato.
Il cofattore di questi enzimi è il glutatione (GSH), costituito da glicina, acido glutammico e cisteina.
Le GSTs catalizzano la reazione tra il gruppo sulfidrico nucleofilo del glutatione e composti
contenenti atomi di carbonio elettrofili. La reazione dell’anione tiolato del glutatione (GS -) porta
alla formazione di un legame tioetere tra il carbonio ed il gruppo sulfidrilico.
I substrati delle GSTs hanno in comune tre caratteristiche: un certo grado di idrofobicità, la
presenza di un atomo di carbonio elettrofilo e la capacità di reagire in qualche misura con il
glutatione.
I prodotti glutatione-coniugati possono essere scissi a derivati cisteinici ad opera di enzimi presenti
soprattutto a livello renale. Questi metaboliti vengono in seguito acetilati formando acidi
mercapturici, cioè coniugati con N-acetil-cisteina. Gli acidi mercapturici sono facilmente escreti
nelle urine. Il distacco dell’acido glutammico dal glutatione-coniugato avviene per mezzo della glutamil transpeptidasi, enzima di membrana presente in quantità elevate nelle cellule deputate a
processi di assorbimento o escrezione. Il distacco della glicina è invece catalizzato dalla cisteinil
glicinasi nella tappa che porta alla formazione del coniugato cisteinico. Nella fase finale
intervengono N-acetil transferasi che, in presenza di acetil-CoA, trasferiscono un acetile sul gruppo
amminico della cisteina per formare il derivato dell’acido mercapturico.
15
Le glutatione S-transferasi favoriscono la coniugazione dei diversi xenobiotici elettrofili con il
glutatione, attenuando l’interazione di questi composti con costituenti essenziali della cellula. In
particolare, le GSTs intervengono nella detossificazione degli intermedi reattivi prodotti dal sistema
del citocromo P450 durante il metabolismo di composti quali bromobenzene, cloroformio e
paracetamolo. Tali intermedi possono stabilire legami covalenti con diverse strutture
macromolecolari dell’epatocita o, in alternativa, possono reagire con il glutatione. Quest’ultima
reazione previene dunque l’attacco chimico a costituenti cellulari vitali.
All’interno delle cellule esiste dunque un delicato equilibrio tra la formazione di metaboliti reattivi
e la loro inattivazione da parte del glutatione. Fattori che alterano questo equilibrio possono
modificare drasticamente il potenziale tossico di quelle sostanze che agiscono attraverso intermedi
reattivi. Gli intermedi reattivi possono al tempo stesso depletare le riserve cellulari di GSH. Dato
che il GSH è il cofattore della glutatione perossidasi, la sua deplezione può promuovere la
perossidazione lipidica, fenomeno che ha deleterie conseguenze per l’integrità delle cellule.
16
METALLI PESANTI
Ci Cianuri
Gli avvelenamenti da acido cianidrico e cianuri non sono più molto frequenti attualmente. L’acido
cianidrico è un liquido altamente volatile (p.e. 26°C) dal caratteristico odore di mandorle amare. I
sali di sodio e di potassio si decompongono all’aria dando acido cianidrico e carbonato di sodio e
potassio. I cianuri sono usati in metallurgia e l’esposizione ad acido cianidrico può avvenire nei
laboratori chimici o nella combustione di materie plastiche (nitrocellulosa e poliuretani)
Effetto: l’azione tossica si esplica attraverso l’inibizione dello stato terminale nel sistema di
ossidazione: il piruvato non si ossida ulteriormente nel ciclo dell’acido citrico, ma piuttosto si
riduce a lattato. Il paziente soffoca perché l’ossigeno non può essere utilizzato. Il colore rosso
brillante del sangue e la colorazione rossa degli organi sono le conseguenze della saturazione del
sangue da parte dell’ossigeno e la sua significativa ridotta utilizzazione da parte dei tessuti. La
saturazione in ossigeno del sangue venoso raggiunge quasi lo stesso livello di quello arterioso.
Sintomatologia: il primo segno è l’acidosi da acido lattico. Si manifesta con emicrania, tachipnea e
sensazione di stanchezza.
Categorie lavoratori a rischio Acido cianidrico e composti. Lavoratori addetti:
a) alla produzione di acido cianidrico, di cianuri e di altri composti del cianogeno;
b) alla derattizzazione e disinfestazione;
c) alla distruzione di parassiti in agricoltura
d) alla depurazione chimica del gas illuminante;
e) alle operazioni di galvanoplastica;
f) alle operazioni di tempera e di cementazione;
g) alla fabbricazione di gomme e resine sintetiche (limitatamente alle operazioni che
espongono all'azione dell'acrilnitrile e dei dissocianati organici).
17
Arsenico
L’arsenico si trova nel suolo, nell’acqua e nell’aria come un comune tossico ambientale. L’impiego
di pesticidi ed erbicidi contenenti arsenico, inoltre, ha fatto aumentare la sua immissione
nell’ambiente; alimenti quali la frutta e gli ortaggi trattati con arseniati, infatti, possono costituire
una fonte di esposizione a questo elemento. Nell’uomo, l’assunzione media giornaliera di arsenico è
di circa 300g e, per lo più, avviene tramite cibi e bevande. La fonte principale di esposizione
professionale a composti contenenti arsenico è rappresentata dalla produzione di erbicidi e pesticidi.
Meccanismo d’azione: La tossicità dei composti arsenicati è in rapporto alla loro velocità di
clearance dall’organismo e, quindi, al grado di accumulo nei tessuti.
Gli arseniati (pentavalenti) sono notoriamente disaccoppianti della fosforilazione ossidativa
mitocondriale. Si ritiene che il meccanismo sia basato sulla sostituzione competitiva dell’arseniato
al posto del fosfato inorganico. Si forma, di conseguenza, un estere arseniato instabile che,
rapidamente, va incontro ad idrolisi. Tale processo viene detto arsenolisi.
Gli arsenicati trivalenti (es arsenito inorganico) sono considerati composti in grado di reagire con i
gruppi sulfidrici. In tal modo inibiscono numerosi enzimi in quanto reagiscono con ligandi biologici
contenenti gruppi –SH disponibili.
Assorbimento, distribuzione ed escrezione: sia le forme trivalenti che quelle pentavalenti vengono
assorbite a livello gastrointestinale. La distribuzione dipende dalla durata della somministrazione e
dal particolare composto arsenicato interessato. Viene immagazzinato soprattutto nel fegato, nel
rene, nel cuore e nel polmone. Minori quantitativi, invece, si ritrovano nel muscolo e nel tessuto
nervoso. La presenza di cheratina (contenente un notevole numero di gruppi –SH) rende ragione
dell’elevata concentrazione di arsenico che si ritrova nelle unghie e nei capelli. L’arsenico
attraversa la barriera placentare provocando danni al feto. Nell’uomo la concentrazione di arsenico
nel sangue del cordone ombelicale sono equivalenti a quelle presenti nella circolazione materna.
Poco si conosce in merito alle biotrasformazioni dei composti arsenicali nell’organismo umano. Le
forme trivalenti, per lo più, vengono ossidate allo stato pentavalente. Sia le forme trivalenti che
quelle pentavalenti, inoltre, vengono metilate: una notevole quota di arsenico viene escreta nelle
urine come metilarsenico.
Effetti farmacologici e tossicologici: Gli effetti sono molteplici a livello di vari sistemi
Sistema cardiovascolare:
piccole dosi di arsenico inorganico inducono una blanda vasodilatazione. Tale effetto può portare ad
edema occulto, particolarmente del viso.
18
Dosi maggiori inducono dilatazione capillare e aumento della permeabilità capillare. Può anche
aversi trasudazione di plasma e la diminuzione del volume vascolare può risultare consistente.
L’esposizione prolungata porta a cancrena delle estremità, soprattutto dei piedi.
Esposizioni ancora più lunghe provocano danno miocardico e ipotensione.
Tratto gastro-intestinale:
Elevate dosi provocano trasudazione di plasma dai capillari e formazione di vescicole al di sotto
della mucosa gastrointestinale. Queste poi si rompono, si distaccano lembi di epitelio e il plasma si
riversa nel lume intestinale dove coagula. Si ha quindi aumento della peristalsi e diarrea.
Reni:
l’azione dell’arsenico sui capillari, sui tubuli e sui glomeruli renali può provocare un grave danno
renale. All’inizio sono colpiti i glomeruli e ne consegue proteinuria. Successivamente si presenta
necrosi e degenerazione tubulare di vario grado.
Cute:
Nell’intossicazione acuta molti composti dell’arsenico hanno sulla cute un effetto vescicante.
L’assunzione cronica di basse dosi, invece, provoca vasodilatazione cutanea, ipercheratosi a livello
delle palme delle mani e dei piedi e iperpigmentazione del tronco e delle estremità.
Sistema nervoso:
L’esposizione sia acuta che cronica può portare ad encefalopatia. Il danno più comune, tuttavia,
consiste in una neuropatia periferica. Questa è seguita da debolezza muscolare e diminuzione dei
riflessi tendinei e atrofia muscolare.
Sangue:
i composti contenenti arsenico colpiscono il midollo osseo e alterano la composizione del sangue.
L’analisi del sangue di solito rileva un’anemia accompagnata da una lieve o modica leucopenia, può
essere anche presente eosinofilia.
Fegato:
nel fegato si producono infiltrazione grassa, necrosi centrale e cirrosi. Il danno può essere lieve o
così grave da portare a morte.
Cancerogenesi e teratogenesi:
l’ingestione cronica di arsenico tramite acqua potabile o l’esposizione a disinfettanti nell’agricoltura
predispongono a carcinomi cutanei, neoplasie polmonari e tumori epatici.
19
Avvelenamento acuto da arsenico:
Entro un’ora dall’ingestione dell’arsenicale di solito si manifesta malessere gastrointestinale,
bruciore alle labbra, senso di costrizione alla gola, difficoltà a deglutire, dolore gastrico atroce,
vomito e diarrea. Possono presentarsi, inoltre, oliguria e ematuria, crampi alla muscolatura
scheletrica e sete molto intensa, convulsioni e, in fine, coma e morte. Se la terapia correttiva è
pronta il paziente può sopravvivere alla fase acuta ma va comunque incontro a neuropatie e altri
danni.
Avvelenamento cronico da arsenico:
I sintomi precoci più usuali sono debolezza e dolenzie muscolari, pigmentazione cutanea,
ipercheratosi e edema. E’ caratteristico il riscontro nelle unghie delle Strie di Mess (righe trasversali
bianche di arsenico depositato, che di solito compaiono 6 settimane dopo l’esposizione). Si può
avere aumento del volume del fegato e ostruzione delle vie biliari con conseguente ittero; cirrosi e
disfunzione renale. Con il progredire dell’intossicazione può insorgere encefalopatia, paralisi
motoria e sensitiva delle estremità, danno a livello midollare.
Categorie lavoratori a rischio:Arsenico, leghe e composti.
Lavoratori addetti:
a) alla produzione dell'arsenico;
b) alla preparazione delle leghe e dei composti;
c) ai lavori di pulitura, verniciatura e smaltatura;
d) alla preparazione delle miscele per la produzione del vetro;
e) alla tintura dei filati e dei tessuti;
f) alla concia delle pelli.
20
Piombo
Il piombo è praticamente ubiquitario nell’ambiente in ragione sia della sua presenza naturale sia
della sua utilizzazione industriale. La fonte più importante di esposizione dell’uomo al piombo è
l’alimentazione. L’assunzione media giornaliera di una persona adulta va da 0.1 a 0.2mg di piombo.
Il piombo assunto si accumula nel sangue e nei tessuti molli ed infine nelle ossa nella forma di
fosfato di piombo. Cibi e bevande a carattere acido (es. succhi di pomodoro, succhi di frutta)
possono solubilizzare il piombo di contenitori inadeguatamente smaltati. Massima parte della
tossicità da piombo, però, deriva dall’esposizione ambientale ed industriale.
Assorbimento, distribuzione ed escrezione: Le vie principali di assorbimento del piombo sono il
tratto gastrointestinale e il sistema respiratorio. Poco è noto circa il trasporto del piombo attraverso
la mucosa gastrointestinale. Si è ipotizzato che vi sia una competizione tra Pb2+ e Ca2+ per un
meccanismo di trasporto comune, in quanto vi è una relazione inversa tra il contenuto del calcio
nella dieta e l’assorbimento del piombo. L’assorbimento attraverso le vie respiratorie varia a
seconda della forma inalata (circa il 90% delle particelle di piombo viene assorbito con l’aria).
Appena assorbito il piombo inorganico si distribuisce nei tessuti molli (soprattutto rene e fegato) e
poi si ridistribuisce nelle ossa nei denti e nei capelli. Il 95% alla fine si trova depositato nel tessuto
osseo. Una piccola percentuale si accumula nel cervello (sostanza grigia e gangli della base)
Avvelenamento acuto da piombo:
è poco frequente, avviene per ingestione di composti acidi solubili del piombo o per inalazione di
vapori di piombo. A livello del cavo orale si riscontrano effetto astringente, sete e sapore metallico,
seguono nausea, vomito e dolore addominale. A carico del SNC si registrano parestesie, dolori e
debolezza muscolare. Può presentarsi anche una crisi emolitica acuta che provoca una grave anemia
ed emoglobinuria. La morte può sopravvenire in 2-3 giorni.
Avvelenamento cronico da piombo:
I sintomi possono essere suddivisi in 6 categorie: gastrointestinali, neuromuscolari, carico del SNC,
ematologici, renali ed altri.
Effetti gastrointestinali: agisce sulla muscolatura liscia dell’intestino. Spesso insorge con sintomi
vaghi quali anoressia, malessere e cefalea. Lo spasmo intestinale, responsabile di forti dolori
addominali, la “colica saturnina” è caratteristica della fase avanzata.
Effetti neuromuscolari: inizialmente si presentano debolezza e affaticabilità; dopo un’intensa
attività muscolare, può presentarsi anche la “paralisi saturnina” che colpisce i gruppi muscolari più
attivi (estensori dell’avambraccio, del polso e delle dita,…)
21
Effetti ematologici: un riscontro comune è l’anemia ipocromica microcitica. Tale anemia si ritiene
causata da riduzione della vita media degli eritrociti e da inibizione della sintesi dell’eme. Il
piombo, infatti, inibisce l’attività di molteplici enzimi deputati alla sintesi dell’eme.
Effetti renali: la tossicità renale si manifesta in due forme: alterazione reversibile dei tubuli renali e
nefropatia interstiziale irreversibile. Clinicamente si rileva proteinuria e ematuria.
Altri effetti: altri segni del saturnismo sono un colorito cinereo del volto e delle labbra, la comparsa
di un “invecchiamento prematuro” con andatura incurvata e scarso tono muscolare.
Meccanismo d’azione: l’effetto del piombo sull’organismo non è facile da spiegare.
Molecolarmente l’effetto del legame ai gruppi –SH diventa manifesto con il danno a quegli enzimi
che sono necessari alla sintesi dell’eme e la produzione di emoglobina e citocromo. Inoltre
danneggia le membrane del globulo rosso e causa un’aumentata fragilità; si sviluppa pertanto
anemia.
Categorie lavoratori a rischio: Piombo,leghe e composti
Lavoratori addetti:
a) alla produzione del piombo;
b) alla preparazione delle leghe e dei composti;
c) alla fabbricazione e preparazione di colori, di vernici e di mastici;
d) alla fabbricazione di lamine, tubi, proiettili ed altri oggetti di piombo o contenenti
piombo; alla cernita e al recupero dei materiali piombiferi;
e) alle operazioni di pittura e di intonaco con mastici o colori di piombo; alla asportazione
di verniciature piombifere;
f) alla saldatura con leghe piombifere e dissaldatura;
g) alla piombatura o smaltatura su superfici metalliche;
p)alla zincatura delle lamiere o alla stagnatura o alla verniciatura dei recipienti con uso di
materiali contenenti piombo;
s) alla preparazione delle miscele per la fabbricazione del vetro piombifero;
22
Mercurio
Per secoli il mercurio è stato un importante costituente di farmaci (diuretici, antibatterici, antisettici,
pomate dermatologiche…), negli ultimi anni, invece, i composti mercuriali sono stati sostituiti da
agenti dotati di maggiore specificità ed efficacia e le manifestazioni di intossicazione da farmaci
mercuriali sono diventate rare. L’avvelenamento da mercurio causato da inquinamento ambientale,
però, è diventato motivo di preoccupazione.
L’uso di combustibili fossili, contenenti mercurio, e l’impiego di mercurio in agricoltura e
nell’industria, hanno provocato un aumento della concentrazione di mercurio nell’aria e nell’acqua.
Fonti di esposizione: Il mercurio elementare è la forma più volatile, l’esposizione ai vapori di
mercurio è di natura professionale (laboratori scientifici). I Sali mercurici, largamente impiegati
nell’industria (es. elettronica), rappresentano la forma più irritante e acutamente tossica del metallo.
Il cloruro mercuroso o calomelano è usato in creme dermatologiche e come antisettico; il nitrato
mercurico era largamente impiegato, in passato, nelle fabbriche di cappelli di feltro e produceva
alterazioni neurologiche e comportamentali. Altri usi del metallo comprendono la produzione di
plastiche, fungicidi e germicidi.
I composti organici del mercurio (come i Sali di alchilmercurio) comprendono un gruppo
eterogeneo di sostanze in grado di produrre differenti effetti tossici. I Sali di alchilmercurio sono
stati largamente impiegati come fungicidi: nel 1971 in Iraq si ebbe la più grande epidemia (6530
persone colpite e 500 morti) dovuta alla messa in commercio di pane contenente cereali trattati con
metilmercurio. La malattia di Minamata fu anche essa causata da metilmercurio: in una piccola
cittadina giapponese, Minamata, un’industria chimica che usava il mercurio lo scaricava nella baia,
ne risultò l’intossicazione di 121 persone e la morte di 46.
Meccanismo d’azione: il mercurio forma legami covalenti con lo zolfo. Nei gruppi sulfidrilici il
mercurio sostituisce l’idrogeno formando dei mercaptidi, X-Hg-SR e Hg(SR)2, dove R è una
proteina e X è un radicale elettronegativo. I mercuriali sono in grado di inattivare gli enzimi
contenenti gruppi –SH e quindi interferire con il metabolismo e le funzioni cellulari. Le varie azioni
terapeutiche e tossiche dei composti mercuriali sono dovute ai sostituenti chimici che influenzano la
solubilità, la dissociazione, la distribuzione, l’affinità per i recettori e l’escrezione dei composti.
Assorbimento e distribuzione: Il mercurio elementare è poco tossico se ingerito perché viene
scarsamente assorbito. L’inalazione di vapori comporta un assorbimento completo attraverso i
polmoni, un’ossidazione a catione bivalente e il raggiungimento rapido dell’encefalo. La tossicità a
carico del SNC è marcata. I Sali organici vengono assorbiti in misura maggiore (90%) rispetto a
quelli inorganici (10%), attraversano la barriera ematoencefalica e la placenta rendendosi perciò
responsabili di effetti neurologici e teratogeni superiori a quelli dei Sali inorganici.
23
Tossicità: i sintomi si manifestano dopo alcune ore, nell’esposizione a vapori di mercurio.
Consistono in debolezza, brividi, sapore metallico, nausea, vomito, diarrea, dispnea, tosse e
sensazione di oppressione toracica. La tossicità polmonare può progredire fino a compromettere la
funzione respiratoria. L’esposizione cronica a vapori di mercurio porta a neurotossicità: tremore,
depressione, irritabilità, timidezza, insonnia, instabilità emotiva, deficit di memoria, disturbi
vasomotori. I Sali di mercurio inorganico, invece, provocano precipitazione delle proteine della
bocca e delle vie gastrointestinali (la mucosa si presenta grigia, dolente), le mucose sono fortemente
danneggiate in assenza di una terapia correttiva. L’effetto sistemico più grave è a carico dei reni:
predomina il danno glomerulare. La sintomatologia associata ai composti organici, invece, è a
carico del sistema nervoso: disturbi visivi, atassia, perdita dell’udito, neurastenia, deterioramento
mentale, tremori muscolari, disturbi del movimento, paralisi e morte.
Diagnosi e trattamento: di fondamentale importanza nella diagnosi da avvelenamento da mercurio è
l’anamnesi di esposizione ambientale o professionale. In assenza di questa si deve cercare conferma
negli esami di laboratorio: una concentrazione di mercurio nel sangue che superi 4g/dl deve essere
considerata anormale. La concentrazione del mercurio nelle urine è stata anche usata per
determinare il carico del metallo nell’organismo (nella popolazione normale il limite massimo è di
25g/litro). Il trattamento prevede: allontanamento dalla fonte di esposizione e terapia chelante
(dimercaprolo o penicilamina). Se assunto per os, si deve cercare di favorire il vomito e limitare
l’assorbimento con carbone vegetale o lavanda gastrica.
24